

Abstract
Over 100 kinds of proteins are expressed as glycosylphosphatidylinositol (GPI)-anchored proteins (GPI-APs) on the cell surface in mammalian cells. GPI-APs possess unique properties in terms of their intracellular trafficking and association with lipid rafts. Although it is clear that GPI-APs play critical roles in various biological phenomena, it is poorly understood how the GPI moiety contributes to these mechanisms. More than 30 genes are involved in the correct biosynthesis of GPI-APs. We here constructed a cell library in which 32 genes involved in GPI biosynthesis were knocked out in human embryonic kidney 293 cells. Using the cell library, the surface expression and sensitivity to phosphatidylinositol-specific phospholipase C of GPI-APs were analyzed. Furthermore, we identified structural motifs of GPIs that are recognized by a GPI-binding toxin, aerolysin. The cell-based GPI-knockout library could be applied not only to basic researches, but also to applications and methodologies related to GPI-APs.
Subject terms: Glycobiology, Glycobiology
Liu et al. construct a knock out cell library of genes involved in the biosynthesis of glycosylphosphatidylinositol (GPI)-anchored protein (GPI-APs) from HEK-293 cells and analyzed the surface expression and sensitivity to phosphatidylinositol-specific phospholipase C. Furthermore, the authors also identify structural motifs of GPIs that are recognized by a GPI-binding toxin, aerolysin.
Introduction
Various types of proteins, including single-membrane-spanning or multi membrane-spanning transmembrane proteins, lipidated proteins, and peripheral proteins, are localized on the cell surface. A number of proteins are covalently bound with a glycolipid, named glycosylphosphatidylinositol (GPI), and are localized at the plasma membrane. GPI anchoring of proteins is one of the common posttranslational modifications in eukaryotes1–4. GPI confers unique properties to modified proteins. GPI-anchored proteins (GPI-APs) are the major proteins that associate with lipid rafts, which are dynamic membrane microdomains composed of sphingolipids and cholesterol5. The association with lipid rafts regulates GPI-APs in polarized transport, endocytosis, and signal transduction in mammalian cells6–9.
The core structure of GPI is conserved and consists of EtNP-6-Man-α1,2-Man-α1,6-Man-α1,4-GlcN-α1,6-inositol-phospholipid (EtNP, ethanolamine-phosphate; Man, mannose; GlcN, glucosamine) among eukaryotic species, whereas the lipid moieties and glycan side chains are different10. In mammalian cells, several side chain modifications on GPI-glycan structures are found. The first Man (Man1) is modified with a side chain, EtNP. The fourth Man (Man4) can be attached to the third Man (Man3) via an α1,2-linkage11. In some proportions of mammalian GPI-APs, N-acetylgalactosamine (GalNAc) is modified to the Man1 through the β1,4-linkage12,13, and it can be further modified by β1,3-galactose (Gal)14 or β1,3-Gal with α2,3-sialic acid15,16. The GPI lipid portion is 1-alkyl-2-acyl-glycerol or diacylglycerol in mammals.
The biosynthesis of GPI is essential for embryonic development, neurogenesis, immune responses, and fertilization. Pathological congenital mutations in GPI biosynthetic genes cause inherited GPI deficiencies (IGDs), which are characterized by intellectual disability, epileptic seizures, hypotonia, and facial dysmorphisms17. Acquired mutations in a GPI biosynthesis gene in hematopoietic stem cells lead to paroxysmal nocturnal hemoglobinuria, in which hemolysis of red blood cells occurs by self-activation of the complement system18. On the other hand, the expression of GPI-APs, such as carcinoembryonic antigen (CEA)19, mesothelin20, folate receptor21, glypican-322, and CD5223 is upregulated in various cancer cells, and such GPI-APs are utilized as biomarkers and therapeutic targets.
GPI biosynthesis and transfer to proteins are carried out in the endoplasmic reticulum (ER). After GPI is transferred to proteins, the lipid and glycan parts of GPI moieties are processed at the ER and the Golgi during GPI-AP transport. Some GPI-APs are cleaved at the GPI moiety by GPI-cleaving enzymes (GPIases)24. There are at least 23 steps in which 33 genes (including 21 phosphatidylinositol-glycan (PIG) genes and 6 post-GPI attachment to proteins (PGAP) genes) are involved for correct GPI-AP biogenesis. Among them, it was reported that defects in 23 GPI biosynthetic genes cause IGDs25. Although the genes responsible for reactions in most steps were identified, it is still not clear how GPI biosynthesis is regulated and which GPI structures are required for these functions.
Here, we performed a systematic genetic disruption of GPI biosynthetic genes in a human embryonic kidney 293 (HEK293) cell line, providing a knockout gene cell library with different GPI-anchor biosynthesis capabilities (GPI-KO cell library). We used the CRISPR-Cas9 system to construct the library and systematically analyzed the expression and sensitivity to phosphatidylinositol-specific phospholipase C (PI-PLC) of GPI-APs. We proved that the GPI-KO cell library can be applied to determine toxin recognition sites and the unique biological characteristics of prion proteins. Our GPI-KO cell library is a sustainable resource for exploring various applications and methodologies of GPI-AP biology.
Results
GPI-AP synthesis capacity of HEK293 cells
HEK293 cells, which are widely used in both basic and applied studies, were used to construct a GPI-KO cell library. We obtained the expression of genes involved in the biosynthesis, processing, and transport of GPI-APs in HEK293 cells using RNA-seq (Supplementary Fig. 1a). All the genes required for GPI-AP biogenesis were expressed in HEK293 cells, whereas the expression of some genes, such as PIGY, PIGZ, and B3GALT4, was limited. In addition, the expression profiles of genes encoding GPI-APs were analyzed. At least 67 GPI-APs were expressed (TPM value ≥ 1) in HEK293 cells (Supplementary Fig. 1b). We detected endogenous GPI-APs expressed on the surface of HEK293 cells by flow cytometry (Supplementary Fig. 2a). Several GPI-APs that were not expressed in HEK293 cells were exogenously expressed, and their surface expression was detected (Supplementary Fig. 2b), suggesting that HEK293 cells could be used for the study of the biological characteristics of GPI-APs.
Genetic disruption of GPI biosynthesis in HEK293 cells
Using similar strategies to those employed for the knockout cell libraries of N-glycosylation26 and glycosaminoglycans (GAGs)27, genes encoding GPI biosynthesis were knocked out using the CRISPR–Cas9 system (Supplementary Fig. 3). We systematically designed knockout constructs targeting genes involved in GPI biosynthesis and validated the guide RNAs (gRNAs) for disruption (Fig. 1 and Supplementary Table 2). Two targets were selected on one exon of each gene and designed to allow knockout confirmation by eliminating the sequence on that exon. Then, using the validated gRNAs, we constructed 32 gene knockout cells (Fig. 1a), generating a GPI-KO cell library.
Fig. 1. The biosynthesis of GPI-APs in mammalian cells.
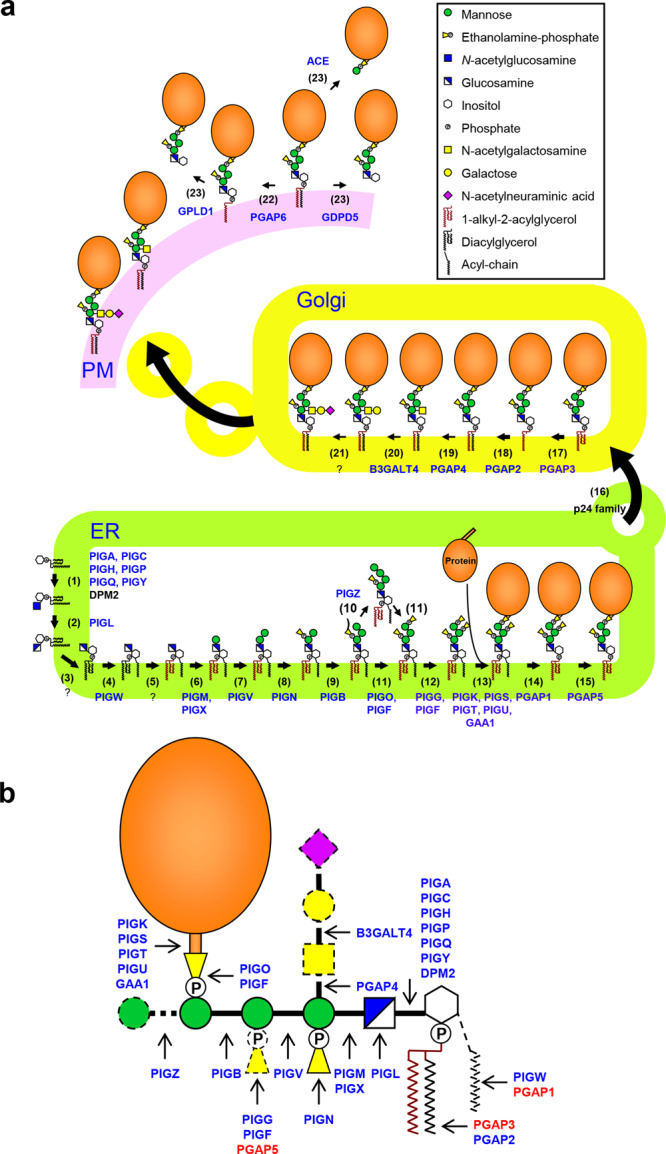
a GPI biosynthesis is carried out in the ER, Golgi, and the cell surface through a series of catalytic reactions. It starts with the transfer of GlcNAc to PI. The first two steps occur on the cytoplasmic side of the ER, and then the modification of polysaccharides and lipids occurs on the luminal side of the ER and Golgi. Proportions of GPI-APs further undergo side chain modification. Gene products that were knocked out in this study are highlighted in blue. b Schematic representation of a human GPI-AP structure. Gene products required for the synthesis of the GPI structure are shown in blue, and gene products that remove the structure are shown in red. The solid lines of the GPI structure indicate the core structure in human cells, whereas the dotted lines indicate accessory structures observed in human GPI-APs.
Initial steps of GPI biosynthesis
GPI biosynthesis starts with the addition of N-acetylglucosamine (GlcNAc) to PI at the cytosolic side of the ER membrane. GPI-GlcNAc transferase (GPI-GnT), composed of six core subunits, PIGA, PIGC, PIGH, PIGP, PIGQ, and PIGY, mediates GlcNAc to PI28 (Fig. 1a, Step 1). KO of PIGA, PIGC, PIGH, and PIGP completely lost GPI-AP expression, whereas KO of PIGQ and PIGY maintained weak expression of GPI-APs (Fig. 2 and Supplementary Fig. 4a), suggesting that KO of regulatory subunits retained GPI-GnT activity.
Fig. 2. GPI-AP expression on the cell surface in the GPI-knockout cell library.

Cell surface expression of three endogenous GPI-APs, CD55, CD59, and prion, was detected by flow cytometry. The expression level of GPI-APs in WT cells was set as 1, and the relative mean ± SD values from three independent experiments were displayed in a bar plot. CD55, blue; CD59, orange; prion, gray. *p < 0.05; **p < 0.01 according to an unpaired Student’s t test. ns not significant.
PIGL is required for the deacetylation of GlcNAc-PI to generate GlcN-PI29 (Fig. 1a, Step 2). GlcN-PI is then flipped into the luminal side of the ER. Inositol acyltransferase, PIGW, catalyzes the addition of an acyl chain to the 2-position of the inositol ring on GlcN-PI to form GlcN-(acyl)PI30 (Fig. 1a, Step 4). KO of these genes completely eliminated the synthesis of GPI-APs (Fig. 2 and Supplementary Fig. 4a).
GPI-mannosyltransferases (GPI-ManTs) and GPI-EtNP transferases
The complex composed of PIGM31 and PIGX32 (GPI-ManT-I) (Fig. 1a, Step 6), PIGV33 (GPI-ManT-II) (Fig. 1a, Step 7), PIGB34 (GPI-ManT-III) (Fig. 1a, Step 9), and PIGZ11 (GPI-ManT-IV) (Fig. 1a, Step 10) catalyzes the transfer of the first, second, third, and fourth Man to the GPI intermediate. PIGM and PIGX make a complex for GPI-ManT-I. The surface expression of GPI-APs was completely removed by KO of PIGM, which encodes the catalytic subunit of GPI-ManT-I. On the other hand, weak expression of GPI-APs remained after KO of PIGX, which encodes the regulatory subunit. KO of PIGV also completely removed the surface expression of GPI-APs. In contrast, KO of PIGB left some GPI-AP expression. The fourth Man modification by PIGZ was nonessential and its KO did not affect the biosynthesis of GPI-APs (Fig. 2). Since both PIGB and PIGZ are α1,2-ManTs, we knocked out PIGZ in PIGB-KO cells to check the redundancy. Even if PIGZ was knocked out in PIGB-KO cells, the expression of CD55 was not changed (Supplementary Fig. 4b), suggesting that PIGB and PIGZ do not have redundancy. Since it was shown that other ER-localized ManTs such as ALG3, 9, and 12, which are required for the biosynthesis of lipid-linked oligosaccharides, have strict specificity35, it would not be possible that those ManTs have redundancy to GPI-MTs. Instead, a GPI-anchor structure, in which a protein is attached to the second EtNP on Man2, was found36. Therefore, it is possible that proteins linked to the second EtNP of GPI are expressed in PIGB-KO cells.
Three EtNPs are added to Man1, Man3, and Man2 mediated by PIGN37 (GPI-EtNP transferase-I) (Fig. 1a, step 8), PIGO and PIGF complex38,39 (GPI-EtNP transferase-III) (Fig. 1a, step 11), and PIGG and PIGF complex40 (GPI-EtNP transferase-II) (Fig. 1a, step 12), generating a complete GPI precursor consisting of EtNP-Man-(EtNP)Man-(EtNP)Man-GlcN-(acyl)PI. GPI-APs were still expressed in PIGN-deficient cells, although the expression level was reduced to approximately 50%. In PIGG-KO cells, the expression of GPI-APs was normal, whereas it was extremely low in PIGO-deficient cells (Fig. 2). Although PIGB and PIGO mediate the addition of Man3 and EtNP to Man3 for protein binding, respectively, the KO cells did not completely lose GPI-AP expression. As mentioned above, we found a GPI-anchor structure in which a protein is attached to the second EtNP on Man236. In PIGO-KO cells, a proportion of proteins utilize the second EtNP for GPI anchoring.
GPI attachment to proteins
GPI-transamidase (GPI-TA) is a multisubunit complex containing five subunits: PIGK41, GAA142, PIGT43, PIGS43, and PIGU44. It recognizes the C-terminal GPI attachment signal of proteins45. PIGK is a catalytic subunit that cleaves the GPI attachment signal and forms an enzyme-substrate intermediate46. GPI is then transferred to the newly exposed C-terminus of the protein3 (Fig. 1a, Step 13). The deletion of every subunit caused the inactivation of GPI-TA, and GPI-APs were not synthesized (Fig. 2).
GPI-anchor remodeling
PGAP1 is a GPI-inositol deacylase that removes an acyl chain from the inositol ring of GPI47 (Fig. 1a, Step 14). Subsequently, a side chain EtNP attached to the second Man is removed by PGAP548 (Fig. 1a, Step 15). Although KO of PGAP1 or PGAP5 causes delayed transport of GPI-APs from the ER to the Golgi47,48, it did not affect the expression of GPI-AP on the cell surface at a steady state (Fig. 2).
In the Golgi apparatus, GPI fatty acid remodeling occurs, in which an unsaturated fatty acid at the sn-2 position of the GPI lipid is replaced with a saturated fatty acid. Elimination of an unsaturated fatty acid and transfer of a saturated fatty acid are mediated by PGAP349,50 (Fig. 1a, Step 17) and PGAP251 (Fig. 1a, Step 18), respectively. The surface expression of GPI-APs in PGAP3-KO cells was only mildly affected, whereas in PGAP2-deficient cells, the surface expression of GPI-APs, except prion, was significantly reduced (Fig. 2). This is because, in PGAP2-deficient cells, lyso-GPI-APs having only one hydrocarbon chain are transported to the cell surface, but they are unstable and released from the plasma membrane. The expression of prion in PGAP2-KO cells was almost comparable with that in WT cells, which was analyzed below.
Glycan moieties in some GPI-APs are further modified in the Golgi. PGAP4 transfers GalNAc to Man1 via 1,4-linkage12,13 (Fig. 1a, Step 19). B3GALT4 could transfer Gal to the GalNAc residue14 (Fig. 1a, Step 20). The KO cells did not affect the expression of GPI-APs (Fig. 2).
Shedding of GPI-APs from the cell membrane
GPI-APs can be cleaved at GPI moieties and released from the membrane as intact proteins (Fig. 1a). Angiotensin-converting enzyme (ACE), glycerol phosphodiesterase 2 (GDE2) (also known as GDPD5), GPI-specific phospholipase D (GPI-PLD/GPLD1), and PGAP6 have GPI-cleaving enzyme (GPIase) activity on specific GPI-APs24. We also constructed gene knockout cells to study the biological function of specific GPI-APs. The surface expression levels of CD59 in PGAP6-KO cells, CD55 in GPLD1-KO, and CD55 and prion in GDPD5-KO and ACE-KO cells were mildly but significantly higher than that in WT cells (Fig. 2). In the WT cells, a fraction of those GPI-APs would be released constantly by those GPIases. By the gene-KO, the surface expression was increased because the GPI-APs are not released.
KO of genes encoding enzymes involved in GPI biosynthesis led to partial PI-PLC resistance
PI-PLC hydrolyzes PI at the site between the phosphate and glycerol backbone10,52. Via treatment with PI-PLC, GPI-APs on the cell surface are cleaved and released (Fig. 3a). When an acyl chain is modified to the inositol ring, the GPI-APs show resistance to PI-PLC.
Fig. 3. The PI-PLC catalytic reaction releases GPI-APs on the cell surface.

a Schematic representation of PI-PLC activity against GPI-APs. The remodeled mature GPI-APs on the cell surface in WT cells are cleaved by PI-PLC and released from the plasma membrane. On the other hand, the inositol-acylated GPI-APs in PGAP1-KO cells show resistance to PI-PLC. b PI-PLC sensitivity of CD55, CD59, and prion. WT and KO cell lines were treated with and without PI-PLC. The residual amounts of CD55, CD59, and prion on the cell surface after PL-PLC treatment were analyzed by flow cytometry. The ratio of remaining GPI-APs was calculated and is shown as the mean ± SD from three independent experiments.
Among the GPI-KO cell library, KO cells that still show GPI-APs on the cell surface were picked up and used to detect the PI-PLC sensitivity of GPI-APs with different GPI structures. In the WT cells, more than 90% of CD55 and CD59 were cleaved by PI-PLC, whereas approximately 50% of the prion proteins showed resistance to PI-PLC treatment (Fig. 3b), suggesting that half of the prion expressed on the cell surface of HEK293 cells has an additional acyl chain. By KO of PGAP1, which encodes GPI-inositol deacylase, GPI-APs showed complete resistance to PI-PLC (Fig. 3b). In the PGAP2-KO cells, the majority of GPI-APs were not cleaved by PI-PLC (Fig. 3b), suggesting that GPI-APs retained on the cell surface in PGAP2-KO cells are inositol-acylated. Since it is known that inositol-acylated GPI-APs cannot be a substrate for PGAP350, GPI-APs that are not processed by PGAP1 may be expressed as inositol-acylated GPI-APs having three hydrocarbon chains in PGAP2-KO cells. In other GPI remodeling-deficient cells, including PGAP5-KO, PGAP3-KO, PGAP4-KO, and B3GALT4-KO cells, GPI-APs are processed by PI-PLC at a similar efficiency in WT cells. Similarly, cells defective in GPI-cleaving enzymes (including PGAP6-KO, GPLD1-KO, GDPD5-KO, and ACE-KO cells) showed that GPI-APs were as sensitive to PI-PLC as WT cells.
To determine the correlation of GPI-inositol deacylation with the presence of side chain EtNPs and Mans on GPI structures, we used cells defective in PIGN, PIGB, PIGO, PIGZ, or PIGG for GPI-AP cleavage by PI-PLC. In PIGZ-KO and PIGG-KO cells, the PI-PLC sensitivity of GPI-APs was normal, suggesting that the fourth Man and the second side chain EtNP on GPI do not affect GPI-inositol deacylation. On the other hand, in PIGO-KO cells, GPI-APs on the cell surface were resistant to PI-PLC treatment. KO of PIGB or PIGN mildly reduced PI-PLC sensitivity. These results suggest that the GPI structure affects GPI-inositol deacylation.
Dissection of inositol-acylated GPI on prion
In the initial analysis of GPI-AP expression and PI-PLC sensitivity in the GPI-KO cell library, the prion protein behaved differently from other GPI-APs, such as CD55 and CD59 (Figs. 2 and 3b). In PGAP2-KO cells, CD55 and CD59 surface expression decreased by more than 90%, whereas prion proteins on the cell surface were only 20% reduced on average (Fig. 4a and b). The PGAP2-rescued cells restored the reduction in CD59 and CD55 (Supplementary Fig. 5). According to western blotting of cell lysates, CD59 and CD55 were not detectable in PGAP2-KO cells, whereas prion proteins could be detected (Fig. 4c). Approximately, 50% of prion proteins were cleaved by PI-PLC in WT cells, whereas in PGAP2-KO cells, 85% of prion proteins on the cell surface showed resistance against PI-PLC (Fig. 3b), suggesting that prion proteins have inositol-acylated GPI anchors.
Fig. 4. Fractions of prion proteins in HEK293 cells possess inositol-acylated GPI.

a Expression of CD55, CD59, and prion on the surface in WT and PGAP2-KO cells was analyzed by flow cytometry. Red shaded areas indicate the expression of GPI-APs in WT and PGAP2-KO cells and dashed lines show the background (stained without 1st antibody). b The mean fluorescence intensities of CD55, CD59, and prion in WT cells were set as 1, and the relative intensities of those in PGAP2-KO cells are displayed as the mean ± SD from three independent experiments with p values (Student’s t test). ns not significant. c Cell lysates prepared from WT and PGAP2-KO cells were analyzed by western blotting. CD55, CD59, and prion were detected. GAPDH was used as a loading control. d Flow cytometric analysis of prion proteins in WT and PGAP2-KO cells overexpressing PGAP1. A plasmid expressing PGAP1 or empty vector was transiently transfected into WT and PGAP2-KO cells together with a pME-BFP plasmid. Three days after transfection, cells were harvested and treated with or without PI-PLC. The BFP-expressing cells were gated, and the surface expression of prion was analyzed. Red shaded areas indicate cells expressing PGAP1, black solid lines indicate cells expressing empty vector, and dashed lines show background (stained without 1st antibody). e Schematic representation of EGFP-FLAG-tagged prion proteins. In the PPP construct, all the parts corresponding to the N-terminal signal, mature part, and GPI attachment signal consist of prion parts. In the CPP, PPC, and PCP constructs, the N-terminal signal, GPI attachment signal, and mature part of the PPP were replaced with CD59 signals. f Cell surface expression of EGFP-FLAG-tagged chimeric proteins was analyzed by flow cytometry. The plasmids pLIB2-PPP-IRES2-mBFP, pLIB2-PPC-IRES2-mBFP, pLIB2-PCP-IRES2-mBFP, or pLIB2-CPP-IRES2-mBFP were stably transfected into HEK293 cells. Cells were harvested and treated with or without PI-PLC. The BFP-expressing cells were gated, and the surface expression of PPP, PPC, PCP, or CPP was analyzed. The mean fluorescence intensities of PPP in HEK293 cells were set as 1, and the relative intensities (mean ± SD values) of other chimeric proteins from five independent experiments were plotted (left panel). HEK293 cells expressing chimeric proteins were treated with and without PI-PLC. The residual amounts of chimeric proteins on the cell surface after PI-PLC treatment were analyzed by flow cytometry. The ratios of remaining proteins (mean ± SD values from five independent experiments) were plotted (right panel). **p < 0.001 according to an unpaired Student’s t test. ns not significant.
To determine whether PI-PLC resistance of prion proteins is caused by inefficient inositol deacylation in HEK293 cells, we transiently overexpressed PGAP1 encoding a GPI-inositol deacylase in WT and PGAP2-KO cells to facilitate inositol deacylation (Fig. 4d). The sensitivity of prion proteins to PI-PLC increased when PGAP1 was overexpressed, and at the same time, prion expression was significantly reduced in PGAP2-KO cells. This confirms that most of the prion protein on the surface of HEK293 cells is present without inositol deacylation. Compared with CD55 and CD59, PGAP1 seems to have a lower affinity with prion protein, which leads to the presence of prion protein without inositol deacylation in PGAP2-deficient cells.
To determine regions of the prion protein that cause inefficient deacylation by PGAP1, we constructed EGFP-FLAG-tagged prion chimera proteins, in which the N-terminal ER insertion signal sequence, mature protein, or C-terminal GPI attachment signal was replaced with corresponding regions of CD59 (Fig. 4e). Compared to endogenous prion proteins, the EGFP-FLAG-tagged chimeric constructs showed high PI-PLC sensitivity (Fig. 4f). Nevertheless, PPP (prion signal sequence/prion mature/prion GPI-attachment signal), CPP (CD59 signal sequence/prion mature/prion GPI-attachment signal), and PPC (prion signal sequence/prion mature/CD59 GPI-attachment signal) constructs showed around 11–15% resistance to PI-PLC in WT cells. On the other hand, the PCP (prion signal sequence/CD59 mature/prion GPI attachment signal) construct on the cell surface was almost completely cleaved by PI-PLC. These results indicate that the mature part of the prion protein determines unique PI-PLC resistance, probably its structure is not efficiently recognized by the GPI-inositol deacylase PGAP1. Alternatively, it is possible that a lower % of PI-PLC sensitivity is caused by the prion dimer formation since it has been reported that prion proteins form dimer structures53.
GPI-KO library for determination of GPI signatures recognized by aerolysin
Aerolysin54 is a member of the pore-forming toxin family secreted from Aeromonas hydrophila, which causes gastroenteritis, deep wound infection, and septicemia. Aerolysin binds to GPI-APs to target cells, and the C-terminal peptide of aerolysin is cleaved off by cell surface proteases, forming an activated heptameric complex, which is inserted into the membrane and acts as a channel55. Therefore, cell lines defective in GPI biosynthesis showed resistance to aerolysin56. Although it has been reported that the GPI-glycan structure and N-glycan structures on GPI-APs are required for toxin binding57,58, the precise GPI signature recognized by aerolysin remains unclear. To determine the GPI structures that aerolysin recognizes, we used a GPI-KO cell library to compare the sensitivity of 15 cell lines to aerolysin (Fig. 5a, b). WT HEK293 cells are sensitive to aerolysin, and almost all cells died at ≥60 nM. On the other hand, the cell lines with weak or no expression of GPI-APs on the cell surface, such as PIGA-, PIGB-, PIGO-, and PIGK-deficient cells, showed resistance to aerolysin treatment (Fig. 5b). In PIGK-deficient cells, although non-protein-linked free GPIs are expressed on the cell surface, aerolysin does not bind with them, showing hyper resistance to aerolysin57,59. The PIGN- and PGAP2-deficient cells also showed resistance to aerolysin, even if GPI-APs were expressed at low levels (Fig. 2). On the other hand, PGAP5-deficient cells express GPI-APs on the cell surface at the same level as WT cells. However, the cells showed resistance to aerolysin (Fig. 5a). PGAP5-KO cells showed resistance to aerolysin at 60 nM and then showed sensitivity at higher concentrations (Fig. 5b). The phenotype was rescued by the expression of PGAP5 in PGAP5-KO cells (Supplementary Fig. 6). The results indicate that the presence of EtNP on Man2 negatively affects the binding of aerolysin to GPI-APs. In addition, PGAP6-deficient cells are highly sensitive to aerolysin, probably due to the increased expression of GPI-APs on the cell surface. The slightly high sensitivity of PGAP4- and B3GALT4-deficient cells to aerolysin means that the glycan side chain modifications of GPI weaken aerolysin binding. These observations indicate that the GPI-KO library is useful to clarify the signature of GPI for toxin recognition.
Fig. 5. Aerolysin recognizes glycan-remodeled GPI structures by PGAP5.

a Fourteen GPI-KO cell lines were treated with different concentrations of proaerolysin (0, 12, 24, 36, 48, and 60 nM), and cell viability was analyzed by Cell Counting Kit-8 (CCK-8, MedChemExpress). HEK293 WT cells were used as a control. The cell viability of cells treated without proaerolysin was set as 1, and the relative viability was plotted as the mean ± SD from three parallel experiments. b Cell lines that showed resistance to aerolysin at low concentrations (60 nM) were selected and treated with high concentrations of proaerolysin (up to 600 nM). The cell viability was plotted as described above.
Discussion
Due to the complex biosynthetic pathway and variable side chain modifications of GPI anchors, it is difficult to compare how gene knockout impacts GPI-AP expression and function. Here, we established and utilized a knockout cell library to analyze the surface expression of GPI-APs and their sensitivity to PI-PLC and to determine the structural signature of GPIs recognized by aerolysin. In addition, we showed that prion proteins have unique properties, the majority of which was resistance to PI-PLC in HEK293 cells. In this study, we constructed a knockout cell library consisting of single-gene knockout cell lines. Since we have already validated a set of gRNAs against each GPI biosynthetic gene, the current knockout cell library could be established using these materials. It is possible to generate complicated GPI structures via a combination of the library with the gRNAs.
When the effects of gene-KO on GPI biosynthesis in mammalian cells are compared with those in yeast cells, similar and different phenomena are observed. It is known that yeast GPI1, which is the mammalian PIGQ homolog, is a non-essential gene among the GPI-GnT genes, suggesting that GPI-GnT is still active in gpi1-deficient yeast cells60. The PIGQ-KO still showed the expression of GPI-APs, consistent with yeast GPI1. On the other hand, KO of mammalian PIGZ, which encodes GPI-ManT-IV, did not affect the surface expression of GPI-APs, whereas the yeast homolog SMP3 is essential for the GPI biosynthetic pathway. The difference would be due to the different substrate recognition of the mammalian and yeast GPI-EtNP transferase-III (mammalian PIGO and PIGF or yeast Gpi13 and Gpi11)61. The GPI-KO cell library could be utilized for the complementation analysis of homologous genes in different species.
The knockout cell library of GPI biosynthetic genes enables us to produce GPIs with unique structures and functions. In particular, when KO cells defective in genes required for GPI glycan and lipid remodeling are used, we can evaluate the functions of GPI side chains and lipid moieties. In PGAP5-KO cells, a side chain EtNP remains attached to the second Man on GPI. Although the expression of GPI-APs was compatible with that in WT cells, the KO cells showed resistance to aerolysin, providing evidence that aerolysin recognizes the second Man without modification. Furthermore, the GPI-KO cell library would be useful to assay the effect of GPI structures on the conversion of prion proteins from cellular forms to scrapie forms. Some reports suggest the importance of GPI-anchors on prion structural changes and pathogenicity62–64. It would be possible to check the efficiency of prion structural changes dependent upon GPI structures by culturing KO cells with the scrapie forms.
When a GPI attachment signal is added to the C-terminus of secretory proteins or extracellular regions of type-I membrane proteins, the proteins can be expressed as GPI-APs. Therefore, the GPI-anchoring system is useful to tether the target proteins on the cell surface (Supplementary Fig. 7a). In addition, it is known that many GPI-APs are incorporated into extracellular vesicles, such as exosomes and virus-like particles (VLPs) (Supplementary Fig. 7b). It has been reported that various immunomodulatory proteins, antigens, and single-chain variable fragments are expressed as GPI-APs and loaded on VLPs to enhance immune responses, vaccination, and targeting, respectively65–67. Another unique feature is that purified GPI-APs can be inserted into the cell membrane through their lipid moieties in vitro. This is called GPI cell painting68 (Supplementary Fig. 7c), which enables the addition of the characteristics into the targeted cells. When recombinant proteins are expressed as GPI forms, the KO cell library produces recombinant proteins with different GPI structures, which might affect protein behaviors, including incorporation into exosomes or VLPs and cell painting. The KO cell library provides opportunities to analyze various phenomena related to GPI-APs.
In short, the knockout cell library of GPI biosynthetic genes can become a sustainable resource for exploring various applications and methods of GPI-AP biology and can provide insights into the genetic and biosynthetic regulation of GPI.
Methods
Cell lines, antibodies, and reagents
HEK293 (ATCC CRL-1573) cells and their derivative cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS) at 37 °C with 5% CO2. PGAP1-KO69 and PGAP2-KO70 cells have been constructed previously. KO cell lines used in this study are listed in Supplementary Table 1. Mouse monoclonal anti-CD55 (clone IA10)50, anti-CD59 (clone 5H8)50, anti-CD230 (14-9230-82; Thermo Fisher Scientific), anti-CD109 (556039; BD Biosciences), anti-GAPDH (60004-1-Ig, clone 1E6D9; Proteintech), anti-FLAG (F3165; M2; Sigma-Aldrich) and rabbit monoclonal anti-HA (3724S; Cell Signaling Technology) were used as primary antibodies. F(ab’)2-goat anti-mouse IgG, PE (12-4010-82; Thermo Fisher Scientific) and F(ab’)2-donkey anti-rabbit IgG, PE (12-4739-81; Thermo Fisher Scientific), and Goat Anti-Mouse IgG, HRP (HS201-1; TransGen Biotech) were used as the secondary antibodies. For flow cytometric analysis, antibodies were used at 10 µg/ml. For western blotting, the primary antibodies and the secondary antibodies were used at 1 and 0.2 µg/ml, respectively. PI-PLC (Thermo Fisher Scientific) and proaerolysin (produced and purified in the laboratory) were used for treatments. Cell Counting Kit-8 (CCK-8, MedChemExpress) was used to detect cell viability.
Plasmids
The gRNA sequences were designed on the E-CRISP website71 and ligated into the pX330-EGFP vector72. All gRNA sequences used for gene KO in this study are listed in Supplementary Table 2. pME-Hyg-3FLAG-rPGAP169 and pME-Hyg-3HA-hPGAP548 were used for rescue experiments. All the primers used in this study are listed in Supplementary Table 3. The DNA fragments corresponding to PGAP2, PGAP6, PGAP4, and B3GALT4 were amplified from cDNA prepared from HEK293 cells using the primers in Supplementary Table 3. pME-Hyg-3HA-hPGAP2 was generated by in-fusion cloning of full-length PGAP2 cDNA into the SalI/NotI site of pME-Hyg-HA. pME-Hyg-hPGAP6-3HA was obtained by in-fusion cloning of full-length PGAP6 cDNA into the XhoI/MluI site of pME-Hyg-HA. pLIB2-BSD-hPGAP4 was obtained by in-fusion cloning of full-length PGAP4 cDNA into the EcoRI/NotI site of pLIB2-BSD. pLIB2-BSD-hB3GALT4 was obtained by in-fusion cloning of full-length B3GALT4 cDNA into the EcoRI/NotI site of pLIB2-BSD. The DNA fragments corresponding to the N-terminal ER-insertion signal, mature protein or C-terminal GPI-attachment signal of prion or CD59 and EGFP-FLAG tag were amplified from prion cDNA and pME-Neo2dH-ssCD59-EGFP-FLAG-CD59. The DNA fragments consisting of PPP (ssprion-EGFP-FLAG-prion-prionss), PPC (ssprion-EGFP-FLAG-prion-CD59ss), PCP (ssprion-EGFP-FLAG-CD59-prionss), or CPP (ssCD59-EGFP-FLAG-prion-prionss) were ligated into the EcoRI/NotI site of the vector pLIB2-IRES2-mBFP to generate pLIB2-PPP-IRES2-mBFP, pLIB2-PPC-IRES2-mBFP, pLIB2-PCP-IRES2-mBFP and pLIB2-CPP-IRES2-mBFP. The DNA fragments corresponding to a mature part and a C-terminal GPI attachment signal of TEX101, GPC3, CRIPTO and SPACA4 were amplified from human cDNA clones by PCR, and they were spliced with ssCD59-HA from pME-puro-ssCD59-HA-CD14. Then, the DNA fragments corresponding to HA-tagged GPI-APs were ligated into the EcoRI/NotI of pLIB2-IRES2-mBFP, generating pLIB2-ssCD59-HA-TEX101-IRES2-mBFP, pLIB2-ssCD59-HA-GPC3-IRES2-mBFP, pLIB2-ssCD59-HA-CRIPTO-IRES2-mBFP and pLIB2-ssCD59-HA-SPACA4-IRES2-mBFP.
Establishment of KO cell lines
The pX330-EGFP plasmids containing gRNA sequences were transfected into HEK293 cells. Three days after transfection, the cells with a high EGFP fluorescence signal were sorted with a cell sorter S3 (Bio-Rad). The collected cells were cultured for more than one week, diluted, and transferred to a 96-well plate for culture to obtain monoclonal knockout cells. Gene knockout was analyzed by Sanger sequencing, and clonal cells without the WT allele were selected.
PI-PLC treatment and flow cytometric analysis
Cells (~106 cells/well) were harvested and washed with 500 μl of PBS. The samples were mixed with reaction buffer (5 U/ml PI-PLC, 0.5% bovine serum albumin (BSA), 5 mM EDTA, and 10 mM HEPES in DMEM without fetal calf serum) and incubated at 37 °C for 1.5 h. After washing the incubated cells with PBS, the cells were stained with primary antibodies (10 µg/ml) (anti-CD55, anti-CD59, or anti-CD230) in FACS buffer (PBS containing 1% BSA and 0.1% NaN3) for 25 min on ice. The samples were then washed twice with FACS buffer and stained with the secondary antibody (10 µg/ml) (F(ab’)2-goat anti-mouse IgG) in FACS buffer for 25 min on ice. After incubation, the samples were washed twice with FACS buffer and analyzed using Accuri C6 (BD). The data were analyzed using Accuri C6 and FlowJo software (BD).
Transfection and retrovirus-based infection
Cells (~106 cells/well) were plated in 6-well plates 1 day before transfection. For transient transfection, 4 μg of plasmids were transfected into cells using polyethyleneimine MAX (PEI-MAX) (Polysciences). Three days after transfection, the transfected cells were analyzed. For retrovirus-based infection, HEK293 cells (~106 cells) were transfected with 1 μg of pGP, 1 μg of pLC-VSVG, and 2 μg of pLIB2-IRES2-mBFP containing the target gene using PEI-MAX. After 12-16 h, the culture medium was changed, and the cells were cultured for 24 h. Then, the culture medium was collected, filtered with a 0.22 μm filter, and mixed with the same amount of DMEM containing 16 μg/ml hexadimethrine bromide (Sigma). The medium containing retrovirus was incubated with receiver cells overnight. One week after infection, cells were used for analysis.
Proaerolysin treatment and cell viability assay
Cells (2 × 104 cells/well) were cultured in a 96-well plate for one day. After removing the medium, 200 μl of prewarmed (at 37 °C) medium containing proaerolysin at different concentrations was added to the 96-well plate. After 8 h of incubation at 37 °C, cell viability was measured using a CCK-8 kit. Each sample was measured in triplicate. Cell viability (%) was calculated as (proaerolysin-treated cells (A450) − background (A450))/(nontreated cells (A450) − background (A450)) × 100.
Cell lysate preparation and analysis
Cells (~106 cells/well) were lysed with 100 μl of RIPA buffer and protein inhibitor cocktail (EDTA-Free, MedChemExpress) on ice for 30 min. After incubation, the samples were centrifuged at 10,000×g for 10 min at 4 °C to remove insoluble fractions. The supernatants were mixed with sample buffer and kept at 4 °C overnight. The protein samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blotting. Original western blotting images are shown in Supplementary Fig. 8.
Statistics and reproducibility
Statistical analyses were done using GraphPad Prism8 (GraphPad Prism Software) and Microsoft Excel 2016 (Microsoft). Source data are deposited as Supplementary data 1. For the statistical analyses, at least three independent or parallel experiments were performed. Unpaired Student’s t test was used to evaluate comparisons between two individual groups. P value < 0.05 were considered statistically significant.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Acknowledgements
We thank Drs. Hideki Nakanishi and Ning Wang (Jiangnan University) for the discussion. This work was supported by grants-in-aid from the National Natural Science Foundation of China 32071278 and 31770853 (M.F.), the Program of Introducing Talents of Discipline to Universities 111-2-06, National first-class discipline program of Light Industry Technology and Engineering LITE2018-015, Top-notch Academic Programs Project of Jiangsu Higher Education Institutions, the International Joint Research Laboratory for Investigation of Glycoprotein Biosynthesis at Jiangnan University, and a grant for Joint Research Project of the Research Institute for Microbial Diseases, Osaka University.
Author contributions
M.F., S.S.L., and T.K. conceptualized and designed the study. With assistance from Y.M., G.Y., and X.D.G., S.S.L., Y.S.L., and X.Y.G. conducted the experiments. S.S.L. and M.F. wrote the paper with input from all authors.
Data availability
The authors declare that data supporting the findings of this study are available within the paper and its supplementary information files.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Elah Pick and Eve Rogers.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-021-02337-1.
References
- 1.Orlean P, Menon AK. Thematic review series: lipid posttranslational modifications. GPI anchoring of protein in yeast and mammalian cells, or: how we learned to stop worrying and love glycophospholipids. J. Lipid Res. 2007;48:993–1011. doi: 10.1194/jlr.R700002-JLR200. [DOI] [PubMed] [Google Scholar]
- 2.Borner GH, Sherrier DJ, Stevens TJ, Arkin IT, Dupree P. Prediction of glycosylphosphatidylinositol-anchored proteins in Arabidopsis. A genomic analysis. Plant Physiol. 2002;129:486–499. doi: 10.1104/pp.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pittet M, Conzelmann A. Biosynthesis and function of GPI proteins in the yeast Saccharomyces cerevisiae. Biochim. Biophys. Acta. 2007;1771:405–420. doi: 10.1016/j.bbalip.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 4.Liu YS, Fujita M. Mammalian GPI-anchor modifications and the enzymes involved. Biochem. Soc. Trans. 2020;48:1129–1138. doi: 10.1042/BST20191142. [DOI] [PubMed] [Google Scholar]
- 5.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 6.Sabharanjak S, Sharma P, Parton RG, Mayor S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev. Cell. 2002;2:411–423. doi: 10.1016/S1534-5807(02)00145-4. [DOI] [PubMed] [Google Scholar]
- 7.Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 1992;68:533–544. doi: 10.1016/0092-8674(92)90189-J. [DOI] [PubMed] [Google Scholar]
- 8.Paladino S, et al. Protein oligomerization modulates raft partitioning and apical sorting of GPI-anchored proteins. J. Cell Biol. 2004;167:699–709. doi: 10.1083/jcb.200407094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tansey MG, Baloh RH, Milbrandt J, Johnson EM., Jr. GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron. 2000;25:611–623. doi: 10.1016/S0896-6273(00)81064-8. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson, M. A. J., et al. Cold Spring Harbor Laboratory Press Copyright 2015–2017 by The Consortium of Glycobiology Editors, La Jolla, California. All rights reserved. (Cold Spring Harbor, NY) 137–150 (2015).
- 11.Taron BW, Colussi PA, Wiedman JM, Orlean P, Taron CH. Human Smp3p adds a fourth mannose to yeast and human glycosylphosphatidylinositol precursors in vivo. J. Biol. Chem. 2004;279:36083–36092. doi: 10.1074/jbc.M405081200. [DOI] [PubMed] [Google Scholar]
- 12.Hirata T, et al. Identification of a golgi GPI-N-acetylgalactosamine transferase with tandem transmembrane regions in the catalytic domain. Nat. Commun. 2018;9:405. doi: 10.1038/s41467-017-02799-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Homans SW, et al. Complete structure of the glycosyl phosphatidylinositol membrane anchor of rat brain Thy-1 glycoprotein. Nature. 1988;333:269–272. doi: 10.1038/333269a0. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, et al. Cross-talks of glycosylphosphatidylinositol biosynthesis with glycosphingolipid biosynthesis and ER-associated degradation. Nat. Commun. 2020;11:860. doi: 10.1038/s41467-020-14678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi A, et al. α2,3 linkage of sialic acid to a GPI anchor and an unpredicted GPI attachment site in human prion protein. J. Biol. Chem. 2020;295:7789–7798. doi: 10.1074/jbc.RA120.013444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stahl N, et al. Glycosylinositol phospholipid anchors of the scrapie and cellular prion proteins contain sialic acid. Biochemistry. 1992;31:5043–5053. doi: 10.1021/bi00136a600. [DOI] [PubMed] [Google Scholar]
- 17.Bellai-Dussault K, Nguyen TTM, Baratang NV, Jimenez-Cruz DA, Campeau PM. Clinical variability in inherited glycosylphosphatidylinositol deficiency disorders. Clin. Genet. 2019;95:112–121. doi: 10.1111/cge.13425. [DOI] [PubMed] [Google Scholar]
- 18.Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Prim. 2017;3:17028. doi: 10.1038/nrdp.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grunnet M, Sorensen JB. Carcinoembryonic antigen (CEA) as tumor marker in lung cancer. Lung Cancer. 2012;76:138–143. doi: 10.1016/j.lungcan.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 20.Jöhrens K, Lazzerini L, Barinoff J, Sehouli J, Cichon G. Mesothelin as a target for cervical cancer therapy. Arch. Gynecol. Obstet. 2019;299:211–216. doi: 10.1007/s00404-018-4933-z. [DOI] [PubMed] [Google Scholar]
- 21.Assaraf YG, Leamon CP, Reddy JA. The folate receptor as a rational therapeutic target for personalized cancer treatment. Drug Resist. Updat. 2014;17:89–95. doi: 10.1016/j.drup.2014.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Zhou F, Shang W, Yu X, Tian J. Glypican-3: a promising biomarker for hepatocellular carcinoma diagnosis and treatment. Med. Res. Rev. 2018;38:741–767. doi: 10.1002/med.21455. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Y, et al. The immunological function of CD52 and its targeting in organ transplantation. Inflamm. Res. 2017;66:571–578. doi: 10.1007/s00011-017-1032-8. [DOI] [PubMed] [Google Scholar]
- 24.Fujihara Y, Ikawa M. GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid Res. 2016;57:538–545. doi: 10.1194/jlr.R063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kinoshita T. Biosynthesis and biology of mammalian GPI-anchored proteins. Open Biol. 2020;10:190290. doi: 10.1098/rsob.190290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Z, et al. Engineered CHO cells for production of diverse, homogeneous glycoproteins. Nat. Biotechnol. 2015;33:842–844. doi: 10.1038/nbt.3280. [DOI] [PubMed] [Google Scholar]
- 27.Chen YH, et al. The GAGOme: a cell-based library of displayed glycosaminoglycans. Nat. Methods. 2018;15:881–888. doi: 10.1038/s41592-018-0086-z. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe R, et al. The first step of glycosylphosphatidylinositol biosynthesis is mediated by a complex of PIG-A, PIG-H, PIG-C and GPI1. Embo J. 1998;17:877–885. doi: 10.1093/emboj/17.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watanabe R, Ohishi K, Maeda Y, Nakamura N, Kinoshita T. Mammalian PIG-L and its yeast homologue Gpi12p are N-acetylglucosaminylphosphatidylinositol de-N-acetylases essential in glycosylphosphatidylinositol biosynthesis. Biochem. J. 1999;339:185–192. doi: 10.1042/bj3390185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murakami Y, et al. PIG-W is critical for inositol acylation but not for flipping of glycosylphosphatidylinositol-anchor. Mol. Biol. Cell. 2003;14:4285–4295. doi: 10.1091/mbc.e03-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maeda Y, et al. PIG-M transfers the first mannose to glycosylphosphatidylinositol on the lumenal side of the ER. Embo J. 2001;20:250–261. doi: 10.1093/emboj/20.1.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashida H, et al. Mammalian PIG-X and yeast Pbn1p are the essential components of glycosylphosphatidylinositol-mannosyltransferase I. Mol. Biol. Cell. 2005;16:1439–1448. doi: 10.1091/mbc.e04-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang JY, et al. PIG-V involved in transferring the second mannose in glycosylphosphatidylinositol. J. Biol. Chem. 2005;280:9489–9497. doi: 10.1074/jbc.M413867200. [DOI] [PubMed] [Google Scholar]
- 34.Takahashi M, et al. PIG-B, a membrane protein of the endoplasmic reticulum with a large lumenal domain, is involved in transferring the third mannose of the GPI anchor. Embo J. 1996;15:4254–4261. doi: 10.1002/j.1460-2075.1996.tb00800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li ST, et al. Reconstitution of the lipid-linked oligosaccharide pathway for assembly of high-mannose N-glycans. Nat. Commun. 2019;10:1813. doi: 10.1038/s41467-019-09752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishida, M., et al. Ethanolamine phosphate on the second mannose as bridge in GPI anchored proteins: towards understanding inherited PIGG deficiency. Preprint at bioRxiv10.1101/2020.11.26.399477 (2020).
- 37.Hong Y, et al. Pig-n, a mammalian homologue of yeast Mcd4p, is involved in transferring phosphoethanolamine to the first mannose of the glycosylphosphatidylinositol. J. Biol. Chem. 1999;274:35099–35106. doi: 10.1074/jbc.274.49.35099. [DOI] [PubMed] [Google Scholar]
- 38.Inoue N, Kinoshita T, Orii T, Takeda J. Cloning of a human gene, PIG-F, a component of glycosylphosphatidylinositol anchor biosynthesis, by a novel expression cloning strategy. J. Biol. Chem. 1993;268:6882–6885. doi: 10.1016/S0021-9258(18)53122-7. [DOI] [PubMed] [Google Scholar]
- 39.Hong Y, et al. Requirement of PIG-F and PIG-O for transferring phosphoethanolamine to the third mannose in glycosylphosphatidylinositol. J. Biol. Chem. 2000;275:20911–20919. doi: 10.1074/jbc.M001913200. [DOI] [PubMed] [Google Scholar]
- 40.Shishioh N, et al. GPI7 is the second partner of PIG-F and involved in modification of glycosylphosphatidylinositol. J. Biol. Chem. 2005;280:9728–9734. doi: 10.1074/jbc.M413755200. [DOI] [PubMed] [Google Scholar]
- 41.Yu J, et al. The affected gene underlying the class K glycosylphosphatidylinositol (GPI) surface protein defect codes for the GPI transamidase. Proc. Natl Acad. Sci. USA. 1997;94:12580–12585. doi: 10.1073/pnas.94.23.12580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hiroi Y, et al. Molecular cloning of human homolog of yeast GAA1 which is required for attachment of glycosylphosphatidylinositols to proteins. FEBS Lett. 1998;421:252–258. doi: 10.1016/S0014-5793(97)01576-7. [DOI] [PubMed] [Google Scholar]
- 43.Ohishi K, Inoue N, Kinoshita T. PIG-S and PIG-T, essential for GPI anchor attachment to proteins, form a complex with GAA1 and GPI8. Embo J. 2001;20:4088–4098. doi: 10.1093/emboj/20.15.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hong Y, et al. Human PIG-U and yeast Cdc91p are the fifth subunit of GPI transamidase that attaches GPI-anchors to proteins. Mol. Biol. Cell. 2003;14:1780–1789. doi: 10.1091/mbc.e02-12-0794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisenhaber B, Bork P, Eisenhaber F. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- 46.Benghezal M, Benachour A, Rusconi S, Aebi M, Conzelmann A. Yeast Gpi8p is essential for GPI anchor attachment onto proteins. Embo J. 1996;15:6575–6583. doi: 10.1002/j.1460-2075.1996.tb01048.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tanaka S, Maeda Y, Tashima Y, Kinoshita T. Inositol deacylation of glycosylphosphatidylinositol-anchored proteins is mediated by mammalian PGAP1 and yeast Bst1p. J. Biol. Chem. 2004;279:14256–14263. doi: 10.1074/jbc.M313755200. [DOI] [PubMed] [Google Scholar]
- 48.Fujita M, et al. GPI glycan remodeling by PGAP5 regulates transport of GPI-anchored proteins from the ER to the Golgi. Cell. 2009;139:352–365. doi: 10.1016/j.cell.2009.08.040. [DOI] [PubMed] [Google Scholar]
- 49.Fujita M, Umemura M, Yoko-o T, Jigami Y. PER1 is required for GPI-phospholipase A2 activity and involved in lipid remodeling of GPI-anchored proteins. Mol. Biol. Cell. 2006;17:5253–5264. doi: 10.1091/mbc.e06-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda Y, et al. Fatty acid remodeling of GPI-anchored proteins is required for their raft association. Mol. Biol. Cell. 2007;18:1497–1506. doi: 10.1091/mbc.e06-10-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tashima Y, et al. PGAP2 is essential for correct processing and stable expression of GPI-anchored proteins. Mol. Biol. Cell. 2006;17:1410–1420. doi: 10.1091/mbc.e05-11-1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Menon AK. Structural analysis of glycosylphosphatidylinositol anchors. Methods Enzymol. 1994;230:418–442. doi: 10.1016/0076-6879(94)30027-5. [DOI] [PubMed] [Google Scholar]
- 53.Meyer RK, et al. A monomer-dimer equilibrium of a cellular prion protein (PrPC) not observed with recombinant PrP. J. Biol. Chem. 2000;275:38081–38087. doi: 10.1074/jbc.M007114200. [DOI] [PubMed] [Google Scholar]
- 54.Diep DB, Nelson KL, Raja SM, Pleshak EN, Buckley JT. Glycosylphosphatidylinositol anchors of membrane glycoproteins are binding determinants for the channel-forming toxin aerolysin. J. Biol. Chem. 1998;273:2355–2360. doi: 10.1074/jbc.273.4.2355. [DOI] [PubMed] [Google Scholar]
- 55.Abrami L, Fivaz M, van der Goot FG. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol. 2000;8:168–172. doi: 10.1016/S0966-842X(00)01722-4. [DOI] [PubMed] [Google Scholar]
- 56.Gordon VM, et al. Clostridium septicum alpha toxin uses glycosylphosphatidylinositol-anchored protein receptors. J. Biol. Chem. 1999;274:27274–27280. doi: 10.1074/jbc.274.38.27274. [DOI] [PubMed] [Google Scholar]
- 57.Abrami L, et al. The glycan core of GPI-anchored proteins modulates aerolysin binding but is not sufficient: the polypeptide moiety is required for the toxin-receptor interaction. FEBS Lett. 2002;512:249–254. doi: 10.1016/S0014-5793(02)02274-3. [DOI] [PubMed] [Google Scholar]
- 58.Hong Y, et al. Requirement of N-glycan on GPI-anchored proteins for efficient binding of aerolysin but not Clostridium septicum alpha-toxin. Embo J. 2002;21:5047–5056. doi: 10.1093/emboj/cdf508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brodsky RA. Paroxysmal nocturnal hemoglobinuria without GPI-anchor deficiency. J. Clin. Investig. 2019;129:5074–5076. doi: 10.1172/JCI131647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Leidich SD, Orlean P. Gpi1, a Saccharomyces cerevisiae protein that participates in the first step in glycosylphosphatidylinositol anchor synthesis. J. Biol. Chem. 1996;271:27829–27837. doi: 10.1074/jbc.271.44.27829. [DOI] [PubMed] [Google Scholar]
- 61.Grimme SJ, Westfall BA, Wiedman JM, Taron CH, Orlean P. The essential Smp3 protein is required for addition of the side-branching fourth mannose during assembly of yeast glycosylphosphatidylinositols. J. Biol. Chem. 2001;276:27731–27739. doi: 10.1074/jbc.M101986200. [DOI] [PubMed] [Google Scholar]
- 62.Chesebro B, et al. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 63.McNally KL, Ward AE, Priola SA. Cells expressing anchorless prion protein are resistant to scrapie infection. J. Virol. 2009;83:4469–4475. doi: 10.1128/JVI.02412-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bate C, Nolan W, Williams A. Sialic acid on the glycosylphosphatidylinositol anchor regulates PrP-mediated cell signaling and prion formation. J. Biol. Chem. 2016;291:160–170. doi: 10.1074/jbc.M115.672394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Skountzou I, et al. Incorporation of glycosylphosphatidylinositol-anchored granulocyte-macrophage colony-stimulating factor or CD40 ligand enhances immunogenicity of chimeric simian immunodeficiency virus-like particles. J. Virol. 2007;81:1083–1094. doi: 10.1128/JVI.01692-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Patel JM, et al. Influenza virus-like particles engineered by protein transfer with tumor-associated antigens induces protective antitumor immunity. Biotechnol. Bioeng. 2015;112:1102–1110. doi: 10.1002/bit.25537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deo VK, et al. A model for targeting colon carcinoma cells using single-chain variable fragments anchored on virus-like particles via glycosyl phosphatidylinositol anchor. Pharm. Res. 2014;31:2166–2177. doi: 10.1007/s11095-014-1316-4. [DOI] [PubMed] [Google Scholar]
- 68.Müller GA. Membrane insertion and intercellular transfer of glycosylphosphatidylinositol-anchored proteins: potential therapeutic applications. Arch. Physiol. Biochem. 2020;126:139–156. doi: 10.1080/13813455.2018.1498904. [DOI] [PubMed] [Google Scholar]
- 69.Liu YS, et al. N-Glycan-dependent protein folding and endoplasmic reticulum retention regulate GPI-anchor processing. J. Cell Biol. 2018;217:585–599. doi: 10.1083/jcb.201706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Matabaro E, et al. Molecular switching system using glycosylphosphatidylinositol to select cells highly expressing recombinant proteins. Sci. Rep. 2017;7:4033. doi: 10.1038/s41598-017-04330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- 72.Hirata T, et al. Post-golgi anterograde transport requires GARP-dependent endosome-to-TGN retrograde transport. Mol. Biol. Cell. 2015;26:3071–3084. doi: 10.1091/mbc.E14-11-1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that data supporting the findings of this study are available within the paper and its supplementary information files.