

Abstract
Objective
To summarize current understanding of diagnostic and post-diagnostic evaluation of common variable immunodeficiency (CVID).
Data sources:
PubMed Central.
Study selections
Original research articles and review articles from 2015 to 2020 along with seminal articles that shaped the diagnostic and post-diagnostic evaluation of CVID were incorporated. This work focuses on initial diagnosis of CVID, genetic evaluations, and post-diagnostic assessment of respiratory, gastrointestinal, hepatobiliary disease as well as spleen and lymph node enlargement.
Results
CVID presents not only with frequent infections, but also with noninfectious complications such as autoimmunity, gastrointestinal disease, chronic lung disease, granulomas, liver disease, lymphoid hyperplasia, splenomegaly, or malignancy. The risk of morbidity and mortality is higher in patients with CVID and noninfectious complications. Detailed diagnostic approaches, which may incorporate genetic testing, can aid characterization of individual CVID cases and shape treatment in some instances. Moreover, continued evaluation after CVID diagnosis is key to optimal management of this complex disorder. These post-diagnostic evaluations include pulmonary function testing, radiologic studies, and laboratory evaluations that may be conducted at frequencies determined by disease activity.
Conclusion
While the diagnosis can be achieved similarly in all CVID patients, those with noninfectious complications have distinct concerns during clinical evaluation. State-of-the-art work-up of CVID with noninfectious complications typically includes genetic analysis, which may shape precision therapy, and thoughtful application of postdiagnostic tests that monitor the presence and progression of disease in the myriad of tissues that may be affected. Even with recent advancements, knowledge gaps in diagnosis, prognosis, and treatment of CVID persist, and continued research efforts are needed.
Keywords: common variable immunodeficiency, CVID, diagnosis, diagnostic testing, laboratory tests, noninfectious complications, pulmonary function testing, radiology
Introduction
Common Variable Immune deficiency (CVID) is a severe form of primary antibody deficiency with heterogeneous phenotypes and etiologies. It is the most prevalent symptomatic primary immunodeficiency estimated to occur in approximately 1 in 25,000.1,2 Although CVID can vary in its presentation, its underlying commonality is hypogammaglobulinemia. Various other abnormalities that accompany this primary antibody deficiency result in a myriad of complications from autoimmunity to lymphoproliferative disorders. In this review, we focus on diagnostic testing and post-diagnostic testing for CVID and conclude with an update on research efforts addressing current knowledge gaps.
CVID Diagnosis
The most recent International Consensus Document (ICON) guidelines list five criteria for CVID diagnosis: (1) IgG level less than 2 standard deviations below age-appropriate references (Table 1) for 2 measurements more than 3 weeks apart unless the level is very low (<100-300 mg/dL depending on the age), (2) either a low IgA or IgM, (3) poor antibody responses to vaccination, (4) greater than 4 years of age, (5) no secondary causes of hypogammaglobulinemia.3 The diagnostic criteria of the European Society for Immunodeficiencies (ESID) has several key differences from the ICON guidelines, (1) decrease of IgA is required, (2) low switched memory B cells (less than 70% of age related normal value) can be used instead of measurement of antibody response to vaccine, (3) no evidence of profound T-cell deficiency, and (4) a clinical manifestation of disease such as an increased susceptibility to infection, autoimmune manifestations, granulomatous disease, or unexplained polyclonal lymphoproliferation, or an affected family member with antibody deficiency (Figure 1).4
Table 1.
Immunoglobulin standards used in evaluation of suspected CVID.
Cut off for Immunoglobulins 2 Standard deviation below the mean by Age and Gender (g/L)
| Male | Female | |||||
|---|---|---|---|---|---|---|
| Age | IgG | IgA | IgM | IgG | IgA | IgM |
| 4 | 5.1 | 0.36 | 0.35 | 5.3 | 0.33 | 0.42 |
| 7 | 6 | 0.48 | 0.37 | 5.9 | 0.44 | 0.45 |
| 10 | 6.6 | 0.57 | 0.38 | 6.4 | 0.52 | 0.48 |
| 14 | 6.6 | 0.64 | 0.4 | 6.8 | 0.62 | 0.5 |
| 18 | 6.5 | 0.68 | 0.41 | 6.9 | 0.69 | 0.52 |
| 20 | 6.5 | 0.71 | 0.41 | 6.9 | 0.71 | 0.53 |
| 30 | 6.5 | 0.8 | 0.43 | 6.8 | 0.75 | 0.57 |
| 40 | 6.6 | 0.87 | 0.42 | 6.7 | 0.78 | 0.55 |
| 50 | 6.6 | 0.93 | 0.4 | 6.6 | 0.81 | 0.49 |
| 60 | 6.7 | 0.98 | 0.39 | 6.5 | 0.85 | 0.45 |
| 70 | 6.8 | 1.03 | 0.38 | 6.5 | 0.9 | 0.42 |
| 80 | 6.9 | 1.07 | 0.37 | 6.5 | 0.96 | 0.39 |
Adapted from Reference distributions for immunoglobulins A, G, and M: A comparison of a large cohort to the world's literature.77
Figure 1.
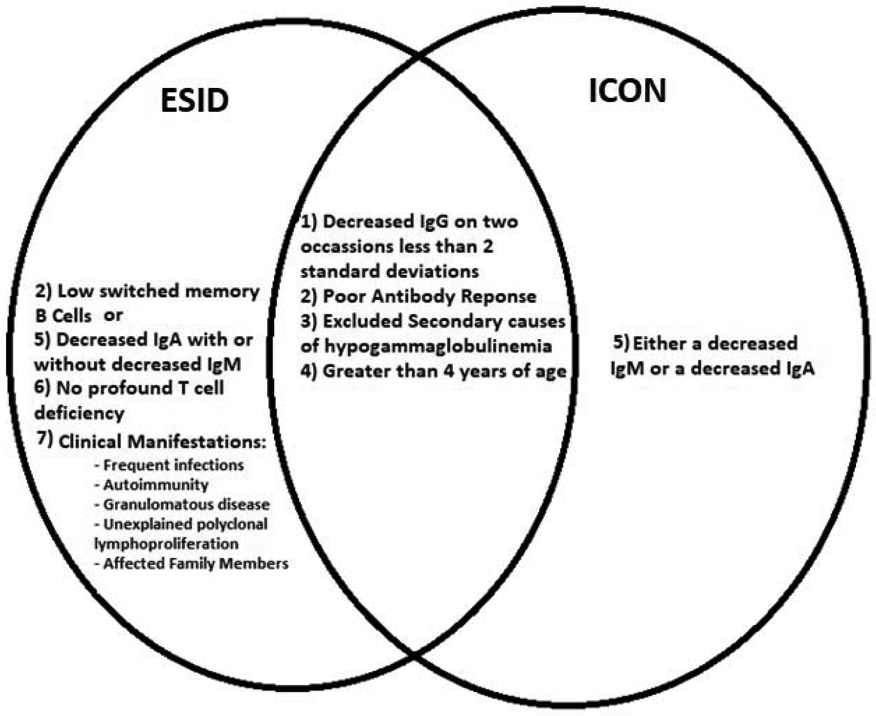
Commonality and differences in key aspects of CVID diagnosis between ESID and ICON.
ESID = European society for immunodeficiencies; ICON = international consensus document.
Next, distinguishing CVID phenotypes is clinically significant because morbidity and mortality can vary. Two seminal works found that CVID patients with noninfectious complications (CVIDc) had worsened survival compared to CVID patients who had infections only (CVIDu) despite usage of immunoglobulin replacement therapy.5,6 The latter paper demonstrated that a simple grouping of CVID patients into those with or without noninfectious complications identified sharp differences in outcome. Given the heterogeneous nature of CVID, such a binary grouping may be an oversimplification. Moreover, as there may be instances where a patient initially appears uncomplicated but later a CVIDc phenotype becomes apparent, flexibility in such designations must be maintained. However, dividing patients into CVIDc and CVIDu is a useful way to identify those likely to need more or less extensive clinical evaluation, respectively. In this manuscript, a patient is classified as having CVIDc if they have a history of autoimmunity, inflammatory bowel disease, interstitial lung disease, significant granulomatous inflammation, chronic liver disease, or CVID-related malignancies like lymphoma.7
History
Hypogammaglobulinemia
On the initial evaluation, it is important that secondary causes of hypogammaglobulinemia be ruled out. A careful medication history will identify agents that may cause secondary hypogammaglobulinemia (Table 2).3 Likewise, clinical history may reveal medical conditions that cause secondary hypogammaglobulinemia, such as significant liver disease, nephrotic syndrome, or severe GI protein loss. Once these secondary causes have been evaluated, a systems-based approach to the patient’s history helps clinching the diagnosis of CVID and enables identification of various complications that may affect these patients.
Table 2.
Medications associated with hypogammaglobulinemia
| Antimalarial |
| Captopril |
| Carbamazepine |
| Glucocorticoids |
| Fenclofenac |
| Gold salts |
| Penicillamine |
| Phenytoin |
| Sulfasalazine |
| Anti-CD20 mAbs (rituximab) |
Adapted from the International Consensus Document (ICON): Common Variable Immunodeficiency Disorders.3
Recurrent Infections
The most common presentation for CVID is recurrent infections, particularly upper and lower respiratory tract infections such as bronchitis, sinusitis, and pneumonia. This is primarily believed to be caused by bacterial pathogens, such as Streptococcus pneumoniae and Haemophilus influenzae, but viral infections likely contribute significantly as well.8,9 Gastrointestinal (GI) tract infections also frequently affect CVID patients and history of diarrhea caused by Giardia, Salmonella, or Campylobacter, among other potential pathogens, is often identified in the initial clinical history of CVID patients.9 Some CVID patients may be identified without a notable infectious history, with immune deficiency work-up initiated on the basis of autoimmune and inflammatory complications characteristic of these patients.
Autoimmunity
The most common noninfectious complication of CVID is likely autoimmunity. Autoimmunity in CVID can involve several organ systems including the gastrointestinal tract, skin, joints, and endocrine system. The most frequent manifestation of autoimmunity is autoimmune cytopenias, typically manifesting as immune thrombocytopenia, autoimmune hemolytic anemia, or, less frequently, autoimmune neutropenia.10 Autoimmune cytopenias occur in approximately 5-20% of CVID patients, depending upon the cohort studied.7,11 Autoimmune manifestations of the GI tract include pernicious anemia, atrophic gastritis, and autoimmune enteropathy.5 Manifestations in the skin include alopecia, lichen planus, vitiligo, and psoriasis.5,7 Endocrine diseases include thyroid involvement from hypothyroidism to thyrotoxicosis as well as insulin-dependent diabetes mellitus.5 Other less common autoimmune complications include systemic lupus erythematosus, vasculitis, antiphospholipid syndrome, anticardiolipin antibody, multiple sclerosis, myasthenia gravis, autoimmune pancreatitis, and severe oral aphthous ulcers.7 Accordingly, patients with autoimmune disease may be screened for hypogammaglobulinemia. Certainly in those with recurrent or persistent autoimmune cytopenias, the diagnosis of CVID should be strongly considered.
Enteropathy
Patients with CVID can experience a variety of GI issues that affect 15% or more of patients.7,11 The most common reported symptom among CVID patients is bowel movement changes; however, the problems can range from recurrent GI infections, such as giardiasis or Campylobacter, to autoimmune GI conditions, as previously mentioned, to malignancy, such as GI cancers and lymphomas.12-14 Enteropathy also encompasses inflammatory bowel disease (Crohn’s disease, ulcerative colitis, ulcerative proctitis), chronic diarrhea of unknown etiology, gastrointestinal bleeding, diverticulitis, irritable bowel syndrome, and esophagitis. Interestingly, eosinophilic esophagitis can manifest in CVID patients despite profound impairment of class-switched antibodies like IgE.15 The presence of significant enteropathy increases non-malignant morbidity in CVID.6
Pulmonary complications
As most common infections in CVID are localized to the upper and lower respiratory tract, it is not surprising that chronic lung disease is a frequent complication of CVID.9 Thirty percent or more of CVID patients suffer from chronic lung disease.16 Asthma may be the most common chronic respiratory manifestation as it is the most often reported.17 Indicating potential for misdiagnosis of lung disease in these patients, one study found only about 30% of CVID patients with obstructive lung disease to have asthma.18 Thus, alternative etiologies of obstructive lung disease should be considered. Bronchiectasis is one of the major manifestations in this category. This complication likely results from tissue damage due to repeated lung infections.19 However, bronchiectasis alone may not increase mortality.6 Interstitial lung disease (ILD), on the other hand, is clearly associated with increased morbidity and mortality.6 While the relationship of ILD with infection is not clear, this complication is associated with autoimmunity and splenomegaly, highlighting a likely role of systemic immune dysregulation in its pathogenesis and its inclusion as a noninfectious complication of CVID.17,20 About 10-20% of CVID patients have ILD.21
Hepatic complications
CVID has been associated with chronic liver disease in about 12% of patients seen at tertiary centers.7,11 The presentation can vary depending on the degree of involvement including granulomas, primary biliary cholangitis, cirrhosis, primary sclerosing cholangitis, nodular regenerative hyperplasia, portal hypertension, esophageal varices with possible variceal bleeding, ascites, spontaneous bacterial peritonitis, hepatopulmonary syndrome, and hepatic encephalopathy.7 The utility of evaluating for hypogammaglobulinemia in all instances of chronic liver disease can be debated. Indeed, chronic liver disease itself can be a cause of hypogammaglobulinemia, complicating the interpretation of such a work-up. Yet, the diagnoses of granulomatous hepatitis and nodular regenerative hyperplasia are rare outside of primary immunodeficiency, and evaluation for CVID should be considered in those with this liver pathology.
Lymphoid hyperplasia and malignancy
CVID can affect the lymph nodes and spleen to varying degrees, typically causing enlargement of these tissues that is detected by physical exam or radiology study. Lymphadenopathy or splenomegaly is benign in most cases, but malignancy does occur and lymphoma is thought to occur more frequently in CVID than the general population, and more specifically in CVIDc. Non-Hodgkin’s lymphomas are the most common hematological malignancies in CVID, and lymphoma is associated with an increased mortality in these patients.6,7 It can be challenging to determine whether a patient has CVID complicated by lymphoma or secondary antibody deficiency due to lymphoma and/or its treatment.
Family history
CVID typically does not run in families, though there are some genetic etiologies linked with CVID that may be identified through family history.22 Importantly, a presumed monogenetic mutation, such as a mutation in CTLA4 or LRBA, should still be considered in those with a family that appears to skip a generation or not affect all individuals the same way. Genetic variants found in CVID patients have been demonstrated to have incomplete penetrance and present as differing phenotypes within the same families.23 We will discuss the utility of genetic testing in CVID in greater detail later in this review.
Laboratory evaluation
Initial evaluation
Immunoglobulin levels are a fundamental component of the diagnosis of CVID as noted earlier. There are additional blood tests that also aid in the work-up of CVID. A basic metabolic panel, liver function testing, and urinalysis can assist in determining the involvement of other organ systems and also rule out liver disease and kidney disease, which can be secondary causes of hypogammaglobulinemia.3 A serum and urine protein electrophoresis can be considered if there is a suspicion for multiple myeloma, another potential secondary cause of hypogammaglobulinemia. Complete blood count with differential and a chemistry panel with liver function testing can be obtained yearly for those with CVIDu, but more frequent labs may be needed for those with CVIDc depending upon the specific complications affecting the patient.24 More frequent evaluation of kidney function is also advisable in those recently started on immunoglobulin replacement therapy, as renal complications have been observed in patients receiving immunoglobulin replacement, particularly sucrose-containing products that may cause osmotic injury in rare instances.3
A complete blood count with differential is important to detect cytopenias including thrombocytopenia, a frequent manifestation of CVID, and lymphopenia, which may also be present.5 Additionally, increased circulating monocytes may be a feature of CVIDc.25 Flow cytometry of the various lymphocyte subsets can be helpful as well. Relative to CVIDu, CVIDc is typically characterized by lower isotype-switched memory B cells (CD19+CD27+IgM− IgD−) and expansion of CD21low or transitional B cells in the blood (Table 3).26 While CVID is often thought of as a B-cell defect, there is T cell involvement in approximately half of patients. T cell lymphopenia of either the CD4+ or CD8+ subsets as well as defective proliferation to specific and non-specific activators are found in CVID, typically those with CVIDc.27 Most clinicians will categorize those with profound T cell lymphopenia (total T cell counts < 500/μL or CD4+ T cells < 200/μL) as combined immunodeficiency rather than CVID. Lastly, measurement of CD45RA and CD45RO may also be considered, as reduction of CD45RA+ naïve T cells and increased proportion of CD45RO+ effector and memory T cells are frequently found in CVIDc.28-31 Profound elevation of CD45RO+ T cells may indicate hyperactivation of mTOR, which can potentially be modulated by rapamycin to improve noninfectious complications of CVID.32,33
Table 3.
CD markers within available B-cell panels
| T cells | CD4 | CD4+ and/or CD8+ T cells may be reduced in CVID, particularly CVIDc. |
| CD8 | ||
| CD45RA | Reduction of CD45RA+ naïve T cells and increased proportion of CD45RO+ T cells are frequently found in CVIDc. | |
| CD45RO | ||
| B cells | CD19 | CVIDc is typically characterized by low isotype-switched memory B cells (CD19+, CD27+) |
| CD27 | ||
| CD21low | CVIDc is typically characterized by expansion of CD21low B cells in the blood |
CVIDc = CVID with noninfectious complications.
It is also vital to determine the patient’s response to vaccines in order to meet diagnostic criteria for CVID.3,4 The most commonly measured antibodies are those against tetanus toxoid, diphtheria toxoid, Haemophilus influenzae, and pneumococcus. These are useful vaccines for diagnostic purposes due to the fact that booster immunizations are needed and these are not live vaccines, so they do not impart the same risks to immunodeficient patients. If the patient has received live viral vaccines like measles, mumps, or rubella in the past, antibody measurements can be considered, but further immunization is not recommended in those with suspected CVID. The 23-valent pneumococcal carbohydrate vaccine is used to evaluate T-cell independent antibodies.3 Additionally, measurement of antibodies against hepatitis A, hepatitis B, influenza virus, or isohemagglutinins may further aid detection of functional impairment of immunoglobulins.34
GI evaluation
Tests that should be considered in CVID patients who present with GI complaints include fecal calprotectin, fecal α1-antitrypsin, and vitamin B12 levels, which has been shown to be low in patients with malabsorption.35 Fecal calprotectin is more sensitive than fecal α1-antitrypsin for detecting mucosal intestinal inflammation and identifying or monitoring disease activity in those with inflammatory bowel disease.35,36 A colonoscopy and/or esophagogastroduodenoscopy can be considered depending on the results of these tests and severity of symptoms. Due to the risk of GI malignancy some have advocated for universal screening endoscopies37,38 for CVID patients while others advise screening only for symptomatic patients, such as those with transient or persistent diarrhea and often weight loss.24
In a small study of 30 CVID patients, upper endoscopy and colonoscopy were performed on all subjects (88% had GI complaints) and abnormalities were detected in approximately 83%.39 Other studies have shown that general screening of CVID patients can lead to early detection of GI pathology in as high as 80%, including intestinal metaplasia, atrophic gastritis, collagenous gastritis, giardiasis, duodenal villous atrophy, and intraepithelial lymphocytosis.40 Biopsies taken from the GI mucosa can show excess intraepithelial lymphocytes, villous blunting, lymphoid aggregates, granulomas, crypt distortion, and a lack of plasma cells.37,41 However, the clinical significance of these findings remains to be fully demonstrated. Currently, there is no universally recommended screening established by consensus. Thus, risks and benefits of such evaluations must be considered with each patient individually.
Pulmonary Evaluation
The most recent ICON recommends CVID patients have a chest CT scan and pulmonary function measurement that includes gas transfer, such as diffusion capacity of the lung for carbon monoxide (DLCO), obtained at some time relatively close to the time of diagnosis and those without evidence of disease may be monitored at least annually with spirometry.3 Abnormalities of these parameters seem to be the earliest markers of significant chronic lung disease.42 Highlighting the prevalence of chronic lung disease in CVID, one study found 80% had radiological evidence of bronchial pathology including bronchiectasis in 61%, bronchial wall thickening in 44%, and mucus plugging in 29%.43
The most common symptom of ILD in CVID patients is dyspnea. Pulmonary function testing may show a restrictive pattern and/or decreased DLCO.44 On CT scan, patients can be noted to have reticular changes, fibrosis, or ground glass appearances.45 Biopsy may be needed to define the specific type of lung disease present. Biopsy of ILD in CVID most typically demonstrates benign lymphoproliferative pathology like lymphocytic interstitial pneumonia (LIP), follicular bronchiolitis, other forms of benign lymphoid hyperplasia, or nonnecrotizing granulomatous lung disease. As granulomatous inflammation is often seen together with benign lymphoproliferation, the term granulomatous-lymphocytic interstitial lung disease (GLILD) is often used for CVID ILD.44 It is important to note that GLILD is an umbrella term for the numerous pathologies identified in CVID, and as some specific pathological diagnosis in CVID can have both granulomatous and lymphocytic inflammation, like LIP, the diagnosis of GLILD may not be utilized by all pathologists.
Hepatic and Splenic Evaluation
CVID liver involvement includes cirrhosis, portal hypertension, and nodular regenerative hyperplasia, an intra-hepatic vasculopathy characterized by alterations in microvascular perfusion leading to hepatocyte injury that can lead to portal hypertension, esophageal varices, and splenomegaly.46,47 If hepatic involvement is suspected, a liver function panel may show an elevation in alkaline phosphatase.20 As this lab value is not specific for a diagnosis, imaging such as ultrasonography, CT scan, or magnetic resonance imaging (MRI) can be considered. An emerging tool is transient elastography, which measures the liver’s stiffness by interpreting the vibrations generated by a specialized ultrasound machine called the FibroScan. This has been used to assess liver involvement without exposing the patient to the risk of infection or bleeding from a biopsy procedure.48 However, biopsy is needed in many cases to ascertain the etiology of liver damage.49 Biopsies typically show nonspecific portal and lobular inflammation, hepatitis, lymphocyte infiltration without plasma cells, granulomas, fibrosis, macrovesicular steatosis, and neogenesis of biliary ducts.49-52
Splenomegaly occurs in 26%-38% of CVID patients.9,20 The extent of splenomegaly can vary. It may be asymptomatic, or it can be linked with chronic liver disease as previously mentioned. Annual or a more frequent evaluation of spleen size and growth rate can be done by ultrasound. Routine splenectomy is not typically indicated as this may increase the risk of infection, but may be considered in severe cases.24 CVID patients with significant splenomegaly appear to be more likely to have autoimmune cytopenias or other types of lymphoproliferation, such as ILD53.
Lymph Node Evaluation
It is not uncommon for patients with CVID to have lymphadenopathy, and while this may be concerning for lymphoma, it is frequently benign in these patients. Such benign lymph node biopsies usually show atypical lymphoid hyperplasia, reactive lymphoid hyperplasia, or granulomatous inflammation with lack of plasma cells.37,54 Yet, the presence of lymphoproliferative disease warrants vigilance in this patient population. One study of CVID patients with lymphoproliferative disease found a 2.5-fold increased odds of having lymphoma than CVID patients without lymphoproliferative disease.55 In most cases, lymphomas are extra nodal and appear in unusual locations such as the lung or mucosal associated tissues.22
Genetic Analysis
Application of next-generation sequencing is most likely to be useful for CVIDc because it offers the opportunity to apply precision medicine to treat autoimmune and inflammatory disease.56 Although many patients may have polygenic or otherwise multifactorial disease, around 15-30% of cases of CVID are now estimated to be monogenic.57,58 Adoption of diagnostic panels of primary immunodeficiency associated genes by commercial and increased ability of medical centers to utilize whole exome or genome sequencing approaches have coincided with reduced cost of these approaches. It is imperative to initiate discussion with the patient prior to this genetic testing to establish goals and expectations. Some patients and doctors may experience anxiety from indeterminate results, including variants of uncertain significance.
Genetic analysis can be used to shape targeted therapy. Examples of where identification of genetic variants may influence treatment in patients with CVID or CVID-like diseases include mutations in LRBA, CTLA4, STAT3, and PIK3CD. Lipopolysaccharide (LPS)-responsive and beige-like anchor protein (LRBA) is a protein encoded by LRBA that assists in the vesicle trafficking and the turnover of the checkpoint molecule cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), an important regulator of T cell activation and mediator of regulatory T cell function.59 The most common clinical manifestations of loss-of-function mutations in LRBA involves splenomegaly and hepatomegaly, autoimmunity, manifesting as immune-mediated cytopenias and organ-specific autoimmunity, and chronic diarrhea associated with lymphocytic infiltration of the GI tract.60 Similarly, mutations of CTLA4 can manifest as hypogammaglobulinemia, diarrhea/enteropathy, ILD, respiratory infections, lymphocytic organ infiltration, and splenomegaly. Abatacept is a CTLA-4 immunoglobulin fusion protein, which is used as a CTLA-4 replacement, and has been shown to be effective to treat noninfectious complications in patients with loss-of-function mutations in LRBA as well as CTLA4.59,61,62
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor tightly controlled by numerous cytokines, growth factors, and hormones. In a study of 13 patients with STAT3 gain-of-function mutation the predominant clinical manifestations were autoimmunity, including autoimmune hemolytic anemia, autoimmune thrombocytopenia, enteropathy, and type 1 diabetes mellitus as well as lymphadenopathy and hepatosplenomegaly.63 Targeted treatments that have shown efficacy in these patients are Janus kinase inhibitors known as jakinibs and tocilizumab, an IL-6 receptor antagonist.63,64 Both IL-6 and Janus kinase are key components of the STAT3 activation cascade. Another gain-of-function defect that can be identified during evaluation of patients with suspected CVID involves PIK3CD. This gene encodes the δ subunit of phosphoinositide-3-kinase, an enzyme involved in growth and proliferation of white blood cells with potent affects upon B and T cell activation. Gain-of-function mutation in PIK3CD causes excessive antigen nonspecific activation of B and T cells, leading to poorly functional or “exhausted” lymphocytes.65 Patients with gain-of-function mutations in PIK3CD, known as the activated PI3K delta syndrome (APDS) often present with recurrent respiratory infections and benign lymphoproliferation with bronchiectasis, GI disease, autoimmune cytopenias, glomerulonephritis, arthritis, and colitis. The lymphoproliferative phenotype of APDS patients increases the risk of lymphoma.66 Rapamycin, which inhibits the mTOR pathway hyper-activated by PI3KD gain-of-function, as well as targeted inhibitors of PI3Kδ have been shown to be efficacious for these patients.66
Emerging approaches in the evaluation of CVID
Serial measurement of serum IgM
Elevated serum IgM has been associated with lymphoproliferative disease and decreased survival in CVID.5,6 Increases in serum IgM of 10 mg/dL or more over 20 months have been associated with progression of ILD as defined by decline in forced vital capacity of 10% or DLCO of 15% predicted.5,6 Additional work identified the degree of serum IgM elevation correlated with the number of ectopic pulmonary B cell follicles and extent of IgM production within them.25 Thus, serial measurement of IgM may be most useful in CVIDc with extensive B cell hyperplasia, particularly in the lungs. Indeed, improvement and recurrence of ILD after rituximab corresponded with decline and elevation of IgM, respectively.
B cell maturation antigen
A frequent challenge in the evaluation of CVID occurs when patients have already begun immunoglobulin replacement therapy but the diagnosis is in question. IgG levels and antibody responses to most vaccines cannot be interpreted in this setting without stopping the immunoglobulin replacement and increasing potential risk of infection. Moreover, evaluation of vaccine responses can last a month or more, delaying treatment. Finally, in those who have received immunosuppression, determining whether hypogammaglobinemia is transient or persistent, and thus requiring immunoglobulin replacement therapy, is challenging. For these reasons, ongoing efforts to improve the diagnosis of CVID are underway.
One such diagnostic approach under evaluation is measurement of serum B cell maturation antigen (BCMA). BCMA is a tumor necrosis factor superfamily receptor for the cytokines a proliferation inducing ligand (APRIL) and B cell activating factor (BAFF) which promote activation and survival. BCMA is almost exclusively expressed on plasma cells, cells that are absent or significantly reduced in CVID. Plasma cells largely reside in the bone marrow or mucosal tissues and their presence cannot be determined without biopsy. As BCMA is endogenously cleaved from the cell surface by γ-secretase into a soluble form that can be measured in the blood, it can act as a rapid, non-invasive surrogate measure of plasma cells. Indeed, serum BCMA was found to be decreased in CVID and X-linked agammaglobulinemia patients and efforts are underway to test this diagnostic tool in challenging clinical scenarios.67
Flow cytometry
As mentioned earlier, application of flow cytometry to assess levels of B cell and T cell subsets has long been incorporated into clinical evaluation of CVID. Recently, expanded application of this approach identified distinct patterns of B cell defects associated with specific forms of primary antibody deficiency.68 While IgA deficiency was defined by loss of IgA-expressing plasma and memory cells, those with CVID show 6 different defects of memory B cells that included IgG- as well as IgA-expressing subsets. Though the clinical application of such detailed immunophenotyping remains to be defined, the extensive subtyping of CVID possible through flow cytometry offers potential to help make sense of observed clinical heterogeneity and improve classification.
Cytokine measurement
Currently, we are unable to predict which CVID patients will develop noninfectious complications or identify when a known complication will progress. All the parameters previously discussed have been associated with the presence or absence of a clinical finding, rather than preceding or predicting clinical manifestations. Thus, we face a “chicken and the egg” phenomenon when interpreting much of the work conducting thus far on CVIDc. Elevation of cytokines has been shown to precede disease activity in systemic lupus erythematosus and other autoimmune diseases. While a similar finding has not yet been uncovered in CVID, the potential exists for a similar prognostic application because similar cytokines are elevated.
Numerous studies have found elevation of T helper type 1 cytokines like IL-12 and IFN-γ in CVIDc, utilizing numerous laboratory approaches such as measurement of RNA expression, ex vivo cell culture, flow cytometry, and measurement of protein in the blood.25,69-75 Recently, application of proteomics, measurement of numerous cytokines and chemokines in the blood simultaneously by array, was included in an unbiased analysis of CVID patients. In this study two distinct CVID groups were identified, with IFN-γ and IL-1β as the most enriched cytokines associated with CVIDc.76 Broad clinical application of proteomics or approaches aimed at measuring RNA cytokine signature have proven useful in uncovering the type 1 interferon signature that precedes disease activity in systemic lupus erythematosus. Ultimately, these advances may help improve our ability to identify CVID patients at risk of development or progression of noninfectious complications.
Conclusion
Though diagnosis of CVID is built upon fundamental evidence of profound antibody deficiency, heterogeneity of this disorder is increasingly appreciated. Chronic noninfectious complications have emerged as the major source of morbidity and mortality and the target of current research efforts. Clinical evaluation of CVID patients can vary depending upon the specific features of their presentation, namely the presence of these noninfectious complications that significantly influence clinical course and justify more extensive evaluation. An example of this individualized approach to CVID is outlined in Figure 2.
Figure 2.
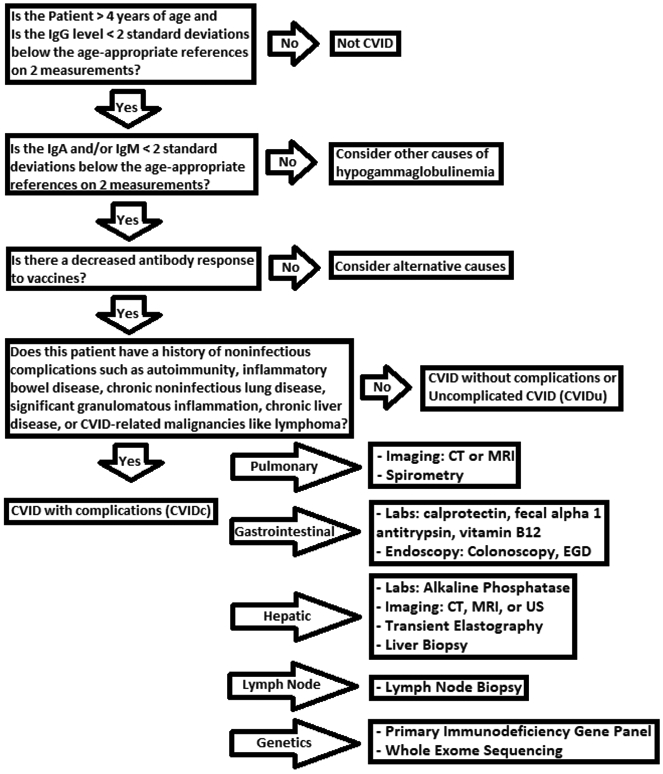
Proposed strategy for clinical evaluation of CVID.
Recently, genetics has uncovered precision treatment approaches offering great potential. New diagnostic tools are being developed and offer hope to further improve management of CVID. With an expanding appreciation of the pathogenic mechanisms underlying noninfectious complications of CVID, the state-of-the-art in clinical evaluation of this immune disorder continues to evolve in exciting ways.
Table 4.
CVID-relevant genes and associated disorders
| Gene defect | Protein function | Clinical Manifestation | Treatment |
|---|---|---|---|
| LRBA Loss of function | Assists in the vesicle trafficking and the turnover of the checkpoint of CTLA4 | splenomegaly and hepatomegaly, autoimmunity, manifesting as immune-mediated cytopenias and organ-specific autoimmunity, and chronic diarrhea | Abatacept |
| CTLA4 Loss of function | An important regulator of T cell activation and mediator of regulatory T cell function | hypogammaglobulinemia, diarrhea/enteropathy, ILD, respiratory infections, lymphocytic organ infiltration, and splenomegaly | Abatacept |
| STAT3 Gain of function | Transcription factor tightly controlled by numerous cytokines, growth factors, and hormones | autoimmunity, including autoimmune hemolytic anemia, autoimmune thrombocytopenia, enteropathy, and type 1 diabetes mellitus as well as lymphadenopathy and hepatosplenomegaly | Jakinibs and Tocilizumab |
| PIK3CD Gain of function | Enzyme involved in growth and proliferation of white blood cells with potent affects upon B and T cell activation | recurrent respiratory infections and benign lymphoproliferation with bronchiectasis, GI disease, autoimmune cytopenias, glomerulonephritis, arthritis, and colitis | Rapamycin |
Adapted from the Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity10
Key Messages.
Recent advancements in our understanding of CVID pathogenesis now shape diagnostic and longitudinal evaluation of this disease.
Work-up of CVID involves thoughtful application of testing to identify and monitor the presence and progression of disease in the myriad of tissues that may be affected.
Genetic testing is potentially useful in CVID, particularly those with noninfectious complications for which it may shape treatment.
Given the extensive heterogeneity of CVID, not all with this diagnosis should be followed with an identical approach. Those with complicated courses warrant closer, more frequent monitoring tailored to their specific manifestation of CVID.
Significant gaps in our knowledge of CVID diagnostic evaluation persist and offer potential to improve identification and management of this primary immunodeficiency if elucidated.
Acknowledgments
Funding Source: PJM received support from National Institutes of Health grants AI137183 and AI151486, a faculty development grant from the American Association of Allergy, Asthma and Immunology Foundation, and a Boston University Career Investment Award.
Abbreviations/Acronyms:
- CTLA-4
cytotoxic T-lymphocyte-associated protein 4
- CVID
common variable immunodeficiency
- CVIDc
CVID with noninfectious complications
- CVIDu
CVID uncomplicated by noninfectious complications
- DLCO
diffusion capacity of the lung for carbon monoxide
- GI
gastrointestinal
- GLILD
granulomatous-lymphocytic interstitial lung disease
- ICON
International Consensus Document
- ILD
interstitial lung disease
- LIP
lymphocytic interstitial pneumonia
- STAT3
signal transducer and activator of transcription 3
Footnotes
Conflicts of Interest: The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Odnoletkova I, Kindle G, Quinti I, et al. The burden of common variable immunodeficiency disorders : A retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis. Published online 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. Published online 2009. doi: 10.1111/j.1365-2141.2009.07669.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonilla FA, Barlan I, Chapel H, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. Published online 2016. doi: 10.1016/j.jaip.2015.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Edgar D, Ehl S. ESID Registry - Working definitions for clinical diagnosis of PID. ESID Regist – Work Defin Clin Diagnosis PID. Published online 2016. [Google Scholar]
- 5.Chapel H, Lucas M, Lee M, et al. Common Variable immunodeficiency disorders: Division into distinct clinical phenotypes. Blood. Published online 2008. doi: 10.1182/blood-2007-11-124545 [DOI] [PubMed] [Google Scholar]
- 6.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. Published online 2012. doi: 10.1182/blood-2011-09-377945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho HE, Cunningham-Rundles C. Non-infectious Complications of Common Variable Immunodeficiency: Updated Clinical Spectrum, Sequelae, and Insights to Pathogenesis. Front Immunol. Published online 2020. doi: 10.3389/fimmu.2020.00149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pulvirenti F, Camilli R, Giufrè M, et al. Risk factors for Haemophilus influenzae and pneumococcal respiratory tract colonization in CVID. J Allergy Clin Immunol. Published online 2018. doi: 10.1016/j.jaci.2018.08.014 [DOI] [PubMed] [Google Scholar]
- 9.Oksenhendler E, Gérard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. Published online 2008. doi: 10.1086/587669 [DOI] [PubMed] [Google Scholar]
- 10.Gereige JD, Maglione PJ. Current Understanding and Recent Developments in Common Variable Immunodeficiency Associated Autoimmunity. Front Immunol. Published online 2019. doi: 10.3389/fimmu.2019.02753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farmer JR, Ong MS, Barmettler S, et al. Common variable immunodeficiency non-infectious disease endotypes redefined using unbiased network clustering in large electronic datasets. Front Immunol. Published online 2018. doi: 10.3389/fimmu.2017.01740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cunningham-Rundles C, Siegal FP, Cunningham-Rundles S, Lieberman P. Incidence of cancer in 98 patients with common varied immunodeficiency. J Clin Immunol. Published online 1987. doi: 10.1007/BF00915550 [DOI] [PubMed] [Google Scholar]
- 13.Kinlen LJ, Webster ADB, Bird AG, et al. PROSPECTIVE STUDY OF CANCER IN PATIENTS WITH HYPOGAMMAGLOBULINAEMIA. Lancet. Published online 1985. doi: 10.1016/S0140-6736(85)91037-2 [DOI] [PubMed] [Google Scholar]
- 14.Mellemkjær L, Hammarström L, Andersen V, et al. Cancer risk among patients with IgA deficiency or common variable immunodeficiency and their relatives: A combined Danish and Swedish study. Clin Exp Immunol. Published online 2002. doi: 10.1046/j.1365-2249.2002.02004.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen M, Ko HM, Riffle ME, et al. Eosinophilic esophagitis diagnosed in a patient with common variable immunodeficiency. J Allergy Clin Immunol Pract. Published online 2016. doi: 10.1016/j.jaip.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maglione PJ. Chronic Lung Disease in Primary Antibody Deficiency: Diagnosis and Management. Immunol Allergy Clin North Am. Published online 2020. doi: 10.1016/j.iac.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weinberger T, Fuleihan R, Cunningham-Rundles C, Maglione PJ. Factors Beyond Lack of Antibody Govern Pulmonary Complications in Primary Antibody Deficiency. J Clin Immunol. Published online 2019. doi: 10.1007/s10875-019-00640-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agondi RC, Barros MT, Rizzo LV, Kalil J, Giavina-Bianchi P. Allergic asthma in patients with common variable immunodeficiency. Allergy Eur J Allergy Clin Immunol. Published online 2010. doi: 10.1111/j.1398-9995.2009.02211.x [DOI] [PubMed] [Google Scholar]
- 19.Schussler E, Beasley MB, Maglione PJ. Lung Disease in Primary Antibody Deficiencies. J Allergy Clin Immunol Pract. Published online 2016. doi: 10.1016/j.jaip.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gathmann B, Mahlaoui N, Gérard L, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. Published online 2014. doi: 10.1016/j.jaci.2013.12.1077 [DOI] [PubMed] [Google Scholar]
- 21.Katsumi T, Murayama K. A case of non-functioning adrenal cortical carcinoma with pulmonary and bony metastases. Acta Urol Jpn. Published online 1989. [PubMed] [Google Scholar]
- 22.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin Immunol. Published online 1999. doi: 10.1006/clim.1999.4725 [DOI] [PubMed] [Google Scholar]
- 23.Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: One diagnosis to rule them all? J Med Genet. Published online 2016. doi: 10.1136/jmedgenet-2015-103690 [DOI] [PubMed] [Google Scholar]
- 24.Cunningham-Rundles C How I treat common variable immune deficiency. Blood. Published online 2010. doi: 10.1182/blood-2010-01-254417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maglione PJ, Gyimesi G, Cols M, et al. BAFF-driven B cell hyperplasia underlies lung disease in common variable immunodeficiency. JCI insight. Published online 2019. doi: 10.1172/jci.insight.122728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arumugakani G, Wood PMD, Carter CRD. Frequency of treg cells is reduced in CVID patients with autoimmunity and splenomegaly and is associated with expanded CD21lo B lymphocytes. J Clin Immunol. Published online 2010. doi: 10.1007/s10875-009-9351-3 [DOI] [PubMed] [Google Scholar]
- 27.Giovannetti A, Pierdominici M, Mazzetta F, et al. Unravelling the Complexity of T Cell Abnormalities in Common Variable Immunodeficiency. J Immunol. Published online 2007. doi: 10.4049/jimmunol.178.6.3932 [DOI] [PubMed] [Google Scholar]
- 28.Azizi G, Kiaee F, Hedayat E, et al. Rheumatologic complications in a cohort of 227 patients with common variable immunodeficiency. Scand J Immunol. Published online 2018. doi: 10.1111/sji.12663 [DOI] [PubMed] [Google Scholar]
- 29.Baumert E, Wolff-Vorbeck G, Schlesier M, Peter HH. Immunophenotypical alterations in a subset of patients with common variable immunodeficiency (CVID). Clin Exp Immunol. Published online 1992. doi: 10.1111/j.1365-2249.1992.tb05826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Genre J, Errante PR, Kokron CM, Toledo-Barros M, Câmara NOS, Rizzo LV. Reduced frequency of CD4+CD25HIGHFOXP3+ cells and diminished FOXP3 expression in patients with Common Variable Immunodeficiency: A link to autoimmunity? Clin Immunol. Published online 2009. doi: 10.1016/j.clim.2009.03.519 [DOI] [PubMed] [Google Scholar]
- 31.Mouillot G, Carmagnat M, Gérard L, et al. B-cell and T-cell phenotypes in CVID patients correlate with the clinical phenotype of the disease. J Clin Immunol. Published online 2010. doi: 10.1007/s10875-010-9424-3 [DOI] [PubMed] [Google Scholar]
- 32.Lucas CL, Kuehn HS, Zhao F, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. Published online 2014. doi: 10.1038/ni.2771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deyaà-Martínez A, Esteve-Solé A, Vélez-Tirado N, et al. Sirolimus as an alternative treatment in patients with granulomatous-lymphocytic lung disease and humoral immunodeficiency with impaired regulatory T cells. Pediatr Allergy Immunol. 2018;29(4). doi: 10.1111/pai.12890 [DOI] [PubMed] [Google Scholar]
- 34.Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. Published online 2012. doi: 10.1016/j.jaci.2012.03.025 [DOI] [PubMed] [Google Scholar]
- 35.Pikkarainen S, Martelius T, Ristimäki A, Siitonen S, Seppänen MRJ, Färkkilä M. A High Prevalence of Gastrointestinal Manifestations in Common Variable Immunodeficiency. Am J Gastroenterol. Published online 2019. doi: 10.14309/ajg.0000000000000140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lewis JD. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology. Published online 2011. doi: 10.1053/j.gastro.2010.11.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agarwal S, Mayer L. Pathogenesis and treatment of gastrointestinal disease in antibody deficiency syndromes. J Allergy Clin Immunol. Published online 2009. doi: 10.1016/j.jaci.2009.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dhalla F, da Silva SP, Lucas M, Travis S, Chapel H. Review of gastric cancer risk factors in patients with common variable immunodeficiency disorders, resulting in a proposal for a surveillance programme. Clin Exp Immunol. Published online 2011. doi: 10.1111/j.1365-2249.2011.04384.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maarschalk-Ellerbroek LJ, Oldenburg B, Mombers IMH, et al. Outcome of screening endoscopy in common variable immunodeficiency disorder and X-linked agammaglobulinemia. Endoscopy. Published online 2013. doi: 10.1055/s-0032-1326078 [DOI] [PubMed] [Google Scholar]
- 40.van der Poorten DK, McLeod D, Ahlenstiel G, et al. Gastric Cancer Screening in Common Variable Immunodeficiency. J Clin Immunol. Published online 2018. doi: 10.1007/s10875-018-0546-3 [DOI] [PubMed] [Google Scholar]
- 41.Daniels JA, Lederman HM, Maitra A, Montgomery EA. Gastrointestinal tract pathology in patients with common variable immunodeficiency (CVID): A clinicopathologic study and review. Am J Surg Pathol. Published online 2007. doi: 10.1097/PAS.0b013e3180cab60c [DOI] [PubMed] [Google Scholar]
- 42.Ilowite J, Spiegler P, Chawla S. Bronchiectasis: New findings in the pathogenesis and treatment of this disease. Curr Opin Infect Dis. Published online 2008. doi: 10.1097/QCO.0b013e3282f4f237 [DOI] [PubMed] [Google Scholar]
- 43.Schütz K, Alecsandru D, Grimbacher B, et al. Imaging of Bronchial Pathology in Antibody Deficiency: Data from the European Chest CT Group. J Clin Immunol. Published online 2019. doi: 10.1007/s10875-018-0577-9 [DOI] [PubMed] [Google Scholar]
- 44.Park JH, Levinson AI. Granulomatous-lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Immunol. Published online 2010. doi: 10.1016/j.clim.2009.10.002 [DOI] [PubMed] [Google Scholar]
- 45.Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol. Published online 2004. doi: 10.1016/j.jaci.2004.05.057 [DOI] [PubMed] [Google Scholar]
- 46.Fuss IJ, Friend J, Yang Z, et al. Nodular regenerative hyperplasia in Common Variable Immunodeficiency. J Clin Immunol. Published online 2013. doi: 10.1007/s10875-013-9873–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song J, Lleo A, Yang GX, et al. Common Variable Immunodeficiency and Liver Involvement. Clin Rev Allergy Immunol. Published online 2018. doi: 10.1007/s12016-017-8638-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crescenzi L, Pecoraro A, Fiorentino A, et al. Liver stiffness assessment by transient elastography suggests high prevalence of liver involvement in common variable immunodeficiency. Dig Liver Dis. Published online 2019. doi: 10.1016/j.dld.2019.05.016 [DOI] [PubMed] [Google Scholar]
- 49.Szablewski V, René C, Costes V. Indolent cytotoxic T cell lymphoproliferation associated with nodular regenerative hyperplasia: a common liver lesion in the context of common variable immunodeficiency disorder. Virchows Arch. Published online 2015. doi: 10.1007/s00428-015-1862-0 [DOI] [PubMed] [Google Scholar]
- 50.Martire B, Gentile A, Francavilla R, De Santis A, De Mattia D. Successful treatment with cyclosporine A of HCV-driven chronic liver disease mimicking autoimmune hepatitis in a patient with common variable immunodeficiency. Immunopharmacol Immunotoxicol. Published online 2005. doi: 10.1080/08923970500416723 [DOI] [PubMed] [Google Scholar]
- 51.Fukushima K, Ueno Y, Kanegane H, et al. A case of severe recurrent hepatitis with common variable immunodeficiency. Hepatol Res. Published online 2008. doi: 10.1111/j.1872-034X.2007.00281.x [DOI] [PubMed] [Google Scholar]
- 52.Daniels JA, Torbenson M, Vivekanandan P, Anders RA, Boitnott JK. Hepatitis in common variable immunodeficiency. Hum Pathol. Published online 2009. doi: 10.1016/j.humpath.2008.09.008 [DOI] [PubMed] [Google Scholar]
- 53.Maglione PJ. Autoimmune and Lymphoproliferative Complications of Common Variable Immunodeficiency. Curr Allergy Asthma Rep. Published online 2016. doi: 10.1007/s11882-016-0597-6 [DOI] [PubMed] [Google Scholar]
- 54.Sander CA, Medeiros LJ, Weiss LM, Yano T, Sneller MC, Jaffe ES. Lymphoproliferative lesions in patients with common variable immunodeficiency syndrome. Am J Surg Pathol. Published online 1992. doi: 10.1097/00000478-199212000-00004 [DOI] [PubMed] [Google Scholar]
- 55.Yakaboski E, Fuleihan RL, Sullivan KE, Cunningham-Rundles C, Feuille E. Lymphoproliferative Disease in CVID: a Report of Types and Frequencies from a US Patient Registry. J Clin Immunol. Published online 2020. doi: 10.1007/s10875-020-00769-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kienzler AK, Hargreaves CE, Patel SY. The role of genomics in common variable immunodeficiency disorders. Clin Exp Immunol. Published online 2017. doi: 10.1111/cei.12947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Valles-Ibáñnez G, Esteve-Solé A, Piquer M, et al. Evaluating the genetics of common variable immunodeficiency: Monogenetic model and beyond. Front Immunol. Published online 2018. doi: 10.3389/fimmu.2018.00636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Maffucci P, Filion CA, Boisson B, et al. Genetic diagnosis using whole exomesequencing in common variable immunodeficiency. Front Immunol. Published online 2016. doi: 10.3389/fimmu.2016.00220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lo B, Zhang K, Lu W, et al. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (80- ). Published online 2015. doi: 10.1126/science.aaa1663 [DOI] [PubMed] [Google Scholar]
- 60.Kostel Bal S, Haskologlu S, Serwas NK, et al. Multiple Presentations of LRBA Deficiency: a Single-Center Experience. J Clin Immunol. Published online 2017. doi: 10.1007/s10875-017-0446-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uzel G, Karanovic D, Su H, et al. Management of Cytopenias in CTLA4 Haploinsufficiency Using Abatacept and Sirolimus. Blood. Published online 2018. doi: 10.1182/blood-2018-99-120185 [DOI] [Google Scholar]
- 62.Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol. Published online 2017. doi: 10.1111/cei.12997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Milner JD, Vogel TP, Forbes L, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood. Published online 2015. doi: 10.1182/blood-2014-09-602763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Forbes LR, Vogel TP, Cooper MA, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. Published online 2018. doi: 10.1016/j.jaci.2018.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Angulo I, Vadas O, Garcon F, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (80- ). Published online 2013. doi: 10.1126/science.1243292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maccari ME, Abolhassani H, Aghamohammadi A, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: The European society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol. Published online 2018. doi: 10.3389/fimmu.2018.00543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Years of Potential Life Lost due to Cancer — United States, 1968–1985. JAMA J Am Med Assoc. Published online 1989. doi: 10.1001/jama.1989.03420020053013 [DOI] [Google Scholar]
- 68.Blanco E, Pérez-Andrés M, Arriba-Méndez S, et al. Defects in memory B-cell and plasma cell subsets expressing different immunoglobulin-subclasses in patients with CVID and immunoglobulin subclass deficiencies. J Allergy Clin Immunol. Published online 2019. doi: 10.1016/j.jaci.2019.02.017 [DOI] [PubMed] [Google Scholar]
- 69.Cambronero R, Sewell WAC, North ME, Webster ADB, Farrant J. Up-Regulation of IL-12 in Monocytes: A Fundamental Defect in Common Variable Immunodeficiency. J Immunol. Published online 2000. doi: 10.4049/jimmunol.164.1.488 [DOI] [PubMed] [Google Scholar]
- 70.Cols M, Rahman A, Maglione PJ, et al. Expansion of inflammatory innate lymphoid cells in patients with common variable immune deficiency. J Allergy Clin Immunol. Published online 2016. doi: 10.1016/j.jaci.2015.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cunill V, Clemente A, Lanio N, et al. Follicular T cells from smB- common variable immunodeficiency patients are skewed toward a Th1 phenotype. Front Immunol. Published online 2017. doi: 10.3389/fimmu.2017.00174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martinez-Pomar N, Raga S, Ferrer J, et al. Elevated serum interleukin (IL)-12p40 levels in common variable immunodeficiency disease and decreased peripheral blood dendritic cells: Analysis of IL-12p40 and interferon-γ gene. Clin Exp Immunol. Published online 2006. doi: 10.1111/j.1365-2249.2006.03063.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Park J, Munagala I, Xu H, et al. Interferon Signature in the Blood in Inflammatory Common Variable Immune Deficiency. PLoS One. Published online 2013. doi: 10.1371/journal.pone.0074893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turpin D, Furudoi A, Parrens M, Blanco P, Viallard JF, Duluc D. Increase of follicular helper T cells skewed toward a Th1 profile in CVID patients with non-infectious clinical complications. Clin Immunol. Published online 2018. doi: 10.1016/j.clim.2018.09.006 [DOI] [PubMed] [Google Scholar]
- 75.Unger S, Seidl M, van Schouwenburg P, et al. The TH1 phenotype of follicular helper T cells indicates an IFN-γ–associated immune dysregulation in patients with CD21low common variable immunodeficiency. J Allergy Clin Immunol. Published online 2018. doi: 10.1016/j.jaci.2017.04.041 [DOI] [PubMed] [Google Scholar]
- 76.Hultberg J, Ernerudh J, Larsson M, Nilsdotter-Augustinsson Å, Nyström S. Plasma protein profiling reflects TH1-driven immune dysregulation in common variable immunodeficiency. J Allergy Clin Immunol. Published online 2020. doi: 10.1016/j.jaci.2020.01.046 [DOI] [PubMed] [Google Scholar]
- 77.Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for immunoglobulins A, G, and M: A practical, simple, and clinically relevant approach in a large cohort. J Clin Lab Anal. Published online 1998. doi: [DOI] [PMC free article] [PubMed] [Google Scholar]