

Abstract
Cyclopentadiene is one of the most reactive dienes in normal electron-demand Diels–Alder reactions. The high reactivities and yields of cyclopentadiene cycloadditions make them ideal as click reactions. In this review, we discuss the history of the cyclopentadiene cycloaddition as well as applications of cyclopentadiene click reactions. Our emphasis is on experimental and theoretical studies on the reactivity and stability of cyclopentadiene and cyclopentadiene derivatives.
Graphical Abstract
1. Introduction
In 2001, Sharpless and co-workers introduced the term “click reaction” to describe chemical transformations that are high-yielding under mild conditions.1 The Diels–Alder reaction is a chemical transformation between a conjugated diene and dienophile that results in the formation of a 6-membered cycloadduct. The most reactive Diels–Alder dienes are 5- or 6-membered cyclic dienes such as 1,2,4,5-tetrazine, ortho-quinone, thiophene dioxide, cyclopentadienone, and cyclopentadiene. Without electron-withdrawing heteroatoms, cyclopentadiene differs from the other Diels–Alder dienes used as click reagents in being able to react rapidly via normal electron-demand Diels–Alder reaction mechanism with electron-deficient dienophiles.
Despite cyclopentadiene being among the first and most widely used Diels–Alder dienes, the cyclopentadiene cycloaddition has yet to be the subject of a review. Here, we cover the history as well as modern applications of cyclopentadiene click reactions with emphasis on experimental and theoretical studies on the reactivities and stabilities of cyclopentadiene and cyclopentadiene derivatives.
2. History of Cyclopentadiene
In 1886, Roscoe observed a C10H12 hydrocarbon from the pyrolysis of phenol and correctly surmised that this compound formed from the spontaneous dimerization of a C5H6 hydrocarbon.2 The structures of these two hydrocarbons were elucidated as cyclopentadiene and its dimer, dicyclopentadiene, by Etard and Lambert in 1891 and by Kraemer and Spilker in 1896.3,4
Albrecht reported the formation of 1:1 and 2:1 adducts3,4 from the reaction of cyclopentadiene with 1,4-benzoquinone in 1906, but the structures of these adducts were incorrectly characterized as if the cyclopentadiene underwent a Michael addition to the α,β-unsaturated ketone, as shown in Scheme 1.5 Diels and Alder correctly characterized the products and recognized the significance of the chemical transformation for the synthesis of 6-membered rings in their seminal 1928 publication (Scheme 2).6 The endo stereochemistry of the Diels–Alder reaction was established by Alder and Stein in 1937.7 Alder’s observation that the diene and dienophile orient to maximize the accumulation of double bonds became known as the Alder endo rule. For this work Diels and Alder would go on to win the 1950 Nobel prize in chemistry, and the Diels–Alder reaction would go on to become one of the most widely used reactions in synthetic organic chemistry.8
Scheme 1.

Mischaracterized 1:1 and 2:1 Adducts from the Reaction of Cyclopentadiene with 1,4-Benzoquinone as Reported by Albrecht in 1906.5
Scheme 2.

Correctly Characterized 1:1 and 2:1 Cycloadducts from the Reaction of Cyclopentadiene with 1,4-Benzoquinone by Diels and Alder in 1928.6
3. Diels–Alder Reactivity and π-Facial Selectivity of Cyclopentadiene Cycloadditions
3.1. Diels–Alder Reactivity
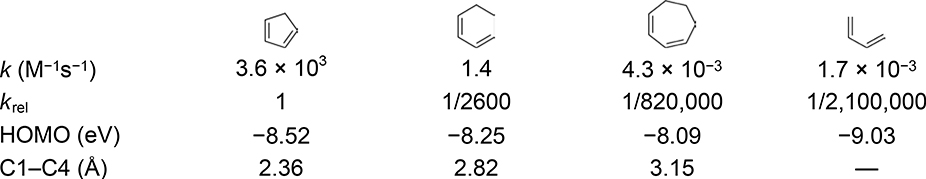
Cyclopentadiene is unique in having high reactivity as a Diels–Alder diene without the presence of any activating electron-withdrawing or electron-donating groups.9 The rate constants for the Diels–Alder reactions of cyclopentadiene, 1,3-cyclohexadiene, 1,3-cycloheptadiene, and butadiene with tetracyanoethylene (TCNE) at 20 °C are listed in Table 1.10 Cyclopentadiene reacts 2,600-fold faster than 1,3-cyclohexadiene, 820,000-fold faster than 1,3-cycloheptadiene, and 2,100,000-fold faster than the acyclic diene, butadiene. The highest-occupied molecular orbital (HOMO) energies measured from the experimental ionization potentials of the dienes shown in Table 1 do not correlate with their Diels–Alder reactivities.11–14 Saur, Susstman, and others related the Diels–Alder reactivity trend of cisoid fused dienes to the distance between the C1 and C4 diene carbons, wherein shorter C1–C4 distances correlated with a faster cycloaddition.10,15,16 Levandowski and Houk expanded upon this analysis by showing that a shorter C1–C4 distance increases the Diels–Alder reactivity by decreasing the out-of-plane distortion required of the conjugated diene double bonds to reach the transition state geometry.17
Table 1.
Rate Constants and Related Parameters for the Diels–Alder Reactions of Cyclopentadiene, 1,3-Cyclohexadiene, 1,3-Cycloheptadiene, and Butadiene with Tetracyanoethylene (TCNE) at 20 °C in CH2Cl2.10
 |
Sauer and co-workers studied the Diels–Alder reactivity of cyclopentadiene toward the acyclic and cyclic electron-deficient dienophiles shown in Scheme 3.18 Cyclic electron-deficient dienophiles, such as N-phenyl maleimide, maleic anhydride, and p-benzoquinone, react significantly faster with cyclopentadiene than do acyclic electron-deficient dienophiles. Consequently, maleimides, maleic anhydride, and p-benzoquinones are choice reaction partners of cyclopentadiene in click chemistry applications.
Scheme 3.

Rate Constants (M−1s−1) for the Diels–Alder Reactions of Various Dienophiles with Cyclopentadiene in Dioxane at 20 °C.18
Solvent can have a significant influence on the rate of Diels–Alder reactions.19–23 The rate of the Diels–Alder reaction of methyl vinyl ketone with cyclopentadiene was measured in various solvents by Breslow and co-workers (Scheme 4).24,25 The reaction in water is nearly 103-fold faster than in isooctane. QM/MM calculations by the Jorgenson group26–29 and others30 suggested that the rate-enhancement in water is the result of hydrophobic aggregation and enhanced hydrogen bonding in the transition state caused by the increased polarization of the carbonyl moiety in the transition state.
Scheme 4.

Reaction Rates for the Diels–Alder Reactions of Cyclopentadiene with Methyl Vinyl Ketone at 20 °C Across Different Solvents.24,25
The ambiphilicity of cyclopentadiene allows cyclopentadiene to partake in both normal and inverse electron-demand Diels–Alder reactions, depending on the electronic nature of the dienophile.31 The Diels–Alder reaction of 1,2,3,4-tetrachlorocyclopentadiene ethylene ketal (TCK) with styrene at 80 °C in dioxane occurs with a rate constant of 3.3 × 10−3 M−1s−1. The electronic nature of the styrene can be tuned through substitution at the para position. As shown in Scheme 5, both electron-withdrawing and electron-donating substituents increase the reactivity of styrene toward TCK. This increase is because of a change in reaction mechanism from a normal electron-demand Diels–Alder reaction when there is an electron-withdrawing group to an inverse electron-demand Diels–Alder reaction when there is an electron-donating group at the para position of styrene. The ambiphilic properties of the cyclopentadiene scaffold allow cyclopentadiene to react with both electron-rich and electron-deficient dienophiles.
Scheme 5.

Rate Constants for the Diels–Alder Reactions of 1,2,3,4-Tetrachlorocyclopentadiene Ethylene Ketal (TCK) with Styrene, p-Methoxystyrene, and p-Nitrostyrene.31
3.2. Hyperconjugative Aromaticity and Antiaromaticity
In 1939, Mulliken observed that cyclopentadiene and 1,3-cyclohexadiene have considerably longer ultraviolet absorption wavelengths than do open-chain dienes.32 The redshift results from the hyperconjugation of the diene π-electrons with the saturated CH2 groups. The heats of hydrogenation for cyclopentadiene, 1,3-cyclohexadiene, and butadiene had previously been reported as −50.9, −55.4, and −57.1 kcal/mol, respectively.33 From these values, Mulliken estimated the strengths of the hyperconjugative interactions to be 6.2 kcal/mol in cyclopentadiene and 1.7 kcal/mol in 1,3-cyclohexadiene. The increased strength of the hyperconjugative interactions in cyclopentadiene led Mulliken to consider if cyclopentadiene is aromatic. Sixty years later, computational studies of cyclopentadiene and 5,5-disubstituted cyclopentadienes by the Schleyer group provided energetic, geometric, and magnetic evidence of aromatic electron delocalization in cyclopentadiene and 5,5-disilylcyclopentadiene, and antiaromatic electron delocalization in 5,5-difluorocyclopentadiene.34,35 Schleyer introduced the terms “hyperconjugative aromaticity” and “hyperconjugative antiaromaticity” to describe, respectively, the aromatic (4N + 2) and antiaromaticity (4N) π-electron delocalization that arise from hyperconjugative interactions of the saturated linkages with the formal π-electrons of cyclopentadiene.
The Diels–Alder reactivity of cyclopentadienes can be tuned through the hyperconjugative aromaticity or antiaromaticity invoked by the substituents at the 5-position.36,37 Frontier molecular orbital theory would predict that 1,2,3,4,5,5,-hexachlorocyclopentadiene with a semiempirically derived HOMO energy of −9.0 eV would react faster than 1,2,3,4-tetrachloro-5,5-difluorocyclopentadiene with a HOMO energy of −9.2 eV in normal electron-demand Diels–Alder reactions.31 Despite the less favorable frontier molecular orbital interactions, the Diels–Alder reaction of 1,2,3,4-tetrachloro-5,5-difluorocyclopentadiene occurs ~100-fold faster than does the Diels–Alder reaction of 1,2,3,4,5,5,-hexachlorocyclopentadiene when the electron-deficient p-nitro-N-phenyl maleic anhydride is the dienophile (Scheme 6). Computations by the Houk group predicted that the hyperconjugative antiaromatic 5-fluorocyclopentadiene will react 4,600-fold faster than cyclopentadiene, whereas the hyperconjugative aromatic 5-silylcyclopentadiene will react 160-fold slower than cyclopentadiene with the electron-deficient dienophile, maleic anhydride.36
Scheme 6.

Rate Constants for the Diels–Alder Reactions of 1,2,3,4,5,5-Hexachlorocyclopentadiene and 1,2,3,4-Tetrachloro-5,5-Difluorocyclopentadiene with p-Nitro-N-Phenyl Maleic Anhydride.31
3.3. π-Facial Selectivity of 5-Substituted Cyclopentadienes
Facially asymmetric cyclopentadienes can undergo stereoselective Diels–Alder reactions. During their seminal studies of 7-norbornyl cations, Winstein, Woodward, and co-workers reported the surprising contrasteric (syn) Diels−Alder reaction of 5-acetoxycyclopentadiene with ethylene shown in Scheme 7.38 Studies on the π-facial selectivity of 5-substituted cyclopentadienes by the Burnell and Fallis groups showed that the π-facial selectivity of the cycloaddition is controlled by the substituent at the 5-position.39–41 For example, with dimethyl acetylenedicarboxylate (DMAD) as the dienophile, 5-fluorocyclopentadiene42 provides exclusively the syn cycloadduct, 5-trimethylsilylcyclopentadiene43 provides exclusively the anti cycloadduct, and 5-chlorocyclopentadiene44 provides a mixture of the syn and anti cyclic adducts (Scheme 8).
Scheme 7.

Contrasteric (syn) π-facial Selectivity in the Diels–Alder Reaction of 5-Acetoxy Cyclopentadiene with Ethylene.38
Scheme 8.

Syn and anti π-Facial Selectivities in Diels–Alder Reactions of Dimethyl Acetylenedicarboxylate (DMAD) with 5-Fluorocyclopentadiene, 5-Chlorocyclopentadiene, and 5-Trimethylsilylcyclopentadiene.42–44
The π-facial selectivity in 5-substituted cyclopentadienes arises from hyperconjugative interactions of the cyclopentadiene π-system with the σ-bond of the C5 substituent.37 When the C5 substituent is a σ-acceptor, the cyclopentadiene puckers into an envelope geometry that minimizes the overlap of the diene π-system with the C5–X antibonding σ* orbital (Figure 1). This puckering minimizes the destabilizing effects of the negative hyperconjugation that lead to hyperconjugative antiaromaticity and predistorts the cyclopentadiene into the envelope-like geometry of the syn transition state. Conversely, when the C5–X substituent is a σ-donor, the cyclopentadiene puckers in the opposite direction to maximize the overlap of the cyclopentadiene π-system with the C5–X σ-bond. This puckering maximizes the stabilizing effects of hyperconjugative aromaticity and favors the anti cycloaddition because the ground state geometry is distorted to resemble the envelope-like transition-state geometry of the anti cycloaddition.
Figure 1.

Syn and anti puckering of the ground and transition state geometries of 5-substituted cyclopentadienes.37
The synthesis of neofinaconitine by Gin and co-workers is an elegant application of π-facial selectivity.45 The key step in the synthesis is the cycloaddition between the 5-methoxycyclopentadiene and cyclopropene shown in Scheme 9 that establishes the stereochemistry of the C14 methoxy group in the neofinaconitine product. The products from the syn cycloaddition were isolated as a 1:1.6 mixture of regioisomers in 85% yield, whereas the products from the anti cycloaddition were isolated as a 1:1.5 mixture of regioisomers in 15% yield. Overall, the desired regioisomer from the syn cycloaddition was isolated with a 54% yield.
Scheme 9.

Control of π-Facial Selectivity in the Synthesis of Neofinaconitine.45
4. Stability of Cyclopentadiene
4.1. Dimerization
Cyclopentadiene is a liquid with a neat concentration of 11.9 M at 25 °C. As both a diene and a dienophile, cyclopentadiene can react with itself to form dicyclopentadiene. The rate constant for the dimerization of cyclopentadiene to endo-dicyclopentadiene at 25 °C has been reported to be 8.3 × 10−7 M−1s−1.46,47 Accordingly, the half-life of neat cyclopentadiene is ~28 h at 25 °C. The dimerization of cyclopentadiene is reversible; at 170 °C, the cyclopentadiene monomer can be distilled from dicyclopentadiene.48–50 Exo-dicyclopentadiene is 0.7 kcal/mol more stable than endo-dicyclopentadiene, but at temperatures between 40 and 120 °C, the formation of exo-dicyclopentadiene occurs two to three orders of magnitude more slowly than does the formation of endo-dicyclopentadiene.49,50 Exo-dicyclopentadiene and other cyclopentadiene oligomers, from the trimer to the hexamer of cyclopentadiene, are observed only when cyclopentadiene is heated at higher temperatures over long periods of time.51–54
Substitution affects the rate of cyclopentadiene dimerization, as shown in Scheme 10. Dialkyl substitution at the 5-positions of cyclopentadiene significantly diminishes dimerization. 5,5-Dimethylcyclopentadiene does not dimerize, even at temperatures as high as 200 °C.55,56 Spiro[2.4]hepta-4,6-diene does not dimerize at room temperature and is commercially available as a monomer. Formation of the spiro[2.4]hepta-4,6-diene dimer requires 2 days of prolonged heating between 70 and 80 °C.57 The computed activation energy for the dimerization of spiro[2.4]hepta-4,6-diene is 4.5 kcal/mol higher than that of cyclopentadiene.58 Monoalkylation has little effect on the rate of cyclopentadiene dimerization. An isomeric mixture of 1-, 2-, and 5-methylcyclopentadiene dimerizes at a rate comparable to cyclopentadiene.49 At 120 °C, the rate constants for the dimerization of cyclopentadiene and methyl cyclopentadiene are 1.13 × 10−3 M−1s−1 and 1.08 × 10−3 M−1s−1, respectively. Methyl and ethylene cyclopentadiene ketals dimerize 790- and 497,000-fold faster, respectively, than does cyclopentadiene.59 The destabilizing effects of hyperconjugative antiaromaticity promote dimerization.
Scheme 10.

Relative Rates for the Dimerization of Cyclopentadiene and Substituted Cyclopentadienes at 25 and 80 °C.31,55,56,59
Introducing substituents across the cyclopentadiene double bonds provides another means to slow the dimerization. 1,2,3,4-Tetrachlorocyclopentadiene dimerizes 3-fold slower than does cyclopentadiene.31 1,2,3,4-Tetraalkyl-substituted cyclopentadienes, such as 1,2,3,4-tetramethyl and 1,2,3,4,5-pentamethylcyclopentadiene (Cp*), do not dimerize and are commercially available as monomers. Collective substitution across the diene double bonds and at the 5-positions is an effective way to slow dimerization. For example, the cyclopentadiene ethylene ketal dimerizes 497,000-fold faster than does cyclopentadiene, but upon chlorination it dimerizes 25-fold slower.31,59 Methoxy groups at the 5-positions of cyclopentadiene accelerate the dimerization by 790-fold, however the tetrachloro derivative, 1,2,3,4-tetrachloro-5,5-dimethoxy cyclopentadiene, does not dimerize.31,59
Though often viewed as an undesirable side reaction, the dimerization of cyclopentadiene has found applications in synthetic organic chemistry. As a graduate student, Schleyer developed a synthesis of adamantane using the cyclopentadiene dimer, dicyclopentadiene, as the starting material (Scheme 11).60 The dicyclopentadiene was hydrogenated with platinum oxide then transformed into adamantane using aluminum chloride as a Lewis acid. Olah and co-workers improved the yield of the second step to 98% through the use of sonication in the presence of the superacid, CF3SO3H·SbF5.61 This simple two-step preparation provides an affordable route to adamantane that is still used today.
Scheme 11.

Two-step Synthesis of Adamantane from the Cyclopentadiene Dimer, Dicyclopentadiene.60
Cubane was thought to be too strained to exist until the ring system was synthesized and shown to be stable by Eaton and Cole in 1964.62–64 The key synthetic step involved the dimerization of 2-bromocyclopentadienone. Chapman simplified the synthesis by switching the diene from 2-bromocyclopentadienone to 2-bromocyclopentadiene ethylene ketal, as shown in Scheme 12.65
Scheme 12.

Dimerization of a Cyclopentadiene Ketal in the Synthesis of Cubane.65
Thiele’s ester was first reported in 1901 from the acid-catalyzed esterification of the dicyclopentadiene dicarboxylic acid (Thiele’s acid) prepared from the treatment of cyclopentadiene with potassium and carbon dioxide.66 An “ester first” synthesis shown in Scheme 13 was later developed as a direct route to Thiele’s ester from the dimerization of the esterified cyclopentadiene.67 Despite the 72 theoretically possible products that could arise from the dimerization of the esterified cyclopentadiene, Thiele’s ester is isolated as the major product in 40% yield along with 1:1 mixture of regioisomers in 22% yield.68 The origin of the product distribution has been of great interest to experimental and theoretical chemists. Explanations for the selectivity enlisting frontier molecular orbital theory,69,70 bent bond theory,71,72 and paralocalization energies73 have been put forth. Photochemical intramolecular [2 + 2] cycloaddition of Thiele’s ester and other endo-dicyclopentadienes allows access to functionalized 1,3-bishomocubane polycyclic cages.74–78
Scheme 13.

Synthesis of Thiele’s Ester from the Dimerization of a Cyclopentadiene Methyl Ester.67
Absinthin is a bitter-tasting naturally occurring triterpene lactone that is isolated from Artemisia absinthium (wormwood)79 and is a key component of a renowned spirit, absinthe.80 The biosynthesis of absinthin is postulated to occur through the dimerization of artabsin, as shown in Scheme 14. A similar dimerization has been shown to occur spontaneously and in good yield over the course of ten days.81 It is not yet known if an enzyme catalyzes the biosynthesis of absinthin.
Scheme 14.

Dimerization of Artabsin to Absinthin.79
4.2. 1,5-Sigmatropic Shifts
In 1939, Drake and Adams reported the unexpected formation of 1,4-diphenylcyclopentadiene from the decarboxylation of 2,4-diphenylcyclopentadiene-1-carboxylate, as shown in Scheme 15.82 The double bonds of the expected 1,3-diphenylcyclopentadiene product migrated to form 1,4-diphenylcyclopentadiene with a continuous chain of conjugated double-bond linkages. Double-bond migration became a general characteristic of substituted cyclopentadienes as other reports followed.83–87
Scheme 15.

Unexpected Formation of the 1,4-Diphenylcyclopentadiene from the Decarboxylation of 2,4-Diphenylcyclopentadiene-1-Carboxylate.82
Csicsery showed that methylcyclopentadiene exists as a mixture of the 5-methyl, 1-methyl, and 2-methyl isomers in a 3:45:52 ratio at thermodynamic equilibrium, as shown in Scheme 16.88 Mironov and Elizarova elucidated the mechanism of the isomerization as a 1,5-hydrogen shift through a series of communications in 1963.89,90 The isomerization occurs via a rapid 1,5-hydrogen shift of 5-methylcyclopentadiene to 1-methylcyclopentadiene that is complete within 3–4 h, followed by a slower 1,5-hydrogen shift of 1-methylcyclopentadiene to 2-methylcyclopentadiene with equilibration occurring over 2–3 days. Kinetic studies by McLean showed that the rearrangement of 5-methylcyclopentadiene to 1-methylcyclopentadiene occurs with a rate constant of 1.8 × 10−4 s−1 at 25 °C.91
Scheme 16.

Isomerization of 5-Methylcyclopentadiene to 1-Methyl and 2-Methyl Cyclopentadiene.88
1,5-Sigmatropic hydrogen migrations occur more readily across the cyclopentadiene scaffold than across other cyclic and acyclic diene scaffolds. In 2-methly-1,3-cycloheptadiene, the 1,5-sigmatropic hydrogen migration proceeds with a rate constant of 3.27 × 10−4 s−1 at 160 °C.92 The rate constant for the 1,5-sigmatropic hydrogen shift in the acyclic cis-1,1-dideutero-1,3-pentadiene at 185 °C is 1.38 × 10−6 s.93 Alabugin and co-workers reported the calculated activation energies at the B3LYP/6–31G(d,p) level of theory for the 1,5-hydrogen migration in cyclopentadiene, 1,3-cycloheptadiene, and cis-1,3-pentadiene to be 27.0, 33.2, and 36.6 kcal/mol, respectively.94
1,5-Sigmatropic shifts are pericyclic reactions that occur through transition states with aromatic electron delocalization.95 The transition-state structures for the 1,5-sigmatropic hydrogen migration in cis-1,3-pentadiene and cyclopentadiene calculated with a 3–21G split valence basis set are shown in Scheme 17.96 The transition state for the 1,5-hydrogen migration in cyclopentadiene with nearly equivalent C–C bond lengths is geometrically more aromatic than in cis-1,3-pentadiene.96,97 The distances across the migrating termini in cis-1,3-pentadiene and cyclopentadiene are 2.6 and 1.5 Å, respectively. The closer termini distance facilitates the transfer of the migrating hydrogen atoms by permitting better overlap between the orbitals of the migrating hydrogen and the carbon atoms.98
Scheme 17.

Optimized Transition-State Structures for the 1,5-Sigmatropic Hydrogen Migration in 1,3-Pentadiene and Cyclopentadiene.96
Mulliken charges show the migrating hydrogen atom bears a partial positive charge of 0.22 and 0.31 in the transition state of cis-1,3-pentadiene and cyclopentadiene, respectively.99 Alabugin and co-workers reported natural bond orbital (NBO) charges on the hydrogen atom in the ground and transition state of cis-1,3-pentadiene and cyclopentadiene.94 The partial charge on the hydrogen atom is 0.25 in both the cis-1,3-pentadiene ground and the transition state. In cyclopentadiene, the partial charge on the hydrogen atom increases from 0.27 in the ground state to 0.37 in the transition state. In the transition state, the cyclopentadiene carbon skeleton resembles that of the aromatic cyclopentadienyl anion and is better able to accommodate the charge transfer that results from the migrating “protonic” hydrogen atom. The increased contribution of the cyclopentadienyl anion structure is also reflected in the NICS(1) values of the transition state.94 The cyclopentadiene transition state is more aromatic, having a NICS(1) value of −9.6 compared to −8.8 in the cis-1,3-pentadiene transition state. The rapid 1,5-sigmatropic hydrogen migration in cyclopentadiene arises from a combination of the conformational rigidity of the five-membered ring that enforces the favorable proximity of the reacting termini and the amplified aromatic cyclopentadienyl anion character of the transition state.
The addition of alkyl groups across the cyclopentadiene double bonds appreciably lowers the rate of the 1,5-hydrogen shift, as shown in Scheme 18. Methyl substitution at the 1-position of a 5-methylcyclopentadiene slows the 1,5-hydrogen shift by an order of magnitude, from a rate constant of 6.2 × 10−4 s−1 in 5-methylcyclopentadiene to 5.5 × 10−5 s−1 in 1,5-dimethylcyclopentadiene at 30 °C.100 The 1,5-hydride shift of a Cp* containing a deuterated methyl group at 30 °C is 3 orders of magnitude slower, having a rate constant of 2.4 × 10−7 s−1.101
Scheme 18.

Rate Constants for 1,5-Hydrogen Migration in 5-Methylcyclopentadiene, 1,5-Dimethylcyclopentadiene, and Cp* at 30 °C.100,101
Electropositive silyl groups migrate faster than does hydrogen in 1,5-sigmatropic shifts. In 1970, Ashe showed that the trimethylsilyl group in 5-trimethylsilylcyclopentadiene migrates a million-fold faster than does the hydrogen atom.102 A year later, Ustynyuk and co-workers reported a rate constant of 50 s−1 for the 1,5-sigmatropic shift of the trimethylsilyl group in 5-trimethylsilylcyclopentadiene at 25 °C.103 1,5-Alkyl migrations are significantly slower than 1,5-hydrogen migrations. 5,5-Dimethylcyclopentadiene isomerizes at temperatures >200 °C.56 At 225 °C, the 1,5-methyl shift in 1,2,3,4,5,5-hexamethylcyclopentadiene occurs with a rate constant of 5.1 × 10−7 s−1.104 This alkyl shift is over a million-fold slower than the 1,5-hydride shift in 1,2,3,4,5-pentamethylcyclopentadiene, which occurs with a rate constant of 1.8 s−1 at a slightly lower temperature of 200 °C.101
Fowler and co-workers calculated the migratory aptitudes of different functionalities across cyclopentadiene.105 The computed activation enthalpies for the migration of a trimethylsilyl group, hydrogen atom, methyl group, and fluorine atom are 13.5, 25.8, 42.6, and 48.0 kcal/mol, respectively. The reactivity parallels the charge of the migratory group in the transition state, which were calculated to be 0.504, 0.387, 0.170, and −0.379 for the trimethylsilyl group, hydrogen atom, methyl group, and fluorine atom, respectively. The π current–density maps of cyclopentadiene and 5-fluorocyclopentadiene supported that the 1,5-hydrogen migration proceeds through a diatropic (aromatic) transition state and the 1,5-fluoro migration proceeds through a paratropic (antiaromatic) transition state. The migratory aptitude is related to the ability of the group to draw positive charge. Groups that migrate with a partial positive charge rapidly migrate through an aromatic transition state that resembles the cyclopentadienyl anion, whereas groups that migrate with a partial negative charge do so slowly through a high-energy antiaromatic transition state that resembles the cyclopentadienyl cation.
In 5-formyl-1,2,3,4,5-pentamethylcyclopenta-1,3-diene, the formyl group migrates with a rate constant of 90 s−1 at 25 °C.106 This rapid migration is a result of secondary orbital interactions. The mixing of the cyclopentadiene HOMO with the low lying lowest-unoccupied molecular orbital (LUMO) of the migrating formyl group to stabilize the transition state is depicted in Scheme 19.107
Scheme 19.

Secondary Orbital Interaction Involving the Overlap of the Cyclopentadiene HOMO with the LUMO of the Migrating Formyl Group in the Transition State for the 1,5-Sigmatropic Shift of the Formyl Group in 5-Formyl-1,2,3,4,5-Pentamethylcyclopenta-1,3-Diene.107
The tricyclo[4.4.01,6.02,8]decane and tricyclo[5.4.01,7.02,9]undecane scaffolds present in bridged sesquiterpene natural products such as sativene,108 longifolene,109 and sinularene110 were initially challenging to access synthetically. Early attempts to synthesize these scaffolds through intramolecular Diels–Alder reactions were unsuccessful because 1,5-hydrogen shifts preceded the intramolecular cycloaddition. From these failed attempts emerged imaginative solutions.
In 1963, Breiger attempted the synthesis of longifolene through an intramolecular cycloaddition of a cyclopentadiene tethered to a dienophile at the 5-position.111 It was, however, the 1-substituted cyclopentadiene isomer that reacted to form the undesired product (Scheme 20). In an effort to shift the equilibrium toward the desired 5-isomer, Glass synthesized a 5-tethered-1,2,3,4-tetrachlorocyclopentadiene.112 The chlorinated cyclopentadiene equilibrated to a mixture containing 30% of the 5-isomer and 70% of the 1- and 2-isomers, but the resulting intramolecular cycloadduct was still that of the 1-substituted cyclopentadiene isomer. It has since been established that the selectivity of the intramolecular cycloadditions involving isomeric mixtures of cyclopentadienes is dependent on the length of the tether between the diene and dienophile.113
Scheme 20.

Early Synthetic Attempts to Access the Tricyclo[5.4.01,7.02,9]undecane Scaffold via Intramolecular Diels–Alder Reactions.111,112
Stille and Grubbs systematically studied the effect of the tether length on the selectivity of cyclopentadiene compounds tethered to α,β-unsaturated ester dienophiles (Scheme 21).113 They observed that two-carbon tethers exclusively form the cycloadduct of the 5-substituted cyclopentadiene with a 53% yield after 18 h of refluxing in toluene. A three-carbon tether forms the 1-substituted cyclopentadiene cycloadduct as the major product in 68% yield and the 2-substituted cyclopentadiene cycloadduct as the minor product in 6% yield after 2 h of refluxing in benzene. After refluxing in benzene for 4 h, the four-carbon tether forms the endo and exo cycloadducts from the 1-substituted cyclopentadiene cycloaddition in 40% and 17% yields, respectively. The intrinsic preference of the 1-isomer to undergo the intramolecular cycloaddition in systems where the length of the tether is three or four carbon atoms makes the synthesis of the tricyclo[4.4.01,6.02,8]decane and tricyclo[5.4.01,7.02,9]undecane scaffolds via an intramolecular Diels–Alder reaction a distinct challenge.
Scheme 21.

Selectivity of Tethered Cyclopentadiene Intramolecular Cycloadditions. aReflux for 18 h in Toluene. bReflux for 2 h in Benzene. cReflux for 4 h in Benzene.113
Instead of trying to prevent the 1,5-hydrogen shift, Fallis considered ways to favor the intramolecular cycloaddition of the 5-substituted cyclopentadiene isomer over the 1- and 2-substituted cyclopentadiene isomers.114 Molecular mechanics calculations showed that encompassing the dienophile in a 6-membered ring increased the strain of the 1- and 2-substituted cyclopentadiene cycloadducts making their formation unfavorable relative to the 5-substituted cyclopentadiene cycloadduct. By encompassing the dienophile in a 6-membered ring as shown in Scheme 22, Fallis and co-workers were able to establish the tricyclic core of (+)-longifolene via intramolecular cyclopentadiene 5,6-dihydro-2H-pyran-2-one Diels–Alder reaction with 97% yield to complete the total synthesis of (+)-longifolene.
Scheme 22.

Successful Intramolecular Diels–Alder Reaction of a 5-Substituted Cyclopentadiene Containing a Four-Carbon Tether in the Synthesis of (+)-Longifolene.114
An alternative synthetic approach developed by Fallis and co-workers involved blocking the 1,5-hydrogen shift by linking the tether to the cyclopropane moiety in spiro[2.4]heptadiene.115,116 Following the intramolecular cycloaddition, the selective cleavage of the cyclopropane ring shown in Scheme 23 allows access to the tricyclo[4.4.01,6.02,8]decane or tricyclo[5.4.01,7.02,9]undecane scaffolds.117 This approach enabled the synthesis of sinularene in 14 steps and 9% overall yield starting from a spiro[2.4]heptadiene.118
Scheme 23.

Use of a Tethered Spiro[2.4]heptadiene to Block the 1,5-Hydrogen Shift. Cleavage of the b Cyclopropane Bond Leads to the Tricyclo[5.4.01,7.02,9]undecane Skeleton of Longifolene; Cleavage of the a or c Bond Leads to the Tricyclo[4.4.01,6.02,8]decane Skeleton of Sinularene or Sativene.117
The Diels–Alder reaction of methylcyclopentadiene with methyl acrylate results in the formation of a complex mixture of endo and exo cycloadducts from the Diels–Alder reactions of the 1- and 2-substituted cyclopentadiene isomers after reacting for 16 h at 18 °C in benzene, as shown in Scheme 24.119 Corey keenly realized that Lewis acids accelerate the Diels–Alder reaction without affecting the rate of the 1,5-sigmatropic shift. The AlCl3-catalyzed Diels–Alder reaction of 5-(benzyloxymethyl)cyclopentadiene with an acrylate ester at −55 °C shown in Scheme 25 results in the formation of only the endo and exo cycloadducts of the 5-substituted cyclopentadiene isomer in 89% and 7% yield, respectively.120 The use of a Lewis acid to obtain Diels–Alder adducts of 5-substituted cyclopentadienes was crucial in Corey’s landmark synthesis of prostaglandin F2α in 1969.121
Scheme 24.

Product Distribution for the Diels–Alder Reaction of Methylcyclopentadiene with Methylacrylate.119
Scheme 25.

Lewis Acid-catalyzed Diels–Alder Reaction of 5-(Benzyloxymethyl)cyclopentadiene with an Acrylate Ester en Route to Prostaglandin F2α.120,121
Tsuji and co-workers observed that Diels–Alder reactions of 2-alkoxycyclopentadienes do not form a mixture of isomeric cycloadducts.122–124 By incorporating an alkoxy functional group at the 2-position, Tsuji and co-workers were able to trap the cycloadducts of 5-substituted cyclopentadienes via intramolecular cycloaddition with a doubly activated dienophile to form the tricyclo[5.2.1.01,5]decane scaffold, as shown in Scheme 26.125 Gleason showed that incorporating a silyl ether at the 2-position of a cyclopentadiene increased the half-life of a 5-substituted cyclopentadiene from ~1 h at 20 °C to ~37 h at 23 °C (Scheme 27).126 Intramolecular Diels–Alder reactions of 2-silyloxycyclopentadienes have since been applied in the synthesis of the antifungal agent Sordarin.127,128
Scheme 26.

In situ Generation and Trapping of a 2-Alkoxy Cyclopentadiene Containing a Three-Carbon Tether at the 5-Position.125
Scheme 27.

Half-lives of 5-Methyl Cyclopentadiene and a 2-Siloxycyclopentadiene Derivative.126
4.3. In Situ Sources of Cyclopentadiene
Bench-stable sources of cyclopentadienes eliminate the need to distill cyclopentadiene from the dimer prior to use. The Richert group developed a crystalline ready-to-use form of cyclopentadiene.129 They showed that cyclopentadiene can be encapsulated as a guest molecule in the crystalline matrix of 1,3,5,7-tetrakis(2,4-dimethoxyphenyl)adamantane (TDA), which is shown in Scheme 28. The crystallization occurs in a stoichiometric ratio of 5 cyclopentadienes for every 4 TDAs. After storing for 4 weeks at 22 °C, more than 85% of the cyclopentadiene remained in monomeric form. Dissolving the crystals in organic solvents results in the release of the encapsulated cyclopentadienes. The Diels–Alder reactions of cyclopentadiene generated from the crystalline form occurred in yields similar to that from freshly distilled dicyclopentadiene.
Scheme 28.

1,3,5,7-Tetrakis(2,4-dimethoxyphenyl)adamantane (TDA) and its Crystal Structure with Cyclopentadiene Encapsulated as a Guest Molecule.129
Norbornadiene can function as a masked cyclopentadiene that is released upon reacting with a 1,2,4,5-tetrazine. This unmasking was first demonstrated by Sauer and co-workers in 1966 when they generated cyclopentadiene as a side product from the Diels–Alder reaction of norbornadiene with 3,6-diphenyl 1,2,4,5-tetrazine en route to the 1,2-diazine (Scheme 29).130 The approach has since been applied to the synthesis of substituted cyclopentadienes. An example of a substituted cyclopentadiene generated from norbornadiene and its subsequent trapping with dimethyl acetylenedicarboxylate is shown in Scheme 30.131,132 If, however, the substituted cyclopentadiene is not trapped with a highly reactive dienophile it will isomerize and dimerize as expected.
Scheme 29.

Mechanism for the Release of Cyclopentadiene from a Norbornadiene after Reacting with a Tetrazine.130
Scheme 30.

Trapping of a 2-Bromo-3-trimethylsilylcyclopentadiene Released from a Norbornadiene with Dimethyl Acetylenedicarboxylate.131,132
5. Polymer Chemistry with Cyclopentadiene
5.1. Synthesis of Cyclopentadiene-Functionalized Polymers
Click chemistry, with its high yields, rapidity, and facility of product-purification, has enriched the field of polymer synthesis. Click reactions involving cyclopentadiene are attractive to polymer chemists because the cyclopentadiene scaffold can be introduced into polymer systems readily via multiple methods. Kennedy and Castner used dimethylcyclopentadienylaluminum as an initiator for the cationic polymerization of tert-butylchloride toward isobutylene monomers, as shown in Scheme 31.133,134 Termination of the isobutylene polymerization occurred by the reaction of the isobutylene cation with the cyclopentadienyl anion, providing polyisobutene polymers containing a terminal cyclopentadiene unit. Termination of 72% of the propagating polymer chains occurred via cyclopentadienylation. The remaining chains were terminated via methylation. When the polymerization was performed at −40 °C, the number of polymer chains terminated by cyclopentadienylation increased to 90%. Heating at 80 °C for 2 h did not result in any dimerization of the terminal cyclopentadiene-terminated polyisobutene polymers. The lack of dimer formation was attributed to the low concentration of cyclopentadiene (0.31% by weight) in the polymer. Whereas the terminal cyclopentadiene polymers were not prone to dimerize, they readily underwent Diels–Alder reactions with maleic anhydride.
Scheme 31.

Initiation, Propagation, and Termination Steps in the Synthesis of a Cyclopentadiene-Capped Isobutylene Polymer.133,134
Grubbs and co-workers prepared cyclopentadiene-functionalized styrene divinylbenzene copolymers by the two routes shown in Scheme 32.135,136 Cyclopentadienes can be prepared from the reaction of cyclopentenones with organolithium or Grignard reagents followed by an acid catalyzed dehydration.137,138 Grubbs and co-workers used this to synthesize a cyclopentadiene-functionalized styrene–divinylbenzene copolymer.136 Bromination followed by lithium–halogen exchange led to the lithiation of the styrene rings in the copolymer. The reaction of the lithiated styrene rings with cyclopentenone and successive acid-catalyzed dehydration provided the cyclopentadiene-functionalized polymer.
Scheme 32.

Cyclopentadienylation of a Styrene–Divinylbenzene Copolymer through the Reaction of the Organolithiated Styrene Motif with Cyclopentenone (top) and through the Reaction of Chloromethylated Styrene Motifs with the Cyclopentadienyl Anion (bottom).135,136
Nucleophilic substitution of alkyl halides or tosylated alcohols with a cyclopentadienyl anion source is the predominant method for the preparation of cyclopentadiene-functionalized polymers.139 The use of this approach for the synthesis of cyclopentadiene-functionalized styrene divinylbenzene copolymers is apparent in Scheme 32.135,136 Treatment of the copolymer with chloromethyl ethyl ether led to the chloromethylation of 14% of the styrene rings. The chloromethylated groups then react with sodium cyclopentadienide to provide cyclopentadiene-functionalized styrene divinylbenzene copolymer linked through a methylene group. Nickelocene can be substituted for sodium cyclopentadienide as a milder source of the nucleophilic cyclopentadienyl anion when potentially reactive electrophilic functionalities are present in the polymer.140,141
5.2. Synthesis of Homopolymers
Homopolymers are polymers with identical repeating units. In 1961, Stille and Plummer prepared high molecular weight homopolymers from the intermolecular Diels–Alder reaction of biscyclopentadienes linked via alkyl tethers.142 The biscyclopentadienes 1,6-bis(cyclopentadienyl)hexane, bis(cyclopentadienyl)nonane, and α,α′-bis(cyclopentadienyl)-p-xylene shown in Scheme 33 were prepared from the reaction of the corresponding dibromo species with 2 equiv of sodium cyclopentadienide. The intermolecular cycloaddition was favored over the intramolecular cycloaddition, resulting in the polymerization of the biscyclopentadiene monomers to form dicyclopentadiene homopolymers under nitrogen gas. Cross-linking trimers from a subsequent Diels–Alder reaction across the strained dicyclopentadiene double bond were observed when the reaction was performed under air. This type of cross-linking has also been observed with dicyclopentadiene diester (Thiele’s ester) cycloadducts formed from the polymerization of a biscyclopentadiene linked via 1,4-butanediyl ester tether.143 The biscyclopentadienes can also copolymerize with highly reactive bis-dienophiles to form homopolymers.142 The 1:1 copolymerization of biscyclopentadienes with p-benzoquinone is shown in Scheme 34.
Scheme 33.

Synthesis of homopolymers via intermolecular Diels–Alder Reaction of Biscyclopentadienes.142
Scheme 34.

Copolymerization of Biscyclopentadienes with p-Benzoquinone.142
5.3. Synthesis of Cyclic Polymers
Cyclic polymers can exhibit unique properties compared to their corresponding acyclic analogues.144 An efficient route to cyclic polymers proceeds through an intramolecular Diels–Alder reaction of a cyclopentadiene tethered to a highly reactive dienophile.145 Barner-Kowollik and co-workers tethered cyclopentadiene to a protected maleimide dienophile through a low-molecular weight poly(methyl methacrylate) polymer, as shown in Scheme 35. Refluxing in toluene for 6 h resulted in the deprotection of the maleimide dienophile, allowing the linear polymer to then cyclize via an intramolecular Diels–Alder reaction of the cyclopentadiene and maleimide termini to generate a cyclic polymer.
Scheme 35.

Formation of a Cyclic Polymer from the Intramolecular Diels–Alder Reaction of a Deprotected Maleimide with Cyclopentadiene.145
5.4. Synthesis of Diblock Copolymers
Block copolymers consist of two polymers held together through a covalent linkage. The coupling of cyclopentadiene-terminated polymers with dienophile-terminated polymers is an effective method for the preparation of diblock copolymers. The synthesis of a diblock copolymer from the Diels–Alder reaction of a cyclopentadiene-terminated poly(2-ethyl-2-oxazoline) polymer with a maleimide-terminated poly(ethylene glycol) (PEG) polymer is shown in Scheme 36.146 The reaction results in the formation of a mixture of block copolymers from the reaction of the 1- and 2-isomers of the cyclopentadiene polymer. The Hawker and Read de Alaniz groups were able to generate pure diblock copolymers from cyclopentadiene maleimide Diels–Alder reactions in which the desired cyclopentadiene polymer was released from the inverse electron-demand Diels–Alder reaction of a norbornadiene with tetrazine.147,148 This strategy, which is depicted in Scheme 37, takes advantage of the orthogonality of the tetrazine–norbornadiene and cyclopentadiene–maleimide cycloadditions.
Scheme 36.

Synthesis of a Diblock Copolymer from the Diels–Alder Reaction of a Cyclopentadiene-Terminated Poly(2-Ethyl-2-Oxazoline) Polymer with a Maleimide-Terminated PEG Polymer.146
Scheme 37.

Diblock Copolymer Synthesis from the Diels–Alder Reaction of a Maleimide-Functionalized Polymer with a Cyclopentadiene-Functionalized Polymer Generated from the Reaction of a Tetrazine and a Norbornadiene.148
The Read de Alaniz group demonstrated the reversible formation of diblock copolymers through the hetero-Diels–Alder reaction of a cyclopentadiene-terminated polystyrene polymer with a nitroso-terminated PEG (Scheme 38).149 The nitroso group can be generated in situ from the copper-catalyzed oxidation of a hydroxyamino group or the retro-Diels–Alder reaction of a 9,10-dimethyl anthracene adduct. The homopolymers were regenerated from the diblock copolymers upon heating. At 95 °C, the nitroso-functionalized PEG was liberated through the retro-Diels–Alder reaction and trapped with benzyl amine. Retro-Diels–Alder reactions of the cycloadducts from peralkylcyclopentadiene and nitroso–carbonyl species have been studied by Yakelis and the Boydston group as thermal triggers for the self-immolation of block copolymers.150
Scheme 38.

Synthesis and Thermal Reversibility of Diblock Copolymers Linked via a Cyclopentadiene Nitroso Cycloadduct.149
Thiocarbonyl compounds have been described as “superdienophiles” because of their rapid reactivity as dienophiles in Diels–Alder reactions.151 The Diels–Alder reaction of thiofluorenone with cyclopentadiene occurs rapidly with a rate constant of 5.1 M−1s−1 in methylene chloride at 20 °C. Barner-Kowollik and co-workers applied the hetero-Diels–Alder reaction of dithioester species with cyclopentadiene to polymer chemistry for the rapid synthesis of diblock copolmyers.152,153 Scheme 39 shows the hetero-Diels–Alder reaction of a cyclopentadiene-functionalized PEG polymer reacting with the diethoxy phosphoryl dithioester-functionalized polymer to form a diblock copolymer under aqueous conditions after 15 min. With the open-chain diene, trans,trans-2,4-hexadien-1-ol, the reaction occurred significantly slower with a required reaction time of 4 h for the quantitative formation of the block copolymer. Cycloadducts from the Diels–Alder reactions of open-chain dienes are less strained and thus more stable than cyclopentadiene cycloadducts.154 The formation of the cyclopentadiene diethoxy phosphoryl dithioester cycloadduct is reversible at temperatures above 90 °C.155,156 Cycloreversion of the diethoxy phosphoryl dithioester cycloadducts of open-chain dienes required temperatures above 160 °C.157,158
Scheme 39.

Reaction Times for the Formation of Diblock Copolymers from the Diel–Alder Reactions of a Diethoxy Phosphoryl Dithioester-Terminated Polymer with Cyclopentadiene and Butadiene-Terminated Polymers.153
Triazoline diones are considered to be the most reactive bench-stable dienophiles.159 The Diels–Alder reaction of 4-phenyl-1,2,4-triazoline-3,5-dione with cyclopentadiene has been reported to occur instantaneously at temperatures as low as −78 °C.160 At 25 °C in toluene, the rate constant for the Diels–Alder reaction 4-phenyl-1,2,4-triazoline-3,5-dione with cyclopentadiene is 1.6 × 105 M−1s−1.161 The Winne and Prez groups prepared a diblock copolymer from the Diels–Alder reaction of a cyclopentadiene-functionalized polyisobornylacralate polymer with 4-butyl-1,2,4-triazoline-3,5-dione bearing a butyl chain (Scheme 40).162 The reaction in THF was complete within 5 s at 20 °C. Triazolines undergo a range of reactions with a wide variety of functional groups. For example, triazoline-3,5-diones undergo ene reactions with polyisoprene polymers.163,164 This reactivity limits the polymers that can be conjugated to the triazoline-3,5-dione to those lacking functionalization.
Scheme 40.

Formation of a Diblock Copolymer via the Diels–Alder Reaction of a Cyclopentadiene-Functionalized Polyisobornylacralate Polymer with 4-Butyl-1,2,4-Triazoline-3,5-Dione.162
5.5. Reversible Cross-Linking of Polymers
Cross-linked polymers are polymers held together through one or more chemical bonds. Kennedy and Castner reported the reversible cross-linking of chlorinated butyl rubber (isobutylene-isoprene) polymers containing randomly distributed pendant cyclopentadienes (Scheme 41).133 After 72 h, the cyclopentadienylated polymers cross-linked to form a gel. Heating to 215 °C led to the thermal dissociation of the dicyclopentadiene cross-linked networks and regeneration of the cyclopentadienylated monomers. Ruckstein and Chen studied the thermal reversibility of chlorinated polymers cross-linked through dicyclopentadiene carboxylates (Thiele’s acid).165–167 Thermally reversible dicyclopentadiene cross-linked networks have potential application as self-healing materials.168,169 The Miura group showed that treating cyclopentadienylated polymers with sodium metal prevented cross-linking by converting the cyclopentadienes to the inert sodium cyclopentadienide species.170
Scheme 41.

Thermally Reversible Cross-linking of Polymers Containing Pendant Cyclopentadienes.133
6. Surface Functionalization of Carbon-Based Materials with Cyclopentadiene
6.1. Fullerene Functionalization
Curl, Kroto, and Smalley shared the 1996 Nobel prize in chemistry for the discovery and synthesis of the C60 fullerene carbon allotrope from soot.171 The size, hydrophobicity, three-dimensionality, electronic configuration, and photophysical properties of fullerenes have led to applications as electron-acceptors in bulk-heterojunction solar cells and as therapeutic agents in medicinal chemistry.172 In 1992, Wudl discovered that C60 reacts with a variety of nucleophiles, dipoles, and dienes.173 This work inspired more detailed studies into the reactivity of C60 toward cyclopentadiene. The Rotello group added cyclopentadiene to a solution of C60 in benzene at 20 °C and observed an immediate color change from a deep purple to golden brown (Scheme 42).174,175 The resulting C60–cyclopentadiene monoadduct was isolated in 74% yield. Nagami and co-workers reacted C60 with cyclopentadiene in a 1:1.5 molar ratio and isolated a mono- and diadduct in 68% and 28% yield, respectively.176 Kroto and co-workers reported that six cyclopentadienes react with C60 in the presence of 10 equiv of cyclopentadiene.177 The ensuing cycloadditions propagate on the norbornene adduct of the previous cycloadduct to form the “ball and chain”178 system shown in Scheme 43.
Scheme 42.

Thermally Reversible Cycloaddition of Cyclopentadiene with C60.174,175
Scheme 43.

Product of the Diels–Alder Reaction of Fullerene with Six Cyclopentadienes.177
The cycloaddition of cyclopentadiene selectively occurs across the [6,6] double bond between two 6-membered rings and not the [5,6] double bond between a 5- and 6-membered ring (Scheme 44).174,177 Computational methods have provided insight into this regioselectivity. With the M06–2X179 functional, the cycloaddition of cyclopentadiene across the [5,6] double bond is predicted to be 15–16 kcal/mol less favorable than across the [6,6] double bond.180–182 This regioselectivity has been attributed to increased orbital overlap between the cyclopentadiene HOMO and the 3-fold degenerate LUMOs of fullerene when the reaction occurs across the [6,6]-double bond.183
Scheme 44.

The cycloaddition of cyclopentadiene across the [6,6] double bond of fullerene is rapid, having a rate constant of 0.6 M−1s−1 at 25 °C.184 This high reactivity is a result of the low-lying 3-fold degenerate LUMOs in C60. At the AM1185 level of theory, the energy of the C60 LUMO is −2.95 eV, which can be compared to 1.44 eV for ethylene.186 The electron-deficient nature of the alkene bonds in C60 leads to poor reactivity with electron-deficient dienes such as 1,2,4,5-tetrazines.187,188
The Diels–Alder reaction of the cyclopentadiene to C60 is thermally reversible at temperatures >75 °C.175 For comparison, the retro-Diels–Alder reaction of norbornene requires temperatures >300 °C.189 The increased lability of the C60–cyclopentadiene adduct relative to norbornene has been attributed to the destabilization that results from the loss of aromaticity in C60.175 At 80 °C, the rate constant of the retro-Diels–Alder reaction is 1.2 × 10−4 s−1 and that of the forward reaction is 3.63 M−1s−1.175,184 The reversible reaction of fullerenes to cyclopentadiene-functionalized silica gels allows for nonchromatic purification of fullerenes.190 Rotello and Nie used a cyclopentadiene-functionalized silica gel to extract C60 selectively from soot at room temperature (Scheme 45). Heating the C60-functionalized silica gel to 100 °C in toluene for 5 h resulted in the release of 93.7% of the bound C60. The increased thermal stability of the cycloadduct with the functionalized cyclopentadiene is the result of stabilizing van der Waals interactions between the alkyl tether that links cyclopentadiene to the silica gel and the C60 surface. This methodology enabled the cost-effective large-scale purification of C60.
Scheme 45.

Thermally Reversible Diels–Alder Reaction of C60 to a Cyclopentadiene-Functionalized Silica Gel for Purification.190
The Rotello group reported the reaction of a cyclopentadiene-functionalized Merrifield resin191 with C60 (Scheme 46).192,193 The resulting cycloadduct exhibited increased stability relative to the free C60–cyclopentadiene adduct. Heating the polymer-functionalized C60 in decalin at 180 °C in the presence of maleic anhydride as a trapping agent resulted in a 48% recovery of fullerene. The increased stability of the fullerene cyclopentadiene resin cycloadduct is the result of stabilizing noncovalent interactions (π-stacking and dispersion) between the aryl groups in the styrene–divinylbenzene copolymer of the resin and the C60 surface. The use of cyclopentadiene-capped styrene–divinylbenzene copolymer resins was implemented by the Dorn group for the purification of endohedral lanthanide trimetallic nitride metallofullerenes.194
Scheme 46.

Reversible Diels–Alder reaction of a Cyclopentadiene-Functionalized Merrifield resin with C60.192,193
Biological applications of C60 are limited by their sparse solubility in polar solvents.195–199 Derivatives of C60 have been developed to increase the solubility across different solvents.200 Barner-Kowollik and Nebhani reacted a cyclopentadiene having a pendant PEG polymer with C60 to prepare a water soluble PEG-functionalized C60 derivative.201 A C60 soluble in fluorous solvents was synthesized by Wilson and Cowers (Scheme 47). They functionalized C60 with a cyclopentadiene having a pendant perfluoroalkyl group.202 The fluorinated cyclopentadiene was less reactive toward C60 than was cyclopentadiene itself and formed a 1.5:1 isomeric mixture of cycloadducts from the reaction of the 1- and 2-cyclopentadiene, respectively. The isomeric cycloadducts were soluble in perfluorohexane. The electron-withdrawing effects of the perfluoroalkyl group decreased the stability of the cycloadduct. At temperatures as low as 60 °C the isomeric cycloadducts began to revert back into starting materials.
Scheme 47.

Functionalization of C60 with a Cyclopentadiene Bearing a Fluorinated Side Chain.202
Since the discovery of C60, a number of fullerene allotropes and endohedral derivatives have been synthesized and treated with cyclopentadiene. The reactivity of cyclopentadiene toward C70, Li+@C60, and La@C82, in particular, has been studied in detail. C70 is an order of magnitude less reactive than C60 toward cyclopentadiene, having a rate constant of 8.8 × 10−2 M−1s−1 at 19 °C.184 In the same study, the rate constant for the reaction of cyclopentadiene with C60 was reported to be 6.4 × 10−1 at 20 °C. The lithium ion encapsulated fullerene, Li+@C60, was more reactive than C60 itself toward cyclopentadiene.203 The reaction of [Li+@C60]PF6− with 1.1 equiv of cyclopentadiene resulted in the formation of the monoadduct in 56% yield along with a regioisomeric mixture of bis cycloadducts. The second cycloaddition occurs on the fullerene cage, suggesting that the double bonds in Li+@C60 monoadduct are more reactive than norbornene double bonds from the initial cycloadduct. Matsuo and co-workers reported that the reaction of Li+@C60 reached equilibrium in 15 s and was too fast for the determination of a rate constant.203 The computationally predicted Gibbs free activation energies for the Diels–Alder reaction of cyclopentadiene with C60 and Li+@C60 are 21.3 and 12.7 kcal/mol, respectively, corresponding to a 3.3 million-fold rate enhancement.182 With the less reactive diene, cyclohexadiene, the experimental rate constant with Li+@C60 was measurable and a 2,400-fold rate enhancement was reported over C60.204 The encapsulated Li+ ion promotes the cycloaddition by lowering the LUMO of C60. The monoadduct of La@C82 and cyclopentadiene readily decomposes at room temperature with a half-life of ~1.8 h.205 1,2,3,4,5-Pentamethylcyclopentadiene fullerene adducts are significantly more stable than the cyclopentadiene fullerene adducts.206 After 12 h at 298 K, 36% of La@C82 Cp* adducts decomposed to La@C82 and Cp*.207 The increased stability of the Cp* cycloadduct is the result of dispersion interactions between the fullerene cage and the methyl substituents of Cp*.208
6.2. Carbon Nanotube Functionalization
The Barner-Kowollik group studied the functionalization of carbon nanotubes with cyclopentadiene-capped poly(methyl methacrylate) polymers via the direct functionalization and prefunctionalization routes shown in Scheme 48. The grafting ratios for direct functionalization of the carbon nanotube with cyclopentadienyl–capped poly(methyl methacrylate) polymers were measured using thermogravimetric analysis, elemental analysis, and x-ray photoelectric spectroscopy.209 Depending on the method of analysis, the grafting ratio was 18–60% higher when the reaction was carried out at 80 °C. This approach has been used to bioconjugate single stranded DNA to carbon nanotubes.210 The alternative route involves prefunctionalizing the carbon nanotube surface with pyridine-based dithioesters that then undergo Diels–Alder reactions with the cyclopentadienyl-functionalized polymers.211 Although this approach leads to higher polymer grafting densities, the prefunctionalization process involves refluxing the carbon nanotubes for 8 h in 8 M nitric acid. The harsh acid treatment results in the oxidation of the carbon nanotube to the detriment of its mechanical and electrical properties.212,213
Scheme 48.

Direct Functionalization and Prefunctionalization of a Carbon Nanotube with Cyclopentadiene-Capped Polymers.209,211
6.3. Graphene Functionalization
Graphene is a two-dimensional material consisting only of sp2-hybridized carbon atoms.214 Diels–Alder reactions have been demonstrated on its surface with graphene acting as either a diene or dienophile.215 The curvature of fullerene and carbon nanotubes introduces a strain that promotes Diels–Alder reactivity.216–218 Graphene lacks any curvature and is less reactive as a dienophile. In 2012, the functionalization of a graphene surface with cyclopentadiene-capped PEG monomethyl ether polymers was carried out to improve the solubility of the material.219 At ambient temperature and 80 °C, grafting ratios of 16.7% and 21.8%, respectively, were observed after 24 h. The functionalized graphene displayed solubility in tetrahydrofuran, ethylene glycol, ethanol, water, acetone, and chloroform.
Houk and co-workers studied computationally the reaction enthalpies for the Diels–Alder reactions of cyclopentadiene across a representative 5 × 5 graphene surface shown in Scheme 49.220 The reaction enthalpies are favorable for the cycloaddition across the double bonds on the edges of the graphene surface and unfavorable across the interior graphene double bonds. Peripheral double bonds a and b have reaction energies of −11.4 and −1.4 kcal/mol, respectively, whereas the cycloaddition across the internal double bond c is unfavorable with a reaction energy of 36.6 kcal/mol. The cycloaddition of a second cyclopentadiene across the norbornene adduct has a reaction enthalpy of −25.8 kcal/mol and is favored over the multisite-functionalization of the graphene surface.
Scheme 49.

5 × 5 Graphene Model Showing the Selectivity of the Cyclopentadiene Diels–Alder Reaction across Bond ‘a’ on the Surface of Graphene.220
7. Bioconjugation with Cyclopentadiene
7.1. Biomolecule Surface Immobilization
Cyclopentadiene Diels–Alder reactions are recognized as an effective method to immobilize biomolecules onto a surface.221–227 In 1999, Yousaf and Mrksich pioneered the use of the Diels–Alder reaction for this purpose.228 They prepared a gold self-assembled monolayer containing unreactive hydroquinone groups. The hydroquinones were oxidized into 1,4-benzoquinones that are reactive toward cyclopentadiene (Scheme 50). The immobilization of biomolecules via the Diels–Alder reaction of bioconjugated cyclopentadienes with the oxidized hydroquinone groups of a self-assembled monolayer allowed the Mrksich group to develop peptide chips for the quantitative evaluation of protein kinase activity and carbohydrate chips for the quantitative evaluation of carbohydrate protein interactions.229–231 These chips are amenable for high-throughput screening applications.
Scheme 50.

Reversible Electrochemical Oxidation of a Hydroquinone-Functionalized Surface to 1,4-Benzoquinone, Which Can Then React with Cyclopentadiene.228
In 2005, the Waldmann group immobilized a cyclopentadiene streptavidin conjugate onto a maleimide-functionalized glass surface.232 Maleimides react an order of magnitude faster than does 1,4-benzoquinone toward cyclopentadiene (see: Scheme 3).18 Ravoo and co-workers made use of the cyclopentadiene maleimide reaction in carbohydrate microarrays.233 The immobilization of the cyclopentadiene-functionalized carbohydrates occurred rapidly on the maleimide surface with reaction times between 5 and 15 min at room temperature.
7.2. Noncanonical Cyclopentadienyl Amino Acids
The Christie and Read de Alaniz groups introduced the cyclopentadiene (CpHK) and spiro[2.4]hepta-4,6-diene (SCpHK) noncanonical amino acids shown in Scheme 51 into antibodies.234–236 Dimerization was observed between antibody side chains bearing CpHK amino acids in close proximity, whereas antibody side chains bearing the less reactive SCpHK amino acids in close proximity did not dimerize. The incorporated CpHK and SCpHK amino acid side chains react toward maleimide-linked drugs with respective rate constant of 77 M−1s−1 and 3 M−1s−1 to form antibody drug conjugates.
Scheme 51.

Noncanonical Cyclopentadiene-Derived Amino Acids CpHK and SCpHK.234–236
7.3. Bioorthogonal Chemistry
Click reactions that can be carried out in biological environments without perturbing any native biological processes are also known as “bioorthogonal” reactions.237 Bioorthogonal reactions enable biological targets functionalized with a chemical handle to be labeled with a chemical probe. Tetrazines,238 cyclopentadiones,239,240 thiophene dioxides,241 and ortho-quinones242,243 have been been explored widely and applied as bioorthogonal dienes. These electron-deficient dienes are generally paired with a trans-cyclooctene (TCO), cyclooctyne, cyclopropene, or norbornene reaction partner. Cyclopentadiene also reacts readily with these strained dienophiles under mild conditions.244,245
Sauers early kinetic studies on the Diels–Alder reactions of tetrazines toward cyclopropene, norbornene, and trans-cyclooctene inspired their use as bioorthogonal reagents.246 Like other bioorthogonal reagents, a cyclopentadiene, 1,2,3,4-tetrachlorocyclopentadiene ethylene ketal (TCK), was revived from the literature and adapted for modern application as a bioorthogonal reagent (Scheme 52).31,247 TCK was the most reactive cyclopentadiene that resisted dimerization from Sauer’s 1997 studies on the Diels–Alder reactivity and dimerization of substituted cyclopentadienes. Houk refers to cyclopentadienes that do not dimerize as being “self-orthogonal” cyclopentadienes.248 The rate constant of the Diels–Alder reaction of TCK with 4-phenyl-1,2,4-triazoline-3,5-dione is 6.0 M−1s−1 in methylene chloride at 20 °C. Although this reaction rate is fast enough for applications in bioorthogonal chemistry, 4-phenyl-1,2,4-triazoline-3,5-dione is a strong Michael acceptor and unstable in physiological contexts.159 Swapping 4-phenyl-1,2,4-triazoline-3,5-dione for the biologically stable dienophile, BCN, resulted in a rate constant of 0.26 M−1s−1 in methanol at 23 °C.248 This rate constant is of the same order of magnitude as the fastest strain promoted azide–alkyne cycloadditions, which are widely used in bioorthogonal chemistry.249 The biocompatibility of the reaction was demonstrated through a labeling study performed in vitro between a TCK–neuropeptide and TCO–fluorophore conjugate. The field of bioorthogonal chemistry is expanding rapidly, driven by active efforts to optimize the stability and reactivity of known bioorthogonal reagents and to develop new bioorthogonal reactions.250 The emergence of cyclopentadiene as a potential bioorthogonal reagent is exciting, as new applications often arise from new reagents.251
Scheme 52.

Rate Constant for the Diels–Alder Reaction of TCK with BCN.248
8. Heterocyclopentadienes
8.1. 1- and 2-Azacyclopentadienes
The Wong and Jung groups explored the potential of 2,3,4,5,5-pentachloro-1-azacyclopentadiene as a Diels–Alder diene.252–254 Heating 2,3,4,5,5-pentachloro-1-azacyclopentadiene in the presence of styrene or vinyl acetate as the dienophile did not afford the expected l-azabicyclo[2.2.1]hept-2-ene cycloadduct but rather the unexpected 2-azabicyclo[2.2.1]hept-2-ene cycloadduct (Scheme 53). The Diels–Alder cycloaddition proceeds through the 2-azacyclopentadiene isomer, which is in equilibrium with the 1-azacyclopentadiene isomer at elevated temperatures. The Houk group computationally studied the reluctance of the 1-azacyclopentadiene isomer to undergo Diels–Alder cycloadditions.255 With ethylene as the dienophile, the computed activation energy for the Diels–Alder reaction with 1-azacyclopentadiene is 11 kcal/mol less favorable than with the 2-azacyclopentadiene isomer. The decreased reactivity of the 1-azacyclopentadiene is related to the decreased overlap of the nitrogen p-orbital, which is contracted relative to a carbon p-orbital, with the p-orbital of ethylene during bond formation.
Scheme 53.

Diels–Alder Reaction of 1,3,4,5,5-Pentachloro-2-azacyclopentadiene Formed from the Thermal Isomerization of 2,3,4,5,5-Pentachloro-1-azacyclopentadiene.252–254
8.2. 2,3-Diazacyclopentadienes
Diels–Alder reactions of 2,3-diazacyclopentadienes have been extensively explored by Hünig, Adam, and others.256–260 5,5-Dimethyl-2,3-diazacyclopentadienes do not readily undergo Diels–Alder reactions, even with strained dienophiles, unless promoted with an acid catalyst (Scheme 54). Fluorination of 2,3-diazacyclopentadienes at the 5-position increases the diene character by lowering the LUMO energy of the pyrazole and inducing hyperconjugative antiaromativity.261,262 The rate constants for the Diels–Alder reactions of 4-fluoro-4-methyl-3,5-diphenyl-4H-pyrazole (MFP) and 4,4-difluoro-3,5-diphenyl-4H-pyrazole (DFP) toward BCN were 0.76 M−1s−1 and 5.2 M−1s−1, respectively. Though more reactive than the tetrazine scaffold toward BCN, the DFP scaffold is unstable in biological settings.
Scheme 54.

Rate Constants for the Diels–Alder Reactions of 4,4-Dimethyl-3,5-Diphenyl-4H-Pyrazole (DMP), MFP, and DFP with BCN.261,262
9. Summary and Perspectives
This review has covered the development and applications of cyclopentadiene as a reagent in click chemistry. The use of monoalkylated cyclopentadiene as a click reagent has been challenging because of the intrinsic propensity of cyclopentadienes to dimerize and isomerize. This challenge can be overcome by generating the cyclopentadiene in situ. Click reactions of cyclopentadiene with electron-deficient dienophiles as reaction partners have limited biological applications because of the instability of electron-deficient dienophiles in biological systems. Finding highly reactive dienophile partners such as BCN that are biologically stable would expand the utility of cyclopentadiene click reactions to biological systems.
Despite the rich structural diversity of the cyclopentadiene scaffold that can be generated through substitution at one of six positions, monoalkyl-substituted cyclopentadienes currently dominate the landscape of cyclopentadiene click chemistry reactions. A number of complex cyclopentadiene scaffolds have been generated as precursors in the synthesis of cyclopentadienyl transition metal complexes. Exploring the Diels–Alder reactivities of these highly substituted cyclopentadienes could reveal new cyclopentadienes with ideal reactivity and stability for applications in click chemistry. We hope this review inspires readers to explore the field of cyclopentadiene click chemistry and to expand upon its scope.
Acknowledgements
B.J.L. was supported by Grant F32 GM137543 (NIH). Work in the Raines laboratory on click chemistry is supported by Grant R01 GM044783 (NIH).
Abbreviations
- BCN
endo-bicyclo[6.1.0]non-4-yn-9-ylmethanol
- Cp
cyclopentadiene
- Cp*
1,2,3,4,5-pentamethylcyclopentadiene
- CpHK
Nε-((2-(cyclopenta-1,3-dien-1-yl)ethoxy)carbonyl)-l-lysine
- DFP
4,4-difluoro-3,5-diphenyl-4H-pyrazole
- DMAD
dimethyl acetylenedicarboxylate
- DMP
4,4-dimethyl-3,5-diphenyl-4H-pyrazole
- HOMO
highest-occupied molecular orbital
- LUMO
lowest-unoccupied molecular orbital
- MFP
4-fluoro-4-methyl-3,5-diphenyl-4H-pyrazole
- μw
microwave
- NICS
nucleus independent chemical shift
- PEG
poly(ethylene glycol)
- NR
no reaction
- rt
room temperature
- SCpHK
Nε-((spiro[2.4]hepta-4,6-dien-1-ylmethoxy)carbonyl)-l-lysine
- TCK
1,2,3,4-tetrachlorocyclopentadiene ethylene ketal
- TCNE
tetracyanoethylene
- TDA
1,3,5,7-tetrakis(2,4-dimethoxyphenyl)adamantane
- TCO
trans-cyclooctene
Biographies
Brian J. Levandowski was born in Edmonds, WA in 1991. He received a B.S. degree in chemistry from Pacific Lutheran University in 2013. His doctoral thesis on the “Diels–Alder Reactivities of Cyclic Dienes and Dienophiles” was completed in 2018 under the supervision of Ken Houk at UCLA. While at UCLA, he was a Christopher Foote and Saul Winstein Fellow. He is now a Kirschstein–NRSA postdoctoral fellow in the Raines laboratory at MIT.
Ronald T. Raines was born in Montclair, NJ. He received B.S. degrees in chemistry and biology from MIT, and A.M. and Ph.D. degrees in chemistry from Harvard University. He was a Helen Hay Whitney postdoctoral fellow at UCSF. In 2017, after nearly 30 years on the faculty at the University of Wisconsin–Madison, he returned to MIT, where he is the Roger and Georges Firmenich Professor of Chemistry. His research group is focused on chemistry to understand and control life processes.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Special Issue Paper
This paper is an additional review for Chem. Rev. 2021, volume 121, issue xx, “Click Chemistry”.
References
- (1).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (2).Roscoe HE Note on the Spontaneous Polymerisation of Volatile Hydrocarbons at the Ordinary Atmospheric Temperature. Liebig’s Ann. 1886, 232, 348–352. [Google Scholar]
- (3).Etard A, P. L. Terpene in the Oil from Compressed Gas. Compt. rend 1891, 112, 945–947. [Google Scholar]
- (4).Spilker A; Kraemer G The Cyclopentadiene of Coal Tar; the Indene of the Aliphatic Series. Chem. Ber. 1896, 29, 552–561. [Google Scholar]
- (5).Albrecht W Additionsproducte von Cyklopentadiën Und Chinonen. Justus Liebigs Ann. Chem. 1906, 348, 31–49. [Google Scholar]
- (6).Diels O; Alder K Synthesen in Der Hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar]
- (7).Alder K; Stein G Untersuchungen Über Den Verlauf Der Diensynthese. Angew. Chem. 1937, 50, 510–519. [Google Scholar]
- (8).Nicolaou KC; Snyder SA; Montagnon T; Vassilikogiannakis G The Diels–Alder Reaction in Total Synthesis. Angew. Chem., Int. Ed. 2002, 41, 1668–1698. [DOI] [PubMed] [Google Scholar]
- (9).Levandowski BJ Diels–Alder Reactivities of Cyclic Dienes and Dienophiles, UCLA, 2018. [Google Scholar]
- (10).Rücker C; Lang D; Sauer J; Friege H; Sustmann R Reaktivität Substituierter 1,3-Butadiene in Diels–Alder-Reaktionen. Chem. Ber. 1980, 113, 1663–1690. [Google Scholar]
- (11).Bischof P; Heilbronner E Photoelektron-Spektren von Cycloalkenen Und Cycloalkadienen. Vorläufige Mitteilung. Helv. Chim. Acta 1970, 53, 1677–1682. [Google Scholar]
- (12).Sustmann R; Schubert R Photoelektronenspektroskopische Bestimmung von Substituenten-Effekten I. Substituierte Butadiene. Tetrahedron Lett. 1972, 13, 2739–2742. [Google Scholar]
- (13).Beez M; Bieri G; Bock H; Heilbronner E The Ionization Potentials of Butadiene, Hexatriene, and Their Methyl Derivatives: Evidence for through Space Interaction between Double Bond π-Orbitals and Non-Bonded Pseudo-π Orbitals of Methyl Groups? Helv. Chim. Acta 1973, 56, 1028–1046. [Google Scholar]
- (14).Asmus P; Klessinger M Photoelectron Spectra of Organic compounds—VI. Tetrahedron 1974, 30, 2477–2483. [Google Scholar]
- (15).Scharf H-D; Plum H; Fleischhauer J; Schleker W Zur Diels–Alder-Reaktivitäts-cis-Fixierter 1,3-Diene. Chem. Ber. 1979, 112, 862–882. [Google Scholar]
- (16).Sustmann R; Böhm M; Sauer J Der Einfluß Des Dien-1,4-Abstandes Auf Die Reaktivität Bei Diels–Alder-Reaktionen. Chem. Ber. 1979, 112, 883–889. [Google Scholar]
- (17).Levandowski BJ; Houk KN Theoretical Analysis of Reactivity Patterns in Diels–Alder Reactions of Cyclopentadiene, Cyclohexadiene, and Cycloheptadiene with Symmetrical and Unsymmetrical Dienophiles. J. Org. Chem. 2015, 80, 3530–3537. [DOI] [PubMed] [Google Scholar]
- (18).Sauer J; Wiest H; Mielert A Eine Studie Der Diels–Alder-Reaktion I Die Reaktivität von Dienophilen Gegenüber Cyclopentadien Und 9.10-Dimethyl-Anthracen. Chem. Ber. 1964, 97, 3183–3207. [Google Scholar]
- (19).Blokzijl W; Engberts JBFN Initial-State and Transition-State Effects on Diels–Alder Reactions in Water and Mixed Aqueous Solvents. J. Am. Chem. Soc. 1992, 114, 5440–5442. [Google Scholar]
- (20).Otto S; Blokzijl W; Engberts JBFN Diels–Alder Reactions in Water. Effects of Hydrophobicity and Hydrogen Bonding. J. Org. Chem. 1994, 59, 5372–5376. [Google Scholar]
- (21).Engberts JBFN Diels–Alder Reactions in Water: Enforced Hydrophobic Interaction and Hydrogen Bonding. Pure Appl. Chem. 1995, 67, 823–828. [Google Scholar]
- (22).Meijer A; Otto S; Engberts JBFN Effects of the Hydrophobicity of the Reactants on Diels–Alder Reactions in Water. J. Org. Chem. 1998, 63, 8989–8994. [Google Scholar]
- (23).Otto S; Engberts JBFN Diels–Alder Reactions in Water. J. Macromol. Sci. Part A Pure Appl. Chem. 2000, 72, 1365–1372. [Google Scholar]
- (24).Rideout DC; Breslow R Hydrophobic Acceleration of Diels–Alder Reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. [Google Scholar]
- (25).Breslow R; Guo T Diels–Alder Reactions in Nonaqueous Polar Solvents. Kinetic Effects of Chaotropic and Antichaotropic Agents and of β-Cyclodextrin. J. Am. Chem. Soc. 1988, 110, 5613–5617. [Google Scholar]
- (26).Blake JF; Jorgensen WL Solvent Effects on a Diels–Alder Reaction from Computer Simulations. J. Am. Chem. Soc. 1991, 113, 7430–7432. [Google Scholar]
- (27).Blake JF; Lim D; Jorgensen WL Enhanced Hydrogen Bonding of Water to Diels–Alder Transition States. Ab Initio Evidence. J. Org. Chem. 1994, 59, 803–805. [Google Scholar]
- (28).Chandrasekhar J; Shariffskul S; Jorgensen WL QM/MM Simulations for Diels−Alder Reactions in Water: Contribution of Enhanced Hydrogen Bonding at the Transition State to the Solvent Effect. J. Phys. Chem. B 2002, 106, 8078–8085. [Google Scholar]
- (29).Acevedo O; Jorgensen WL Understanding Rate Accelerations for Diels–Alder Reactions in Solution Using Enhanced QM/MM Methodology. J. Chem. Theory Comput. 2007, 3, 1412–1419. [DOI] [PubMed] [Google Scholar]
- (30).Yang Z; Doubleday C; Houk KN QM/MM Protocol for Direct Molecular Dynamics of Chemical Reactions in Solution: The Water-Accelerated Diels–Alder Reaction. J. Chem. Theory Comput. 2015, 11, 5606–5612. [DOI] [PubMed] [Google Scholar]
- (31).Eibler E; Höcht P; Prantl B; Roßmaier H; Schuhbauer HM; Wiest H; Sauer J Transitions of Electron Demand in Pericyclic Reactions: Normal, Neutral, and Inverse Diels–Alder Reactions of Polyhalogenated Cyclopentadienes. Liebigs Ann./Recl. 1997, 1997, 2471–2484. [Google Scholar]
- (32).Mulliken RS Intensities of Electronic Transitions in Molecular Spectra IV. Cyclic Dienes and Hyperconjugation. J. Chem. Phys. 1939, 7, 339–352. [Google Scholar]
- (33).Conant JB; Kistiakowsky GB Energy Changes Involved in the Addition Reactions of Unsaturated Hydrocarbons. Chem. Rev. 1937, 20, 181–194. [Google Scholar]
- (34).Nyulászi L; Schleyer P v. R. Hyperconjugative π-Aromaticity: How To Make Cyclopentadiene Aromatic. J. Am. Chem. Soc. 1999, 121, 6872–6875. [Google Scholar]
- (35).Fernández I; Wu JI; Schleyer P. v. R. Substituent Effects on “Hyperconjugative” Aromaticity and Antiaromaticity in Planar Cyclopolyenes. Org. Lett. 2013, 15, 2990–2993. [DOI] [PubMed] [Google Scholar]
- (36).Levandowski BJ; Zou L; Houk KN Schleyer Hyperconjugative Aromaticity and Diels–Alder Reactivity of 5-Substituted Cyclopentadienes. J. Comput. Chem. 2016, 37, 117–123. [DOI] [PubMed] [Google Scholar]
- (37).Levandowski BJ; Zou L; Houk KN Hyperconjugative Aromaticity and Antiaromaticity Control the Reactivities and π-Facial Stereoselectivities of 5-Substituted Cyclopentadiene Diels–Alder Cycloadditions. J. Org. Chem. 2018, 83, 14658–14666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Winstein S; Shatavsky M; Norton C; Woodward RB 7-Norbornenyl and 7-Norbornyl Cations. J. Am. Chem. Soc. 1955, 77, 4183–4184. [Google Scholar]
- (39).Wellman MA; Burry LC; Letourneau JE; Bridson JN; Miller DO; Burnell DJ Facial Selectivity in the Diels−Alder Reactions of 5-Chloro-, 5-Bromo-, and 5-Iodo-1,3-Cyclopentadiene and Derivatives. J. Org. Chem. 1997, 62, 939–946. [Google Scholar]
- (40).Macaulay JB; Fallis AG π-Facial Selectivity: Heteroatom Directed Syn/Anti Stereoselection in Diels–Alder Cycloadditions of Plane-Nonsymmetric Cyclopentadienes. J. Am. Chem. Soc. 1988, 110, 4074–4076. [Google Scholar]
- (41).Xidos JD; Poirier RA; Pye CC; Burnell DJ An Ab Initio Study of Facial Selectivity in the Diels–Alder Reaction. J. Org. Chem. 1998, 63, 105–112. [DOI] [PubMed] [Google Scholar]
- (42).McClinton MA; Sik V 5-Fluorocyclopentadiene: Synthesis and Utility. J. Chem. Soc. Perkin Trans. I 1992, 1891–1895. [Google Scholar]
- (43).Kraihanzel CS; Losee ML Ethynylsilanes. IV. The Effect of Temperature on the Diels–Alder Addition of Acetylenic Dienophiles to 1-Trimethylsilylcyclopentadiene. J. Am. Chem. Soc. 1968, 90, 4701–4705. [Google Scholar]
- (44).Franck-Neumann M; Sedrati M Studies on the Effect of Remote Substituents on Stereoreactivity. III. Influence of Direct Electronic Activation of the Dipolarophilic Double-Bond on the Course of Diazoalkanes Cycloaddition to 7-Halonorbornadienes. Tetrahedron Lett. 1983, 24, 1391–1394. [Google Scholar]
- (45).Shi Y; Wilmot JT; Nordstrøm LU; Tan DS; Gin DY Total Synthesis, Relay Synthesis, and Structural Confirmation of the C18-Norditerpenoid Alkaloid Neofinaconitine. J. Am. Chem. Soc. 2013, 135, 14313–14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Raistrick B; Sapiro RH; Newitt DM 360. Liquid-Phase Reactions at High Pressures. Part V. The Polymerisation of Cyclopentadiene and α-Dicyclopentadiene. J. Chem. Soc. 1939, 1761–1769. [Google Scholar]
- (47).Turnbull AG; Hull HS A Thermodynamic Study of the Dimerization of Cyclopentadiene. Aust. J. Chem. 1968, 21, 1789–1797. [Google Scholar]
- (48).Alder K; Stein G Zur Polymerisation Cyclischer Kohlenwasserstoffe V Der Abbau Der Polymeren Cyclopentadiene Mit Seleniger Säure. Justus Liebigs Ann. Chem. 1933, 504, 205–215. [Google Scholar]
- (49).Krupka J Kinetics of Thermal Dimerization of Cyclopentadiene and Methylcyclopentadienes and Their Codimerization. Pet. Coal 2010, 52, 290–306. [Google Scholar]
- (50).Narayan A; Wang B; Nava Medina IB; Mannan MS; Cheng Z; Wang Q Prediction of Heat of Formation for Exo-Dicyclopentadiene. J. Loss Prev. Process Indust. 2016, 44, 433–439. [Google Scholar]
- (51).Staudinger H; Bruson HA Hochpolymere Verbindungen. 7. Mitteilung. Über Das Dicyclopentadien Und Weitere Polymere Cyclopentadiene. Justus Liebigs Ann. Chem. 1926, 447, 97–110. [Google Scholar]
- (52).Staudinger H Hochpolymere Verbindungen. Über Die Konstitution Des Dicyclopentadiens. Justus Liebigs Ann. Chem. 1928, 467, 73–75. [Google Scholar]
- (53).Alder K; Stein G Zur Polymerisation Cyclischer Kohlenwasserstoffe. VI. Die Stereochemie Der Cyclopentadien-Polymerisation. Justus Liebigs Ann. Chem. 1933, 504, 216–257. [Google Scholar]
- (54).Herndon WC; Grayson CR; Manion JM Retro-Diels–Alder Reactions. III. Kinetics of the Thermal Decompositions of Exo- and Endo-Dicyclopentadiene. J. Org. Chem. 1967, 32, 526–529. [Google Scholar]
- (55).Rouse R; Tyler W III. 5,5-Dimethylcyclopentadiene. J. Org. Chem. 1961, 26, 3525–3527. [Google Scholar]
- (56).McLean S; Findlay DM Thermal and Photochemical Rearrangements of Methyl-Substituted Cyclopentadienes. Can. J. Chem. 1970, 48, 3107–3109. [Google Scholar]
- (57).Alder K; Ache H-J; Flock FH Über Einige Versuche Mit Spiro-[2.4]-Heptadien-(1.3). Chem. Ber. 1960, 93, 1888–1895. [Google Scholar]
- (58).Froese RDJ; Organ MG; Goddard JD; Stack TDP; Trost BM Theoretical and Experimental Studies of the Diels–Alder Dimerizations of Substituted Cyclopentadienes. J. Am. Chem. Soc. 1995, 117, 10931–10938. [Google Scholar]
- (59).Eaton PE; Hudson RA Cyclopentadienone Ketals. J. Am. Chem. Soc. 1965, 87, 2769–2771. [Google Scholar]
- (60).Schleyer P. v. R. A Simple Preparation of Adamantane. J. Am. Chem. Soc. 1957, 79, 3292–3292. [Google Scholar]
- (61).Farooq O; Farnia SMF; Stephenson M; Olah GA Superacid-Catalyzed near-Quantitative Isomerization of C4n+6H4n+12 (n = 1–3) Polycyclic Precursors to Diamondoid Cage Hydrocarbons Promoted by 1-Haloadamantanes and Sonication. J. Org. Chem. 1988, 53, 2840–2843. [Google Scholar]
- (62).Eaton PE; Cole TW The Cubane System. J. Am. Chem. Soc. 1964, 86, 962–964. [Google Scholar]
- (63).Eaton PE; Cole TW Cubane. J. Am. Chem. Soc. 1964, 86, 3157–3158. [Google Scholar]
- (64).Biegasiewicz KF; Griffiths JR; Savage GP; Tsanaktsidis J; Priefer R Cubane: 50 Years Later. Chem. Rev. 2015, 115, 6719–6745. [DOI] [PubMed] [Google Scholar]
- (65).Chapman NB; Key JM; Toyne KJ Preparations and Properties of Caged Polycyclic Systems. 1. Pentacyclo[5.3.0.02,5.03,9.04,8]decane and Pentacyclo[4.3.0.02,5.03,8.04,7]nonane Derivatives. J. Org. Chem. 1970, 35, 3860–3867. [Google Scholar]
- (66).Thiele J Ueber Abkömmlinge Des Cyclopentadiëns. Ber. Dtsch. Chem. Ges. 1901, 34, 68–71. [Google Scholar]
- (67).Dive G; Robiette R; Chenel A; Ndong M; Meier C; Desouter-Lecomte M Laser Control in Open Quantum Systems: Preliminary Analysis toward the Cope Rearrangement Control in Methyl-Cyclopentadienylcarboxylate Dimer. Theor. Chem. Acc. 2012, 131, 075008. [Google Scholar]
- (68).Chen J; Kilpatrick B; Oliver AG; Wulff JE Expansion of Thiele’s Acid Chemistry in Pursuit of a Suite of Conformationally Constrained Scaffolds. J. Org. Chem. 2015, 80, 8979–8989. [DOI] [PubMed] [Google Scholar]
- (69).Spino C; Pesant M; Dory Y A New Look at the Diels–Alder Transition State. Angew. Chem., Int. Ed. 1998, 37, 3262–3265. [DOI] [PubMed] [Google Scholar]
- (70).Fleming I Molecular Orbitals and Organic Chemical Reactions; John Wiley & Sons: Chichester, UK, 2009; pp 241–242. [Google Scholar]
- (71).Deslongchamps G; Deslongchamps P Bent Bonds and the Antiperiplanar Hypothesis as a Simple Model to Predict Diels–Alder Reactivity: Retrospective or Perspective? Tetrahedron 2013, 69, 6022–6033. [Google Scholar]
- (72).Chen J; Lu L; Wulff J Radical Stabilization Algorithm as a Predictive Tool for Novel and Reported Noncanonical Thiele’s Acid Analogues. Synlett 2017, 28, 2777–2782. [Google Scholar]
- (73).Spino C; Crawford J; Cui Y; Gugelchuk M The Dimerisation of 2-Methoxycarbonylbuta-1,3-Diene: The Importance of Paralocalisation Energy in Assessing Diene Reactivity. J. Chem. Soc. Perkin Trans. 2 1998, 1499–1506. [Google Scholar]
- (74).Cookson RC; Hudec J; Williams RO Rearrangements of Oxo-Dicyclopentadienes. Tetrahedron Lett. 1960, 1, 29–32. [Google Scholar]
- (75).Schenck GO; Steinmetz R Photofragmentierung von Dehydronorcampher in Cyclopentadien Und Keten Und Photosensibilisierte Synthese Des Pentacyclo[5.2.1.02.6.03.9.04.8] Decans. Chem. Ber. 1963, 96, 520–525. [Google Scholar]
- (76).Dunn GL; Donohue JK The Structure of Thiele’s Ester, a Dimethyl Dicyclopentadienedicarboxylate. Tetrahedron Lett. 1968, 9, 3485–3487. [Google Scholar]
- (77).Marchand AP Synthesis and Chemistry of Homocubanes, Bishomocubanes, and Trishomocubanes. Chem. Rev. 1989, 89, 1011–1033. [Google Scholar]
- (78).Minter DE; Marchand AP; Lu S-P Structural Analysis and Complete Assignment of the 1H and 13C NMR Spectra of Thiele’s Ester. Magn. Reson. Chem. 1990, 28, 623–627. [Google Scholar]
- (79).Vokáč K; Samek Z; Herout V; Šorm F The Structure of Artabsin and Absinthin. Tetrahedron Lett. 1968, 9, 3855–3857. [Google Scholar]
- (80).Walker EE The Effects of Absinthe. Med. Rec. 1906, 30, 568–572. [Google Scholar]
- (81).Zhang W; Luo S; Fang F; Chen Q; Hu H; Jia X; Zhai H Total Synthesis of Absinthin. J. Am. Chem. Soc. 2005, 127, 18–19. [DOI] [PubMed] [Google Scholar]
- (82).Drake NL; Adams JR Some 1,4-Diaryl-1,3-Cyclopentadienes. J. Am. Chem. Soc. 1939, 61, 1326–1329. [Google Scholar]
- (83).Alder K; Flock FH; Hausweiler A; Reeber R Über Die Konstitution Der Cyclopentadien-Carbonsäure Aus Cyclopentadien-Kalium. Chem. Ber. 1954, 87, 1752–1759. [Google Scholar]
- (84).Alder K; Muders R Über Die Konstitution Des Kohlenwasserstoffes von Damsky Und Seine Überführung in 1-Methyl-Santen („ɛ-Fenchen”). Chem. Ber. 1958, 91, 1083–1092. [Google Scholar]
- (85).Peters D 350. Cyclopentadienecarboxylic Acid. The Structure of the Monomer and the Dimers. J. Chem. Soc. 1959, 1761–1765. [Google Scholar]
- (86).Neureiter N Pyrolysis of 1,1-Dichloro-2-Vinylcyclopropane Synthesis of 2-Chlorocyclopentadiene. J. Org. Chem. 1959, 24, 2044–2046. [Google Scholar]
- (87).Riemschneider R; Nerin R Substitutionsprodukte Des Cyclopentadiens, 9. Mitt.: Phenylcyclopentadien. Monatsh. Chem. 1960, 91, 829–835. [Google Scholar]
- (88).Csicsery SM Methylcyclopentadiene Isomers. J. Org. Chem. 1960, 25, 518–521. [Google Scholar]
- (89).Mironov VA; Sobolev EV; Elizarova AN Substituted Cyclopent Adienes and Related Compounds: Communication 10. The Three Isomeric Methylcyclopentadienes. Russ. Chem. Bull. 1963, 12, 1467–1474. [Google Scholar]
- (90).Mironov VA; Sobolev EV; Elizarova AN Some General Characteristic Properties of Substituted Cyclopentadienes. Tetrahedron 1963, 19, 1939–1958. [Google Scholar]
- (91).McLean S; Webster CJ; Rutherford RJD Kinetic Isotope Effect for the Thermally-Induced Migration of Hydrogen in Cyclopentadienes. Can. J. Chem. 1969, 47, 1555–1559. [Google Scholar]
- (92).Mironov VA; Akhrem AA Kinetics of the Thermal Isomerization of 2-Methyl-1,3-Cyclopentadiene. Russ. Chem. Bull. 1972, 21, 1791–1792. [Google Scholar]
- (93).Roth WR; König J [Hydrogen Displacements. IV. Kinetic Isotope Effect of the 1.5 Hydrogen Displacement in cis-Pentadiene-(1.3)]. Justus Liebigs Ann. Chem. 1966, 699, 24–32. [DOI] [PubMed] [Google Scholar]
- (94).Alabugin IV; Manoharan M; Breiner B; Lewis FD Control of Kinetics and Thermodynamics of [1,5]-Shifts by Aromaticity: A View through the Prism of Marcus Theory. J. Am. Chem. Soc. 2003, 125, 9329–9342. [DOI] [PubMed] [Google Scholar]
- (95).Jiao H; Schleyer P. v. R. Aromaticity of Pericyclic Reaction Transition Structures: Magnetic Evidence. J. Phys. Org. Chem 1998, 11, 655–662. [Google Scholar]
- (96).Rondan NG; Houk KN Transition Structures for 1,5-Sigmatropic Hydrogen Shifts of 1,3-Pentadiene and Cyclopentadiene. Tetrahedron Lett. 1984, 25, 2519–2522. [Google Scholar]
- (97).Jiao H; Schleyer P. v. R. Introductory Lecture. Electrostatic Acceleration of the 1,5-H Shifts in Cyclopentadiene and in Penta-1,3-Diene by Li Complexation: Aromaticity of the Transition Structures. J. Chem. Soc. Faraday Trans. 1994, 90, 1559–1567. [Google Scholar]
- (98).Houk KN; Li Y; Evanseck JD Transition Structures of Hydrocarbon Pericyclic Reactions. Angew. Chem., Int. Ed. Engl. 1992, 31, 682–708. [Google Scholar]
- (99).Kahn SD; Hehre WJ; Rondan NG; Houk KN Nature of the Migrating Group in 1,5-Sigmatropic Hydrogen Shifts. J. Am. Chem. Soc. 1985, 107, 8291–8292. [Google Scholar]
- (100).McLean S; Haynes P Hydrogen Migration in Cyclopentadienes. Tetrahedron 1965, 21, 2329–2342. [Google Scholar]
- (101).Borodkin GI; Susharin ER; Shakirov MM; Shubin VG Degenerate Sigmatropic Rearrangement of 1,2,3,4,5-Pentamethyl-1,3-Cyclopentadiene. Zh. Org. Khim. 1985, 21, 1809–1819. [Google Scholar]
- (102).Ashe AJ Hydrogen and Trimethylsilyl Migrations in 5-(Trimethylsilyl) Cyclopentadiene. J. Am. Chem. Soc. 1970, 92, 1233–1235. [Google Scholar]
- (103).Sergeyev NM; Avramenko GI; Kisin AV; Korenevsky VA; Ustynyuk YA Nuclear Magnetic Resonance Spectroscopy of Metal Cyclopentadienyls V. Metallotropic Rearrangement in Silicon Cyclopentadienyls: 1,2 Shift and Spectral Regularities in σ-Cyclopentadienyls. J. Organomet. Chem. 1971, 32, 55–77. [Google Scholar]
- (104).Borodkin GI, Susharin ER, and Shubin VG. Degenerate Rearrangement of 1,2,3,4,5,5-Hexamethyl-1,3-Cyclopentadiene. Zh. Org. Khim. 19, 1004–1010. [Google Scholar]
- (105).Clarke J; Fowler PW; Gronert S; Keeffe JR Effect of Ring Size and Migratory Groups on [1,n] Suprafacial Shift Reactions. Confirmation of Aromatic and Antiaromatic Transition-State Character by Ring-Current Analysis. J. Org. Chem. 2016, 81, 8777–8788. [DOI] [PubMed] [Google Scholar]
- (106).Bushby RJ; Jones DW Rapid Circumambulation of the Formyl Group in 5-Formyl-1,2,3,4,5-Pentamethylcyclopenta-1,3-Diene. J. Chem. Soc., Chem. Commun. 1979, 688–690. [Google Scholar]
- (107).Minkin VI, Minyaev RM, Yudilevich IA, Kletskii ME. Theoretical Study of the Carousel Rearrangement of 5-Formyl-1,3-Cyclopentadiene. Zh. Org. Khim. 1988, 24, 2241. [Google Scholar]
- (108).de Mayo P; Williams RE Sativene, Parent of the Toxin from Helminthosporium sativum. J. Am. Chem. Soc. 1965, 87, 3275. [DOI] [PubMed] [Google Scholar]
- (109).Dev S Aspects of Longifolene Chemistry. An Example of Another Facet of Natural Products Chemistry. Acc. Chem. Res. 1981, 14, 82–88. [Google Scholar]
- (110).Beechan CM; Djerassi C; Eggert H Terpenoids–LXXIV. Tetrahedron 1978, 34, 2503–2508. [Google Scholar]
- (111).Brieger G Synthetic Transformation of Natural Products. I. 1-Methylene-2,10-Methano-5,5,9-Trimethyldecahydronaphthalene. J. Am. Chem. Soc. 1963, 85, 3783–3784. [Google Scholar]
- (112).Glass RS; Herzog JD; Sobczak RL Preparation and Reactions of 5-Alkylpentachloro-1,3-Cyclopentadienes. Application to Sesquiterpene Synthesis. J. Org. Chem. 1978, 43, 3209–3214. [Google Scholar]
- (113).Stille JR; Grubbs RH Intramolecular Diels–Alder Reaction of α,β-Unsaturated Ester Dienophiles with Cyclopentadiene and the Dependence on Tether Length. J. Org. Chem. 1989, 54, 434–444. [Google Scholar]
- (114).Bo L; Fallis AG Direct Total Synthesis of (+)-Longifolene via an Intramolecular Diels–Alder Strategy. J. Am. Chem. Soc. 1990, 112, 4609–4610. [Google Scholar]
- (115).Antczak K; Kingston JF; Fallis AG Stereoselective Total Syntheses of (±)-Sinularene and (±)-5-Epi-Sinularene: A General Intramolecular Diels–Alder Approach to Tricyclic Sesquiterpenes. Can. J. Chem. 1985, 63, 993–995. [Google Scholar]
- (116).Gallacher G; Ng AS; Attah-Poku SK; Antczak K; Alward SJ; Kingston JF; Fallis AG Studies of Intramolecular Diels–Alder Reactions: Preparation of Methyl spiro[2,4]hepta-4,6-Dien-1-yl Esters and Their Internal Cycloaddition Reactivity. Can. J. Chem. 1984, 62, 1709–1716. [Google Scholar]
- (117).Attah-Poku SK; Antczak K; Alward SJ; Fallis AG A General Synthesis of the tricyclo[4.4.01,6.05,9]decane (sinularene) and tricyclo[5.4.01,7.06,10]undecane (longifolene) Ring Systems by Selective Cleavage of Bridged-Ring Cyclopropyl Ketones. Can. J. Chem. 1984, 62, 1717–1721. [Google Scholar]
- (118).Antczak K; Kingston JF; Fallis AG; Hanson AW A General Intramolecular Diels–Alder Approach to Tricyclic Sesquiterpenes: Stereoselective Total Syntheses of (±)-Sinularene and (±)-5-Epi-Sinularene. Can. J. Chem. 1987, 65, 114–123. [Google Scholar]
- (119).Mellor JM; Webb CF Stereochemistry of the Diels–Alder Reaction: Reaction of Dienophiles with Cyclopentadiene and Methylcyclopentadiene. J. Chem. Soc., Perkin Trans. 2 1974, 26–31. [Google Scholar]
- (120).Corey EJ; Weinshenker NM; Schaaf TK; Huber W Stereo-Controlled Synthesis of Prostaglandins F2α and E2 (dl). J. Am. Chem. Soc. 1969, 91, 5675–5677. [DOI] [PubMed] [Google Scholar]
- (121).Corey EJ; Ensley HE Letter: Preparation of an Optically Active Prostaglandin Intermediate via Asymmetric Induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [DOI] [PubMed] [Google Scholar]
- (122).Ohkita M; Tsuji T; Nishida S Reaction of Cyclopent-2-En-1-One Ethylene Acetal with Dienophiles via Its Ring-Opened Enol Ether Form. Single Step Synthesis of Norbornan-2-One Ethylene Acetals. J. Chem. Soc., Chem. Commun. 1991, 37–38. [Google Scholar]
- (123).Ohkita M; Nishizawa O; Tsuji T; Nishida S Single Step Synthesis of Norbornan-2-One Dithioacetals from Cyclopent-2-En-1-One Dithioacetals and Dienophiles. J. Chem. Soc., Chem. Commun. 1993, 1676–1677. [Google Scholar]
- (124).Ohkita M; Nishizawa O; Tsuji T; Nishida S A One-Step Synthesis of 2-Norbornanone Ethylene Acetals from 2-Cyclopenten-1-One Ethylene Acetals and Dienophiles via [2 + 4] Cycloaddition of in Situ Generated 2-(2-Hydroxyethoxy)cyclopenta-1,3-Dienes and Intramolecular Reacetalization. J. Org. Chem. 1993, 58, 5200–5208. [Google Scholar]
- (125).Ohkita M; Kawai H; Tsuji T Intramolecular Diels–Alder Reaction of Cyclopenta-1,3-Diene Derivatives Generated in Situ from 4-(Pent-4-Enyl)cyclopent-2-Enone Ethylene Ketals. J. Chem. Soc. Perkin 1 2002, 366–370. [Google Scholar]
- (126).Hudon J; Cernak TA; Ashenhurst JA; Gleason JL Stable 5-Substituted Cyclopentadienes for the Diels–Alder Cycloaddition and Their Application to the Synthesis of Palau’amine. Angew. Chem., Int. Ed. 2008, 47, 8885–8888. [DOI] [PubMed] [Google Scholar]
- (127).Schulé A; Liang H; Vors J-P; Ciufolini MA Synthetic Studies toward Sordarin: Building Blocks for the Terpenoid Core and for Analogues Thereof. J. Org. Chem. 2009, 74, 1587–1597. [DOI] [PubMed] [Google Scholar]
- (128).Wu Y; Dockendorff C Synthesis of a Novel Bicyclic Scaffold Inspired by the Antifungal Natural Product Sordarin. Tetrahedron Lett. 2018, 59, 3373–3376. [Google Scholar]
- (129).Richert C; Krupp F; He S; Frey W A Crystalline Ready-to-Use Form of Cyclopentadiene. Synlett 2018, 29, 1707–1710. [Google Scholar]
- (130).Sauer J; Heinrichs G Kinetik Und Umsetzungen von 1.2.4.5-Tetrazinen Mit Winkelgespannten Und Elektronenreichen Doppelbindungen. Tetrahedron Lett. 1966, 7, 4979–4984. [Google Scholar]
- (131).Dalkılıç E; Güney M; Daştan A; Saracoglu N; Lucchi OD; Fabris F Novel and Versatile Protocol for the Preparation of Functionalized Benzocyclotrimers. Tetrahedron Lett. 2009, 50, 1989–1991. [Google Scholar]
- (132).Dalkılıç E; Daştan A Synthesis of Cyclopentadiene Derivatives by Retro-Diels–Alder Reaction of Norbornadiene Derivatives. Tetrahedron 2015, 71, 1966–1970. [Google Scholar]
- (133).Kennedy JP; Castner KF Thermally Reversible Polymer Systems by Cyclopentadienylation. I. A Model for Termination by Cyclopentadienylation of Olefin Polymerization. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 2039–2054. [Google Scholar]
- (134).Kennedy JP; Castner KF Thermally Reversible Polymer Systems by Cyclopentadienylation. II. The Synthesis of Cyclopentadiene-Containing Polymers. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 2055–2070. [Google Scholar]
- (135).Grubbs RH; Gibbons C; Kroll LC; Bonds WD; Brubaker CH Activation of Homogeneous Catalysts by Polymer Attachment. J. Am. Chem. Soc. 1973, 95, 2373–2375. [Google Scholar]
- (136).Bonds WD; Brubaker CH; Chandrasekaran ES; Gibbons C; Grubbs RH; Kroll LC Polystyrene Attached Titanocene Species. Preparation and Reactions. J. Am. Chem. Soc. 1975, 97, 2128–2132. [Google Scholar]
- (137).Freeman BH; Gagan JMF; Lloyd D Preparation of Cyclopentadienes and Diazocyclopentadienes via Cyclopentenolones and Cyclopentenones. Tetrahedron 1973, 29, 4307–4312. [Google Scholar]
- (138).Nishina Y; Tatsuzaki T; Tsubakihara A; Kuninobu Y; Takai K Synthesis of Multisubstituted Cyclopentadienes from Cyclopentenones Prepared via Catalytic Double Aldol Condensation and Nazarov Reaction Sequence. Synlett 2011, 2585–2589. [Google Scholar]
- (139).Shearer AS; de Miguel YR The Synthesis of Resin-Bound Tetramethylcyclopentadienes: An Evaluation of Two Methodologies. Tetrahedron Lett. 2006, 47, 447–450. [Google Scholar]
- (140).Leadbeater NE Facile Synthesis of Polymer-Supported Cyclopentadienes. Tetrahedron Lett. 2002, 43, 691–693. [Google Scholar]
- (141).Inglis AJ; Paulöhrl T; Barner-Kowollik C Ambient Temperature Synthesis of a Versatile Macromolecular Building Block: Cyclopentadienyl-Capped Polymers. Macromolecules 2010, 43, 33–36. [Google Scholar]
- (142).Stille JK; Plummer L Polymerization by the Diels–Alder Reaction. J. Org. Chem. 1961, 26, 4026–4029. [Google Scholar]
- (143).Murphy EB; Bolanos E; Schaffner-Hamann C; Wudl F; Nutt SR; Auad ML Synthesis and Characterization of a Single-Component Thermally Remendable Polymer Network: Staudinger and Stille Revisited. Macromolecules 2008, 41, 5203–5209. [Google Scholar]
- (144).Haque FM; Grayson SM The Synthesis, Properties and Potential Applications of Cyclic Polymers. Nat. Chem. 2020, 12, 433–444. [DOI] [PubMed] [Google Scholar]
- (145).Glassner M; Blinco JP; Barner-Kowollik C Diels–Alder Reactions as an Efficient Route to High Purity Cyclic Polymers. Macromol. Rapid Commun. 2011, 32, 724–728. [DOI] [PubMed] [Google Scholar]
- (146).Glassner M; Kempe K; Schubert US; Hoogenboom R; Barner-Kowollik C One-Pot Synthesis of Cyclopentadienyl Endcapped poly(2-Ethyl-2-Oxazoline) and Subsequent Ambient Temperature Diels–Alder Conjugations. Chem. Commun. 2011, 47, 10620–10622. [DOI] [PubMed] [Google Scholar]
- (147).Amant AHS; St. Amant AH; Discekici EH; Bailey SJ; Zayas MS; Song J-A; Shankel SL; Nguyen SN; Bates MW; Anastasaki A; Hawker CJ; de Alaniz JR Norbornadienes: Robust and Scalable Building Blocks for Cascade “Click” Coupling of High Molecular Weight Polymers. J. Am. Chem. Soc. 2019, 141, 13619–13624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Bailey SJ; Discekici EH; Barbon SM; Nguyen SN; Hawker CJ; Read de Alaniz J Norbornadiene Chain-End Functional Polymers as Stable, Readily Available Precursors to Cyclopentadiene Derivatives. Macromolecules 2020, 53, 4917–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Samoshin AV; Hawker CJ; de Alaniz JR Nitrosocarbonyl Hetero-Diels–Alder Cycloaddition: A New Tool for Conjugation. ACS Macro Lett. 2014, 3, 753–757. [DOI] [PubMed] [Google Scholar]
- (150).Peterson GI; Church DC; Yakelis NA; Boydston AJ 1,2-Oxazine Linker as a Thermal Trigger for Self-Immolative Polymers. Polymer 2014, 55, 5980–5985. [Google Scholar]
- (151).Rohr U; Schatz J; Sauer J Thio- and Selenocarbonyl Compounds as “Superdienophiles” in [4 + 2] Cycloadditions. Eur. J. Org. Chem. 1998, 1998, 2875–2883. [Google Scholar]
- (152).Inglis AJ; Sinnwell S; Stenzel MH; Barner-Kowollik C Ultrafast Click Conjugation of Macromolecular Building Blocks at Ambient Temperature. Angew. Chem., Int. Ed. 2009, 48, 2411–2414. [DOI] [PubMed] [Google Scholar]
- (153).Glassner M; Delaittre G; Kaupp M; Blinco JP; Barner-Kowollik C (Ultra)fast Catalyst-Free Macromolecular Conjugation in Aqueous Environment at Ambient Temperature. J. Am. Chem. Soc. 2012, 134, 7274–7277. [DOI] [PubMed] [Google Scholar]
- (154).Wiberg KB The Concept of Strain in Organic Chemistry. Angew. Chem., Int. Ed. Engl. 1986, 25, 312–322. [Google Scholar]
- (155).Paulöhrl T; Inglis AJ; Barner-Kowollik C Reversible Diels–Alder Chemistry as a Modular Polymeric Color Switch. Adv. Mater. 2010, 22, 2788–2791. [DOI] [PubMed] [Google Scholar]
- (156).Langer M; Brandt J; Lederer A; Goldmann AS; Schacher FH; Barner-Kowollik C Amphiphilic Block Copolymers Featuring a Reversible Hetero Diels–Alder Linkage. Polym. Chem. 2014, 5, 5330–5338. [Google Scholar]
- (157).Inglis AJ; Sinnwell S; Davis TP; Barner-Kowollik C; Stenzel MH Reversible Addition Fragmentation Chain Transfer (RAFT) and Hetero-Diels–Alder Chemistry as a Convenient Conjugation Tool for Access to Complex Macromolecular Designs. Macromolecules 2008, 41, 4120–4126. [Google Scholar]
- (158).Sinnwell S; Synatschke CV; Junkers T; Stenzel MH; Barner-Kowollik C A Study into the Stability of 3,6-Dihydro-2H-Thiopyran Rings: Key Linkages in the RAFT Hetero-Diels–Alder Click Concept. Macromolecules 2008, 41, 7904–7912. [Google Scholar]
- (159).De Bruycker K; Billiet S; Houck HA; Chattopadhyay S; Winne JM; Du Prez FE Triazolinediones as Highly Enabling Synthetic Tools. Chem. Rev. 2016, 116, 3919–3974. [DOI] [PubMed] [Google Scholar]
- (160).Cookson RC; Gilani SSH; Stevens IDR 4-Phenyl-1,2,4-Triazolin-3,5-Dione: A Powerful Dienophile. Tetrahedron Lett. 1962, 3, 615–618. [Google Scholar]
- (161).Kiselev VD; Kornilov DA; Lekomtseva II; Konovalov AI Reactivity of 4-Phenyl-1,2,4-Triazoline-3,5-Dione and Diethylazocarboxylate in [4 + 2]-Cycloaddition and Ene Reactions: Solvent, Temperature, and High-Pressure Influence on the Reaction Rate. Int. J. Chem. Kinet. 2015, 47, 289–301. [Google Scholar]
- (162).Billiet S; De Bruycker K; Driessen F; Goossens H; Van Speybroeck V; Winne JM; Du Prez FE Triazolinediones Enable Ultrafast and Reversible Click Chemistry for the Design of Dynamic Polymer Systems. Nat. Chem. 2014, 6, 815–821. [DOI] [PubMed] [Google Scholar]
- (163).Butler GB; Williams AG Low-Temperature Modification of Dienic Polymers via the “Ene” Reaction with 4-Substituted-1,2,4-Triazoline-3,5-Diones. J. Polym. Sci. Polym. Chem. Ed. 1979, 17, 1117–1128. [Google Scholar]
- (164).de Lucca Freitas L; Jacobi MM; Gonçalves G; Stadler R Microphase Separation Induced by Hydrogen Bonding in a Poly(1,4-Butadiene)-Block-poly(1,4-Isoprene) Diblock Copolymer–An Example of Supramolecular Organization via Tandem Interactions. Macromolecules 1998, 31, 3379–3382. [Google Scholar]
- (165).Chen X; Ruckenstein E Thermally Reversible Linking of Halide-Containing Polymers by Potassium Dicyclopentadienedicarboxylate. J. Polym. Sci. Pol. Chem. 1999, 37, 4390–4401. [Google Scholar]
- (166).Ruckenstein E; Chen X Crosslinking of Chlorine-Containing Polymers by Dicyclopentadiene Dicarboxylic Salts. J. Polym. Sci. Pol. Chem. 2000, 38, 818–825. [Google Scholar]
- (167).Chen X; Ruckenstein E Thermally Reversible Covalently Bonded Linear Polymers Prepared from a Dihalide Monomer and a Salt of Dicyclopentadiene Dicarboxylic Acid. J. Polym. Sci. Pol. Chem. 2000, 38, 1662–1672. [Google Scholar]
- (168).Murphy EB; Wudl F The World of Smart Healable Materials. Prog. Polym. Sci. 2010, 35, 223–251. [Google Scholar]
- (169).Liu Y-L; Chuo T-W Self-Healing Polymers Based on Thermally Reversible Diels–Alder Chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar]
- (170).Miura M; Akutsu F; Usui T; Ikebukuro Y; Nagakubo K Soluble Cyclopentadienylated Polymers. Makromol. Chem. 1985, 186, 473–481. [Google Scholar]
- (171).Kroto HW; Heath JR; O’Brien SC; Curl RF; Smalley RE C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar]
- (172).Brabec CJ; Gowrisanker S; Halls JJM; Laird D; Jia S; Williams SP Polymer–Fullerene Bulk-Heterojunction Solar Cells. Adv. Mater. 2010, 22, 3839–3856. [DOI] [PubMed] [Google Scholar]
- (173).Wudl F; Hirsch A; Khemani KC; Suzuki T; Allemand P-M, P.; Koch A; Eckert H; Srdanov G; Webb HM Survey of Chemical Reactivity of C60, Electrophile and Dieno—Polarophile Par Excellence. ACS Symp. Ser. 1992, 161–175. [Google Scholar]
- (174).Rotello VM; Howard JB; Yadav T; Morgan Conn M; Viani E; Giovane LM; Lafleur AL Isolation of Fullerene Products from Flames: Structure and Synthesis of the C60-Cyclopentadiene Adduct. Tetrahedron Lett. 1993, 34, 1561–1562. [Google Scholar]
- (175).Giovane LM; Barco JW; Yadav T; Lafleur AL; Marr JA; Howard JB; Rotello VM Kinetic Stability of the Fullerene C60–Cyclopentadiene Diels–Alder Adduct. J. Phys. Chem. 1993, 97, 8560–8561. [Google Scholar]
- (176).Tsuda M; Ishida T; Nogami T; Kurono S; Ohashi M Isolation and Characterization of Diels–Alder Adducts of C60 with Anthracene and Cyclopentadiene. J. Chem. Soc., Chem. Commun. 1993, 1296–1298. [Google Scholar]
- (177).Meidine MF; Roers R; John Langley G; Avent AG; Darwish AD; Firth S; Kroto HW; Taylor R; Walton DRM Formation and Stabilisation of the Hexa-Cyclopentadiene Adduct of C60. J. Chem. Soc., Chem. Commun. 1993, No. 17, 1342. [Google Scholar]
- (178).Khan SI; Oliver AM; Paddon-Row MN; Rubin Y Synthesis of a Rigid “Ball-and-Chain” Donor–Acceptor System through Diels–Alder Functionalization of Buckminsterfullerene (C60). J. Am. Chem. Soc. 1993, 115, 4919–4920. [Google Scholar]
- (179).Zhao Y; Truhlar DG The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- (180).Cui C-X; Liu Y-J Role of Encapsulated Metal Cation in the Reactivity and Regioselectivity of the C60 Diels–Alder Reaction. J. Phys. Chem. A. 2015, 119, 3098–3106. [DOI] [PubMed] [Google Scholar]
- (181).Yang T; Fukuda R; Cammi R; Ehara M Diels–Alder Cycloaddition of Cyclopentadiene and C60 at the Extreme High Pressure. J. Phys. Chem. A. 2017, 121, 4363–4371. [DOI] [PubMed] [Google Scholar]
- (182).Villegas-Escobar N; Poater A; Solà M; Schaefer HF; Toro-Labbé A Decomposition of the Electronic Activity in Competing [5,6] and [6,6] Cycloaddition Reactions between C60 and Cyclopentadiene. Phys. Chem. 2019, 21, 5039–5048. [DOI] [PubMed] [Google Scholar]
- (183).Fernández I; Solà M; Bickelhaupt FM Why Do Cycloaddition Reactions Involving C60 Prefer [6,6] over [5,6] Bonds? Chemistry 2013, 19, 7416–7422. [DOI] [PubMed] [Google Scholar]
- (184).Pang LSK; Wilson MA Reactions of Fullerenes C60 and C70 with Cyclopentadiene. J. Phys. Chem. 1993, 97, 6761–6763. [Google Scholar]
- (185).Dewar MJS; Zoebisch EG; Healy EF; Stewart JJP Development and Use of Quantum Mechanical Molecular Models. 76. AM1: A New General Purpose Quantum Mechanical Molecular Model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar]
- (186).Manoharan M; De Proft F; Geerlings P Aromaticity Interplay between Quinodimethanes and C60 in Diels–Alder Reactions: Insights from a Theoretical Study. J. Org. Chem. 2000, 65, 6132–6137. [DOI] [PubMed] [Google Scholar]
- (187).Miller GP; Tetreau MC Facile, Completely Regioselective 1,4-Hydrogenations of C60-Diaryltetrazine Monoadducts. Org. Lett. 2000, 2, 3091–3094. [DOI] [PubMed] [Google Scholar]
- (188).Tetreau MC Diels–Alder Reactions of [60]Fullerene with 1,2,4,5-Tetrazines and Additions to [60]Fullerene–Tetrazine Monoadducts, Ph.D. Thesis, University of New Hampshire, 2002. [Google Scholar]
- (189).Herndon WC; Cooper WB Jr.; Chambers MJ Retro-Diels–Alder Reactions. II. Gas-Phase Thermal Decomposition of Bicyclo[2.2.1]hept-2-Ene. J. Phys. Chem. 1964, 68, 2016–2018. [Google Scholar]
- (190).Nie B; Rotello VM Nonchromatographic Purification of Fullerenes via Reversible Addition to Silica-Supported Dienes. J. Org. Chem. 1996, 61, 1870–1871. [DOI] [PubMed] [Google Scholar]
- (191).Merrifield RB Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]
- (192).Guhr KI; Greaves MD; Rotello VM Reversible Covalent Attachment of C60 to a Polymer Support. J. Am. Chem. Soc. 1994, 116, 5997–5998. [Google Scholar]
- (193).Nie B; Rotello V Attachment of Fullerenes to Materials: The Importance of Backbone–Fullerene Interactions. J. Phys. Chem. Solids 1997, 58, 1897–1899. [Google Scholar]
- (194).Ge Z; Duchamp JC; Cai T; Gibson HW; Dorn HC Purification of Endohedral Trimetallic Nitride Fullerenes in a Single, Facile Step. J. Am. Chem. Soc. 2005, 127, 16292–16298. [DOI] [PubMed] [Google Scholar]
- (195).Ruoff RS; Tse DS; Malhotra R; Lorents DC Solubility of Fullerene (C60) in a Variety of Solvents. J. Phys. Chem. 1993, 97, 3379–3383. [Google Scholar]
- (196).Heymann D Solubility of C60 in Alcohols and Alkanes. Carbon 1996, 34, 627–631. [Google Scholar]
- (197).Beck MT; Mándi G Solubility of C60. Fuller. Sci. Technol. 1997, 5, 291–310. [Google Scholar]
- (198).Bezmel’nitsyn VN; Eletskii AV; Okun’ MV Fullerenes in Solutions. PHYS-USP 1998, 41, 1091–1114. [Google Scholar]
- (199).Nakamura E; Isobe H Functionalized Fullerenes in Water. The First 10 Years of Their Chemistry, Biology, and Nanoscience. Acc. Chem. Res. 2003, 36, 807–815. [DOI] [PubMed] [Google Scholar]
- (200).Hansen CM; Smith AL Using Hansen Solubility Parameters to Correlate Solubility of C60 Fullerene in Organic Solvents and in Polymers. Carbon 2004, 42, 1591–1597. [Google Scholar]
- (201).Nebhani L; Barner-Kowollik C Functionalization of Fullerenes with Cyclopentadienyl and Anthracenyl Capped Polymeric Building Blocks via Diels–Alder Chemistry. Macromol. Rapid Commun. 2010, 31, 1298–1305. [DOI] [PubMed] [Google Scholar]
- (202).Wilson SR; Yurchenko ME; Schuster DI; Khong A; Saunders M Synthesis of Fluorous Fullerene Adducts: Reversible Solubilization of Fullerenes in Perfluorinated Solvents. J. Org. Chem. 2000, 65, 2619–2623. [DOI] [PubMed] [Google Scholar]
- (203).Kawakami H; Okada H; Matsuo Y Efficient Diels–Alder Addition of Cyclopentadiene to Lithium Ion Encapsulated [60]Fullerene. Org. Lett. 2013, 15, 4466–4469. [DOI] [PubMed] [Google Scholar]
- (204).Ueno H; Kawakami H; Nakagawa K; Okada H; Ikuma N; Aoyagi S; Kokubo K; Matsuo Y; Oshima T Kinetic Study of the Diels–Alder Reaction of Li+@C60 with Cyclohexadiene: Greatly Increased Reaction Rate by Encapsulated Li+. J. Am. Chem. Soc. 2014, 136, 11162–11167. [DOI] [PubMed] [Google Scholar]
- (205).Maeda Y; Miyashita J; Hasegawa T; Wakahara T; Tsuchiya T; Nakahodo T; Akasaka T; Mizorogi N; Kobayashi K; Nagase S; Kato T; Ban N; Nakajima H; Watanabe Y Reversible and Regioselective Reaction of La@C82 with Cyclopentadiene. J. Am. Chem. Soc. 2005, 127, 12190–12191. [DOI] [PubMed] [Google Scholar]
- (206).Meidine MF; Avent AG; Darwish AD; Kroto HW; Ohashi O; Taylor R; Walton DRM Pentamethylcyclopentadiene Adducts of [60]- and [70]-Fullerene. J. Chem. Soc. Perkin Trans. 2 1994, No. 6, 1189–1193. [Google Scholar]
- (207).Maeda Y; Sato S; Inada K; Nikawa H; Yamada M; Mizorogi N; Hasegawa T; Tsuchiya T; Akasaka T; Kato T; Slanina Z; Nagase S Regioselective Exohedral Functionalization of La@C82 and Its 1,2,3,4,5-Pentamethylcyclopentadiene and Adamantylidene Adducts. Chemistry 2010, 16, 2193–2197. [DOI] [PubMed] [Google Scholar]
- (208).Garcia-Borràs M; Luis JM; Swart M; Solà M Diels–Alder and Retro-Diels–Alder Cycloadditions of (1,2,3,4,5-Pentamethyl)cyclopentadiene to La@C2v-C82: Regioselectivity and Product Stability. Chemistry 2013, 19, 4468–4479. [DOI] [PubMed] [Google Scholar]
- (209).Zydziak N; Hübner C; Bruns M; Barner-Kowollik C One-Step Functionalization of Single-Walled Carbon Nanotubes (SWCNTs) with Cyclopentadienyl-Capped Macromolecules via Diels−Alder Chemistry. Macromolecules 2011, 44, 3374–3380. [Google Scholar]
- (210).Yameen B; Rodriguez-Emmenegger C; Ahmed I; Preuss CM; Dürr CJ; Zydziak N; Trouillet V; Fruk L; Barner-Kowollik C A Facile One-Pot Route to Poly(Carboxybetaine Acrylamide) Functionalized SWCNTs. Chem. Commun. 2013, 49, 6734–6736. [DOI] [PubMed] [Google Scholar]
- (211).Zydziak N; Preuss CM; Winkler V; Bruns M; Hübner C; Barner-Kowollik C Hetero Diels–Alder Chemistry for the Functionalization of Single-Walled Carbon Nanotubes with Cyclopentadienyl End-Capped Polymer Strands. Macromol. Rapid Commun. 2013, 34, 672–680. [DOI] [PubMed] [Google Scholar]
- (212).Marshall MW; Popa-Nita S; Shapter JG Measurement of Functionalised Carbon Nanotube Carboxylic Acid Groups Using a Simple Chemical Process. Carbon 2006, 44, 1137–1141. [Google Scholar]
- (213).Pumera M; Smíd B; Veltruská K Influence of Nitric Acid Treatment of Carbon Nanotubes on Their Physico-Chemical Properties. J. Nanosci. Nanotechnol. 2009, 9, 2671–2676. [DOI] [PubMed] [Google Scholar]
- (214).Allen MJ; Tung VC; Kaner RB Honeycomb Carbon: A Review of Graphene. Chem. Rev. 2010, 110, 132–145. [DOI] [PubMed] [Google Scholar]
- (215).Sarkar S; Bekyarova E; Niyogi S; Haddon RC Diels–Alder Chemistry of Graphite and Graphene: Graphene as Diene and Dienophile. J. Am. Chem. Soc. 2011, 133, 3324–3327. [DOI] [PubMed] [Google Scholar]
- (216).Haddon RC Chemistry of the Fullerenes: The Manifestation of Strain in a Class of Continuous Aromatic Molecules. Science 1993, 261, 1545–1550. [DOI] [PubMed] [Google Scholar]
- (217).Chen Z; Thiel W; Hirsch A Reactivity of the Convex and Concave Surfaces of Single-Walled Carbon Nanotubes (SWCNTs) towards Addition Reactions: Dependence on the Carbon-Atom Pyramidalization. ChemPhysChem 2003, 4, 93–97. [DOI] [PubMed] [Google Scholar]
- (218).Willocq B; Lemaur V; El Garah M; Ciesielski A; Samorì P; Raquez J-M; Dubois P; Cornil J The Role of Curvature in Diels–Alder Functionalization of Carbon-Based Materials. Chem. Commun. 2016, 52, 7608–7611. [DOI] [PubMed] [Google Scholar]
- (219).Yuan J; Chen G; Weng W; Xu Y One-Step Functionalization of Graphene with Cyclopentadienyl-Capped Macromolecules via Diels–Alder “click” Chemistry. J. Mater. Chem. 2012, 22, 7929–7936. [Google Scholar]
- (220).Bian S; Scott AM; Cao Y; Liang Y; Osuna S; Houk KN; Braunschweig AB Covalently Patterned Graphene Surfaces by a Force-Accelerated Diels–Alder Reaction. J. Am. Chem. Soc. 2013, 135, 9240–9243. [DOI] [PubMed] [Google Scholar]
- (221).Love KR; Seeberger PH Carbohydrate Arrays as Tools for Glycomics. Angew. Chem., Int. Ed. 2002, 41, 3583–3586. [DOI] [PubMed] [Google Scholar]
- (222).Shin I; Park S; Lee M-R Carbohydrate Microarrays: An Advanced Technology for Functional Studies of Glycans. Chemistry 2005, 11, 2894–2901. [DOI] [PubMed] [Google Scholar]
- (223).Monzo A; Guttman A Immobilization Techniques for Mono- and Oligosaccharide Microarrays. QSAR Comb. Sci. 2006, 25, 1033–1038. [Google Scholar]
- (224).Wong LS; Khan F; Micklefield J Selective Covalent Protein Immobilization: Strategies and Applications. Chem. Rev. 2009, 109, 4025–4053. [DOI] [PubMed] [Google Scholar]
- (225).Lin P-C; Weinrich D; Waldmann H Protein Biochips: Oriented Surface Immobilization of Proteins. Macromol. Chem. Phys. 2010, 211, 136–144. [Google Scholar]
- (226).Palomo JM Diels–Alder Cycloaddition in Protein Chemistry. Eur. J. Org. Chem. 2010, 2010, 6303–6314. [Google Scholar]
- (227).Escorihuela J; Marcelis ATM; Zuilhof H Click Chemistry: Metal-Free Click Chemistry Reactions on Surfaces. Adv. Mater. Interfaces 2015, 2, 1500135. [Google Scholar]
- (228).Yousaf MN; Mrksich M Diels–Alder Reaction for the Selective Immobilization of Protein to Electroactive Self-Assembled Monolayers. J. Am. Chem. Soc. 1999, 121, 4286–4287. [Google Scholar]
- (229).Houseman BT; Huh JH; Kron SJ; Mrksich M Peptide Chips for the Quantitative Evaluation of Protein Kinase Activity. Nat. Biotechnol. 2002, 20, 270–274. [DOI] [PubMed] [Google Scholar]
- (230).Houseman BT; Mrksich M Carbohydrate Arrays for the Evaluation of Protein Binding and Enzymatic Modification. Chem. Biol. 2002, 9, 443–454. [DOI] [PubMed] [Google Scholar]
- (231).Houseman BT; Mrksich M Towards Quantitative Assays with Peptide Chips: A Surface Engineering Approach. Trends Biotechnol. 2002, 20, 279–281. [DOI] [PubMed] [Google Scholar]
- (232).de Araújo AD; Palomo JM; Cramer J; Köhn M; Schröder H; Wacker R; Niemeyer C; Alexandrov K; Waldmann H Diels–Alder Ligation and Surface Immobilization of Proteins. Angew. Chem., Int. Ed. 2005, 45, 296–301. [DOI] [PubMed] [Google Scholar]
- (233).Wendeln C; Heile A; Arlinghaus HF; Ravoo BJ Carbohydrate Microarrays by Microcontact Printing. Langmuir 2010, 26, 4933–4940. [DOI] [PubMed] [Google Scholar]
- (234).St Amant AH; Lemen D; Florinas S; Mao S; Fazenbaker C; Zhong H; Wu H; Gao C; Christie RJ; Read de Alaniz J Tuning the Diels–Alder Reaction for Bioconjugation to Maleimide Drug-Linkers. Bioconjugate Chem. 2018, 29, 2406–2414. [DOI] [PubMed] [Google Scholar]
- (235).St Amant AH; Huang F; Lin J; Lemen D; Chakiath C; Mao S; Fazenbaker C; Zhong H; Harper J; Xu W; Patel N; Adams L; Vijayakrishnan B; Howard PW; Marelli M; Wu H; Gao C; Read de Alaniz J; Christie RJ A Reactive Antibody Platform for One-Step Production of Antibody–Drug Conjugates through a Diels–Alder Reaction with Maleimide. Bioconjugate Chem. 2019, 30, 2340–2348. [DOI] [PubMed] [Google Scholar]
- (236).St Amant AH; Huang F; Lin J; Rickert K; Oganesyan V; Lemen D; Mao S; Harper J; Marelli M; Wu H; Gao C; Read de Alaniz J; Christie RJ A Diene-Containing Noncanonical Amino Acid Enables Dual Functionality in Proteins: Rapid Diels–Alder Reaction with Maleimide or Proximity-Based Dimerization. Angew. Chem., Int. Ed. 2019, 58, 8489–8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Sletten EM; Bertozzi CR Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Blackman ML; Royzen M; Fox JM Tetrazine Ligation: Fast Bioconjugation Based on Inverse-Electron-Demand Diels–Alder Reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Wang D; Viennois E; Ji K; Damera K; Draganov A; Zheng Y; Dai C; Merlin D; Wang B A Click-and-Release Approach to CO Prodrugs. Chem. Commun, 2014, 50, 15890–15893. [DOI] [PubMed] [Google Scholar]
- (240).Levandowski BJ; Svatunek D; Sohr B; Mikula H; Houk KN Secondary Orbital Interactions Enhance the Reactivity of Alkynes in Diels–Alder Cycloadditions. J. Am. Chem. Soc. 2019, 141, 2224–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Wang W; Ji X; Du Z; Wang B Sulfur Dioxide Prodrugs: Triggered Release of SO via a Click Reaction. Chem. Commun. 2017, 53, 1370–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Bruins JJ; Westphal AH; Albada B; Wagner K; Bartels L; Spits H; van Berkel WJH; van Delft FL Inducible, Site-Specific Protein Labeling by Tyrosine Oxidation-Strain-Promoted (4 + 2) Cycloaddition. Bioconjugate Chem. 2017, 28, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Bruins JJ; Blanco-Ania D; van der Doef V; van Delft FL; Albada B Orthogonal, Dual Protein Labelling by Tandem Cycloaddition of Strained Alkenes and Alkynes to ortho-Quinones and Azides. Chem. Commun. 2018, 54, 7338–7341. [DOI] [PubMed] [Google Scholar]
- (244).Jendralla H The Influence of Endocyclic Oxygen Atoms on the Cycloaddition Reactivity of trans-Cyclo-Octene. Tetrahedron 1983, 39, 1359–1363. [Google Scholar]
- (245).Binger P; Wedemann P; Goddard R; Brinker UH Cyclopropene: A New Simple Synthesis and Diels–Alder Reactions with Cyclopentadiene and 1,3-Diphenylisobenzofuran. J. Org. Chem. 1996, 61, 6462–6464. [DOI] [PubMed] [Google Scholar]
- (246).Thalhammer F; Wallfahrer U; Sauer J Reaktivität Einfacher Offenkettiger Und Cyclischer Dienophile Bei Diels–Alder-Reaktionen Mit Inversem Elektronenbedarf. Tetrahedron Lett. 1990, 31, 6851–6854. [Google Scholar]
- (247).Eibler E; Burgemeister T; Höcht P; Prantl B; Roßmaier H; Schuhbauer HM; Wiest H; Sauer J Polyhalogenated Cyclopentadienes in [4 + 2] Cycloadditions: Preparative Aspects. Liebigs Ann./Recl. 1997, 1997, 2451–2469. [Google Scholar]
- (248).Levandowski BJ; Gamache RF; Murphy JM; Houk KN Readily Accessible Ambiphilic Cyclopentadienes for Bioorthogonal Labeling. J. Am. Chem. Soc. 2018, 140, 6426–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Harris T; Alabugin IV Strain and Stereoelectronics in Cycloalkyne Click Chemistry. Mendeleev Commun. 2019, 29, 237–248. [Google Scholar]
- (250).Row RD; Prescher JA Constructing New Bioorthogonal Reagents and Reactions. Acc. Chem. Res. 2018, 51, 1073–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Devaraj NK The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Daniels PH; Wong JL; Atwood JL; Canada LG; Rogers RD Unreactive 1-Azadiene and Reactive 2-Azadiene in Diels–Alder Reaction of Pentachloroazacyclopentadienes. J. Org. Chem. 1980, 45, 435–440. [Google Scholar]
- (253).Jung ME; Shapiro JJ Synthesis and Structure Determination of 2-azabicyclo[2.2.1]hept-2-Enes and Their Derivatives. W Effect on Chemical Shifts. J. Am. Chem. Soc. 1980, 102, 7862–7866. [Google Scholar]
- (254).Rammash BK; Gladstone CM; Wong JL Azadiene Chemistry. 6. Competitive Diene and Dienophilic Character of 2,3,4,5,5-Pentachloro-1-Azacyclopentadiene in Diels–Alder Reaction with Conjugated Dienes. J. Org. Chem. 1981, 46, 3036–3040. [Google Scholar]
- (255).Fell JS; Martin BN; Houk KN Origins of the Unfavorable Activation and Reaction Energies of 1-Azadiene Heterocycles Compared to 2-Azadiene Heterocycles in Diels–Alder Reactions. J. Org. Chem. 2017, 82, 1912–1919. [DOI] [PubMed] [Google Scholar]
- (256).Beck K; Höhn A; Hünig S; Prokschy F Azobrücken Aus Azinen, I. Isopyrazole Als Elektronenarme Diene Zur Synthese von 2,3-Diazabicyclo[2.2.1]heptenen. Chem. Ber. 1984, 117, 517–533. [Google Scholar]
- (257).Beck K; Hünig S Protonengesteuerte Gleichgewichtseinstellung Zwischen [4+ + 2]- Und [4 + 2+]-Cycloaddukten, Ein Charakteristisches Beispiel Für Die Sonderstellung Der Diels–Alder-Reaktion Zwischen Zwei 1,3-Dienen. Angew. Chem., Int. Ed. 1987, 99, 694–695. [Google Scholar]
- (258).Beck K; Hünig S; Klärner F-G; Kraft P; Artschwager-Perl U Azobrücken Aus Azinen, VII Diels–Alder-Reaktionen Mit Inversem Elektronenbedarf Zwischen Isopyrazolen Und Cycloalkenen Sowie Cycloalkadienen.—Ein Vergleich von Säure-Katalyse Und Beschleunigung Durch Druck. Chem. Ber. 1987, 120, 2041–2051. [Google Scholar]
- (259).Adam W; Harrer HM; Nau WM; Peters K Electronic Substituent Effects on the Acid-Catalyzed [4+ + 2] Cycloaddition of Isopyrazoles with Cyclopentadiene and the Photochemical and Thermal Denitrogenation of the Resulting 1,4-Diaryl-7,7-Dimethyl-2,3-Diazabicyclo[2.2.1]hept-2-Ene Azoalkanes to Bicyclo[2.1.0]pentanes. J. Org. Chem. 1994, 59, 3786–3797. [Google Scholar]
- (260).Adam W; Ammon H; Nau WM; Peters K 4-Halo-4H-Pyrazoles: Cycloaddition with Cyclopentadiene to Azoalkanes of the 2,3-Diazabicyclo[2.2.1]hept-2-Ene Type versus Electrophilic Addition with Cyclopentene. J. Org. Chem. 1994, 59, 7067–7071. [Google Scholar]
- (261).Levandowski BJ; Abularrage NS; Houk KN; Raines RT Hyperconjugative Antiaromaticity Activates 4-Pyrazoles as Inverse-Electron-Demand Diels–Alder Dienes. Org. Lett. 2019, 21, 8492–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Abularrage NS; Levandowski BJ; Raines RT Synthesis and Diels–Alder Reactivity of 4-Fluoro-4-Methyl-4H-Pyrazoles. Int. J. Mol. Sci. 2020, 21, 3964. [DOI] [PMC free article] [PubMed] [Google Scholar]