

Abstract
Background
Methylmalonic aciduria (MMA) combined with homocystinuria, cobalamin(cbl)C deficiency type (OMIM 277400), is the most common autosomal recessive inherited disorder of intracellular cobalamin metabolism caused by mutations in the MMACHC gene (OMIM 609831), of which more than 100 mutations have been identified to date. In this study, we only identified a coding mutation in one allele at the MMACHC gene locus, and no large fragments deletion or duplication were found. Up to now, only three epimutation cblC cases were reported. We hypothesized whether the MMACHC was hypermethylated.
Methods
To address this hypothesis, the entire coding region and adjacent splice sites of the panel genes involved in metabolic diseases were sequenced using the Illumina HiSeq X platform, followed by confirmation via Sanger sequencing in their parents and brothers. Methylation analysis of the MMACHC was performed using an EpiTect Bisulfite Kit and methylation‐specific PCR (MSP) to investigate the role of epimutations in cblC disease.
Results
We identified a clearly pathogenic single heterozygous c.658_660del, p. (K220del) mutation, which was also identified in the mother. Analysis of the MMACHC indicated a heterozygous epimutation consisting of 34 hypermethylated CpG sites in a CpG island encompassing the promoter and first exon of the MMACHC, which was also identified in the father. Furthermore, we identified a single heterozygous c.*2C>T mutation in the sixth exon of the PRDX1 (OMIM 176763) in patients and their fathers, which was the only sequence variation that segregated with the MMACHC methylation. Neither c.658_660del and epimutation in MMACHC nor c.*2C>T in PRDX1 was discovered in her brother.
Conclusion
We report compound heterozygotes in MMACHC for a genetic mutation and an epimutation in cblC cases. To our best knowledge, this is the first report of two cblC cases from China caused by compound heterozygous mutations with a coding mutation in one allele and an epimutation in the other at the MMACHC locus.
Keywords: cblC, epimutation, hypermethylated, MMACHC
This is the first report of two cblC cases from China caused by compound heterozygous mutations with a coding mutation in one allele and an epimutation in the other at the MMACHC gene locus.

1. INTRODUCTION
MMA is the most common type of organic acidemia in China. According to the type of enzyme defect, it can be divided into methylmalonyl‐CoA mutase deficiency and disorder of the metabolism of its coenzyme cobalamin. Based on the blood Hcy level, MMA can be divided into isolated MMA or combined methylmalonic aciduria and homocystinuria, whereby the latter consists of cblC, cblD, cblF, cblJ, and cblX (Huemer et al., 2017; Yu et al., 2013). In China, patients with combined methylmalonic acidemia and homocystinuria account for approximately 80% of all MMA cases. Among the subtypes, the cblC defect is the most common (Liu et al., 2012). The incidence of cblC defect in China appears about 1:3500 in previous reports(Guo et al., 2018; Han et al., 2016).
The gene associated with the cblC defect is the MMACHC, which maps to chromosome 1p34.1. It comprises 4 coding exons and its full‐length cDNA contains 846 base pairs encoding a protein of 282 amino acids (Lerner‐Ellis et al., 2006). To date, more than a hundred MMACHC gene mutations have been deposited in the human gene mutation database (HGMD, www.hgmd.cf.ac.uk/ac/index.php), most of which are missense/nonsense mutations, followed by small deletions. The c.80A>G, c.609G>A, c.482G>A, c.394C>T, and c.658_660del mutations in the MMACHC gene were the most common mutations in Chinese Patients (Liu et al., 2010). In 2018, Guéant et al. have for the first time reported three “epi‐cblC” cases, which are compound heterozygotes for a genetic mutation and a promoter epimutation. The secondary epimutation of their epi‐cblC cases is located in a trio of genes which was a forced antisense transcription of MMACHC resulted from the skipping of the last exon of PRDX1. This skipping was the causative defect that produced the epimutation. They found that silencing of PRDX1 decreased the methylation of exon 1 of MMACHC and the promoter, and restored the transcription of MMACHC (Gueant et al., 2018). DNA methylation is the earliest discovered and the best understood epigenetic modification (Feng et al., 2010). Methylation of cytosine, usually at CpG dinucleotides, is involved in epigenetic regulation of gene expression. Promoter methylation is typically associated with repression, whereas gene methylation correlates with transcriptional activity (Jones, 2012).
In this study, paternal hypermethylation of the promoter of MMACHC was identified. We report two cases of a compound heterozygous case with a genetic mutation and an epimutation at the MMACHC gene locus.
2. MATERIALS AND METHODS
2.1. Ethical compliance and informed consent
These cases study were approved by the Institutional Review Board of Zhengzhou children's hospital. Informed consent was obtained from all patients’ parents for being included in the study.
2.2. DNA extraction
Peripheral blood samples were collected from patients and their families. Genomic DNA from 250 μl of EDTA‐treated peripheral blood was isolated using the Chemagic 360 kit (CMG‐536; PerkinElmer) according to the manufacturer's instructions, and was quantified using a NANODROP ONE (Thermo Fisher Scientific). The extracted DNA was stored at −20°C until further use.
2.3. Gene sequencing
NGS was performed using our standard protocols at the WE‐HEALTH Biomedical Technology Company. A library was constructed by extracting genomic DNA from the proband. 175 target genes, including MMADHC, LMBRD1, ABCD4, HCFC1, THAP11, ZNF143, CD320, TCN1, TCN2 and so on, exons and the adjacent splicing regions (about 50 bp) were captured by probe hybridization and enriched. The enriched genes were subjected to quality control, and sequenced using the Illumina HiSeq X System sequencer with paired‐end 150‐bp read length. The data interpretation rules were in accordance with the American College of Medical Genetics and Genomics (ACMG, PMID: 25741868) classification criteria and guidelines for genetic variation (Richards et al., 2015), including well‐defined genetic variations related to genetic diseases and some relevant journal reports or database entries. Sanger sequencing was used to verify the genetic variation and the primers were designed using Oligo7 (Table 1).
TABLE 1.
PCR primers used for mutation analysis of MMACHC a
| Primer | Targeted fragment | Sequence (5′–3′) | Length (bp) |
|---|---|---|---|
| MMACHC‐1F | Exon 1 | ACTGAGTCTTCCTGATTTCGTGT | 423 |
| MMACHC‐1R | CACTGAAACTCTGTCCCTGTTG | ||
| MMACHC‐2F | Exon 2 | TGCATCACATAGCGTCAGTG | 467 |
| MMACHC‐2R | AGCCTGGCTTTAGGGTATCA | ||
| MMACHC‐3F | Exon 3 | TCATGTTTTCCCTTCTGAGGA | 395 |
| MMACHC‐‐3R | CAAAGCTAATTTGTTCTGGGTTG | ||
| MMACHC‐4F | Exon 4 | AGGCCTAGCTTGCAATGATG | 694 |
| MMACHC‐4R | GAAGGCAGATGGGAATTCTG | ||
| PRDX1‐1F | Exon 1 | TTCAGGGACACTGGACAATG | 300 |
| PRDX1‐1R | TCTAACAAGCCACCCCTAGA | ||
| PRDX1‐2F | Exon 2 | GAGCATCTTGGCAGTTAGTGG | 424 |
| PRDX1‐2R | CAGGAGAATGGCGTGAAACTG | ||
| PRDX1‐3F | Exon 3 | GCAGTTGAGTGTACCCTAGTG | 334 |
| PRDX1‐3R | GCCAACTGGATACTTGTCCTG | ||
| PRDX1‐4F | Exon 4 | GACCAGTCCATTTCCTTCAGG | 325 |
| PRDX1‐4R | GTCTCTCATTCCACCTAACGAG | ||
| PRDX1‐5F | Exon 5 | TCCATAGGAGAATGGTGGTGC | 283 |
| PRDX1‐5R | GCCTGCCTTGTAAGACACCACA | ||
| MSP‐F | Exon 1 | TCGTCGGGTTTAAAATTTC | 103 |
| MSP‐R | CCGATTATATTCGCAAAAAA | ||
| UM‐F | Exon 1 | GTTGTTGGGTTTAAAATTTT | 103 |
| UM‐R | CCAATTATATTCACAAAAAAC |
Abbreviations: F, forward primer;MSP, methylation specific PCR; R, reverse primer; UM, un‐methylation.
The GenBank reference sequence used for studying MMACHC was NM_015506.3 and PRDX1 was NM_181697.3(GRCh37).
2.4. Determination of DNA methylation
The methylation status of the CpG islands in the 5′‐region of the MMACHC (RefSeq: −433 to +67 of the promoter region from NCBI) was determined by bisulfite conversion and methylation‐specific polymerase chain reaction (PCR). One microgram of genomic DNA was converted by bisulfite using the EpiTect® Bisulfite Kit (QIAGEN). The bisulfite‐treated DNA was amplified by PCR using primers were designed using the Methyl Primer Express software and Taq DNA Polymerase (Zymo CWBIO Research). The amplification products were detected by agarose gel electrophoresis.
2.5. Real‐time PCR
In case 006 and her parents, 3 ml fresh peripheral blood were collected and used for total RNA isolation. Two‐hundred nanograms of RNA was reverse‐transcribed in a 20μL reaction volume using StarScript Ⅱ First‐strand cDNA Synthesis Mix With gDNA Remover (GenStar) according to the manufacturer's recommendations; 2 μl of cDNA was used for qPCR with SYBR® Premix Ex Taq™(TAKARA) in a 20 μl reaction volume according to the manufacturer's instructions, with reverse and forward primers at a concentration of 0.2 μM. Specific amplifications were performed using the following primers:
MMACHC forward 5′‐CAGCAAGCTCAGCGTGTAAC‐3′, reverse 5′‐ CATGCCACCTGGAAGGGGTAA‐3′. Quantification was performed using the housekeeping gene GAPDH as an internal standard with the following primers: forward 5′‐GGAGCGAGATCCCTCCAAAAT‐3′, reverse 5′‐GGCTGTTGTCATACTTCTCATGG‐3′. Temperature cycling for MMACHC was 10 min at 95℃ followed by 40 cycles consisting of 95℃ for 10 s, 58℃ for 30 s, and 72℃ for 40 s. Results were expressed as arbitrary units by calculating the ratio of crossing points of amplification curves of MMACHC and the internal standard by using the δδCt method.
3. RESULTS
3.1. Clinical symptoms and laboratory auxiliary examinations
The case‐006, a 2‐month‐old girl, was the second child of healthy, non‐consanguineous Chinese parents. She was born at term with a birth weight of 2.7 kg and a birth length of 48 cm. After 2 months of breastfeeding, she displayed markedly poor feeding, poor mental reaction, developmental delay, malnutrition, pale lips, susceptibility to viral infections, lethargy, and cortical blindness. Metabolic screening by GC‐MS revealed methylmalonic aciduria was 153 mmol/mol Cr (normal range 0–5.3 mmol/mol Cr). Analysis of plasma acylcarnitine by Applied Biosystems API 3200 tandem mass spectrometric (LC‐MS/MS) analyzer demonstrated a significantly elevated ratio of propionyl carnitine/acetyl carnitine with reduced methionine. Plasma total homocysteine was 138.6 μmol/L (normal range <15 μmol/L). Serum vitamin B12 and folate were normal. The mother had no history of anemia or long‐term vegetarian diet, and her homocysteine was normal. In combination with the complete blood count and no anemia in the mother, secondary MMA probably is referring to maternal B12 deficiency was ruled out. After intravenous administration of 2.5 ml po bid L‐carnitine to improve the metabolism, intramuscular supplementation of 2.5 mg qod vitamin B12 (hydroxo‐cobalamin) and oral 500 mg po bid betaine treatment, the ratios of propionylcarnitine/acetylcarnitine (C3/C2) and propionylcarnitine/free carnitine (C3/C0) close to normal. The clinical diagnosis was consistent with MMA and homocystinuria.
The case‐007, a 14 days boy, also was the second child of healthy, non‐consanguineous Chinese parents. Neonatal screening showed elevated levels of C3 and a significantly elevated ratio of propionyl C3/C2. Metabolic screening by GC‐MS revealed methylmalonic aciduria was 20 mmol/mol Cr (normal range 0–5.3 mmol/mol Cr). Plasma total homocysteine was 175 μmol/l (normal range <15 μmol/L). Methionine levels were 21.31 μmol/L (normal range 0–15 μmol/L). Since birth, he displayed poor mental reaction and poor feeding. After intravenous administration of 2.5 ml po bid L‐carnitine, intramuscular supplementation of 0.6 mg qod vitamin B12 (hydroxo‐cobalamin), and oral 750 mg po bid betaine supplement, the ratios of C3/C2 and C3/C0 close to normal. The clinical diagnosis was consistent with MMA and homocystinuria.
3.2. A single heterozygous mutation in the MMACHC was identified in the cblC patients
The cblC diagnosis was based on clinical and metabolic analyses. Sequencing of the exons and flanking intronic regions of the genes involved in metabolic diseases in the case‐006 was done using the NGS Illumina panel. A single heterozygous mutation c.658_660del, p.(Lys220del) was identified in the MMACHC (Figure 1a), which was reported as a hotspot mutation in the Chinese population(13). At the same time, a splice region variant c.*2C>T was identified in the PRDX1 which was 3′ to MMACHC gene. No additional gene mutations were identified in the patient.
FIGURE 1.
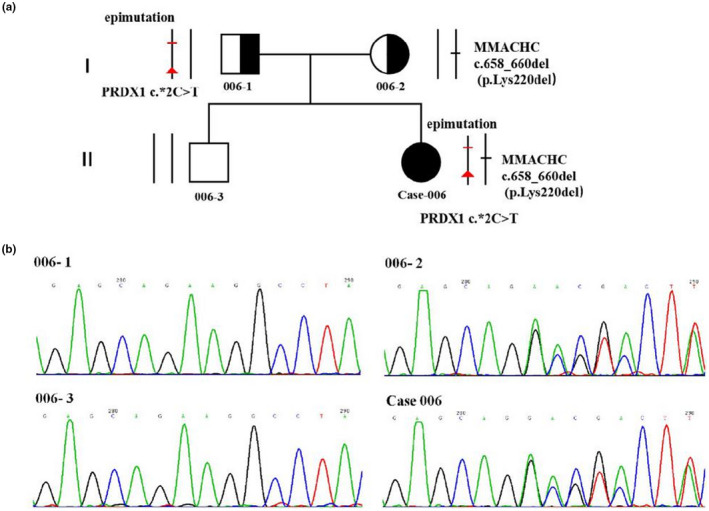
Pedigree derived from NGS and Sanger sequencing. (a) Pedigree of the case‐006; Circles, female; squares, male; filled, patients; half of filled, mutation carrier. black bar, mutation (heterozygous c.658_660del, p. Lys220del) found in the case‐006 and her mother; red bar, epimutation encompassing the MMACHC promoter/exon 1 found in the case‐006 and her father; red arrow, a single heterozygous c.*2C>T mutation in PRDX1, that segregated with the phenotype across the proband and her father. (b) Sanger sequencing of the single heterozygous c.658_660del(p. Lys220del) mutation in MMACHC among family members. 006–1, father; 006–2, mother; 006–3, brother; case‐006, the patient
Sanger sequencing of the 4 coding exons and splicing regions of the MMACHC of her family members revealed the single heterozygous c.658_660del, p.(Lys220del) inherited from her mother who with normal phenotype (006‐2, Figure 1a), while no variation was identified in her father (006‐1) or brother (006‐3) (Figure 1b). The splice region variant c.*2C>T in the PRDX1 was identified, which also existed in her father and no variation in her mother and brother.
In case‐007, through sanger sequencing of the 4 coding exons and splicing regions of the MMACHC, we identified a single heterozygous substitution c.609G>A, p.(Trp203Ter) in the MMACHC, which was also identified in his mother(007‐2) and brother(007‐3). No variation was identified in his father(007‐1) (Figure 2). His mother (007‐2) and brother (007‐3) are therefore carriers of methylmalonic acidemia with a heterozygous c.609G>A, p.(Trp203Ter) substitution of the MMACHC (Figure 2a). The sequencing primers are listed in Table 1. There is no large fragments deletion or duplication in MMACHC. Sanger sequencing was performed for the PRDX1 exons and exon/intron junctions and no variants were identified in case‐007 and her family members. No additional studies were performed due to lack of sample availability.
FIGURE 2.
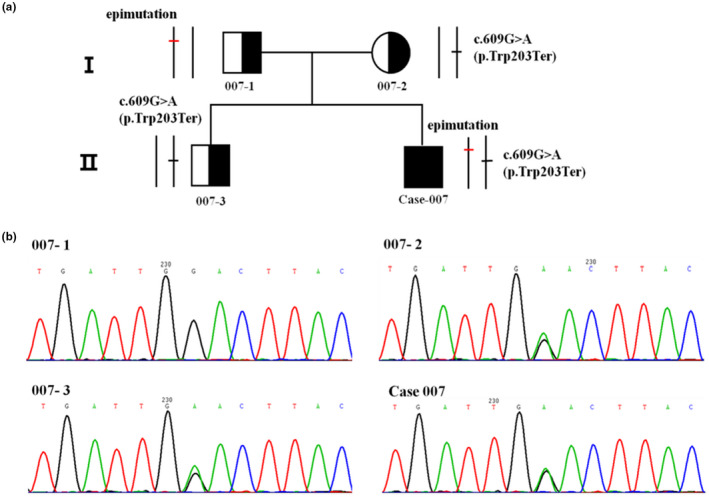
Pedigree derived from Sanger sequencing. (a) Pedigree of the case‐007; Circles, female; squares, male; filled, patients; half of filled, mutation carrier. black bar, mutation (heterozygous c.609G>A, p.(Trp203Ter)) found in the case‐007 and his mother and brother; red bar, epimutation encompassing the MMACHC promoter/exon 1 found in the case‐007 and his father. (b) Sanger sequencing of the single heterozygous c.609G>A, p.(Trp203Ter) mutation in MMACHC among family members. 007–1, father; 007–2, mother; 007–3, brother; case‐007, the patient
3.3. Identification of an epimutation in the MMACHC promoter region
We performed methylation analysis of the MMACHC by PCR amplification of bisulfite‐treated DNA from case‐006 and case‐007 and their family members. This revealed a heterozygous epimutation consisting of 34 hypermethylated CpG sites in a CpG island encompassing the promoter and first exon of MMACHC (Chr1:45,965,621–45,966,071) (Figure 3a,b), which was present in both the patients and their fathers. The epimutation was absent in the DNA from their mothers and brothers (Figure 3c,d). The epimutation in MMACHC of the patients was therefore inherited from their fathers (Figure 1a).
FIGURE 3.

Methylation analysis of the promoter and first exon of MMACHC. (a) The promoter and first exon of MMACHC (Chr1:45,965,621–45,966,071); pink long‐line, a CpG island; pink bar, 34 hypermethylated CpG sites. (b) The sequence of the CpG island; yellow base, bisulfite conversion site; all non‐methylated cytosines (C) were converted to uracil (U), while methylated cytosines (C) were left unchanged. (c) Methylation‐specific PCR products were detected by agarose gel electrophoresis of case‐006 and her family members; M, methylated; U, not methylated. (d) Methylation‐specific PCR products were detected by agarose gel electrophoresis of case‐007 and his family members
Taken together, these results showed an epimutation causing promoter hypermethylation and MMACHC silencing in the paternally transmitted allele, together with genetic mutations in the alleles inherited from mothers.
4. DISCUSSION
The cblC type of combined MMA and homocystinuria is the most frequent inherited metabolic disease in China, mainly causing nervous system damage. Cases of the cblC disorder are usually caused by homozygous or compound heterozygous mutations in the MMACHC gene (Lerner‐Ellis et al., 2006; Watkins & Rosenblatt, 2011). Our cblC case was diagnosed as combined MMA and homocystinuria according to the metabolic screening and biochemical tests. She manifested compound heterozygosity, with an epimutation in one allele of the MMACHC gene, which is a rare genetic cause, and a genetic mutation in the other one. Here, we first identified cblC cases that are compound heterozygotes for a genetic mutation and a promoter epimutation in Chinese population.
DNA methylation is one of the most intensely studied epigenetic modifications, which involves the transfer of a methyl group onto the C5 position of a cytosine to form 5‐methylcytosine (Demond & Kelsey, 2020). Commonly, DNA methylation occurs on cytosines that precede a guanine nucleotide, the so‐called CpG sites (Figure 2b). This modification is essential for the regulation of tissue‐specific gene expression and genomic imprinting by recruiting proteins involved in gene repression or by inhibiting the binding of transcription factors to DNA (Jones, 2012; Pensold et al., 2020). CpG islands are stretches of DNA roughly 1000 base pairs in length that have a higher CpG density than the rest of the genome which are not methylated (Deaton & Bird, 2011) (Bird et al., 1985). The majority of gene promoters, roughly 70%, reside within CpG islands (Saxonov et al., 2006). The location and preservation of CpG islands throughout evolution implies that these regions have a functional role. Previous reports have described DNA methylation of genes that mimics recessive mutations by creating a transcriptional haploinsufficiency with tissue‐specific epigenetic silencing (Zhou et al., 2006).
In mammals, the paternal genome is demethylated during the events of fertilization and re‐methylated in the somatic cells of the next generation. An epimutation causing promoter hypermethylation and MMACHC silencing in the paternally transmitted allele is different from the spermatozoa erasure previously observed in families with epigenetic silencing of the MLH1 (Hitchins et al., 2011). To detect methylation, the genomic DNA was subjected to bisulfite modification, which converts all cytosines (C) that are not methylated into uracils (U), while methylated cytosines remain unchanged. This difference can be detected by PCR with different primers to determine whether the gene is methylated at the CpG island. The results revealed hypermethylation abnormalities in the promoter and exon 1 region of the MMACHC in case‐006, case‐007 and their fathers, which were not found in their mothers or brothers. Epimutations inherited from fathers may affect the normal expression of the MMACHC. Together with genetic mutations, epimutations are the molecular genetic cause of the disease in the two patients. Thus, this study enriches the mutation spectrum of the cblC, providing help for precision diagnosis and genetic counseling.
In case‐006, we identified the only sequence variation, a single heterozygous c.*2C>T substitution, that segregated with the phenotype across the patient and her father in the PRDX1 gene, based on the panel of genes involved in metabolic diseases. This substitution in the 3’UTR of PRDX1 has an extremely low frequency (p < 0.0005) in the normal population database(ESP database、1000 Genomes Project、EXAC database). According to the ACMG guide, this mutation is judged as uncertain significance. Previous research demonstrated that the silencing of PRDX1 gene restored the expression of MMACHC gene in cultured cells, and that it produced 10–15% hypomethylation of the allele initially fully methylated in the clones generated from bisulfite‐treated DNA(Gueant et al., 2018). We found the epimutation and the c.*2C>T PRDX1 gene substitution, and confirmed the paternal transmission of the PRDX1 gene mutation along with the epimutation (Figure 1a). The c.*2C>T substitution is a 3’ UTR variation of the PRDX1 gene and a 500 bp downstream variation of the MMACHC (Figure 4). In case‐006 and her father, MMACHC expression was down‐regulated (Figure 5), so we speculated that the PRDX1 c.*2C>T substitution might silence of MMACHC expression through the methylation of its promoter. However, the exact mechanism needs further study. In case‐007 and his family members, no mutations were identified in the PRDX1 coding region and adjacent splice sites. Maybe PRDX1 gene has non‐coding region variation or large fragment deletion or duplication, which we did not find. Thus, how this epimutation arose remains to be studied.
FIGURE 4.
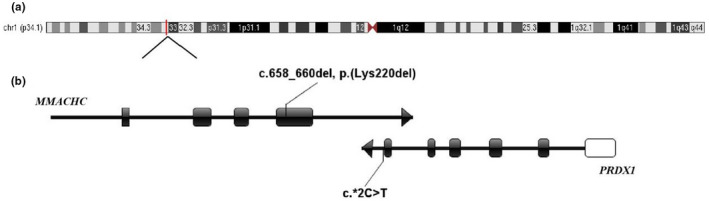
Location of the c.*2C>T variant in PRDX1 gene. (a) The location of MMACHC and PRDX1 gene in chromosome. (b) The location of the variants. Black boxes represent exons, between exon and exon are introns. White boxes represent non‐coding regions. Arrows represent the direction of gene transcription
FIGURE 5.
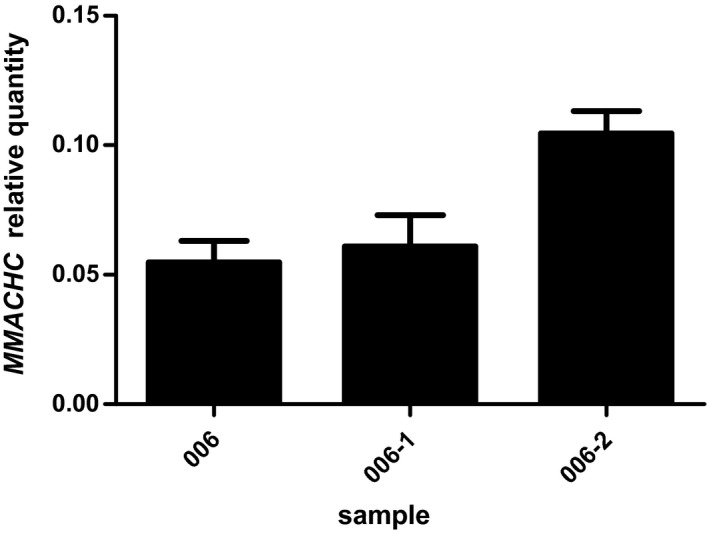
Expression test for the MMACHC gene in case 006. Error bars represent the standard deviations from three parallel experiments
5. CONCLUSIONS
This is the first report of two cblC cases from China caused by compound heterozygous mutations with a coding mutation in one allele and an epimutation in the other at the MMACHC gene locus. Based on the results of this study, it can be suggested to search for MMACHC methylation in cblC patients with typical severe presentation despite only the presence of a single heterozygous genetic mutation. Epimutations enrich the mutation spectrum of the MMACHC gene, providing a broader perspective for clinical diagnosis.
CONFLICT OF INTEREST
Xiaoman Zhang, Qiong Chen, Yinsen Song, Pengbo Guo, Yanhong Wang, Shuying Luo, Yaodong Zhang, Chongchen Zhou, Dongxiao Li, Yongxing Chen and Haiyan Wei declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
ZXM, GPB, WYH, SYS, and ZYD performed experiments and drafted the manuscript. CQ, LDX, CYX, LSY, and ZCC provided clinical evaluations and drafted manuscript. WHY and LDX supervised and drafted manuscript. All authors approved the final manuscript.
ACKNOWLEDGEMENTS
The authors thank the patient with MMA and her family for participating in this study. This research project was supported by Henan Children’s Hospital. The authors also thank all the involved persons for their kind help.
Xiaoman Zhang and Qiong Chen have contributed equally to this work.
Contributor Information
Dongxiao Li, Email: li_dongxiao@sina.com.
Haiyan Wei, Email: weihaiyan2008@yeah.net.
DATA AVAILABILITY STATEMENT
Raw data were not deposited but may be available by contacting the authors.
REFERENCES
- Bird, A. , Taggart, M. , Frommer, M. , Miller, O. J. , & Macleod, D. (1985). A fraction of the mouse genome that is derived from islands of nonmethylated, CpG‐rich DNA. Cell, 40(1), 91–99. 10.1016/0092-8674(85)90312-5 [DOI] [PubMed] [Google Scholar]
- Deaton, A. M. , & Bird, A. (2011). CpG islands and the regulation of transcription. Genes & Development, 25(10), 1010–1022. 10.1101/gad.2037511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demond, H. , & Kelsey, G. (2020). The enigma of DNA methylation in the mammalian oocyte. F1000Research, 9, 146. 10.12688/f1000research.21513.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng, S. , Jacobsen, S. E. , & Reik, W. (2010). Epigenetic reprogramming in plant and animal development. Science, 330(6004), 622–627. 10.1126/science.1190614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guéant, J.‐L. , Chéry, C. , Oussalah, A. , Nadaf, J. , Coelho, D. , Josse, T. , Flayac, J. , Robert, A. , Koscinski, I. , Gastin, I. , Filhine‐Tresarrieu, P. , Pupavac, M. , Brebner, A. , Watkins, D. , Pastinen, T. , Montpetit, A. , Hariri, F. , Tregouët, D. , Raby, B. A. , … Rosenblatt, D. S. (2018). APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nature Communications, 9(1), 67. 10.1038/s41467-017-02306-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, K. , Zhou, X. , Chen, X. , Wu, Y. , Liu, C. , & Kong, Q. (2018). Expanded newborn screening for inborn errors of metabolism and genetic characteristics in a Chinese population. Frontiers in Genetics, 9, 122. 10.3389/fgene.2018.00122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, B. , Cao, Z. , Tian, L. , Zou, H. , Yang, L. , Zhu, W. , & Liu, Y. (2016). Clinical presentation, gene analysis and outcomes in young patients with early‐treated combined methylmalonic acidemia and homocysteinemia (cblC type) in Shandong province, China. Brain and Development, 38(5), 491–497. 10.1016/j.braindev.2015.10.016 [DOI] [PubMed] [Google Scholar]
- Hitchins, M. P. , Rapkins, R. W. , Kwok, C.‐T. , Srivastava, S. , Wong, J. J. L. , Khachigian, L. M. , Polly, P. , Goldblatt, J. , & Ward, R. L. (2011). Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer‐affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell, 20(2), 200–213. 10.1016/j.ccr.2011.07.003 [DOI] [PubMed] [Google Scholar]
- Huemer, M. , Diodato, D. , Schwahn, B. , Schiff, M. , Bandeira, A. , Benoist, J.‐F. , Burlina, A. , Cerone, R. , Couce, M. L. , Garcia‐Cazorla, A. , la Marca, G. , Pasquini, E. , Vilarinho, L. , Weisfeld‐Adams, J. D. , Kožich, V. , Blom, H. , Baumgartner, M. R. , & Dionisi‐Vici, C. (2017). Guidelines for diagnosis and management of the cobalamin‐related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. Journal of Inherited Metabolic Disease, 40(1), 21–48. 10.1007/s10545-016-9991-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P. A. (2012). Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nature Reviews Genetics, 13(7), 484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Lerner‐Ellis, J. P. , Tirone, J. C. , Pawelek, P. D. , Doré, C. , Atkinson, J. L. , Watkins, D. , Morel, C. F. , Fujiwara, T. M. , Moras, E. , Hosack, A. R. , Dunbar, G. V. , Antonicka, H. , Forgetta, V. , Dobson, C. M. , Leclerc, D. , Gravel, R. A. , Shoubridge, E. A. , Coulton, J. W. , Lepage, P. , … Rosenblatt, D. S. (2006). Identification of the gene responsible for methylmalonic aciduria and homocystinuria, cblC type. Nature Genetics, 38(1), 93–100. 10.1038/ng1683 [DOI] [PubMed] [Google Scholar]
- Liu, M.‐Y. , Yang, Y.‐L. , Chang, Y.‐C. , Chiang, S.‐H. , Lin, S.‐P. , Han, L.‐S. , Qi, Y. U. , Hsiao, K.‐J. , & Liu, T.‐T. (2010). Mutation spectrum of MMACHC in Chinese patients with combined methylmalonic aciduria and homocystinuria. Journal of Human Genetics, 55(9), 621–626. 10.1038/jhg.2010.81 [DOI] [PubMed] [Google Scholar]
- Liu, Y. P. , Ma, Y. Y. , Wu, T. F. , Qiao, W. , & Yang, Y. L. (2012). Abnormal findings during newborn period of 160 patients with early‐onset methylmalonic aciduria. Zhonghua Er Ke Za Zhi Chinese Journal of Pediatrics, 50(6), 410–414. [PubMed] [Google Scholar]
- Pensold, D. , Reichard, J. , Van Loo, K. M. J. , Ciganok, N. , Hahn, A. , Bayer, C. , Liebmann, L. , Groß, J. , Tittelmeier, J. , Lingner, T. , Salinas‐Riester, G. , Symmank, J. , Halfmann, C. , González‐Bermúdez, L. , Urbach, A. , Gehrmann, J. , Costa, I. , Pieler, T. , Hübner, C. A. , … Zimmer‐Bensch, G. (2020). DNA methylation‐mediated modulation of endocytosis as potential mechanism for synaptic function regulation in murine inhibitory cortical interneurons. Cerebral Cortex, 30(7), 3921–3937. 10.1093/cercor/bhaa009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxonov, S. , Berg, P. , & Brutlag, D. L. (2006). A genome‐wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proceedings of the National Academy of Sciences of the United States of America, 103(5), 1412–1417. 10.1073/pnas.0510310103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins, D. , & Rosenblatt, D. S. (2011). Inborn errors of cobalamin absorption and metabolism. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 157C(1), 33–44. 10.1002/ajmg.c.30288 [DOI] [PubMed] [Google Scholar]
- Yu, H.‐C. , Sloan, J. L. , Scharer, G. , Brebner, A. , Quintana, A. M. , Achilly, N. P. , Manoli, I. , Coughlin, C. R. , Geiger, E. A. , Schneck, U. , Watkins, D. , Suormala, T. , Van Hove, J. L. K. , Fowler, B. , Baumgartner, M. R. , Rosenblatt, D. S. , Venditti, C. P. , & Shaikh, T. H. (2013). An X‐linked cobalamin disorder caused by mutations in transcriptional coregulator HCFC1. American Journal of Human Genetics, 93(3), 506–514. 10.1016/j.ajhg.2013.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H. , Brockington, M. , Jungbluth, H. , Monk, D. , Stanier, P. , Sewry, C. A. , Moore, G. E. , & Muntoni, F. (2006). Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. American Journal of Human Genetics, 79(5), 859–868. 10.1086/508500 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw data were not deposited but may be available by contacting the authors.