

Abstract
Objective
We determined the yield, genetic spectrum, and actual origin of de novo mutations (DNMs) for infantile spasms (ISs) in a Chinese cohort. The efficacy of levetiracetam (LEV) for STXBP1‐related ISs was explored also.
Methods
Targeted sequencing of 153 epilepsy‐related candidate genes was applied to 289 Chinese patients with undiagnosed ISs. Trio‐based amplicon deep sequencing was used for all DNMs to distinguish somatic/mosaic mutations from germline ones.
Results
Total of 26 DNMs were identified from 289 recruited Chinese patients with undiagnosed ISs. Among them, 24 DNMs were interpreted as pathogenic mutations based on American College of Medical Genetics and Genomics guidelines, contributing to 8.3% (24/289) of diagnosis yield in the Chinese IS cohort. CDKL5 and STXBP1 are the top genes with recurrent DNMs, accounting for 3.1% (9/289) of yield. Further deep resequencing for the trio members showed that 22.7% (5/22) of DNMs are actually somatic in the proband or a parent. These somatic carriers presented milder seizure attacks than those with true germline DNMs. After treatment with LEV for half a year, three patients with DNM in STXBP1 showed improved clinical symptoms, including seizure‐free and normal electroencephalogram, except for a patient with a second DNM in DIAPH3.
Significance
Our study confirmed the contribution and genetic spectrum of DNMs in Chinese IS patients. Somatic mutation account for a quarter of DNMs in IS cases. Treatment with LEV improved the prognosis of STXBP1‐related ISs.
Keywords: de novo mutation, infantile spasms, levetiracetam, somatic or mosaicism mutation, STXBP1
The contribution and genetic spectrum of de novo mutations (DNMs) was confirmed in infantile spasms (ISs). Somatic mutation accounted for 22.7% of DNMs in IS patients. Treatment with levetiracetam improved the prognosis of STXBP1‐related ISs.
1. INTRODUCTION
Infantile spasms (ISs) are a common form of epileptic encephalopathy (EE) in infancy, with an incidence of 2–5/10,000 live births (Lux & Osborne, 2004). Hypsarrhythmia in the electroencephalogram (EEG) is the key diagnosable sign of ISs, which usually occur in clusters. For infants and young children with ISs, the neurodevelopmental delay is the primary concern of both neurologists and parents, as it has been shown that the majority of patients (85%) with ISs have developmental delay (Yuskaitis et al., 2018). However, long‐term follow‐up has also shown that improved neurodevelopmental outcomes can be achieved in up to 23.6% of IS patients with early diagnosis and intervention (Widjaja et al., 2014), indicating that early diagnosis improves the neurodevelopmental status of ISs.
As a subtype of EE, ISs have a high degree of clinical and genotypic heterogeneity. Clinically, ISs show highly dynamic evolution between different age groups. For example, Ohtahara syndrome evolves to West syndrome or to Lennox–Gastaut syndrome as the age of onset changes (McTague et al., 2015). Genetically, approximately 70 genes including 43 dominant and 22 recessive genes involving ion channels, synaptic regulation, and developmental functions, have been identified as definitive causative genes of human EE (Carvill et al., 2013; EuroEPINOMICS‐RES Consortium et al., 2014; Lindy et al., 2018; McTague et al., 2015). With rapid advances in high‐throughput genomic sequencing techniques, the diagnostic yield for monogenic mutations has increased 10%–50%, depending on the number of genes assayed, case inclusion, and age of onset (Allen et al., 2013; Carvill et al., 2013; Helbig et al., 2016; Lemke et al., 2012; Lindy et al., 2018; McTague et al., 2015; Trump et al., 2016). However, each gene is individually responsible for less than 1% of EE cases (Noebels, 2015; Ottman et al., 2010), implying complex genotypic heterogeneity of ISs. Besides the germline mutations, somatic mosaicism has recently emerged as an important cause of EE or ISs (Depienne et al., 2010; Jdila et al., 2017; Kato et al., 2015; Masliah‐Plachon et al., 2010; Milh et al., 2015; Saitsu et al., 2010).
Another important advance in our understanding of the genetic basis of ISs is the identification of de novo mutations (DNMs). These include both copy number variants (CNVs) and single‐nucleotide variants (SNVs), which have been recognized as important causes of childhood EE or ISs (Allen et al., 2013; Claes et al., 2001; Epi4K Consortium, 2016; EuroEPINOMICS‐RES Consortium et al., 2014; Hamdan et al., 2017; Saitsu et al., 2008), also explaining the sporadic occurrence of ISs. Chromosomal microarray analyses performed in patients with ISs have shown that up to 6.8% of IS cases carry de novo pathogenic or likely pathogenic CNVs (Mefford et al., 2011). In 2013, the Epi4K Consortium performed the first trio‐based whole‐exome sequencing (WES) study for 264 families affected by ISs or Lennox–Gastaut syndrome. Their data showed that 7% of analyzed patients had a definitive causal DNM (Allen et al., 2013). Their further study on 356 trio‐based WES cases demonstrated a 12% yield (EuroEPINOMICS‐RES Consortium et al., 2014). Another cohort study from 8565 Western patients with both epilepsy and neurodevelopmental disorders tested via 70‐gene targeted sequencing showed that 72% of 417 deleterious variants were DNMs in dominant genes (Lindy et al., 2018).
The monogenic diagnostic yield of Chinese patients with EE has been reported (Miao et al., 2018; Yang et al., 2018; Zhang et al., 2016). The yields of causative mutations varied between 18% and 42% depending on the age of onset, sample, and panel size. However, DNM was reported only in very small cohorts with single gene assayed (Xie et al., 2019), or sporadically for individual case presentations (Li et al., 2018). Therefore, the actual contribution of DNMs in Chinese IS patients are still not well clarified. Studying the contribution and genetic spectrum of DNMs to ISs can facilitate the genetic diagnostic strategy, leading to patients with a negative family history of EE being referred to genetic testing.
In this study, we applied the targeted sequencing to cover the coding regions of 153 epilepsy‐related genes, quickly screening the pathogenic DNMs across a Chinese IS cohort. In addition, we used trio‐based deep sequencing to confirm the origin of the DNMs, distinguishing somatic/mosaic mutations from germline ones. Finally, we explored the efficacy of levetiracetam (LEV) therapy for STXBP1‐related ISs.
2. MATERIALS AND METHODS
2.1. Patient recruitment
Children with ISs referred to Children's Medical Center of the Chinese People's Liberation Army General Hospital were recruited from June 2015 to December 2017. All patients fulfilled the diagnostic criteria of the West Delphi Group (Lux & Osborne, 2004). The clinical manifestations, EEG, and brain MRI of each recruited patient were collected. All participants completed the routine genetic screening procedure (including karyotyping, TSC1/TSC2 sequencing and MLPA, metabolic disease screening). Only patient with negative results were recruited.
2.2. Target panel sequencing and variant calling
Genomic DNA was extracted from peripheral blood leukocytes of all probands and their parents using a DNA extraction kit (TIANGEN, Beijing, China). The target genes included the published gene list of the Epi4K Consortium (EuroEPINOMICS‐RES Consortium et al., 2014) and the curated genes in six databases (OMIM, GeneCard, MelaCard, Orphanet, HPO, and HGMD) with the following query term: “epilepsy/seizure/IS”. A total of 153 genes including 45 AR genes, 64 AD genes, 18 X‐linked genes, and 26 candidate risk genes were chosen for sequencing (see Table S1). Target libraries were enriched by the GenCap custom enrichment kit (MyGenostics, Beijing, China) and were sequenced on an Illumina HiSeq2000 (Illumina, San Diego, USA) for paired‐end reads of 150 bp. Quality controls were performed for raw data and the clean reads were mapped to the UCSC hg19 human reference genome using BWA. The SNP and indel variants were detected by GATK Haplotype Caller. Following this, SNVs with minor allele frequency (MAF) >0.1% or >1% (for dominant and recessive genes, respectively) reported in our in‐house database, the dbSNP database, 1000 Genomes dataset, or gnomAD database were not recognized as rare SNVs. The functional effects of missense mutations were predicted by four algorithms (PolyPhen, Sorting Intolerant from Tolerant, Protein Analysis Through Evolutionary Relationships, and Pathogenic Mutation Prediction). Three disease‐related databases (ClinVar, HGMD, COSMIC) were used to assess whether it is novel or reported pathogenic SNV.
The CNVkit tool of NextGENe software (version 2.4.1.2; SoftGenetics, PA. USA) was used to analyze and visualize the candidate CNVs. Only CNVs spanning more than five amplicons were chosen for validation using quantitative PCR (7500 Fast Real‐Time PCR System, Thermo Fisher Scientific, see Table S2 for designed primers) or Agilent 4 × 180 K array‐CGH chip (Agilent Technologies, CA, USA) (Figure S1).
2.3. Mutation validation and family segregation analysis
All candidate SNVs detected from each proband were re‐sequenced in their available core family members on an ABI PRISM 3730 genetic analyzer (Thermo Fisher Scientific, MA, USA) after touchdown PCR. In order to avoid PCR dropout, we used two sets of primer pairs to validate each DNM. The segregation status of candidate variants was then used as the inheritance evidence during the process of interpreting the pathogenicity. We strictly followed the guidelines of the American College of Medical Genetics and Genomics (ACMG) to interpret the pathogenicity of SNVs (Richards et al., 2015) and CNVs (Riggs et al., 2019).
2.4. Amplicon‐based deep sequencing (ADS) for DNM
Amplicon‐based deep sequencing (ADS) was used to detect somatic mutations, as described previously (Jiang et al., 2017). Genomic DNA from all 21 trios with 22 DNMs was amplified to generate the DNM‐specific amplicon (Table S3). Only the primer pair which have been validated without amplification bias in Sanger sequencing was used. Using Ion‐Plus Fragment Library Kit (Thermo Fisher Scientific), a sequencing library was prepared, purified, quantified, and enriched in Ion Sphere Particles (ISPs). Finally, enriched ISPs were loaded onto the 316 V2 chip and sequenced in an Ion Torrent Personal Genome Machine (Thermo Fisher Scientific, MA USA). Sequencing data from PGM were analyzed through the Integrative Genomics Viewer Version 2.3.25. The variant ratio was defined using the proportion of variant reads relative to the total sequencing reads.
Using the unpublished data of alleles with inherited germline heterozygous variants, we established the normal distribution curve of the variant ratio for heterozygous germline SNVs at appropriate sequencing coverage. With the mean and standard deviation (SD) of these normally distributed heterozygous germline SNVs, any DNM whose variant ratio lower than 2SD of Mean was automatically recognized as somatic mutation.
2.5. Protein–protein interaction network analyses
We uploaded the gene list with de novo SNVs combined with the genes covered in the de novo CNVs to the online Ingenuity Pathway Analysis platform (IPA; Qiagen, CA, USA) to identify potential networks, related disorders, and canonical pathways.
2.6. Treatment evaluation of LEV
Four patients with STXBP1 DNMs were included to evaluate the efficacy of LEV. Their treatment history, category of routine anti‐epilepsy drugs (AEDs), the duration and dosage of LEV, the frequency of seizure attack, and EEG during treatment were recorded. We definite “effective to levetiracetam” when patient shows decreased seizure attack (≥50%). LEV was prescribed from 10 to 20 mg/kg/day (initial dosage) and increased gradually to 50 mg/kg/day (maximum dosage). The time of follow‐up was from 7.3 to 19.77 months.
2.7. Statistical analysis
Chi‐squared/Fisher's exact test or Kruskal–Wallis test was used to compare the qualitative variables between two groups, and independent t‐test was used to compare the quantitative variables between two groups. p< 0.05 was considered statistically significant.
3. RESULTS
3.1. General information on the Chinese IS cohort
A total of 289 undiagnosed patients were recruited for this study based on primary routine examinations (Table 1). The ratio of males to females was 1.39:1. The median age of enrolled children was 8.7 months (range: 0.07–36.8 months). The median seizure frequency was 35 times/day (0–905 times/day) and the first attack occurred at a median age of 4 months (range: 0.16–15 months). In addition, 18.7% of patients developed other seizure types in the progression of their disease, including focal seizures, tonic seizures, generalized tonic–clonic seizures, atonic seizures, and/or myoclonic seizures. Brain magnetic resonance imaging (MRI) was normal in 220 patients (76.1%). And 49 patients (17.0%) revealed nonspecific changes including nonfocal cortical and subcortical atrophy, delayed myelination, thinning of the corpus callosum, and/or T2 hyperintensity of basal ganglia.
TABLE 1.
General information on the Chinese IS cohort
| General information | Number (%) |
|---|---|
| Sex | |
| Male | 168 (58.1%) |
| Female | 121 (41.9%) |
| Age at admission (months) | |
| Median | 8.7 |
| Range | 0.07–36.8 |
| Age at first attack (months) | |
| Median | 4 |
| Range | 0.16–15 |
| ≥ two seizure type (%) | 54 (18.7%) |
| Seizure frequency (times/day) | |
| Median | 35 |
| Range | 0–905 |
| MRI | |
| Normal | 220 (76.1%) |
| Other changes | 49 (17.0%) |
| NA | 20 (6.9%) |
| Number of Antiepileptic drugs (AED) | |
| 0 or NA | 1 (0.3%) |
| 1 | 87 (30.1%) |
| 2 | 84 (29.1%) |
| 3 | 75 (26.0%) |
| ≥4 | 42 (14.5%) |
| Median | 2 |
| Range | 0–8 |
| Effects on discharged AED | |
| Seizure free | 157 (54.3%) |
| Effective (≥50% attack) | 62 (21.5%) |
| Invalid (<50% attack) | 46 (15.9%) |
| NA | 24 (8.3%) |
Abbreviation: NA, not available.
3.2. The diagnostic yield and DNM spectrum of the Chinese IS cohort
The mean coverage of the targeted sequencing was 250× and the region with >20× constituted 96% of the total. Null variants (nonsense, frameshift, canonical splice sites, initiation codon, single exon, or multiexon deletion) and deleterious missense variants predicted by at least two software programs were preferentially selected for Sanger validation and family segregation analysis. With this strategy, a total of 889 candidate SNVs and 4 CNVs were chosen for Sanger sequencing, real‐time PCR, or aCGH. Paternity testing was performed for all probands with DNM using 12 STR alleles.
In this Chinese IS cohort, six patients carried homozygous or complex trans heterozygous mutations in recessive genes, and 28 patients carried 29 DNMs in epilepsy‐related genes, including 25 de novo SNVs (Patient 29 carried two DNMs: one in DIAPH3 and the other in STXBP1) and 4 de novo CNVs. Further PCR experiments with the second set primer validated one false de novo SNV due to PCR dropout, and biological parental testing validated a non‐parental relationship for another two de novo SNVs, leaving 22 SNVs and 4 CNVs in 25 patients. 24 DNMs except for DIAPH3 and CACNA1A were evaluated as pathogenic or likely pathogenic in accordance with the ACMG guidelines (Richards et al., 2015; Riggs et al., 2019). In contrast, no autosomal recessive variants were evaluated as pathogenic or likely pathogenic. Thus, the yield of pathogenic DNM from this undiagnosed Chinese IS cohort was 8.3% (24/289), indicating that DNMs of dominant genes rather than biallelic mutations of recessive genes contribute to the ISs in this Chinese cohort. The clinical and genetic information of these IS patients with DNMs were presented in Tables 2, 3 and S4. The validation and parental inheritance of de novo CNVs were described in Figure S1.
TABLE 2.
Detailed clinical information and genetic characteristics of patients with de novo SNVs detected in the Chinese IS cohort
| ID/age gender | Onset age of seizure and type | Onset age of spasms | NDD comorbid a | EEG | Gene | Mutation (GRCh37/hg 19) | Zygosity | Reported or Novel | Classification |
|---|---|---|---|---|---|---|---|---|---|
| 30/9 m/M | 7 m | 7 m | DD, ASD | Hypsarrhythmia | ARX | chrX:25023007 G>A; c.1469C>T; p.Pro490Leu | Hemi. | Novel | Likely pathogenic (PS2+PM2+PP3) |
| IS | NM_139058 | ||||||||
| 44/6 m/M | 4 m | 4 m | DD, ASD | Multifocal | ARX | chrX:25025525 C>T; c.1151G>A; p.Arg384His | Hemi. | Reported, conflicting interpretations | Likely pathogenic (PS2+PM2+PP3) |
| IS | NM_139058 | ||||||||
| 24/6 m/F | 3 m | 3 m | ASD | Hypsarrhythmia | CACNA1A | chr19:13318443 G>T; c.7205C>A; p.Pro2402Gln | Het. | Novel | VOUS (PS2+PM2) |
| IS | NM_001127222 | ||||||||
| 2/8 m/F | 0.25 m | 7 m | DD, ASD | Multifocal | CDKL5 | chrX:18528955T>C; c.80T>C; p.Val27Ala | Het. | Novel | Likely pathogenic (PM1+PS2+PM2+PP3) |
| Focal | NM_003159 | ||||||||
| 50/24 m/M | 14 m | 14 m | DD, ASD | Multifocal | CDKL5 | chrX:18528976 C>A; c.99+2C>A; splicing site | Hemi. | Novel | Pathogenic (PVS1+PS2+PM2) |
| IS | NM_003159 | ||||||||
| 36/8 m/F | 8 m | 8 m | DD, ASD | Hypsarrhythmia | CDKL5 | chrX:18598089 G>T; c.403+1G>T; splicing site | Het. | Reported as pathogenic, different nucleotide in same location | Pathogenic (PVS1+PS2+PM5+PM2) |
| IS | NM_003159 | ||||||||
| 39/5 m/M | 2.5 m | 4 m | DD, ASD | Multifocal | CDKL5 | chrX:18622719 C>T; c.1675C>T; p.Arg559X | Hemi. | Reported as pathogenic | Pathogenic (PVS1+PS2+PS4_moderate+PM2) |
| Tonic | NM_003159 | ||||||||
| 34/17 m/F | 0.7 m | 4 m | DD, ASD | Hypsarrhythmia | CDKL5 | chrX:18622774‐18622776, del GG; c.1731_1732del GG; p.Met577fs | Het. | Novel | Pathogenic (PVS1+PS2+PM2) |
| Focal | NM_003159 | ||||||||
| 29/12 m/F | 0.16 m | 5 m | ASD | Hypsarrhythmia | DIAPH3 | chr13:60565358 C>T; c.1295G>A; p.Arg432Lys | Het. | Novel | VUS (PM2+PS2_moderate) |
| Focal | NM_001042517 | ||||||||
| 47/13 m/F | 4 m | 4 m | DD, ASD | Multifocal | GRIN2B | chr12:13761698 A>T; c.1849T>A; p. Ser617Thr | Het. | Novel | Likely pathogenic (PS2+PM2+PP3) |
| IS and Tonic | NM_000834 | ||||||||
| 46/15 m/M | 4 m | 4 m | DD, ASD | Hypsarrhythmia | KCNB1 | chr20:47990350 G>A;.2 c.1747C>T; p. Arg583* | Het. | Reported as Pathogenic | Pathogenic (PVS1+ PS2+PS4_supporting+PM2) |
| IS | NM_004975 | ||||||||
| 32/17 m/F | 14 m | 14 m | DD, ASD | Hypsarrhythmia | KCNB1 | chr20:47991468 G>A; c.629C>T; p.Thr210Met | Het. | Reported as Pathogenic | Likely pathogenic (PS2+PS4_supporting+PM2+PP3) |
| IS | NM_004975 | ||||||||
| 33/13 m/F | 6 m | 6 m | DD, ASD | Hypsarrhythmia | KCNQ2 | chr20:62076109 C>T; c.593G>A; p.Arg198Gln | Het. | Reported, conflicting interpretations | Likely pathogenic (PS2+PM2+PS4_moderate+PP3) |
| IS | NM_004518 | ||||||||
| 42/23 m/F | 6 m | 13 m | DD, ASD | Multifocal | MEF2C | chr5:88119562 C>G; c.44G>C; p.Arg15Pro | Het. | Reported as pathogenic in same amino acid | Likely Pathogenic (PM5+PS2+PM2+PP3) |
| Tonic | NM_001131005 | ||||||||
| 37/2 m/F | 1 m | 1 m | DD | Hypsarrhythmia | SCN2A | chr2:166245202 G>A; c.4886G>A; p.Arg1629His | Het. | Reported, conflicting interpretations | Likely pathogenic (PS2+PM2+PP3) |
| IS | NM_001040142 | ||||||||
| 45/13 m/M | 11 m | 11 m | NA | Multifocal | SCN2A | chr2:166245511‐166245512 delC; c.5196delC; p.Pro1733Lfs*36 | Het. | Novel | Pathogenic (PVS1+PS2+PM2) |
| IS | NM_001040142 | ||||||||
| 38/6 m/F | 6.3 m | 6.3 m | DD, ASD | Hypsarrhythmia | SCN8A | chr12:52082568 G>A; c.641G>A; p.Gly214D | Het. | Reported as pathogenic | Likely pathogenic (PS2+PS4_supporting+PM2+PP3) |
| IS | NM_014191 | ||||||||
| 51/4 m/M | 2.5 m | 2.5 m | DD | Hypsarrhythmia | STXBP1 | chr9:130420701 G>C; c.217G>C; p.Ala73Pro | Het. | Novel | Likely pathogenic (PS2+PM2+PP3) |
| IS | NM_003165 | ||||||||
| 31/3 m/F | 2.5 m | 2.5 m | DD | Hypsarrhythmia | STXBP1 | chr9:130438177‐130438178 insG; c.1205_1206insG; p.Tyr402_D403delinsX | Het. | Novel | Pathogenic (PVS1+PS2+PM2) |
| Focal | NM_003165 | ||||||||
| 35/3 m/F | 1.7 m | 1.7 m | DD, ASD | Hypsarrhythmia | STXBP1 | chr9:130438970‐130438970 delC; c.1297delC; p.Pro433fs | Het. | Novel | Pathogenic (PVS1+PS2+PM2) |
| IS | NM_003165 | ||||||||
| 29/12 m/F | 0.16 m | 5 m | ASD | Hypsarrhythmia | STXBP1 | chr9:130440783 G>A; c.1433G>A; p.Trp478X | Het. | Novel | Pathogenic (PVS1+PS2+PM2) |
| Focal | NM_003165 | ||||||||
| 49/3 m/F | 1 m | 1 m | DD, ASD | Multifocal | TCF4 | chr18:53018122 A>G; c.482T>C; p.Leu161Pro | Het. | Novel | Likely pathogenic (PS2+PM2+PP3) |
| IS | NM_001083962NM_001083962 |
Abbreviation: NA, not available.
Reported: have reported in the public disease‐related databases including HGMD/ClinVar/ClinGen database.
TABLE 3.
Detailed clinical information and genetic characteristics of patients with de novo CNVs detected in the Chinese IS cohort
| ID/age gender | Onset age of seizure and type | Onset age of spasms | NDD 4 | EEG | Cytogenic band (GRCh37/hg19) | Type | Number of involved gene | Reported or novel a | Classification |
|---|---|---|---|---|---|---|---|---|---|
| 103/15 m/M | 2 m | 3 m | DD | Multifocal | 1p36.33‐p36.32 (752721‐3645052) x1 | Deletion | 82 genes including GABRD | 1p36 deletion syndrome | Pathogenic |
| Focal | |||||||||
| 202/8.9 m/M | 4 m | 4 m | DD | NA | 7q21.11 (77645000‐79083000) x1 | Deletion | 8 genes including MAGI2 | Reported as Pathogenic in literature b | Likely pathogenic |
| IS | |||||||||
| 201/7 m/M | 3 m | 4 m | DD, ASD | Multifocal | 15q11.1‐q13.3 (20095481‐32880457) x4 | Triplication | 240 genes including UBE3A, MAGEL1, GABRB3, SNRPN | PWS/AS region duplication syndrome | Pathogenic |
| Focal | |||||||||
| 72/10 m/F | 2 m | 2 m | DD, ASD | Multifocal | 16p13.11 (14910205‐16561151) x3 | Duplication | 32 genes including MYH11 | 16p13.11 recurrent duplication | Likely pathogenic |
| IS |
Abbreviation: NA, not available.
Reported: have reported in the public disease‐related databases including HGMD/ClinVar/ClinGen database.
Marshall CR, Young EJ, Pani AM, Freckmann ML, et al. Infantile spasms is associated with deletion of the MAGI2 gene on chromosome 7q11.23‐q21.11. Am J Hum Genet. 2008 Jul;83(1):106–11.
The DNMs spectrum comprised nine null SNVs, 13 deleterious missense SNVs on 12 genes. The genes with recurrent DNMs included CDKL5 (n = 5), STXBP1 (n = 4), SCN2A (n = 2), ARX (n = 2), and KCNB1 (n = 2). DNMs of CDKL5 and STXBP1 account for 3.1% of the IS cohort. After reviewing the ClinVar and HGMD professional database, we confirmed nine recurrent pathogenic SNVs at specific nuclide acid sites as well as two recurrent CNVs which have previously been reported as pathogenic CNVs in Western and Chinese early‐onset EE cohorts (Li et al., 2019; Michaud et al., 2014). In total, recurrent DNMs accounted for 46% (11/24) of the causative variants in the Chinese IS cohort. Among them, 1p36.33 deletion and 15q11.2–13.1 triplication are known syndromic genomic disorders. For 16p13.11 duplication in patient 72, although little evidence of triplosensitivity (TS=1) was reported on the ClinGen website, one recent large case‐control study has proved 16p13.1 duplication is significantly associated with series of neurodevelopmental disorders with incomplete penetrance and variable expressivity (Allach El Khattabi et al., 2018).
Recruited patients were divided into two groups based on whether DNM was identified. Neither the sex (10/25 vs. 158/264, p=0.055) nor onset age (4.42 vs. 4.89, p=0.564) was significantly different in two groups. Although patients with DNM showed other seizure more often (36.0% vs. 17.0%, p=0.03), and showed more seizure frequency (128 vs. 69, p=0.192) than patient without DNM, the difference did not reach significance. The efficacy of routine AEDs in two groups was not significantly different also (48.0% vs. 60.4%, p=0.572).
The Gene Ontology functional analysis showed DNMs significant enriches in neurotransmission or the morphogenesis of neurons (Table 4). There are 13 de novo SNVs involve in this pathway, and seven de novo SNVs involved in ion channel. A close but direct connection was seen between genes affected by DNMs (Figure 1). Moreover, we found that 21/26 DNMs occurred in strongly conserved genes that are intolerant of loss‐of‐function (LOF) mutations (Table 4, pLI > 0.9), implying that, for these genes with DNMs, possibly it is haploinsufficiency via LOF rather than gain of function (GOF) contributes to ISs. In addition, we compared the enriched pathway of DNM genes with that of remained genes without DNM, no differential enriched pathway was seen in DNM genes.
TABLE 4.
Pathways involved and the intolerant score of DNMs detected in the Chinese IS cohort
| Diseases or functions annotation | p value | Gene name | Sample count in this IS cohort |
|---|---|---|---|
| Neurotransmission | 9.73E‐07 | CACNA1A, GABRD, GRIN2B, KCNB1, KCNQ2, SCN2A, SCN8A, STXBP1 | 13 |
| Development of neurons | 2.05E‐04 | ARX, CACNA1A, CDKL5, GABRB3, MAGI2, MEF2C, NDN, UBE3A | 13 |
| Presynaptic compartments | – | STXBP1 | 3 |
| Post‐synaptic compartments | – | GRIN2B, GABRD, GABRB3 | 2 |
| Ion channel | – | CACNA1A, KCNB1, SCN2A, SCN8A, KCNQ2 | 7 |
| Intolerant score in ExAC database | Gene name | Sample count in this IS cohort | |
|---|---|---|---|
| pLI > 0.98 | GRIN2B, STXBP1, KCNB1, SCN2A, SCN8A, CACNA1A, KCNQ2, UBE3A, TCF4, CDKL5, GABRB3, MYH11 | 20 | |
| 0.98 ≥ pLI > 0.9 | GABRD | 1 | |
| 0.9 > pLI > 0.75 | ARX. MAGI2, SNRPN | 4 |
The default Fisher's exact test was used to calculate a p value that determined the probability of the genes being involved in each pathway.
FIGURE 1.
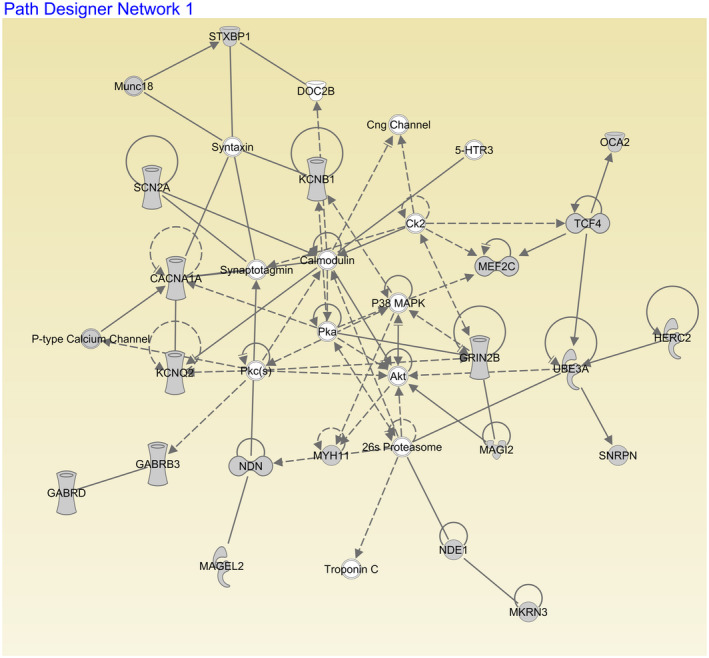
Protein–protein interaction network of DNM genes identified from the Chinese IS cohort. Genes are represented as nodes and only solid nodes indicate DNM genes in our cohort. Edges indicate known interactions between proteins (solid lines for direct interactions, dashed lines for mean indirect interactions). The gene shapes are indicative of molecular class, as defined by IPA: enzyme, rhombus; ion channel, vertical rectangle; kinase, inverted triangle; ligand‐dependent nuclear receptor, horizontal rectangle; phosphatase, triangle; transcription regulator, horizontal oval; transmembrane receptor, vertical oval; transporter, trapezoid; and unknown, circle
The main clinical features of CDKL5‐related encephalopathy include early‐onset seizures (usually occurring within the first three months) and severe neurodevelopmental problems (Fehr, Wong, et al., 2016). Male carriers have much more severe neurodevelopmental impairment than female carriers (Fehr, Downs, et al., 2016). In addition, the clinical outcome was found to be directly associated with the location of the CDKL5 mutation. Mutations in the N‐terminal kinase domain (aa 13 to aa 297) were associated with more severe clinical symptoms than mutations in the C‐terminal region (Fehr, Downs, et al., 2016; Fehr, Wong, et al., 2016). As CDKL5 is the gene most commonly showing DNMs (n = 5) in our Chinese IS cohort, we compared the relation between domain of DNM location and clinical phenotypes. The ratio of males to females was 2:3. The median age at spasms onset was 7 months (range: 4–14 months) and the median frequency of seizures was 55 times/day (range: 6–905 times/day). All patients had developmental delay and ASD. Three patients (Patients 2, 36, and 50) were found to carry mutation in the N‐terminal kinase domain. Surprisingly, one male patient (Patient 50, c.99+2C>A) did not have a more severe clinical phenotype despite carrying a germline DNM in the N‐terminal kinase domain, and he did not present IS attach until 14 months of age and became seizure‐free with one anti‐epileptic drug (AED). We assumed that some appropriately spliced transcript was possibly produced from this mutant allele. In addition, another male patient (Patient 39, c.1675C>T) also exhibited a moderate clinical phenotype (we will discuss this case later when considering the somatic mutation status).
3.3. Somatic and germline mosaicism status of four de novo SNVs
A previous deep sequencing study of DNMs proved that a 39% mutant ratio is a reasonable cut‐off to deviate somatic mutation from germline heterozygous mutations (Acuna‐Hidalgo et al., 2015). With ADS for our germline heterozygous mutations (range: 405–600×, mean: 497.7×), the variant cut‐off of somatic mutations in our ASD platform was 44%. To minimize the false identification of somatic mutations, we chose 40% as the cut‐off to definite the somatic mutations. The mutation with allelic ratio higher than 1% in tested parents was considered as germline mosaicism.
The mean resequencing coverage of 22 de novo SNVs was 4633× (329–18,484×, Table S5). We found that four presumed germline DNMs were actually mosaic mutations in the blood of the offspring (Table 5), including two STXBP1 null DNMs (Patient 29 and Patient 35), one CDKL5 null DNM (Patient 39), and one KCNQ2 missense DNM (Patient 33). We also found that Patient 44 with a germline missense DNM in ARX inherited this as a consequence of low‐level (only 2%) mosaicism present in his unaffected mother (Table 5). In total, the somatic or germline mosaic mutation explained 22.7% (5/22) of de novo SNVs in our Chinese IS patients with the remainder having true germline DNMs. We also compared the phenotype between germline and somatic mutations in our cohorts. Patient 39 (CDKL5, 65% mutation) presented with a lower seizure rate compared with patient 50 with germline DNM (20 vs. 55 times/day).
TABLE 5.
Somatic or germline mosaicism of DNM SNVs identified in the Chinese IS cohort
| Sample ID | Tested sample | Gene | Mutation (GRCh37/hg 19) | Total coverage a,b | Read of alternate allele (%) a,b |
|---|---|---|---|---|---|
| 29 | Blood | STXBP1 (NM_003165) | chr9:130440783 G>A; c.1433G>A; p.Trp478X | 8173 | 3307 (40%) |
| 29F | Blood | STXBP1 (NM_003165) | chr9:130440783 G>A; c.1433G>A; p.Trp478X | 2880 | 1 (0%) |
| 29M | Blood | STXBP1 (NM_003165) | chr9:130440783 G>A; c.1433G>A; p.Trp478X | 10,021 | 2 (0%) |
| 33 | Blood | KCNQ2 (NM_004518) | chr20:62076109 C>T; c.593G>A; p.Arg198Gln | 15,892 | 5943 (37%) |
| 33F | Blood | KCNQ2 (NM_004518) | chr20:62076109 C>T; c.593G>A; p.Arg198Gln | 17,410 | 7 (0%) |
| 33M | Blood | KCNQ2 (NM_004518) | chr20:62076109 C>T; c.593G>A; p.Arg198Gln | 18,189 | 7 (0%) |
| 35 | Blood | STXBP1 (NM_003165) | chr9:130438970‐130438970 delC; c.1297delC; p.P433fs | 4055 | 1599 (39%) |
| 35F | Blood | STXBP1 (NM_003165) | chr9:130438970‐130438970 delC; c.1297delC; p.P433fs | 1724 | 9 (0.5%) c |
| 35M | Blood | STXBP1 (NM_003165) | chr9:130438970‐130438970 delC; c.1297delC; p.P433fs | 5557 | 39 (0.7%) c |
| 39 | Blood | CDKL5 (NM_003159) | chrX:18622719 C>T; c.1675C>T; p.Arg559X | 1020 | 661 (65%) |
| 39F | Blood | CDKL5 (NM_003159) | chrX:18622719 C>T; c.1675C>T; p.Arg559X | 2652 | 4 (0%) |
| 39M | Blood | CDKL5 (NM_003159) | chrX:18622719 C>T; c.1675C>T; p.Arg559X | 1689 | 1 (0%) |
| 44 | Blood | ARX (NM_139058) | chrX:25025525 C>T; c.1151G>A; p.Arg384His | 575 | 559 (97%) |
| 44F | Blood | ARX (NM_139058) | chrX:25025525 C>T; c.1151G>A; p.Arg384His | 841 | 1 (0%) |
| 44M | Blood | ARX (NM_139058) | chrX:25025525 C>T; c.1151G>A; p.Arg384His | 646 | 13 (2%) |
Abbreviations: F, father; M, mother.
Both total coverage and the alternate‐allele reads were calculated according to the amplicon‐based deep sequencing results.
It was supposed to be sequencing error as mutant allele frequency was lower than 1% and it was identified in both of the parents.
3.4. Efficiency of LEV treatment in patients with DNM in STXBP1
Three patients carry STXBP1 null mutations (Patient 29, 31 and 35) and one patient carry missense mutation (Patient 51), in which two patients (Patient 29 and 35) presented somatic mutations. The median onset age of spasms was 2.5 months (1.7–5 months) and the median number of epileptic spasms was 242.5 times/day (129–650 times/day). EEG consistently showed hypsarrhythmia. All patients had various degrees of developmental delay, although the brain MRI results were normal except for patient 29. These clinical phenotypes were similar to those in other Western or Chinese EE cohorts (Li et al., 2018; Stamberger et al., 2016). Patients with germline null mutation (Patient 31, c.1205‐1206insG) did not present a higher spasms rate (320 vs. 650 times/day) or earlier onset age (2.5 vs. 2.5 m) than one with germline missense mutation (Patient 51, c.217G>A). But patient with somatic mutation (Patient 35, c.1297delC, 39% mutation) did present a lower seizure rate than two patients with a germline mutation (Patient 31 and 51) (165 vs. 320 or 650 times/day). Patient 29 carried a second DNM in DIAPH3. Lesca Gaetan previously described partial deletion of DIAPH3 in a patient with EE of the Landau–Kleffner and continuous spikes and waves (Lesca et al., 2012). The epilepsy of Patient 29 was not under control with the routine AEDs including topiramate, valproate, low‐dose LEV and corticotropin plus MgSO4.
The remission effect of levetiracetam (LEV) to early‐onset EE has been reported sporadically in a patient with STXBP1 mutation (Li et al., 2018; Milh et al., 2011; Vatta et al., 2012) since the first case was reported by Dilena et al. (2015). Meanwhile, the invalid efficacy of LEV to epilepsy also has been reported in a patient carrying STXBP1 mutation (Deprez et al., 2010; Li et al., 2016; Romaniello et al., 2013). In our cohort, LEV was prescribed to four patients with DNMs in STXBP1. The AEDs for the four patients before LEV treatment was shown in Table S6. It showed when the dosage of Levetiracetam was increased to 50 mg/kg/day, three patients (Figure 2, Patients 31, 35, and 51) except for Patient 29 showed seizure‐free within 40 days (range: 3–40 days). Consist, they all presented normal EEG within six months (range: 0.2–6 months). Further EEG interview still showed normal. Patient 29, who carries second DNM in DIAPH3 beside STXBP1, presented intractable seizures and abnormal EEG even with the maximum dosage of LEV for three months. Our pilot retrospective analysis of LEV therapy proved that LEV is effective for Chinese patients with STXBP1 mutation.
FIGURE 2.
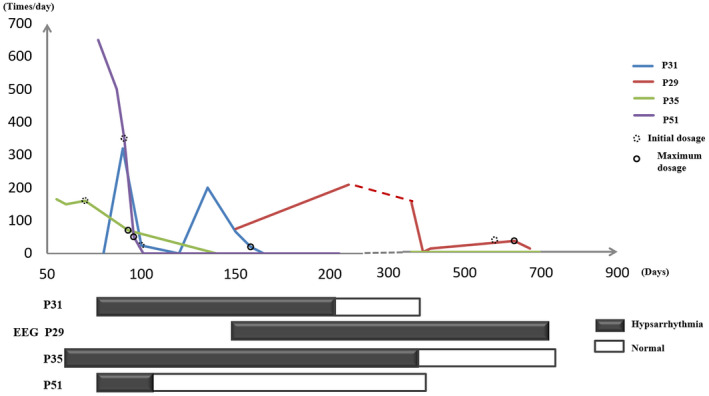
IS episode and treatment of Levetiracetam for four IS patients with STXBP1 DNM. Different lines were used to represent the developmental trajectory of IS attacks in four patients with STXBP1 DNM. The X‐axis represented the review point of EEG, treatment after birth, and the Y‐axis represented the number of daily episodes of IS attack. Levetiracetam was gradually increased from 10 to 20 mg/kg/day (initial dosage, dashed circle) to 50 mg/kg/day (maximal dosage, solid circle). The progresses of their electroencephalograms (EEG) were illuminated different rectangles during 1.5 years after birth (black for hypsarhythmia and white for normal EEG)
4. DISCUSSION
With the rapid development of sequencing technology, epilepsy panels or WES are now widely used as a first‐tier genetic diagnostic strategy for pediatric EE. The diagnostic yield varies from 10% to 48.5% depending on the number of target genes (Carvill et al., 2013; Helbig et al., 2016; Lemke et al., 2012; Lindy et al., 2018; McTague et al., 2015; Trump et al., 2016). The diagnostic yield varies according to the age of onset also. For example, the yield is much higher among those in whom onset occurs under 2 months of age compared with those with onset <2 years (39% vs. 14%) (Trump et al., 2016). The diagnostic yield increased to 43% or 52% when the onset was in the neonatal period (Helbig et al., 2016; Trump et al., 2016).
The diagnostic yield of Chinese early‐onset EE has previously been explored (Miao et al., 2018; Wang et al., 2019; Yang et al., 2018; Zhang et al., 2016). One recent publication on a Chinese pediatric epilepsy cohort showed that the genetic diagnostic rate was 26.7% (Yang et al., 2018). However, a similar study had not been carried out for an IS cohort. ISs occur in the first year after birth, so identifying the diagnostic yield and genetic spectrum of Chinese IS patients would improve diagnostic logistics. In this study, we revealed that the diagnostic yield of a Chinese undiagnosed IS cohort using 153‐gene target sequencing was only 8.7%, which is much lower than those of Western IS/LGS and previous Chinese EE cohorts. We assumed that the true contribution of all monogenic disorders in this Chinese IS cohort could have been underestimated because all recruited patients had suffered diagnosis odyssey before being referred to Beijing Children's Hospital. More important, we strictly followed the ACMG guidelines to interpret the pathogenicity of each variant and excluded all uncertain variants from the monogenic diagnostic yield (DNM in DIAPH3 and CACNA1A). In addition, for the inherited variant, although negative family history was recorded, the reduced penetrance of some inherited deleterious variants cannot excluded because we did not evaluate parents’ phenotypes in detailed, and this also explained the low diagnostic yield in our IS cohort.
As noted above, recent studies using trio‐based exome sequencing have confirmed that DNMs are a major etiological factor in Caucasian patients with EE (Allen et al., 2013; Carvill et al., 2013; Claes et al., 2001; Epi4K Consortium, 2016; EuroEPINOMICS‐RES Consortium et al., 2014; Saitsu et al., 2008; Shen et al., 2016; Trump et al., 2016; Yu et al., 2019). The 356 trio‐based WES data of the Epi4K Consortium and Epilepsy Phenome/Genome Project showed that at least 12% of individuals with ISs or Lennox–Gastaut syndrome have a definitive disease‐causing DNM (EuroEPINOMICS‐RES Consortium et al., 2014). They also revealed that the top genes with recurrent DNMs are SCN1A, STXBP1, GABRB3, CDKL5, SCN8A, SCN2A, ALG13, DNM1, and HDAC4 in descending order of frequency. Study of another Western early‐onset seizure cohort proved that DNMs in SCN2A are predominant (10/400), followed by those in CDKL5 and STXBP1, as revealed by sequencing of 46 genes (Trump et al., 2016). In contrast, a study of a medium‐sized cohort of Western infants with EE (n = 500) with the sequencing of 65 genes showed that CHD2 and SYNGAP1, rather than SCN2A, rank as hotspot genes with DNMs, accounting for 2.2% of referred patients (Carvill et al., 2013).
DNMs have been sporadically reported in Chinese EE cohorts (Miao et al., 2018; Xie et al., 2019; Zhang et al., 2016). However, neither the yield nor the architecture of DNMs has been distinctly characterized due to the small sample sizes. In our study, we revealed that pathogenic DNMs contributed to the causation of 8.3% (24/289) of Chinese patients with IS. This yield of DNM was similar to the yield (7.6%) of one Caucasian IS study with target sequencing (Muir et al., 2019), but lower than that of Epl4K Consortium (12%) with WES (EuroEPINOMICS‐RES Consortium et al., 2014). More important, we found that the DNMs of dominant genes accounted for 100% of the monogenic diagnostic yield of ISs, indicating that DNMs of dominant genes rather than biallelic mutations of recessive genes contribute to ISs in this Chinese cohort.
The top‐ranked genes with DNM in the Chinese IS cohort are CDKL5 and STXBP1, rather than SCN1A found in Caucasian EE cohorts (EuroEPINOMICS‐RES Consortium et al., 2014; Trump et al., 2016), which accounted for 3.1% of cases in our IS cohort. Meanwhile, five of the DNM genes identified in the Caucasian EE cohort (STXBP1, CDKL5, SCN2A, GABRB3, SCN8A) were also detected in this Chinese IS cohort (EuroEPINOMICS‐RES Consortium et al., 2014). The pathways/network of our DNMs are enriched in neurotransmission, similar to the network of DNMs in the Caucasian early‐onset EE cohort (McTague et al., 2015), supporting the overlapping mechanism of ISs between different ethnic groups. Our study also supports the findings of Zhang et al., who identified CDKL5 as the most common DNM‐affected gene in their small Chinese early‐onset EE cohort (17 genes, 175 patients), and DNMs in CDKL5 accounting for 13.1% of tested patients (23/175) (Zhang et al., 2016). Moreover, in 2018, with a medium‐sized Chinese early‐onset epilepsy cohort, Yang identified some hotspot genes with recurrent variants (ABCC8, CDKL5, DEPDC5, KCNQ2, MECP2, MUT, PCDH19, PRRT2, SCN1A, SCN2A, STXBP1, and TSC2) (Yang et al., 2018). Notably, they found KCNQ2 and STXBP1 mutations were enriched in the neonatal group, but SCN1A mutation significantly occurred in the first‐year group. Unfortunately, Yang did not perform inheritance analysis for these hotspot genes. In contrast to Yang's work, we completed parental validation for each pathogenic variant. We confirmed that their three genes (CDKL5, STXBP1, SCN2A) in neonatal group are hotspot genes with DNM in our IS patients. Inversely, none of SCN1A mutation was detected in our IS cohort, this suggested that SCN1A is not major causal gene carrying DNM in Chinese IS patients. Recently, another study of a large Western cohort also confirmed 100% DNM rates of epilepsy genes in CDKL5, STXBP1, SCN8A, GABRA1, and FOXG1, which is higher than the rates for SCN2A (90%), GRIN2A (88%), KCNQ2 (82%), and SCN1A (76%) (Lindy et al., 2018). Considering the above, we assumed that the unidentified mutations in CDKL5, STXBP1, and SCN2A in Yang's Chinese cohort should be DNMs.
Recently, both germline mosaicism and somatic mosaicism have also been reported as important mechanisms for early‐onset EE (Masliah‐Plachon et al., 2010). Examples of somatic mosaicism include KCNQ2 mutations in neonatal EE (Milh et al., 2015), CDKL5 mutations in West syndrome (Jdila et al., 2017; Kato et al., 2015; Masliah‐Plachon et al., 2010), paternal gonadal mosaicism of SCN1A mutations in Dravet syndrome (Depienne et al., 2010; Morimoto et al., 2006), and STXBP1‐related Ohtahara syndrome (Saitsu et al., 2010). Hence, through deep sequencing, we identified that 22.7% (5/22) of the “de novo mutations in Sanger sequencing” are not really DNMs.
A previous study showed that carriers or patients with the mosaic mutation have milder symptoms than those with germline mutations, and that there is a positive relationship between mutant ratio and affected status (Stosser et al., 2017). For example, a low somatic mutation level (16.9%) in STXBP1 was associated with normal neurodevelopment (Saitsu et al., 2010), SCN1A mosaic mutation level was directly related to the severity of Dravet syndrome (Depienne et al., 2010), and patients with 20%–30% mutant ratio in KCNQ2 presented with mild neonatal epilepsy but normal neurological development (Milh et al., 2015). As seen in our data, the patients with the somatic mutation, on either CDKL5 or STXBP1, showed lower seizure attacks compared with ones with the germline mutation. However, due to limit sample size, further validation study with more Chinese IS patients carrying different somatic frequency in identical gene is needed.
A number of studies have shown that the seizure symptom of IS patients, carrying either null or missense STXBP1 mutation, can be controlled by levetiracetam (Dilena et al., 2015). One Chinese study described the efficacy of levetiracetam on early‐onset EE in 7 patients with DNM of STXBP1 (Li et al., 2018). Among them, four patients showed good response to LEV alone or in combination with other routine AEDs, and were free from seizure for 4–18 months, and one patient showed reduced seizure (twice a year). Besides, in a recent study of 15 Chinese children with STXBP1 mutations, a good response to levetiracetam in six of 8 children treated (Cao et al., 2020). Consistent, three of four patients with DNM of STXBP1 in our study, also showed good response to LEV alone or in combination with other routine AEDs. Our study provides more evidence that levetiracetam can specifically reverse the STXBP1‐related IS symptom regardless of null or missense mutation. However, as per the clinical phenotype of Patient 29 with a second DNM that we reported in this paper, modifying factors may nullify the efficiency of levetiracetam for some cases with STXBP1 mutation. The clinical phenotype also influences the efficiency of Levetiracetam. In the future, more cases with STXBP1 mutation and a second pathogenic mutation are needed to confirm the factors modifying treatment efficacy.
In conclusion, our study confirmed the contribution and genetic spectrum of DNMs, with an 8.3% yield in a Chinese IS cohort. Somatic mutations underlie the origin of 22.7% of DNMs in IS patients. Treatment with levetiracetam improved the prognosis of STXBP1‐related ISs.
ETHICAL COMPLIANCE
This study was approved by the Ethics Committee of Chinese People's Liberation Army General Hospital and informed consent for publication was obtained from all parents/legal guardians for case presentation and publication.
CONFLICT OF INTEREST
The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
AUTHOR CONTRIBUTIONS
XLC and LZ designed the work. XLC finished final pathogenic interpretation for SNVs/CNVs and most of intellectual revision for manuscript. LL and FL are responsible for target gene design, data acquisition and manuscript drafting. FL, HX and YZ performed most of bench works including the target sequencing, amplicon‐based deep sequencing, paternity testing, real‐time PCR and aCGH experiment. LL and ZL performed most of day works including bioinformatics analysis, primer design, public mutation database researching and other statistical analyses. QW, QL, YW and MZ all were responsible for clinical works including patient recruitment, clinical data collection, patient review and treatment follow‐up. PJ offered some genetic suggestion and language edition. XDC gave key suggestions for design and manuscript writing. All authors read and approved the final manuscript.
Supporting information
Fig S1
Table S1‐6
ACKNOWLEDGMENTS
We sincerely thank all of the patients and their parents for their support and contributions to the study. This work was supported by grants from CAMS Innovation Fund for Medical Sciences (2016‐I2M‐1‐008), the Beijing Natural Science Foundation (7202019 to Xiaoli Chen), the Chinese National Nature Science Fund (31671310 to Xiaoli Chen, 81471329 to Liping Zou), National Science and Technology Major Project (2016YFC1000707 to Liping Zou), the Public Health Program of Capital (Z141100002114001 to Liping Zou), and Capital Health Research and Development of Special (2020‐2‐1131) and the Advanced Personnel Training Program of Beijing Municipal Health Bureau to Xiaoli Chen.
Liying Liu and Fang Liu contributed equally to this work.
Contributor Information
Liping Zou, Email: zouliping21@hotmail.com.
Xiaoli Chen, Email: xiaolichen@pumc.edu.cn, Email: cxlwx@sina.com.
DATA AVAILABILITY STATEMENT
All variant data supporting the conclusions of this manuscript were submitted to DECIPHER database.
REFERENCES
- Acuna‐Hidalgo, R. , Bo, T. , Kwint, M. P. , van de Vorst, M. , Pinelli, M. , Veltman, J. A. , Hoischen, A. , Vissers, L. E. , & Gilissen, C. (2015). Post‐zygotic point mutations are an underrecognized source of de novo genomic variation. American Journal of Human Genetics, 97(1), 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allach El Khattabi, L. , Heide, S. , Caberg, J. H. , Andrieux, J. , Doco Fenzy, M. , Vincent‐Delorme, C. , Callier, P. , Chantot‐Bastaraud, S. , Afenjar, A. , Boute‐Benejean, O. , Cordier, M. P. , Faivre, L. , Francannet, C. , Gerard, M. , Goldenberg, A. , Masurel‐Paulet, A. , Mosca‐Boidron, A. L. , Marle, N. , Moncla, A. , … Pipiras, E. (2018). 16p13.11 microduplication in 45 new patients: Refined clinical significance and genotype‐phenotype correlations. Journal of Medical Genetics, 57(5), 301–307. [DOI] [PubMed] [Google Scholar]
- Allen, A. S. , Berkovic, S. F. , Cossette, P. , Delanty, N. , Dlugos, D. , Eichler, E. E. , Epstein, M. P. , Glauser, T. , Goldstein, D. B. , Han, Y. , Heinzen, E. L. , Hitomi, Y. , Howell, K. B. , Johnson, M. R. , Kuzniecky, R. , Lowenstein, D. H. , Lu, Y. F. , Madou, M. R. , Marson, A. G. , … Winawer, M. R. (2013). De novo mutations in epileptic encephalopathies. Nature, 501(7466), 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, J. , Ji, X. , Mao, Y. , Zhang, P. , Liu, W. , Zhang, H. , Ding, N. , & Chen, Q. (2020). Clinical and genetic characteristics of children with STXBP1 encephalopathy. Zhonghua Er Ke Za Zhi, 58(6), 493–498. [DOI] [PubMed] [Google Scholar]
- Carvill, G. L. , Heavin, S. B. , Yendle, S. C. , McMahon, J. M. , O'Roak, B. J. , Cook, J. , Khan, A. , Dorschner, M. O. , Weaver, M. , Calvert, S. , Malone, S. , Wallace, G. , Stanley, T. , Bye, A. M. , Bleasel, A. , Howell, K. B. , Kivity, S. , Mackay, M. T. , Rodriguez‐Casero, V. , … Mefford, H. C. (2013). Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature Genetics, 45(7), 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes, L. , Del‐Favero, J. , Ceulemans, B. , Lagae, L. , Van Broeckhoven, C. , & De Jonghe, P. (2001). De novo mutations in the sodium‐channel gene SCN1A cause severe myoclonic epilepsy of infancy. American Journal of Human Genetics, 68(6), 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depienne, C. , Trouillard, O. , Gourfinkel‐An, I. , Saint‐Martin, C. , Bouteiller, D. , Graber, D. , Barthez‐Carpentier, M. A. , Gautier, A. , Villeneuve, N. , Dravet, C. , Livet, M. O. , Rivier‐Ringenbach, C. , Adam, C. , Dupont, S. , Baulac, S. , Heron, D. , Nabbout, R. , & Leguern, E. (2010). Mechanisms for variable expressivity of inherited SCN1A mutations causing Dravet syndrome. Journal of Medical Genetics, 47(6), 404–410. [DOI] [PubMed] [Google Scholar]
- Deprez, L. , Weckhuysen, S. , Holmgren, P. , Suls, A. , Van Dyck, T. , Goossens, D. , Del‐Favero, J. , Jansen, A. , Verhaert, K. , Lagae, L. , Jordanova, A. , Van Coster, R. , Yendle, S. , Berkovic, S. F. , Scheffer, I. , Ceulemans, B. , & De Jonghe, P. (2010). Clinical spectrum of early‐onset epileptic encephalopathies associated with STXBP1 mutations. Neurology, 75(13), 1159–1165. [DOI] [PubMed] [Google Scholar]
- Dilena, R. , Striano, P. , Traverso, M. , Viri, M. , Cristofori, G. , Tadini, L. , Barbieri, S. , Romeo, A. , & Zara, F. (2015). Dramatic effect of levetiracetam in early‐onset epileptic encephalopathy due to STXBP1 mutation. Brain and Development, 38(1), 128–131. [DOI] [PubMed] [Google Scholar]
- Epi4K Consortium . (2016). De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. American Journal of Human Genetics, 99(2), 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EuroEPINOMICS‐RES Consortium; Epilepsy Phenome/Genome Project; Epi4K Consortium . (2014). De Novo Mutations in Synaptic Transmission Genes Including DNM1 Cause Epileptic Encephalopathies. American Journal of Human Genetics, 95(4), 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, S. , Downs, J. , Ho, G. , de Klerk, N. , Forbes, D. , Christodoulou, J. , Williams, S. , & Leonard, H. (2016). Functional abilities in children and adults with the CDKL5 disorder. American Journal of Medical Genetics: Part A, 170(11), 2860–2869. [DOI] [PubMed] [Google Scholar]
- Fehr, S. , Wong, K. , Chin, R. , Williams, S. , de Klerk, N. , Forbes, D. , Krishnaraj, R. , Christodoulou, J. , Downs, J. , & Leonard, H. (2016). Seizure variables and their relationship to genotype and functional abilities in the CDKL5 disorder. Neurology, 87(21), 2206–2213. [DOI] [PubMed] [Google Scholar]
- Hamdan, F. F. , Myers, C. T. , Cossette, P. , Lemay, P. , Spiegelman, D. , Laporte, A. D. , Nassif, C. , Diallo, O. , Monlong, J. , Cadieux‐Dion, M. , Dobrzeniecka, S. , Meloche, C. , Retterer, K. , Cho, M. T. , Rosenfeld, J. A. , Bi, W. , Massicotte, C. , Miguet, M. , Brunga, L. , … Michaud, J. L. (2017). High rate of recurrent de novo mutations in developmental and epileptic encephalopathies. American Journal of Human Genetics, 101(5), 664–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helbig, K. L. , Farwell Hagman, K. D. , Shinde, D. N. , Mroske, C. , Powis, Z. , Li, S. , Tang, S. , & Helbig, I. (2016). Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genetics in Medicine, 18(9), 898–905. [DOI] [PubMed] [Google Scholar]
- Jdila, M. B. , Issa, A. B. , Khabou, B. , Rhouma, B. B. , Kamoun, F. , Ammar‐Keskes, L. , Triki, C. , & Fakhfakh, F. (2017). Novel mutations in the CDKL5 gene in complex genotypes associated with West syndrome with variable phenotype: First description of somatic mosaic state. Clinica Chimica Acta, 473, 51–59. [DOI] [PubMed] [Google Scholar]
- Jiang, Q. , Liu, F. , Miao, C. , Li, Q. , Zhang, Z. , Xiao, P. , Su, L. , Yu, K. , Chen, X. , Zhang, F. , Chakravarti, A. , & Li, L. (2017). RET somatic mutations are underrecognized in Hirschsprung disease. Genetics in Medicine, 20(7), 770–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, T. , Morisada, N. , Nagase, H. , Nishiyama, M. , Toyoshima, D. , Nakagawa, T. , Maruyama, A. , Fu, X. J. , Nozu, K. , Wada, H. , Takada, S. , & Iijima, K. (2015). Somatic mosaicism of a CDKL5 mutation identified by next‐generation sequencing. Brain and Development, 37(9), 911–915. [DOI] [PubMed] [Google Scholar]
- Lemke, J. R. , Riesch, E. , Scheurenbrand, T. , Schubach, M. , Wilhelm, C. , Steiner, I. , Hansen, J. , Courage, C. , Gallati, S. , Burki, S. , Strozzi, S. , Simonetti, B. G. , Grunt, S. , Steinlin, M. , Alber, M. , Wolff, M. , Klopstock, T. , Prott, E. C. , Lorenz, R. , … Biskup, S. (2012). Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia, 53(8), 1387–1398. [DOI] [PubMed] [Google Scholar]
- Lesca, G. , Rudolf, G. , Labalme, A. , Hirsch, E. , Arzimanoglou, A. , Genton, P. , Motte, J. , de Saint Martin, A. , Valenti, M. P. , Boulay, C. , De Bellescize, J. , Keo‐Kosal, P. , Boutry‐Kryza, N. , Edery, P. , Sanlaville, D. , & Szepetowski, P. (2012). Epileptic encephalopathies of the Landau‐Kleffner and continuous spike and waves during slow‐wave sleep types: Genomic dissection makes the link with autism. Epilepsia, 53(9), 1526–1538. [DOI] [PubMed] [Google Scholar]
- Li, D. , Bhoj, E. , McCormick, E. , Wang, F. , Snyder, J. , Wang, T. , Zhao, Y. , Kim, C. , Chiavacci, R. , Tian, L. , Falk, M. J. , & Hakonarson, H. (2016). Early infantile epileptic encephalopathy in an STXBP1 patient with lactic acidemia and normal mitochondrial respiratory chain function. Case Reports in Genetics, 2016, 4140780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. Y. , Ye, J. , Huang, Z. Y. , Lin, Y. C. , Liu, A. H. , Li, L. P. , Chen, J. , & Wang, Y. P. (2019). Clinical analysis of five cases of autism spectrum disorder complicated with epilepsy with chromosome copy number variation. Zhonghua Yi Xue Za Zhi, 99(33), 2615–2618. [DOI] [PubMed] [Google Scholar]
- Li, T. , Cheng, M. , Wang, J. , Hong, S. , Li, M. , Liao, S. , Xie, L. , & Jiang, L. (2018). De novo mutations of STXBP1 in Chinese children with early onset epileptic encephalopathy. Genes, Brain, and Behavior, 17(8), e12492. [DOI] [PubMed] [Google Scholar]
- Lindy, A. S. , Stosser, M. B. , Butler, E. , Downtain‐Pickersgill, C. , Shanmugham, A. , Retterer, K. , Brandt, T. , Richard, G. , & McKnight, D. A. (2018). Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia, 59(5), 1062–1071. [DOI] [PubMed] [Google Scholar]
- Lux, A. L. , & Osborne, J. P. (2004). A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: Consensus statement of the West Delphi group. Epilepsia, 45(11), 1416–1428. [DOI] [PubMed] [Google Scholar]
- Masliah‐Plachon, J. , Auvin, S. , Nectoux, J. , Fichou, Y. , Chelly, J. , & Bienvenu, T. (2010). Somatic mosaicism for a CDKL5 mutation as an epileptic encephalopathy in males. American Journal of Medical Genetics: Part A, 152A(8), 2110–2111. [DOI] [PubMed] [Google Scholar]
- McTague, A. , Howell, K. B. , Cross, J. H. , Kurian, M. A. , & Scheffer, I. E. (2015). The genetic landscape of the epileptic encephalopathies of infancy and childhood. The Lancet Neurology, 15(3), 304–316. [DOI] [PubMed] [Google Scholar]
- Mefford, H. C. , Yendle, S. C. , Hsu, C. , Cook, J. , Geraghty, E. , McMahon, J. M. , Eeg‐Olofsson, O. , Sadleir, L. G. , Gill, D. , Ben‐Zeev, B. , Lerman‐Sagie, T. , Mackay, M. , Freeman, J. L. , Andermann, E. , Pelakanos, J. T. , Andrews, I. , Wallace, G. , Eichler, E. E. , Berkovic, S. F. , & Scheffer, I. E. (2011). Rare copy number variants are an important cause of epileptic encephalopathies. Annals of Neurology, 70(6), 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, P. , Feng, J. , Guo, Y. , Wang, J. , Xu, X. , Wang, Y. , Li, Y. , Gao, L. , Zheng, C. , & Cheng, H. (2018). Genotype and phenotype analysis using an epilepsy‐associated gene panel in Chinese pediatric epilepsy patients. Clinical Genetics, 94(6), 512–520. [DOI] [PubMed] [Google Scholar]
- Michaud, J. L. , Lachance, M. , Hamdan, F. F. , Carmant, L. , Lortie, A. , Diadori, P. , Major, P. , Meijer, I. A. , Lemyre, E. , Cossette, P. , Mefford, H. C. , Rouleau, G. A. , & Rossignol, E. (2014). The genetic landscape of infantile spasms. Human Molecular Genetics, 23(18), 4846–4858. [DOI] [PubMed] [Google Scholar]
- Milh, M. , Lacoste, C. , Cacciagli, P. , Abidi, A. , Sutera‐Sardo, J. , Tzelepis, I. , Colin, E. , Badens, C. , Afenjar, A. , Coeslier, A. D. , Dailland, T. , Lesca, G. , Philip, N. , & Villard, L. (2015). Variable clinical expression in patients with mosaicism for KCNQ2 mutations. American Journal of Medical Genetics: Part A, 167A(10), 2314–2318. [DOI] [PubMed] [Google Scholar]
- Milh, M. , Villeneuve, N. , Chouchane, M. , Kaminska, A. , Laroche, C. , Barthez, M. A. , Gitiaux, C. , Bartoli, C. , Borges‐Correia, A. , Cacciagli, P. , Mignon‐Ravix, C. , Cuberos, H. , Chabrol, B. , & Villard, L. (2011). Epileptic and nonepileptic features in patients with early onset epileptic encephalopathy and STXBP1 mutations. Epilepsia, 52(10), 1828–1834. [DOI] [PubMed] [Google Scholar]
- Morimoto, M. , Mazaki, E. , Nishimura, A. , Chiyonobu, T. , Sawai, Y. , Murakami, A. , Nakamura, K. , Inoue, I. , Ogiwara, I. , Sugimoto, T. , & Yamakawa, K. (2006). SCN1A mutation mosaicism in a family with severe myoclonic epilepsy in infancy. Epilepsia, 47(10), 1732–1736. [DOI] [PubMed] [Google Scholar]
- Muir, A. M. , Myers, C. T. , Nguyen, N. T. , Saykally, J. , Craiu, D. , De Jonghe, P. , Helbig, I. , Hoffman‐Zacharska, D. , Guerrini, R. , Lehesjoki, A. E. , Marini, C. , Moller, R. S. , Serratosa, J. , Sterbova, K. , Striano, P. , von Spiczak, S. , Weckhuysen, S. , & Mefford, H. C. (2019). Genetic heterogeneity in infantile spasms. Epilepsy Research, 156, 106181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels, J. (2015). Pathway‐driven discovery of epilepsy genes. Nature Neuroscience, 18(3), 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottman, R. , Hirose, S. , Jain, S. , Lerche, H. , Lopes‐Cendes, I. , Noebels, J. L. , Serratosa, J. , Zara, F. , & Scheffer, I. E. (2010). Genetic testing in the epilepsies—Report of the ILAE Genetics Commission. Epilepsia, 51(4), 655–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs, E. R. , Andersen, E. F. , Cherry, A. M. , Kantarci, S. , Kearney, H. , Patel, A. , Raca, G. , Ritter, D. I. , South, S. T. , Thorland, E. C. , Pineda‐Alvarez, D. , Aradhya, S. , & Martin, C. L. (2019). Technical standards for the interpretation and reporting of constitutional copy‐number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genetics in Medicine, 22(2), 245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romaniello, R. , Zucca, C. , Tenderini, E. , Arrigoni, F. , Ragona, F. , Zorzi, G. , Bassi, M. T. , & Borgatti, R. (2013). A novel mutation in STXBP1 gene in a child with epileptic encephalopathy and an atypical electroclinical pattern. Journal of Child Neurology, 29(2), 249–253. [DOI] [PubMed] [Google Scholar]
- Saitsu, H. , Hoshino, H. , Kato, M. , Nishiyama, K. , Okada, I. , Yoneda, Y. , Tsurusaki, Y. , Doi, H. , Miyake, N. , Kubota, M. , Hayasaka, K. , & Matsumoto, N. (2010). Paternal mosaicism of an STXBP1 mutation in OS. Clinical Genetics, 80(5), 484–488. [DOI] [PubMed] [Google Scholar]
- Saitsu, H. , Kato, M. , Mizuguchi, T. , Hamada, K. , Osaka, H. , Tohyama, J. , Uruno, K. , Kumada, S. , Nishiyama, K. , Nishimura, A. , Okada, I. , Yoshimura, Y. , Hirai, S. , Kumada, T. , Hayasaka, K. , Fukuda, A. , Ogata, K. , & Matsumoto, N. (2008). De novo mutations in the gene encoding STXBP1 (MUNC18‐1) cause early infantile epileptic encephalopathy. Nature Genetics, 40(6), 782–788. [DOI] [PubMed] [Google Scholar]
- Shen, D. , Hernandez, C. C. , Shen, W. , Hu, N. , Poduri, A. , Shiedley, B. , Rotenberg, A. , Datta, A. N. , Leiz, S. , Patzer, S. , Boor, R. , Ramsey, K. , Goldberg, E. , Helbig, I. , Ortiz‐Gonzalez, X. R. , Lemke, J. R. , Marsh, E. D. , & Macdonald, R. L. (2016). De novo GABRG2 mutations associated with epileptic encephalopathies. Brain, 140(1), 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamberger, H. , Nikanorova, M. , Willemsen, M. H. , Accorsi, P. , Angriman, M. , Baier, H. , Benkel‐Herrenbrueck, I. , Benoit, V. , Budetta, M. , Caliebe, A. , Cantalupo, G. , Capovilla, G. , Casara, G. , Courage, C. , Deprez, M. , Destree, A. , Dilena, R. , Erasmus, C. E. , Fannemel, M. , … Weckhuysen, S. (2016). STXBP1 encephalopathy: A neurodevelopmental disorder including epilepsy. Neurology, 86(10), 954–962. [DOI] [PubMed] [Google Scholar]
- Stosser, M. B. , Lindy, A. S. , Butler, E. , Retterer, K. , Piccirillo‐Stosser, C. M. , Richard, G. , & McKnight, D. A. (2017). High frequency of mosaic pathogenic variants in genes causing epilepsy‐related neurodevelopmental disorders. Genetics in Medicine, 20(4), 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trump, N. , McTague, A. , Brittain, H. , Papandreou, A. , Meyer, E. , Ngoh, A. , Palmer, R. , Morrogh, D. , Boustred, C. , Hurst, J. A. , Jenkins, L. , Kurian, M. A. , & Scott, R. H. (2016). Improving diagnosis and broadening the phenotypes in early‐onset seizure and severe developmental delay disorders through gene panel analysis. Journal of Medical Genetics, 53(5), 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta, M. , Tennison, M. B. , Aylsworth, A. S. , Turcott, C. M. , Guerra, M. P. , Eng, C. M. , & Yang, Y. (2012). A novel STXBP1 mutation causes focal seizures with neonatal onset. Journal of Child Neurology, 27(6), 811–814. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Wen, Y. , Zhang, Q. , Yu, S. , Chen, Y. , Wu, X. , Zhang, Y. , & Bao, X. (2019). Gene mutational analysis in a cohort of Chinese children with unexplained epilepsy: Identification of a new KCND3 phenotype and novel genes causing Dravet syndrome. Seizure, 66, 26–30. [DOI] [PubMed] [Google Scholar]
- Widjaja, E. , Go, C. , McCoy, B. , & Snead, O. C. (2014). Neurodevelopmental outcome of infantile spasms: A systematic review and meta‐analysis. Epilepsy Research, 109, 155–162. [DOI] [PubMed] [Google Scholar]
- Xie, H. , Su, W. , Pei, J. , Zhang, Y. , Gao, K. , Li, J. , Ma, X. , Wu, X. , & Jiang, Y. (2019). De novo SCN1A, SCN8A, and CLCN2 mutations in childhood absence epilepsy. Epilepsy Research, 154, 55–61. [DOI] [PubMed] [Google Scholar]
- Yang, L. , Kong, Y. , Dong, X. , Hu, L. , Lin, Y. , Chen, X. , Ni, Q. , Lu, Y. , Wu, B. , Wang, H. , Lu, Q. R. , & Zhou, W. (2018). Clinical and genetic spectrum of a large cohort of children with epilepsy in China. Genetics in Medicine, 21(3), 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, X. , Yang, L. , Li, J. , Li, W. , Li, D. , Wang, R. , Wu, K. , Chen, W. , Zhang, Y. , Qiu, Z. , & Zhou, W. (2019). De novo and inherited SETD1A variants in early‐onset epilepsy. Neuroscience Bulletin, 35(6), 1045–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuskaitis, C. J. , Ruzhnikov, M. R. Z. , Howell, K. B. , Allen, I. E. , Kapur, K. , Dlugos, D. J. , Scheffer, I. E. , Poduri, A. , & Sherr, E. H. (2018). Infantile spasms of unknown cause: Predictors of outcome and genotype‐phenotype correlation. Pediatric Neurology, 87, 48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Li, J. , Zhao, Y. , Bao, X. , Wei, L. , & Wang, J. (2016). Gene mutation analysis of 175 Chinese patients with early‐onset epileptic encephalopathy. Clinical Genetics, 91(5), 717–724. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Table S1‐6
Data Availability Statement
All variant data supporting the conclusions of this manuscript were submitted to DECIPHER database.