

Abstract
Laminin polymerization is a key step of basement membrane assembly that depends on the binding of α, β and γ N-terminal LN domains to form a polymer node. Nodal assembly can be divided into two steps consisting of β- and γ-LN dimerization followed by calcium-dependent addition of the α-LN domain. The assembly and structural organization of laminin-111 LN-LEa segments was examined by size-exclusion chromatography (SEC) and electron microscopy. Triskelion-like structures were observed in negatively-stained images of purified α1/β1/γ1 LN-LEa trimers. Image averaging of these revealed a heel-to-toe organization of the LN domains with angled outward projections of the LEa stem-like domains. A series of single-amino acid substitutions was introduced into the polymerization faces of the α1, β1 and γ1 LN domains followed by SEC analysis to distinguish between loss of β-γ mediated dimerization and loss of α-dependent trimerization (with intact β-γ dimers). Dimer-blocking mutations were confined to the γ1-toe and the β1-heel, whereas the trimer-only-blocking mutations mapped to the γ1-heel, β1-toe and the α1-toe and heel. Thus, in the polymer node the γ1-toe pairs with the β1-heel, the β1-toe pairs with the α1-heel, and the α1-toe pairs with the γ1-heel.
Keywords: basement membrane, self-assembly, triskelion, LN mutations, image averaging
Introduction
Basement membranes (BMs) are cell-adherent extracellular matrices (ECMs) essential for cell polarization, differentiation, and maintenance of compartmentalized animal tissues [1-3]. They serve as supportive cell scaffolds that act as signaling platforms, with the matrix anchored to transmembrane receptors linked to the cytoskeleton. Signaling through cell-surface receptors to modulate cell and tissue shape, differentiation and specific functions depends upon the mechanical properties of the BM, dictated by its structural organization and crosslinking [2, 4]. Key components of BMs are members of the laminin and type IV collagen family, nidogens and heparan sulfate (HS) proteoglycans perlecan and agrin. These components are organized into polymers of laminin and collagen, the latter cross-linked, with inter-polymer links provided by nidogen and possibly also HS chains [5]. Each laminin molecule is a heterotrimer consisting of α, β and γ subunits joined through a long coiled-coil. There are five α-, four β-, and three γ mammalian subunits as well as splice variants (α3A, α3B) that combine together to give at least 15 laminins heterotrimers [6], most of which are cross-shaped and capable of polymerization [7, 8]. Genetic loss of the α1, β1, γ1 and α5 subunits results in failure of BM assembly during early embryogenesis [9-12]. In contrast, mutations in the genes coding for late-expressing laminin subunits cause congenital defects in kidney/eye (β2 in Pierson syndrome) [13], muscle/peripheral nerve/brain (α2 in LAMA2-muscular dystrophy) [14], and skin (α3, β3, or γ2 in epidermolysis bullosa) [15]. A subset of laminin gene mutations in the region coding for the LN domains cause selective inactivation of polymerization, resulting in partial disruptions of BM assembly and milder forms of the above diseases [16-19]. One consequence of polymer loss is laminin matrix softening and attenuation in which receptor-mediated tensions between receptor and polymer are reduced [20].
Laminins contain a tandem organization of globular and rod-like domains in each of the three subunits (Fig. 1). Most laminins (i.e. β1/γ1 and β2/γ1 laminins paired with the α1, α2, α3B and α5 subunits) have three short arms (α,β,γ) each tipped by an LN domain and one long arm (coiled-coil) from which extends the α-subunit LG domains. The LN domains mediate polymerization, the γ1LEb3 domain mediates binding to nidogen, the coiled-coil mediates binding to agrin, the LG1-3 domains and coiled-coil terminus mediate integrin-binding, and the LG4-5 domains mediate binding to α-dystroglycan (αDG) [1]. Intervening LE domains (EGF-like domains containing an additional cysteine pair) are thought to mostly act as spacers between the binding domains and also provide short-arm flexibility. BM assembly is initiated by laminins which bind to cognate receptors and polymerize, forming a sheet-like cell-attached matrix (“receptor-facilitated assembly”). This initial matrix provides the sites for nidogen, collagen-IV, perlecan and agrin binding needed to complete the core BM architecture [1].
Figure 1. Laminin-111 assembly, protein domains and LN-LEa segments.

A. Model of laminin polymerization based on previous studies. Laminin first assembles into a β-γ dimer through LN domain interactions. The α-LN domain of a third laminin molecule binds to the β-γ dimer in a calcium-dependent interaction, forming the polymer node. Additional laminin molecules bind to the initial complex, forming a sheet-like polymer (cc, critical concentration of polymerization). B. Domain organization of laminin-111 with corresponding LN-LEa segments. C. Coomassie Blue-stained gel (SDS-PAGE, 7.5%, reduced) of recombinant. α1-, β1- and γ1-LN-LEa glycoproteins. D. PNGaseF de-glycosylation of α1-LN-LEa. After enzyme treatment, a single band, migrating at 55 kDa, is seen.
The biological significance of polymerization has been addressed, in part, by determining what happens to tissues and cells with laminins unable to self-assemble. It was found that the muscular dystrophy and peripheral neuropathy of the dy2J/dy2J mouse results from an in-frame deletion-driven loss of the α2 LN domain that prevents polymerization [21]. This mouse serves as a model for the ambulatory variant of LAMA2-muscular dystrophy in which a cluster of α2LN missense mutations has been identified as causative of the disease. The muscular weakness, accompanied by a reduction in nerve conduction in the hindlimbs, is accompanied by a reduction of laminin density in the myofiber and Schwann cell basement membranes and an increase in solubilized laminin in these tissues [19]. Similarly, a cluster of β2 LN mutations has been found to cause Pierson syndrome [13]. Both the α2 and β2 mutated residues are highly conserved in LN domains. Disease substitution of two residues found in LAMA2-MD and Pierson syndrome into the α1 and β1 chains, respectively, prevented polymerization [18].
Laminin polymerization was first described for laminin-111 [22] and later for other laminins containing the α2, β2, α3B and α5 subunits [7, 8, 23]. The process was found to be a nucleation-propagation self-assembly with an initial calcium-independent oligomerization step that is followed by a calcium-dependent aggregation step. Subsequent studies revealed that the polymer is held together by reversible bonds that could be dissociated with fragments containing the terminal LN domains, that the polymer has a mesh-like appearance when visualized in freeze-etch Pt/C replicas by electron microscopy [24], that assembly requires all three short arm LN domains, that the relevant calcium is located in the γ1 subunit [25, 26], and that the trimer-forming step has a dissociation constant of ~1.4 μM as measured by equilibrium gel filtration [25]. A later study revealed the crystal structure of the α5 LN-LEa domains (homologous to those of α1) in which three conserved polymer-forming residues were identified on a central portion of one face of the LN domain (“front” face) [27]. The opposite “back” face contained an N-linked glycan thought to sterically prevent binding access. The crystal structures of the homologous β1 and γ1 LN-LEa domains were also described and found to similarly contain N-linked glycans on their LN back faces [26]. It was then shown that residues on the β1 and γ1 LN front face are involved in polymerization [28]. Only one calcium site exists among the LN domains, positioned in the mid-portion of the γ1 LN domain [26]. Another feature of the three structures is an angle bend (~140°) between the LN oblong globule and LEa rod [26, 27]. Dimer and trimer association of soluble LN-LEa segments has been studied by surface plasmon resonance [27] and size-exclusion chromatography (SEC) [23]. The data strongly support a two-step polymerization model: β-γ dimerization followed by incorporation of α.
In order to understand the distribution of interacting residues and organization of the laminin-111 polymer node, we combined extensive structure-based mutagenesis with SEC analysis and negative-staining electron microscopy. We report that the N-terminal segments of the α1, β1 and γ1 subunits assemble into a flat triskelion-like complex in which the LN domains contact each other in a heel-to-toe arrangement with the rod-like LEa domains extending out from the LN domains at the corners of the triskelion.
Results
Protein analysis of wild-type laminin LN-LEa proteins.
Recombinant laminin-111 α1, β1 and γ1 glycoprotein segments consisting of the LN through LEa4 domains were expressed and secreted by stable clones of HEK293 cells (Fig. 1). These proteins, following purification by epitope tag affinity chromatography, were characterized by SDS-PAGE. Single bands were identified for human β1LN-LEa and human γ1LN-LEa (apparent molecular mass of 75 kDa and 63 kDa, respectively) but mouse α1-LNLEa migrated as two band with apparent molecular masses of 68 kDa and 65 kDa. Following treatment with PNGaseF in SDS to remove N-linked glycan, α1LN-LEa migrated as a single band with an apparent molecular mass of 55 kDa, the predicted protein mass.
SEC analysis of wild-type LN-LEa.
SEC was used to analyze α1-, β1- and γ1-LN-LEa glycoprotein monomers, and the dimers and trimers that assemble, based on a method in which LN-LEa elution profiles and complex composition were established [23]. A fast flow rate of 1 ml/min was found to limit dissociation of complexes during chromatography. The single LN-LEa proteins, β1-γ1 dimers, and α1-β1-γ1 trimers were separated from each other and eluted at the indicated volumes (Fig. 2). In agreement with the earlier study [23], trimer assembly required all three proteins. An elevated absorbance “valley” was noted between the trimer and dimer peaks, particularly at higher incubation concentrations, that was considered to represent trimer-to-dimer dissociation during chromatography. Interestingly, when γ1-LN-LEa bearing a single amino substitution (D266R) located at the LN tip was co-incubated with the other wild-type (WT) proteins, the elevated valley was no longer observed. As discussed ahead, this substitution was found to stabilize the trimer fraction. Reducing SDS-PAGE of the fractions confirmed that the trimer peak contained all three proteins, with lower amounts of these proteins in later eluting peaks, as expected given the reversible nature of the assembly process. SDS-PAGE of the dimer peak (following incubation of β1- with γ1-LN-LEa) revealed the presence of both proteins.
Figure 2. Size-exclusion chromatography of WT LN-LEa monomers, dimers and trimers.

A. α1-LN-LEa, β1-LN-LEa and γ1-LN-LEa were analyzed by SEC (1 ml/min flow rate) each alone, following incubation of the β1 and γ1 components, or following incubation of the α1, β1 and γ1 components together, at 27°C for 1 hr. Monomer (-M-), dimer (D) and trimer (T) elution positions are marked. B, C. Aliquots of 0.5 ml fractions were analyzed by SDS-PAGE (7.5%, reducing). Gels were stained with Coomassie Blue. These confirmed the monomer, dimer and trimer assignments. D, E. SEC profiles for trimer (D) and dimer (E) fractions shown for different initial concentrations (three lower concentration plots use right-sided y-axis values). The “sum” is the arithmetic sum of β1 and γ1 monomers. F, G. Concentration-dependency for trimer and dimer assembly (data points were fitted for simple binding).
The concentration dependency of trimer and dimer assembly was examined (Fig. 2, D-G). For calculations, the trimer fraction was considered as all protein eluting before the dimer fraction, while the dimer fraction was considered as all protein eluting before the sum of the β1 and γ1 monomers. The apparent dissociation constants (KD) were estimated by fitting for simple ligand binding to be 2.0 μM for trimer and 14 μM for dimer assembly. These apparent dissociation constants were within two-fold of those previously determined by equilibrium gel filtration for trimer (1.4 μM) [25] and dimer by plasmon resonance (22 μM) [23]. Nonetheless, these values were considered approximations that are inherently inaccurate because of the problem of dissociation during chromatography. The purpose of these determinations here was to establish the concentration range that was needed to identify those mutated LN-LEa segments that were defective in their ability to form dimers and/or trimers.
Negative stain electron microscopy.
Conditions for adequate resolution of LN-LEa proteins required the production of very thin films of uranyl acetate with further improvement obtained by the stabilizing addition of trehalose [29]. Monomer samples were examined by electron microscopy following negative staining of diluted aliquots on carbon-coated grids (Fig. 3). The α1, β1 and γ1 LN-LEa particles had the appearance of elongated entities, thickened at one end. Image averaging revealed tadpole-like structures with either a wide or narrower head. Specific bends were generally not appreciated. Negative staining of the WT β1-/γ1-dimer fraction was heterogeneous in appearance, rich in monomer-sized particles. The particles were not well-resolved by image averaging (data not shown). It is thought heterogeneity may have resulted from rapid dissociation of the complexes occurring during and after gel filtration.
Figure 3. Negative-stain images of α1-, β1- and γ1-LN-LEa monomers.
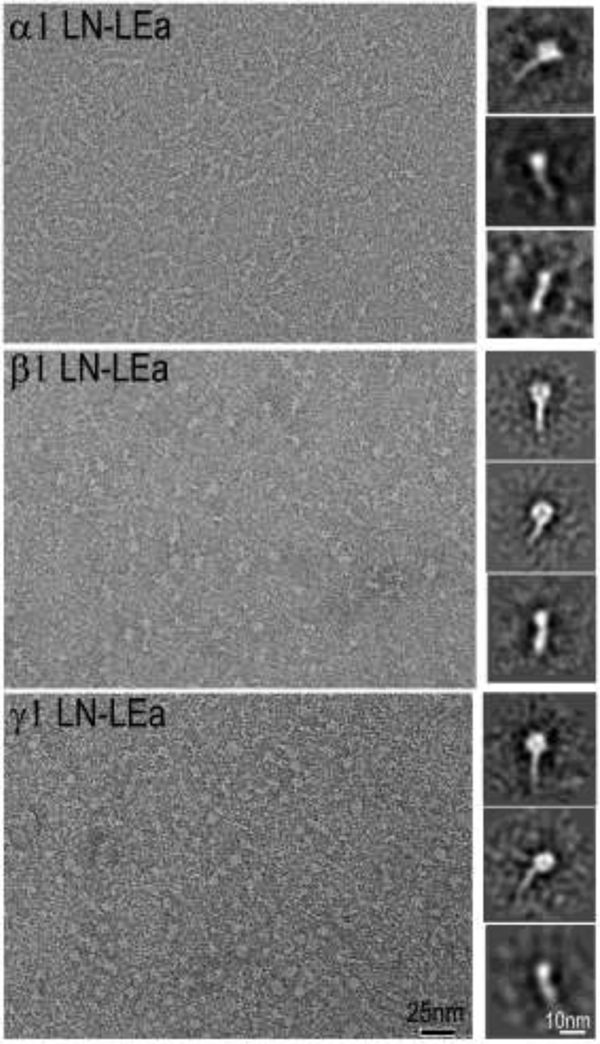
Representative micrograph field shown in left panels. Class-averaged images shown in right side panels. Note the monomer consist of an oblong globule attached to a narrow stem-like structure. The oblong globules were noted to be positioned as wider and narrower presentations, thought to correspond to the known flat shape as determined by crystallography [26, 47].
The α1/β1/γ1 LN-LEa trimer fraction (~10 ml elution volume) was also analyzed by negative staining (Fig. 4 and Suppl. Fig. 1). Larger triangular particles, many with small areas of central clearing, were noted. Image averaging (32 class averages) revealed symmetrical triskelion-like structures with three spokes projecting out from the corners. Three components could be identified in many of the class averages, each consisting of an internal oblong structure connected at an angle to a narrower outward projecting stem. The three foot-like structures are organized together heel-to-toe.
Fig. 4. Negative-stain images of LN-LEa trimer complexes.

Top panel: Negative stain electron micrograph of a SEC-purified trimer fraction resulting from the incubation of γ1-D266R-LN-LEa with WT α1- and β1-LN-LEa (60,000 x magnification). Fields contained trimeric particles with narrow projections from the corners and central clearing. Bottom panels. Full set of class averages derived from analysis of 11,721 particles (74x74 pixel box size) that reveal the triskelion consists of three segments that contact each other. Each segment consists of a foot-like inner portion in a heel-to-toe arrangement attached to rod-like ankle projecting out.
Side orientations of the triskelions were not observed, preventing the generation of 3D reconstructions. Further, inspection of class averages also revealed that in some triskelions, the heel-to-toe orientations were clockwise (upper row), while in others the triskelion heel-to-toe orientations were counter-clockwise (lower row) (Fig. 5). This suggests that the triskelion is flat, with either one face or the other lying on the carbon surface. While the trimeric triskelion was generated in initial experiments using the WT γ1-LN-LEa (Suppl. Fig. 1), the accidental discovery that the γ1D266R mutation stabilized the trimer led us to use the latter for most triskelion analysis (Fig. 4,5). While the γ1D266R trimers subjected to image averaging were more plentiful and homogenous in appearance, we observed no discernable morphological difference between the two types of trimer. Further, it was not possible with either to determine which subunit toe bound to which heel because of the triskelion pseudo-symmetry and because the LN-LEa domain components could not be distinguished from each other within the triskelion. SEC analysis of targeted mutations in the three LN domains was used instead to make this determination.
Figure 5. LN orientations.

Triskelions can be arranged in either a clockwise heel-to-toe (upper row) or a counter-clockwise (lower row) orientation. This suggests that these structures are flat, lying either on one side or the other on the carbon support film.
SEC analysis of dimer/trimer assembly with LN-LEa proteins bearing single amino acid LN substitutions.
Substantial sequence identity is found among the α, β and γ1 LN-LEa segments, particularly in the front polymerization face [18, 27] of the three LN domains [18]. Homology models were prepared for mouse α1-LN-LEa, human β1-LN-LEa and α1-LN-LEa from the atomic coordinates of the mouse α5, β1 and γ1 LN-LEa crystal structures [26, 27]. Conserved solvent-exposed residues were identified on the polymerization face of the LN domains, located either in the toe region (the upper part and tip of the front face) or the heel region (the lower part of the front face, near the juncture with the LEa rod) regions. In order to determine which residues were likely involved in LN dimer- and trimer-forming interactions, potentially disruptive single amino acid substitutions were designed (charge reversals or substitution of hydrophobic residues to charged ones). The expectation, given the apparent toe-to-heel organization of class average images of the trimer and the known order of assembly in which the α-LN domain can only bind to a β-γ dimer [18, 23, 28], was that mutations that prevent or greatly reduce both dimer and trimer assembly identify the surfaces that mediate β-γ binding interactions, whereas mutations that prevent only trimer assembly identify the surfaces that mediate interactions with the α subunit. All mutants were obtained as soluble proteins from the supernatant of transfected HEK293 cells, with comparable yields as the corresponding wild-type proteins. This suggests that the mutations do not affect folding and secretion.
Seven amino acid substitutions were introduced into the LN domain of γ1LN-LEa protein. The modified proteins (containing D261R, S213R, R149E, Y147R, R285E, D266R, and K268A), purified from conditioned medium, were analyzed by SEC alone or following incubation with WT β1-LN-LEa and WT α1-LN-LEa proteins (Fig. 6). The toe mutations D261R and S213R prevented both dimer and trimer assembly. In contrast, the heel mutations R149E, Y147R, and R285E prevented α-β-γ trimer assembly but not β-γ dimer assembly. These results indicate the γ1-toe binds to the β1-heel. Two mutations, D266R and K268A (Suppl. Fig. 2), map to locations above and posterior to the toe portion of the polymerization face. These mutations did not prevent dimer and trimer assembly.
Fig. 6. SEC analysis of dimer and trimer assembly involving γ1-LN-LEa mutants.

A. The front face of the γ1 LN domain was modified with the indicated single amino acid substitutions. The mutated γ1-LN-LEa proteins (labeled “γ1M” in graphs) were analyzed by SEC in combination with WT α1-LN-LEa and β1-LN-LEa or with β1-LN-LEa alone to determine whether trimer or dimer assembly was altered. Elution volumes of trimers (T), dimers (D) and monomers (-M-) marked in red above peaks. The mutated γ1 monomers eluted at the same volume as WT γ1 monomer. B, C. γ1-D261R produced neither dimer nor trimer. D, E. γ1-S213R produced neither dimer nor trimer. F, G. γ1-Y147R produced dimer but no trimer. H, I. γ1-R149E produced dimer but no trimer. J, K. γ1-R285E produced dimer but no trimer.
Previously it has been shown that S68R, located in the toe of the LN domain of β1-LN-LEa (Fig. 7), yields a dimer upon incubation with WT α1 and γ1 LN-LEa proteins [23]. This modification corresponds to the human β2S80R mutation found to cause Pierson syndrome [13]. Four new amino acid substitutions were introduced into the LN domain of β1LN-LEa. The modified proteins (containing F77A, S200R, E204R, R208E, and D218R) were evaluated alone, after incubation with WT α1 and α1 LN-LEa by SEC. F77A and S200R β1LN-LEa, like S68R, when incubated with the WT proteins, formed dimers but not trimers. These mutations were clustered in the toe region. In contrast, when β1-LN-LEa containing either E204R, R208E or D218R was incubated with WT γ1-LN-LEa, the proteins remained almost entirely monomeric, consistent with these β1 residues binding to the α1-toe. The β1-D218R protein and, to a considerably lesser extent, β1-R208E still yielded trimer when incubated with WT α1-LN-LEa and α1-LN-LEa. However, the trimer fraction was reduced compared to WT trimer, most notably at lower concentration (Fig. 7, E,I,K). Careful examination of the chromatograms revealed that the D218R mutation did not completely disrupt β1-γ1 dimer assembly (Fig. 7, J,L): the β1-D218R/γ1 incubation mixture eluted ~0.2 ml earlier than the leading-edge of the arithmetic sum of monomeric WT β1 and γ1 proteins, indicating a weak interaction of β1-D218R with γ1 that was not of sufficient strength, however, to result in a distinct dimer peak. It was concluded that a small surviving fraction of transient dimer can still associate with the α1 subunit and become incorporated into a stable trimer. Trimer formation is least-affected for the β1 mutation (D218R) that is farthest from the α1-binding toe region.
Fig. 7. SEC analysis of dimer and trimer assembly involving β1-LN-LEa mutants.

A. The front face of the β1 LN domain was modified with the indicated single amino acid substitutions. The mutated β1 proteins (labeled “β1M” in graphs) were analyzed by SEC in combination with WT α1-LN-LEa and γ1-LN-LEa or with γ1-LN-LEa alone to determine whether trimer or dimer assembly was altered. Elution volumes of trimers (T), dimers (D) and monomers (-M-) marked in red above peaks. The mutated β1 monomers eluted at the same volume as WT β1 monomer. B. β1-F77A produced dimer and small amounts of trimer. C. Concentration-dependency of trimer containing β1-F77A (the WT data are the same as in Fig. 2F). D. β1-S200R produced dimer but no trimer. E, F. β1-E204R produced neither dimer nor trimer. G, H. β1-R208E produced little or no dimer and small amounts of trimer. I, J. β1-D218R produced very little dimer and reduced amounts of trimer. In (F), (H) and (J), the arithmetic sums of β1 and γ1 monomers are shown for comparison. K,L. Concentration-dependency of trimer and dimer containing β1-R208E and β1-D218R (the WT data are the same as in Fig. 2F).
Seven amino acid substitutions were introduced in the LN domain of α1-LN-LEa. Of these, five prevented trimer but not dimer assembly (Fig. 8). The residues are located in the toe region (K58E, E61R, and R263D) and the heel region (Y128R and E203R) of the polymerization face of α1 LN. Two additional substitutions (N210R, R212E) had no adverse effect on trimer assembly. Interestingly, the α1R263D mutation targets the analogous position of α5R291L that is found to cause a group of congenital defects [16].
Fig. 8. SEC analysis of dimer and trimer assembly involving β1-LN-LEa mutants.

A. The front face of the β1 LN domain was modified with the indicated single amino acid substitutions. The mutated β1 proteins (labeled “β1M” in graphs) were analyzed by SEC in combination with WT β1-LN-LEa and γ1-LN-LEa to determine whether trimer assembly was altered. Elution volumes of trimers (T), dimers (D) and monomers (-M-) marked in red above peaks. The mutated α1 monomers eluted at the same volume as WT α1 monomer. All mutants produced dimer but no trimer: B. α1-R263D C. α1-R263L D. α1-E61R. E. α1-Y128R F. α1-E203R.
Several amino acid substitutions in the γ1 and α1 LN domains did not prevent dimer or trimer assembly (Suppl. Fig. 2). β1D196R and γ1K268A, the latter located in a toe region loop posterior to the front face, were indistinguishable from WT γ1. γ1D266R, located immediately adjacent to K268, also did not prevent dimer or trimer assembly. However, the SEC profiles of γ1D266R differed from that of WT in that the elevated valley normally lying between the trimer and dimer peaks was absent at all concentrations examined. This WT inter-peak zone, containing all three proteins and eluting larger than dimer, is presumed to represent trimer in the process of dissociating during the chromatography run. This finding suggested that γ1D266R stabilized the trimer, a potential benefit for negative stain and future crystallographic or cryo-EM analysis. The γ1D266R LN-LEa protein interactions were evaluated further. The trimer apparent dissociation constant was nearly identical to those of WT, whereas the dissociation was somewhat slower, suggesting that both forward and reverse rates were slowed. Slowing the flow rate reduced WT trimer while increasing the inter-peak protein species while slowing the flow rate had almost no effect on the γ1D266R trimer profile. Further, while WT trimer was completely prevented with EDTA, γ1D266R trimer was only partially prevented by EDTA. Finally, γ1D266R trimer and dimer assembly were not observed when incubated with β1E204R or β1R208E, just like WT. Two α1 LN mutations (N210R and R212E), located to one side of the heel region of the front face, behaved similarly if not identically to WT dimers and trimers. It was thought that these residues lie to the periphery of the heel polymerization patch.
The consequences of amino acid substitutions in all three subunit LN-LEa proteins are summarized in Fig. 9. The only LN modifications that eliminated both trimers and dimers are those located in the toe of the γ1 LN domain and the heel of the β1 LN domain. Substitutions that eliminated only trimers but retained dimers map to the heel of γ1, toe of β1, and both toe and heel of α1. From this it is deduced that the γ1-toe bind to the β1-heel, the γ1-heel binds to the α1-toe, and the β1-toe binds to the α1-heel.
Fig. 9. Summary of mutations that prevented or greatly reduced trimer or dimer assembly.

The LN-LEa segments of the laminin α1, β1 and γ1 chains have the shape of a foot, with the toes corresponding to the tip of the LN domain and the heel corresponding to the LN region near the juncture with the first LEa domain. Modifications in the γ1-toe and the β1-heel produce only monomers. All other modifications produce β1-γ1 dimers but no trimers. From this it is deduced that the γ1-toe bind to the β1-heel, the γ1-heel binds to the α1-toe, and the β1-toe binds to the α1-heel. This pseudo-symmetric arrangement places the front faces of the three LN domains in the center of the triskelion, with polymerization-critical residues facing inward towards each other.
Discussion
The organization of the laminin polymer node has eluded resolution since the discovery of polymerization more than three decades ago. This is the case despite the resolution of other laminin domains and domain complexes by various research groups [30]. The laminin polymer node is important for several reasons. First, it provides a key structural element of BMs. Loss of polymerization results in BM attenuation with decreased laminin. Second, a series of muscle/nerve and kidney/eye disorders result from mutations within the LN polymerization domains. Third, the LN domain can bind to netrin-4, an LN-LEa containing protein that can vary the degree of laminin polymerization, stiffness and cell behavioral responses by inhibiting node formation [31, 32]. Fourth, a LaNT splice-variant protein of the α3 laminin subunit can bind to the node, altering the polymer and cell functions [8, 33].
In this study we have used a combination of negative staining electron microscopy with image averaging and SEC of WT and mutated LN-LEa glycoproteins to visualize the trimer node and gain insights into its molecular organization. The trimer nodes had a highly symmetrical triskelion appearance with the thicker LN domains touching each other in a heel-to-toe arrangement. The narrower rod segments project out from the central ring-like structure, roughly every 120°. From the SEC experiments with mutated LN-LEa proteins we deduced that the γ1-toe pairs with the β1-heel, the β1-toe pairs with the α1-heel, and the α1-toe pairs with the γ1-heel. The flat nature of the triskelion suggests that in the intact laminin polymer, the polymer node is the same distance from the cell surface as the short arms themselves, producing an almost sheet-like layer above and parallel to that of the cell surface.
An intriguing consequence of the polymer node organization is that the polymer layer is expected to be anisotropic: Since the short arm vectors of a given chain type are parallel in the polymer, there are three non-equivalent principal directions related by a ~120° rotation. It is possible that such anisotropy would affect cell behavior such as migration through receptor-α-short arm interactions. Ligands for integrin α1β1, α2β1 and α3β1, detected in α1, α2 and α5 LN domain-containing short arm fragments might serve this function [34-37].
One question that arises is whether netrin-4, which binds strongly to the γ1-LN-LEa segment and which inhibits laminin polymerization, uses a closely related set of interactions to form a dimer [31]. Netrin-4 possesses a similar LN backbone structure and shares a number of conserved residues with β1-LN in the front face. For example, mouse netrin-4 LN domain mutations E195R and R199A substantially contribute to loss of laminin binding [31]. These residues correspond to E204 and R208 in mouse β1, both shown here to be involved in polymerization. Other residues in the polymerization face are shared as well, suggesting that netrin-4 and β1 bind γ1 in a similar manner. In contrast, the toe regions of β1 and netrin-4 are dissimilar. There is a serine in the position corresponding to β1S68/β2S80 in netrin-4, but β1F77 and β1S200 are replaced non-conservatively by an aspartic acid and a disulfide-forming cysteine, respectively, in netrin-4. Therefore, netrin-4 is predicted not to be able to bind to laminin α1.
It has been proposed that netrin-4 is a tunable regulator of laminin polymerization [31]. A recent study has also proposed that BM stiffness, notably resulting from the state of the laminin polymer, is an important determinant of metastases formation in various tissues [32]. Further, netrin-4 was identified as a modulator of laminin polymer stiffness in a concentration-dependent manner. Netrin-4, in binding to laminin, caused a reduction of stiffness that resulted in a decrease in malignant tumor spread in vitro, with clinical evidence that netrin-4 levels correlate with tumor metastases and patient survival. Another netrin-like protein, α3-LaNT, is a splice variant of the α3 laminin subunit and consists of an LN with two adjacent LE domains [33]. Given that a similar α3B LN-LEa fragment has been found to inhibit laminin-111 polymerization at lower concentrations compared to α1 and α5 [8], one can hypothesize that α3-LaNT should block conversion of polymer node dimers to trimer, thus acting as another inhibitor of laminin polymerization. In vitro studies revealed that α3-LaNT alters cell adhesion and wound closure, perhaps operating through such a mechanism [33]. Given the wide tissue distribution of the protein, it might serve as another general regulator of laminin polymerization [38].
Autosomal recessive missense mutations in the genes coding for LN domains have been reported for the α2 and β2 subunits and identified as causes of laminin-deficient muscular dystrophy/neuropathy and Pierson syndrome glomerulonephropathy/retinopathy respectively [14, 16, 18, 39-44]. These mutations alter conserved residues that map either to the front face or the interior of the LN domain. Mutations that remove one member of a cysteine pair (e.g. α2C79R, β2C182Y) or that alter the LN interior (e.g. α2S146F, β2L139P) are most likely to disrupt the LN fold while others (e.g. α2G284R, β2S80R, β2H147R) are more likely to directly affect a binding residue. Mutations affecting the LN domains of other laminin subunits in humans are beginning to emerge as well. One, α5R286L (α5R291L in a corresponding mouse model), was associated with kidney reflux, craniofacial malformations, and syndactyly of the toes [16]. The mutation, evaluated in vitro in laminin with homologous α1R263L protein, was found to ablate laminin polymerization. New mutations in laminin LN domains continue to be identified. The ability to predict whether these mutations adversely affect polymerization may be improved both by the available crystal structures and the systematic topographical mapping of the interacting LN faces.
A final comment is that the actual residue interactions between the LN domains remain to be identified. Higher resolution methods will be required to work out these molecular details. The trimeric complexes used in the current study are likely suitable to allow for either crystallization or cryo-electron microscopy to determine this structural detail.
Experimental Procedures:
Plasmid construction and purification of LN-LEa proteins.
Two step overlapping PCR was used to generate a 1.6 Kb NheI-XhoI insert of N-flag α1LNLEa from the N-flag α1LNNd pcDNA3.1 Zeocin [45]. Briefly, a primary 5’pcr product was produced using β1F1 1F-5’GGCGTGGATAGCGGTTTGAC3’ and α1LNLEa-2R-5’ CCCTCTAGACTCGAGCTACTCGGAGCAGCCCTC3’. The 3’ product was generated with α1LNLEa-2F-5’ GAGGGCTGCTCCGAGTAGCTCGAGTCTAGAGGG3’ and α1 LNLEa Xho1R-5’ GAAGCCATAGAGCCCACCGCATC3’. The PCR products were sewn together with the β1F1 and α1 LNLEa Xho1R primers and inserted in to the NheI –XhoI site of N-flag α1LNNd pcDNA3.1 Zeocin. All subsequent α1 point mutations were made with 2F and 2R primers (Suppl. Table I) and inserted into αLNLEa N-flag pcDNA3.1 Zeocin with HindIII-XagI (1489bp). A C-flag tagged β1LNLEa was derived from N-HA βLNNd pcDNA3.1 Zeocin (McKee 2018). PCR of 5’ (β1F1 1F-5’GGCGTGGATAGCGGTTTGAC3’ and β1LNLEa 2F-5’ GATTTGGATGGATGTCGAGACTACAAGGACGACGATG3’) and 3’ (β1LNLEa-2R-5’ CATCGTCGTCCTTGTAGTCTCGACATCCATCCAAATC3’ and β1LNLEa Xba1R-5’ GCCATAGAGCCCACCGCATC3’) was sewn together using 1F and 1R followed by NheI XbaI digest (1695 bp) and insertion into N-HA βLNNd pcDNA3.1 Zeocin. Point mutations of β1LNLEa were made with the 2F and 2R primers and inserted into N-HA βLNLEa c-flag pcDNA3.1 Zeocin through HindIII-XagI (826 bp). The γ1LNLEa was produced with overlapping PCR of 5’ (γ1F1 1F −5’CGGTAGGCGTGTACGGTGGGAG3’ and γ12R-5’-GTCGTCCTTGTAGTCGTCTGAGATCACGGCG3’) and 3’ (γ12F-5’CGCCGTGATCTCAGACGACTACAAGGACGAC3’ and γ1Apa1R-5’-GACACCTACTCAGACAATGCGATGC3’) products using 1F and 1R, followed by ApaI (1105 bp) digest of hγ1 c-Flag pRC/CMV G418 [28]. Point mutations were made with the 2F and 2R primers and inserted into ApaI (1105 bp) hγ1 pRC/CMVG418 or HindIII-Eco32I (1152) of γLNNd pcDNA3.1 Zeocin (mutants R147E and Y145R).
Mammalian Cell Culture and protein purification.
Human embryonic kidney cells (HEK293 cells) were cultured in DMEM (Invitrogen) supplemented with 10% Fetal Bovine Serum (Atlanta Biological), 200mM L-Glutamine and Penicillin-Streptomycin (1,000 u/ml Penicillin and 1,000 μg/ml Streptomycin, Invitrogen). Plasmids were stably transfected into HEK293 cells with Lipofectamine LTX (Invitrogen) according to manufacture instructions. Stable cell lines expressing were supplemented with Puromycin, Zeocin or G418 at a final concentration of 1, 100, and 500 μg/ml, respectively. Anti-FLAG beads (A2220, Sigma) were used in immunoprecipitation followed by SDS-Page and Coomassie blue staining to screen stable cell lines. The recombinant proteins were purified from media on anti-FLAG M2-agarose (Sigma) and eluted with 100ug/ml FLAG peptide in wash buffer (150mM NaCl, 50mM Tris pH 7.4, 1mMEDTA), concentration in an Amicon Ultra-15 filter (30K MWCO), and dialyzed in TBS50 (90 mM NaCl, 50 mM Tris pH 7.4, 0.125mM EDTA). Protein concentrations were determined by absorbance at 280nm. Proteins were solubilized in Laemmli sample buffer and evaluated under reducing 7.5% SDS-Polyacrylamide gel electrophoresis. Gels were stained with Coomassie Brilliant Blue R-250, imaged with BioRad Gel Doc 2000 and analyzed with Quantity One software (BioRad). For FPLC analysis of trimer and dimer complexes, proteins were concentrated in Amicon Ultra 0.5ml (Millipore, 10K) to 150ul to desired concentrations and incubated with 1mM calcium at 27C for 1 hour.
The calculated protein masses and extinction coefficients for the wild-type (WT) α1, β1 and γ1 proteins from sequence are 55.6 kDa (ε=82,260 M−1cm−1), 57.61 (ε=67,250 M−1cm−1) and 52.04 (ε=62,320 M−1cm−1) respectively. The sequences of the WT proteins are provided in Suppl. Table II. These factors were used to convert measured absorbance values (280 nm) into molar concentrations. WT and amino acid-substituted recombinant α1, β1 and γ1 LN-LEa proteins with amino acid substitutions were similarly prepared and purified. The modified proteins were found to migrate with same as their WT proteins by SDS-PAGE.
LN-LEa protein domain self-assembly into β1-γ1 dimers and α1-β1-γ1 trimers.
The proteins, following concentration, were analyzed alone or in combination at concentrations ranging from 0.04 to 2 mg/ml in Tris-buffered saline (0.05 M Tris-HCl, 0.1 M NaCl, pH 7.4) with 2 mM CaCl2 or (if indicated) 5 mM EDTA. Incubations were performed at 27°C for 1 hour prior to analysis.
Size-exclusion chromatography.
Size-exclusion chromatography was performed with a Superdex 200 Increase 10/300 GL (GE Healthcare) column connected to an AKTA FPLC system (Pharmacia/GE Healthcare) controlled by a computer using Unicorn 5.3 software. The column was maintained in a buffer of 20 mM HEPES, 150 mM NaCl, 20 mM imidazole, 2 mM CaCl2, and 0.02% NaN3, pH 7.5 (at RT). Aliquots (0.15 ml) of proteins were injected into the column through a 2 ml injector loop with a general flow rate of 1.0 ml/min at room temperature. 0.5 ml fractions were collected for analysis by reducing SDS-PAGE (7.5% acrylamide gels). For evaluation of the concentration dependency of trimer assembly, the fraction of protein eluting as trimer was calculated as the integrated sum of absorbance values eluting slower than dimer (trimer peak + tail) using Unicorn software. For evaluation of the concentration dependency of dimer assembly, the fraction of protein eluting as dimer was calculated as the integrated sum of absorbance values eluting earlier than the mathematical sum of monomeric proteins at the same concentration.
Negative staining electron microscopy.
Aliquots (100 μl) of protein samples at ~ 3-7 μg/ml were pipetted onto a paraffin sheet. Glow discharged carbon-coated grids were placed face-down onto the protein solution for 5 min. The grids were then lifted off of the drop, wicked (Whatman #1 paper), and placed on a separate drop of freshly prepared 1.5% uranyl acetate containing 0.125% trehalose for 30 sec. The grids were then lifted off of the stain, wicked, and allowed to dry face up on a paraffin sheet sitting on ice. Grids were examined in a CM12 electron microscope (Philips/FEI) at 100 kV and a magnification of 60,000 or 75,000. Multiple images were recorded with a digital camera (3.0 or 3.5 Å/pixel). Images were then subjected to analysis in the program Eman 2.2. [46]. Following particle selection (“boxing”), 16 – 32 bispectrum-based class averages were generated.
Supplementary Material
Highlights:
Laminin-111 short arm LN domains front faces bind to each other to form a polymer node organized as a symmetrical flat triskelion.
The polymer node LN domains have a toe-to-heel organization.
The gamma LN toe binds to the beta LN heel.
The gamma LN heel and the beta LN toe bind to the alpha LN toe and heel respectively.
Acknowledgments
Research in the laboratory of P.D.Y was funded by N.I.H. grant R01-DK36425. Research in the laboratory of E.H. was funded by a Wellcome Trust Senior Investigator Award (101748/Z/13/Z).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Yurchenco PD, Basement membranes: cell scaffoldings and signaling platforms, Cold Spring Harb Perspect Biol 3(2) (2011) a004911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Pozzi A, Yurchenco PD, Iozzo RV, The nature and biology of basement membranes, Matrix Biol 57-58 (2017) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Morrissey MA, Sherwood DR, An active role for basement membrane assembly and modification in tissue sculpting, J Cell Sci 128(9) (2015) 1661–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ramos-Lewis W, Page-McCaw A, Basement membrane mechanics shape development: Lessons from the fly, Matrix Biol 75-76 (2019) 72–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hohenester E, Yurchenco PD, Laminins in basement membrane assembly, Cell Adh Migr 7(1) (2013) 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Macdonald PR, Lustig A, Steinmetz MO, Kammerer RA, Laminin chain assembly is regulated by specific coiled-coil interactions, J Struct Biol 170(2) (2010) 398–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cheng YS, Champliaud MF, Burgeson RE, Marinkovich MP, Yurchenco PD, Self-assembly of laminin isoforms, J Biol Chem 272(50) (1997) 31525–32. [DOI] [PubMed] [Google Scholar]
- [8].Garbe JH, Gohring W, Mann K, Timpl R, Sasaki T, Complete sequence, recombinant analysis and binding to laminins and sulphated ligands of the N-terminal domains of laminin [alpha]3B and [alpha]5 chains, Biochem J 362(Pt 1) (2002) 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Miner JH, Cunningham J, Sanes JR, Roles for laminin in embryogenesis: exencephaly, syndactyly, and placentopathy in mice lacking the laminin alpha5 chain, J Cell Biol 143(6) (1998) 1713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miner JH, Li C, Mudd JL, Go G, Sutherland AE, Compositional and structural requirements for laminin and basement membranes during mouse embryo implantation and gastrulation, Development 131(10) (2004) 2247–56. [DOI] [PubMed] [Google Scholar]
- [11].Smyth N, Vatansever HS, Murray P, Meyer M, Frie C, Paulsson M, Edgar D, Absence of basement membranes after targeting the LAMC1 gene results in embryonic lethality due to failure of endoderm differentiation, J Cell Biol 144(1) (1999) 151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Alpy F, Jivkov I, Sorokin L, Klein A, Arnold C, Huss Y, Kedinger M, Simon-Assmann P, Lefebvre O, Generation of a conditionally null allele of the laminin alpha1 gene, Genesis 43(2) (2005) 59–70. [DOI] [PubMed] [Google Scholar]
- [13].Matejas V, Hinkes B, Alkandari F, Al-Gazali L, Annexstad E, Aytac MB, Barrow M, Blahova K, Bockenhauer D, Cheong HI, Maruniak-Chudek I, Cochat P, Dotsch J, Gajjar P, Hennekam RC, Janssen F, Kagan M, Kariminejad A, Kemper MJ, Koenig J, Kogan J, Kroes HY, Kuwertz-Broking E, Lewanda AF, Medeira A, Muscheites J, Niaudet P, Pierson M, Saggar A, Seaver L, Suri M, Tsygin A, Wuhl E, Zurowska A, Uebe S, Hildebrandt F, Antignac C, Zenker M, Mutations in the human laminin beta2 (LAMB2) gene and the associated phenotypic spectrum, Hum Mutat 31(9) (2010) 992–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jimenez-Mallebrera C, Brown SC, Sewry CA, Muntoni F, Congenital muscular dystrophy: molecular and cellular aspects, Cell Mol Life Sci 62(7-8) (2005) 809–23. [DOI] [PubMed] [Google Scholar]
- [15].McGowan KA, Marinkovich MP, Laminins and human disease Microsc Res Tech 51(3) (2000) 262–79. [DOI] [PubMed] [Google Scholar]
- [16].Jones LK, Lam R, McKee KK, Aleksandrova M, Dowling J, Alexander SI, Mallawaarachchi A, Cottle DL, Short KM, Pais L, Miner JH, Mallett AJ, Simons C, McCarthy H, Yurchenco PD, Smyth IM, A mutation affecting laminin alpha 5 polymerisation gives rise to a syndromic developmental disorder, Development (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Funk SD, Bayer RH, McKee KK, Okada K, Nishimune H, Yurchenco PD, Miner JH, A deletion in the N-terminal polymerizing domain of laminin beta2 is a new mouse model of chronic nephrotic syndrome, Kidney Int (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].McKee KK, Aleksandrova M, Yurchenco PD, Chimeric Protein Identification of Dystrophic, Pierson and Other Laminin Polymerization Residues, Matrix Biol 67 (2018) 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].McKee KK, Crosson SC, Meinen S, Reinhard JR, Ruegg MA, Yurchenco PD, Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype, J Clin Invest 127(3) (2017) 1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Anguiano M, Morales X, Castilla C, Pena AR, Ederra C, Martinez M, Ariz M, Esparza M, Amaveda H, Mora M, Movilla N, Aznar JMG, Cortes-Dominguez I, Ortiz-de-Solorzano C, The use of mixed collagen-Matrigel matrices of increasing complexity recapitulates the biphasic role of cell adhesion in cancer cell migration: ECM sensing, remodeling and forces at the leading edge of cancer invasion, PLoS One 15(1) (2020) e0220019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Colognato H, Yurchenco PD, The laminin alpha2 expressed by dystrophic dy(2J) mice is defective in its ability to form polymers, Curr Biol 9(22) (1999) 1327–1330. [DOI] [PubMed] [Google Scholar]
- [22].Yurchenco PD, Tsilibary EC, Charonis AS, Furthmayr H, Laminin polymerization in vitro. Evidence for a two-step assembly with domain specificity, J.Biol.Chem. 260 (1985) 7636–7644. [PubMed] [Google Scholar]
- [23].Purvis A, Hohenester E, Laminin network formation studied by reconstitution of ternary nodes in solution, J Biol Chem 287(53) (2012) 44270–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yurchenco PD, Cheng YS, Colognato H, Laminin forms an independent network in basement membranes, J Cell Biol 117(5) (1992) 1119–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yurchenco PD, Cheng YS, Self-assembly and calcium-binding sites in laminin. A three-arm interaction model, J. Biol. Chem. 268 (1993) 17286–17299. [PubMed] [Google Scholar]
- [26].Carafoli F, Hussain SA, Hohenester E, Crystal structures of the network-forming short-arm tips of the laminin beta1 and gamma1 chains, PLoS One 7(7) (2012) e42473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hussain SA, Carafoli F, Hohenester E, Determinants of laminin polymerization revealed by the structure of the alpha5 chain amino-terminal region, EMBO Rep 12(3) (2011) 276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].McKee KK, Harrison D, Capizzi S, Yurchenco PD, Role of laminin terminal globular domains in basement membrane assembly, J Biol Chem 282(29) (2007) 21437–47. [DOI] [PubMed] [Google Scholar]
- [29].De Carlo S, Harris JR, Negative staining and cryo-negative staining of macromolecules and viruses for TEM, Micron 42(2) (2011) 117–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hohenester E, Structural biology of laminins, Essays Biochem 63(3) (2019) 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Reuten R, Patel TR, McDougall M, Rama N, Nikodemus D, Gibert B, Delcros JG, Prein C, Meier M, Metzger S, Zhou Z, Kaltenberg J, McKee KK, Bald T, Tuting T, Zigrino P, Djonov V, Bloch W, Clausen-Schaumann H, Poschl E, Yurchenco PD, Ehrbar M, Mehlen P, Stetefeld J, Koch M, Structural decoding of netrin-4 reveals a regulatory function towards mature basement membranes, Nat Commun 7 (2016) 13515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Reuten R, Zendehroud S, Nicolau M, Fleischhauer L, Laitala A, Kiderlen S, Nikodemus D, Wullkopf L, Nielsen SR, McNeilly S, Prein C, Rafaeva M, Schoof EM, Furtwangler B, Porse BT, Kim H, Won KJ, Sudhop S, Zornhagen KW, Suhr F, Maniati E, Pearce OMT, Koch M, Oddershede LB, Van Agtmael T, Madsen CD, Mayorca- Guiliani AE, Bloch W, Netz RR, Clausen-Schaumann H, Erler JT, Basement membrane stiffness determines metastases formation, Nat Mater (2021). [DOI] [PubMed] [Google Scholar]
- [33].Hamill KJ, Langbein L, Jones JC, McLean WH, Identification of a novel family of laminin N-terminal alternate splice isoforms: structural and functional characterization, J Biol Chem 284(51) (2009) 35588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Colognato-Pyke H, O'Rear JJ, Yamada Y, Carbonetto S, Cheng YS, Yurchenco PD, Mapping of network-forming, heparin-binding, and alpha 1 beta 1 integrin-recognition sites within the alpha-chain short arm of laminin- 1, J. Biol. Chem. 270(16) (1995) 9398–406. [DOI] [PubMed] [Google Scholar]
- [35].Colognato H, MacCarrick M, O'Rear JJ, Yurchenco PD, The laminin alpha2-chain short arm mediates cell adhesion through both the alpha1beta1 and alpha2beta1 integrins, J Biol Chem 272(46) (1997) 29330–6. [DOI] [PubMed] [Google Scholar]
- [36].Ettner N, Gohring W, Sasaki T, Mann K, Timpl R, The N-terminal globular domain of the laminin alpha1 chain binds to alpha1beta1 and alpha2beta1 integrins and to the heparan sulfate- containing domains of perlecan FEBS Lett 430(3) (1998) 217–21. [DOI] [PubMed] [Google Scholar]
- [37].Nielsen PK, Yamada Y, Identification of cell-binding sites on the Laminin alpha 5 N-terminal domain by site-directed mutagenesis, J Biol Chem 276(14) (2001) 10906–12. [DOI] [PubMed] [Google Scholar]
- [38].Troughton LD, Reuten R, Sugden CJ, Hamill KJ, Laminin N-terminus alpha31 protein distribution in adult human tissues, PLoS One 15(12) (2020) e0239889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Matejas V, Al-Gazali L, Amirlak I, Zenker M, A syndrome comprising childhood-onset glomerular kidney disease and ocular abnormalities with progressive loss of vision is caused by mutated LAMB2, Nephrol Dial Transplant 21(11) (2006) 3283–6. [DOI] [PubMed] [Google Scholar]
- [40].Funk SD, Lin MH, Miner JH, Alport syndrome and Pierson syndrome: Diseases of the glomerular basement membrane, Matrix Biol 71-72 (2018) 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gavassini BF, Carboni N, Nielsen JE, Danielsen ER, Thomsen C, Svenstrup K, Bello L, Maioli MA, Marrosu G, Ticca AF, Mura M, Marrosu MG, Soraru G, Angelini C, Vissing J, Pegoraro E, Clinical and molecular characterization of limb-girdle muscular dystrophy due to LAMA2 mutations, Muscle Nerve 44(5) (2011) 703–9. [DOI] [PubMed] [Google Scholar]
- [42].Di Blasi C, Piga D, Brioschi P, Moroni I, Pini A, Ruggieri A, Zanotti S, Uziel G, Jarre L, Della Giustina E, Scuderi C, Jonsrud C, Mantegazza R, Morandi L, Mora M, LAMA2 Gene Analysis in Congenital Muscular Dystrophy: New Mutations, Prenatal Diagnosis, and Founder Effect, Arch Neurol 62(10) (2005) 1582–6. [DOI] [PubMed] [Google Scholar]
- [43].Lehnhardt A, Lama A, Amann K, Matejas V, Zenker M, Kemper MJ, Pierson syndrome in an adolescent girl with nephrotic range proteinuria but a normal GFR, Pediatr Nephrol (2012). [DOI] [PubMed] [Google Scholar]
- [44].Mohney BG, Pulido JS, Lindor NM, Hogan MC, Consugar MB, Peters J, Pankratz VS, Nasr SH, Smith SJ, Gloor J, Kubly V, Spencer D, Nielson R, Puffenberger EG, Strauss KA, Morton DH, Eldahdah L, Harris PC, A novel mutation of LAMB2 in a multigenerational mennonite family reveals a new phenotypic variant of Pierson syndrome, Ophthalmology 118(6) (2011) 1137–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].McKee KK, Capizzi S, Yurchenco PD, Scaffold-forming and adhesive contributions of synthetic laminin-binding proteins to basement membrane assembly, J Biol Chem 284(13) (2009) 8984–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ, EMAN2: an extensible image processing suite for electron microscopy, J Struct Biol 157(1) (2007) 38–46. [DOI] [PubMed] [Google Scholar]
- [47].Harrison D, Hussain SA, Combs AC, Ervasti JM, Yurchenco PD, Hohenester E, Crystal structure and cell surface anchorage sites of laminin alpha 1LG4-5, J Biol Chem 282 (2007) 11573–11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.