

Abstract
Background
DNA double‐strand breaks (DSBs) are harmful to the cell as it could lead to genomic instability and cell death when left unrepaired. Homologous recombination and nonhomologous end‐joining (NHEJ) are two major DSB repair pathways, responsible for ensuring genome integrity in mammals. There have been multiple efforts using small molecule inhibitors to target these DNA repair pathways in cancers. SCR7 is a very well‐studied anticancer molecule that blocks NHEJ by targeting one of the critical enzymes, Ligase IV.
Recent findings
In this review, we have highlighted the anticancer effects of SCR7 as a single agent and in combination with other chemotherapeutic agents and radiation. SCR7 blocked NHEJ effectively both in vitro and ex vivo. SCR7 has been used for biochemical studies like chromosomal territory resetting and in understanding the role of repair proteins in cell cycle phases. Various forms of SCR7 and its derivatives are discussed. SCR7 is also used as a potent biochemical inhibitor of NHEJ, which has found its application in improving genome editing using a CRISPR‐Cas system.
Conclusion
SCR7 is a potent NHEJ inhibitor with unique properties and wide applications as an anticancer agent. Most importantly, SCR7 has become a handy aid for improving genome editing across different model systems.
Keywords: chemotherapy, DNA double‐strand break, DNA repair inhibitors, genome editing, homologous recombination, nonhomologous DNA end‐joining
1. INTRODUCTION
DNA repair pathways play a significant role in maintaining genome integrity. Defects in the DNA repair pathways inside cells can predispose them to cancer. 1 , 2 DNA repair proteins, when overexpressed in cancers, promote their growth and provide resistance to therapy. 3 , 4 One of the cancer therapeutic modalities involves targeting the repair of DNA double‐strand breaks (DSBs), resulting in the inhibition of the repair machinery. This results in the accumulation of DSBs and cell death. 5 , 6 Among different DNA damages, DSBs are considered as most deleterious as a failure of their repair results in cell death.
Three different DNA repair pathways homologous recombination (HR), nonhomologous end‐joining (NHEJ), and alternative NHEJ, are responsible for the repair of DNA DSBs. 7 , 8 , 9 HR is known to operate during the S‐phase of the cell cycle, whereas NHEJ is functional throughout the cell cycle. HR utilizes the information from a template to mend the breaks and is precise without errors. 10 , 11 , 12 NHEJ is an error‐prone pathway involving insertion or deletion at the site of repair as it involves a template‐independent mode of joining. When there is a DSB, the KU proteins (KU70/KU80) localize to the site of the break to protect them from degradation. KU70/80 then recruit other proteins like Artemis and DNA‐PKcs, which are endonucleases and bring about end resection. 12 , 13 , 14 , 15 PAXX, another protein recently been known to stabilize the repair machinery. 16 Pol μ and Pol λ, belonging to the Pol X family, fill the gap, thereby carrying out the polymerization at the repair site. The final step of NHEJ being ligation involves DNA ligase IV/XRCC4/XLF complex to seal the ends and repair the broken DNA 11 , 17 (Figure 1). Another pathway known as alternative NHEJ (A‐NHEJ), which is a back‐up pathway to classical NHEJ (c‐NHEJ) is present in cells and use a short terminal microhomology region for sealing the DNA break. 9 , 12 , 14 , 18
FIGURE 1.
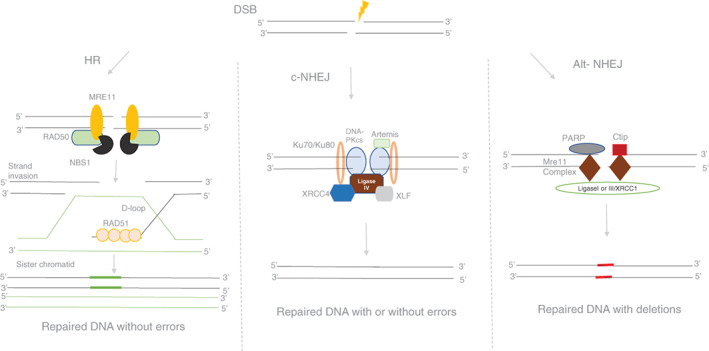
Schematic representation of DSB repair pathways that operates in a mammalian cell. There are mainly three pathways to take care of the repair of DSBs in mammalian cells; homologous recombination (HR), classical nonhomologous end‐joining (c‐NHEJ), and alternative NHEJ (A‐NHEJ). As shown in the figure, each pathway has its own repertoire of proteins, which aids in repairing the broken DNA. HR repairs DNA breaks based on the homology and occurs at the replicative phase of the cell cycle and is error‐free. c‐NHEJ occurs throughout the cell cycle and is error‐prone. Ligation of broken ends is mediated by Ligase IV/XRCC4/XLF complex. Alternative NHEJ is also error‐prone, leading to extensive deletions, and repair occurs based on the microhomology regions
Several small‐molecule inhibitors targeting DSB repair pathways have been developed. 2 , 19 One such inhibitor is SCR7, which interferes with the sealing of the nick, which is the last step of the NHEJ. 6 , 12 , 20 Ligase IV, the enzyme dedicated to the final ligation of broken ends, is one of the most critical proteins associated with NHEJ. SCR7 binds to the DNA binding domain of Ligase IV to block NHEJ and leave the ends unrepaired, resulting in the accumulation of DNA breaks and thus leading to activation of cell death pathways. 20 The activity of SCR7 has been demonstrated both in vitro in several cancer cell lines and in vivo in multiple animal tumor models. 20
SCR7 was first reported in 2012 and has made an impact across different fields. 12 , 19 SCR7 has been used in biochemical studies and, most importantly, to improve genome editing using CRISPR‐Cas technology. 6 , 12 The recent advances in genome engineering technology are due in part to the use of small‐molecule inhibitors like SCR7, which has enhanced the efficiency of genome editing. However, in this short review, we discuss the evolution of SCR7 as a cancer therapeutic agent and its other applications.
2. THERAPEUTIC ROLE OF SCR7
Cancer cells alter DNA repair protein expression to gain a survival advantage. 21 Harnessing the defects in DNA repair genes and utilizing differential expression of repair proteins in cancer cells compared to normal cells can be a great strategy for inhibiting the progression of cancer. 22 , 23 One such example that has given successful results is the FDA‐approved PARP inhibitor Olaparib that can bring about synthetic lethality in breast and ovarian cancers that are deficient in BRCA1/2. 24 , 25 ATM inhibitor KU‐55933 has shown promising results in Fanconi anemia deficient pancreatic cancer cell line. 26
NHEJ is a rapid and quick fixing DNA repair process that takes care of 70% DSBs within the cells particularly operating when homology is absent. 8 , 11 When NHEJ is inhibited at the initial steps of damage recognition or end resection, there is a chance of them being rerouted to the back‐up alternative NHEJ pathway. 9 , 18 , 27 Cancer cells escape cell death signals as they can shift from one repair pathway to the other efficiently. Therefore, blocking a protein like Ligase IV at the final step of the repair process does not allow for shift easily and lead to efficient cell death signaling. 2
Chen et al developed L189, a pan‐ligase inhibitor, which inhibits all three ligases, Ligase IV, III, and I. However, this molecule was not extensively used as anticancer therapeutics. 28
The most effective and widely studied DNA Ligase IV/XRCC4 inhibitor is SCR7. 12 Chemically, SCR7 is 5,6‐bis(benzylidene amino)‐2‐mercapto‐pyrimidin‐4‐ol that binds to the DNA‐binding domain of Ligase IV and exerts its action. In the cell‐free repair assays, SCR7 showed concentration‐dependent inhibition of NHEJ. 20 The sensitivity of cancer cell lines to SCR7 correlated with the expression levels of Ligase IV. Knockdown studies elucidated the specificity of SCR7. 20 Western blotting analysis of MCF7 cells treated with SCR7 showed a decrease in anti‐apoptotic proteins that coincided with an increase in pro‐apoptotic proteins. Induction of the intrinsic pathway of apoptosis in mouse tumors was evident in the form of DNA fragmentation using TUNEL assay. In three different mouse models including xenografts, significant tumor regression was observed even leading to an increase in lifespan 20 (Figure 2).
FIGURE 2.
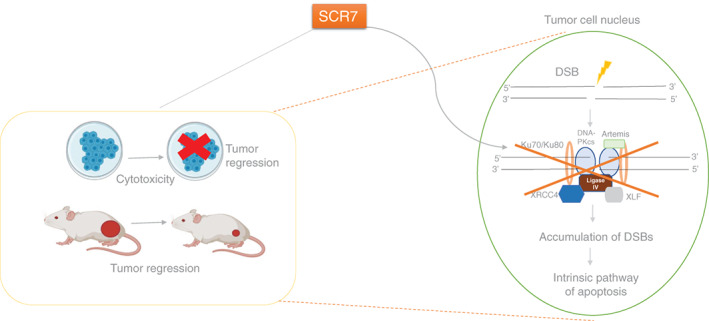
Mechanism of SCR7‐mediated cell death during tumor regression. SCR7, an inhibitor of NHEJ, is known to induce cytotoxicity in cancer cells leading to tumor regression in mice. Once administered, SCR7 is diffused inside the nucleus and binds to the DNA binding domain of Ligase IV. This prevents the recruitment of Ligase IV to DSBs, resulting in inhibition of NHEJ, thus, culminating in the accumulation of DSBs inside the cells. Finally, activation of the intrinsic apoptotic pathway led to cancer cell death and thus tumor regression
The significant effect of SCR7 that is of additional interest is that it sensitizes the cancer cells to chemo and radiotherapeutic agents when given in combination. 20 Studies by Gkotzamanidou et al 29 revealed that in patients with multiple myeloma, combination therapy involving DNA repair (SCR7, NU7026, and RS‐1) inhibitors along with an alkylating agent, melphalan showed better cytotoxicity. 29 Another chemotherapeutic drug, doxorubicin, when administered along with SCR7, showed a 15% to 50% higher cytotoxicity in cervical cancer. 30 Gopalakrishnan et al 31 showed that SCR7 enhances ionizing radiation's effects in one of the most common types of non‐Hodgkin lymphoma, diffuse large B cell lymphoma. 31 Further, inhibition of the NHEJ pathway by SCR7 overcomes HSP110 conferred resistance to oxaliplatin in the SW480 colorectal adenocarcinoma cell line. 32
3. DIFFERENT FORMS OF SCR7
Vartak et al showed that parental SCR7 get autocyclized to cyclized‐SCR7 that possess the same molecular weight (334.09), formula (C18H14N4OS), and melting temperature (221°C‐225°C). 33 Although parental SCR7 and cyclized version are the same in molecular formula and other properties, SCR7‐pyrazine is an entirely different molecule with a molecular weight of 332.07, formula C18H12N4OS, and melting point 194°C to 196°C. Studies have shown that SCR7‐cyclized and SCR7‐pyrazine exhibits a Ligase IV‐dependent inhibition of NHEJ; however, the latter shows less specificity inside cells (Figure 3). The nanopolymer‐encapsulated version of SCR7, E‐SCR7, induced not only 5‐fold higher cytotoxicity in the cells but also increased its bioavailability. 34 , 35 SCR7 and its pyrazine form were further developed as water‐soluble versions that regressed tumors in mice. 36 , 37 , 38 However, water‐soluble SCR7 was Ligase IV specific, whereas water‐soluble SCR7‐pyrazine had pan‐ligase activity. 6 , 36 , 37
FIGURE 3.
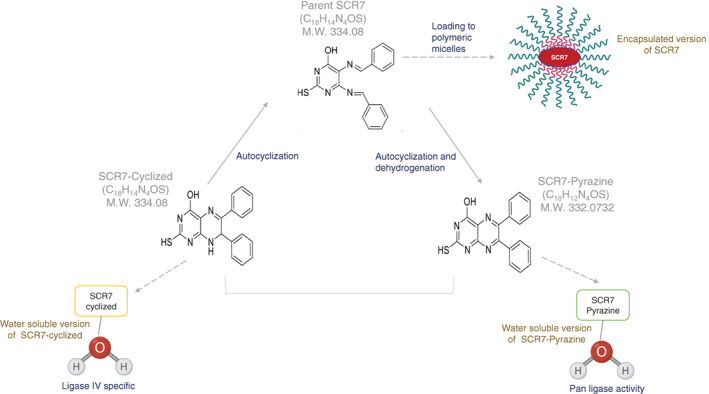
Different forms of SCR7. Parental SCR7 gets autocyclized to form a cyclized version of SCR7 having the same molecular weight, formula, and melting temperature as that of parental SCR7. SCR7‐pyrazine is the oxidized version of parental SCR7. It is a different entity compared to parental SCR7 with a molecular weight of 332.07 and melting point 194°C–196°C. When parent SCR7 loaded onto polymeric micelles, called encapsulated SCR7, it showed a better bioavailability. The water‐soluble version of SCR had ligase IV activity, whereas SCR7‐pyrazine had pan ligase activity. Part of the figures made using biorender.com
One of the drawbacks of SCR7 is its high IC50 in cancer cells. Recently, SCR130, a spiro derivative of SCR7, was shown to induce cytotoxicity at 2 μM in Nalm6 cells, 20 times higher than that of SCR7 (40 μM) and exerts its effect in Ligase IV dependent manner. 39 It induces cytotoxicity by activating both the intrinsic and extrinsic apoptotic pathways. Like SCR7, SCR130 also sensitizes cancer cells to γ‐radiation when used in combination. 39
4. SCR7 IS A BIOCHEMICAL INHIBITOR OF NHEJ
Apart from being an excellent anticancer agent, SCR7 has also been known for its biochemical role. From previous studies, it is evident that SCR7 can block the ligation of DSBs with either compatible or noncompatible ends. 20 SCR7 can inhibit the amalgamation of breaks present in both plasmid and oligomers 6 , 12 , 19 , 20 [in press]. These results have given a new dimension to SCR7, focussing on roles other than anticancer activity. To capture the dynamics of NHEJ in the cell, single‐molecule FRET along with super‐resolution localization microscopy was used. In this study, 100 μM of SCR7 was used as a control to elucidate the mechanism and dynamics of NHEJ. 40 , 41 Tripathi et al 42 used SCR7 as a biochemical inhibitor to understand the DNA repair protein's role at different phases of the cell cycle. 42 SCR7 has also been used to understand the resetting of chromosome territory upon DNA damage. 43 The Zebrafish as a model system has been developed to screen for inhibitors of NHEJ, owing to the high frequency of NHEJ in early embryonic development. SCR7 alleviated SV40 fragment‐induced embryonic lethality. SCR7‐induced disruption in NHEJ established Zebrafish as a model system for screening targets against NHEJ, which might be useful for the development of novel anticancer drugs. 44
5. ROLE OF SCR7 IN GENOME EDITING
Another vital role of SCR7 is its use in genome editing. 6 , 12 , 38 In CRISPR/Cas9 mediated genome editing, after the induction of DSBs by Cas9, endogenous DNA repair pathways, HR or NHEJ are activated. NHEJ being error‐prone in its action results in gene disruption by generating indels at the target; on the other hand, HR can integrate the donor DNA into the target site. 45 Efforts have been put in the direction of shifting the balance toward HR during the editing process. 6 Various studies had shown that the addition of SCR7 showed a 2‐19‐fold enhancement in the efficiency of genome editing in mammalian cells and mice embryos. Inhibition of NHEJ by SCR7 shifts the mode of repair to HR 46 , 47 , 48 (Figure 4). The efficiency of CRISPR/Cas mediated gene editing increased by 2 to 3‐folds in porcine fetal fibroblast and cancer cells in the presence of SCR7. 49 , 50 Apart from mammalian systems, SCR7 also increased HDR by 40% when transfection studies were performed after cloning genes of interest from Saccharomyces cerevisiae. 51 , 52 One of the gene inactivation methods involves the integration of markers (resistance cassette) for productive editing. 53 showed that SCR7 increased the efficacy of productive (integration) editing. Similarly, enhanced knock‐in efficiency using CRISPR/Cas in the presence of SCR7‐pyrazine in Xenopus oocyte has been reported, 54 suggesting the use of SCR7 in inhibiting NHEJ independent of the model system and gene editing mechanisms using CRISPR/Cas.
FIGURE 4.
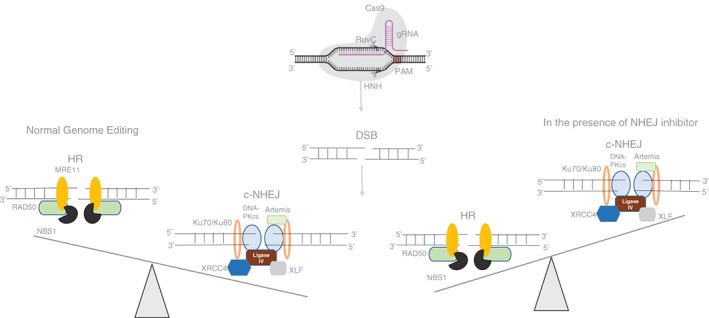
SCR7 tilts the balance toward HR during genome editing. Incubation of SCR7 and CRISPR/Cas9 mixture with mammalian cells leads to inhibition of nonhomologous end‐joining and thus, the DSB repair occurs primarily only through HR. In the absence of c‐NHEJ, the cell is dependent on HDR for mending the broken ends that result in an error‐free repair of the target site
6. CONCLUSION
It has become evident that inhibiting DNA DSB repair pathways has a broad range of applications. Among that, NHEJ inhibitor SCR7 and its novel derivative SCR130 are the most potent to date as they block NHEJ efficiently both in vitro and ex vivo. Not only as a monotherapy, in combination with radiation but SCR7 has also shown promising results. Both autocyclized and oxidized forms of SCR7 inhibit NHEJ efficiently, although the oxidized pyrazine version is less efficient inside the cells. The broad spectrum of SCR7 utility is evident in the form of cancer therapeutics to its use as a biochemical inhibitor. Besides, SCR7 is also an attractive tool for improving genome editing using CRISPR/Cas across model systems.
7. FUTURE DIRECTIONS
The use of DNA repair inhibitors, combined with DNA‐damaging drugs, has revealed its therapeutic importance both in laboratories and clinical trials. Olaparib is one of the successful examples of DNA repair, which is currently used in clinics. NHEJ inhibitor, SCR7 in combination with chemotherapeutic agents and radiation, has been tested in preclinical settings; however, more studies are required to assess its clinical relevance. Although SCR130 showed improved IC50 and NHEJ inhibition, itrelevance in vivo tumor regression is yet to be seen. More potent derivatives of SCR7 need to be explored in combination with chemotherapy to achieve efficient tumor regression at lower doses. Besides, to classify cancers that would respond to Ligase IV inhibitors based on the expression of Ligase IV is essential for its use as therapeutics. Patients with low levels of ligase are generally sensitive to radiation. Cataloging of patients based on polymorphisms and Ligase IV expression can be used as a standard in deciding the therapeutic regimen using NHEJ inhibitors. Also, it would be interesting to see how new SCR7 derivatives contribute toward biochemical inhibition of NHEJ and their impact on improving CRISPR/Cas mediating gene editing, the future of cancer therapeutics as well as other genetic diseases.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHORS CONTRIBUTION
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization, B.C., S.C.R.; Writing ‐ Original Draft, M.M.; Writing ‐ Review & Editing, B.C., S.C.R.; Supervision, B.C., S.C.R.
ETHICAL STATEMENT
Not applicable.
ACKNOWLEDGMENTS
We thank Ujjayinee Ray for critical reading and comments on the manuscript. This work was supported by grants from CEFIPRA (IFC/5203‐4/2015/131), DAE (January 21, 2016‐BRNS/35074) to Sathees C. Raghavan and DBT Glue‐Grant (BT/PR23078/MED/29/1253/2017 to Bibha Choudhary. Meghana Manjunath is supported by a Senior Research Fellowship (SRF) from DST‐INSPIRE, India.
Manjunath M, Choudhary B, Raghavan SC. SCR7, a potent cancer therapeutic agent and a biochemical inhibitor of nonhomologous DNA end‐joining. Cancer Reports. 2021;4:e1341. 10.1002/cnr2.1341
Contributor Information
Bibha Choudhary, Email: vibha@ibab.ac.in.
Sathees C. Raghavan, Email: sathees@iisc.ac.in.
DATA AVAILABILITY STATEMENT
Data sharing does not apply to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Lieberman HB. DNA damage repair and response proteins as targets for cancer therapy. Curr Med Chem. 2008;15(4):360‐367. [DOI] [PubMed] [Google Scholar]
- 2. Srivastava M, Raghavan SC. DNA double‐strand break repair inhibitors as cancer therapeutics. Chem Biol. 2015;22(1):17‐29. [DOI] [PubMed] [Google Scholar]
- 3. Pucci S, Mazzarelli P, Rabitti C, et al. Tumor specific modulation of KU70/80 DNA binding activity in breast and bladder human tumor biopsies. Oncogene. 2001;20(6):739‐747. [DOI] [PubMed] [Google Scholar]
- 4. Shintani S, Mihara M, Li C, et al. Up‐regulation of DNA‐dependent protein kinase correlates with radiation resistance in Oral squamous cell carcinoma. Cancer Sci. 2003;94(10):894‐900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trenner A, Sartori AA. Harnessing DNA double‐strand break repair for cancer treatment. Front Oncol. 2019;9:1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vartak SV, Raghavan SC. Inhibition of nonhomologous end joining to increase the specificity of CRISPR/Cas9 genome editing. FEBS J. 2015;282(22):4289‐4294. [DOI] [PubMed] [Google Scholar]
- 7. Kanaar R, Wyman C. DNA repair by the MRN complex: break it to make it. Cell. 2008;135(1):14‐16. [DOI] [PubMed] [Google Scholar]
- 8. Pandey M, Raghavan SC. DNA double‐strand break repair in mammals. J Radiat Cancer Res. 2017;8(2):93. [Google Scholar]
- 9. Rooney S, Chaudhuri J, Alt FW. The role of the non‐homologous end‐joining pathway in lymphocyte development. Immunol Rev. 2004;200(1):115‐131. [DOI] [PubMed] [Google Scholar]
- 10. Burma S, Chen BPC, Chen DJ. Role of non‐homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair. 2006;5(9–10):1042‐1048. [DOI] [PubMed] [Google Scholar]
- 11. Lieber MR. The mechanism of double‐strand DNA break repair by the nonhomologous DNA end‐joining pathway. Annu Rev Biochem. 2010;79:181‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ray U, Raghavan SC. Modulation of DNA double‐strand break repair as a strategy to improve precise genome editing. Oncogene. 2020;39:1‐13. [DOI] [PubMed] [Google Scholar]
- 13. Bunting SF, Nussenzweig A. End‐joining, translocations and cancer. Nat Rev Cancer. 2013;13(7):443‐454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deriano L, Roth DB. Modernizing the nonhomologous end‐joining repertoire: alternative and classical NHEJ share the stage. Annu Rev Genet. 2013;47:433‐455. [DOI] [PubMed] [Google Scholar]
- 15. Zhao B, Watanabe G, Lieber MR. Polymerase μ in non‐homologous DNA end joining: importance of the order of arrival at a double‐strand break in a purified system. Nucleic Acids Res. 2020;48(7):3605‐3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ochi T, Blackford AN, Coates J, et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double‐strand break repair. Science. 2015;347(6218):185‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Davis AJ, Chen DJ. DNA double strand break repair via non‐homologous end‐joining. Transl Cancer Res. 2013;2(3):130‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC. Homology and enzymatic requirements of microhomology‐dependent alternative end joining. Cell Death Dis. 2015;6(3):e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ray U, Raghavan SC. Inhibitors of DNA double‐strand break repair at the crossroads of cancer therapy and genome editing. Biochem Pharmacol. 2020;182:114195. [DOI] [PubMed] [Google Scholar]
- 20. Srivastava M, Nambiar M, Sharma S, et al. An inhibitor of nonhomologous end‐joining abrogates double‐strand break repair and impedes cancer progression. Cell. 2012;151(7):1474‐1487. [DOI] [PubMed] [Google Scholar]
- 21. McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37‐55. [DOI] [PubMed] [Google Scholar]
- 22. Gavande NS, VanderVere‐Carozza PS, Hinshaw HD, et al. DNA repair targeted therapy: the past or future of cancer treatment? Pharmacol Ther. 2016;160:65‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jekimovs C, Bolderson E, Suraweera A, Adams M, O'Byrne KJ, Richard DJ. Chemotherapeutic compounds targeting the DNA double‐Strand break repair pathways: the good, the bad, and the promising. Front Oncol. 2014;4:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly (ADP‐ribose) polymerase. Nature. 2005;434(7035):913‐917. [DOI] [PubMed] [Google Scholar]
- 25. Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917‐921. [DOI] [PubMed] [Google Scholar]
- 26. Kennedy RD, Chen CC, Stuckert P, et al. Fanconi anemia pathway–deficient tumor cells are hypersensitive to inhibition of ataxia telangiectasia mutated. J Clin Invest. 2007;117(5):1440‐1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ceccaldi R, Rondinelli B, D'Andrea AD. Repair pathway choices and consequences at the double‐strand break. Trends Cell Biol. 2016;26(1):52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X, Zhong S, Zhu X, et al. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008;68(9):3169‐3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gkotzamanidou M, Terpos E, Bamia C, Munshi NC, Dimopoulos MA, Souliotis VL. DNA repair of myeloma plasma cells correlates with clinical outcome: the effect of the nonhomologous end‐joining inhibitor SCR7. Blood. 2016;128(9):1214‐1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kumar A, Shetake NG, Ali M, et al. Recent advances in radiation research for human health and environment. J Radiat Cancer Res. 2017;8(4):186. [Google Scholar]
- 31. Gopalakrishnan V, Radha G, Raghavan SC, Choudhary B. Inhibitor of nonhomologous end joining can inhibit proliferation of diffuse large B‐cell lymphoma cells and potentiate the effect of ionization radiation. J Radiat Cancer Res. 2018;9(2):93. [Google Scholar]
- 32. Causse SZ, Marcion G, Chanteloup G, et al. HSP110 translocates to the nucleus upon Genotoxic chemotherapy and promotes DNA repair in colorectal cancer cells. Oncogene. 2019;38(15):2767‐2777. [DOI] [PubMed] [Google Scholar]
- 33. Vartak SV, Swarup HA, Gopalakrishnan V, et al. Autocyclized and oxidized forms of SCR 7 induce cancer cell death by inhibiting nonhomologous DNA end joining in a ligase IV dependent manner. FEBS J. 2018;285(21):3959‐3976. [DOI] [PubMed] [Google Scholar]
- 34. John F, George J, Srivastava M, et al. Pluronic copolymer encapsulated SCR7 as a potential anticancer agent. Faraday Discuss. 2015;177:155‐161. [DOI] [PubMed] [Google Scholar]
- 35. John F, George J, Vartak SV, et al. Enhanced efficacy of pluronic copolymer micelle encapsulated SCR7 against cancer cell proliferation. Macromol Biosci. 2015;15(4):521‐534. [DOI] [PubMed] [Google Scholar]
- 36. Pandey M, Gopalakrishnan V, Swarup HA, et al. Water‐soluble version of SCR7‐pyrazine inhibits DNA repair and abrogates tumor cell proliferation. J Radiat Cancer Res. 2019;10(1):27. [Google Scholar]
- 37. Ray U, Jose AE, Suresh R, et al. Water‐soluble SCR7 can abrogate DNA end joining and induce cancer cell death. Clin Oncol Res. 2020;2020(7):1‐7. 10.31487/j.COR.2020.07.09. [DOI] [Google Scholar]
- 38. Ray U, Vartak SV, Raghavan SC. NHEJ inhibitor SCR7 and its different forms: promising CRISPR tools for genome engineering. Gene. 2020;763:144997. 10.1016/j.gene.2020.144997. [DOI] [PubMed] [Google Scholar]
- 39. Ray U, Raul SK, Gopinatha VK, et al. Identification and characterization of novel SCR7‐based small‐molecule inhibitor of DNA end‐joining, SCR130 and its relevance in cancer therapeutics. Mol Carcinog. 2020;59(6):618‐628. [DOI] [PubMed] [Google Scholar]
- 40. Reid DA, Conlin MP, Yin Y, et al. Bridging of double‐stranded breaks by the nonhomologous end‐joining ligation complex is modulated by DNA end chemistry. Nucleic Acids Res. 2017;45(4):1872‐1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reid DA, Keegan S, Leo‐Macias A, et al. Organization and dynamics of the nonhomologous end‐joining machinery during DNA double‐strand break repair. Proc Natl Acad Sci. 2015;112(20):E2575–E2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tripathi V, Agarwal H, Priya S, et al. MRN complex‐dependent recruitment of ubiquitylated BLM helicase to DSBs negatively regulates DNA repair pathways. Nat Commun. 2018;9(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kulashreshtha M, Mehta IS, Kumar P, Rao BJ. Chromosome territory relocation during DNA repair requires nuclear myosin 1 recruitment to chromatin mediated by ϒ‐H2AX signaling. Nucleic Acids Res. 2016;44(17):8272‐8291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yang Z, Chen S, Xue S, et al. Injection of an SV40 transcriptional terminator causes embryonic lethality: a possible Zebrafish model for screening nonhomologous end‐joining inhibitors. Onco Targets Ther. 2018;11:4945‐4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR‐Cas9 for genome engineering. Cell. 2014;157(6):1262‐1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chu HW, Rios C, Huang C, et al. CRISPR–Cas9‐mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015;22(10):822‐829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Maruyama T, Dougan SK, Truttmann MC, Bilate AM, Ingram JR, Ploegh HL. Increasing the efficiency of precise genome editing with CRISPR‐Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol. 2015;33(5):538‐542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Singh P, Schimenti JC, Bolcun‐Filas E. A mouse geneticist's practical guide to CRISPR applications. Genetics. 2015;199(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu Z, Shi Z, Guo X, et al. Ligase IV inhibitor SCR7 enhances gene editing directed by CRISPR–Cas9 and SsODN in human cancer cells. Cell Biosci. 2018;8(1):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lin S, Staahl BT, Alla RK, Doudna JA. Enhanced homology‐directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. elife. 2014;3:e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Robert F, Barbeau M, Éthier S, Dostie J, Pelletier J. Pharmacological inhibition of DNA‐PK stimulates Cas9‐mediated genome editing. Genome Med. 2015;7(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Shao Y, Guan Y, Wang L, et al. CRISPR/Cas‐mediated genome editing in the rat via direct injection of one‐cell embryos. Nat Protoc. 2014;9(10):2493‐2512. [DOI] [PubMed] [Google Scholar]
- 53. Killian T, Dickopf S, Haas AK, Kirstenpfad C, Mayer K, Brinkmann U. Disruption of diphthamide synthesis genes and resulting toxin resistance as a robust technology for quantifying and optimizing CRISPR/Cas9‐mediated gene editing. Sci Rep. 2017;7(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aslan Y, Tadjuidje E, Zorn AM, Cha S‐W. High‐efficiency non‐mosaic CRISPR‐mediated knock‐in and indel mutation in F0 Xenopus. Development. 2017;144(15):2852‐2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing does not apply to this article as no new data were created or analyzed in this study.