

Abstract
Background
Early‐onset osteoporosis (EOOP) is defined by low bone mineral density (BMD), which increases the risk of fracture. Although the prevalence of osteoporosis at a young age is unknown, low BMD is highly linked to genetic background. Heterozygous pathogenic variants in low‐density lipoprotein receptor‐related protein 5 (LRP5) are associated with EOOP. This study aimed to investigate the genetic profile in patients with EOOP to better understand the variation in phenotype severity by using a targeted gene sequencing panel associated with bone fragility.
Method and Results
We used a sequencing panel with 17 genes reported to be related to bone fragility for analysis of 68 patients with EOOP. We found a high positivity rate of EOOP with LRP5 variants (14 patients, 20.6%). The remaining 79.4% of patients with EOOP but without LRP5 variants showed variable disease severity, as observed in patients with at least one variant in this gene. One patient, with multiple fractures and spine L1‐L4 BMD Z‐score −2.9, carried a novel pathogenic homozygous variant, c.2918T>C, p.(Leu973Pro), without any pseudoglioma. In addition to carrying the LRP5 variant, 2 other patients carried a heterozygous variant in Wnt signaling pathway genes: dickkopf WNT signaling pathway inhibitor 1 (DKK1) [NM_012242.4: c.359G>T, p.(Arg120Leu)] and Wnt family member 3A (WNT3A) [NM_033131.3: c.377G>A, p. (Arg126His)]. As compared with single‐variant LRP5 carriers, double‐variant carriers had a significantly lower BMD Z‐score (−4.1 ± 0.8) and higher mean number of fractures (6.0 ± 2.8 vs. 2.2 ± 1.9). Analysis of the family segregation suggests the inheritance of BMD trait.
Conclusion
Severe forms of EOOP may occur with carriage of 2 pathogenic variants in genes encoding regulators of the Wnt signaling pathway. Two‐variant carriers of Wnt pathway genes had severe EOOP. Moreover, DKK1 and WNT3A genes should be included in next‐generation sequence analyses of bone fragility.
Keywords: DKK1, LRP5, osteoporosis, Wnt signaling pathway, WNT3A
Short abstract
Gene association may occur in the same signaling pathway and can generate a severe bone phenotype in early‐onset osteoporosis. Recessive form associated with lipoprotein receptor‐related protein 5 could be responsible for a stronger phenotype. Interestingly this recessive form is not associated with ocular problems as observed in pseudoglioma osteoporosis or vitreoretinopathy. Assessment of genetics based on an next generation sequencing panel should include WNT3A and DKK1.
1. INTRODUCTION
Early‐onset osteoporosis (EOOP) is a rare form of primary osteoporosis occurring at a young age and is based on low bone mineral density (BMD) and impaired bone structure, which leads to increased risk of fracture and the exclusion of secondary causes of osteoporosis (Kauffman et al., 2001). EOOP is characterized by skeletal fractures often associated with low BMD. Bone mass accrual is determined by genetic factors, which explains 60% to 80% of the variability (Boudin & Van Hul, 2017). Analysis of monogenic bone diseases allowed for identifying several genes, thus shedding light on major pathways in bone metabolism and in human diseases. Studies have reported a pathogenic heterozygous variant in low‐density lipoprotein receptor‐related protein 5 (LRP5) OMIM 603506 (Collet et al., 2017; Hartikka et al., 2005), proto‐oncogene Wnt‐1 (WNT1) OMIM 164820, and plastin 3 (PLS3) in X‐linked osteoporosis, OMIM 300131 (Kämpe et al., 2017; Laine et al., 2013; van Dijk et al., 2013).
LRP5 encodes for a co‐receptor of the Wnt signaling pathway involved in BMD regulation (Gong et al., 2001); its activation promotes bone formation. Loss‐of‐function mutations in LRP5 result in failure to transmit signals downstream of the WNT canonical pathway. Consequently, the inhibition of Wnt signaling impairs proper bone acquisition, thus causing low BMD and osteoporosis at a young age (Cui et al., 2011; Gong et al., 2001). Reported cases associated with LRP5 variants were described as exhibiting dominant inheritance; however, the variable severity of osteoporotic phenotypes remains elusive.
Two other candidate genes, dickkopf WNT signaling pathway inhibitor 1 (DKK1) OMIM 605189 and Wnt family member 3A (WNT3A) OMIM 606359, were proposed to be associated with EOOP (Korvala, Löija, et al., 2012). Indeed, these 2 genes are considered key factors that maintain normal BMD levels and prevent fractures (Boland et al., 2004; Pinzone et al., 2009).
To better understand the variations in phenotype severity, we explored the molecular etiology in EOOP by using a sequencing panel related to bone fragility. We found that variants in 2 different genes involved in Wnt signaling might lead to lower BMD and/or more fractures. Also, we highlighted that homozygous variants in LRP5 could lead to severe osteoporosis, similar to that observed with 2 heterozygous variants in 2 different genes. Thus, homozygous variants or the combination of heterozygous variants could result in a more severe osteoporotic phenotype as compared with a single LRP5 variant.
2. METHODS AND PATIENTS
Our cohort included 68 patients referred to the bone rare diseases reference center at Lariboisière hospital (Paris, France) for evaluation of primary osteoporosis. Clinical examination showed no dysmorphia and no clinical abnormalities in eyes, ears or vessels. By imaging and biochemical analysis, we excluded any secondary causes of osteoporosis, including malignant disease, Paget disease, malabsorption, hypogonadism, hemochromatosis, hyperthyroidism, hypercortisolism, vitamin D deficiency or mastocytosis. We also excluded patients taking medications that interfere with bone metabolism, particularly bisphosphonates, denosumab or teriparatide. We analyzed bone biomarkers and BMD at the lumbar spine and the total hip by using a Lunar device and expressed as Z‐scores. Most of the patients had low BMD Z‐score < −2 SD at the spine or hip or a history of fragility fractures without any associated diseases. Spinal radiography was used to assess vertebral fractures. We excluded patients with a diagnosis or clinical signs of osteogenesis imperfecta.
2.1. Genetic analysis
This study, including the molecular analysis, was approved by a French ethics committee from Lariboisière hospital (Paris, France) and was performed after written informed consent from patients for genetic testing. DNA was extracted from whole blood samples by using the Qiasymphony instrument (Qiagen, Venlo, The Netherlands), according to the manufacturer's protocol. DNA was screened by targeted next generation sequencing (NGS) with a panel of 17 genes associated with bone fragility (BMP1 (NG_029659.1), COL1A1 (NG_007400.1), COL1A2 (NG_007405.1), CRTAP (NG_008122.1), CREB3L1 (NG_033264.1), DKK1(NC_000010.11), FKBP10 (NG_015860.1), IFITM5 (NG_032892.1), LRP5 (NG_015835.2), LRP6 (NG_016168.2), PLS3 (NG_012518.2), P3H1 (NG_008123.1), SERPINF1 (NG_028180.1), SP7 (NG_023391.2), WNT1 (NG_033141.1), WNT3A (NC_000001.11), and WNT16 (NG_029242.1)). The NGS analysis involved using the surelectQXT kit (Agilent, Les Ulis, France) for library preparation and the hybrid capture system for sequencing on a Miseq sequencer (Illumina, Paris, France). Sequencing results were obtained after aligning fastqs, mapping and variant calling by using the SeqNext software (JSI Medical Systems, Ettenheim, Germany). The SeqNext software is based on Smith‐Waterman (Shpaer et al., 1996) and BWA (Burrows Wheeler Aligner) algorithms (Li & Durbin, 2012). Copy number variations were also determined by using this software. For each exon, the coverage was 100% at 30×. The highest minor allele frequency of variants was investigated by using 1000 Genomes phase 3 (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/data) and gnomAD (https://gnomad.broadinstitute.org/); the filtering criteria was minor allele frequency <0.05% in one of the databases, corresponding to the definition of a rare disease in the European Union. (Orphanet: an online database of rare diseases and orphan drugs. Copyright, INSERM 1997. Available at http://www.orpha.net Accessed (March 16, 2021)).
Sanger sequencing by using Life Technologies reagents and software on an ABI3130 sequencer (ThermoFischer, Les Ulis, France) confirmed the potentially pathogenic variants identified in the panel. Variant pathogenicity was evaluated by using Alamut (SOPHiA Genetics, Lausanne, Switzerland) including variable in silico predictive software (SIFT, MutationTaster and Poly‐Phen 2) and Combined Annotation Dependent Depletion (https://cadd.gs.washington.edu).
3. RESULTS
3.1. Characteristics of patients
Our cohort of 68 patients presented EOOP with low BMD or a history of fractures, with variable severity among patients. We excluded 3 patients because they carried different variants in genes responsible for osteogenesis imperfecta (COL1A1, COL1A2) and also one patient carried a variant in WNT1 (NM_005430.3:c.999_1021del, p. Thr336Alafs*125). Although this gene has been reported in EOOP (Laine et al., 2013), the patient presented a similar phenotype: one fracture at age 43 years and low BMD (Z‐score −1.7). We focused on only patients carrying at least one variant in LRP5.
Of the remaining 65 patients, 14 carried a variant in LRP5 of class 4 (95–99% likelihood of pathogenicity) or class 5 (probability of being pathogenic >99%) according to sequence variant classification from Plon et al. (2008) (Tables 1 and 2). The positivity rate was 20.5% (14/68) for patients with variants in LRP5 or 21.5% (14/65) after excluding the 3 patients with COL1A1, COL1A2 and WNT1 variants. All patients with one or two variants in LRP5 had low BMD at the spine (mean Z‐score −2.6 ± 1.1). The mean age at diagnosis was 43.8 ± 12.9 years, but the first osteoporotic fracture occurred at a mean age of 27.2 ± 18.1 years (Table 1). Among the 14 patients with a mutation in LRP5, 12 experienced at least one fracture, with a high variable number of fractures per patient. The most common fracture site was the spine, followed by the wrist and ribs.
TABLE 1.
Clinical and genetic aspects of patients with idiopathic osteoporosis
| n° | Sex | Age at diagnosis | BMD | No. of fractures | Types of fractures | Age at first fracture | Pathogenic variants LRP5 HTZ | Other genes HTZ | Highest population MAF |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 17 | Spine L1‐L4 Z‐score ‐4.7 | 8 | Spine | 10 | c.1759G>A, p.(Asp587Asn) | DKK1 c.359G>T, p. Arg120Leu | LRP5 <0.01/DKK1 0.02 |
| Total hip Z‐score −3.4 | |||||||||
| 2 | F | 51 | Spine L1‐L4 Z‐score −3.6 | 4 | Ischial/ribs | 8 | c.4616C>T, p.(Pro1539Leu) | WNT3A c.377G>A, p. Arg126His | LRP5 <0.01/WNT3A <0.01 |
| Total hip Z‐score −2.1 | |||||||||
| 3 | M | – | Spine L1‐L4 Z‐score −2.9 | 5 | Wrist/pelvis/acetabulum/femur/spine | 13 | c.2918T>C, p. Leu973Pro HMZ | – | <0.01 |
| Total hip Z‐score −3.7 | |||||||||
| 4 | F | 46 | Spine L1‐L4 Z‐score −3.7 | 8 | Spine | 46 | c.3107G>A, p. Arg1036Gln | – | 0.01/splicing prediction high |
| Total hip Zscore −1.5 | c.2409_2503+79del | ||||||||
| 5 | M | 53 | Spine L1‐L4 Z‐score −3.0 | 6 | Wrist/clavicle/humerus/spine | 10 | c.1418T>C, p. Met473Thr c.1999G>A, p.(Val667Met) | – | <0.01/0.09 |
| Total hip Z‐score −1.2 | |||||||||
| 6 | M | 57 | Spine L1‐L4 Z‐score −1.1 | 2 | Ribs/spine | 38 | c.3107G>A, p. Arg1036Gln c.1999G>A, p.(Val667Met) | – | 0.01/0.09 |
| Total hip Z‐score −0.6 | |||||||||
| 7 | M | 60 | Spine L1‐L4 Z‐score −2.2 | 1 | External condyle of the knee | 58 | c.4252del.p. Ala1418Profs*21 | – | <0.01/0.09 |
| Total hip Z‐score −1.2 | c.1999G>A, p.(Val667Met) | ||||||||
| 8 | M | 37 | Spine L1‐L4 Z‐score −2.9 | 3 | Spine | 35 | c.(533G>A; 1057C>T), p.(Arg178Gln,. Arg353Trp) | – | <0.01/<0.01 |
| Total hip Z‐score −1.6 | |||||||||
| 9 | F | 47 | Spine L1‐L4 Z‐score −2.3 | 0 | – | – | c.3883T>C, p. Cys1295Arg | – | <0.01 |
| Total hip Z‐score −1.4 | |||||||||
| 10 | F | 52 | Spine L1‐L4 Z‐score −0.4 | 4 | Spine/ribs/pelvis | 47 | c.3050G>A, p. Ser1017Asn | – | <0.01 |
| Total hip Z‐score −1.1 | |||||||||
| 11 | F | 38 | Spine L1‐L4 Z‐score −3.3 | 3 | Spine | 37 | c.3107G>A, p. Arg1036Gln | – | 0.01 |
| Total hip Z‐score −1.3 | |||||||||
| 12 | F | 36 | Spine L2‐L4 Z‐score −3.0 | 0 | – | – | c.408C>A, p. Asn136Lys | – | <0.01 |
| Total hip Z‐score −2.4 | |||||||||
| 13 | M | 23 | Spine L1‐L4 Z‐score −2.6 | 1 | Wrist | 10 | c.2362C>T, p. Arg788Trp | – | 0.02 |
| 14 | M | 52 | Spine L1‐L4 Z‐score −2.1 | 2 | Wrist/spine | 14 | c.3863A>G, p. Asp1288Gly | – | <0.01 |
| Total hip Z‐score −1.2 |
MAF disposition: mean allele frequency first variant/second variant, when 2 variants are present. LRP5 (NG_015835.2), DKK1(NC_000010.11), WNT3A (NC_000001.11).
Abbreviations: F, female; HMZ, homozygous; HTZ, heterozygous; M, male.
TABLE 2.
In silico prediction of variants in patients with idiopathic osteoporosis
| n° | Pathogenic variants LRP5 HTZ | CADD | SIFT | PolyPhen‐2 HumDiv | MutationTaster |
|---|---|---|---|---|---|
| 1 | c.1759G>A, p.(Asp587Asn) | PHRED:23.4 | Deleterious (score: 0) | Possibly damaging, score 0.844 (sensitivity: 0.83; specificity: 0.93) | Disease causing (prob: 1) |
| 2 | c.4616C>T, p.(Pro1539Leu) | PHRED:24.8 | Deleterious (score: 0) | Possibly damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| rs148725079 | |||||
| 3 | c.2918T>C, p.Leu973Pro HMZ | PHRED:27.2 | Deleterious (score: 0.01) | Possibly damaging, score 0.913 (sensitivity: 0.81; specificity: 0.94) | Disease causing (prob: 1) |
| 4 | c.3107G>A, p.Arg1036Gln | PHRED:24.1 | Deleterious (score: 0.04) | Possibly damaging, score 0.658 (sensitivity: 0.86; specificity: 0.91) | Disease causing (prob: 0.984) |
| c.2409_2503+79del p.(Gly804Serfs*34) | |||||
| 5 | c.1418T>C, p.Met473Thr rs1023949893 + PM | PHRED:25.1 | Deleterious (score: 0) | Possibly damaging, score 0.935 (sensitivity: 0.80; specificity: 0.94) | Disease causing (prob: 1) |
| 6 | c.3107G>A, p.Arg1036Gln rs61889560 + PM | PHRED:24.1 | Deleterious (score: 0.04) | Possibly damaging, score 0.658 (sensitivity: 0.86; specificity: 0.91) | Disease causing (prob: 0.984) |
| 7 | c.4252del.(p.Ala1418Profs*21) + PM | ||||
| 8 | c.(533G>A; 1057C>T), p.(Arg178Gln,. Arg353Trp) rs371514699 | PHRED:27.5 | Tolerated (score: 0.09) | Probably damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| PHRED:27.2 | Deleterious (score: 0.01) | Probably damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) | ||
| 9 | c.3883T>C, p.Cys1295Arg | PHRED:28.5 | Deleterious (score: 0) | Probably damaging, score 0.999 (sensitivity: 0.14; specificity: 0.99) | Disease causing (prob: 1) |
| 10 | c.523C>T, p.Arg175Trp | PHRED:26.6 | Deleterious (score: 0) | Probably damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| 11 | c.3107G>A, p.Arg1036Gln | PHRED:24.1 | Deleterious (score: 0.04) | Possibly damaging, score 0.658 (sensitivity: 0.86; specificity: 0.91) | Disease causing (prob: 0.984) |
| rs61889560 | |||||
| 12 | c.408C>A, p.Asn136Lys | PHRED:20.4 | Deleterious (score: 0.04) | Possibly damaging, score 0.864 (sensitivity: 0.83; specificity: 0.93) | Disease causing (prob: 1) |
| 13 | c.2362C>T, p.Arg788Trp | PHRED:28.4 | Deleterious (score: 0) | Probably damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| rs1000296899 | |||||
| 14 | c.3863A>G, p. Asp1288Gly | PHRED:28.1 | Deleterious (score: 0) | Probably damaging, score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| rs762014835 |
| n° | Other genes HTZ | CADD | SIFT | PolyPhen‐2 | MutationTaster |
|---|---|---|---|---|---|
| 1 | DKK1 c.359G>T, p.Arg120Leu | PHRED: 32 | Deleterious (score: 0) | Probably damaging with a score 1.000 (sensitivity: 0.00; specificity: 1.00) | Disease causing (prob: 1) |
| 2 | WNT3A c.377G>A, p.Arg126His | PHRED: 23.3 | Deleterious (score: 0.02) | Benign with a score of 0.148 (sensitivity: 0.92; specificity: 0.86) | Disease causing (prob: 1) |
LRP5*c.1999G>A, p.(Val667Met) at heterozygous level rs4988321. LRP5 (NG_015835.2), DKK1(NC_000010.11), WNT3A (NC_000001.11).
Abbreviations: HMZ, homozygosis; HTZ, heterozygosis; PM, polymorphism.
3.2. Gene association in the WNT signaling pathway
Among the 14 patients carrying LRP5 variants, 7 presented 1 variant, 5 had 2 variants and 2 had a combination of LRP5 variant with a variant of another gene. Patients 1 and 2 carried a class 4 heterozygous variant in LRP5, associated with a class 4 heterozygous variant in DKK1 [p.(Arg120Leu)] and WNT3A [p.(Arg126His)], respectively (Tables 1 and 2). Patient 2 had no history of osteoporosis in her family; we only had information on only her brother. The brother's molecular analysis confirmed the c.4616C>T, p.(Pro1539Leu) LRP5 heterozygous variant, classified as class 4 (probably pathogenic). The presence of the variant was associated with low BMD: the brother's Z‐score at the spine was −2.1, but he did not have the WNT3A variant. Both patients 1 and 2 were severely affected by osteoporosis, without vision disturbance from childhood, as was patient 3, with a novel homozygous variant in LRP5, p.[(Leu973Pro)];[(Leu973Pro)].
Patient 3, with low BMD at the lumbar spine (Z‐score −2.9) and total hip (Z‐score −3.7) and 5 atypical fractures (wrist, pelvis, acetabulum, femur and spine), has a daughter with the same variant at a heterozygous level. The daughter presented a low BMD at the lumbar spine (Z‐score −2.9) and total hip (Z‐score −2.4). Patients with only one heterozygous variant in LRP5 had similar values. Patient 4 presented 2 different variants in LRP5, c.[3107G>A]; [2409_2503+79del] in different alleles. The splicing donor in c.2503 is abolished in the presence of the variant c.2409_2503+79del according to MaxEnt and NNSPLICE values: with a severe phenotype, maximal median scores are 12 (range 0–12) and 1 (range 0–1), respectively. Patients 5, 6, and 7 carried a class 4 variant in LRP5 associated with the LRP5 variant p.(Val667Met), considered a risk factor. Patient 8 presented 2 different variants in the same allele in LRP5, and patient 9 had a novel heterozygous variant in LRP5, c.3883T>C, p.(Cys1295Arg). The variants of patients 10 to 14 were previously reported and can be found in The Human Gene Mutation Database (HGMD®).
We then analyzed the bone phenotype according to 3 variant groups: patients with monogenic heterozygous variants in LRP5, patients with monogenic homozygous variants or the heterozygous compound in LRP5 and either combination of LRP5 variant associated with another gene from the Wnt pathway. Carrying 2 LRP5 variants was associated with a lower spine or hip Z‐score and higher number of fractures (Table 3). In addition, patients with 2 gene variants were younger at diagnosis and a lower BMD and higher number of fractures than those with 2 LRP5 variants.
TABLE 3.
Patients with monogenic, monogenic homozygous/heterozygous compound and gene association profiles in EOOP
| Monogenic heterozygous variants in LRP5 (patients 5 to 14) | Monogenic homozygous variants/heterozygous compound variants (patients 3 and 4) | Gene association (patients 1 and 2) | |
|---|---|---|---|
| Age at diagnosis, years | 45.5 ± 11.5 | 46.0 ± 0.0 | 34.0 ± 24.0 |
| Age at first fracture, years | 31.1 ± 17.9 | 29.5 ± 23.3 | 9.0 ± 1.4 |
| Spine L1‐L4 BMD Z‐score | −2.2 ± 0.8 | −3.3 ± 0.6 | −4.1 ± 0.8 |
| Total hip BMD Z‐score | −1.1 ± 1.0 | −2.6 ± 1.5 | −2.7 ± 0.9 |
| Number of fractures | 2.2 ± 1.9 | 6.5 ± 2.1 | 6.0 ± 2.8 |
Data are mean ± SD.
Abbreviation: BMD, bone mineral density.
3.3. Family segregation
To better understand the pathogenic effect of the gene association, we analyzed the family members of 2 EOOP patients (Figure 1). The BMD Z‐score for patient 1 was −4.7, much lower than that for her father and brother (Figure 1a, Tables 1 and 2). She carried one variant in LRP5 [p.(Asp587Asn)] and one variant in DKK1 [p.(Arg120Leu)]. Her father had a heterozygous variant in LRP5, with Z‐score −1.9, and did not experience any fractures. Her 45‐year‐old brother had low spine BMD (Z‐score −2.6), no fractures and no variant from our panel.
FIGURE 1.
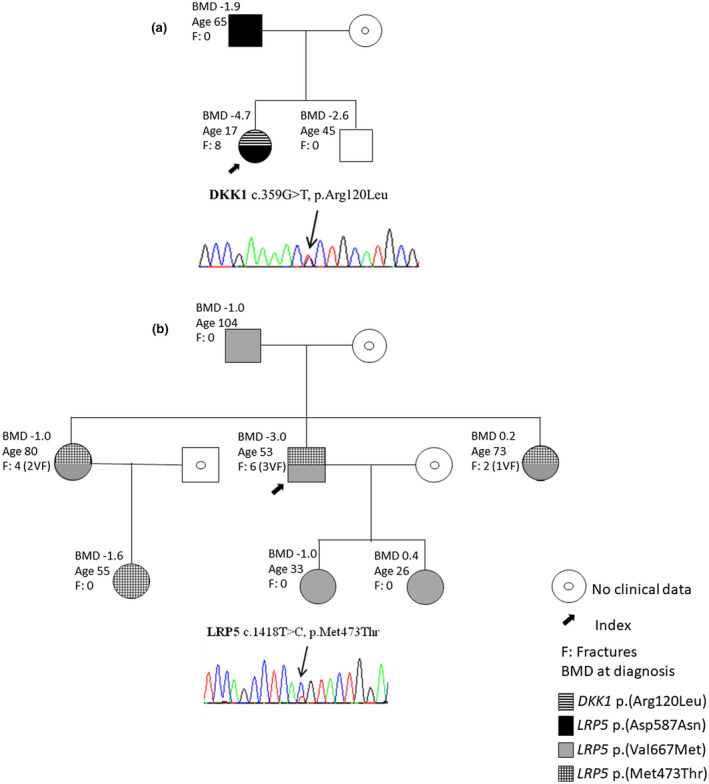
Family segregation and bone mineral density (BMD) Z‐scores. (a) Pedigree of patient 1, with a chromatogram showing the DKK1 missense variant c.359G > T, p.(Arg120Leu). Patient 1 presented a stronger phenotype than her father, with only one variant in LRP5, and as compared with a previous description of the same variant in DKK1 (Korvala, Löija, et al. 2012). (b) Pedigree of patient 5. He carried 2 variants in LRP5: p.(Val667Met) and c.1418T>C, p.Met473Thr. The chromatogram shows the LRP5 p.Met473Thr variant. BMD Z‐scores vary among family members. To confirm the pathogenicity effect of the variants, functional studies are required. Age and vertebral fractures (VFs) are displayed in the figure; VFs erroneously increase the BMD
Patient 5 presented one p.(Val667Met) variant and one variant, p.(Met473Thr), in LRP5. His Z‐score was low (−3.0), and he experienced multiple fractures. The 2 sisters had both variants as well and had multiple fractures of the vertebrae, humerus and wrist. However, their spine Z‐scores were −1.0 and 0.2 (Figure 1b), related to the lumbar vertebral fractures; both had more than 10 years’ treatment for osteoporosis before the BMD was measured.
4. DISCUSSION
Our study highlights the major role of LRP5 and the possible association with a synergic inactivation of the Wnt pathway in EOOP. Pathogenic variants in LRP5 as well as in DKK1 and WNT3A have been reported separately in EOOP (Korvala, Jüppner, et al., 2012; Korvala, Löija, et al., 2012). Here, we report an association of pathogenic variants in genes that could account for osteoporosis severity. In this cohort, we found 2 young patients whose first fractures occurred between age 8 and 10 years; they had a class 4 (probably pathogenic) heterozygous variant in LRP5 and also carried another variant in a gene coding for proteins in the Wnt signaling pathway. Patient 1 presented a class 4 (probably pathogenic) variant in LRP5 and in DKK1. DKK1 is a gene‐coding protein that downregulates the Wnt signaling pathway. The p.(Arg120Leu) DKK1 variant was previously described by Korvala, Löija, et al. (2012) in a family with a less severe phenotype than our patient 1. Korvala, Löija, et al. (2012) described 2 siblings who carried the p.(Arg120Leu) variant in DKK1 and presented a similar bone phenotype as patients with a heterozygous pathogenic variant in LRP5. The DKK1 p.(Arg120Leu) variant in addition to an LRP5 variant, p.(Asp587Asn), could have led to more severe osteoporosis, as illustrated by a high number of fractures and low BMD as compared with patients with a single variant. This variant might be a gain‐of‐function variant because DKK1 inhibits binding to the LRP5 co‐receptor, which results in inhibition of β‐catenin–dependent Wnt signaling.
Patient 2 had a severe bone phenotype, as shown by a very low BMD. She carried 2 variants, one in LRP5, p.(Pro1539Leu), and one novel variant in WNT3A, p. Arg126His. Her brother only carried the LRP5 variant, and his mild phenotype (low BMD without fractures) confirmed our findings that the gene association might be responsible for the severity of the phenotype of patient 2. The WNT3A novel variant—the protein encoding for a ligand activator of the WNT signaling pathway (Działo et al., 2019). Indeed, patients carrying 2 variants in 2 different genes had lower BMD and fractures occurring in childhood, which indicate a more severe osteoporosis than patients with a single LRP5 variant. Of note, for these patients, the bone phenotype (mean BMD Z‐score −4.1 ± 0.8, with number of fractures 6.0 ± 2.8) was similar to patients 4 and 5 (mean BMD Z‐score −3.3 ± 0.6, with number of fractures 6.5 ± 2.1), who carried a homozygous variant or 2 LRP5 variants, respectively.
Recently, analysis of a large EOOP cohort revealed relevant LRP5 and LRP6 variants that contribute to severe EOOP characterized by a high number of fractures and reduced BMD (Stürznickel et al., 2020). Confirming our previous findings, the authors also mentioned a large heterogeneity in severity of osteoporosis. Therefore, the variability of the phenotype could be explained by the presence of 2 or more variants in the same gene or different genes but in the same pathway, even if one of those variants is considered a polymorphism.
Patient 5 had multiple osteoporotic fractures and two variants in different alleles p.[Val667Met],[Met473Thr]. His 2 sisters also carried both variants; they had vertebral fractures despite higher BMD Z‐score than for patient 5. This finding could be explained by the presence of scoliosis, which results in falsely elevated BMD values (Tenne et al., 2013). The niece of patient 5 carried only the variant p.(Met473 Thr) at a heterozygous level, with a BMD Z‐score of −1.6, which suggests that this variant could have pathogenicity. Normal and low BMD values were observed in the two daughters of patient 5, in the presence of a unique p. Val667Met variant at a heterozygous level (Figure 1b). This variant could be considered a polymorphism, although it has been described as associated with low BMD (Ferrari et al., 2005; van Meurs et al., 2008).
Carriage of a homozygous variant leads to low BMD (Collet et al., 2017; Stürznickel et al., 2020). The presence of the recessive LRP5 form is responsible for osteoporosis pseudogliomia syndrome or vitreoretinopathy (Ai et al., 2005). Our study showed that the recessive form of osteoporosis can occur without any blindness or any effect on both the retina and vitreous body. In this case or in gene association cases including LRP5 and DKK1 or LRP5 and WNT3A, the clinical and radiological diagnosis is close to the osteogenesis imperfecta moderate form. Digenic profiles related to the Wnt signaling pathway have been described (He et al., 2013; Waschk et al., 2016) but never related to bone fragility. When combined, variants in different genes can lead to a more complex phenotype, and overlapping disease phenotypes are likely to occur with variants of 2 genes encoding proteins from the common pathway (Posey et al., 2017).
Our study has limitations, such as a small cohort of 68 with EOOP. In addition, 79.4% of patients did not have any pathogenic variants explained by the targeted NGS, which suggests that some other genes or environmental factors could be involved. Whole‐genome sequencing or epigenetic approaches would be necessary to confirm the cause of EOOP. However, the positivity rate for LRP5 remained high (20.6%) in our cohort. Our study showed that the recessive disease form associated with LRP5 could be responsible for the severe osteoporotic phenotype and that gene association may occur in the same signaling pathway and can generate a severe bone phenotype in EOOP revealed in young adults. Consequently, assessment of genetics based on an NGS panel should include WNT3A and DKK1.
ETHICAL STATEMENT
This study was approved by a French ethics committee from Lariboisière hospital (Paris, France).
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest in relation to the work.
AUTHOR CONTRIBUTIONS
CCS, MR, CC, and MCS gathered clinical information. CCS, CC, and MCS performed the literature review and drafted the manuscript. MR performed molecular genetic analysis. CC supervised the molecular genetic analysis. PO, SF, TFB, and MCS followed the patients and their family members. The manuscript was approved by all authors.
ACKNOWLEDGMENTS
We thank the patients for giving their consent to take part in our study. We acknowledge Dr. Agnes Ostertag for help in finding the Z‐score for some patients and Laura Smales (BioMedEditing, Toronto, Canada) for editing the manuscript.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Ai, M. , Heeger, S. , Bartels, C. F. , & Schelling, D. K. , & Osteoporosis‐Pseudoglioma Collaborative Group . (2005). Clinical and molecular findings in osteoporosis‐pseudoglioma syndrome. American Journal of Human Genetics, 77(5), 741–753. 10.1086/497706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland, G. M. , Perkins, G. , Hall, D. J. , & Tuan, R. S. (2004). Wnt 3a promotes proliferation and suppresses osteogenic differentiation of adult human mesenchymal stem cells. Journal of Cellular Biochemistry, 93(6), 1210–1230. 10.1002/jcb.20284 [DOI] [PubMed] [Google Scholar]
- Boudin, E. , & Van Hul, W. (2017). Mechanisms in endocrinology: Genetics of human bone formation. European Journal of Endocrinology, 177(2), R69–R83. 10.1530/EJE-16-0990 [DOI] [PubMed] [Google Scholar]
- Collet, C. , Ostertag, A. , Ricquebourg, M. , Delecourt, M. , Tueur, G. , Isidor, B. , Guillot, P. , Schaefer, E. , Javier, R. M. , Funck‐Brentano, T. , Orcel, P. , Laplanche, J. L. , & Cohen‐Solal, M. (2017). Primary osteoporosis in young adults: Genetic basis and identification of novel variants in causal genes. JBMR Plus, 2(1), 12–21. 10.1002/jbm4.10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui, Y. , Niziolek, P. J. , MacDonald, B. T. , Zylstra, C. R. , Alenina, N. , Robinson, D. R. , Zhong, Z. , Matthes, S. , Jacobsen, C. M. , Conlon, R. A. , Brommage, R. , Liu, Q. , Mseeh, F. , Powell, D. R. , Yang, Q. M. , Zambrowicz, B. , Gerrits, H. , Gossen, J. A. , He, X. I. , … Robling, A. G. (2011). Lrp5 functions in bone to regulate bone mass. Nature Medicine, 17(6), 684–691. 10.1038/nm.2388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Działo, E. , Rudnik, M. , Koning, R. I. , Czepiel, M. , Tkacz, K. , Baj‐Krzyworzeka, M. , Distler, O. , Siedlar, M. , Kania, G. , & Błyszczuk, P. (2019). WNT3a and WNT5a transported by exosomes activate WNT signaling pathways in human cardiac fibroblasts. International Journal of Molecular Sciences, 20(6), 1436. 10.3390/ijms20061436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, S. L. , Deutsch, S. , Baudoin, C. , Cohen‐Solal, M. , Ostertag, A. , Antonarakis, S. E. , Rizzoli, R. , & de Vernejoul, M. C. (2005). LRP5 gene polymorphisms and idiopathic osteoporosis in men. Bone, 37(6), 770–775. 10.1016/j.bone.2005.06.017 [DOI] [PubMed] [Google Scholar]
- Gong, Y. , Slee, R. B. , Fukai, N. , Rawadi, G. , Roman‐Roman, S. , Reginato, A. M. , Wang, H. , Cundy, T. , Glorieux, F. H. , Lev, D. , Zacharin, M. , Oexle, K. , Marcelino, J. , Suwairi, W. , Heeger, S. , Sabatakos, G. , Apte, S. , Adkins, W. N. , Allgrove, J. , … Warman, M. L. (2001). LDL receptor‐related protein 5 (LRP5) affects bone accrual and eye development. Cell, 107(4), 513–523. 10.1016/s0092-8674(01)00571-2 [DOI] [PubMed] [Google Scholar]
- Hartikka, H. , Mäkitie, O. , Männikkö, M. , Doria, A. S. , Daneman, A. , Cole, W. G. , Ala‐Kokko, L. , & Sochett, E. B. (2005). Heterozygous mutations in the LDL receptor‐related protein 5 (LRP5) gene are associated with primary osteoporosis in children. Journal of Bone and Mineral Research, 20(5), 783–789. 10.1359/JBMR.050101 [DOI] [PubMed] [Google Scholar]
- He, H. , Han, D. , Feng, H. , Qu, H. , Song, S. , Bai, B. , & Zhang, Z. (2013). Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS ONE, 8(11), e80393. 10.1371/journal.pone.0080393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämpe, A. J. , Costantini, A. , Mäkitie, R. E. , Jäntti, N. , Valta, H. , Mäyränpää, M. , Kröger, H. , Pekkinen, M. , Taylan, F. , Jiao, H. , & Mäkitie, O. (2017). PLS3 sequencing in childhood‐onset primary osteoporosis identifies two novel disease‐causing variants. Osteoporosis International, 28(10), 3023–3032. 10.1007/s00198-017-4150-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman, R. P. , Overton, T. H. , Shiflett, M. , & Jennings, J. C. (2001). Osteoporosis in children and adolescent girls: Case report of idiopathic juvenile osteoporosis and review of the literature. Obstetrical & Gynecological Survey, 56(8), 492–504. 10.1097/00006254-200108000-00023 [DOI] [PubMed] [Google Scholar]
- Korvala, J. , Jüppner, H. , Mäkitie, O. , Sochett, E. , Schnabel, D. , Mora, S. , Bartels, C. F. , Warman, M. L. , Deraska, D. , Cole, W. G. , Hartikka, H. , Ala‐Kokko, L. , & Männikkö, M. (2012). Mutations in LRP5 cause primary osteoporosis without features of OI by reducing Wnt signaling activity. BMC Medical Genetics, 13, 26. 10.1186/1471-2350-13-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korvala, J. , Löija, M. , Mäkitie, O. , Sochett, E. , Jüppner, H. , Schnabel, D. , Mora, S. , Cole, W. G. , Ala‐Kokko, L. , & Männikkö, M. (2012). Rare variations in WNT3A and DKK1 may predispose carriers to primary osteoporosis. European Journal of Medical Genetics, 55(10), 515–519. 10.1016/j.ejmg.2012.06.011 [DOI] [PubMed] [Google Scholar]
- Laine, C. M. , Joeng, K. S. , Campeau, P. M. , Kiviranta, R. , Tarkkonen, K. , Grover, M. , Lu, J. T. , Pekkinen, M. , Wessman, M. , Heino, T. J. , Nieminen‐Pihala, V. , Aronen, M. , Laine, T. , Kröger, H. , Cole, W. G. , Lehesjoki, A. E. , Nevarez, L. , Krakow, D. , Curry, C. J. , Cohn, D. H. , … Mäkitie, O. (2013). WNT1 mutations in early‐onset osteoporosis and osteogenesis imperfecta. The New England Journal of Medicine, 368(19), 1809–1816. 10.1056/NEJMoa1215458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzone, J. J. , Hall, B. M. , Thudi, N. K. , Vonau, M. , Qiang, Y. W. , Rosol, T. J. , & Shaughnessy, J. D., Jr. (2009). The role of Dickkopf‐1 in bone development, homeostasis, and disease. Blood, 113(3), 517–525. 10.1182/blood-2008-03-145169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plon, S. E. , Eccles, D. M. , Easton, D. , Foulkes, W. D. , Genuardi, M. , Greenblatt, M. S. , Hogervorst, F. B. , Hoogerbrugge, N. , Spurdle, A. B. , Tavtigian, S. V. , & IARC Unclassified Genetic Variants Working Group . (2008). Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Human Mutation, 29(11), 1282–1291. 10.1002/humu.20880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey, J. E. , Harel, T. , Liu, P. , Rosenfeld, J. A. , James, R. A. , Coban Akdemir, Z. H. , Walkiewicz, M. , Bi, W. , Xiao, R. , Ding, Y. , Xia, F. , Beaudet, A. L. , Muzny, D. M. , Gibbs, R. A. , Boerwinkle, E. , Eng, C. M. , Sutton, V. R. , Shaw, C. A. , Plon, S. E. , Yang, Y. , … Lupski, J. R. (2017). Resolution of disease phenotypes resulting from multilocus genomic variation. The New England Journal of Medicine, 376(1), 21–31. 10.1056/NEJMoa1516767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpaer, E. G. , Robinson, M. , Yee, D. , Candlin, J. D. , Mines, R. , & Hunkapiller, T. (1996). Sensitivity and selectivity in protein similarity searches: A comparison of Smith‐Waterman in hardware to BLAST and FASTA. Genomics, 38(2), 179–191. 10.1006/geno.1996.0614 [DOI] [PubMed] [Google Scholar]
- Stürznickel, J. , Rolvien, T. , Delsmann, A. , Butscheidt, S. , Barvencik, F. , Mundlos, S. , Schinke, T. , Kornak, U. , Amling, M. , & Oheim, R. (2020). Clinical phenotype and relevance of LRP5 and LRP6 variants in patients with early‐onset osteoporosis (EOOP). Journal of Bone and Mineral Research, Advance online publication. 10.1002/jbmr.4197 [DOI] [PubMed] [Google Scholar]
- Tenne, M. , McGuigan, F. , Besjakov, J. , Gerdhem, P. , & Åkesson, K. (2013). Degenerative changes at the lumbar spine—Implications for bone mineral density measurement in elderly women. Osteoporosis International, 24(4), 1419–1428. 10.1007/s00198-012-2048-0 [DOI] [PubMed] [Google Scholar]
- van Dijk, F. S. , Zillikens, M. C. , Micha, D. , Riessland, M. , Marcelis, C. L. , de Die‐Smulders, C. E. , Milbradt, J. , Franken, A. A. , Harsevoort, A. J. , Lichtenbelt, K. D. , Pruijs, H. E. , Rubio‐Gozalbo, M. E. , Zwertbroek, R. , Moutaouakil, Y. , Egthuijsen, J. , Hammerschmidt, M. , Bijman, R. , Semeins, C. M. , Bakker, A. D. , Everts, V. , … Pals, G. (2013). PLS3 mutations in X‐linked osteoporosis with fractures. The New England Journal of Medicine, 369(16), 1529–1536. 10.1056/NEJMoa1308223 [DOI] [PubMed] [Google Scholar]
- van Meurs, J. B. , Trikalinos, T. A. , Ralston, S. H. , Balcells, S. , Brandi, M. L. , Brixen, K. , Kiel, D. P. , Langdahl, B. L. , Lips, P. , Ljunggren, O. , Lorenc, R. , Obermayer‐Pietsch, B. , Ohlsson, C. , Pettersson, U. , Reid, D. M. , Rousseau, F. , Scollen, S. , Van Hul, W. , Agueda, L. , Akesson, K. , … GENOMOS Study . (2008). Large‐scale analysis of association between LRP5 and LRP6 variants and osteoporosis. JAMA, 299(11), 1277–1290. 10.1001/jama.299.11.1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waschk, D. E. , Tewes, A. C. , Römer, T. , Hucke, J. , Kapczuk, K. , Schippert, C. , Hillemanns, P. , Wieacker, P. , & Ledig, S. (2016). Mutations in WNT9B are associated with Mayer‐Rokitansky‐Küster‐Hauser syndrome. Clinical Genetics, 89(5), 590–596. 10.1111/cge.12701 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.