

Abstract
Background & Aims:
Mothers against decapentaplegic homolog 7 (SMAD7) is an antagonist of the transforming growth factor β (TGF-β) signaling. In the present investigation, we sought to determine the relevance of SMAD7 in liver carcinogenesis using in vitro and in vivo approaches.
Approach & Results:
We found that SMAD7 is upregulated in a subset of human hepatocellular carcinoma (HCC) samples with poor prognosis. Gene set enrichment analysis (GSEA) revealed that SMAD7 expression correlates with activated YAP/NOTCH pathway and cholangiocellular signature genes in HCCs. These findings were substantiated in human HCC cell lines. In vivo, overexpression of Smad7 alone was unable to initiate HCC development, but it significantly accelerated c-Myc/MCL1 induced mouse HCC formation. Consistent with human HCC data, c-Myc/MCL1/Smad7 liver tumors exhibited an increased cholangiocellular gene expression along with Yap/Notch activation and epithelial-mesenchymal transition (EMT). Intriguingly, blocking of the Notch signaling did not affect c-Myc/MCL1/Smad7-induced hepatocarcinogenesis while preventing cholangiocellular signature expression and EMT, whereas ablation of Yap abolished c-Myc/MCL1/Smad7-driven HCC formation. In mice overexpressing a myristoylated/activated form of AKT, co-expression of SMAD7 accelerated carcinogenesis and switched the phenotype from HCC to intrahepatic cholangiocarcinoma (iCCA) lesions. In human iCCA, SMAD7 expression was robustly upregulated, especially in the most aggressive tumors and directly correlated with the levels of YAP/NOTCH targets as well as cholangiocellular and EMT markers.
Conclusions:
The present data indicate that SMAD7 contributes to liver carcinogenesis by activating the YAP/NOTCH signaling cascade and by inducing a cholangiocellular and EMT signature.
Keywords: SMAD7, Hepatocellular Carcinoma, YAP, NOTCH, Epithelial Mesenchymal Transition, HCC, EMT
Introduction
Primary liver cancer, mainly consisting of hepatocellular carcinoma (HCC) and intrahepatic cholangiocarcinoma (iCCA), is one of the most common and lethal cancers worldwide (1). Noticeably, a subset of HCCs exhibits an iCCA molecular signature (2). Currently, curative treatments are limited to only ~20% HCC patients with early stage tumors. Targeted therapies and, more recently, immunotherapies have demonstrated some efficacy against advanced HCC. However, virtually all HCC patients eventually progress under these regimens (3, 4). The treatment options for iCCA remain very limited (5). The overall 5-year survival rate for HCC and iCCA is low, at ~20% (6), and 7~20% (7), respectively. Therefore, a better understanding of the molecular pathogenesis of these tumor entities is urgently needed for more effective treatments.
Previous studies have established that the transforming growth factor (TGF)-β superfamily plays a critical role in the regulation of liver fibrosis (8), inflammation (9) as well as HCC (10) and iCCA (11) tumorigenesis. TGF-β has three isoforms, behaving as bi-functional regulators that either inhibit or stimulate cell proliferation (12). In most cell types, TGF-β family cytokines initiate signal transduction by binding to the receptor complex, wherein the SMAD proteins are pivotal intracellular effectors (13, 14). SMAD7 belongs to the subclass of inhibitory SMADs that function as antagonists of TGF-β signaling. Upon activation, SMAD7 forms a heteromeric complex with activated SMAD2/3 and interferes with SMAD2/3-SMAD4 complex formation (15). SMAD7 also facilitates the ubiquitylation and degradation of phosphorylated SMAD2/3 by recruiting E3 ligase NEDD4–2 to the SMAD2/3-SMAD7 heteromeric complex (16). In addition, SMAD7 may function as a transcriptional repressor (17).
Previous work indicated that SMAD7 may exert pro- or anti-tumorigenic effects depending on the tumor type or cellular context (18). In human HCC, the expression profile of SMAD7 remains controversial. Park et al. reported much higher SMAD7 levels in advanced tumors than in dysplastic nodules and early HCCs (19). In contrast, Xia et al. identified decreased expression of SMAD7 in HCC samples, particularly in patients with early recurrence and poor prognosis (20). Moreover, Sun et al. demonstrated that a reduced Smad7 expression in HCC compared to matched non-tumorous tissue is associated with malignant clinicopathological features and unfavorable clinical outcome of patients after surgery (21). In iCCA, it has been found that SMAD7 levels are elevated and significantly associated with lymph node metastasis and perineural invasion as well as with shorter overall survival (22).
In the current study, we extensively investigated the role of SMAD7 in HCC and iCCA using tumor derived cell lines, mouse models, and human specimens. Collectively, our study indicates that SMAD7 promotes the growth of HCC and iCCA cells in vitro and accelerates liver carcinogenesis in vivo by activating the YAP/NOTCH pathway, promoting cholangiocellular gene expression, and inducing EMT.
Material and methods
Constructs and reagents
Constructs applied in this study include pT3-EF1α, pT3-EF1α-HA-Smad7 (Human), pT3-EF1α-c-Myc (Human), pT3-EF1α-MCL1 (Mouse), pT3-EF1α-dnRBP-J (Human), pT3-EF1α-myr-Akt-HA (Mouse), pCMV, pCMV-Cre and pCMV-sleeping beauty (SB) transposase. All plasmids were purified using the Endotoxin Free Maxiprep kit (Sigma-Aldrich, St. Louis, MO).
Cell culture and in vitro studies
HLE, MHCC97H, SNU449, and Huh7 human HCC cell lines, and KKU156, KKU213, and RBE iCCA cell lines were used for the in vitro studies. The HLE, KKU156, and KKU213 cell lines were purchased from the JCRB Cell Bank, whereas SNU449, Huh7, and RBE cell lines were purchased from ATCC. The MHCC97H cell line was a kind gift from Dr. Binbin Liu from Fudan University, Shanghai, China. Cell lines were validated (Genetica DNA Laboratories, Burlington, NC, USA) and maintained as monolayer cultures in Dulbecco’s modified Eagle medium with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 100 U/mL penicillin, and 100 g/mL streptomycin (Gibco). Transfection with lentivirus and colony formation assay were performed as described before in detail (23).
Statistical analysis
The Prism 7.0 software (GraphPad, San Diego, CA) was used to analyze the data, which are presented as Means ± SD. Comparisons between two groups were performed with two-tailed unpaired t test when dataset achieve Gaussian distribution, or non-parametric test when sample size was small. Welch correction and linear regression were applied when necessary. Kaplan–Meier method and log-rank test were used for survival analysis. Multi-variate Cox regression analysis using Statistical Package for Social Science software (SPSS, version 16.0, Chicago, IL, USA) was applied to compare the hazards ratios of the two groups with confounding variables taken into account. P values < 0.05 were considered statistically significant.
Please refer to Supporting Materials for more detailed information.
Results
Deregulated SMAD7 expression in human HCC samples
First, we analyzed SMAD7 genomic abnormalities in HCC samples. Using the cBioPortal for Cancer Genomics (24), we identified 3 cohorts of 1017 HCC patients with SMAD7 genetic alterations. In total, 3 HCC patients displayed SMAD7 gene amplification mutation and 1 exhibited a SMAD7 deep deletion. Missense mutations were identified in 5 patients (Supporting Fig. S1). Thus, SMAD7 aberrant genetic events are relatively rare in human HCC.
Next, we analyzed SMAD7 mRNA expression in human HCC samples (Supporting Table S1) in comparison with normal liver tissues using the TCGA LIHC data (25). Overall, SMAD7 mRNA levels were decreased in HCC samples when compared to normal liver samples (Fig. 1A). However, subgroup analysis of SMAD7 expression revealed three distinct groups in human HCC: SMAD7-high, SMAD7-medium and SMAD7-low (Fig. 1B–D). No statistical difference concerning the etiology (hepatitis and alcohol consumption) of HCCs among the three groups was noted (Supporting Fig. S2). Kaplan-Meier and linear regression analysis indicated that SMAD7-high patients displayed a poor survival outcome when compared to the other two groups (Fig. 1E). However, multi-variate Cox analysis indicated that hepatitis and tumor differentiation stages were confounding variables (Supporting Table S2). In an independent human HCC cohort (n=64), the mRNA levels of SMAD7 did not differ significantly when comparing non-tumorous surrounding liver tissues with corresponding HCC specimens with better outcome (HCCB; survival longer than 3 years following partial liver resection), whereas highest levels of SMAD7 were detected in the most aggressive tumors (HCCP; survival shorter than 3 years) (Supporting Fig. S3A). On the other hand, no significant differences in overall survival were observed in the HCC collection based on SMAD7 expression using the long-rank test. However, a SMAD7 mRNA level above the 75th percentile in HCC specimens was associated with significantly shorter survival (Supporting Tables S3–S7 and Supporting Fig. S3B,C). The deregulated mRNA expression pattern was also consistent with SMAD7 protein expression trend based on the Human Protein Atlas dataset (Fig. 1F). The results are consistent with the previous studies supporting upregulation as well as downregulation of TGF-β signaling members in distinct subgroups of human HCCs (26).
Fig. 1. SMAD7 expression in human HCC samples.
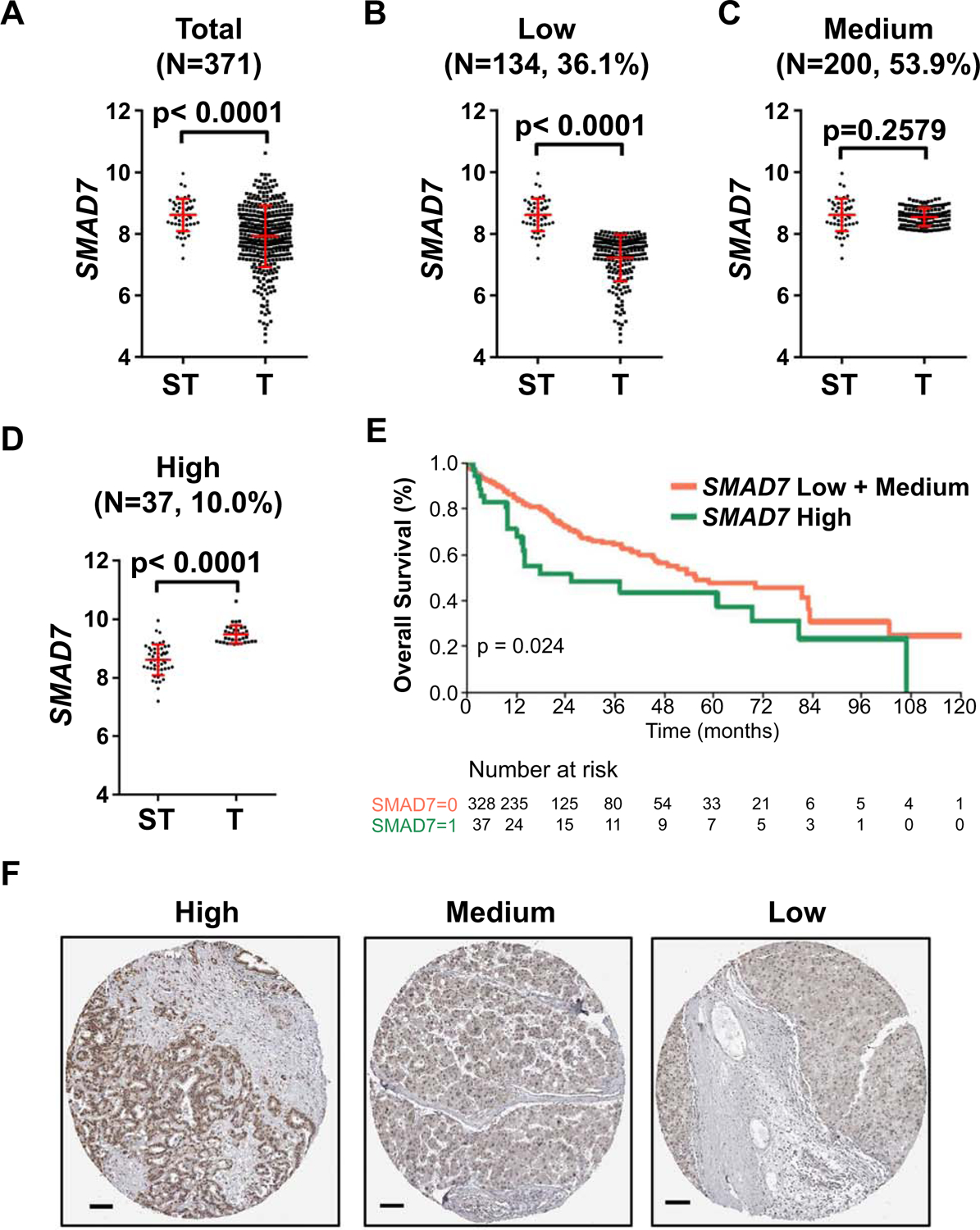
(A) SMAD7 expression was downregulated in HCC samples (T; Mean ± SEM = 7.924 ± 0.05192, N=371) compared to surrounding tumor normal liver samples (ST; Mean ± SEM =8.616 ± 0.07457, N=50). Unpaired t test was applied for statistical analysis, P <0.0001. (B, C, D) Subgroup analysis of SMAD7 expression showing comparison of SMAD7 expression in surrounding tumor normal liver samples (ST, Mean=8.616, 95%CI: 8.466 −8.766; N=50) and SMAD7 low expression (B) HCC samples (T, Mean=7.218, 95%CI: 7.113 −7.324; N=200), SMAD7 medium expression (C) HCC samples (T, Mean=8.547, 95%CI: 8.496 −8.598; N=134), and SMAD7 high expression (D) samples (T, Mean=9.482, 95%CI: 9.533 −9.431; N=37). Mann Whitney test was used for statistical analysis, P values are indicated in the figures. (E) Kaplan-Meier survival curves for the 3 HCC subgroups indicating that SMAD7 high group has a poor survival outcome. (F) Representative immunohistochemical staining images of SMAD7 high/medium/low expression pattern among human HCC samples (https://www.proteinatlas.org/). Scale bars: 100μm. Abbreviations: T, tumor; ST, surrounding non-tumorous tissue; SEM, standard error of mean; CI, confidence intervals.
To dissect further the functional variations of SMAD7 among HCC samples, we identified genes whose expression patterns correlated with that of SMAD7 in the TCGA LIHC dataset and performed gene set enrichment analysis (GSEA) (Supporting Table S8). We found that high expression of SMAD7 was positively associated with NOTCH signaling pathway and YAP signature (Supporting Fig. S4A,B). Consistently, mRNA levels of NOTCH signaling targets and YAP along with YAP targets were all found to be positively correlated with mRNA levels of SMAD7 in our independent HCC cohort (Supporting Fig. S4C,D). As NOTCH and YAP are pivotal players in cholangiocyte fate determination (27, 28), we examined the correlation between SMAD7 and liver cell fate, including hepatocyte and cholangiocyte, signature genes. Interestingly, SMAD7 expression was positively correlated with the expression of hepatic stem/progenitor signatures genes (Supporting Fig. S5) and cholangiocyte markers such as EPCAM and KRT19 and other cholangiocellular signature genes (Supporting Fig. S6). In contrast, SMAD7 expression was negatively correlated with that of hepatocellular signature genes (Supporting Fig. S7) in HCC.
Given that increased YAP/NOTCH axis has been linked to EMT (29, 30), we investigated whether SMAD7 expression correlates with major EMT transcriptional factors and EMT markers in human HCC. Based on the TCGA data, SMAD7 expression was positively correlated with EMT-activating transcription factors (SNAI1, TWIST1, ZEB1, and ZEB2) as well as the mesenchymal marker VIM (Supporting Fig. S8).
Altogether, the present findings indicate that high expression of SMAD7 correlates with poor patient survival as well as YAP/NOTCH cascade activation, increased hepatic stem/progenitor and cholangiocellular gene signature and EMT in human HCC.
SMAD7 overexpression activates YAP and increases HCC cell growth in vitro
The correlative analysis in human HCC samples suggested that high SMAD7 expression might promote YAP activation. To test this hypothesis in vitro, we transfected three human HCC cell lines (HLE, MHCC97H, and SNU449) with HA-tagged SMAD7. The three HCC cell lines were selected based on their low SMAD7 levels reported in the Liver Cancer Model Repository (https://www.picb.ac.cn/limore/feature). We found that overexpression of SMAD7 increased HCC cell growth by augmenting proliferation and decreasing apoptosis (Fig. 2A and 2B, respectively). Similar results were obtained when assessing colony formation in three cell lines (Supporting Fig. S9). SMAD7 overexpression also led to significant increase of HCC cell invasion (Supporting Fig. S10). Western blot analysis revealed increased YAP protein levels upon SMAD7 transfection (Fig. 2C). Importantly, there was a significant increase in the levels of nuclear YAP (Fig. 2D), and qRT-PCR analysis demonstrated augmented expression of YAP (CTGF, CYR61, JAG1) and NOTCH (HES1, SOX9; not shown) target genes as well as stemness (EPCAM, CD24, BMI1, and DLK1) and cholangiocyte markers (KRT7, KRT19, CD133, and SOX9) upon SMAD7 overexpression (Fig. 2E; Supporting Fig. S11 and S12). Furthermore, transient transfection of HA-tagged SMAD7 led to a significant increase of TEAD reporter activity (a surrogate marker of YAP dependent activation) in the HLE and SNU449 cell lines (Fig. 2F). Conversely, reduction of proliferation and increase of apoptosis, decrease in TEAD reporter activity, and decline of YAP (CTGF, CYR61, JAG1) and NOTCH (HES1, SOX9) target genes, without affecting YAP expression, was detected following SMAD7 silencing in the Huh7 cell line (Supporting Fig. S13).
Fig. 2. SMAD7 overexpression promotes cell proliferation and activates YAP/NOTCH signaling.
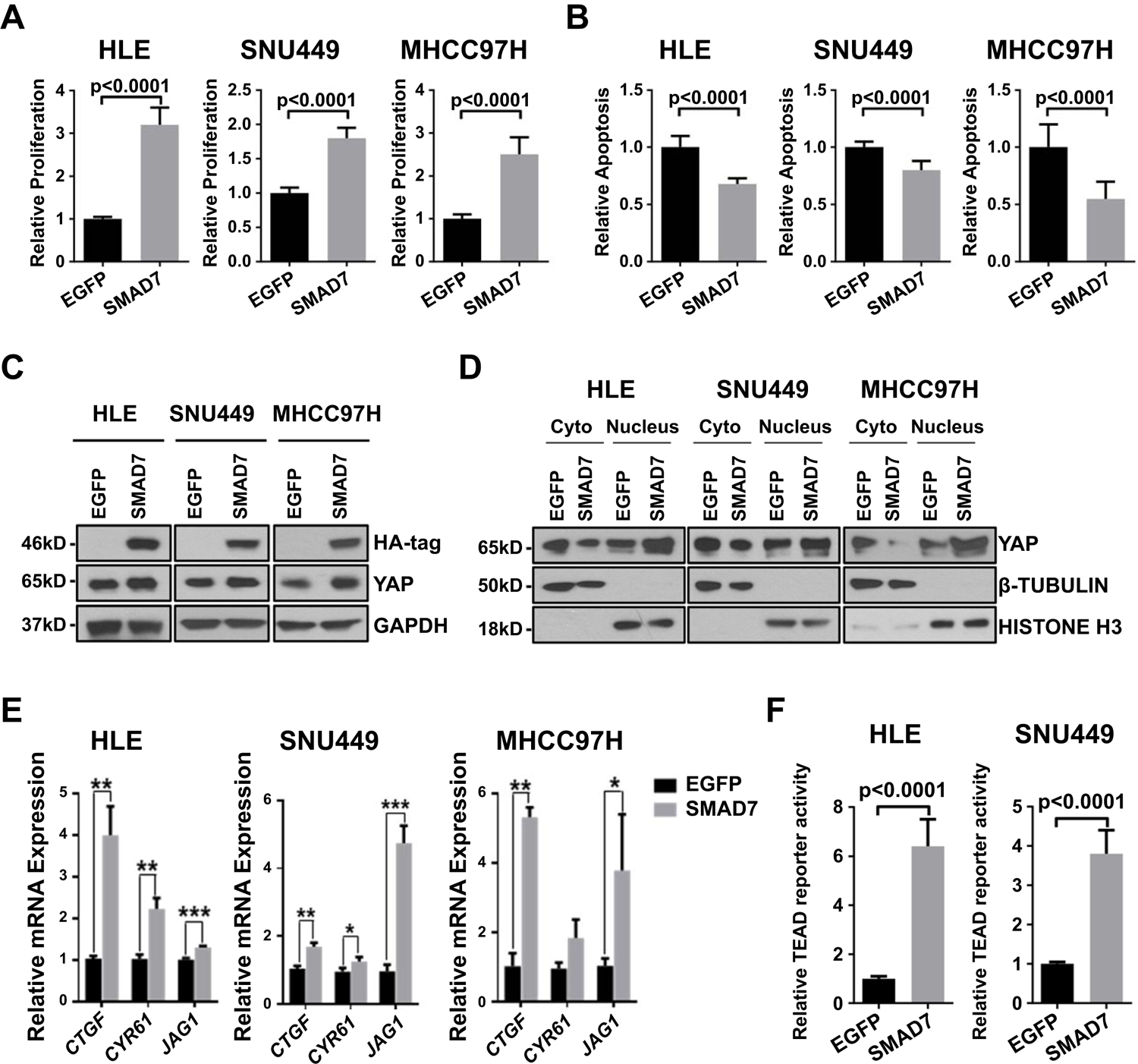
(A) Effect of SMAD7 transfection on proliferation of HLE, SNU449 and MHCC97H cell lines. (B) Effect of SMAD7 transfection on apoptosis of HLE, SNU449 and MHCC97H cell lines. (C) Western blot results confirming expression of HA-tag and upregulation of YAP, SMAD7 in SMAD7-transfected cell lines. (D) Western blot results showing cytoplasmic (Cyto) and nuclear Yap expression of EGFP-transfected and SMAD7-transfected human HCC cell lines (HLE, SNU449 and MHCC-97H). β-Tubulin was used as a cytoplasmic protein loading control, HISTONE-H3 was used as a nuclear protein loading control. (E) mRNA expression of YAP and NOTCH targets (CTGF, CYR61 and JAG1). (F) Effect of SMAD7 transfection on TEAD reporter activity of HLE and SNU449 cell lines. * P <0.05, ** P <0.01, *** P <0.001.
Altogether, these data indicate that overexpression of SMAD7 leads to YAP signaling activation and increases in vitro growth of HCC cells.
Overexpression of Smad7 accelerates c-Myc/MCL1 driven HCC development in mice
Our analysis suggested the oncogenic potential of high SMAD7 expression in hepatocarcinogenesis. To test this hypothesis in vivo, we first examined whether SMAD7 overexpression alone suffices to initiate liver tumor development. Thus, the HA-tagged Samd7 plasmid along with sleeping beauty (SB) transposon was delivered into mouse hepatocytes by using hydrodynamic tail vein injection (HTVi). Mouse livers were harvested at 20 weeks post injection (p.i). Neither preneoplastic nor neoplastic lesions were detected by H&E staining in SMAD7-injected mouse livers. In addition, cell proliferation remained at an extremely low level as indicated by few Ki67 (+) cells. Sporadic HA-tagged Smad7 staining was present in pericentral hepatocytes, while Ck19 (+) cells were only detected in the bile ducts (Supporting Fig. S14). Thus, the data indicate that overexpression of Smad7 alone is not sufficient to induce mouse liver tumorigenesis. In light of these findings, we hypothesized that Smad7 acts as a tumor modifier rather than a bona fide oncogene. To validate our hypothesis, we applied a well-characterized murine HCC model induced by hydrodynamic co-expression of c-Myc and MCL1 oncogenes (c-Myc/MCL1) in the liver (31, 32). In brief, mice were hydrodynamically injected with c-Myc, MCL1 and Smad7 plasmids (c-Myc/MCL1/Smad7). Additional mice were injected with c-Myc, MCL1, and pT3-EF1α empty vector (c-Myc/MCL1/pT3) (Fig. 3A). Noticeably, overexpression of Smad7 accelerated tumor growth in the c-Myc/MCL1 mouse HCC model. While c-Myc/MCL1/pT3 injected mice developed large tumor burden and were required to be euthanized ~5 weeks post injection, c-Myc/MCL1/Smad7 injected mice exhibited large abdominal masses and needed to be sacrificed between 2 to 3 weeks post injection (Fig. 3B). Histopathological analysis revealed that undistinguishable tumors developed in c-Mc/MCL1/pT3 and c-Myc/MCL1/Smad7 mice, consisting of small, highly basophilic malignant hepatocytes (Fig. 3C). Immunohistochemistry (IHC) and Western blot analysis confirmed the ectopically injected c-Myc in c-Myc/MCL1/pT3 and c-Myc/MCL1/Smad7 mouse HCC. HA-tagged Smad7, leading to decreased p-Smad2/3, was also readily detected in c-Myc/MCL1/Smad7 mouse HCC (Fig. 3C,D).
Fig. 3. Smad7 enhances c-Myc/MCL1 induced hepatocarcinogenesis.
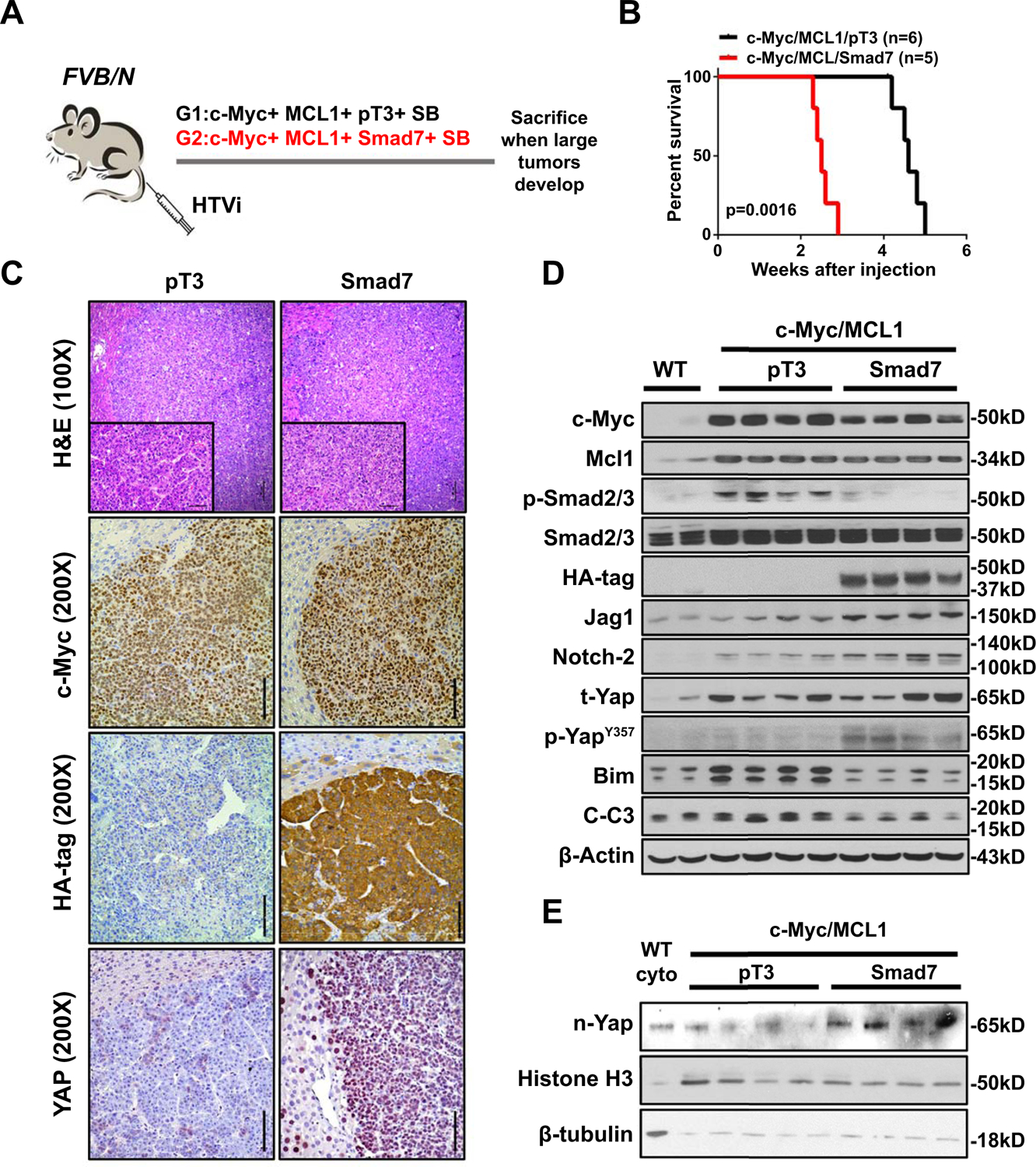
(A) Study design. FVB/N mice were injected with c-Myc/MCL1/pT3/SB (N=6) or c-Myc/MCL1/Smad7/SB (N=5) plasmids, respectively. Mice were monitored and sacrificed when moribund. (B) Survival curve showing that Smad7 accelerates c-Myc/MCL1 tumor development. (C) Representative images of H&E, c-Myc, HA-tag staining in c-Myc/MCL1/pT3 and c-Myc/MCL1/Smad7 mouse HCCs. Original magnifications: 100X and 200X. Scale bars: 200μm for 100X, 100μm for 200X. (D) Western blot results showing protein expression of wild-type (WT) normal liver, c-Myc/MCL1/pT3 and c-Myc/MCL1/Smad7 tumor tissues. β-Actin was used as a loading control. (E) Western blot results of nuclear expression of Yap (n-Yap) in the two mouse groups. Histone was used as a loading control. Abbreviations: HTVi, hydrodynamic tail vein injection; n, nuclear; t, total.
Next, we investigated the Yap/Notch pathway status in c-Myc/MCL1/pT3 and c-Myc/MCL1/Smad7 tumors. While Yap protein levels did not change significantly in tumor tissues from the two mouse cohorts (Fig. 3D), p-YapY357, which was required for full activation of Yap (33), as well as nuclear Yap levels significantly increased in c-Myc/MCL1/Smad7 HCCs (Fig. 3C–E). Consistently, qRT-PCR analysis demonstrated the increased expression of Yap target genes, including Ctgf, Cyr61, Jag1, and Notch2 in c-Myc/MCL1/Smad7 HCCs (Supporting Fig. S15A). The elevated expression of Jag1 and Notch2 was also validated at protein level by Western blotting (Fig. 3D). Increased Jag1/Notch2 promoted canonical Notch activation, as substantiated by higher mRNA expression of Nrap and Hes1 target genes (Supporting Fig. S15B). YAP has been shown to function as a transcriptional repressor to inhibit the expression of BIM, a pro-apoptosis gene (34). Consistent with this report, Bim protein expression was downregulated by Smad7 overexpression (Fig. 3D), leading to decreased cleaved Caspase 3 and cleaved Parp levels as analyzed by Western blot analysis and/or IHC (Fig. 3D and Supporting Fig. S16A). Consequently, apoptosis was lower in c-Myc/MCL1/Smad7 tumors when compared with c-Myc/MCL1/pT3 corresponding lesions (Supporting Fig. S16B,C). The decreased apoptosis imposed by Smad7 overexpression may be one of the major mechanisms whereby Smad7 overexpression accelerates HCC development in vivo. Furthermore, Myc/MCL1/Smad7 tumors exhibited higher proliferative activity than c-Myc/MCL1/pT3 lesions, as assessed by the percentage of Ki67 (+) cells (Supporting Fig. S16D).
Given that YAP and its mediated NOTCH activation are associated with cholangiocellular gene expression and EMT in HCC (35), we examined whether these phenotypes occur in c-Myc/MCL1/Smad7 mouse HCC cells. IHC of cholangiocellular marker Ck19 revealed that c-Myc/MCL1/pT3 HCCs were completely Ck19 (−). Ck19 (+) cells consisted only of biliary epithelial cells in non-tumor regions. In striking contrast, ~60% of c-Myc/MCL1/Smad7 tumor lesions showed moderate to strong Ck19 (+), and this was accompanied by decreased immunoreactivity for the hepatocyte marker Hnf4α expression in these cells (Fig. 4A and 4B). Consistently, increased Epcam (+) cells, another cholangiocyte marker, was detected in c-Myc/MCL1/Smad7 HCC by immunofluorescence (IF) (Fig. 4C). Increased expression of hepatic progenitor genes in c-Myc/MCL1/Smad7 liver tumors was also observed (Supporting Fig. S15C). As concerns EMT, we performed double IF. Using Ck19 IF for the cholangiocellular-like tumor cells, and E-Cadherin as epithelial cell marker, we discovered that Ck19(+) tumor cells in c-Myc/MCL1/Smad7 HCC lesions were E-Cadherin (−) (Supporting Fig. S17A). In contrast, in the non-tumorous regions of c-Myc/MCL1/pT3 mice, Ck19 (+) and E-Cadherin (+) bile duct epithelial cells could be readily detected. Next, we co-stained Ck19 with the mesenchymal marker Vimentin and observed the concomitant staining of Ck19 with Vimentin in c-Myc/MCL1/Smad7 tumor lesions (Supporting Fig. S17B). The results indicate that these Ck19 (+) tumor cells lose E-Cadherin while gaining Vimentin expression, consistent with EMT.
Fig. 4. Smad7 induces Ck19 positive cells in c-Myc/MCL1 mouse HCCs.
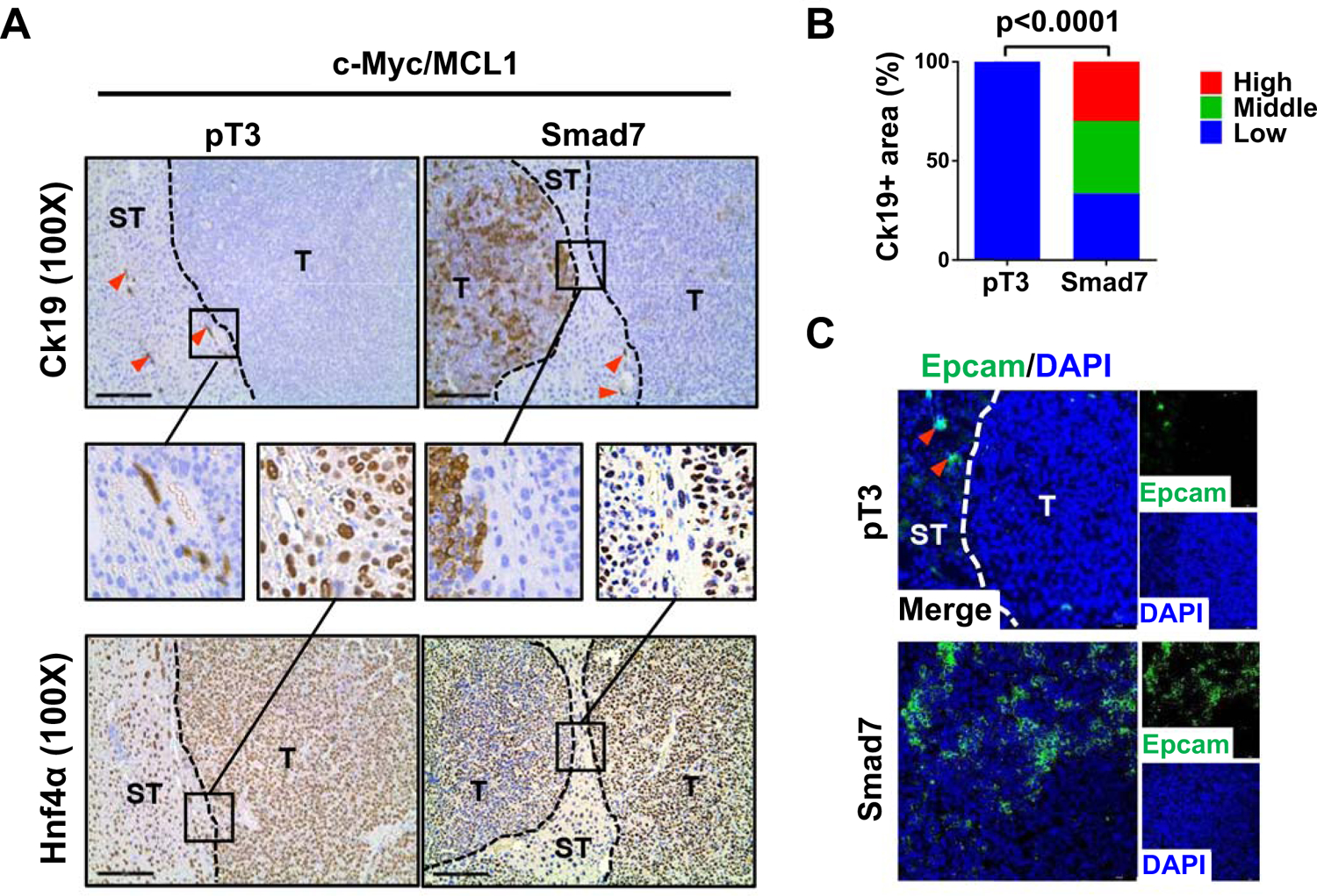
(A) Representative immunohistochemical staining images of cholangiocyte marker (Ck19) and hepatocyte marker (Hnf4a). Red arrows indicate Ck19 positive stained intrahepatic biliary cells. T: tumor, ST: Surrounding tissue. Small black boxes inside figures indicate areas of interest, large black boxes linked by black line denote enlarged views for better visualization. Original magnification: 330X. Scale bar: 200μm. (B) Percentages of Ck19 high/medium/low expression areas in tumor samples. (C) Representative immunofluorescence staining images of Epcam in c-Myc/MCL1/pT3 and c-Myc/MCL1/Smad7 mouse liver tissues. Red arrows indicate Epcam positive stained intrahepatic biliary cells. Scale bars: 50 mm.
Finally, we investigated the cellular origin of Ck19 (+) cells in liver tumor samples. Previous studies have established that the hydrodynamic transfection technique delivers the gene/s of interest specifically into pericentral hepatocytes (36, 37). To further validate this observation, we conducted IHC staining for c-Myc and glutamine synthetase (Gs), a well-known marker for pericentral hepatocytes (38), on serial sections of liver tissues 3–4 days after plasmid transfection in wild-type mice. We found that c-Myc positive hepatocytes were located around the Gs(+) pericentral region, but not in the periportal region (Supporting Fig. S18). Next, we applied a stochastic multicolor Cre-reporter R26R-confetti homozygous mice (will be referred as “Confetti mice”) for lineage tracing (39, 40). Hepatocyte specific AAV8-TBG-Cre (41) was injected into 4-week-old Confetti mice to trigger the expression of fluorescent proteins. Plasmids mixture of c-Myc/MCL1/Smad7/SB was delivered into mouse liver 2 weeks later (Supporting Fig. S19A). Upon Cre recombination, the multicolor construct recombines randomly to result in multiple possible outcomes with different fluorescent proteins being expressed in mouse hepatocytes (Supporting Fig. S19B). If the labeled hepatocytes developed into tumors, the tumors cells would maintain the original fluorescent protein expression, although these tumors may have evolved into different cell lineages (Supporting Fig. S19C). Therefore, as long as the tumor cells demonstrated the expression of the fluorescent protein, they must derive from hepatocytes. In the c-Myc/MCL-1/Smad7 tumors, we found that a number of Ck19 (+) tumor cells co-stained with cell membrane expressed Cyan Fluorescent Protein (CFP) signals, while other Ck19 (+) tumor cells were labeled with cytoplasm GFP/YFP signals (Supporting Fig. S20). Altogether, these data support the hypothesis that these Ck19 (+) cells originated from hepatocytes. It is worth to note that the AAV8-TBG-Cre system only labels a subset of mouse cells; thus, some Ck19 positive tumor cells were negative for the fluorescence signal (Supporting Fig. S19C).
Overall, our data demonstrate that Smad7 overexpression does not modify the tumor histological phenotype but accelerates c-Myc/MCL1 driven HCC development by enhancing Yap/Notch activation and increasing cholangiocellular gene expression and EMT.
Notch blockade inhibits EMT and cholangiocellular gene expression without affecting tumor progression
As the Notch pathway is activated in c-Myc/MCL1/Smad7 mouse HCC, we investigated the functional contribution of the canonical Notch signaling in Smad7 dependent hepatocarcinogenesis. Thus, we co-delivered the c-Myc/MCL1/Smad7 constructs with the dominant negative form or RBP-J (dnRBP-J), which effectively blocks the canonical Notch pathway (27), to the mouse liver. Additional mice were injected with c-Myc/MCL1/Smad7 and pT3 empty vector as control (Fig. 5A). Interestingly, blocking of the Notch signaling did not affect tumor development, with all c-Myc/MCL1/Smad7/dnRBP-J and c-Myc/MCL1/Smad7/pT3 mice developing lethal burden of liver tumors by 4 weeks post injection (Fig. 5B). Expression of c-Myc, MCL1, HA-tagged Smad7, and V5-tageed dnRBP-J was validated by Western blotting in both mouse cohorts. No differences were detected in the expression of p-Smad2/3, Bim, Cyclin D1 (Ccnd1), Pcna or Cleaved caspase-3 as well as in proliferation and apoptosis rates between the two groups, indicating that Notch does not regulate TGF-β signaling or cell proliferation and apoptosis processes in c-Myc/MCL1/Smad7 mice (Supporting Fig. S21A–D).
Fig. 5. Blockade of the Notch signaling does not affect c-Myc/MCL1/Smad7 driven mouse HCC development.
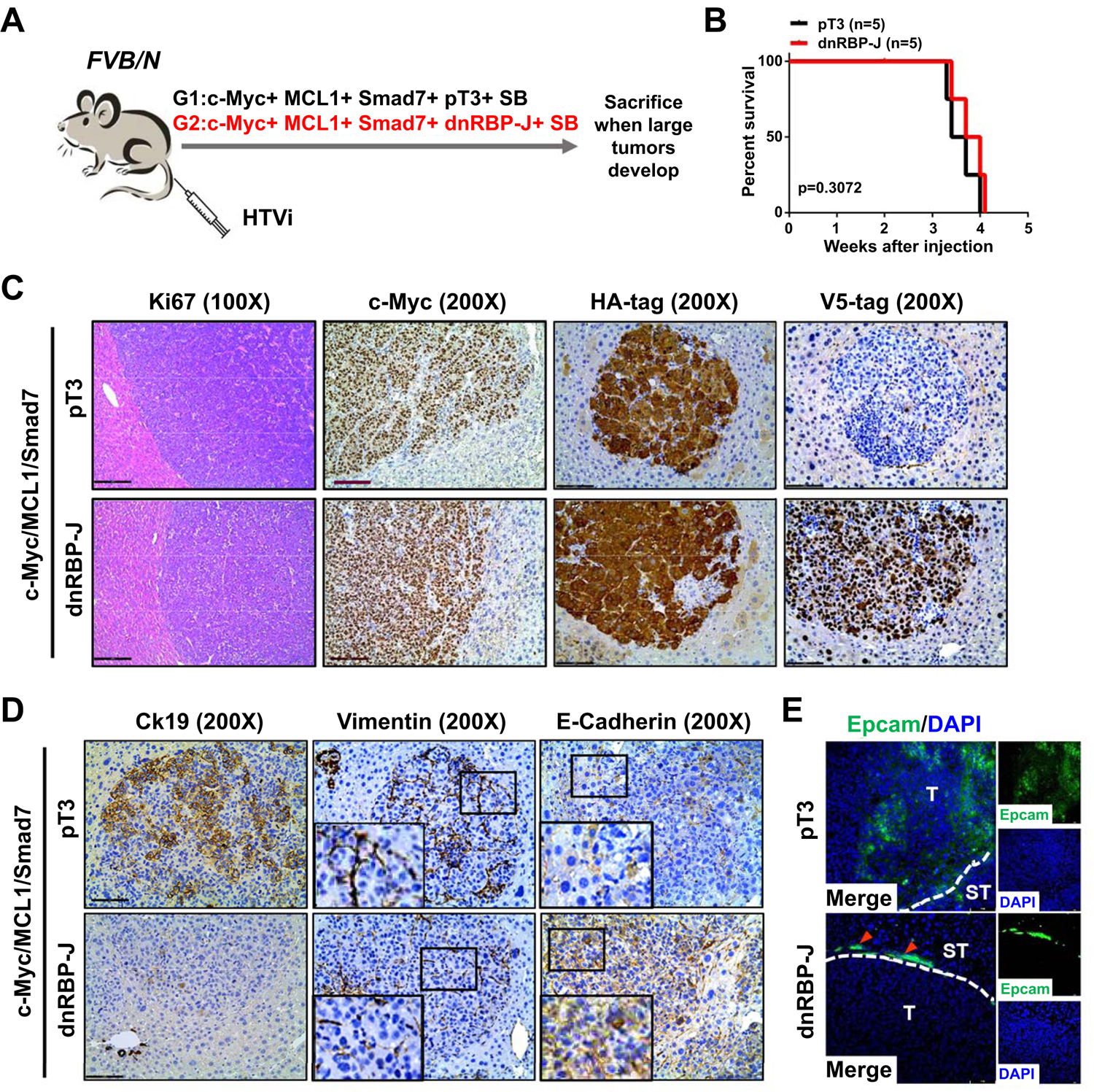
(A) Study design. FVB/N mice were injected with c-Myc/MCL1/Smad7/pT3/SB (N=5) or c-Myc/MCL1/Smad7/dnRBP-J/SB (N=5) plasmids, respectively. Mice were monitored and sacrificed when moribund. (B) Survival curve showing that suppression of the Notch signaling does not prolong c-Myc/MCL1/Smad7 mouse survival. (C) Representative H&E and immunohistochemical staining images of c-Myc, HA-tag and V5-tag in both groups. Scale bars: 200 μm for 100X, 100μm for 200X. (D) Representative immunohistochemical staining images of Ck19, Vimentin, and E-cadherin in both groups. Small black boxes inside figures indicate areas of interest, large black boxes on left lower corner denote enlarged views for better visualization. Magnification: 1000X. Scale bar: 100μm. (E) Representative immunofluorescence staining images of Epcam in both groups. Red arrows indicate normal bile duct cells. Scale bar: 50 μm, Abbreviations: H&E, hematoxylin and eosin; HTVi, hydrodynamic tail vein injection.
Next, we investigated whether Notch pathway modulates EMT or cholangiocellular gene expression patterns in tumor cells. Histopathological evaluation revealed the presence of highly similar HCC lesions in the two cohorts of mice (Fig. 5C). Interestingly, immunohistochemistry and immunofluorescence showed a significant decrease of Ck19 (+) and/or Epcam (+) tumor cells in c-Myc/MCL1/Smad7/dnRBP-J mouse liver tumors (Fig. 5D,E). In addition, while loss of E-Cadherin and increased Vimentin expression were detected in c-Myc/MCL1/Smad7/pT3 tumor tissues, similar to what we have described previously (Fig. 5D), this phenotype was significantly attenuated in c-Myc/MCL1/Smad7/dnRBP-J HCCs.
Altogether, our results demonstrate that the canonical Notch signaling drives EMT and cholangiocellular gene signatures along c-Myc/MCL1/Smad7 driven HCC, without affecting hepatocarcinogenesis.
Oncogenic Smad7 depends on Yap to promote HCC formation
Since both in vitro and in vivo studies indicate that overexpression of SMAD7 leads to YAP activation, we hypothesized that Yap activity is required for the accelerated hepatocarcinogenesis induced by Smad7 overexpression in c-Myc/MCL1/Smad7 mice. To confirm this hypothesis, Yap knockout mice (Yapflox/flox) were hydrodynamically injected with c-Myc/MCL1/Smad7/pCMV-Cre plasmids, allowing the expression of c-Myc/MCL1/Smad7 oncogenes in Yap null hepatocytes. Additional Yapflox/flox mice were injected with c-Myc/MCL1/Smad7/pCMV (empty vector control of pCMV-Cre) as control (Fig. 6A). Consistent with our previous results, all c-Myc/MCL1/Smad7/pCMV injected mice became moribund and had to be euthanized by 4 weeks post injection. In contrast, deletion of Yap completely blocked Smad7 oncogenic function in the c-Myc/MCL1 model. Indeed, all c-Myc/MCL1/Smad7/pCMV-Cre injected Yapflox/flox mice were healthy even at 25 weeks post injection (Fig. 6B). Grossly, numerous nodules were detected in the livers of c-Myc/MCL1/Smad7/pCMV injected Yapflox/flox mice, whereas no tumors could be found in c-Myc/MCL1/Smad7/pCMV-Cre injected Yapflox/flox mice (Fig. 6C). Microscopically, c-Myc/MCL1/Smad7/pCMV mice exhibited numerous tumor lesions, while c-Myc/MCL1/Smad7/pCMV-Cre mouse liver tissues were completely normal (Fig. 6D). Ectopically injected c-Myc and HA-tagged Smad7 were observed in sporadic hepatocytes in c-Myc/MCL1/Smad7/pCMV-Cre mouse livers, implying the successful delivery of the oncogenes (Fig. 6D).
Fig. 6. Depletion of Yap completely abolishes c-Myc/MCL1/Smad7 induced mouse HCC development.
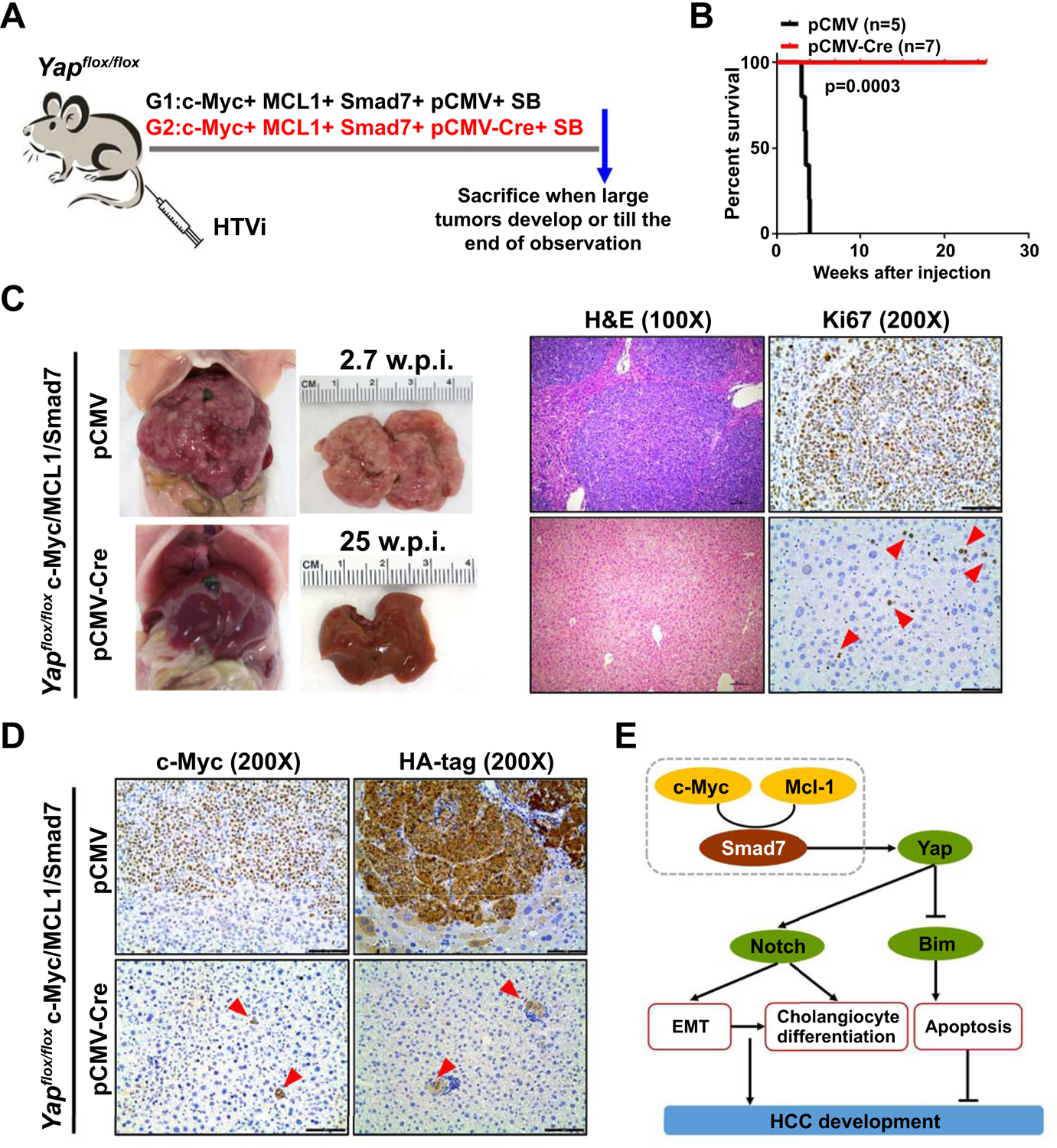
(A) Study design. Yapflox/flox mice were injected with c-Myc/MCL1/Smad7/pCMV/SB (N=5) and c-Myc/MCL1/Smad7/pCMV-Cre/SB (N=7) plasmids, respectively. Mice were monitored and sacrificed when moribund. (B) Survival curve showing that depletion of Yap completely blocks c-Myc/MCL1/Smad7 induced mouse HCC development. (C) Representative gross images, H&E staining, and immunohistochemical staining images of Ki67 in c-Myc/MCL1/Smad7/pCMV (2.7 weeks post injection) and c-Myc/MCL1/Smad7/pCMV-Cre (25 weeks post injection) groups. (D) Representative immunohistochemical staining images of c-Myc and HA-tag in both groups. Scale bars: 200 μm for 100X, 100μm for 200X. (E) Scheme showing the role of Smad7 in promoting c-Myc/MCL1 mouse hepatocarcinogenesis. Abbreviations: H&E, hematoxylin and eosin; HTVi, hydrodynamic tail vein injection.
Altogether, our results indicate that Yap is required for Smad7 induced HCC growth in vivo.
SMAD7 is upregulated in human iCCA and leads to in vivo cholangiocarcinoma development in association with AKT
Next, we assessed the functional role of SMAD7 in iCCA. In a human iCCA collection (n=50; Supporting Table S9), levels of SMAD7 were significantly more elevated in tumors than in corresponding non-tumorous counterparts and normal livers from healthy patients (Fig. 7A). Higher SMAD7 levels in CCA samples than in non-tumorous surrounding tissues were also detected in the TCGA CHOL (cholangiocarcinoma) cohort (Fig. 7B). To determine a possible prognostic role of SMAD7 in this cancer type, SMAD7 mRNA levels in tumors were related to the length of the patients’ survival. Importantly, higher SMAD7 gene expression correlated with lower iCCA survival rate, as assessed by both Kaplan–Meier and linear regression analysis (p < 0.0001; Fig. 7C and Supporting Table S10). Of note, this association remained strongly significant after multivariate Cox regression analysis (p < 0.0001; Supporting Tables S11 and S12), thus suggesting SMAD7 mRNA levels as a possible independent prognostic factor for iCCA. No significant differences were detected neither in residual resection nor in treatments received after surgery in iCCA patients with better or poorer prognosis (not shown). In the same tumor samples, SMAD7 expression directly correlated with levels of NOTCH (HES1, SOX9) and YAP (CYR61, CTGF) targets as well as with cholangiocellular (EPCAM, KRT19) and EMT markers (SNAI1, VIM) (Supporting Fig. S22). Consistent results were provided by the TCGA CHOL dataset (Supporting Fig. S23). By IHC, we found that SMAD7 is not expressed in normal hepatocytes and cholangiocytes, whereas intense SMAD7 was detected in stellate cells, in accordance with the Human Protein Atlas data (https://www.proteinatlas.org/) (Fig. 7D). Noticeably, iCCA specimens displayed a strong nuclear immunoreactivity for SMAD7 in 42 of 50 (84%) samples, whereas low/absent staining for SMAD7 was detected in the remaining iCCA samples and in all matching non-neoplastic liver tissues. Positive nuclear staining for YAP and the NOTCH target HES1 was detected in 49 (98%) and 39 (78%) tumors, respectively (Fig. 7D). Similar to that reported in HCC cell lines, transient overexpression of HA-tagged SMAD7 in human KKU156 and KKU213 CCA cell lines led to increase proliferation and decrease of apoptosis, which were paralleled by induction of NOTCH (HES1, SOX9), YAP (CYR61, CTGF), cholangiocellular (KRT19, EPCAM), and EMT (SNAI1, VIM) targets, as well as by increased TEAD activity (Supporting Fig. S24). Opposite results were obtained when SMAD7 was silenced in the RBE cell line by specific small interfering siRNA (Supporting Fig. S25).
Fig. 7. SMAD7 and its downstream effectors are overexpressed in human intrahepatic cholangiocarcinoma (iCCA) specimens.
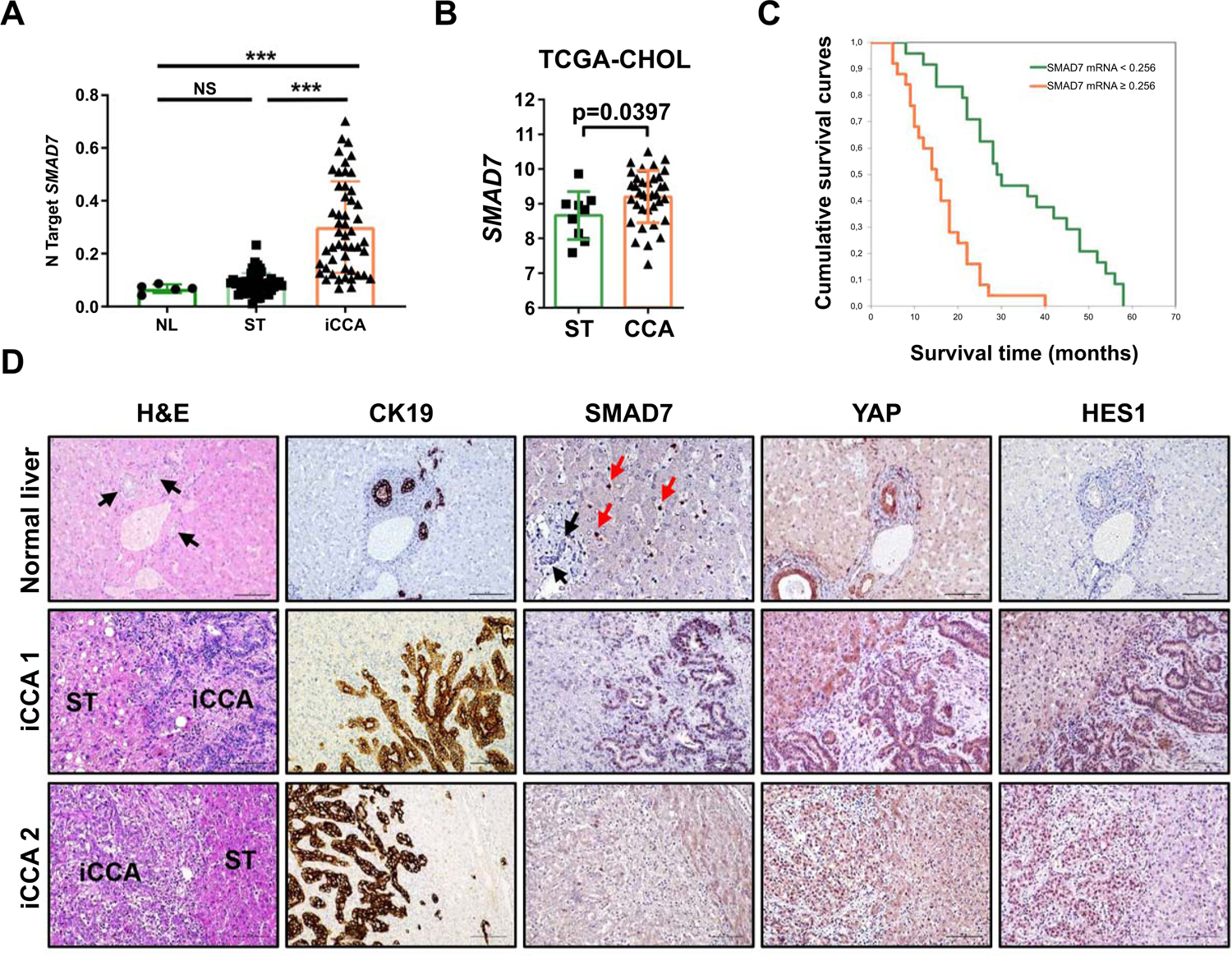
(A) Quantitative real-time RT-PCR analysis of SMAD7 mRNA levels in normal livers (n = 5), iCCA (n = 50), and corresponding non-tumorous surrounding liver tissues (ST; n = 50). Quantitative values were calculated by using the PE Biosystems Analysis software and expressed as number target (N Target). N Target = 2−ΔCt, wherein the ΔCt value of each sample was calculated by subtracting the average Ct value of the SMAD7 gene from the average Ct value of the β-Actin gene. P-value was calculated using Mann–Whitney U test. *** P < 0.0001 when compared to Normal Liver (NL); *** P < 0.0001 when compared to Normal liver (NL) and Surrounding Tissue (ST). Abbreviation: NS, not significant. (B) SMAD7 expression in non-tumorous surrounding liver tissues (ST; n = 9) and cholangiocarcinoma tumor tissues (CCA; n = 36) from the TCGA CHOL cohort. (C) Kaplan–Meier survival curve of human iCCA with high and low SMAD7 mRNA levels, showing the unfavorable outcome of patients with elevated expression of this gene. (D) Representative immunohistochemical patterns of SMAD7 and YAP and NOTCH (HES1) downstream effectors in normal livers and two human intrahepatic cholangiocarcinoma specimens (iCCA 1 and 2). In normal liver, bile ducts composed of cholangiocytes (indicated by black arrows and being positive for CK19, YAP, and HES1 immunoreactivity) as well as normal hepatocytes display the absence of SMAD7 staining. Immunoreactivity for SMAD7 is limited to stellate cells (indicated by red arrows). Equivalent results were detected in non-tumorous surrounding liver tissues (not shown). In contrast, iCCA 1 exhibits concomitantly strong nuclear staining for SMAD7, YAP, and HES1 proteins, whereas weak/absent immunoreactivity is displayed by the non-tumorous surrounding counterpart (ST). iCCA 2 shows instead weak nuclear staining for SMAD7, limited to few tumor cells, whereas YAP and HES1 proteins display strong nuclear immunoreactivity. The CK19 staining was used as a marker of biliary differentiation of the tumors. Original magnification: 200X or 400x (for SMAD7 in normal liver); scale bar: 100 μm or 50 μm (for SMAD7 in normal liver). Abbreviation: H&E, hematoxylin and eosin staining.
Finally, we assessed the role of SMAD7 in iCCA in vivo. Given that both SMAD7 targets, activated Notch and Yap, are able to convert normal hepatocytes into iCCA cells in AKT overexpressing livers (37, 42, 43), we overexpressed SMAD7 and myristoylated/activated AKT in the mouse liver via HTVi to determine whether the same phenotype occurs (Fig. 8A). Noticeably, co-expression of SMAD7 and AKT led to development of pure iCCA by 13.8–15 weeks post injection (Fig. 8A,B), a time point when AKT only overexpressing mice display exclusively clusters of hepatocellular preneoplastic lesions (Supporting Fig. S26). Immunoreactivity for the injected constructs as well as markers of Yap and Notch pathway activation, together with cholangiocellular markers Ck19 and Epcam was detected in these tumors (Fig. 8C,D).
Fig. 8. Smad7 cooperates with AKT to induce cholangiocarcinoma development in mice.
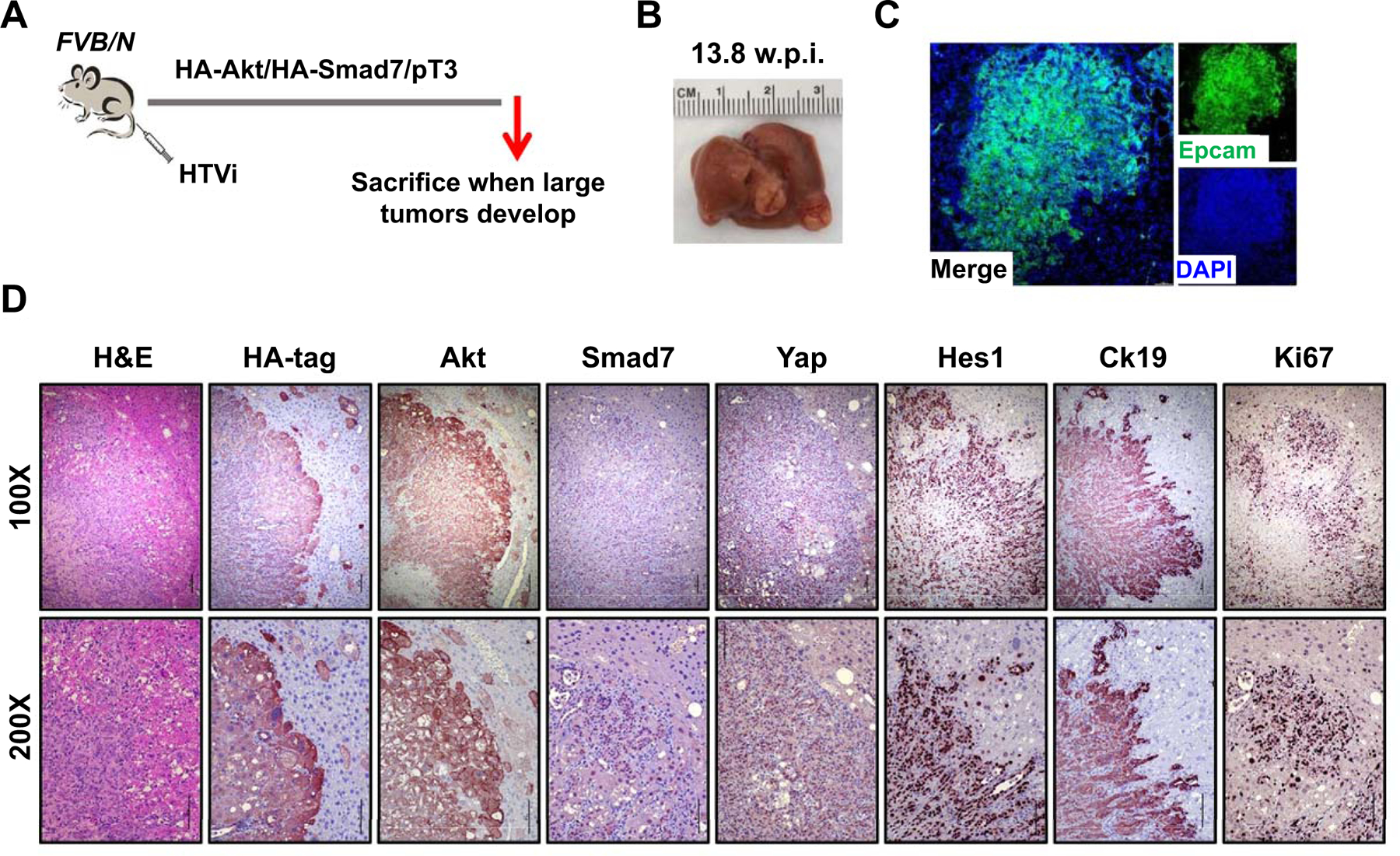
(A) Study design. FVB/N mice were injected with Akt/Smad7/SB (N=8) plasmids. Mice were monitored and sacrificed when moribund. (B) Macroscopic image of an Akt/Smad7 mouse showing the presence of large tumor nodules on the liver surface. (C, D) Immunofluorescence (C) and immunohistochemistry images (D) showing that Akt/Smad7 mice express the injected transgenes (HA-tag; Akt, and Smad7) as well as markers of Yap (Yap) and Notch (Hes1) signaling activation. Ck19 and Ki67 staining indicate the cholangiocellular differentiation and the proliferative features of Akt/Smad7 tumor lesions. Scale bars: 200 μm for 100X, 100μm for 200X. Abbreviation: HTVi, hydrodynamic tail vein injection.
Overall, the present data indicate that SMAD7 is induced in human iCCA and drives cholangiocarcinogenesis in the mouse in association with AKT.
Discussion
In the present study, we demonstrate that SMAD7, a canonical antagonist of the TGF-β signaling (15), contributes to both HCC and iCCA tumorigenesis, thus indicating that inhibition of TGF-β signaling plays a pivotal role in promoting HCC and iCCA formation and growth. Importantly, our results imply that SMAD7 is not a bona fide oncogene, but rather a tumor modifier. When overexpressed, SMAD7 is unable to drive malignant conversion, but supports HCC and iCCA malignant properties, including proliferation, survival, and invasion. It is important to emphasize SMAD7 possesses both pro- and anti-tumorigenic activities. Accordingly, subgroups of HCC with high and low SMAD7 expression were detected. These data suggest that the functional contribution of SMAD7 to HCC development might be context and oncogene dependent.
We discover SMAD7 overexpression led to hepatocyte-malignant cholangiocyte conversion in the context of AKT, but not c-Myc/MCL1, overexpression in vivo, implying that other factors cooperate with SMAD7 to modulate liver cell fate. In light of our data, it appears clear that SMAD7 augments the possibility of hepatocyte-malignant cholangiocyte conversion in the context of a “pro-cholangiocellular” environment. Previously, we have shown that AKT overexpression alone triggers the development of both HCC and iCCA in the mouse liver, with the predominance of hepatocellular over cholangiocellular lesions (43). Thus, the present results indicate that SMAD7 co-expression favors a selective pressure toward the development of malignant cholangiocellular lesions in AKT mice.
The cellular origin of primary liver tumors in humans, including HCC and iCCA, is highly debated (44). Indeed, studies have demonstrated a high degree of plasticity of cells within the liver, including hepatocytes, cholangiocytes, and HPCs. All these cells have the capability to transdifferentiate and contribute to liver regeneration and tumorigenesis (44, 45). As human primary liver tumors may develop from different cellular lineages, modeling of tumor development from different cell types within the liver is critical and has major translational implications.
At the molecular level, we found that SMAD7 activates the YAP/NOTCH cascade in vitro and in vivo, resulting in more aggressive tumors with cholangiocellular, stemness, and EMT molecular features. Specifically, we discovered that overexpression of SMAD7 promotes YAP dependent oncogenic activity, and ablation of Yap completely abolishes tumor accelerating properties of Smad7 in vivo. These data, together with the induction of the same changes in human HepG2 and Hep293TT hepatoblastoma cell lines following overexpression of SMAD7 (not shown) identify SMAD7 as a major inducer of the NOTCH and YAP pathways in liver cancer. Nonetheless, our study demonstrates that YAP and NOTCH possess distinct function downstream of SMAD7, at least in mice. Indeed, while depletion of Yap completely inhibited c-Myc/MCL1/Smad7 tumor development, Notch blockade only diminished cholangiocellular gene expression and EMT, thus suggesting that Yap is the pivotal player along c-Myc/MCL1/Smad7 hepatocarcinogenesis, whereas Notch acts downstream of Yap to control EMT and cholangiocellular gene expression. Furthermore, we show that SMAD7 enhances p-YapY357 expression, leading to YAP nuclear translocation. Interestingly, this regulation is likely to be independent of Hippo, as our preliminary data show the absence of significant suppression of the Hippo kinases in c-Myc/MCL1/Smad7 mouse HCCs. Consistently, no changes in the levels of p-YapS127, the major YAP phosphorylation site for Hippo kinases, were detected in these mice (Supporting Fig. S27). Although previous data suggest that SMAD7 directly binds to YAP (46), we failed to detect the direct interaction of SMAD7 and YAP in liver tumor cells. SRC family kinases, such as FAK, have been shown to induce p-YapY357 (47). However, whether FAK is able to regulate p-YapY357 in HCC and iCCA remains to be determined. Thus, it would be important to identify the major kinases regulating p-YapY357 in liver tumor cells and to define how SMAD7 interacts with these kinases to regulate YAP activation.
Finally, these data might have important translational implications. It is now recognized that liver cancer, including both HCC and iCCA, is a highly heterogeneous disease with multiple molecular subgroups. It is important to model the distinct molecular subgroups with combination of different oncogenic events. As we found that SMAD7 fully depends on YAP to exert its oncogenic potential, targeting YAP could represent a promising treatment option for human HCC and iCCA patients displaying SMAD7 activation. The SMAD7 overexpressing mouse tumors will serve as excellent preclinical models to test these therapies.
Supplementary Material
Financial Support
This study was supported by NIH grants R01CA190606 and R01CA239251 to XC, P30DK026743 for UCSF Liver Center, UCSF Program for Breakthrough Biomedical Research and Sandler Foundation to MR, and National Natural Science Foundation (grant number 82002967) to HW.
List of Abbreviations
- SMAD7
Mothers against decapentaplegic homolog 7
- TGF-β
Transforming growth factor β
- HCC
Hepatocellular carcinoma
- GSEA
Gene set enrichment analysis
- EMT
Epithelial-mesenchymal transition
- iCCA
Intrahepatic cholangiocarcinoma
- TCGA
The Cancer Genome Atlas
- NES
Normalized enrichment score
- SB
Sleeping beauty
- Cre
Cyclization recombination
- CMV
Cytomegalovirus
- HTVi
Hydrodynamic tail vein injection
- IHC
Immunohistochemistry
- Gs
Glutamine synthetase
- GFP
Green fluorescent protein
- YFP
Yellow fluorescent protein
- RFP
Red florescent protein
- CFP
Cyan fluorescent protein
- Ccnd1
Cyclin D1
- BIM
Bcl-2 interacting mediator of cell death
- MCL1
Myeloid cell leukemia 1
- YAP
Yes-associated protein
- dnRBP-J
Dominant negative recombination signal binding protein for immunoglobulin kappa J region
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to disclose.
Author names in bold designate shared co-first authors.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2.Brunt E, Aishima S, Clavien P-A, Fowler K, Goodman Z, Gores G, Gouw A, et al. cHCC-CCA: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 2018;68:113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kudo M, Finn RS, Qin S, Han K-H, Ikeda K, Piscaglia F, Baron A, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. The Lancet 2018;391:1163–1173. [DOI] [PubMed] [Google Scholar]
- 4.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, Kudo M, et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med 2020;382:1894–1905. [DOI] [PubMed] [Google Scholar]
- 5.Personeni N, Lleo A, Pressiani T, Colapietro F, Openshaw MR, Stavraka C, Pouptsis A, et al. Biliary Tract Cancers: Molecular Heterogeneity and New Treatment Options. Cancers 2020; 11:3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llovet JM, Montal R, Villanueva A. Randomized trials and endpoints in advanced HCC: Role of PFS as a surrogate of survival. J Hepatol. 2019;70:1262–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Banales JM, Marin JJG, Lamarca A, Rodrigues PM, Khan SA, Roberts LR, Cardinale V, et al. Cholangiocarcinoma 2020: the next horizon in mechanisms and management. Nat Rev Gastroenterol Hepatol. 2020;17:557–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD pathway and its regulation in hepatic fibrosis. J Histochem Cytochem. 2016;64:157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan W, Liu T, Chen W, Hammad S, Longerich T, Hausser I, Fu Y, et al. ECM1 Prevents Activation of Transforming Growth Factor beta, Hepatic Stellate Cells, and Fibrogenesis in Mice. Gastroenterology 2019;157:1352–1367.e1313. [DOI] [PubMed] [Google Scholar]
- 10.Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, Liang X, et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 2015;64:1593–1604. [DOI] [PubMed] [Google Scholar]
- 11.Moon H, Ju H-L, Chung SI, Cho KJ, Eun JW, Nam SW, Han K-H, et al. Transforming Growth Factor-β Promotes Liver Tumorigenesis in Mice via Up-regulation of Snail. Gastroenterology 2017;153:1378–1391.e1376. [DOI] [PubMed] [Google Scholar]
- 12.Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol 2016;8:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wakefield LM, Hill CS. Beyond TGFβ: roles of other TGFβ superfamily members in cancer. Nat. Rev. Cancer 2013;13:328–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Macias MJ, Martin-Malpartida P, Massague J. Structural determinants of Smad function in TGF-beta signaling. Trends Biochem Sci 2015;40:296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan X, Liao H, Cheng M, Shi X, Lin X, Feng X-H, Chen Y-G. Smad7 Protein Interacts with Receptor-regulated Smads (R-Smads) to Inhibit Transforming Growth Factor-β (TGF-β)/Smad Signaling. J Biol Chem. 2016;291:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miyazawa K, Miyazono K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb Perspect Biol 2017;9:a022095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Fei T, Zhang L, Zhang R, Chen F, Ning Y, Han Y, et al. Smad7 antagonizes transforming growth factor β signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol Cell Biol 2007;27:4488–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stolfi C, Marafini I Fau, De Simone V, Pallone F Fau, Monteleone G. The dual role of Smad7 in the control of cancer growth and metastasis. Int J Mol Sci 2013;14:23774–23790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park YN, Chae KJ, Oh BK, Choi J, Choi KS, Park C. Expression of Smad7 in hepatocellular carcinoma and dysplastic nodules: resistance mechanism to transforming growth factor-beta. Hepatogastroenterology 2004;51:396–400. [PubMed] [Google Scholar]
- 20.Xia H, Ooi Ll, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets PTEN and SMAD7 to promote drug resistance and recurrence of liver cancer. Hepatology 2013;58:629–641. [DOI] [PubMed] [Google Scholar]
- 21.Sun H, Peng Z, Tang H, Xie D, Jia Z, Zhong L, Zhao S, et al. Loss of KLF4 and consequential downregulation of Smad7 exacerbate oncogenic TGF-beta signaling in and promote progression of hepatocellular carcinoma. Oncogene 2019;36:2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Q, Liu L, Liu CH, Shao F, Xie F, Zhang CH, Hu SY. Expression of Smad7 in cholangiocarcinoma: prognostic significance and implications for tumor metastasis. Asian Pac J Cancer Prev 2012;13:5161–5165. [DOI] [PubMed] [Google Scholar]
- 23.Wang J, Wang H, Peters M, Ding N, Ribback S, Utpatel K, Cigliano A, et al. Loss of Fbxw7 synergizes with activated Akt signaling to promote c-Myc dependent cholangiocarcinogenesis. J Hepatol 2019;71:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cerami E, Gao J, Dogrusoz U, Gross B, Sumer S, Aksoy B, Jacobsen A, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012; 2: 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173:400–416.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen J, Zaidi S, Rao S, Chen JS, Phan L, Farci P, Su X, et al. Analysis of Genomes and Transcriptomes of Hepatocellular Carcinomas Identifies Mutations and Gene Expression Changes in the Transforming Growth Factor-beta Pathway. Gastroenterology 2018;154:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Dong M, Xu Z, Song X, Zhang S, Qiao Y, Che L, et al. Notch2 controls hepatocyte-derived cholangiocarcinoma formation in mice. Oncogene 2018;37:3229–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pepe-Mooney BJ, Dill MT, Alemany A, Ordovas-Montanes J, Matsushita Y, Rao A, Sen A, et al. Single-Cell Analysis of the Liver Epithelium Reveals Dynamic Heterogeneity and an Essential Role for YAP in Homeostasis and Regeneration. Cell Stem Cell 2019;25:23–38.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim CL, Choi SH, Mo JS. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019;8:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kar R, Jha N, Jha SK, Sharma A, Dholpuria S, Asthana N, Chaurasiya K, et al. A “NOTCH” Deeper into the Epithelial-To-Mesenchymal Transition (EMT) Program in Breast Cancer. Genes (Basel) 2019;10:pii: E961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendez-Lucas A, Li X, Hu J, Che L, Song X, Jia J, Wang J, et al. Glucose Catabolism in Liver Tumors Induced by c-MYC Can Be Sustained by Various PKM1/PKM2 Ratios and Pyruvate Kinase Activities. Cancer Res 2017;77:4355–4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu Z, Xu M, Liu P, Zhang S, Shang R, Qiao Y, Che L, et al. The mTORC2-Akt1 Cascade Is Crucial for c-Myc to Promote Hepatocarcinogenesis in Mice and Humans. Hepatology 2019;70:1600–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, Lubrano S, et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 2019;35:457–472.e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao WB, Lu Q, Nguyen MN, Su Y, Ziemann M, Wang LN, Kiriazis H, et al. Stimulation of beta-adrenoceptors up-regulates cardiac expression of galectin-3 and BIM through the Hippo signalling pathway. Br J Pharmacol 2019;176:2465–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M, Wang J, Xu Z, Li R, Wang P, Shang R, Cigliano A, et al. SNAI1 Promotes the Cholangiocellular Phenotype, but not Epithelial-Mesenchymal Transition, in a Murine Hepatocellular Carcinoma Model. Cancer Res 2019;79:5563–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang G, Gao X, Song YK, Vollmer R, Stolz DB, Gasiorowski JZ, Dean DA, et al. Hydroporation as the mechanism of hydrodynamic delivery. Gene Therapy 2004;11:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest. 2012;122:2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Planas-Paz L, Orsini V, Boulter L, Calabrese D, Pikiolek M, Nigsch F, Xie Y, et al. The RSPO–LGR4/5–ZNRF3/RNF43 module controls liver zonation and size. Nat Cell Biol 2016;18:467–479. [DOI] [PubMed] [Google Scholar]
- 39.de Roo JJ, Vloemans SA, Vrolijk H, de Haas EF, Staal FJ. Development of an in vivo model to study clonal lineage relationships in hematopoietic cells using Brainbow2.1/Confetti mice. Future Sci OA 2019;5:FSO427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon-Veenboer C, Barker N, et al. Intestinal Crypt Homeostasis Results from Neutral Competition between Symmetrically Dividing Lgr5 Stem Cells. Cell 2010;143:134–144. [DOI] [PubMed] [Google Scholar]
- 41.Greig JA, Limberis MP, Bell P, Chen SJ, Calcedo R, Rader DJ, Wilson JM. Nonclinical Pharmacology/Toxicology Study of AAV8.TBG.mLDLR and AAV8.TBG.hLDLR in a Mouse Model of Homozygous Familial Hypercholesterolemia. Hum Gene Ther Clin Dev 2017;28:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, Song X, Cao D, Xu Z, Fan B, Che L, Hu J, et al. Pan-mTOR inhibitor MLN0128 is effective against intrahepatic cholangiocarcinoma in mice. J Hepatol 2017;67:1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, et al. Increased Lipogenesis, Induced by AKT-mTORC1-RPS6 Signaling, Promotes Development of Human Hepatocellular Carcinoma. Gastroenterology 2011;140:1071–1083.e1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu W-Y, Bird TG, Boulter L, Tsuchiya A, Cole AM, Hay T, Guest RV, et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat Cell Biol 2015;17:971–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raven A, Lu W-Y, Man TY, Ferreira-Gonzalez S, O’Duibhir E, Dwyer BJ, Thomson JP, et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017;547:350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferrigno O, Lallemand F, Verrecchia F, L’Hoste S, Camonis J, Atfi A, Mauviel A. Yes-associated protein (YAP65) interacts with Smad7 and potentiates its inhibitory activity against TGF-β/Smad signaling. Oncogene 2002;21:4879–4884. [DOI] [PubMed] [Google Scholar]
- 47.Sugihara T, Werneburg NW, Hernandez MC, Yang L, Kabashima A, Hirsova P, Yohanathan L, et al. YAP Tyrosine Phosphorylation and Nuclear Localization in Cholangiocarcinoma Cells Are Regulated by LCK and Independent of LATS Activity. Molecular Cancer Res 2018;16:1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.