

Abstract
Domain classifications are a useful resource for computational analysis of the protein structure, but elements of their composition are often opaque to potential users. We perform a comparative analysis of our classification ECOD against the SCOPe, SCOP2, and CATH domain classifications with respect to their constituent domain boundaries and hierarchal organization. The coverage of these domain classifications with respect to ECOD and to the PDB was assessed by structure and by sequence. We also conducted domain pair analysis to determine broad differences in hierarchy between domains shared by ECOD and other classifications. Finally, we present domains from the major facilitator superfamily (MFS) of transporter proteins and provide evidence that supports their split into domains and for multiple conformations within these families. We find that the ECOD and CATH provide the most extensive structural coverage of the PDB. ECOD and SCOPe have the most consistent domain boundary conditions, whereas CATH and SCOP2 both differ significantly.
Introduction
Proteins and protein complexes contain domains, evolutionarily distinct subunits which confer function either solely or in concert with other domains.1−3 As such, domains represent the building blocks of proteins that guide their evolution. Both sequence and structure similarities between domains are useful in determining their evolutionary relationships. Structural similarity can be used to infer homology over a greater evolutionary distance than the sequence. However, the difficulty in obtaining protein structures for known sequences led to discrete types of classifications, those that focused principally on the sequence (and the utility of deep multiple sequence alignments for homology detection)4−7 and those that classify structures (and the utility of structural similarity for the detection of distant homology).8−12 Recent developments in protein structure prediction, exemplified by the recent CASP14 results and the performance of the DeepMind predictor: AlphaFold2, signal an incoming change for sequence- and structure-based domain classifications alike.13 When near-native predictions of globular protein domains are readily (if not easily) available, both sequence and structural classifications may adapt to incorporate predicted structures of sequence families lacking experimental structures. If this were to come to pass, the distinction between structure and sequence classifications would become purely historical. Due to the nature of experimental methods used to solve protein structures, well-behaved proteins and fragments corresponding to their protein domains that easily form crystals (X-ray) or are relatively small (NMR) have dominated protein structure databases since their inception.14
Such methodology tends to exclude large, multidomain proteins and disordered or flexible regions of proteins that contribute to both their function and their evolution.15,16 With recent improvements in cryo-EM techniques to determine protein structure, the growth of the field is revolutionizing structural biology.17,18 This technique is producing an ever-increasing number of larger and more complete protein structures that are not limited by their ability to crystallize. Categories of proteins such as those that span the membrane and exist as dynamic macromolecular complexes or fibrous assemblies are increasingly dominating newly released structures. Thus, a more complete picture of structure space is emerging that includes nondomain sequences not easily classified in the ECOD hierarchy.
Proteins function as dynamic entities that can adopt multiple functional conformations.19,20 Many of these conformations have been captured in static forms in the existing experimental structures, and they often involve flexible interactions between domains.21,22 Theoretically, the increasing availability of large multidomain structure examples should also expand examples of alternate conformations between domains. With its classification of sequence-related protein domains that function similarly, the family level of ECOD classification is poised to provide large-scale examples of protein conformation change. The view that proteins exist as an ensemble of multiple substructures whose dynamic behavior contributes to their evolvability can begin to be addressed with the sequence to structure ensemble relationships in ECOD families.
Domains are defined at an intersection of multiple competing concepts. Sequence continuity, structural compactness, and functional considerations can each be taken into account to different degrees by separate classifications. These alternate definitions lead to observable differences between similar types of classifications. Examining inconsistencies between proteins classified in multiple classifications can provide insights toward improving classification and provide a foundation for analysis into function. Additionally, examining classifications for consistency can also lead to biological insights. We recently published an account of our analysis of domains containing a minimal Rossmann-like motif (RLM). Using structural motif analysis, we reorganized the two largest X-groups containing Rossmann domains in ECOD: “Rossmann-related” (ECOD id: 2003) and former X-group “other Rossmann-like domains” (ECOD id: 2111). Manual justification, based on structure and sequence similarity, did not reaffirm uniting more than 100 of the homology groups within the previous “other Rossmann-like domains” X-group. Its reclassification provided 102 new X-groups, which constitute about 4% of overall X-groups number in ECOD v275. Moreover, 44 H-groups changed their location and were moved or merged to other H-groups (e.g., “Rossmann-related” and “Flavodoxin-like”), representing 22% of the initial number of RLM-containing H-groups in previous versions of ECOD.23 Overall, this resulted in the unification of the two largest X-groups containing Rossmann motifs in ECOD and the generation of numerous novel X-groups where, despite containing a common RLM, the homology with other X-groups could not be justified.
Our structural domain classification, evolutionary classification of protein domains (ECOD), is nearing a decade of active development and 7 years of public release.12 The details of the ECOD classification have been published elsewhere; briefly, the pilot version of ECOD was developed from a previous version of SCOP (v1.75) that sorted existing SCOP domains into a novel hierarchy that de-emphasized fold similarity and emphasized distant homology. ECOD, like other extant structural domain classifications, relies on a combination of manual curation to define new domains and groups of domains and automated methods to assign newly observed proteins to the existing groups. A core challenge of maintaining ECOD is keeping pace with the ever-growing rate of structure release and the complexity of these structures. Consequently, all structural domain classifications are incomplete. They only partially cover the known set of experimentally determined protein structures. Luckily, this set of protein structure is highly redundant, both because of the concentration of investigator interest and the biophysical properties of protein structures. Here, we assess the completeness of ECOD and other structural domain classifications (CATH,24 SCOPe,10 and SCOP29) with respect to the deposited set of structures in the PDB and a curated set of reference proteins in the UniProt sequence database. We find generally that recently deposited structures, especially those of viral and other fast-evolving proteins, are not categorized in structural domain classifications and offer some potential strategies to accelerate their classification in the future. We also present an analysis of major facilitator transporter proteins that justifies the split of their duplication into separate domains, as well as investigate the separation of structures of these proteins into open and closed conformations. Together, we think that this provides a view of where domain classifications stand now and a vision of what they may need to encompass in the future.
Results and Discussion
Completeness and Coverage of Major Structural Domain Classifications
Domain classifications of protein structures are necessarily incomplete. Insofar as structures are still being determined whose homology is indeterminate, we suspect that additional structures are required to achieve a covering set of domains. Even so, it is likely that we have achieved a largely covering set and that we will only asymptotically approach completion with the release of more structures.25 We compared the coverage of current versions of ECOD, CATH, SCOPe, and SCOP2 with a recent release of the PDB in order to compare their coverage of the known protein structural space. ECOD, CATH, and SCOPe pursue similar classification strategies wherein a smaller number of structures are manually curated and used to seed further classification by automated alignment methods. Additionally, both SCOPe and CATH release stable versions along with more frequent periodic versions which are subject to subsequent error checking before being incorporated into a stable release. We compared the total deposited domains from each classification against a version of the PDB current to November 2020 (see details in Methods). Depositions consisting solely of nucleic acids were not considered. Both ECOD and SCOPe used an earlier version of SCOP (v1.75) in their derivation and are expected to share some domains due to that shared ancestry.
Comparing the number of observed structure depositions in each classification by the presence of at least one domain classified in that structure compared to the release date of that deposition, we derived a running cumulative total of structures observed in each classification at each release date compared to the structure observed in each classification. We observe that ECOD and SCOP classify the most PDB structures and UniProt sequences. SCOP2, although classifying fewer structures, classifies roughly the same amount of sequences as SCOPe over time (Figure 1).
Figure 1.

Completeness of domain classifications with respect to the sequence and structure. (left) Cumulative sum of structures released prior to a release date with at least one domain in ECOD (green), CATH (blue), SCOPe (cyan), and SCOP2 (green). The overall sum of PDB release dates (red) provided for comparison. (right) Comparison of PDB chains with known UniProt annotation by SIFTs with at least one domain defined in each domain classification. Where multiple chains with the same UniProt accession are available, the structure with the earliest release date is used.
Comparison of ECOD Domains to Other Major Structural Domain Comparisons
We also compared the consistency of ECOD domain classifications with respect to other major domain classifications. Domain classifications differ both in how they divide proteins into domains and how these domains are organized into hierarchies. We evaluated ECOD with respect to each of the major domain classifications discussed above in terms of domain partition. Where two classifications’ definition of the domain overlapped significantly, we denote that these domains are “shared” between the classifications. Where two domains are not shared, but are defined from the same peptide chain, we label these domains as from a shared chain. Domains that are from chains unique to a classification are called “unique domains.” This terminology allows us to distinguish domains that differ in partition from those that differ due to the protein content of individual classifications.
We compared the domain partition between ECOD v278 to SCOPe v2.07-2020-07-16 by examining the bidirectional coverage of possible shared domains. This analysis reveals the extent to which classifications divide proteins into similar domains. The domain coverage of ECOD by SCOPe and SCOPe by ECOD is shown in Figure 2A (left and right, respectively) as the population of matched, unmatched, and unique (see the Methods Section) domains for each of the 15 most populated ECOD H-groups and SCOPe folds. As both SCOPe and ECOD were derived from SCOP v1.75, we find that their domain boundaries are largely similar: 83% of SCOPe domains map to an ECOD domain. Although only 29% of ECOD domains map to a SCOPe domain, the 63% of ECOD domains which could not be mapped are due to the inclusion of structures not found in SCOPe. This is likely a consequence of the more aggressive automatic update procedure of ECOD compared to the more conservative protocol of SCOPe.10,26 Among the most populated ECOD H-groups, we find that the majority of domains are either mapped directly to an SCOPe domain or are from PDB chains not yet incorporated into SCOPe. For example, in ECOD H-group 11.1, “IgG beta sandwich domains”, 38% of ECOD domains map directly to a SCOPe domain, 60% occur in chains not contained in SCOPe, and only 2% of ECOD domains come from a PDB chain contained in SCOPe where no domain mapping could be found. Similarly, for SCOPe fold b.1 “immunoglobulin-like beta-sandwich”, 98% of SCOPe domains are mapped to ECOD, 1% of domains in this fold cannot be mapped to an ECOD domain despite sharing a chain with ECOD, and 1% of domains are from chains unique to SCOPe. This suggests that the criteria for domain boundary selection are very similar for ECOD and SCOPe and that a major source of difference between the classifications is simply the structures considered. We note that these calculations were carried out over the full set of domains in both classifications, which are known to be highly sequence redundant. In order to compare the classifications in terms of hierarchal organization, in addition to domain boundary selection, we used domain pair analysis as described previously.12,27,28
Figure 2.

ECOD and SCOPe domain partition comparison. (left) Domain population of 15 most populated H-groups in ECOD v278 stratified by shared (blue), novel partition on shared chain (orange), and new domains from unshared chains (gray). (right) SCOPe 15 most populated folds stratified by shared (blue), unmatched (orange), and unique (gray) domains. (inset) Total unique, unmatched, and shared domains from SCOPe and ECOD.
We compared ECOD v278 to CATH v4.3 using domain boundary correspondence. ECOD and CATH differ fundamentally in methods for domain boundary selection and conceptually in domain hierarchy. Although ECOD (and SCOPe) relies principally on sequence alignment methods for automatic extension of classification, earlier versions of CATH contained an automated structural search method involved in domain parsing.24,29 The 4.3 version of CATH introduced a substantial rework to the methods used for automatic domain parsing.30 The results of the domain boundary analysis are summarized in Figure 3. Many ECOD H-groups contain domains from chains contained in CATH but where there is no definitive mapping. This lack of mapping suggests that substantive differences in domain boundary choices exist between ECOD and CATH. For example, among domains belonging to the SH3 H-group in ECOD, less than 45% map to domains in CATH.
Figure 3.

Well-populated ECOD H-group domain comparison with CATH topologies. (A) (left) Top 15 most populated ECOD H-groups composed of “matched” domains with 90% bidirectional sequence coverage (blue), “unmatched” domains from chains in both SCOP and CATH that have no corresponding domain (orange), and “unique” domains defined from chains that occur in only one classification. (right) The top 15 most populated CATH topologies with matched (blue), unmatched (orange), and unique (gray) domains. (inset) Total matched, unmatched, and unique domains from SCOP and CATH. (B) Trypsin (PDB: 3otj) complexed with BPTI (partial peptide in the magenta stick) at the interface of duplicated RIFT-barrel subunits (ECOD 1.1, cyan and green). Interacting residues from both subunits are on the transparent surface. Active-site residues (spheres) contribute from both subunits. The domain schematic (below) highlights the sequence discontinuous CATH domain definition. (C) First RIFT barrel colored as in panel B except for the N-terminal RIFT β-strands (blue) and C-terminal RIFT β-strands (red). An active-site residue is in the RIFT crossover loop (black sphere marks the Ca position). The N-terminal sequence discontinuous loop (orange) is connected by dashes (green) where the duplicated RIFT barrel is inserted. (D) Second RIFT-barrel is colored as in panel C with active-site residues (black spheres), intervening loops (green), and dashes (cyan, representing the first RIFT barrel position) connecting the C-terminal sequence discontinuous helix (orange).
Among the top populated ECOD H-groups, ECOD classification is less consistent with CATH than with SCOPe (Figure 3A, left, orange bars). The differences between several of these H-groups encompass folds with a Rossmann-like motif (e.g., ECOD H-groups: 2003.1, 2004.1, 2007.1, and 7523.1) and have been compared previously.23,31 Inconsistencies among other large H-groups include examples from ECOD immunoglobulin-related (11.1; 92% overlap), TIM barrel (2002.1; 86% overlap), SH3 barrel (4.1; 71% overlap), RIFT-related barrels (1.1; 54% overlap), HTH (101.1; 57% overlap), and protein kinases (206.1: 2.3% overlap), among others. Among the most populated β-barrels, the RIFT-related barrel group tends to be less consistent than the SH3 group. A large portion of the inconsistent RIFT-related domains come from the CATH topology thrombin H (Figure 4, **), which includes trypsin-like proteases. Trypsin-like proteases consist of duplicated RIFT-related barrel subdomains. The barrels pack compactly together to form the catalytic core, with the active site cleft running alongside the subdomain boundary. Given the shared active site and the reliance of ECOD on manual sequence-based classification, we do not split trypsin-like serine proteases into independent domains. However, CATH considers them as independent. For comparison, the increased consistency of the SH3 domain definition between the two classifications may be due to their function as protein scaffold domains that bind to proline-rich or other motifs.32 One of the main discrepancies for SH3 barrel domains stems from their presence in the ribosome complex. ECOD includes a C-terminal extended tail in the SH3 definition, while CATH limits the domain definition to the compact unit.
Figure 4.

Plasticity of distantly related protein kinase homologs. (A) Cki1 (PDB: 1csn) with bound Mg-ATP (gray sphere-black stick) in the catalytic cleft between the N-lobe (green, CATH 3.30.200) and the conserved core of the C-lobe (cyan, CATH 1.10.510) with additional elaborated helices (white). (B) Minimal kinase domain from OspG (PDB: 4q5hA) bound to MG-AMPPNP is colored as above. (C) Core protein kinase C-lobe SSEs (colored in rainbow from the N-terminus to the C-terminus) are common to Cki1 (left) and OspG (right). (D) ATP-grasp GART (PDB: 1kjqB; CATH 3.30.1 and 3.30.470) in complex with Mg-ADP is colored by the subdomain. An insertion in the N-lobe (white) replaces the N-terminal strands in protein kinases. The C-subdomain includes a pronounced β-sheet with similar topology to (E) SAICAR (PDB: 2gqrA, CATH 3.30.200 and 3.30.470) bound to Mg-ADP. (F) Common C-subdomain from SAICAR (rainbow) lacks the C-terminal helix (left), but a related SAICAR Itpkb includes the C-helix (PDB: 2aqx).
The large superfamily of kinases and related homologs shows a notable inconsistency between ECOD domain definitions (ECOD 206.1) and CATH definitions (CATH 3.30.200 and 1.10.510). ECOD combines the protein kinase fold together with that of SAICAR and ATP-grasp based on previously noted homology.33−35 Each of these groups include two subdomains that combine to form an obligate active site, as exemplified by the active-site cleft formed between the N-lobe and C-lobe of the protein kinases (Figure 4A). ECOD does not separate these subdomains, keeping the protein kinase, SAICAR, and ATP-grasp folds as single units. Alternately, CATH separates the subdomains and classifies them based on topology of extant proteins: including the protein kinase N-lobe (3.30.200) and C-lobe (1.10.510), the SAICAR N-lobe (3.30.200) and C-domain (3.30.470), and the ATP-grasp domains (3.30.1490 and 3.30.470). CASP distinguishes the mainly helical protein kinase C-lobe (1.10.510) from the distantly related SAICAR and ATP-grasp C-domains (3.3.470). Instead, ECOD uses core structure motifs that are common among homologs to define the relationship. For example, the shigella effector kinase adopts a minimal kinase domain that excludes most of the C-terminal helices (Figure 4B). The limited structure includes three conserved helices and three short β-strands that surround the active site (Figure 4C).
Similar subdomains from glycinamide ribonucleotide transformylase (GART) ATP-grasp (Figure 4D) and from SAICAR phosphoribosylaminoimidazole-succinocarboxamide synthase (Figure 4E) contribute to a central active site that binds ligand is a similar orientation as in protein kinases. However, the C-terminal domains from GART and SAICAR exhibit a pronounced β-sheet when compared to the protein kinase C-lobe. This difference leads to an alternate topology definition in CATH, which considers the protein kinase C-lobe as a helical topology (1.10.510) and the others as alpha beta two-layered sandwiches (3.30.470). While the sheets differ in lengths, they all include a similar set of secondary structure elements (Figure 4F). The SAICAR C-subdomain lacks the protein kinase C-terminal helix. However, some SAICAR homologs such as inositol 1,4,5-trisphophate 3-kinase (Itpkb) possess this element. CATH classifies the inositol kinase as a single domain together with the protein kinase C-lobes (1.10.510). The pronounced difference in protein kinase/SAICAR/ATP-grasp classification in ECOD and CATH stem from substantial plasticity of the folds. The evolutionary considerations in ECOD lead to a unified group of homologs that exhibit alternate topologies, while the topology-based classification scheme in CATH distinguishes among the diverse scaffolds.
Finally, we conducted the domain boundary comparison to the most recent version of SCOP2. SCOP2 contains a redesigned protein classification schema that clusters families and superfamilies of domains into networks. SCOP2 domains can be defined both by a “family range” and “superfamily range.” For the purposes of this analysis, we compared the SCOP2 family domain ranges to ECOD domain ranges. Of the classifications considered, SCOP2 contains the fewest domains, containing 33,845 domains in 1502 folds and 2706 superfamilies. Nearly all SCOP2 domains are included in ECOD, while more than 95% of ECOD domains are not present in SCOP2 (Figure 5, right).
Figure 5.

Well-populated ECOD H-groups compared to SCOP2 folds. (A) ECOD H-groups with matched (blue), unmatched (orange), and unique (gray) ECOD domains. (B) SCOP2 folds with matched (blue), unmatched (orange), and unique (gray) SCOP2 domains.
The domain coverage of ECOD by SCOP2 and SCOP2 by ECOD is shown in Figure 5 (left and right, respectively). SCOP2 and ECOD exhibited larger differences in domain partition compared to those between SCOPe and ECOD. A total of 11 out of the top 15 most populated SCOP folds have greater than 40% unmatched domains and four of them have more than 50% unmatched domains (ferredoxin-like, immunoglobulin-like, OB-fold, and ribonuclease-like), suggesting significantly different domain boundary definitions. One possible reason could be the much smaller sample sizes of SCOP2 domains. For PLP-dependent transferase-like fold, all of the SCOP2 domains are unmatched compared to ECOD, as ECOD separated the N-terminal catalytic domain and the C-terminal domain of these PLP-dependent enzymes into two X groups, while SCOP2 kept them together as single-domain units.
Distribution of Equivalent Domain Pairs among Structural Domain Classifications
We analyzed the distribution of homologous domain pairs between ECOD and other domain classifications: SCOP, SCOP2, and CATH. This is similar to a domain pair analysis we previously presented,12 although in that case, the domain pair needed to be shared among all involved classifications, whereas in this case, we performed a series of pairwise analyses. Domain pair analysis reports the overall similarity between two hierarchal levels of differing domain classifications. We compared ECOD H-groups to SCOPe folds, CATH topologies, and SCOP2 folds (Figure 6). This analysis generally describes the relative breadth of classification levels. ECOD H-groups have a similar breadth of classification to CATH topology groups and SCOP2 folds but encompass a slightly broader definition of homology than SCOPe folds.
Figure 6.
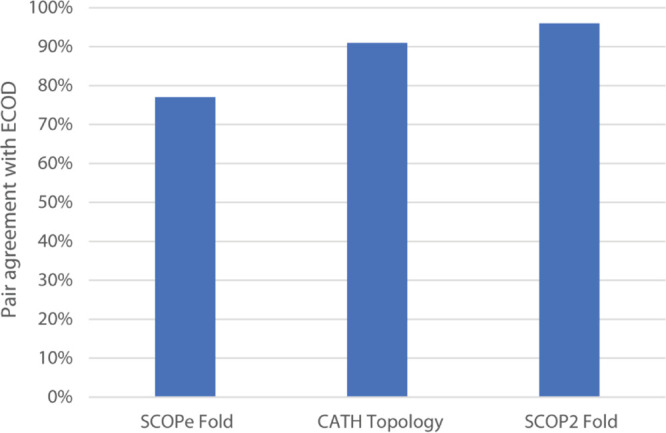
Fraction of ECOD H-group domain pairs found in other classifications. For every domain pair (i.e., pair of matched domains between classifications sharing an H-group in ECOD), we assess whether these domain pairs are found in the same SCOPe fold, CATH topology, or SCOP2 fold. This broadly measures the average evolutionary distance encompassed by other classifications and their hierarchical levels compared to ECOD homology groups (H-groups).
Comparison of Domain Partition between ECOD and Other Classifications
Domain classifications vary not only based on how they organize domains into hierarchies by the sequence and structure but also in how they partition proteins into domains. We analyzed the domain partition of chains shared by ECOD with other domain classifications to quantitate the degree to which domain partition varies among single-domain proteins in ECOD. We chose to limit this analysis to proteins with a single domain because they are the most populated in all classifications and it is a concise way to investigate and illustrate differences between classifications. Classifications vary in how many domains they define because of a variety of factors: some prefer not to split duplications unless independent instances of a duplicated domain can be found. Classifications can vary on the degree to which the function should be considered in the partition of domains, especially when structural factors of that function are provided by sequence-distant residues. The protein kinase/ATP-grasp/SAICAR H-group is a canonical example of a protein that some classifications (ECOD and SCOPe) choose to define as a single domain (despite its clear dual-lobe structure) for functional considerations, whereas others (CATH) choose to divide the protein into two structurally compact domains.33 The previously discussed RIFT-barrel H-group is another set of domains where partition strategies can vary significantly between domain classifications.
SCOPe and ECOD are both descendants of SCOP v1.75 and so contain some of the same implicit assumptions regarding domain partition. ECOD differs from SCOPe in a number of ways that impact domain partition. We compared ECOD single-domain proteins (i.e., domains from PDB chains containing only that domain) from chains present in pairs of classifications. Our goal was to identify whether (1) there are single-domain proteins that do not achieve our bidirectional coverage and (2) where ECOD over- or undersplits single-domain proteins with respect to other classifications. ECOD classifies 533,057 PDB chains, 66% of which are single-domain proteins. Of these 354,861 ECOD single-domain proteins, 148,305 (41%) are from chains/proteins also classified by SCOPe. A total of 145,757 ECOD single-domain proteins (98%) are matched by coverage to a SCOPe domain, and 144,106 (97%) of these cases that match in SCOPe are also single-domain. This leaves 2181 (1.4%) cases where an (1) ECOD single-domain protein is from a SCOPe chain but matches no SCOPe domain and (2) SCOPe defines that protein as single domain (i.e., both ECOD and SCOPe classify a PDB chain as single domain but vary sufficiently by coverage to prevent a match). Conversely, of 167,118 SCOPe single-domain proteins from chains also classified by ECOD, 144,572 (86%) match an ECOD domain where 144,106 (86%) of those are single-domain matches. Finally, 22,546 (13%) SCOPe single-domain proteins from chains shared with ECOD do not match any ECOD domain. The low percentage of unmapped ECOD single domains compared with the relatively higher level of unmapped SCOPe single domains indicates that ECOD tends to split proteins into smaller domains when there is disagreement between classifications. The high rate of the overall single-domain matches indicates a correspondence between ECOD and SCOPe single-domain proteins, while the SCOPe results indicated a small fraction of SCOPe single-domain proteins (generally unsplit duplications) that do not match. In conclusion, SCOPe and ECOD still retain a high degree of similarity in their classification and boundary partition of single-domain proteins.
We also compared the domain partition of ECOD to CATH using this method. CATH varies more fundamentally from ECOD on the strength of structural compactness on determining domain boundaries and so we expected more varied results. We compared the same set of ECOD single-domain proteins to CATH. A total of 210,386 (59%) ECOD domains are from chain/proteins that are also classified by CATH. By the coverage threshold, 175,168 ECOD (83%) single-domain proteins match a CATH domain and 172,950 (82%) of these matches are to a CATH single-domain protein. A total of 32,993 (5%) ECOD single-domain proteins are defined on chains present in CATH, yet those ECOD domains are unmatched. The top two most populated ECOD H-groups containing these unmatched ECOD domains are the PK-like and RIFT barrel domains. Conversely, 204,555 (62%) of PDB chains classified by CATH are single domains, and of these, 197,619 (96%) are derived from chains also classified by ECOD. Finally, 172,950 (87%) of CATH single-domain proteins also match to an ECOD single-domain protein definition, whereas 21,433 (10%) of CATH-single domain proteins match no domain. In contrast to the SCOPe, the higher degree of mismatch among single-domain proteins here indicates that both ECOD and CATH oversplit some domains with respect to the opposing classification.
Finally, we compared the domain partition of SCOP2 to ECOD. A total of 21,382 ECOD single-domain proteins are from PDB chains defined by both ECOD and SCOP2. Of these ECOD single-domains, 20,178 (94%) match by coverage to a SCOP2 domain, of which 20,033 (93%) are SCOP2 single-domain proteins. A total of 1114 (5%) ECOD single-domain proteins are from chains classified in SCOP2 but match no SCOP2 domain by coverage. Conversely, 24,297 (98%) SCOP2 single-domain proteins are from chains classified in both SCOP2 and ECOD. A total of 20,190 (83%) SCOP single-domains match by coverage to some ECOD domain, and 20,019 (82%) of these matches are to ECOD single-domains. A total of 4107 SCOP (2%) single-domain proteins do not match to any ECOD domain, indicative of ECOD splitting these chains into multidomain architectures.
Major Facilitator Superfamily General Substrate Transporter Ensembles
Major facilitator superfamily (MFS) transporters contain 12 transmembrane helices (TMH) arranged around a single cavity formed between a domain duplication of 6 TMH.36 Thus, the MFS transporters likely arose from an ancient 6TMH transporter that duplicated and fused to produce present day 12 TMH MFS topology. Because the peptide binding site is located at the intersection of these two domains, sequence-based classifications such as PFAM define the entire 12TMH entity as a single functional unit, with binding contributed by residues from both domains. ECOD splits the MFS structures into two domains based on their observed duplication and their structural compactness and independence. Sequence and structure evidence further supports splitting of the 6TMH domain into two primordial 3TMH units that form an interdigitated complex that would interact from alternate sides of the membrane.37−39 In support of primitive MFS-like 6TMH-containing domains, a recent expansion of the MFS family to include novel transporters without a known structure includes several families with a single 6TMH domain.40 Splitting of duplications is one question on which domain classifications, sequence and structure, differ.
ECOD splits the duplicated domains from the MFS transporters prior to their classification into families. As such, the N-terminal and C-terminal halves of the largest and most diverse MFS superfamily (defined as MFS_1 or PF07690 by PFAM) group into several different families (MFS_1, MFS_1_1, MFS_1_2, 5050.1.1.19_develop277_1 and 5050.1.1.20_develop277_2). This separation reflects sequence divergence that can result from duplication events. Alternatively, the N-terminal and C-terminal domains from the remaining ECOD families reflect simple duplications that classify into the same group (PTR2, MFS_2, FPN1, and LacY_symp). These groupings roughly reflect previously defined relationships in the transporter classification database.40,41
MFS transporters follow an alternating access model where the central cavity opens to the outside to allow substrate binding and then transitions to an inward-facing conformation to release the substrate on the other side of the membrane.42,43 This conformation change occurs at the interface of the two duplicated 6TMH domains defined in ECOD. We compared structures from one of the smaller and less diverse MFS family represented by E. coli lactose permease (LacY) with structures from both the inward (Figure 7A) and outward (Figure 6B) conformations.44−46 LacY symporters utilize an ion gradient to transport β-galactosides across the membrane against a concentration gradient. The LacY structure ensemble can be depicted as a tree generated from scores of structure comparisons (Figure 7C), which separates the inward facing (left) and outward facing (right) conformations. Closer inspection of the inward facing conformations highlight flexibility in the loop connecting the two domains and the C-terminal helix that line the cytoplasmic side of the structure (Figure 7D). Alternatively, flexibility in the outward facing conformations highlights a periplasmic gate that leads to partial occlusion of two substrate-bound structures (Figure 7C, indicated by * in the tree). Interestingly, the outward facing conformations require either stabilization with a nanobody (6c9w and 5gxb) or mutation of two residues (4zyr and 4oaa), supporting the idea that conformational diversity is an evolvable trait. The presence of multiple observed conformations in structures reflects a known limitation of structural domain classifications, including ECOD, in that recording these conformations is not supported by their taxonomies.
Figure 7.
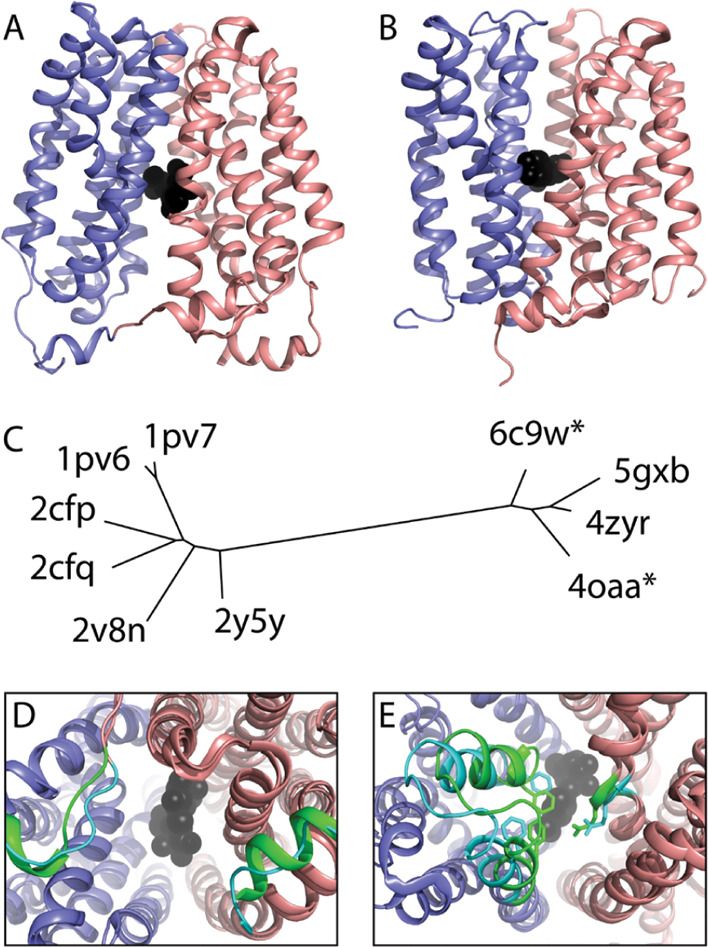
MFS Transporter LacY family structure ensemble. The LacY structure is composed of a 6TMH domain duplication (colored slate and salmon) with a central cleft that binds the substrate (black spheres). LacY adopts (A) an inward facing and (B) an outward facing conformation. (C) Conformations are separated in a tree depicting distances between all LacY structures. (D) Inward-facing structure superpositions highlight flexibility (colored cyan and green) on the cytoplasmic side. (E) Outward facing structure superpositions highlight flexibility in the periplasmic gate (cyan), leading to partial occlusion of the substrate (green).
Comparison of Equivalent Hierarchal Levels by Domain Pair Analysis
Where matched domains were found between ECOD and other domain classifications, we analyzed differences in their classification by domain pair analysis. For each pair of shared domains occupying a taxonomic level in one classification (e.g., ECOD H-group), we examined whether the domain pair was also paired on an equivalent level in another classification (e.g., SCOPe fold). For the purposes of this analysis, we considered ECOD H-groups, SCOP2 and SCOPe folds, and CATH topologies to be equivalent. These equivalencies are based on our experience with these classifications: generally speaking, ECOD H-groups tend to be quite broad, so we chose the broadest level possible from other comparisons in order to generate the closest possible comparison.
Conclusions
Domain classifications provide useful community resources for the study of the protein structure. By curating and analyzing the ever-expanding exploration of the protein sequence and structure space, milestones for further analysis are created. Analogous to how the protein structural space is explored nonrandomly in congruence with investigator interest, domain classifications of this space also develop in potentially unexpected ways based on the choices of their curators. Since there is no “first principles” analysis of protein domains, periodic comparative analysis can be used to determine how domain classifications are changing with respect to each other and with respect to the overall set of known structures. Here, we present an update on the state of protein domain classification 7 years subsequent to the release of our protein domain classification, ECOD. By domain boundaries, our classification continues to match well with SCOPe, while classifying many more structures. Both CATH and SCOP2 differ more in boundaries and are likely interesting alternative sources of boundaries for users. Determining when and how to split domain duplications is a key conceptual point on which domain classifications may differ. We present an analysis and structural comparison of ECOD domains from the MFS superfamily and offer supporting evidence for having them split into multiple domains. For potential users, we anticipate that ECOD provides the broadest coverage of the known structures with the most distant homologous relationships. SCOPe is also a descendant from SCOP v1.75 and is the most similar for those looking for a benchmark. CATH provides a distinct perspective on domain definition more focused on structural compactness and has a rigorous treatment of protein families and their function that was beyond the scope of this work. SCOP2 provides the most efficient coverage of protein space by focusing on classifying only nonredundant representatives. We suspect that advent of cheap near-native protein structural predictions will inevitably lead to the necessity for domain classifications to explicitly label and organize multiple native domain conformations in the near future.
Methods
Generation of Unrooted Structure Similarity Trees
We batch downloaded structures defined in the ECOD LacY_symp family from the PDB47 and limited those structures to chain A. We compared all against all structures using Dalilite48,49 and transformed the Dali Z scores to distances using the following transformation: −ln(DaliZAB/min(DaliZAA or DaliZBB)) where DaliZAB is the Dali Z-score between two structures and DaliZAA and DaliZBB are the scores of the self-alignments. We generated a nonrooted tree using the fitch program (with global rearrangements) with an input matrix of the calculated structure-based distances.50
Calculating Domain Classification Completeness by the Sequence and Structure against the PDB
Completeness of domain classifications was calculated against a version of the PDB current to November 2020. This PDB reference contains 171,447 depositions with 566,056 peptide chains. A total of 878 obsolete PDB entries were retained where necessary to incorporate all domains from the classifications being studied. ECOD v278 was downloaded from http://prodata.swmed.edu/ecod, containing 789,634 domains from 2460 X-groups (possible homology) and 3716 H-groups. CATH v4.3 was downloaded from https://cathdb.info containing 500,238 domains from 1472 topology groups and 6631 homologous superfamilies. A periodic update of SCOPe (v2.07-2020-07-16) was downloaded from https://scop.berkeley.edu/ containing 317,172 domains from 1457 folds and 2323 superfamilies, and noncanonical classes were excluded from analysis. UniProt accessions were associated with PDB entries using the SIFTS database.51 PDB release dates were retrieved from the mmCIF records for each deposition.
Comparison of Domain Partition by the Overall Coverage
We compared the domain partition of classifications by residue coverage of these domains from shared peptide chains. PDB ranges were translated into internal “seq_id” ranges (as in the pdbx_poly_seq_scheme records from the mmCIF representation) in order to make more accurate comparisons. These seq_id ranges uniquely identify individual residues (without the need for insertion codes) and always range from 1 to N where N is the length of the protein. They also disambiguate problems between consistent numbering and unresolved residues in the structure, which can be difficult using standard author-provided PDB residue numbers. Where domain residue ranges overlapped by 90% or more, they were deemed shared domains. Where domains were defined on a shared peptide chain but they did not satisfy our overlap criteria, they were described as “unmatched domains from shared chains” or simply “unmatched.” Finally, where domains were defined on peptide chains that could only be found in one classification, these domains were deemed “unique” to that classification. ECOD, SCOPe, and CATH each record the locations of noncanonical domains or regions that do not properly satisfy their own domain criteria (e.g., expression tags, peptides, and synthetic or de novo designed domains). For SCOPe, we only conducted domain comparisons for SCOPe domains from classes [a–g]. For CATH, only domains from classes 1–4 were considered. SCOP2 does not record noncanonical domain regions at this time.
Acknowledgments
The study is supported in part by the grants (to N.V.G.) from the National Institutes of Health (GM127390) and the Welch Foundation (I-1505).
The authors declare no competing financial interest.
References
- Majumdar I.; Kinch L. N.; Grishin N. V. A database of domain definitions for proteins with complex interdomain geometry. PLoS One 2009, 4, e5084 10.1371/journal.pone.0005084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chothia C.; Gough J.; Vogel C.; Teichmann S. A. Evolution of the protein repertoire. Science 2003, 300, 1701–1703. 10.1126/science.1085371. [DOI] [PubMed] [Google Scholar]
- Lees J. G.; Dawson N. L.; Sillitoe I.; Orengo C. A. Functional innovation from changes in protein domains and their combinations. Curr. Opin. Struct. Biol. 2016, 38, 44–52. 10.1016/j.sbi.2016.05.016. [DOI] [PubMed] [Google Scholar]
- Mistry J.; Chuguransky S.; Williams L.; Qureshi M.; Salazar G. A.; Sonnhammer E. L. L.; Tosatto S. C. E.; Paladin L.; Raj S.; Richardson L. J.; Finn R. D.; Bateman A. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M.; Derbyshire M. K.; Yamashita R. A.; Marchler-Bauer A. NCBI’s Conserved Domain Database and Tools for Protein Domain Analysis. Curr. Protoc. Bioinf. 2020, 69, e90 10.1002/cpbi.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; Attwood T. K.; Babbitt P. C.; Bateman A.; Bork P.; Bridge A. J.; Chang H.-Y.; Dosztányi Z.; El-Gebali S.; Fraser M.; Gough J.; Haft D.; Holliday G. L.; Huang H.; Huang X.; Letunic I.; Lopez R.; Lu S.; Marchler-Bauer A.; Mi H.; Mistry J.; Natale D. A.; Necci M.; Nuka G.; Orengo C. A.; Park Y.; Pesseat S.; Piovesan D.; Potter S. C.; Rawlings N. D.; Redaschi N.; Richardson L.; Rivoire C.; Sangrador-Vegas A.; Sigrist C.; Sillitoe I.; Smithers B.; Squizzato S.; Sutton G.; Thanki N.; Thomas P. D.; Tosatto S. C. E.; Wu C. H.; Xenarios I.; Yeh L.-S.; Young S.-Y.; Mitchell A. L. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017, 45, D190–D199. 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinegger M.; Mirdita M.; Söding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat. Methods 2019, 16, 603–606. 10.1038/s41592-019-0437-4. [DOI] [PubMed] [Google Scholar]
- Jürgens C.; Strom A.; Wegener D.; Hettwer S.; Wilmanns M.; Sterner R. Directed evolution of a (beta alpha )8-barrel enzyme to catalyze related reactions in two different metabolic pathways. Proc. Natl. Acad. Sci. 2000, 97, 9925–9930. 10.1073/pnas.160255397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva A.; Kulesha E.; Gough J.; Murzin A. G. The SCOP database in 2020: expanded classification of representative family and superfamily domains of known protein structures. Nucleic Acids Res. 2020, 48, D376–D382. 10.1093/nar/gkz1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandonia J.-M.; Fox N. K.; Brenner S. E. SCOPe: classification of large macromolecular structures in the structural classification of proteins-extended database. Nucleic Acids Res. 2019, 47, D475–D481. 10.1093/nar/gky1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox N. K.; Brenner S. E.; Chandonia J. M. SCOPe: Structural Classification of Proteins--extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Res. 2014, 42, D304–D30. 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Schaeffer R. D.; Liao Y. X.; Kinch L. N.; Pei J. M.; Shi S. Y.; Kim B. H.; Grishin N. V. ECOD: An Evolutionary Classification of Protein Domains. PLoS Comput. Biol. 2014, 10, e1003926 10.1371/journal.pcbi.1003926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. “It will change everything”: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. 10.1038/d41586-020-03348-4. [DOI] [PubMed] [Google Scholar]
- Ziegler S. J.; Mallinson S. J. B.; St. John P. C.; Bomble Y. J. Advances in integrative structural biology: Towards understanding protein complexes in their cellular context. Comput. Struct. Biotechnol. J. 2021, 19, 214–225. 10.1016/j.csbj.2020.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Tawfik D. S. Protein dynamism and evolvability. Science 2009, 324, 203–207. 10.1126/science.1169375. [DOI] [PubMed] [Google Scholar]
- James L. C.; Tawfik D. S. Conformational diversity and protein evolution - a 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. 10.1016/s0968-0004(03)00135-x. [DOI] [PubMed] [Google Scholar]
- Henderson R. From Electron Crystallography to Single Particle CryoEM (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 2018, 57, 10804–10825. 10.1002/anie.201802731. [DOI] [PubMed] [Google Scholar]
- Cheng Y. Single-particle cryo-EM-How did it get here and where will it go. Science 2018, 361, 876–880. 10.1126/science.aat4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauenfelder H.; Sligar S.; Wolynes P. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Wei G.; Xi W.; Nussinov R.; Ma B. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. 10.1021/acs.chemrev.5b00562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim W. A. The modular logic of signaling proteins: building allosteric switches from simple binding domains. Curr. Opin. Struct. Biol. 2002, 12, 61–68. 10.1016/s0959-440x(02)00290-7. [DOI] [PubMed] [Google Scholar]
- Ma B.; Shatsky M.; Wolfson H. J.; Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002, 11, 184–197. 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev K. E.; Kinch L. N.; Dustin Schaeffer R.; Pei J.; Grishin N. V. A Fifth of the Protein World: Rossmann-like Proteins as an Evolutionarily Successful Structural unit. J. Mol. Biol. 2021, 433, 166788. 10.1016/j.jmb.2020.166788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I.; Bordin N.; Dawson N.; Waman V. P.; Ashford P.; Scholes H. M.; Pang C. S. M.; Woodridge L.; Rauer C.; Sen N.; Abbasian M.; Le Cornu S.; Lam S. D.; Berka K.; Varekova I. H.; Svobodova R.; Lees J.; Orengo C. A. CATH: increased structural coverage of functional space. Nucleic Acids Res. 2021, 49, D266–D273. 10.1093/nar/gkaa1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kihara D.; Skolnick J. The PDB is a covering set of small protein structures. J. Mol. Biol. 2003, 334, 793–802. 10.1016/j.jmb.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Chandonia J.-M.; Fox N. K.; Brenner S. E. SCOPe: Manual Curation and Artifact Removal in the Structural Classification of Proteins - extended Database. J. Mol. Biol. 2017, 429, 348–355. 10.1016/j.jmb.2016.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer R. D.; Jonsson A. L.; Simms A. M.; Daggett V. Generation of a consensus protein domain dictionary. Bioinformatics 2011, 27, 46–54. 10.1093/bioinformatics/btq625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley C.; Jones D. T. A systematic comparison of protein structure classifications: SCOP, CATH and FSSP. Structure 1999, 7, 1099–1112. 10.1016/s0969-2126(99)80177-4. [DOI] [PubMed] [Google Scholar]
- Sillitoe I.; Dawson N.; Thornton J.; Orengo C. The history of the CATH structural classification of protein domains. Biochimie 2015, 119, 209–217. 10.1016/j.biochi.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentzsch R.; Orengo C. A. Protein function prediction using domain families. BMC Bioinf. 2013, 14, S5. 10.1186/1471-2105-14-s3-s5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev K. E.; Kinch L. N.; Schaeffer R. D.; Grishin N. V. Functional analysis of Rossmann-like domains reveals convergent evolution of topology and reaction pathways. PLoS Comput. Biol. 2019, 15, e1007569 10.1371/journal.pcbi.1007569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah N. H.; Amacher J. F.; Nocka L. M.; Kuriyan J. The Src module: an ancient scaffold in the evolution of cytoplasmic tyrosine kinases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 535–563. 10.1080/10409238.2018.1495173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin N. V. Phosphatidylinositol phosphate kinase: a link between protein kinase and glutathione synthase folds. J. Mol. Biol. 1999, 291, 239–247. 10.1006/jmbi.1999.2973. [DOI] [PubMed] [Google Scholar]
- Galperin M. Y.; Koonin E. V. Divergence and Convergence in Enzyme Evolution*. J. Biol. Chem. 2012, 287, 21–28. 10.1074/jbc.r111.241976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreelatha A.; Kinch L. N.; Tagliabracci V. S. The secretory pathway kinases. Biochim. Biophys. Acta 2015, 1854, 1687–1693. 10.1016/j.bbapap.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marger M. D.; Saier M. H. Jr A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem. Sci. 1993, 18, 13–20. 10.1016/0968-0004(93)90081-w. [DOI] [PubMed] [Google Scholar]
- Västermark Å.; Saier M. H. Major Facilitator Superfamily (MFS) evolved without 3-transmembrane segment unit rearrangements. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E1162–E1163. 10.1073/pnas.1400016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Västermark Å.; Lunt B.; Saier M. Major Facilitator Superfamily Porters, LacY, FucP and XylE of Escherichia coli Appear to Have Evolved Positionally Dissimilar Catalytic Residues without Rearrangement of 3-TMS Repeat Units. J. Mol. Microbiol. Biotechnol. 2014, 24, 82–90. 10.1159/000358429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej M. G.; Dang S.; Yan N.; Kaback H. R. Evolutionary mix-and-match with MFS transporters. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5870–5874. 10.1073/pnas.1303538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S. C.; Davejan P.; Hendargo K. J.; Javadi-Razaz I.; Chou A.; Yee D. C.; Ghazi F.; Lam K. J. K.; Conn A. M.; Madrigal A.; Medrano-Soto A.; Saier M. H. Jr. Expansion of the Major Facilitator Superfamily (MFS) to include novel transporters as well as transmembrane-acting enzymes. Biochim. Biophys. Acta, Biomembr. 2020, 1862, 183277. 10.1016/j.bbamem.2020.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saier M. H. Jr; Reddy V. S.; Tsu B. V.; Ahmed M. S.; Li C.; Moreno-Hagelsieb G. The Transporter Classification Database (TCDB): recent advances. Nucleic Acids Res. 2016, 44, D372–D379. 10.1093/nar/gkv1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N. Structural Biology of the Major Facilitator Superfamily Transporters. Annu. Rev. Biophys. 2015, 44, 257–283. 10.1146/annurev-biophys-060414-033901. [DOI] [PubMed] [Google Scholar]
- Quistgaard E. M.; Löw C.; Guettou F.; Nordlund P. Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat. Rev. Mol. Cell Biol. 2016, 17, 123–132. 10.1038/nrm.2015.25. [DOI] [PubMed] [Google Scholar]
- Smirnova I.; Kasho V.; Kaback H. R. Lactose permease and the alternating access mechanism. Biochemistry 2011, 50, 9684–9693. 10.1021/bi2014294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramson J.; Smirnova I.; Kasho V.; Verner G.; Kaback H. R.; Iwata S. Structure and mechanism of the lactose permease of Escherichia coli. Science 2003, 301, 610–615. 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- Kumar H.; Finer-Moore J. S.; Jiang X.; Smirnova I.; Kasho V.; Pardon E.; Steyaert J.; Kaback H. R.; Stroud R. M. Crystal Structure of a ligand-bound LacY-Nanobody Complex. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 8769–8774. 10.1073/pnas.1801774115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodsell D. S.; Zardecki C.; Di Costanzo L.; Duarte J. M.; Hudson B. P.; Persikova I.; Segura J.; Shao C.; Voigt M.; Westbrook J. D.; Young J. Y.; Burley S. K. RCSB Protein Data Bank: Enabling biomedical research and drug discovery. Protein Sci. 2020, 29, 52–65. 10.1002/pro.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Kaariainen S.; Rosenstrom P.; Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics 2008, 24, 2780–2781. 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L.; Park J. DaliLite workbench for protein structure comparison. Bioinformatics 2000, 16, 566–567. 10.1093/bioinformatics/16.6.566. [DOI] [PubMed] [Google Scholar]
- Fitch W. M.; Margoliash E. Construction of phylogenetic trees. Science 1967, 155, 279–284. 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- Dana J. M.; Gutmanas A.; Tyagi N.; Qi G.; O’Donovan C.; Martin M.; Velankar S. SIFTS: updated Structure Integration with Function, Taxonomy and Sequences resource allows 40-fold increase in coverage of structure-based annotations for proteins. Nucleic Acids Res. 2019, 47, D482–D489. 10.1093/nar/gky1114. [DOI] [PMC free article] [PubMed] [Google Scholar]