

Abstract
Schizophrenia is a complex neuropsychiatric syndrome with a heterogeneous genetic, neurobiological and phenotypic profile. Presently, no objective biological measures, i.e. biomarkers, are available to inform diagnostic or treatment decisions. Neuroimaging is well positioned for biomarker development in schizophrenia, as it may capture phenotypic variations in molecular and cellular disease targets, or in brain circuits. These mechanistically based biomarkers may represent a direct measure of the pathophysiological underpinnings of the disease process, and thus could serve as true intermediate or surrogate endpoints. Effective biomarkers could validate new treatment targets or pathways, predict response, aid in selection of patients for therapy, determine regimens, and provide rationale for personalized treatments.
In this review, we discuss a range of mechanistically plausible neuroimaging biomarker candidates, including dopamine hyperactivity, N-methyl-d-aspartate receptor hypofunction, hippocampal hyperactivity, immune dysregulation, dysconnectivity, and cortical gray matter volume loss. We then focus on the putative neuroimaging biomarkers for disease risk, diagnosis, target engagement and treatment response in schizophrenia. Finally, we highlight areas of unmet needs and discuss strategies to advance biomarker development.
INTRODUCTION
Schizophrenia is a complex neuropsychiatric syndrome with a heterogeneous genetic, neurobiological and phenotypic profile. Presently, no objective biological measures, i.e. biomarkers are available that inform diagnostic or treatment decisions. A biomarker, as outlined by the FDA/NIH Biomarker Working group, is “a characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions” (1). Neuroimaging is a strong candidate for biomarker development in schizophrenia. Imaging can capture phenotypic variations in molecular and cellular disease targets, or in brain circuits that are a unique representation of gene-environment interactions and are associated with behavioral alterations (2). It offers versatility in terms of measuring multiple pathophysiological mechanisms, including brain structural integrity deficits, functional dysconnectivity, and altered neurotransmitter systems (Figure 1) (3). For a biomarker to be practically useful, it must be a proxy of a clinically relevant measure. It needs to have an acceptable level of sensitivity, specificity and predictive value (4). Ideally, it will also be easily quantifiable and cost effective. In a 2012 consensus report, the American Psychiatric Association Work Group on Neuroimaging Markers of Psychiatric Disorders suggested a number of criteria that should be met in order to establish validity of a neuroimaging biomarker. A diagnostic biomarker should have a sensitivity of >80% for detecting a particular psychiatric disorder, a specificity of >80% to distinguish this disorder from other psychiatric disorders and a positive predictive value that approaches about 90%. Further, the data used to establish a biomarker should require confirmation by at least two independent sets of qualified investigators with results published in peer-reviewed journals (5). To date, no neuroimaging biomarker has met these criteria. Nonetheless, a number of studies have made progress towards the goal of biomarker development in schizophrenia.
Figure 1:

A brief introduction to relevant terms in neuroimaging.
Mechanistically plausible targets for biomarker development in schizophrenia
Mechanistically based biomarkers represent a direct measure of the pathophysiological underpinnings of the disease process (6) and thus can serve as true intermediate or surrogate endpoints (Figure 2). These biomarkers can validate new treatment targets or pathways, predict treatment response, aid in selection of patients for therapy, determine therapeutic regimens and provide rationale for personalized treatments (7). Ideally, biomarker development targets would reflect fundamental neurobiological alterations, have analogues in preclinical models, correlate with measures of clinical symptom severity, and be consistent with models of disease pathology (8). Here, we selectively review mechanistically plausible biomarker development targets meeting these criteria (Table).
Figure 2: Mechanistically plausible neuroimaging biomarker development targets.
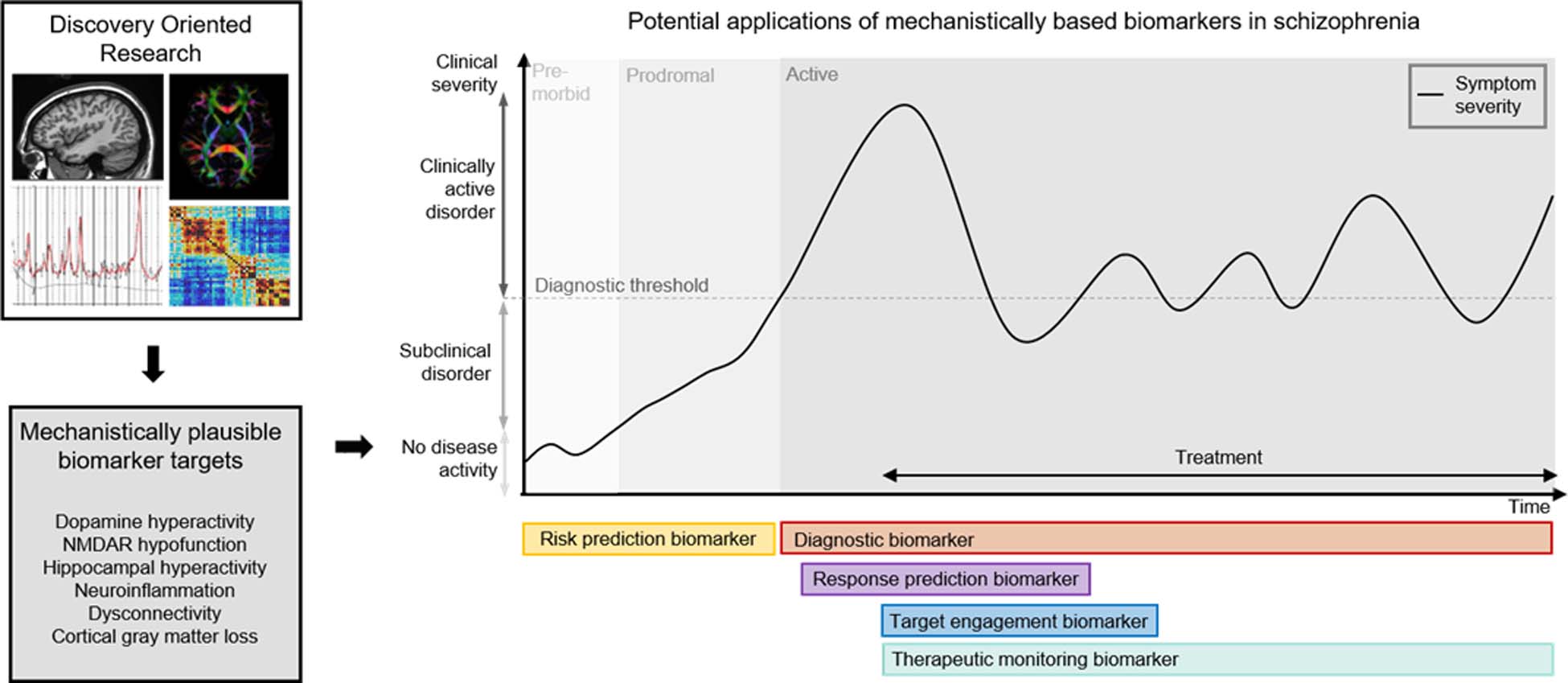
Based on discovery oriented research a number of mechanistically plausible neuroimaging biomarker development targets have been identified. Diagnostic biomarkers aid in the detection of the presence of a disease state. Target engagement biomarkers confirm therapeutic properties of drugs and can be used for determination of appropriate dosing. Treatment response predication biomarkers guide the appropriate choice of treatment. Therapeutic monitoring biomarkers monitor the response to a specific treatment or monitor relapse of a disorder.
Table:
Mechanistically plausible targets for biomarker development in schizophrenia
| Putative Biomarker target | Model of disease pathology | Preclinical model | Association with clinical variables |
|---|---|---|---|
| Dopamine hyperactivity | Dopaminergic hyperactivity in subcortical and limbic brain regions results in positive symptoms | Amphetamine model | Associations are reported between dopamine dysregulation and psychosis severity, D12 receptor occupancy predicts antipsychotic treatment response |
| NMDA receptor hypofunction | NMDA receptor hypofunction GABAergic interneurons causes disinhibition of the glutamatergic pyramidal cell | NMDA receptor antagonist (ketamine, PCP, MK-801) models | Associations are reported between glutamate levels and subsequent treatment response |
| Hippocampal hyperactivity | A hyperglutamatergic state causes hippocampal hyperactivity, which then results in downstream dopamine circuit dysregulation | Methylazoxymethanol acetate (MAM) model | Associations are reported between hippocampal regional cerebral blood flow (rCBF) and psychosis symptom severity as well as treatment response |
| Neuroinflammation/ immune dysregulation | Microglia are primed to act in a hyper-responsive manner and shift to a pro-inflammatory state in response to stress resulting in aberrant neurotransmission and structural injury | Maternal immune activation (MIA) models | Inflammation appears more prominent in early illness stages and in patients with acute psychosis symptom exacerbations |
| Dysconnectivity | Disturbances in the excitation/ inhibition balance result in altered functional network architecture | NMDA receptor antagonist models, fractalkine knockout model | Associations are reported between dysconnectivity and psychosis symptom severity as well as treatment response |
| Cortical gray matter loss | Abnormalities in brain maturational processes mediated by environmental factors results in gray matter loss | Sub-chronic PCP antagonism, MIA models | Associations are reported between gray matter loss and disease severity and are linked to overall poor outcomes |
Dopamine hyperactivity has a long history as a prominent pathophysiologic hypothesis of schizophrenia (9, 10), as medications that treat psychosis are dopamine D2 receptor antagonists (11), and dopamine enhancing drugs like stimulants are psychotomimetic (12). In rodent models, amphetamine administration induces locomotor sensitization that is accompanied by an increase in dopamine efflux from the nucleus accumbens and dorsal striatum (13). In patients, a link between dopamine dysregulation and psychosis severity (14) and a relationship between baseline dopamine D2 receptor occupancy and antipsychotic treatment response are reported (15–17).
N-methyl-d-aspartate receptor (NMDAR) hypofunction is widely hypothesized to be a central neurobiological alteration in schizophrenia (18, 19). Experimental evidence supports NMDAR hypofunction as a high priority target for biomarker development. NMDAR hypofunction on the ɣ-aminobutyric acid (GABAergic) interneuron causes disinhibition of the glutamatergic pyramidal cell (20–22). The presence of a hyperglutamatergic state in different brain areas in patients with schizophrenia has been empirically confirmed and replicated in a number of magnetic resonance spectroscopy (MRS) studies (23–29). Preclinical data further support this target by demonstrating that experimentally induced NMDAR hypofunction results in increased firing of glutamatergic neurons in animal models (30) and produces psychosis like behavioral phenotypes (31–33) and glutamatergic excess in healthy human subjects (34–36). Unfortunately, no validated positron emission tomography (PET) ligand visualizing NMDAR function in vivo is available to date (37).
Another biomarker development target is hippocampal hyperactivity. Here, the model suggests that a hyperglutamatergic state causes hippocampal hyperactivity, which then may result in downstream dopamine circuit dysregulation and psychotic symptoms (38, 39). Several studies reported hippocampal hyperactivity in patients with schizophrenia (40–43), and found a relationship between hippocampal regional cerebral blood flow (rCBF) and psychosis symptom severity (44) as well as antipsychotic treatment response (45). Linking cellular-level mechanisms and neuroimaging findings, Schobel and colleagues reported a series of experiments in a preclinical model of psychosis, showing that ketamine administration causes an increase in extracellular glutamate and hippocampal hyperactivity (46). Experiments in a methylazoxymethanol acetate (MAM) model, which recapitulates a developmental disruption leading to neurophysiological and behavioural deficits that resemble components of schizophrenia, further demonstrate that hippocampal hyperactivity results in increased dopaminergic signalling which can be reversed by inactivating the ventral hippocampus (47), suggesting possible translational utility of this marker.
Neuroinflammation or immune dysregulation as plausible biomarkers for schizophrenia are rooted in the observation of a link between autoimmune processes and development of psychosis (48). In this model, microglia are primed during early development into a hyper-responsive mode, then shift to a pro-inflammatory state in response to stress during critical developmental periods. This in turn can result in aberrant neurotransmission, synaptic pruning and structural injury of neurons and glia (49). Maternal immune activation (MIA) models in rodents show a post pubertal symptom onset and have structural, neurochemical and behavioral abnormalities recapitulating the human clinical picture (50). Evidence of immune dysregulation has also been reported in postmortem studies (51) and in studies examining cytokines in cerebrospinal fluid of patients (52). A meta-analysis of PET studies using translocator protein (TSPO) binding, which is expressed during the inflammatory response, found significantly elevated tracer binding in patients with schizophrenia compared to controls with a small-to-moderate effect size (Hedge’s g= 0.31), but no difference in volume of distribution was detected (53), but also see (54) who did not report evidence of neuroinflammation in the dorsolateral prefrontal cortex or hippocampus in unmedicated patients after controlling for relevant genetic polymorphisms. Two studies report evidence of neuroinflammation in recent onset and acutely ill psychosis spectrum patients (55, 56). Interestingly, these alterations are not found in medicated chronic patients (57, 58), suggesting a dynamic imbalance. This is further supported by a number of diffusion imaging studies reporting increased extracellular free water, a proxy of inflammation, which appears more prominent in the early illness compared to chronic stages (59–62). Taken together, biomarkers related to immune dysregulation may be dynamic in that they capture pathological processes present only during illness onset or psychosis exacerbations.
Human connectome studies have provided data concordant with the hypothesis that brain network dysconnection is fundamental to psychosis (63–65). The dysconnectivity model proposes that NMDAR mediated disturbances in the excitation/ inhibition balance are correlated with altered functional network architecture, which results in the symptoms of schizophrenia (66, 67). A number of studies have demonstrated a relationship between brain network dysconnectivity and the development of psychotic symptoms (62), symptom severity (68) and response to antipsychotic pharmacotherapy (69–71). Lending additional support to this model are studies that report disruption in functional brain networks following experimentally induced NMDAR hypofunction in healthy human subjects (35, 72, 73). While dysconnectivity classically is explained by changes in neurotransmitter systems and aberrant modulation of synaptic efficiency (66), contemporary theories also consider the underlying anatomical connections. Prominent neurodevelopmental models postulate that genetic and environmental factors may affect early white matter developmental trajectories followed by the onset of psychosis, which then results in further white matter integrity reductions (74–77). Those perturbations result in white matter development disruptions and altered behavioral phenotypes, supporting this neurodevelopmental model (78). For example, mice lacking the chemokine receptor fractalkine exhibit a transient reduction of microglia and a deficit in synaptic pruning, which results in decreased connectivity between the frontal cortex and hippocampus and deficits in social interactions (79). Importantly, diffusion imaging studies report associations between white matter abnormalities and disease severity across symptom dimensions (80–84), poor response to antipsychotic treatment (85), and worse overall outcomes (86, 87).
Similarly, cortical gray matter volume loss, which may be most prominent in fronto-temporal regions, is consistently found as a hallmark feature that already is present at illness onset and may become progressively worse with longer illness duration (88, 89). Abnormalities in brain maturational processes mediated by environmental factors are hypothesized to underlie gray matter alterations (90). Sub-chronic NMDA receptor antagonism (91, 92) and MIA models (93) have resulted in gray matter reductions in preclinical studies. Gray matter loss has been linked to disease severity across symptom dimensions (94–96) and overall poor clinical outcomes (97), suggesting it may be a viable candidate for diagnostic and prognostic biomarker development.
To recapitulate, we discussed a number of mechanistically plausible biomarker development targets. However, it needs to be acknowledged that this is not an exhaustive list. Other targets like oxidative stress or GABA dysfunction, where a disruption in the fast-spiking GABAergic interneurons has strong evidentiary support in the postmortem schizophrenia literature and parvalbumin-positive interneuron dysfunction has been identified as common factor behind several of the relevant animal models (98, 99), are also plausible. As we next discuss, the specific utility of any of these markers may depend on the phase of illness and the specific clinical question to be answered.
Biomarkers for risk prediction
Correct identification of individuals at risk for psychosis in the prodromal phase provides a major opportunity for early intervention and disease prevention. However, when solely relying on clinically high-risk criteria, correct disease prediction is estimated to be 15–30% (100). In a two-center study examining structural MRI for predicting transition risk in patients at high risk for developing schizophrenia, Koutsouleris and colleagues demonstrated a statistically significant improvement in prognostic certainty over clinical assessments alone (101). The anatomical features associated with schizophrenia included gray matter reductions in the prefrontal, cingulate, striatal and cerebellar cortices. However, their pooled sample was unusual in the fact that approximately 45% of the enrolled subjects transitioned to a psychotic illness, which is substantially higher than in a typical sample for this type of study (101); replication in independent samples will therefore be important.
In a small study, Schobel and colleagues found that cerebral blood volume in the CA1 subfield of the hippocampus predicted clinical progression with a positive predictive value of 71% and a negative predictive value of 82% (102). The same group later reported that left anterior CA1 cerebral blood volume also predicted time to psychosis onset in high-risk patients, and demonstrated this to be a more sensitive marker of clinical outcomes compared to subthreshold psychotic symptoms (46).
Thalamic glutamate measurements with MRS in a relatively small sample of ultra-high risk subjects predicted the clinical course with an odds ratio 0.52, such that higher baseline glutamate levels were associated with subsequent remission of prodromal symptoms (103). In contrast, high risk subjects who later developed a frank psychotic episode showed higher glutamate levels in the dorsal caudate compared to the non-transition group and controls. That study reported a surprisingly large effect size of Cohen’s d= 1.39 (104). Another small study showing that clinical high-risk individuals who later became psychotic had higher hippocampal glutamate levels compared to those who did not transition; the effect size was Hedges g= 0.57 (105), suggesting brain regions are an important consideration when developing spectroscopy based biomarkers.
Diagnostic Biomarkers
Diagnostic biomarkers have the goal of detecting the presence of a disease state and establishing objective disease signatures. A meta-analysis of multivariate pattern recognition studies of neuroimaging-based diagnostic biomarkers found that these markers separate patients from controls with an overall sensitivity and specificity of approximately 80%. Interestingly, they reported that sensitivity was higher in chronic patients compared to first episode patients, and found potential effects of symptom severity and antipsychotic dose on specificity, suggesting that these biomarkers may be better in correctly classifying those with a higher overall disease burden (106). Leveraging the Functional Biomedical Informatics Research Network Data Repository (fBIRN), Calhoun and colleagues demonstrated a multimodal classifier, which combined structural MRI and resting state functional MRI, improved classification accuracy when compared to each modality used separately (107). This suggests that an aggregate approach to biomarker development may be fruitful.
Target Engagement Biomarkers
Target engagement biomarkers are meant to confirm desired therapeutic properties or determine individualized dosing for a specific treatment. To qualify as a target engagement biomarker it must measure a direct interaction between the treatment and the intended molecular/functional target in the central nervous system, which for medications is typically accomplished with PET. However, any neuroimaging outcome measure is theoretically an engageable target, as long as it has the potential to change measurably with a targeted intervention.
PET studies of the dopaminergic system provided the classic demonstration of target engagement for antipsychotic medications through several lines of converging evidence. These included establishment of abnormal dopaminergic function in patients with schizophrenia (17, 108), discovery of a link between dopamine dysregulation and psychosis severity (14), and demonstration of a reduction in dopamine stimulation of D2 receptors with antipsychotic medication treatment (109).
In the absence of a specific PET ligand targeting the glutamatergic system, a number of MRI markers are being used as a proxy for glutamate. The Fast-Fail Trials in Psychotic Spectrum Disorders (FAST-PS) biomarker project, based on the hypothesis of glutamatergic neurotransmitter system disturbances as a core pathological feature in schizophrenia (18), approached validation of target engagement in two phases. In the first stage, three candidate biomarkers that are putatively sensitive to glutamatergic alterations were evaluated for their power to detect changes resulting from experimentally induced NMDAR hypofunction using a ketamine challenge (110). The effect size was most robust and consistent across sites for a resting-state blood oxygen level dependent signal (BOLD) response in the dorsal anterior cingulate cortex, suggesting possible utility of this measure as a biomarker for glutamatergic change. Importantly, a mean signal change of 0.5% fully separated subjects receiving ketamine from those receiving placebo. In the second stage, this marker was used to enrich the sample by eliminating subjects failing to demonstrate a ketamine induced BOLD signal change before randomizing healthy volunteers to pomaglumetad, a partial mGluR-2/3 agonist, or placebo. Here, the goal was to test the hypothesis that the drug would blunt the ketamine induced BOLD response, indicating its glutamate target engagement, and to determine dosing of the medication. This stage of the study has now completed data collection; pending results will inform the decision for pomaglumetad to be advanced into further trials.
Biomarkers predicting response to treatment/ aid in therapeutic monitoring
This type of biomarker is intended to characterize patients in context of a given treatment before it is started, and it may be used for patient stratification into biomarker positive and negative groups. In addition, these biomarkers may also be used to monitor effectiveness of treatment or to predict deleterious side effect occurrence and probability of relapse (111).
Antipsychotic medications principally act on dopamine D2 receptors, which informed the dopamine hyperactivity hypothesis of schizophrenia. A number of studies reported a relationship between dopamine D2 receptor occupancy at baseline and subsequent clinical response to antipsychotic medications (15, 17), and a relationship between D2 receptor occupancy with antipsychotic medication treatment and the extent of clinical improvement (15, 16). Interestingly, the level of haloperidol induced D2 receptor blockage has also been found to be associated with the degree of extrapyramidal symptom severity, and prolactin elevation (15). Taken together, dopamine D2 receptor occupancy appears to be both a mediator and a moderator of antipsychotic treatment response.
The schizophrenia literature links increased gray matter volume reductions to worse long term clinical outcomes. In first episode psychosis patients, a machine learning classifier based on gray matter morphometry found baseline gray matter volumes in the bilateral parahippocampal gyri to be most important for predicting remission status after six months of antipsychotic treatment, with an accuracy of 79% (112). Similarly, a multivariate morphometry study found that lower gray matter volume in the bilateral inferior frontal gyrus and anterior insula predicted a lack of improvement in symptoms over one year (113). The gyrification index, a gray matter morphometric feature acting as a surrogate of neurodevelopmental trajectories, was altered in schizophrenia (114) and possibly sensitive to antipsychotic exposure (115). In first episode psychosis, reduced gyrification in frontal, insular, and temporal cortices has been associated with subsequent poor response to antipsychotic treatment (116), suggesting it may be a useful predictor of treatment response.
A growing body of evidence suggests the potential to predict treatment response using glutamatergic neurometabolites measured with MRS. OPTiMiSE, a European multicenter study, found that higher baseline glutamate levels in the anterior cingulate cortex in first episode psychosis patients were associated with greater symptom severity and lower likelihood of remission after four weeks of antipsychotic treatment (117). This is consistent with a prior report by the same group finding 11% higher glutamate levels in the same region in a small group of medication-resistant compared to medication-responsive patients (Cohen’s d= 0.76), however accurate classification of response status was not possible with this measure (118). In a 2017 review of longitudinal spectroscopy studies, Egerton and colleagues reported an overall mean reduction in brain Glx (glutamate+ glutamine) of 6.5% following antipsychotic treatment and suggested that antipsychotic treatment response may be associated with lower glutamate levels before treatment and greater reductions in glutamate with treatment. However, it is possible that effects are region specific, as others reported that in the hippocampus, higher, as opposed to lower baseline Glx, may be associated with favorable treatment response (25).
A promising resting state fMRI based putative biomarker has recently been described in a series of experiments examining functional connectivity of the striatum, a principal site of antipsychotic drug action. First, Sarpal and colleagues demonstrated an increase in striatal connectivity after sixteen weeks of antipsychotic treatment, and this change was correlated with the degree of clinical symptom improvement (119). Following this, they created a baseline “striatal connectivity index” that predicted subsequent treatment response in first episode psychosis patients. This index was then tested for its utility to separate good from poor treatment responders in an independent cohort of hospitalized chronic schizophrenia patients. Notably, this index showed a significant separation between good and poor responders, with a positive predictive value of 76% and a negative predictive value of 79% (71). Extending this work, others later reported lower striatal connectivity in antipsychotic treatment-resistant compared to non-refractory schizophrenia patients (120).
A number of resting state fMRI studies suggest that connectivity and topology of several other known brain networks (69, 70, 121–124) may also have utility in predicting response to antipsychotic treatment, though the performance of the majority of these putative markers remains to be established at the single subject level. The only study to date investigating the accuracy of resting state cortical connectivity at the single subject level reported that baseline functional connectivity between the superior temporal cortex and other cortical regions predicted clinical response after 10 weeks of antipsychotic treatment, with a balanced accuracy of 82% (125). Another group used a non-connectivity based resting state index, ALFF (the amplitude of low frequency fluctuations), to build a machine learning classifier predicting treatment response in recent onset schizophrenia patients. The authors reported that ALFF in the left postcentral gyrus/ inferior parietal lobule predicted response status with an accuracy of 72.7%. A major strength of this study was the independent replication sample (using a different scanner) that showed a similar accuracy of 75% in predicting remission status (126).
Challenges in biomarker development
Three challenges have limited the development of neuroimaging biomarkers in schizophrenia: internal heterogeneity, analytic approaches, and clinical utility. First, one of the principal challenges of diagnostic biomarker research is that the standard nosology in schizophrenia is based on symptoms alone. There is an inherent tension between the effort to develop biomarkers for classic DSM-based diagnoses and the growing sense that it may be more fruitful to develop biomarkers that identify more biologically homogeneous subgroups of patients (127). In an effort to overcome these inherent limitations, the Research Domain Criteria (RDoC) paradigm was introduced as an alternative framework for the investigation of psychiatric disorders (128), where disorders are considered in terms of disruptions of normal-range operation systems. Alternatively, the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) initiative has incorporated a dimensional approach in an effort to analyze biomarker outcomes for disease definition and neuropathology in psychosis spectrum disorders (129). Findings support the idea that neuroimaging-based biomarkers do not obey the boundaries set by symptom based nosology, underscoring the limitations of diagnostic biomarker development studies conducted within the context of traditional diagnostic boundaries.
Closely related is the complexity of the underlying pathophysiology. Due to the high degree of intricate associations between neurotransmitter systems, immune systems, functional brain network architecture and brain structure, a biomarker may reflect the result of multiple modulatory inputs rather than the primary etiological factor (130). Unfortunately, multivariate approaches capitalizing on the vast amount of multimodal neuroimaging data available to detect representative pathway biomarkers assessing key pathological features such as NMDAR hypofunction, inflammation or neuronal plasticity have yet to be developed. It also remains unclear if putative biomarkers assessed in a clinically stable state or in a more actively psychotic state will be most informative. Furthermore, the cross-sectional assessment of the candidate marker may not be the most relevant because the change of a given marker over time in an individual may be most predictive. Additionally, it is possible that certain relevant biological signatures such as alterations in spine density or synaptic integrity (131, 132) may simply be below the resolution to be captured by existing imaging methods.
The second challenge lies in defining standard best practices for analytics, both in pre-processing of the original data and in the development of prediction rules. To the first point, the majority of conventional imaging indices are affected by scanner specific measurement errors, which can significantly impact cross-site reliability. Furthermore, there is a lack of consensus in the field on how to harmonize imaging sequences or mitigate measurement error in the post-processing stage across sites, which greatly reduces biomarker accuracy and reliability in multi-site studies. To the second point, many biomarker studies do not report their results in the most rigorous fashion. The ultimate goal of every biomarker is to predict an unseen future: conversion to first-episode psychosis, response to a specific medication, long-term prognosis, etc. The most rigorous approach to assessing that prediction performance is replication of a biomarker on a new dataset, collected by researchers independent of the original team. Since that type of replication generally takes years, a close alternative is cross validation: reporting a prediction model’s performance on new data it has never seen before (as opposed to the data originally used to build the model) (133, 134). Without cross validation, biomarker studies are susceptible to the problem of “overfitting” - building models that work very well on one specific sample, but that do not generalize to the larger clinical population. Unfortunately, cross validation remains rare in schizophrenia biomarker studies, as it does in psychiatric biomarker research more generally.
The third challenge lies in determining the clinical utility of a biomarker. The decision to use a biomarker in clinical practice should be based on an expectation that it will have a positive health impact, therefore measuring biomarker performance in generic terms is not sufficient for confirmation of clinical utility (135). Demonstration of clinical utility is typically a two-step process that first determines the accuracy of a biomarker and then shows that using the biomarker information in managing patients, given the benefits and risks associated with the assessment, improves outcomes in a clinically meaningful way (136). Prospective, confirmatory multicenter studies or decision modeling approaches could be used to demonstrate clinical utility (135, 136), but these types of studies are clearly a missing element in the evaluation of neuroimaging biomarkers in the field.
Of course, these challenges are not just specific to neuroimaging biomarker development in schizophrenia, but largely apply to neuroimaging biomarker research in general. Similarly, the following section where we discuss next generation biomarker development studies and reflect on best practices is also applicable to the broader field.
Next generation biomarker development studies
Several lines of research are well positioned to contribute to the advancement of neuroimaging biomarker discovery (Figure 3).
Figure 3:
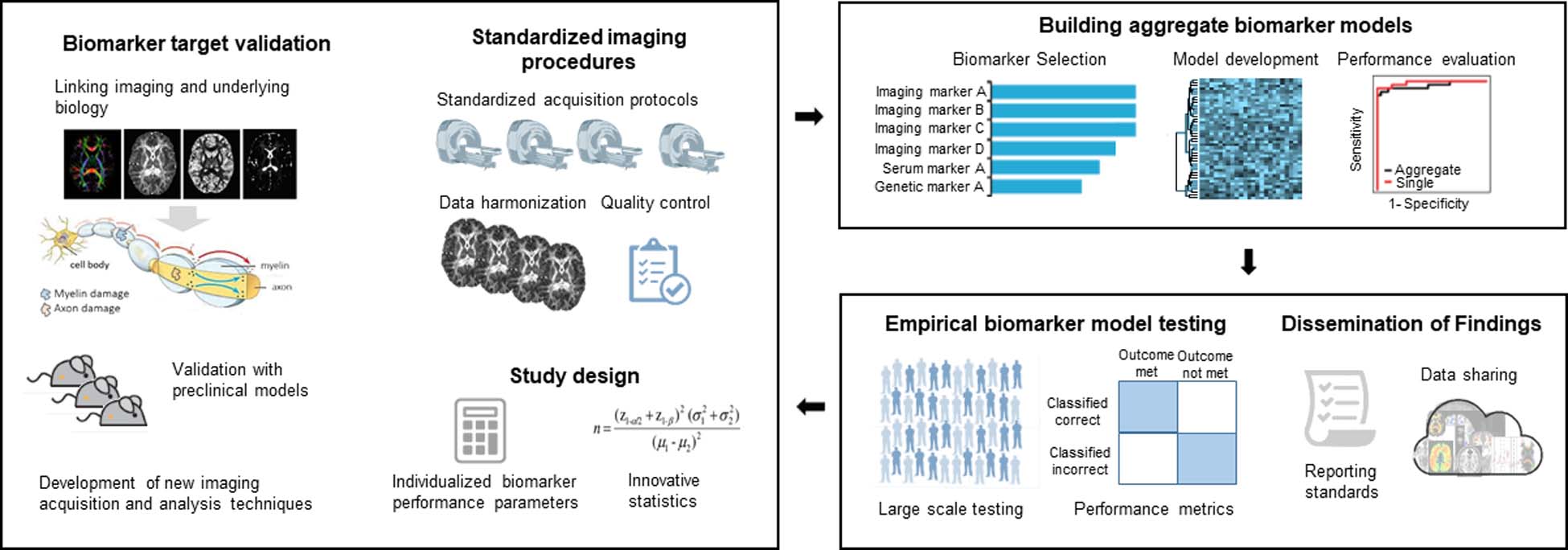
Next generation of neuroimaging biomarker studies in schizophrenia.
Many of the routinely assessed imaging markers are difficult to directly link to relevant pathological processes. For example, glutamate levels measured with spectroscopy do not equal the amount of glutamatergic neurotransmission, but rather reflect the amount of neuronal, glial and synaptic glutamate present in a voxel. BOLD imaging does not measure neural activity directly. It is a signal that stems from a complex interaction between blood flow, blood volume, and oxygen consumption related to neural activity. The field has demonstrated a commitment to the long term goal of creating the next generation of imaging tools that allow us to map the brain and relevant pathological signatures on a much more granular level (137), which will likely accelerate biomarker discovery. Continuing development of innovative technologies that are biologically validated and capture relevant features of complex neural circuits will provide unique opportunities. Here, radiotracer development holds distinct promise, as novel pharmacotherapeutics will likely be targeted at modulation of neurotransmitter systems. Development of specific ligands that capture principal pathophysiological processes such as NMDAR hypofunction or immune dysregulation could result in significant advancement of diagnostic and target engagement biomarker research. Another strategy that may have greater short-term feasibility is to capitalize on already existing imaging techniques and leverage advances in computational modeling to increase specificity of measures. For this strategy to be successful, vital components are to test the biological accuracy by rigorous histological validation and confirmation of disease relevance in preclinical models.
It will be critical to design studies that follow sound methodology to minimize bias and maximize precision (138). Unfortunately, this is not a trivial undertaking with neuroimaging data, as factors such as magnetic field strength, sequence acquisition parameters, scanner variability and data analysis methods affect measurements. Additionally, head movement has a negative impact on data quality in virtually all neuroimaging modalities (139). A number of these problems can be mitigated by implementation of standardized image acquisition protocols, use of imaging phantoms as external reference if feasible, rigorous data quality control protocols, retrospective sequence harmonization (140), as well as standardized data processing pipelines and data analytics.
Future studies would benefit from including efforts for cross-validation of biomarkers in independent samples not only in those with comparable clinical characteristics, but also with various clinical presentations (acutely psychotic, clinically stable), at early and late disease stages, and with differential antipsychotic medication exposure. This type of data would be invaluable in delineating the temporal evolution and scope of a candidate biomarker.
Because an aggregate approach, either as a combination of multiple imaging modalities or the addition of information from serological or genetic markers, could improve biomarker performance, an important next step beyond independent replication will be to embed a number of biomarkers in a predictive model and to test overall performance, discrimination, calibration and clinical utility of the model (141). An excellent example of the important considerations of predictive models, in this case clinical risk prediction models for conversion to psychosis (141), could also serve as a road map for building analogous models for disease risk or therapeutic response that are neuroimaging biomarker based.
Conventional statistical approaches for three dimensional neuroimaging data rely on mass-univariate analyses (142). Machine learning, a field in artificial intelligence, is designed to detect patterns in data with multivariate statistics aimed at making predictions at the single subject level. To date, the majority of machine learning studies have focused on correctly classifying the presence of a disease state. However, the practical value here is limited, as those patients are already “correctly classified” through clinical assessments. Studies geared towards building models that can accurately predict the risk for disease development, clinical course, or a patient’s likelihood to respond to a given treatment may be more clinically useful but have been less common (143). There are also a number of technical limitations that constrain interpretability of machine learning studies in schizophrenia. Because overfitting of data is a major source of bias, it is important that sample sizes are adequate when developing a model and that the model is then cross-validated as noted above (144). Another limitation of machine learning approaches is that many studies use a “black box” approach, reporting sensitivity, specificity and accuracy of an imaging classifier, without providing spatial maps outlining relevant features of classifiers. Even though these types of complementary analyses are computationally expensive, they would make models more interpretable and add face validity to them. This idea of adding “explanability” to artificial intelligence techniques is a major priority for Federal funders whose investments often shape a field (145). Computer science holds immense promise to provide solutions for these technical limitations in the next generation of biomarker discovery studies, not only in the optimization of informatics infrastructures or design of user-friendly software interfaces geared towards neuroscientists with limited training in command-line based coding, but also as interdisciplinary collaborators guiding appropriate implementation of multivariate statistics.
The literature on best practices on how to objectively and effectively evaluate the clinical utility of a neuroimaging biomarker in schizophrenia is sparse. Defining biomarker performance standards based on established decision theory concepts has shown potential to inform clinically relevant levels of sensitivity and specificity in cancer research. The authors argue against arbitrary preset targets in favor of calculating target levels for a given intended use of a biomarker, e.g. screening, vs. confirmatory testing. These calculations consider variables such as prevalence of the disease, which affects the true positive and negative predictive values, and the cost/ benefit ratio of such tests (146). Additionally, performance of a biomarker in one context may not be relevant to the setting of interest (147). Development of calculators for estimation of individualized minimally acceptable biomarker performance indicators and necessary sample size based on these considerations could be invaluable in the design of future biomarker studies.
Lastly, to accelerate biomarker development it is important to address transparency and reproducibility of research findings. On a policy level, many funding agencies now mandate sharing of generated data with the scientific community. Unfortunately, the sharing process is quite laborious at this time. Refining electronic data capture systems to allow highly automated sharing and integration of data sharing and analyses tools will improve reproducibility of imaging findings (148). It will also be important to develop reporting standards containing the minimum recommendations for biomarker development studies, similar to those for randomized clinical trials (149), and systematic reviews (150) which are widely recognized in the scientific community. These guidelines help to improve the quality of reporting in scientific journals by outlining essential items necessary for a clear and transparent account of research methods and results. Another interesting idea that could address reproducibility issues for future studies is the creation of a preregistration database for neuroimaging biomarker studies with the original hypothesis and its justification, similar to what is done in clinical trials (151).
Moving forward, nationwide or multinational initiatives supporting large-scale studies that are developing targeted biomarkers have real potential to individualize clinical care in this complex neuropsychiatric syndrome. In the field of Alzheimer’s disease, a private public partnership, the Alzheimer’s Disease Neuroimaging Initiative (ADNI) was designed to develop biomarkers for the early detection and tracking of the disease. The investment of $218 million to date has enabled ADNI to discover biomarkers for use in clinical trial subject selection and as surrogate outcome measures, to develop standardized protocols for use across multiple centers, to create platforms allowing open data access, and to make advances in the understanding of the relationship between biomarkers and disease progression (152). This initiative could serve as a road-map for the design of and indicator for the likely necessary financial commitment needed in biomarker development for schizophrenia spectrum disorders.
Acknowledgements
This work was supported by NIMH grants K23MH106683 and R01MH118484 to Dr. Kraguljac. We would like to thank Dr. John Krystal for his thoughtful feedback on the first draft of the manuscript, and Dr. Anthony Vernon for sharing his expertise on animal models.
Disclosure of competing interests and financial support
This article is derived from work done by the Workgroup for Biomarkers and Novel Treatments of the APA Council of Research on behalf of the American Psychiatric Association (APA) and remains the property of the APA. It has been altered only in response to the requirements of peer review. Copyright © [year] American Psychiatric Association. Published with permission.
The findings, opinions, and conclusions of this report do not necessarily represent the views of the officers, trustees, or all members of the American Psychiatric Association. The views expressed are those of the authors of the manuscript.
Dr. Kraguljac serves as consultant for Neurocrine Biosciences, Inc..
Dr. McDonald reports research supported by the National Institute of Mental Health, National Institute of Neurological Disease and Stroke, National Institute of Aging, Patient-Centered Outcomes Research Institute, Stanley Foundation, Soterix, Neuronetics, NeoSync and Cervel Neurotherapeutics. He has a contract with Oxford University Press to co-edit a book on the Clinical Guide to Transcranial Magnetic Stimulation in the Treatment of Depression and is a consultant for Signant Health.
Dr. Widge acknowledges support from the National Institutes of Health (R01 MH119384), from the Minnesota’s Discovery, Research, and InnoVation Economy (MnDRIVE) initiative, and the Minnesota Medical Discovery Team on Addictions. He also reports device donations from Medtronic and consulting income from Medtronic, Cyberonics, and Circuit Therapeutics. He has multiple patent applications in the area of brain stimulation and circuit modification to improve cognition.
Dr. Rodriguez reports serving as a consultant to Blackthorn and Epiodyne, receiving grant support from Biohaven Pharmaceuticals, and receiving remuneration from APA Publishing for her duties as Deputy Editor of the journal The American Journal of Psychiatry.
Dr Tohen is a former full time employee at Lilly (1997 to 2008). He has been a consultant for AstraZeneca, Abbott, BMS, Lilly, GSK, J&J, Otsuka, Roche, Lundbeck, Elan, Alkermes, Merck, Pamlab, Alexza, Forest, Teva, Sunovion, Gedeon Richter, and Wyeth. His spouse is a former employee at Lilly (1998-2013)
Dr. Nemeroff has received grants or research support from NIH and the Stanley Medical Research Institute; he has served as a consultant for Bracket (Clintara), Dainippon Pharma, Fortress Biotech, Intra-Cellular Therapies, Janssen Research and Development, Magstim, Prismic Pharmaceuticals, Sumitomo Navitor Pharmaceuticals, Sunovion, Taisho Pharmaceutical, Takeda, TC MSO, and Xhale; he has served on scientific advisory boards for the American Foundation for Suicide Prevention (AFSP), the Anxiety Disorders Association of America (ADAA), Bracket (Clintara), the Brain and Behavior Research Foundation, the Laureate Institute for Brain Research, Skyland Trail, and Xhale and on directorial boards for ADAA, AFSP, and Gratitude America; he is a stockholder in AbbVie, Antares, BI Gen Holdings, Celgene, Corcept Therapeutics, OPKO Health, Seattle Genetics, and Xhale; he receives income or has equity of $10,000 or more from American Psychiatric Publishing, Bracket (Clintara), CME Outfitters, Intra-Cellular Therapies, Magstim, Takeda, and Xhale; and he holds patents on a method and devices for transdermal delivery of lithium (patent 6,375,990B1) and a method of assessing antidepressant drug therapy via transport inhibition of monoamine neurotransmitters by ex vivo assay (patent 7,148,027B2).
REFERENCES
- 1.Group F-NBW: in BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring (MD)2016. [Google Scholar]
- 2.Abi-Dargham A, Horga G. The search for imaging biomarkers in psychiatric disorders. Nat Med. 2016;22:1248–1255. [DOI] [PubMed] [Google Scholar]
- 3.Marquez F, Yassa MA. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol Neurodegener. 2019;14:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraemer HC, Schultz SK, Arndt S. Biomarkers in psychiatry: methodological issues. Am J Geriatr Psychiatry. 2002;10:653–659. [PubMed] [Google Scholar]
- 5.Botteron K, Carter CS, Castellanos FX, Dickstein DP, Drevets W, Kim KL, Pescosolido MF, Rauch S, Seymour KE, Sheline Y, Zubieta J-K: Consensus report of the APA work group on neuroimaging markers of psychiatric disorders. Edited by Association AP. Arlington, VA2012. [Google Scholar]
- 6.Bough KJ, Amur S, Lao G, Hemby SE, Tannu NS, Kampman KM, Schmitz JM, Martinez D, Merchant KM, Green C, Sharma J, Dougherty AH, Moeller FG. Biomarkers for the development of new medications for cocaine dependence. Neuropsychopharmacology. 2014;39:202–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Park JW, Kerbel RS, Kelloff GJ, Barrett JC, Chabner BA, Parkinson DR, Peck J, Ruddon RW, Sigman CC, Slamon DJ. Rationale for biomarkers and surrogate end points in mechanism-driven oncology drug development. Clin Cancer Res. 2004;10:3885–3896. [DOI] [PubMed] [Google Scholar]
- 8.Tregellas JR. Neuroimaging biomarkers for early drug development in schizophrenia. Biol Psychiatry. 2014;76:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, Weinberger DR, Weisenfeld N, Malhotra AK, Eckelman WC, Pickar D. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci U S A. 1997;94:2569–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hietala J, Syvalahti E. Dopamine in schizophrenia. Ann Med. 1996;28:557–561. [DOI] [PubMed] [Google Scholar]
- 11.Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci U S A. 2000;97:7673–7675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl). 1987;91:415–433. [DOI] [PubMed] [Google Scholar]
- 13.Jones CA, Watson DJ, Fone KC. Animal models of schizophrenia. Br J Pharmacol. 2011;164:1162–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. [DOI] [PubMed] [Google Scholar]
- 15.Kapur S, Zipursky R, Jones C, Remington G, Houle S. Relationship between dopamine D(2) occupancy, clinical response, and side effects: a double-blind PET study of first-episode schizophrenia. Am J Psychiatry. 2000;157:514–520. [DOI] [PubMed] [Google Scholar]
- 16.Nordstrom AL, Farde L, Wiesel FA, Forslund K, Pauli S, Halldin C, Uppfeldt G. Central D2-dopamine receptor occupancy in relation to antipsychotic drug effects: a double-blind PET study of schizophrenic patients. Biol Psychiatry. 1993;33:227–235. [DOI] [PubMed] [Google Scholar]
- 17.Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, Gorman JM, Laruelle M. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry. 1995;52:998–1007. [DOI] [PubMed] [Google Scholar]
- 19.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. [DOI] [PubMed] [Google Scholar]
- 20.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37:4–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coyle JT. NMDA receptor and schizophrenia: a brief history. Schizophr Bull. 2012;38:920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, Gil R, Slifstein M, Abi-Dargham A, Lisanby SH, Shungu DC. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2012;69:449–459. [DOI] [PubMed] [Google Scholar]
- 24.de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Stephano S, Favila R, Diaz-Galvis L, Alvarado-Alanis P, Ramirez-Bermudez J, Graff-Guerrero A. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry. 2013;70:1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kraguljac NV, Morgan CJ, Reid MA, White DM, Jindal RD, Sivaraman S, Martinak BK, Lahti AC. A longitudinal magnetic resonance spectroscopy study investigating effects of risperidone in the anterior cingulate cortex and hippocampus in schizophrenia. Schizophr Res. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kraguljac NV, White DM, Reid MA, Lahti AC. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA Psychiatry. 2013;70:1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Plitman E, de la Fuente-Sandoval C, Reyes-Madrigal F, Chavez S, Gomez-Cruz G, Leon-Ortiz P, Graff-Guerrero A. Elevated Myo-Inositol, Choline, and Glutamate Levels in the Associative Striatum of Antipsychotic-Naive Patients With First-Episode Psychosis: A Proton Magnetic Resonance Spectroscopy Study With Implications for Glial Dysfunction. Schizophr Bull. 2016;42:415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull. 2013;39:120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK. Nature of Glutamate Alterations in Schizophrenia: A Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry. 2016;73:665–674. [DOI] [PubMed] [Google Scholar]
- 30.Jackson ME, Homayoun H, Moghaddam B. NMDA receptor hypofunction produces concomitant firing rate potentiation and burst activity reduction in the prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. [DOI] [PubMed] [Google Scholar]
- 32.Kraguljac NV, Carle M, Frolich MA, Tran S, Yassa MA, White DM, Reddy A, Lahti AC. Mnemonic Discrimination Deficits in First-Episode Psychosis and a Ketamine Model Suggests Dentate Gyrus Pathology Linked to N-Methyl-D-Aspartate Receptor Hypofunction. Biol Psychiatry Cogn Neurosci Neuroimaging. 2018;3:231–238. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB Jr., Charney DS. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. [DOI] [PubMed] [Google Scholar]
- 34.Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–396. [DOI] [PubMed] [Google Scholar]
- 35.Kraguljac NV, Frolich MA, Tran S, White DM, Nichols N, Barton-McArdle A, Reid MA, Bolding MS, Lahti AC. Ketamine modulates hippocampal neurochemistry and functional connectivity: a combined magnetic resonance spectroscopy and resting-state fMRI study in healthy volunteers. Mol Psychiatry. 2017;22:562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van der Aart J, Golla SSV, van der Pluijm M, Schwarte LA, Schuit RC, Klein PJ, Metaxas A, Windhorst AD, Boellaard R, Lammertsma AA, van Berckel BNM. First in human evaluation of [(18)F]PK-209, a PET ligand for the ion channel binding site of NMDA receptors. EJNMMI Res. 2018;8:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heckers S, Konradi C. GABAergic mechanisms of hippocampal hyperactivity in schizophrenia. Schizophr Res. 2015;167:4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lieberman JA, Girgis RR, Brucato G, Moore H, Provenzano F, Kegeles L, Javitt D, Kantrowitz J, Wall MM, Corcoran CM, Schobel SA, Small SA. Hippocampal dysfunction in the pathophysiology of schizophrenia: a selective review and hypothesis for early detection and intervention. Mol Psychiatry. 2018;23:1764–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talati P, Rane S, Skinner J, Gore J, Heckers S. Increased hippocampal blood volume and normal blood flow in schizophrenia. Psychiatry Res. 2015;232:219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–550. [DOI] [PubMed] [Google Scholar]
- 42.Tregellas JR, Smucny J, Harris JG, Olincy A, Maharajh K, Kronberg E, Eichman LC, Lyons E, Freedman R. Intrinsic hippocampal activity as a biomarker for cognition and symptoms in schizophrenia. Am J Psychiatry. 2014;171:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molina V, Sanz J, Sarramea F, Benito C, Palomo T. Prefrontal atrophy in first episodes of schizophrenia associated with limbic metabolic hyperactivity. J Psychiatr Res. 2005;39:117–127. [DOI] [PubMed] [Google Scholar]
- 44.Lahti AC, Weiler MA, Holcomb HH, Tamminga CA, Carpenter WT, McMahon R. Correlations between rCBF and symptoms in two independent cohorts of drug-free patients with schizophrenia. Neuropsychopharmacology. 2006;31:221–230. [DOI] [PubMed] [Google Scholar]
- 45.Lahti AC, Weiler MA, Holcomb HH, Tamminga CA, Cropsey KL. Modulation of limbic circuitry predicts treatment response to antipsychotic medication: a functional imaging study in schizophrenia. Neuropsychopharmacology. 2009;34:2675–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, Inbar BP, Corcoran CM, Lieberman JA, Moore H, Small SA. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32:507–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK, Rosenfeld MR, Balice-Gordon R, Lynch DR. Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008;7:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Howes OD, McCutcheon R. Inflammation and the neural diathesis-stress hypothesis of schizophrenia: a reconceptualization. Transl Psychiatry. 2017;7:e1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mattei D, Schweibold R, Wolf SA. Brain in flames - animal models of psychosis: utility and limitations. Neuropsychiatr Dis Treat. 2015;11:1313–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trepanier MO, Hopperton KE, Mizrahi R, Mechawar N, Bazinet RP. Postmortem evidence of cerebral inflammation in schizophrenia: a systematic review. Mol Psychiatry. 2016;21:1009–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gallego JA, Blanco EA, Husain-Krautter S, Madeline Fagen E, Moreno-Merino P, Del Ojo-Jimenez JA, Ahmed A, Rothstein TL, Lencz T, Malhotra AK. Cytokines in cerebrospinal fluid of patients with schizophrenia spectrum disorders: New data and an updated meta-analysis. Schizophr Res. 2018;202:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marques TR, Ashok AH, Pillinger T, Veronese M, Turkheimer FE, Dazzan P, Sommer IEC, Howes OD. Neuroinflammation in schizophrenia: meta-analysis of in vivo microglial imaging studies. Psychol Med. 2019;49:2186–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hafizi S, Tseng HH, Rao N, Selvanathan T, Kenk M, Bazinet RP, Suridjan I, Wilson AA, Meyer JH, Remington G, Houle S, Rusjan PM, Mizrahi R. Imaging Microglial Activation in Untreated First-Episode Psychosis: A PET Study With [(18)F]FEPPA. Am J Psychiatry. 2017;174:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Berckel BN, Bossong MG, Boellaard R, Kloet R, Schuitemaker A, Caspers E, Luurtsema G, Windhorst AD, Cahn W, Lammertsma AA, Kahn RS. Microglia activation in recent-onset schizophrenia: a quantitative (R)-[11C]PK11195 positron emission tomography study. Biol Psychiatry. 2008;64:820–822. [DOI] [PubMed] [Google Scholar]
- 56.Doorduin J, de Vries EF, Willemsen AT, de Groot JC, Dierckx RA, Klein HC. Neuroinflammation in schizophrenia-related psychosis: a PET study. J Nucl Med. 2009;50:1801–1807. [DOI] [PubMed] [Google Scholar]
- 57.Takano A, Arakawa R, Ito H, Tateno A, Takahashi H, Matsumoto R, Okubo Y, Suhara T. Peripheral benzodiazepine receptors in patients with chronic schizophrenia: a PET study with [11C]DAA1106. Int J Neuropsychopharmacol. 2010;13:943–950. [DOI] [PubMed] [Google Scholar]
- 58.Kenk M, Selvanathan T, Rao N, Suridjan I, Rusjan P, Remington G, Meyer JH, Wilson AA, Houle S, Mizrahi R. Imaging neuroinflammation in gray and white matter in schizophrenia: an in-vivo PET study with [18F]-FEPPA. Schizophr Bull. 2015;41:85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oestreich LK, Lyall AE, Pasternak O, Kikinis Z, Newell DT, Savadjiev P, Bouix S, Shenton ME, Kubicki M, Australian Schizophrenia Research B, Whitford TJ, McCarthy-Jones S. Characterizing white matter changes in chronic schizophrenia: A free-water imaging multi-site study. Schizophr Res. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oestreich LK, Pasternak O, Shenton ME, Kubicki M, Gong X, Australian Schizophrenia Research B, McCarthy-Jones S, Whitford TJ. Abnormal white matter microstructure and increased extracellular free-water in the cingulum bundle associated with delusions in chronic schizophrenia. Neuroimage Clin. 2016;12:405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lyall AE, Pasternak O, Robinson DG, Newell D, Trampush JW, Gallego JA, Fava M, Malhotra AK, Karlsgodt KH, Kubicki M, Szeszko PR. Greater extracellular free-water in first-episode psychosis predicts better neurocognitive functioning. Mol Psychiatry. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kraguljac NV, Anthony T, Monroe WS, Skidmore FM, Morgan CJ, White DM, Patel N, Lahti AC. A longitudinal neurite and free water imaging study in patients with a schizophrenia spectrum disorder. Neuropsychopharmacology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pettersson-Yeo W, Allen P, Benetti S, McGuire P, Mechelli A. Dysconnectivity in schizophrenia: where are we now? Neurosci Biobehav Rev. 2011;35:1110–1124. [DOI] [PubMed] [Google Scholar]
- 64.van den Heuvel MP, Fornito A. Brain networks in schizophrenia. Neuropsychol Rev. 2014;24:32–48. [DOI] [PubMed] [Google Scholar]
- 65.Baker JT, Holmes AJ, Masters GA, Yeo BT, Krienen F, Buckner RL, Ongur D. Disruption of cortical association networks in schizophrenia and psychotic bipolar disorder. JAMA Psychiatry. 2014;71:109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Friston K, Brown HR, Siemerkus J, Stephan KE. The dysconnection hypothesis (2016). Schizophr Res. 2016;176:83–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anticevic A, Cole MW, Repovs G, Savic A, Driesen NR, Yang G, Cho YT, Murray JD, Glahn DC, Wang XJ, Krystal JH. Connectivity, pharmacology, and computation: toward a mechanistic understanding of neural system dysfunction in schizophrenia. Front Psychiatry. 2013;4:169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Palaniyappan L, Simmonite M, White TP, Liddle EB, Liddle PF. Neural primacy of the salience processing system in schizophrenia. Neuron. 2013;79:814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hadley JA, Kraguljac NV, White DM, Ver Hoef L, Tabora J, Lahti AC. Change in brain network topology as a function of treatment response in schizophrenia: a longitudinal resting-state fMRI study using graph theory. NPJ Schizophr. 2016;2:16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kraguljac NV, White DM, Hadley JA, Visscher K, Knight D, ver Hoef L, Falola B, Lahti AC. Abnormalities in large scale functional networks in unmedicated patients with schizophrenia and effects of risperidone. Neuroimage Clin. 2016;10:146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sarpal DK, Argyelan M, Robinson DG, Szeszko PR, Karlsgodt KH, John M, Weissman N, Gallego JA, Kane JM, Lencz T, Malhotra AK. Baseline Striatal Functional Connectivity as a Predictor of Response to Antipsychotic Drug Treatment. Am J Psychiatry. 2016;173:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Driesen NR, McCarthy G, Bhagwagar Z, Bloch M, Calhoun V, D’Souza DC, Gueorguieva R, He G, Ramachandran R, Suckow RF, Anticevic A, Morgan PT, Krystal JH. Relationship of resting brain hyperconnectivity and schizophrenia-like symptoms produced by the NMDA receptor antagonist ketamine in humans. Mol Psychiatry. 2013;18:1199–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fleming LM, Javitt DC, Carter CS, Kantrowitz JT, Girgis RR, Kegeles LS, Ragland JD, Maddock RJ, Lesh TA, Tanase C, Robinson J, Potter WZ, Carlson M, Wall MM, Choo TH, Grinband J, Lieberman J, Krystal JH, Corlett PR. A multicenter study of ketamine effects on functional connectivity: Large scale network relationships, hubs and symptom mechanisms. Neuroimage Clin. 2019;22:101739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Carletti F, Woolley JB, Bhattacharyya S, Perez-Iglesias R, Fusar Poli P, Valmaggia L, Broome MR, Bramon E, Johns L, Giampietro V, Williams SC, Barker GJ, McGuire PK. Alterations in white matter evident before the onset of psychosis. Schizophr Bull. 2012;38:1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bloemen OJ, de Koning MB, Schmitz N, Nieman DH, Becker HE, de Haan L, Dingemans P, Linszen DH, van Amelsvoort TA. White-matter markers for psychosis in a prospective ultra-high-risk cohort. Psychol Med. 2010;40:1297–1304. [DOI] [PubMed] [Google Scholar]
- 76.Karlsgodt KH, Niendam TA, Bearden CE, Cannon TD. White matter integrity and prediction of social and role functioning in subjects at ultra-high risk for psychosis. Biol Psychiatry. 2009;66:562–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kochunov P, Hong LE. Neurodevelopmental and neurodegenerative models of schizophrenia: white matter at the center stage. Schizophr Bull. 2014;40:721–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Xu H, Li XM. White matter abnormalities and animal models examining a putative role of altered white matter in schizophrenia. Schizophr Res Treatment. 2011;2011:826976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhan Y, Paolicelli RC, Sforazzini F, Weinhard L, Bolasco G, Pagani F, Vyssotski AL, Bifone A, Gozzi A, Ragozzino D, Gross CT. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci. 2014;17:400–406. [DOI] [PubMed] [Google Scholar]
- 80.Wolkin A, Choi SJ, Szilagyi S, Sanfilipo M, Rotrosen JP, Lim KO. Inferior frontal white matter anisotropy and negative symptoms of schizophrenia: a diffusion tensor imaging study. Am J Psychiatry. 2003;160:572–574. [DOI] [PubMed] [Google Scholar]
- 81.Skelly LR, Calhoun V, Meda SA, Kim J, Mathalon DH, Pearlson GD. Diffusion tensor imaging in schizophrenia: relationship to symptoms. Schizophr Res. 2008;98:157–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bopp MHA, Zollner R, Jansen A, Dietsche B, Krug A, Kircher TTJ. White matter integrity and symptom dimensions of schizophrenia: A diffusion tensor imaging study. Schizophr Res. 2017;184:59–68. [DOI] [PubMed] [Google Scholar]
- 83.Kochunov P, Rowland LM, Fieremans E, Veraart J, Jahanshad N, Eskandar G, Du X, Muellerklein F, Savransky A, Shukla D, Sampath H, Thompson PM, Hong LE. Diffusion-weighted imaging uncovers likely sources of processing-speed deficits in schizophrenia. Proc Natl Acad Sci U S A. 2016;113:13504–13509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nestor PG, Kubicki M, Niznikiewicz M, Gurrera RJ, McCarley RW, Shenton ME. Neuropsychological disturbance in schizophrenia: a diffusion tensor imaging study. Neuropsychology. 2008;22:246–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reis Marques T, Taylor H, Chaddock C, Dell’acqua F, Handley R, Reinders AA, Mondelli V, Bonaccorso S, Diforti M, Simmons A, David AS, Murray RM, Pariante CM, Kapur S, Dazzan P. White matter integrity as a predictor of response to treatment in first episode psychosis. Brain. 2014;137:172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mitelman SA, Newmark RE, Torosjan Y, Chu KW, Brickman AM, Haznedar MM, Hazlett EA, Tang CY, Shihabuddin L, Buchsbaum MS. White matter fractional anisotropy and outcome in schizophrenia. Schizophr Res. 2006;87:138–159. [DOI] [PubMed] [Google Scholar]
- 87.Luck D, Buchy L, Czechowska Y, Bodnar M, Pike GB, Campbell JS, Achim A, Malla A, Joober R, Lepage M. Fronto-temporal disconnectivity and clinical short-term outcome in first episode psychosis: a DTI-tractography study. J Psychiatr Res. 2011;45:369–377. [DOI] [PubMed] [Google Scholar]
- 88.Cropley VL, Klauser P, Lenroot RK, Bruggemann J, Sundram S, Bousman C, Pereira A, Di Biase MA, Weickert TW, Weickert CS, Pantelis C, Zalesky A. Accelerated Gray and White Matter Deterioration With Age in Schizophrenia. Am J Psychiatry. 2017;174:286–295. [DOI] [PubMed] [Google Scholar]
- 89.Gur RE, Turetsky BI, Bilker WB, Gur RC. Reduced gray matter volume in schizophrenia. Arch Gen Psychiatry. 1999;56:905–911. [DOI] [PubMed] [Google Scholar]
- 90.Thompson PM, Vidal C, Giedd JN, Gochman P, Blumenthal J, Nicolson R, Toga AW, Rapoport JL. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci U S A. 2001;98:11650–11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Doostdar N, Kim E, Grayson B, Harte MK, Neill JC, Vernon AC. Global brain volume reductions in a sub-chronic phencyclidine animal model for schizophrenia and their relationship to recognition memory. J Psychopharmacol. 2019;33:1274–1287. [DOI] [PubMed] [Google Scholar]
- 92.Barnes SA, Sawiak SJ, Caprioli D, Jupp B, Buonincontri G, Mar AC, Harte MK, Fletcher PC, Robbins TW, Neill JC, Dalley JW. Impaired limbic cortico-striatal structure and sustained visual attention in a rodent model of schizophrenia. Int J Neuropsychopharmacol. 2014;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Crum WR, Sawiak SJ, Chege W, Cooper JD, Williams SCR, Vernon AC. Evolution of structural abnormalities in the rat brain following in utero exposure to maternal immune activation: A longitudinal in vivo MRI study. Brain Behav Immun. 2017;63:50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dempster K, Norman R, Theberge J, Densmore M, Schaefer B, Williamson P. Cognitive performance is associated with gray matter decline in first-episode psychosis. Psychiatry Res Neuroimaging. 2017;264:46–51. [DOI] [PubMed] [Google Scholar]
- 95.Wible CG, Anderson J, Shenton ME, Kricun A, Hirayasu Y, Tanaka S, Levitt JJ, O’Donnell BF, Kikinis R, Jolesz FA, McCarley RW. Prefrontal cortex, negative symptoms, and schizophrenia: an MRI study. Psychiatry Res. 2001;108:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Padmanabhan JL, Tandon N, Haller CS, Mathew IT, Eack SM, Clementz BA, Pearlson GD, Sweeney JA, Tamminga CA, Keshavan MS. Correlations between brain structure and symptom dimensions of psychosis in schizophrenia, schizoaffective, and psychotic bipolar I disorders. Schizophr Bull. 2015;41:154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.van Haren NE, Hulshoff Pol HE, Schnack HG, Cahn W, Brans R, Carati I, Rais M, Kahn RS. Progressive brain volume loss in schizophrenia over the course of the illness: evidence of maturational abnormalities in early adulthood. Biol Psychiatry. 2008;63:106–113. [DOI] [PubMed] [Google Scholar]
- 98.Steullet P, Cabungcal JH, Coyle J, Didriksen M, Gill K, Grace AA, Hensch TK, LaMantia AS, Lindemann L, Maynard TM, Meyer U, Morishita H, O’Donnell P, Puhl M, Cuenod M, Do KQ. Oxidative stress-driven parvalbumin interneuron impairment as a common mechanism in models of schizophrenia. Mol Psychiatry. 2017;22:936–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gonzalez-Burgos G, Fish KN, Lewis DA. GABA neuron alterations, cortical circuit dysfunction and cognitive deficits in schizophrenia. Neural Plast. 2011;2011:723184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, Barale F, Caverzasi E, McGuire P. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Arch Gen Psychiatry. 2012;69:220–229. [DOI] [PubMed] [Google Scholar]
- 101.Koutsouleris N, Riecher-Rossler A, Meisenzahl EM, Smieskova R, Studerus E, Kambeitz-Ilankovic L, von Saldern S, Cabral C, Reiser M, Falkai P, Borgwardt S. Detecting the psychosis prodrome across high-risk populations using neuroanatomical biomarkers. Schizophr Bull. 2015;41:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schobel SA, Lewandowski NM, Corcoran CM, Moore H, Brown T, Malaspina D, Small SA. Differential targeting of the CA1 subfield of the hippocampal formation by schizophrenia and related psychotic disorders. Arch Gen Psychiatry. 2009;66:938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Egerton A, Stone JM, Chaddock CA, Barker GJ, Bonoldi I, Howard RM, Merritt K, Allen P, Howes OD, Murray RM, McLean MA, Lythgoe DJ, O’Gorman RL, McGuire PK. Relationship between brain glutamate levels and clinical outcome in individuals at ultra high risk of psychosis. Neuropsychopharmacology. 2014;39:2891–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Favila R, Stephano S, Graff-Guerrero A. Striatal glutamate and the conversion to psychosis: a prospective 1H-MRS imaging study. Int J Neuropsychopharmacol. 2013;16:471–475. [DOI] [PubMed] [Google Scholar]
- 105.Bossong MG, Antoniades M, Azis M, Samson C, Quinn B, Bonoldi I, Modinos G, Perez J, Howes OD, Stone JM, Allen P, McGuire P. Association of Hippocampal Glutamate Levels With Adverse Outcomes in Individuals at Clinical High Risk for Psychosis. JAMA Psychiatry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kambeitz J, Kambeitz-Ilankovic L, Leucht S, Wood S, Davatzikos C, Malchow B, Falkai P, Koutsouleris N. Detecting neuroimaging biomarkers for schizophrenia: a meta-analysis of multivariate pattern recognition studies. Neuropsychopharmacology. 2015;40:1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ulloa A, Plis S, Calhoun V: Improving Classification Rate of Schizophrenia Using a Multimodal Multi-Layer Perceptron Model with Structural and Functional MR2018. [Google Scholar]
- 108.Kegeles LS, Abi-Dargham A, Frankle WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry. 2010;67:231–239. [DOI] [PubMed] [Google Scholar]
- 109.Frankle WG, Gil R, Hackett E, Mawlawi O, Zea-Ponce Y, Zhu Z, Kochan LD, Cangiano C, Slifstein M, Gorman JM, Laruelle M, Abi-Dargham A. Occupancy of dopamine D2 receptors by the atypical antipsychotic drugs risperidone and olanzapine: theoretical implications. Psychopharmacology (Berl). 2004;175:473–480. [DOI] [PubMed] [Google Scholar]
- 110.Javitt DC, Carter CS, Krystal JH, Kantrowitz JT, Girgis RR, Kegeles LS, Ragland JD, Maddock RJ, Lesh TA, Tanase C, Corlett PR, Rothman DL, Mason G, Qiu M, Robinson J, Potter WZ, Carlson M, Wall MM, Choo TH, Grinband J, Lieberman JA. Utility of Imaging-Based Biomarkers for Glutamate-Targeted Drug Development in Psychotic Disorders: A Randomized Clinical Trial. JAMA Psychiatry. 2018;75:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Weickert CS, Weickert TW, Pillai A, Buckley PF. Biomarkers in schizophrenia: a brief conceptual consideration. Dis Markers. 2013;35:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bodnar M, Harvey PO, Malla AK, Joober R, Lepage M. The parahippocampal gyrus as a neural marker of early remission in first-episode psychosis: a voxel-based morphometry study. Clin Schizophr Relat Psychoses. 2011;4:217–228. [DOI] [PubMed] [Google Scholar]
- 113.Li M, Li X, Das TK, Deng W, Li Y, Zhao L, Ma X, Wang Y, Yu H, Meng Y, Wang Q, Palaniyappan L, Li T. Prognostic Utility of Multivariate Morphometry in Schizophrenia. Front Psychiatry. 2019;10:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palaniyappan L, Liddle PF. Aberrant cortical gyrification in schizophrenia: a surface-based morphometry study. J Psychiatry Neurosci. 2012;37:399–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nelson EA, White DM, Kraguljac NV, Lahti AC. Gyrification Connectomes in Unmedicated Patients With Schizophrenia and Following a Short Course of Antipsychotic Drug Treatment. Front Psychiatry. 2018;9:699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Palaniyappan L, Marques TR, Taylor H, Handley R, Mondelli V, Bonaccorso S, Giordano A, McQueen G, DiForti M, Simmons A, David AS, Pariante CM, Murray RM, Dazzan P. Cortical folding defects as markers of poor treatment response in first-episode psychosis. JAMA Psychiatry. 2013;70:1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Egerton A, Broberg BV, Van Haren N, Merritt K, Barker GJ, Lythgoe DJ, Perez-Iglesias R, Baandrup L, During SW, Sendt KV, Stone JM, Rostrup E, Sommer IE, Glenthoj B, Kahn RS, Dazzan P, McGuire P. Response to initial antipsychotic treatment in first episode psychosis is related to anterior cingulate glutamate levels: a multicentre (1)H-MRS study (OPTiMiSE). Mol Psychiatry. 2018. [DOI] [PubMed] [Google Scholar]
- 118.Mouchlianitis E, Bloomfield MA, Law V, Beck K, Selvaraj S, Rasquinha N, Waldman A, Turkheimer FE, Egerton A, Stone J, Howes OD. Treatment-Resistant Schizophrenia Patients Show Elevated Anterior Cingulate Cortex Glutamate Compared to Treatment-Responsive. Schizophr Bull. 2016;42:744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sarpal DK, Robinson DG, Lencz T, Argyelan M, Ikuta T, Karlsgodt K, Gallego JA, Kane JM, Szeszko PR, Malhotra AK. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry. 2015;72:5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.White TP, Wigton R, Joyce DW, Collier T, Fornito A, Shergill SS. Dysfunctional Striatal Systems in Treatment-Resistant Schizophrenia. Neuropsychopharmacology. 2016;41:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kraguljac NV, White DM, Hadley N, Hadley JA, Ver Hoef L, Davis E, Lahti AC. Aberrant Hippocampal Connectivity in Unmedicated Patients With Schizophrenia and Effects of Antipsychotic Medication: A Longitudinal Resting State Functional MRI Study. Schizophr Bull. 2016;42:1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hadley JA, Nenert R, Kraguljac NV, Bolding MS, White DM, Skidmore FM, Visscher KM, Lahti AC. Ventral tegmental area/midbrain functional connectivity and response to antipsychotic medication in schizophrenia. Neuropsychopharmacology. 2014;39:1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Doucet GE, Moser DA, Luber MJ, Leibu E, Frangou S. Baseline brain structural and functional predictors of clinical outcome in the early course of schizophrenia. Mol Psychiatry. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang LX, Guo F, Zhu YQ, Wang HN, Liu WM, Li C, Wang XR, Cui LB, Xi YB, Yin H. Effect of second-generation antipsychotics on brain network topology in first-episode schizophrenia: A longitudinal rs-fMRI study. Schizophr Res. 2019;208:160–166. [DOI] [PubMed] [Google Scholar]
- 125.Cao B, Cho RY, Chen D, Xiu M, Wang L, Soares JC, Zhang XY. Treatment response prediction and individualized identification of first-episode drug-naive schizophrenia using brain functional connectivity. Mol Psychiatry. 2018. [DOI] [PubMed] [Google Scholar]
- 126.Cui LB, Cai M, Wang XR, Zhu YQ, Wang LX, Xi YB, Wang HN, Zhu X, Yin H. Prediction of early response to overall treatment for schizophrenia: A functional magnetic resonance imaging study. Brain Behav. 2019;9:e01211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cuthbert BN, Insel TR. Toward the future of psychiatric diagnosis: the seven pillars of RDoC. BMC Med. 2013;11:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Insel TR. The NIMH Research Domain Criteria (RDoC) Project: precision medicine for psychiatry. Am J Psychiatry. 2014;171:395–397. [DOI] [PubMed] [Google Scholar]
- 129.Tamminga CA, Pearlson GD, Stan AD, Gibbons RD, Padmanabhan J, Keshavan M, Clementz BA. Strategies for Advancing Disease Definition Using Biomarkers and Genetics: The Bipolar and Schizophrenia Network for Intermediate Phenotypes. Biol Psychiatry Cogn Neurosci Neuroimaging. 2017;2:20–27. [DOI] [PubMed] [Google Scholar]
- 130.Goff DC, Romero K, Paul J, Mercedes Perez-Rodriguez M, Crandall D, Potkin SG. Biomarkers for drug development in early psychosis: Current issues and promising directions. Eur Neuropsychopharmacol. 2016;26:923–937. [DOI] [PubMed] [Google Scholar]
- 131.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. [DOI] [PubMed] [Google Scholar]
- 132.Berdenis van Berlekom A, Muflihah CH, Snijders G, MacGillavry HD, Middeldorp J, Hol EM, Kahn RS, de Witte LD. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr Bull. 2020;46:374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Grzenda A, Widge AS. Electroencephalographic Biomarkers for Predicting Antidepressant Response: New Methods, Old Questions. JAMA Psychiatry. 2020. [DOI] [PubMed] [Google Scholar]
- 134.Widge AS, Bilge MT, Montana R, Chang W, Rodriguez CI, Deckersbach T, Carpenter LL, Kalin NH, Nemeroff CB. Electroencephalographic Biomarkers for Treatment Response Prediction in Major Depressive Illness: A Meta-Analysis. Am J Psychiatry. 2019;176:44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Pletcher MJ, Pignone M. Evaluating the clinical utility of a biomarker: a review of methods for estimating health impact. Circulation. 2011;123:1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Parkinson DR, McCormack RT, Keating SM, Gutman SI, Hamilton SR, Mansfield EA, Piper MA, Deverka P, Frueh FW, Jessup JM, McShane LM, Tunis SR, Sigman CC, Kelloff GJ. Evidence of clinical utility: an unmet need in molecular diagnostics for patients with cancer. Clin Cancer Res. 2014;20:1428–1444. [DOI] [PubMed] [Google Scholar]
- 137.Insel TR, Landis SC, Collins FS. Research priorities. The NIH BRAIN Initiative. Science. 2013;340:687–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gosho M, Nagashima K, Sato Y. Study designs and statistical analyses for biomarker research. Sensors (Basel). 2012;12:8966–8986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Makowski C, Lepage M, Evans AC. Head motion: the dirty little secret of neuroimaging in psychiatry. J Psychiatry Neurosci. 2019;44:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cetin Karayumak S, Bouix S, Ning L, James A, Crow T, Shenton M, Kubicki M, Rathi Y. Retrospective harmonization of multi-site diffusion MRI data acquired with different acquisition parameters. Neuroimage. 2019;184:180–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fusar-Poli P, Hijazi Z, Stahl D, Steyerberg EW. The Science of Prognosis in Psychiatry: A Review. JAMA Psychiatry. 2018;75:1289–1297. [DOI] [PubMed] [Google Scholar]
- 142.Calhoun VD, Lawrie SM, Mourao-Miranda J, Stephan KE. Prediction of Individual Differences from Neuroimaging Data. Neuroimage. 2017;145:135–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Davatzikos C Machine learning in neuroimaging: Progress and challenges. Neuroimage. 2019;197:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Woo CW, Chang LJ, Lindquist MA, Wager TD. Building better biomarkers: brain models in translational neuroimaging. Nat Neurosci. 2017;20:365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Health NIoM: Explainable artificial intelligence for decoding and modulating neural circuit activity linked to behavior. 2019. pp. https://grants.nih.gov/grants/guide/pa-files/PAR-19-344.html.
- 146.Pepe MS, Janes H, Li CI, Bossuyt PM, Feng Z, Hilden J. Early-Phase Studies of Biomarkers: What Target Sensitivity and Specificity Values Might Confer Clinical Utility? Clin Chem. 2016;62:737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. J Natl Cancer Inst. 2008;100:1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Poline JB, Breeze JL, Ghosh S, Gorgolewski K, Halchenko YO, Hanke M, Haselgrove C, Helmer KG, Keator DB, Marcus DS, Poldrack RA, Schwartz Y, Ashburner J, Kennedy DN. Data sharing in neuroimaging research. Front Neuroinform. 2012;6:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schulz KF, Altman DG, Moher D, Group C. CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340:c332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Scherer A Reproducibility in biomarker research and clinical development: a global challenge. Biomark Med. 2017;11:309–312. [DOI] [PubMed] [Google Scholar]
- 152.Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, Donohue MC, Green RC, Harvey D, Jack CR Jr., Jagust W, Morris JC, Petersen RC, Saykin AJ, Shaw L, Thompson PM, Toga AW, Trojanowski JQ, Alzheimer’s Disease Neuroimaging I. Impact of the Alzheimer’s Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement. 2015;11:865–884. [DOI] [PMC free article] [PubMed] [Google Scholar]