

Abstract
Background
Collagen VI-related dystrophy spans a clinical continuum from severe Ullrich congenital muscular dystrophy to milder Bethlem myopathy. This disease is caused by causative variants in COL6A1, COL6A2, or COL6A3. Most reported causative variants are de novo; therefore, to identify possible associated causative variants, comprehensive large cohort studies are required for different ethnicities.
Methods
We retrospectively reviewed clinical information, muscle histology, and genetic analyses from 147 Japanese patients representing 130 families, whose samples were sent for diagnosis to the National Center of Neurology and Psychiatry between July 1979 and January 2020. Genetic analyses were conducted by gene-based resequencing, targeted panel resequencing, and whole exome sequencing, in combination with cDNA analysis.
Results
Of a total of 130 families with 1–5 members with collagen VI-related dystrophy, 120 had mono-allelic and 10 had bi-allelic variants in COL6A1, COL6A2, or COL6A3. Among them, 60 variants were in COL6A1, 57 in COL6A2, and 23 in COL6A3, including 37 novel variants. Mono-allelic variants were classified into four groups: missense (69, 58%), splicing (40, 33%), small in-frame deletion (7, 6%), and large genomic deletion (4, 3%). Variants in the triple helical domains accounted for 88% (105/120) of all mono-allelic variants.
Conclusions
We report the causative variant profile of a large set of Japanese cases of collagen VI-related dystrophy. This dataset can be used as a reference to support genetic diagnosis and variant-specific treatment.
Keywords: Collagen VI-related dystrophy, Ullrich congenital muscular dystrophy, Bethlem myopathy, Sarcolemma-specific collagen VI deficiency, cDNA analysis
Background
Collagen VI is an important component of the interstitium in skeletal muscles, and consists of three chains, alpha 1, 2, and 3, which are encoded by COL6A1, COL6A2, and COL6A3 genes, respectively [1]. Causative variants in COL6A1, COL6A2, or COL6A3 cause a clinical continuum collectively called ‘collagen VI-related dystrophy’. At the more severe end of the continuum is Ullrich congenital muscular dystrophy (UCMD; OMIM 254090), and patients may have de novo variants or show autosomal recessive inheritance [2–4]. Bethlem myopathy (BM; OMIM 158810) is at the milder end, and patients mostly show autosomal dominant inheritance [4] although autosomal recessive inheritance has been reported [5, 6]. UCMD is the second- and the third- most common CMD in Japan [7] and in the UK [8]. In a study of the population in northern England, prevalence of UCMD was 0.13 cases per 100,000, whilst the prevalence of BM was 0.77 cases per 100,000 [9].
Collagen VI-related dystrophy shows characteristic clinical phenotypes, which include proximal muscle weakness, skin and joint changes, scoliosis, and respiratory failure [1, 10, 11]. Muscle pathology encompasses variable histological changes including fiber size variation, an increased number of internal nuclei, and disproportionately prominent endomysial connective tissue considering the relative scarceness of necrotic and regenerating fibers [4, 12]. We have previously reported two patterns of collagen VI distribution in muscles among patients: completely deficient (CD) or deficient on the sarcolemma but with deposits in the interstitium (sarcolemma-specific collagen VI deficiency: SSCD) [7, 13].
The eventual diagnosis of this disease is made by genetic analysis. Before and in the era of next-generation sequencing (NGS), several studies have demonstrated a genetic spectrum in collagen VI-related dystrophy, showing that a distribution of variants is common across several ethnic backgrounds [7, 11, 14–16]: the most common glycine substitution in the triple helical domain (THD), other missense variants, nonsense variants, splicing variants causing exon-skipping, small in-frame deletion/insertions, and small deletion/insertions causing a premature stop codon. Large genomic deletions spanning multiple exons are rare [10, 17–19]. Recently, a highly recurrent intronic variant in COL6A1 has been identified [20].
The aim of the present study was to elucidate the causative variant profile of collagen VI-related dystrophy in Japan by comprehensive genetic analysis including cDNA analysis, and to correlate the findings with immunostaining for collagen VI on muscle biopsies.
Results
We identified pathogenic variants in a total of 130 families with collagen VI-related dystrophy, which represented 1–5 members per family, seen at the National Center of Neurology and Psychiatry (NCNP) between July 1979 and January 2020, among them 120 families carried mono-allelic and 10 bi-allelic pathogenic variants (Table 1). One hundred and forty variants were identified, including 37 novel variants in 40 families, and these consisted of 60 allelic variants in COL6A1, 57 allelic variants in COL6A2, and 23 allelic variants in COL6A3 (Fig. 1). In 94 families with a mono-allelic variant, this was sporadic without family history (94/130, 72%). Among the 37 novel variants, we identified 24 missense variants, six splicing variants, three small in-frame deletions, three large deletions, and one nonsense variant (Fig. 2).
Table 1.
Causative variant profile of collagen VI-related dystrophy
| Family | Gene | Mono or Bi-allelic | Category | Domain | Nucleotide change | Protein change | Phenotype | COL6 IHC | Inheritance | Report |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | COL6A1 | BA | Missense | C1 |
c.1879G>C homozygous |
p.G627R | BM | Normal | Recessive | Novel |
| 2-1a | COL6A1 | MA | Splicing | N1 | c.428+1G>T | p.Y77_G143del | BM | SSCD | Dominant | [12] |
| 2-2a | COL6A1 | MA | Splicing | N1 | c.428+1G>T | p.Y77_G143del | BM | Normal | Dominant | [12] |
| 3 | COL6A1 | MA | Large deletion | THD | c.589-7_804+490del | p.E197_E285del | Intermediate | SSCD | Dominant | Novel |
| 4 | COL6A1 | MA | Large deletion | THD | c.589-7_804+490del | p.E197_E285del | UCMD | SSCD | de novo | Novel |
| 5 | COL6A1 | MA | Large deletion | THD | c.765_903+26del | p.P254_K301del | UCMD | SSCD | de novo | Novel |
| 6 | COL6A1 | MA | Glycine substitution | THD | c.806G>A | p.G269E | UCMD | SSCD | de novo | [28] |
| 7 | COL6A1 | MA | Glycine substitution | THD | c.833G>A | p.G278E | Intermediate | SSCD | de novo | [11] |
| 8 | COL6A1 | MA | Glycine substitution | THD | c.841G>A | p.G281R | Intermediate | Normal | de novo | [14] |
| 9 | COL6A1 | MA | Small deletion | THD | c.845_847del | p.E282del | UCMD | SSCD | de novo | Novel |
| 10 | COL6A1 | MA | Glycine substitution | THD | c.849G>A | p.G284R | UCMD | SSCD | Dominant | [14] |
| 11 | COL6A1 | MA | Glycine substitution | THD | c.850G>A | p.G284R | UCMD | SSCD | de novo | [14] |
| 12 | COL6A1 | MA | Glycine substitution | THD | c.850G>A | p.G284R | UCMD | SSCD | de novo | [14] |
| 13 | COL6A1 | MA | Glycine substitution | THD | c.850G>A | p.G284R | UCMD | SSCD | de novo | [14] |
| 14 | COL6A1 | MA | Glycine substitution | THD | c.850G>A | p.G284R | UCMD | SSCD | de novo | [14] |
| 15 | COL6A1 | MA | Glycine substitution | THD | c.850G>A | p.G284R | UCMD | SSCD | de novo | [14] |
| 16 | COL6A1 | MA | Glycine substitution | THD | c.859G>C | p.G287R | UCMD | NA | de novo | [35] |
| 17 | COL6A1 | MA | Glycine substitution | THD | c.860G>A | p.G287E | UCMD | SSCD | de novo | Novel |
| 18 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | UCMD | SSCD | de novo | [38] |
| 19 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | UCMD | Normal | Dominant | [38] |
| 20 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | Intermediate | SSCD | de novo | [38] |
| 21 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | UCMD | SSCD | de novo | [38] |
| 22 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | Intermediate | NA | de novo | [14] |
| 23 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | Intermediate | SSCD | de novo | [14] |
| 24 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | UCMD | SSCD | de novo | [14] |
| 25 | COL6A1 | MA | Glycine substitution | THD | c.868G>A | p.G290R | Intermediate | SSCD | de novo | [14] |
| 26-1b | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | Intermediate | NA | Dominant | [35] |
| 26-2b | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | Intermediate | NA | Dominant | [35] |
| 26-3b | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | Intermediate | NA | Dominant | [35] |
| 27 | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | UCMD | SSCD | de novo | [35] |
| 28 | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | UCMD | NA | de novo | [35] |
| 29 | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | Intermediate | SSCD | de novo | [35] |
| 30-1c | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | BM | NA | Dominant | [35] |
| 30-2c | COL6A1 | MA | Glycine substitution | THD | c.877G>A | p.G293R | BM | NA | Dominant | [35] |
| 31 | COL6A1 | MA | Glycine substitution | THD | c.895G>A | p.G299R | Intermediate | SSCD | de novo | [35] |
| 32 | COL6A1 | MA | Glycine substitution | THD | c.896G>A | p.G299E | Intermediate | SSCD | de novo | [23] |
| 33 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | UCMD | SSCD | de novo | [20] |
| 34 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | Intermediate | SSCD | de novo | [20] |
| 35 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | UCMD | SSCD | de novo | [20] |
| 36 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | UCMD | SSCD | de novo | [20] |
| 37 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | Intermediate | SSCD | de novo | [20] |
| 38 | COL6A1 | MA | Splicing | THD | c.930+189C>T | p.K310_G311insTRSTAPRRPLHLEGQGQPPRHPAK | UCMD | SSCD | de novo | [20] |
| 39 | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | Intermediate | SSCD | de novo | Novel |
| 40-1d | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | SSCD | Dominant | Novel |
| 40-2d | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | NA | Dominant | Novel |
| 41-1e | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | SSCD | Dominant | Novel |
| 41-2e | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | NA | Dominant | Novel |
| 41-3e | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | NA | Dominant | Novel |
| 41-4e | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | NA | Dominant | Novel |
| 41-5e | COL6A1 | MA | Missense | THD | c.956A>G | p.K319R | BM | NA | Dominant | Novel |
| 42 | COL6A1 | MA | Missense | THD | c.957G>T | p.K319N | UCMD | SSCD | de novo | [7] |
| 43 | COL6A1 | MA | Splicing | THD | c.958-2A>T | p.G320_K334del | UCMD | SSCD | de novo | Novel |
| 44 | COL6A1 | MA | Small deletion | THD | c.958_966del | p.G320_E322del | UCMD | SSCD | de novo | [7] |
| 45 | COL6A1 | MA | Small deletion | THD | c.958_966del | p.G320_E322del | UCMD | SSCD | de novo | [7] |
| 46 | COL6A1 | MA | Small deletion | THD | c.958_966del | p.G320_E322del | UCMD | SSCD | de novo | [7] |
| 47 | COL6A1 | MA | Small deletion | THD | c.967_975del | p.K324_G326del | UCMD | SSCD | de novo | [7] |
| 48 | COL6A1 | MA | Splicing | THD | c.1003-1G>A | p.G335_D352del | BM | SSCD | de novo | [39] |
| 49 | COL6A1 | MA | Glycine substitution | THD | c.1022G>A | p.G341D | Intermediate | SSCD | de novo | [2] |
| 50-1f | COL6A1 | MA | Glycine substitution | THD | c.1022G>A | p.G341D | BM | SSCD | Dominant | [2] |
| 50-2f | COL6A1 | MA | Glycine substitution | THD | c.1022G>A | p.G341D | BM | NA | Dominant | [2] |
| 51 | COL6A1 | MA | Glycine substitution | THD | c.1022G>T | p.G341V | BM | Normal | Dominant | [18] |
| 52 | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | Intermediate | SSCD | de novo | [30] |
| 53 | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | Intermediate | SSCD | de novo | [30] |
| 54 | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | Intermediate | SSCD | de novo | [30] |
| 55-1g | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | BM | Normal | Dominant | [30] |
| 55-2g | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | BM | NA | Dominant | [30] |
| 56-1h | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | BM | SSCD | Dominant | [30] |
| 56-2h | COL6A1 | MA | Splicing | THD | c.1056+1G>A | p.G335_D352del | BM | NA | Dominant | [30] |
| 57 | COL6A1 | MA | Splicing | THD | c.1056+3A>C | p.G335_D352del | BM | SSCD | de novo | [32] |
| 58 | COL6A1 | MA | Glycine substitution | THD | c.1138G>A | p.G380R | UCMD | SSCD | de novo | Novel |
| 59 | COL6A1 | MA | Glycine substitution | THD | c.1255G>A | p.G419S | BM | Normal | Dominant | Novel |
| 60 | COL6A1 | MA | Splicing | THD | c.1461+4A>G | p.G467_E487del | UCMD | Normal | de novo | [28] |
| 61 | COL6A2 | BA |
Splicing Splicing |
THD THD-C1 |
c.1270-1G>C c.1771-3C>G |
p.G424_K444del p.G591fs |
UCMD | CD | Recessive | [7] |
| 62 | COL6A2 | BA | Splicing | THD |
c.1572+1G>C homozygous |
p.G508_P524del | UCMD | CD | Recessive | [7] |
| 63 | COL6A2 | BA |
Splicing Small deletion |
THD C1 |
c.1770+5G>A c.2267_2272del |
p.G562_T573del p.A756_I757del |
UCMD | SSCD | Recessive |
[13] [13] |
| 64 | COL6A2 | BA |
Splicing Small deletion |
THD-C1 C1 |
c.1771-2A>T c.2279_2280del |
p.G591fs p.D761fs |
BM | CD | Recessive | [7] |
| 65-1i | COL6A2 | BA |
Missense Missense |
C1 C2 |
c.2093C>T c.2927T>C |
p.A698V p.L976S |
BM | SSCD | Recessive |
Novel Novel |
| 65-2i | COL6A2 | BA |
Missense Missense |
C1 C2 |
c.2093C>T c.2927T>C |
p.A698V p.L976S |
BM | NA | Recessive |
Novel Novel |
| 66 | COL6A2 | BA |
PTC Missense |
C2 |
c.2386A>T c.2584C>T |
p.K796X p.R862W |
UCMD | SSCD | Recessive |
Novel, [40] |
| 67 | COL6A2 | BA | Small deletion |
C1-C2 THD |
c.2678_2700del homozygous |
p.P893fs | UCMD | CD | Recessive | [7] |
| 68 | COL6A2 | BA |
Missense Small deletion |
N1 |
c.2488C>T c.1487_1512del |
p.R830W p.R498fs |
BM | SSCD | Recessive | [5, 41] |
| 69 | COL6A2 | MA | Missense | N1 | c.167G>A | p.S56N | BM | SSCD | de novo | Novel |
| 70 | COL6A2 | MA | Missense | THD | c.565G>A | p.A189T | BM | Normal | de novo | Novel |
| 71 | COL6A2 | MA | Splicing | THD | c.736-1G>A | p.C246_K267del | UCMD | SSCD | de novo | [42] |
| 72 | COL6A2 | MA | Glycine substitution | THD | c.785G>T | p.G262V | BM | SSCD | Dominant | Novel |
| 73 | COL6A2 | MA | Splicing | THD | c.801+1G>T | p.C246_K267del | UCMD | SSCD | de novo | [16] |
| 74 | COL6A2 | MA | Splicing | THD | c.801+2T>C | p.C246_K267del | UCMD | SSCD | de novo | [3] |
| 75 | COL6A2 | MA | Glycine substitution | THD | c.802G>T | p.G268C | UCMD | SSCD | de novo | Novel |
| 76 | COL6A2 | MA | Glycine substitution | THD | c.812G>A | p.G271D | Intermediate | SSCD | de novo | [7] |
| 77 | COL6A2 | MA | Glycine substitution | THD | c.820G>A | p.G274S | Intermediate | SSCD | Dominant | Novel |
| 78 | COL6A2 | MA | Glycine substitution | THD | c.821G>A | p.G274D | Intermediate | SSCD | de novo | Novel |
| 79 | COL6A2 | MA | Glycine substitution | THD | c.838G>C | p.G280R | Intermediate | SSCD | de novo | Novel |
| 80 | COL6A2 | MA | Glycine substitution | THD | c.839G>A | p.G280D | BM | SSCD | de novo | Novel |
| 81 | COL6A2 | MA | Splicing | THD | c.855+1G>A | p.G268_Q285del | UCMD | SSCD | de novo | [35] |
| 82 | COL6A2 | MA | Splicing | THD | c.856-2A>G | p.G286_K309del | UCMD | SSCD | Dominant | [7] |
| 83-1j | COL6A2 | MA | Splicing | THD | c.856-2A>G | p.G286_K309del | BM | SSCD | Dominant | [7] |
| 83-2j | COL6A2 | MA | Splicing | THD | c.856-2A>G | p.G286_K309del | BM | NA | Dominant | [7] |
| 84 | COL6A2 | MA | Glycine substitution | THD | c.866G>A | p.G289D | BM | SSCD | Dominant | [8] |
| 85 | COL6A2 | MA | Glycine substitution | THD | c.875G>T | p.G292V | UCMD | SSCD | de novo | [7] |
| 86 | COL6A2 | MA | Glycine substitution | THD | c.893G>A | p.G298E | UCMD | SSCD | de novo | Novel |
| 87 | COL6A2 | MA | Large deletion | THD | c.900+102_1000-43del | p.G301_K333del | UCMD | SSCD | de novo | Novel |
| 88 | COL6A2 | MA | Glycine substitution | THD | c.901G>T | p.G301C | UCMD | SSCD | de novo | [7] |
| 89 | COL6A2 | MA | Glycine substitution | THD | c.902G>T | p.G301V | Intermediate | SSCD | de novo | Novel |
| 90 | COL6A2 | MA | Glycine substitution | THD | c.902G>A | p.G301D | UCMD | SSCD | de novo | [7] |
| 91 | COL6A2 | MA | Glycine substitution | THD | c.902G>A | p.G301D | UCMD | SSCD | de novo | [7] |
| 92 | COL6A2 | MA | Glycine substitution | THD | c.911G>T | p.G304V | BM | SSCD | de novo | Novel |
| 93 | COL6A2 | MA | Missense | THD | c.943G>A | p.D315N | UCMD | SSCD | de novo | Novel |
| 94 | COL6A2 | MA | Missense | THD | c.943G>A | p.D315N | BM | Normal | Dominant | Novel |
| 95 | COL6A2 | MA | Splicing | THD | c.950_954+8del | p.G310_K318del | UCMD | SSCD | de novo | Novel |
| 96 | COL6A2 | MA | Splicing | THD | c.955-2A>C | p.G319_K333del | UCMD | NA | de novo | Novel |
| 97 | COL6A2 | MA | Splicing | THD | c.955-2A>G | p.G319_K333del | UCMD | SSCD | de novo | [14] |
| 98 | COL6A2 | MA | Splicing | THD | c.955-2A>G | p.G319_K333del | Intermediate | SSCD | de novo | [14] |
| 99-1k | COL6A2 | MA | Splicing | THD | c.1053+1G>A | p.G334_ R351del | BM | SSCD | Dominant | Novel |
| 99-2k | COL6A2 | MA | Splicing | THD | c.1053+1G>A | p.G334_ R351del | BM | NA | Dominant | Novel |
| 100 | COL6A2 | MA | Glycine substitution | THD | c.1664G>A | p.G555E | UCMD | SSCD | de novo | Novel |
| 101 | COL6A2 | MA | Small deletion | C1 | c.1858_1860del | p.I620del | UCMD | SSCD | de novo | Novel |
| 102 | COL6A2 | MA | Missense | C1 | c.1861G>A | p.D621N | BM | SSCD | Dominant | [2] |
| 103 | COL6A2 | MA | Missense | C1 | c.1870G>A | p.E624K | BM | Normal | de novo | [42] |
| 104 | COL6A2 | MA | Missense | C1 | c.2192C>G | p.T731R | BM | SSCD | de novo | Novel |
| 105 | COL6A2 | MA | Glycine substitution | C1 | c.2197G>A | p.G733R | BM | SSCD | de novo | [43] |
| 106 | COL6A2 | MA | Missense | C1 | c.2271C>G | p.I757M | BM | Normal | de novo | Novel |
| 107 | COL6A2 | MA | Small deletion | C2 | c.2741_2743del | p.F914del | BM | SSCD | de novo | Novel |
| 108 | COL6A2 | MA | Missense | C2 | c.2978G>A | p.R993H | BM | SSCD | de novo | Novel |
| 109 | COL6A3 | BA |
Small deletion Small deletion |
N1 C3 |
c.5692delG c.8737delG |
p.V1898fs p.A2913fs |
UCMD | CD | Recessive | [7] |
| 110 | COL6A3 | MA | Missense | N1 | c.5525G>A | p.G1842E | BM | SSCD | Dominant | [12] |
| 111 | COL6A3 | MA | Missense | N1 | c.5525G>A | p.G1842E | BM | SSCD | Dominant | [12] |
| 112-1l | COL6A3 | MA | Missense | N1 | c.5525G>A | p.G1842E | BM | NA | Dominant | [12] |
| 112-2l | COL6A3 | MA | Missense | N1 | c.5525G>A | p.G1842E | BM | NA | Dominant | [12] |
| 113 | COL6A3 | MA | Missense | THD | c.5525G>A | p.G1842E | BM | Normal | Dominant | [12] |
| 114 | COL6A3 | MA | Splicing | THD | c.6157-2A>G | p.G2053_P2070del | UCMD | SSCD | de novo | [7] |
| 115 | COL6A3 | MA | Splicing | THD | c.6157-2A>G | p.G2053_P2070del | Intermediate | SSCD | de novo | [7] |
| 116 | COL6A3 | MA | Glycine substitution | THD | c.6158G>T | p.G2053V | UCMD | SSCD | de novo | [15] |
| 117-1m | COL6A3 | MA | Missense | THD | c.6199G>A | p.E2067K | BM | SSCD | Dominant | [28] |
| 117-2m | COL6A3 | MA | Missense | THD | c.6199G>A | p.E2067K | BM | Normal | Dominant | [28] |
| 118 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | UCMD | SSCD | de novo | [25] |
| 119 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | UCMD | SSCD | de novo | [25] |
| 120 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | UCMD | SSCD | de novo | [25] |
| 121 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | UCMD | SSCD | de novo | [25] |
| 122 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | Intermediate | SSCD | de novo | [25] |
| 123 | COL6A3 | MA | Splicing | THD | c.6210+1G>A | p.G2053_P2070del | UCMD | SSCD | de novo | [25] |
| 124 | COL6A3 | MA | Splicing | THD | c.6210+2T>A | p.G2053_P2070del | UCMD | SSCD | de novo | [7] |
| 125 | COL6A3 | MA | Glycine substitution | THD | c.6212G>A | p.G2071D | UCMD | SSCD | de novo | [8] |
| 126 | COL6A3 | MA | Glycine substitution | THD | c.6247G>T | p.G2083C | Intermediate | SSCD | de novo | Novel |
| 127 | COL6A3 | MA | Splicing | THD | c.6309G>A | p.G2095_K2103del | UCMD | SSCD | de novo | Novel |
| 128 | COL6A3 | MA | Splicing | THD | c.6309+1G>A | p.G2095_K2103del | UCMD | SSCD | de novo | [35] |
| 129 | COL6A3 | MA | Splicing | THD | c.6310-2A>G | p.G2104_D2118del | UCMD | SSCD | de novo | Novel |
| 130 | COL6A3 | MA | Splicing | THD | c.6283-1G>Tn c.6310-2A>Tn | p.G2095_K2103delinsNSFLYLPVRLIPSL | Intermediate | SSCD | de novo | [37, 44] |
IHC, immunohistochemistry; MA, mono-allelic; BA, bi-allelic; PTC, premature stop codon; THD, triple helical domain; NA, not available; UCMD, Ullrich congenital muscular dystrophy; BM, Bethlem myopathy; CD, complete deficiency; SSCD, sarcolemma-specific collagen VI deficiency
abrothers; b26-2 and 26-3 are the sons of 26-1; d40-2 is the father of 40-1; e41-2 and 41-3 are the cousins of 41-1, and 41-4 and 41-5 are the sons of 41-2; fsisters; g55-2 is the son of 55-1; h56-2 is the daughter of 55-1; ibrothers; j83-2 is the mother of 83-1; kbrothers; lbrothers; m117-2 is the mother of 117-1; ntwo variants on one allele. We used the following transcripts: COL6A1, NM_001848; COL6A2, NM_001849; COL6A3, NM_004369)
Fig. 1.
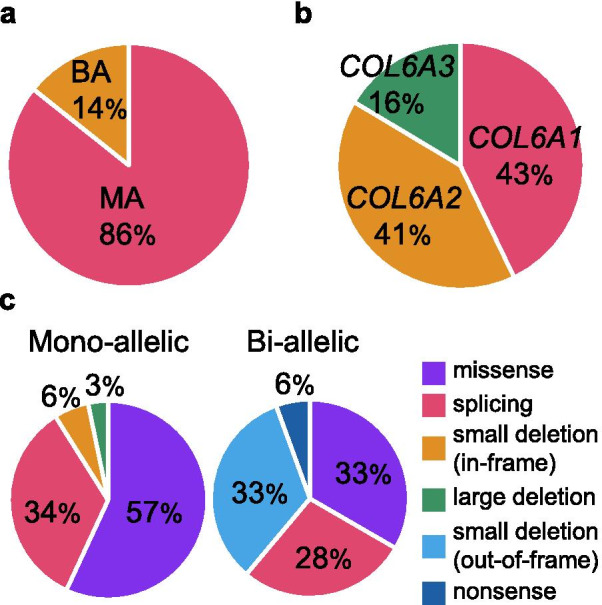
Type and frequency of variants in collagen VI-related dystrophy. The proportion of (a) bi-allelic (BA) and mono-allelic (MA) variants, and (b) variants in COL6A1, COL6A2, and COL6A3. The frequencies of various types of (c) mono-allelic and (d) bi-allelic variants.
Fig. 2.

Schematic domain structure of collagen VI polypeptide chains and localization of the identified mono-allelic variants. The identified missense variants and small in-frame deletions are indicated by triangles. Large genomic deletions, exonic deletions by splicing variants, and pseudoexon insertions are indicated by rectangles. Previously reported variants are shown in pink and novel ones in yellow. A single cysteine residue (C) in each triple helical domain (THD) is important for molecular assembly. Most mono-allelic variants are clustered in the N-terminal side of or around the cysteine residue in the THD. (Figure is
modified from Lampe et al. [14]).
Among the ten families with bi-allelic variants, in eight the variants were in COL6A2, while the other two each had variants in COL6A1, or in COL6A3. Six of these ten families had variants producing a premature termination codon or causing aberrant splicing, which leads to in-frame exon skipping in both alleles, and all had UCMD phenotypes. One of the ten families, #66, had a nonsense and a missense variant and also exhibited a UCMD phenotype. The affected individuals of the remaining three families had single nucleotide variants causing non-glycine substitutions and all showed BM phenotypes, although family #68 had a 26 bp-deletion causing a premature termination codon in one allele.
In the 120 families carrying a mono-allelic variant, the variants were as follows: missense (69, 58%), splicing (40, 33%), small in-frame deletion (7, 6%), and large deletion (4, 3%; Table 1). Variants in the THD accounted for 88% (105/120) and glycine substitution accounted for 48% (50/120). The variant c.868G>A (p.G290R) in COL6A1 was found in eight families, while in 64 (53%) of the mono-allelic variant was unique. With respect to the genotype-phenotype correlation, the majority (82%, 86/105) of families having variants in the THD showed UCMD or intermediate phenotypes, while the majority (93%, 14/15) of families harboring variants outside the THD showed milder phenotypes. It is important to note that all seven families showing the skipping of exon 14 in the THD of COL6A1 had BM or intermediate phenotypes.
Three novel heterozygous multiple exon deletions were detected in four families (Fig. 3). The deletions spanned from exon 5 to exon 8 in COL6A1 (Family #3 and #4), from exon 8 to exon 10 in COL6A1 (Family #5), and from exon 8 to exon 10 in COL6A2 (Family #87). All these large deletions were in-frame and distributed in the THD.
Fig. 3.
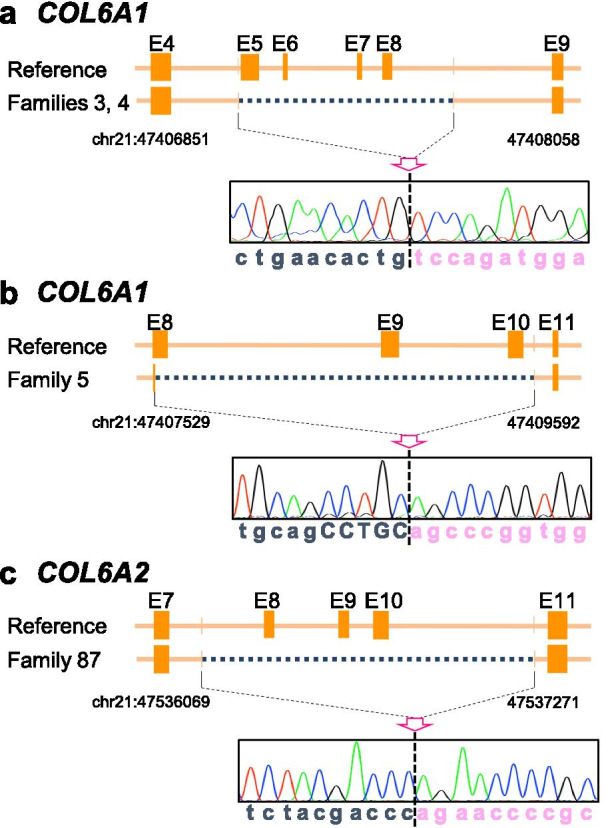
Schematic diagrams and electropherograms at breakpoints of large genomic deletions in COL6A1 and COL6A2. We found a deletion of 216 bp (COL6A1) in transcripts in Family #3 and #4, and a deletion of 144 bp (COL6A1) and 99 bp (COL6A2) in transcripts in Family #5 and #87, respectively. At the genomic level, Family #3 and #4 carried a deletion of 1.2 kb spanning from IVS4-7 to IVS8+490 in COL6A1 (a). The 5′ breakpoint of the 2.1 kb deletion found in Family #5 was located at the sixth base of exon 8 of COL6A1 and its 3′ breakpoint was at − 43 of intron 10 (b). One of the COL6A2 alleles of Family #87 contained a 1.2 kb deletion extending from IVS7+102 to IVS10-43 (c). E: exon sequence. The numbering of genomic positions at the breakpoints are based on the sequence from the Gene Reference Consortium GRCh37/hg19.
We performed immunostaining for collagen VI in muscle biopsies from 125 affected individuals in 123 families. In 115 patients with a mono-allelic variant, 91% (92/101) with the variant within and 71% (10/14) with the variant outside the THD showed SSCD. Even the biopsies from families harboring multiple exon deletions showed the typical SSCD staining pattern, suggesting dominant-negative effect of those variants (Fig. 4). Among the ten families having bi-allelic variants, five showed a CD pattern, while the five families carrying missense variant(s) showed a SSCD or a normal pattern. Observation at high magnification using immunofluorescence staining revealed trace amounts of extracellular collagen VI in the muscle biopsies of three families with CD (Family #64, #67, and #109), while collagen VI was retained within the mesenchymal cells in two families (#61 and #62; Fig. 5).
Fig. 4.
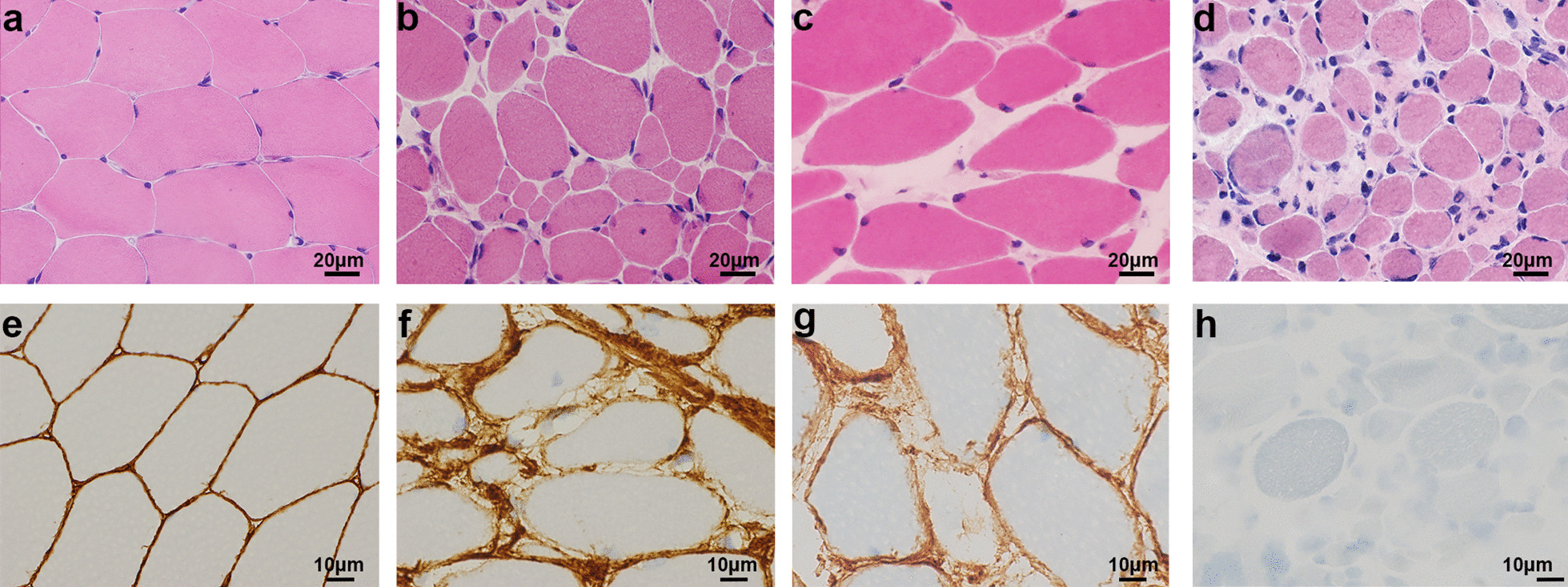
Representative muscle pathology of patients with pathogenic variants in triple helical domains. Histology of muscle from a control individual (a, e), a member of Family #87 with UCMD and a large genomic deletion (b, f), a member of Family #72 with BM with a glycine substitution in the triple helical domain (c, g), and a member of Family #109 with UCMD with bi-allelic small deletions in COL6A3 (d, h). Hematoxylin and eosin, scale bar 20 μm. (a–d) Immunostaining for collagen VI, scale bar 10 μm (e–h).
Fig. 5.
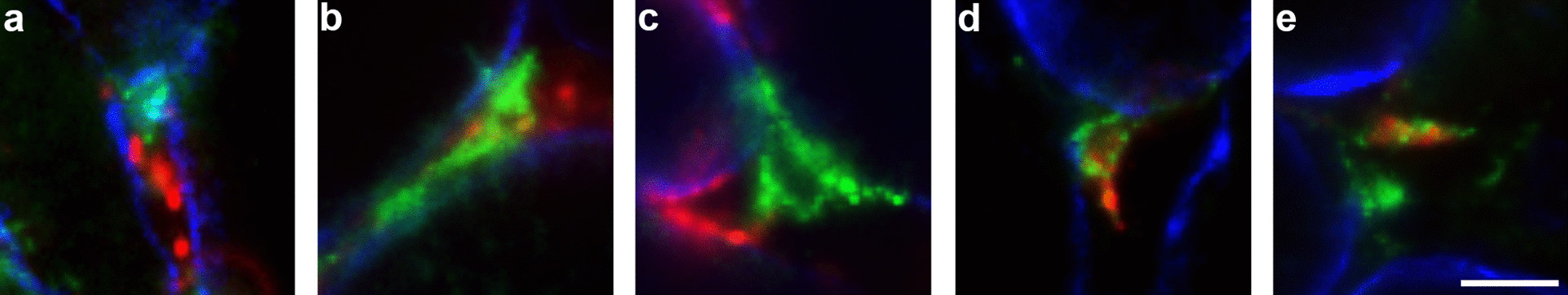
The highly sensitive detection of collagen VI in patients’ muscles showing complete deficiency by routine immunostaining. The highly sensitive immunofluorescence staining for collagen VI (green), PDGFRα (red), and laminin α2 (blue) in muscles of patients showing complete collagen VI deficiency (a, Family #64; b, Family #67; c, Family #109; d, Family #61; e, Family #62). Scale bar, 10 μm. Highly magnified immunofluorescence images showed that collagen VI formed small deposits in the extracellular space in muscles from patients with truncated variants in both alleles (a–c), while in patients with an in-frame deletion in at least one allele, the collagen VI was retained within mesenchymal cells (d, e).
We reviewed all available muscle imaging data (34 families including 23 cases and 24 cases tested by MRI and CT, respectively. Thirteen cases were tested by both modalities). At least one of three typical findings in collagen VI-related dystrophy (tigroid or outside in pattern in the vastus lateralis; target sign in the rectus femoris; a hyperintense rim between the soleus and gastrocnemius) [21] was seen in 85% (29/34) of the families. Among 29 families had mono-allelic variants in the THD, 86% (25/29) of these had typical imaging findings. Three in four families (75%) with a mono-allelic variant outside the THD. In families with bi-allelic variants, the imaging data was available in only family, who showed typical imaging findings.
Discussion
We have elucidated the causative variant profile of collagen VI-related dystrophy in Japan (Table 1). Furthermore, we report 37 novel variants in 40 families, comprising 24 missense, six splicing, three small in-frame deletion, three large genomic deletion, and one nonsense. From the genetic information, we have established the causative variant profile of the largest cohort at a single center as far as we are aware. The majority of the variants were mono-allelic (86%, 120/140), and 67% (94/140) of them were likely to be de novo because the parents of the patients were not apparently affected and their DNAs were not available, as has previously been described [11, 14, 15, 22–24]. Therefore, our causative variant profile may be useful as a reference for diverse ethnicities. Given that all cases with collagen VI-related dystrophy in this cohort were sent to our center from hospitals in Japan, we calculated the occurrence of severe UCMD in Japan as 1.63 cases per year and estimated that about 70% of collagen VI-related dystrophy were diagnosed at our center, which is an estimated incidence of 0.20 in 100,000 births, higher than that found for northern England (0.13/100,000) [9]. This is most likely because of the difference of the diagnostic system between the two countries.
Among the mono-allelic variants, 88% (105/120) were located in the THD. The association between mono-allelic variants in the THD and the SSCD staining pattern (91%, 92/101) may be explained by the fact that tetramers containing dominant mutations in the THD are secreted but cause the impaired ability to form microfibrils and the reduced binding of collagen VI to extracellular matrix [25, 26]. Furthermore, those mono-allelic variants in the THD are associated with UCMD or intermediate phenotype (82%, 86/105). In contrast, mono-allelic variants outside the THD were also associated with SSCD (71%, 10/14) but a BM phenotype (93%, 14/15) (Table 2). However, as shown in the literatures, genotypes cannot be associated with specific phenotypes, with some variants reported to cause both UCMD and BM phenotypes [14–16, 24]. In fact, in our cohort, the families with c.877G>A in COL6A1, c.856-2A>G in COL6A2, or c.943G>A in COL6A2 showed a wide range of phenotypes from milder BM to severer UCMD, while conversely the variation in phenotypes of families with c.956A>G or c.1022G>A in COL6A1 was quite narrow and those families showed BM or intermediate phenotypes.
Table 2.
Genotype-phenotype correlation of collagen VI-related dystrophy in this study
| Domain | Phenotype | IHC | |
|---|---|---|---|
| Mono-allelic | THD | UCMD (55%) | SSCD (91%) |
| Intermediate (26%) | |||
| Outside of the THD | BM (93%) | SSCD (71%) | |
| Bi-allelic | PTC in both alleles | UCMD (100%) | CD (100%) |
| Missense/in-frame deletion in at least one allele | UCMD/BM | SSCD (86%) |
IHC, immunohistochemistry; PTC, premature stop codon; THD, triple helical domain; UCMD, Ullrich congenital muscular dystrophy; BM, Bethlem myopathy; CD, complete deficiency; SSCD, sarcolemma-specific collagen VI deficiency
In addition, we found four heterozygous large deletions in families with UCMD phenotype. All the deletions were located in the N-terminal side of the cysteine residue important for the assembly of the collagen VI tetramer. This is in accordance with all the reported multiple exon deletions [17, 19, 25, 27–29]. Intriguingly, the deletion in the region containing the cysteine residue caused relatively mild phenotypes in our cohort and in those of previous reports [11, 30–32]. This may be explainable by the mechanism that the loss of the distinctive cysteine residue causes the failure in dimer formation of the mutant COL6A1, resulted in the reduced normal COL6A1 dimer production into 1/4 in amount [31]. On the contrary, deletions of the entire COL6A2 are reported to show recessively acting loss of function variants [33]. Thus, collagen VI proteins with large genomic deletions in the N-terminal side of the THD, which have the deletions no more than 72 amino acid residues, may act in a dominant-negative fashion and show UCMD or intermediate phenotypes.
In this study, we identified ten families having bi-allelic variants and five and four families showed CD and SSCD collagen VI staining patterns in muscles, respectively. We can presume that families with truncated variants in both alleles will be associated with CD and severe UCMD phenotypes, whilst those with missense variants or in-frame deletions at least in one allele will be associated with SSCD and milder BM phenotypes. In fact, three families with truncated variants in both alleles (CD) and five families with missense or in-frame deletion at least in one allele (SSCD) displayed compatible patterns with the aforementioned presumption, regardless of causative genes. Interestingly, the other two bi-allelic families had in-frame deletion(s) in one and in two alleles, but they showed CD and severe UCMD phenotypes. To explore the mechanism causing the loss of collagen VI in muscles in these families, we observed the trace of collagen VI remaining in their biopsied muscles. In muscles from patients with truncated variants in both alleles, collagen VI formed small deposits in the extracellular space, while in patients with an in-frame deletion in at least one allele, the collagen VI was retained within mesenchymal cells. Thus, we hypothesized that, from those cases with extracellular deposits visible, the truncated collagen VI molecules can form tetramers and be secreted, but the secreted collagen VI will be unstable and degraded extracellularly. On the other hand, in the cases with a retained trace, the in-frame deleted molecules failed to make a tetramer and be secreted. Additional detailed molecular analyses are required to understand the precise mechanism.
The multiple analyses (RNA analysis and immunostaining, reviewing the clinical information) were used for validation of pathogenicity of novel variants. For example, the patients with mono allelic THD variants showed missense or in-frame deletion in transcripts and SSCD staining pattern of collagen VI in muscles, and severe UCMD phenotype. In contrast, the patients with extra-THD variants showed SSCD staining pattern of collagen VI in muscles, and typically milder BM-phenotypes. This information is essentially compatible to the genotype-phenotype correlation in collagen VI-related dystrophy shown in previous reports and adds many examples. The cumulative information further contributes the establishment of the genotype-phenotype database in collagen VI-related dystrophy.
Conclusion
Our report provides a large causative variant catalog of collagen VI-related dystrophy in Japan, which can be used as a reference for genetic diagnosis and will also be helpful in variant-specific therapy in the future. The majority of causal variants of collagen VI-related dystrophy was mono-allelic de novo, and most of them were located in the THD and associated with SSCD and UCMD or intermediate phenotypes.
Methods
Clinical information
This retrospective cohort study was performed on patients seen at the NCNP, a major referral center for muscle disease in Japan, between July 1979 and January 2020. Frozen muscle and blood samples from patients were sent for diagnosis to the NCNP from all over Japan.
Clinically or pathologically suspected collagen VI-related dystrophy with possible pathogenic variants in COL6A1, COL6A2, or COL6A3 was identified in 147 affected individuals in 130 families. Patients with collagen VI-related dystrophy were classified into three categories, UCMD, intermediate and BM, according to phenotypic stratification as previously described [4, 28, 34, 35].
This study was approved by the institutional review boards of the NCNP. All the human materials used in this study were obtained for diagnostic purposes. The patients or their parents provided written informed consent for use of the samples for research.
Muscle histology
Muscle biopsy samples for histological examination were frozen in isopentane cooled in liquid nitrogen. A set of routine histochemical analyses was performed for diagnosis. When the patients were suspected of having collagen VI-related dystrophy or had elevated serum creatine kinase, immunohistochemistry was performed using standard procedures with an antibody against collagen type VI (VI-26, 1:1000; MP Biomedicals, LLC, Irvine, CA) as previously described [7]. Immunofluorescence staining using standard procedures was performed with antibodies against collagen type VI (VI-26, 1:500; MP Biomedicals), PDGFRα (1:500, Cell Signaling Technology, Danvers MA), and laminin α2 (4H8-2, 1:500; Santa Cruz, Dallas TX)[36].
Genetic analysis
Genomic DNA was isolated from peripheral blood lymphocytes or muscle specimens using standard techniques. All exons and their flanking intronic regions in COL6A1, COL6A2, and COL6A3 were amplified and sequenced directly in 52 families using an ABI PRISM 3130xl Genetic Analyzer (Applied Biosystems, Waltham, MA). Sixty-five families were analyzed using the target resequencing panel for muscular dystrophy because we developed a method for screening gene causative variant in our laboratory since 2014 using Ion PGM NGS [37]. Thirteen families were analyzed by whole exome sequencing because they were initially suspected of having other types of muscular disease.
The splice site-creating variant Chr21:47,409,881 C>T in intron 11 of COL6A1, was manually screened by the Sanger method [20].
cDNA analysis
Total RNA was extracted from frozen muscle using a Total RNA Kit (Nippon Gene, Tokyo, Japan) and cDNA was synthesized with oligo (dT)20 primer using SuperScript IV Reverse Transcriptase (Thermo Fisher Scientific, Waltham, MA) using standard techniques [13].
Identification of pathogenic variants
Novel pathogenic variants were identified using a previously described method [37] with modifications. Briefly, the likely pathogenic variants were defined according to the following criteria: (1) a glycine substitution in the THD; (2) causes exon skipping in the THD; (3) a large genomic deletion; (4) produces a nonsense codon or small insertion/deletion causing a premature stop codon in patients with bi-allelic variants; (5) a missense variant (except a glycine substitution or a substitution outside the THD). If outside the THD, the predicted amino acid substitution was a) predicted to be pathogenic by more than one in silico tool (PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), MutationTaster (http://www.mutationtaster.org/), or CADD (http://cadd.gs.washington.edu/)), and/or b) co-segregated with the phenotype within a family. Missense variants were filtered with an allele frequency threshold of < 0.01 in gnomAD (https://gnomad.broadinstitute.org/), NHLBI GO Exome Sequencing Project (http://evs.gs. washington.edu/EVS/), or the integrative Japanese Genome Variation Database (https://ijgvd.megabank.tohoku.ac.jp). The variants identified by target resequencing or whole exome sequencing were confirmed by Sanger sequencing.
Acknowledgements
The authors would like to thank the study patients, their families, and physicians.
Abbreviations
- BM
Bethlem myopathy
- CD
Complete deficiency
- SSCD
Sarcolemma-specific collagen VI deficiency
- THD
Triple helical domain
- UCMD
Ullrich congenital muscular dystrophy
Authors' contributions
MI and SN contributed to the conception and design of this study. MI, YS, TY, AI, MO, and SN analyzed and interpreted the data, and MI and SN wrote the manuscript. IN supervised the study. All authors read and approved the final manuscript.
Funding
This study was supported partly by an Intramural Research Grant (30-9, 2-5, and 2-6) for Neurological and Psychiatric Disorders of the NCNP and AMED under Grant Number JP20ek0109490h0001, and by Joint Usage and Joint Research Programs of the Institute of Advanced Medical Sciences, Tokushima University grant number 2020, 2R19.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Declarations
Ethics approval and consent to participate
All clinical information and materials used in the present study were obtained for diagnostic purposes with written informed consent. The study was approved by the Ethics Committee of the National Center of Neurology and Psychiatry (NCNP).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bönnemann CG. The collagen VI-related myopathies: muscle meets its matrix. Nat Rev Neurol. 2011;7(7):379–90. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scacheri PC, Gillanders EM, Subramony SH, Vedanarayanan V, Crowe CA, Thakore N, et al. Novel mutations in collagen VI genes: expansion of the Bethlem myopathy phenotype. Neurology. 2002;58(4):593–602. doi: 10.1212/WNL.58.4.593. [DOI] [PubMed] [Google Scholar]
- 3.Peat RA, Smith JM, Compton AG, Baker NL, Pace RA, Burkin DJ, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology. 2008;71(5):312–21. doi: 10.1212/01.wnl.0000284605.27654.5a. [DOI] [PubMed] [Google Scholar]
- 4.Yonekawa T, Nishino I. Ullrich congenital muscular dystrophy: clinicopathological features, natural history and pathomechanism(s) J Neurol Neurosurg Psychiatry. 2015;86(3):280–7. doi: 10.1136/jnnp-2013-307052. [DOI] [PubMed] [Google Scholar]
- 5.Foley AR, Hu Y, Zou Y, Columbus A, Shoffner J, Dunn DM, et al. Autosomal recessive inheritance of classic Bethlem myopathy. Neuromuscul Disord. 2009;19(12):813–7. doi: 10.1016/j.nmd.2009.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gualandi F, Urciuolo A, Martoni E, Sabatelli P, Squarzoni S, Bovolenta M, et al. Autosomal recessive Bethlem myopathy. Neurology. 2009;73(22):1883–91. doi: 10.1212/WNL.0b013e3181c3fd2a. [DOI] [PubMed] [Google Scholar]
- 7.Okada M, Kawahara G, Noguchi S, Sugie K, Murayama K, Nonaka I, et al. Primary collagen VI deficiency is the second most common congenital muscular dystrophy in Japan. Neurology. 2007;69(10):1035–42. doi: 10.1212/01.wnl.0000271387.10404.4e. [DOI] [PubMed] [Google Scholar]
- 8.Sframeli M, Sarkozy A, Bertoli M, Astrea G, Hudson J, Scoto M, et al. Congenital muscular dystrophies in the UK population: clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord. 2017;27(9):793–803. doi: 10.1016/j.nmd.2017.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132(Pt 11):3175–86. doi: 10.1093/brain/awp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Allamand V, Brinas L, Richard P, Stojkovic T, Quijano-Roy S, Bonne G. ColVI myopathies: where do we stand, where do we go? Skelet Muscle. 2011;1:30. doi: 10.1186/2044-5040-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fan Y, Liu A, Wei C, Yang H, Chang X, Wang S, et al. Genetic and clinical findings in a Chinese cohort of patients with collagen VI-related myopathies. Clin Genet. 2018;93(6):1159–71. doi: 10.1111/cge.13230. [DOI] [PubMed] [Google Scholar]
- 12.Deconinck N, Richard P, Allamand V, Behin A, Lafôret P, Ferreiro A, et al. Bethlem myopathy: long-term follow-up identifies COL6 mutations predicting severe clinical evolution. J Neurol Neurosurg Psychiatry. 2015;86(12):1337–46. doi: 10.1136/jnnp-2013-307245. [DOI] [PubMed] [Google Scholar]
- 13.Ishikawa H, Sugie K, Murayama K, Awaya A, Suzuki Y, Noguchi S, et al. Ullrich disease due to deficiency of collagen VI in the sarcolemma. Neurology. 2004;62(4):620–3. doi: 10.1212/01.WNL.0000113023.84421.00. [DOI] [PubMed] [Google Scholar]
- 14.Lampe AK, Dunn DM, von Niederhausern AC, Hamil C, Aoyagi A, Laval SH, et al. Automated genomic sequence analysis of the three collagen VI genes: applications to Ullrich congenital muscular dystrophy and Bethlem myopathy. J Med Genet. 2005;42(2):108–20. doi: 10.1136/jmg.2004.023754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butterfield RJ, Foley AR, Dastgir J, Asman S, Dunn DM, Zou Y, et al. Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 2013;34(11):1558–67. doi: 10.1002/humu.22429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim SY, Kim WJ, Kim H, Choi SA, Lee JS, Cho A, et al. Collagen VI-related myopathy: expanding the clinical and genetic spectrum. Muscle Nerve. 2018;58(3):381–8. doi: 10.1002/mus.26093. [DOI] [PubMed] [Google Scholar]
- 17.Pan TC, Zhang RZ, Sudano DG, Marie SK, Bönnemann CG, Chu ML. New molecular mechanism for Ullrich congenital muscular dystrophy: a heterozygous in-frame deletion in the COL6A1 gene causes a severe phenotype. Am J Hum Genet. 2003;73(2):355–69. doi: 10.1086/377107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lucioli S, Giusti B, Mercuri E, Vanegas OC, Lucarini L, Pietroni V, et al. Detection of common and private mutations in the COL6A1 gene of patients with Bethlem myopathy. Neurology. 2005;64(11):1931–7. doi: 10.1212/01.WNL.0000163990.00057.66. [DOI] [PubMed] [Google Scholar]
- 19.Pepe G, Lucarini L, Zhang RZ, Pan TC, Giusti B, Quijano-Roy S, et al. COL6A1 genomic deletions in Bethlem myopathy and Ullrich muscular dystrophy. Ann Neurol. 2006;59(1):190–5. doi: 10.1002/ana.20705. [DOI] [PubMed] [Google Scholar]
- 20.Cummings BB, Marshall JL, Tukiainen T, Lek M, Donkervoort S, Foley AR, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;1792:14. doi: 10.1126/scitranslmed.aal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mercuri E, Lampe A, Allsop J, Knight R, Pane M, Kinali M, et al. Muscle MRI in Ullrich congenital muscular dystrophy and Bethlem myopathy. Neuromuscul Disord. 2005;15(4):303–10. doi: 10.1016/j.nmd.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Jimenez-Mallebrera C, Foley AR, Fernandez-Fuente M, Brown SC, Torelli S, et al. Flow cytometry analysis: a quantitative method for collagen VI deficiency screening. Neuromuscul Disord. 2012;22(2):139–48. doi: 10.1016/j.nmd.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Armaroli A, Trabanelli C, Scotton C, Venturoli A, Selvatici R, Brisca G, et al. Paternal germline mosaicism in collagen VI related myopathies. Eur J Paediatr Neurol. 2015;19(5):533–6. doi: 10.1016/j.ejpn.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 24.Zanoteli E, Soares PS, Silva A, Camelo CG, Fonseca A, Albuquerque MAV, et al. Clinical features of collagen VI-related dystrophies: a large Brazilian cohort. Clin Neurol Neurosurg. 2020;192:105734. doi: 10.1016/j.clineuro.2020.105734. [DOI] [PubMed] [Google Scholar]
- 25.Baker NL, Mörgelin M, Peat R, Goemans N, North KN, Bateman JF, et al. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 2005;14(2):279–93. doi: 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- 26.Kawahara G, Okada M, Morone N, Ibarra CA, Nonaka I, Noguchi S, et al. Reduced cell anchorage may cause sarcolemma-specific collagen VI deficiency in Ullrich disease. Neurology. 2007;69(10):1043–9. doi: 10.1212/01.wnl.0000271386.89878.22. [DOI] [PubMed] [Google Scholar]
- 27.Bozorgmehr B, Kariminejad A, Nafissi S, Jebelli B, Andoni U, Gartioux C, et al. Ullrich congenital muscular dystrophy (UCMD): clinical and genetic correlations. Iran J Child Neurol. 2013;7(3):15–22. [PMC free article] [PubMed] [Google Scholar]
- 28.Foley AR, Quijano-Roy S, Collins J, Straub V, McCallum M, Deconinck N, et al. Natural history of pulmonary function in collagen VI-related myopathies. Brain. 2013;136(Pt 12):3625–33. doi: 10.1093/brain/awt284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma S, Goyal P, Guglani L, Peinhardt C, Pelzek D, Barkhaus PE. COL6A and LAMA2 mutation congenital muscular dystrophy: a clinical and electrophysiological study. J Clin Neuromuscul Dis. 2018;19(3):108–16. doi: 10.1097/CND.0000000000000198. [DOI] [PubMed] [Google Scholar]
- 30.Lamandé SR, Shields KA, Kornberg AJ, Shield LK, Bateman JF. Bethlem myopathy and engineered collagen VI triple helical deletions prevent intracellular multimer assembly and protein secretion. J Biol Chem. 1999;274(31):21817–22. doi: 10.1074/jbc.274.31.21817. [DOI] [PubMed] [Google Scholar]
- 31.Lampe AK, Zou Y, Sudano D, O'Brien KK, Hicks D, Laval SH, et al. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum Mutat. 2008;29(6):809–22. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- 32.Koppolu AA, Madej-Pilarczyk A, Rydzanicz M, Kosińska J, Gasperowicz P, Dorszewska J, et al. A novel de novo COL6A1 mutation emphasizes the role of intron 14 donor splice site defects as a cause of moderate-progressive form of ColVI myopathy—a case report and review of the genotype-phenotype correlation. Folia Neuropathol. 2017;55(3):214–20. doi: 10.5114/fn.2017.70486. [DOI] [PubMed] [Google Scholar]
- 33.Foley AR, Hu Y, Zou Y, Yang M, Medne L, Leach M, et al. Large genomic deletions: a novel cause of Ullrich congenital muscular dystrophy. Ann Neurol. 2011;69(1):206–11. doi: 10.1002/ana.22283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quijano-Roy S, Allamand V, Riahi N, Gartioux C, Briñas L, Leclair-Richard D, et al. C.P.2.03 Predictive factors of severity and management of respiratory and orthopaedic complications in 16 Ullrich CMD patients. Neuromuscul Disord. 2007;17(9–10):844. doi: 10.1016/j.nmd.2007.06.279. [DOI] [Google Scholar]
- 35.Briñas L, Richard P, Quijano-Roy S, Gartioux C, Ledeuil C, Lacène E, et al. Early onset collagen VI myopathies: genetic and clinical correlations. Ann Neurol. 2010;68(4):511–20. doi: 10.1002/ana.22087. [DOI] [PubMed] [Google Scholar]
- 36.Noguchi S, Ogawa M, Malicdan MC, Nonaka I, Nishino I. Muscle weakness and fibrosis due to cell autonomous and non-cell autonomous events in collagen VI deficient congenital muscular dystrophy. EBioMedicine. 2017;15:193–202. doi: 10.1016/j.ebiom.2016.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishikawa A, Mitsuhashi S, Miyata N, Nishino I. Targeted massively parallel sequencing and histological assessment of skeletal muscles for the molecular diagnosis of inherited muscle disorders. J Med Genet. 2017;54(2):104–10. doi: 10.1136/jmedgenet-2016-104073. [DOI] [PubMed] [Google Scholar]
- 38.Giusti B, Lucarini L, Pietroni V, Lucioli S, Bandinelli B, Sabatelli P, et al. Dominant and recessive COL6A1 mutations in Ullrich scleroatonic muscular dystrophy. Ann Neurol. 2005;58(3):400–10. doi: 10.1002/ana.20586. [DOI] [PubMed] [Google Scholar]
- 39.Hicks D, Lampe AK, Barresi R, Charlton R, Fiorillo C, Bonnemann CG, et al. A refined diagnostic algorithm for Bethlem myopathy. Neurology. 2008;70(14):1192–9. doi: 10.1212/01.wnl.0000307749.66438.6d. [DOI] [PubMed] [Google Scholar]
- 40.Elmas M, Yıldız H, Erdoğan M, Gogus B, Avcı K, Solak M. Comparison of clinical parameters with whole exome sequencing analysis results of autosomal recessive patients; a center experience. Mol Biol Rep. 2019;46(1):287–99. doi: 10.1007/s11033-018-4470-7. [DOI] [PubMed] [Google Scholar]
- 41.Higuchi I, Shiraishi T, Hashiguchi T, Suehara M, Niiyama T, Nakagawa M, et al. Frameshift mutation in the collagen VI gene causes Ullrich's disease. Ann Neurol. 2001;50(2):261–5. doi: 10.1002/ana.1120. [DOI] [PubMed] [Google Scholar]
- 42.Zhang RZ, Zou Y, Pan TC, Markova D, Fertala A, Hu Y, et al. Recessive COL6A2 C-globular missense mutations in Ullrich congenital muscular dystrophy: role of the C2a splice variant. J Biol Chem. 2010;285(13):10005–15. doi: 10.1074/jbc.M109.093666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nallamilli BRR, Chakravorty S, Kesari A, Tanner A, Ankala A, Schneider T, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574–87. doi: 10.1002/acn3.649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shimomura H, Lee T, Tanaka Y, Awano H, Itoh K, Nishino I, et al. Two closely spaced mutations in cis result in Ullrich congenital muscular dystrophy. Hum Genome Var. 2019;6:21. doi: 10.1038/s41439-019-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.