

Abstract

In this study, seven derivatives of salicylaldehyde thiosemicarbazones (1–7) were synthesized by refluxing substituted thiosemicarbazide and salicylaldehyde in an ethanol solvent. Different spectral techniques (UV–vis, IR, and NMR) were used to analyze the prepared compounds (1–7). Accompanied by the experimental study, quantum chemical studies were also carried out at the M06/6-311G(d,p) level. A comparative analysis of the UV–visible spectra and vibrational frequencies between computational and experimental findings was also performed. These comparative data disclosed that both studies were observed to be in excellent agreement. Furthermore, natural bond orbital investigations revealed that nonbonding transitions were significant for the stability of prepared molecules. In addition, frontier molecular orbital (FMO) findings described that a promising charge transfer phenomenon was found in 1–7. The energies of FMOs were further used to determine global reactivity parameters (GRPs). These GRP factors revealed that all synthesized compounds (1–7) contain a greater hardness value (η = 2.1 eV) and a lower softness value (σ = 0.24 eV), which indicated that these compounds were less reactive and more stable. Nonlinear optical (NLO) evaluation displayed that compound 5 consisted of greater values of linear polarizability ⟨α⟩ and third-order polarizability ⟨γ⟩ of 324.93 and 1.69 × 105 a.u., respectively, while compound 3 exhibited a larger value of second-order polarizability (βtotal) of 508.41 a.u. The NLO behavior of these prepared compounds may be significant for the hi-tech NLO applications.
1. Introduction
Thiosemicarbazones (TSCs) correspond to a versatile class of Schiff-based ligands having sulfur and nitrogen as donor atoms with considerable interest due to their ion-sensing ability, structural diversity, bonding modes, and biological implications.1 Heterocyclic thiosemicarbazones due to their biological properties like antimicrobial,2 anticonvulsant,3 anti-HIV,4 antihypertensive,5 hypoglycemic,6 trypanocidal,7 local anesthetic,8 and cytotoxic activities9,10 have been focused on. A heterocyclic analogue has applications in drug development such as bacterial infection, central nervous system disorders, and anti-allergic and analgesic agents. Thiosemicarbazones also exhibit a variety of scientifically promising procedures such as anticorrosion, antifouling,11 and plant growth-promoting activities12 and are employed for making electrodes.13 Thiosemicarbazones can undergo tautomerism and form planar, rigid Schiff bases that are proficient to interact with metal cations that have attracted the attention of researchers to consider thiosemicarbazones as interesting ligands to synthesize metal complexes.14 Another application of such analogues is to coordinate with transition metal ions in N,S-bidentate mode; more diversified binding modes also become possible when an additional coordinating group is present.15 Transition metal complexes having soft or hard donor moieties have been extensively used in organometallic and coordination chemistry.16 Salicylaldehyde thiosemicarbazone derivatives are remarkable materials for their second harmonic generation (SHG)17 and are applicable in the field of material sciences such as nonlinear optics (NLO).18 NLO materials have been extensively applied in broad practical fields as these are essential frequency conversion sources in real-time laser instruments19−26 and exhibit outstanding advantages in comparison to conventional inorganic materials such as low absorption loss, lower dielectric constants, higher electro-optic coefficients, and easiness to modify and process.27−31 Second-order NLO displays a wide range of applications in the field of biological imaging, optical switches, optical data processing,32 communication technology, and photonics industries.
Recently, computational analysis along with the experimental work provides complete perception regarding the properties of materials. Therefore, the newly synthesized compounds are theoretically characterized via infrared (IR), UV–vis, NBO, FMO analyses, etc. The principal aim of the current work is to present the complete structural and spectroscopic description of these novel compounds with the support of combined theoretical techniques and experimental data.
2. Results and Discussion
All thiosemicarbazone derivatives were prepared according to the modified procedure as described earlier. The IR spectra gave important information about the skeleton of structures. The infrared spectrum of novel compounds showed a significant absorption band in the functional group region. Intense bands at frequencies of 3322–3000 and 3770–3640 cm–1 were responsible for NH and OH group stretching, respectively. The characteristic band at 1680 cm–1 corresponding to C=O disappeared, and bands of the azomethine linkage (C=N) appeared in the frequency range of 1650–1519 cm–1, which verified the existence of the carbonyl group. The significant strong absorption peak of C=S and the free aliphatic CH group appeared in the regions of 1296–1221 and 3077–2970 cm–1, respectively.
1H NMR and 13C NMR spectra also helped determine the structure of entitled products. The triplet peak appeared in the region of 9.061–9.176 ppm, corresponding to one proton of the CS–NH group. The 1H NMR singlet signals of the hydrazine proton (N–NH) and the azomethine proton (—CH=N) appeared at the more deshielded regions of 11.551–11.695 and 10.261–10.286 ppm, respectively. In the aromatic region, the multiple absorption bands at 7.017–7.373 ppm were responsible for protons 2–6. The 13 and 15 H of 1–4 showed a singlet peak at 8.139–8.334 ppm and the singlet peak at 8.296–8.327 ppm corresponds to 14 and 16 H for 5–7. The doublet signal for 12 H for 1–4 appeared at 8.132–8.161 ppm, whereas the doublet peak at 8.135–8.148 ppm was responsible for 13 H for 5–7. The signals that appeared in the region of 6.776–6.815 ppm correspond to phenolic hydroxy hydrogen (OH proton) for 1–7. Compounds 5–6 showed a singlet peak in the shielded region of 2.285–2.231 ppm as attributed to the three protons of phenyl-CH3. Compound 7 has a methyl group attached to benzene whose peak appeared in a more shielded region of 2.247 ppm. The signal observed at the upfield region (shielded proton) of 4.762–4.834 ppm corresponds to CH2–N for compounds 1–7. The 13C NMR spectra of 1–7 showed that the peak in the upfield region in the range of 68.042–99.997 ppm corresponds to CH2–NH. The large shifts arise for C=S and N=CH at 177.914–206.626 and 126.832–178.182 ppm, respectively. The other characteristic signals resonated in good agreement through the proposed structures.
2.1. Natural Bond Orbital (NBO) Analysis
NBO analysis is significantly adopted for the explanation of inter- and intramolecular transitions and transfer of charges from an electron donor to acceptor orbitals of studied molecules.33 The NBO analysis of 1–7 was performed at the M06/6-311G(d,p) level, and their calculated outcomes are tabulated in Tables S1–S7. For the evaluation of interactions, the second-order Fock matrix was utilized.34
| 1 |
In eq 1, E(2) stands for the stabilization energy, qi refers to the donor–orbital occupancy, Fi,j indicates the off-diagonal NBO Fock matrix elements, and εi and εj point out the diagonal elements.
Generally, four kinds of molecular interactions, π → π*, σ → σ*, LP → π*, and LP → σ*, are found for the investigated compounds. Among them, π → π* exhibited a higher value of stabilization energy; hence, it is considered as a significant transition, while σ → σ* transitions presented the smallest stability values. However, interactions LP → π* and LP → σ* are examined with substantial energy of stabilization. Among the studied compounds transitions, π(C25-C27) → π*(C28-C30), π(C24-C26) → π*(C27-C29), π(C1-C6) → π*(C4-C5), π(C23-C25) → π*(C26-C28), π(C21-C23) → π*(N18-C19), π(C22-C24) → π*(C22-C24), and π(C24-C26) → π*(C27-C29) displayed higher values of stabilization energies of 26.69, 26.68, 25.42, 26.75, 54.54, 26.68, and 26.71 kcal/mol in 1–7, respectively. Moreover, some other kinds of significant π → π* donor–acceptor intramolecular charge transfer (ICT) interplays are also studied such as π(C28-C30) → π*(C24-C26), π(C3-C4) → π*(C1-C2), π(C2-C3) → π*(C1-C6), π(C1-C6) → π*(C2-C3), π(C22-C14) → π*(C25-C27), π(C5-C6) → π*(C1-C2), and π(C2-C3) → π*(C4-C5) with 24.23, 25.66, 24.46, 22.84, 26.78, 24.71, and 21.89 kcal/mol stabilization energies in 1–7, respectively. The least values of stability are determined as 4.87, 0.54, 0.51, 1.09, 0.54, 0.53, and 1.13 kcal/mol for π(N21-C22) → π*(C24-C26), π(C24-C26) → π*(C24-C26), π(C4-C5) → π*(C4-C5), π(N19-C20) → π*(N19-C20), π(C22-C24) → π*(C22-C24), π(C24-C26) → π*(C24-C26), and π(N20-C21) → π*(N20-C21) transitions for compounds 1–7, respectively (see Tables S1–S7).
In σ → σ* transitions, greater values of stabilization energies are found to be 9.06, 9.10, 9.06, 9.03, 8.89, 4.74, and 9.01 kcal/mol for σ(C22-H23) → σ*(N19-N21), σ(C21-H22) → σ*(N18-N20), σ(C21-H22) → σ*(N18-N20), σ(C20-H21) → σ*(N17-N19), σ(C19-H20) → σ*(N16-N18), σ(O32-H33) → σ*(C23-C24), and σ(C21-H22) → σ*(N18-N20) interactions in 1–7, respectively. Similarly, some other transitions such as σ(C12-H14) → σ*(C3-C12), σ(C2-F35) → σ*(C1-C2), σ(C16-N18) → σ*(N18-H19), σ(C6-F34) → σ*(C5-C6), σ(C34-H37) → σ*(C4-C34), σ(C29-H31) → σ*(C26-H30), and σ(C11-H13) → σ*(C3-C11) with the lowest stabilization energies of 0.51, 0.51, 0.54, 0.50, 0.52, 0.81, and 0.51 kcal/mol, respectively, are also obtained for 1–7 as shown in Tables S1–S7. These least values of energy are examined due to the existence of weak interactions between the donor and accepter species.
Similar kinds of transitions are also examined in accordance with resonance. LP1(N15) → σ*(C17-S18), LP1(N14) → π*(C16S17), LP1(C27) → π*(C23C25), LP1(N13) → σ*(C15-S16), LP1(N12) → σ*(C14-S15), LP1(N14) → σ*(C16-S17), and LP1(N14) → σ*(C16-S17) with high stabilization energies, i.e., 49.93, 31.93, 70.69, 50.52, 68.04, 55.03, and 54.60 kcal/mol, are studied for 1–7, respectively. On the other hand, LP1(N15) → σ*(C3-C12), LP1(S17) → σ*(C16-S17), LP1(N14) → σ*(C3-C11), LP1(N13) → σ*(C3-C10), LP1(N12) → σ*(C14-N16), LP2(S17) → σ*(C3-C11), and LP1(N14) → σ*(C3-C11) produced 0.52, 0.88, 0.71, 0.66, 0.66, 0.91, and 0.55 kcal/mol, respectively, which demonstrated the least energies of electron-donating interactions in 1–7 (Tables S1–S7). Finally, based on the NBO study, it can be inferred that the intramolecular charge transfer, hyperconjugative interactions, and extended conjugations are the primary causes that offer stability to these prepared products.
2.2. UV–Visible Analysis
To evaluate the absorption properties, charge transfer, and molecular orbital transitions in 1–7, UV–vis analysis was performed at the TD-DFT/M06/6-311G(d,p) level of theory. A comparative analysis was carried out between experimental and DFT-computed works to investigate molecular orbital contributions, oscillator strengths (f), absorption wavelengths (λ), and excitation energies of 1–7 as tabulated in Table S8. The simulated wavelength (λ) is noticed in the ranges of 291–393, 277–389, 275–386, and 276–387 nm for 1–4, respectively, which is found to be in accordance with the experimentally determined λmax values: 296, 222, 222, and 219 nm. Similarly, the experimental λmax values for 5–7 are found at 296, 223, and 244 nm, respectively, while their DFT absorption bands are examined in the ranges of 278–392, 273–390, and 273–390 nm, respectively. Overall, DFT-calculated λmax of 1–7 showed reasonable agreement with the experimentally determined λmax as manifested in Table S8.
2.3. Vibrational Analysis
The simulated and experimental vibrational analyses were performed to explore the nature of vibrational modes of 1–7. The absorption frequencies are listed in Tables S9–S15 and the experimental spectra are shown in Figures S3–S9.
2.3.1. C–H Vibrations
In heteroaromatic and aromatic stretching, absorption frequencies for C–H were determined at 3100–3000 cm–1.35 In compounds 1–7, simulated asymmetric and symmetric absorption bands are found to be at 3174, 3192, 3010, 3188, 3144, 3147, and 3110 cm–1, respectively (Tables S9–S15). These aforesaid vibrations are strongly in agreement with experimental vibrational frequencies at 3150, 3170, 3150, 3155, 3175, 3140, and 3130 cm–1, respectively (see Figures S3–S9).
2.3.2. C–C Vibrations
The absorption bands for C=C vibrations were investigated at 1650–1400 cm–1.36 In compound 1, simulated C–C stretching frequencies in the benzene ring are found at 1643–1627 cm–1, which closely matched with experimentally calculated data in the region of 1450–1600 cm–1. Meanwhile, in compound 4, simulated C–C absorption vibrations are found at 1668–1626 cm–1, which significantly matched with the experimental data (1550–1650 cm–1). Similarly, in compound 7, carbon–carbon stretching absorption peaks emerged at 1647–1626 cm–1 (see Tables S9–S15), which are in excellent resemblance with experimental data (see Figure S9).
2.3.3. N–H Vibrations
Usually, nitrogen–hydrogen wavenumbers were located at 3450–3250 cm–1.37 In compounds 1–7, N–H simulated stretching vibrations for NH2 are located at the ranges of 3564–3466, 3563–3465, 3561–3453, 3562–3454, 3564–3451, 3560–3469, and 3565–3451 cm–1, respectively. Furthermore, the experimental spectra showed the symmetric stretching vibrations at 3450–3400, 3460–3450, and 3500–3450 cm–1 in compounds 1, 2, and 7, respectively (Figures S3, S4, and S9). Moreover, in 1–7, the rocking vibrations for NH2 appeared at 1688, 1550, 1546, 1693, 1689, 1694, and 1692 cm–1, respectively. Alternatively, the experimentally noticed rocking vibrations for NH2 are found at 1600, 1650, 1550, 1545, 1585, 1600, and 1630 cm–1, respectively, for 1–7. Furthermore, some other N–H vibrational frequencies are determined experimentally at 3002, 3114, 3006, 3117, 3045, 3000, and 3117 cm–1 in compounds 1–7, respectively.
2.3.4. C=N Vibration Bands
Vibrational bands for C=N were studied at 1382–1266 cm–1.38 In aromatic nitro compounds, stretching (symmetric and asymmetric) mode of vibrations becomes the main reason for strong absorption at 1700–1500 cm–1. In 1–7, C=N vibrational mode is examined at 1543, 1689, 1693, 1546, 1692, 1686, and 1534 cm–1, respectively (Tables S9–S15), showing good agreement with experimental values of 1542, 1548, 1548, 1550, 1530, 1548, and 1547 cm–1, respectively (see Figures S3–S9). Furthermore, in C=N, some other vibrations are found at 1380–1361, 1379–1360, and 1381–1376 cm–1 for compounds 1, 4, and 7, respectively.
2.3.5. C=S Vibrations
The C=S absorption peaks are analyzed in the region of 1500–1200 cm–1.39 Considering compounds 1–7, the experimentally determined C=S vibrations are found at 1229, 1229, 1226, 1275, 1223, 1225, and 1196 cm–1, respectively, while simulated values are observed at 1361, 1360, 1369, 1360, 1357, 1381, and 1381 cm–1, respectively; both are in excellent agreement with each other (see Tables S9–S15 and Figures S3–S9).
2.3.6. O–H Vibrations
The O–H vibrational mode is also considered, a significant stretching absorption frequency examined in entitled compounds. The O–H absorption bands emerged at 3700–3550 cm–1.40 The simulated modes for the aforesaid wavenumber occurred at 3889, 3888, 3887, 3887, 3887, 3888, and 3887 cm–1 for molecules 1–7, respectively, which are excellently matched with the experimental band. Apart from this, some other simulated modes of vibrations appeared at 1643, 1626, and 1647 cm–1 in 1, 4, and 7, respectively (Tables S9–S14).
2.3.7. C–X Vibrations
The C–Cl group existed in our compound 4. The stretching bands for C–Cl usually appeared at 800 cm–1.41,42 The simulated modes for 4 occurred at 880 cm–1. Additionally, the C–Br group is also presented in our synthesized compounds; hence, DFT-based vibrational modes occurred at 910–864, 910–880, 947–863, 914–880, 923–810, 920–810, and 948–913 cm–1 in 1–7, respectively (Tables S9–S15), which are excellently correlated with experimental values of 850, 800, 830, 810, 815, 800, and 840 cm–1, respectively (see Figures S3–S9).
2.4. Frontier Molecular Orbital (FMO) Analysis
Frontier molecular orbital (FMO) theory is considered a significant model to evaluate the optoelectronic behavior, light absorption potential, and reactivity of a molecule.43,44 The bandgap between HOMO and LUMO is in direct relation with the kinetic stability and chemical reactivity of compounds. The smaller energy gap of molecules indicated the more possibilities of intramolecular charge transfer; hence, it enhanced their reactivity as well as polarizability and vice versa.45 The HOMO/LUMO, HOMO-1/LUMO+1, and HOMO-2/LUMO+2 energies of 1–7 were explored and their results are tabulated in Table 1 and Table S16, while their pictorial description of charge densities is displayed in Figure 1 and Figures S1 and S2.
Table 1. Computed Values of EHOMO, ELUMO, and Energy Gap (ELUMO – EHOMO) for 1–7a.
| compound | EHOMO | ELUMO | ΔE |
|---|---|---|---|
| 1 | –5.76 | –1.59 | 4.17 |
| 2 | –5.76 | –1.58 | 4.18 |
| 3 | –5.84 | –1.62 | 4.22 |
| 4 | –5.82 | –1.62 | 4.20 |
| 5 | –5.78 | –1.65 | 4.13 |
| 6 | –5.72 | –1.57 | 4.15 |
| 7 | –5.71 | –1.55 | 4.16 |
Energy gap (ΔE) = ELUMO – EHOMO; units in eV.
Figure 1.

Frontier molecular orbitals of studied compounds 1–7.
The calculated energy gap values of HOMO/LUMO for 1–7 are 4.17, 4.18, 4.22, 4.20, 4.13, 4.15, and 4.16 eV, respectively. This energy difference (ΔE) between HOMO and LUMO orbitals for all compounds (1–7) is found to be nearly equal magnitude. The highest value of ΔE revealed that all compounds have high chemical stability and low reactivity. Accompanying the energies of molecular orbitals, FMO analysis also explained the ICT transitions in compounds. The pictographs of charge densities for HOMO/LUMO are displayed in Figure 1, while those for HOMO-1/LUMO+1 and HOMO-2/LUMO+2 are shown in Figures S1 and S2. The charge densities for HOMO are located over the N-methyl-2-methylenehydrazinecarbothioamide part in 1–4, while those for LUMO are concentrated at (Z)-2-(5-bromo-2 hydroxybenzylidene) hydrazine carbothioamide. Similarly, for 5–7, charges for HOMO exist at the (Z)-N-(but-2-en-1-yl)-2-methylenehydrazinecarbothioamide portion, while those for LUMO are located at (Z)-2-(5-bromo-2 hydroxybenzylidene) hydrazinecarbothioamide (see Figure 1).
2.5. Global Reactivity Parameters
The reactive nature of compounds can be determined by global reactivity parameters (GRPs), global hardness (η), softness (σ), electron affinity (EA), electronegativity (X), chemical potential (μ), ionization potential (IP), and global electrophilicity (ω) by using HOMO/LUMO energies.46,47 The ionization potential and electron affinity might be utilized to depict the electron gaining and losing capacity of molecules, which interrelated with HOMO/LUMO energies.48,49 The data tabulated in Table S17 indicate that the values of ionization potential are noticed to be higher than electron affinity, which disclosed that all compounds have better capability to accept an electron. Furthermore, the molecules with the least energy gap were soft exhibiting tunable character, least kinetic stability, and high reactivity and vice versa.50 The observed value of chemical hardness for 1–7 is higher (3.73 eV) than their softness (0.24 eV), which illustrates the less reactivity and more stability of the synthesized compounds (see Table S17).
2.6. Nonlinear Optical Properties
NLO materials are recognized as significant due to their applicability in the fields of medicine, electronics, modulation of optical signals, and telecommunication.51,52 Organic compounds are appreciably able to exhibit strong NLO properties.53,54 The NLO response in organic molecules is observed due to asymmetric polarization. The stronger NLO response might be noticed owing to the existence of electron-withdrawing or electron-donating groups in the compounds at the appropriate site that creates strong push–pull architecture. Moreover, these electron-withdrawing or electron-donating groups are connected with the conjugated system of the π framework that enhances the NLO response.55 Herein, the linear optical response (polarizability) and nonlinear optical response including first hyperpolarizability (βtotal) and second hyperpolarizability (⟨γ⟩) were carried out by using the M06/6-311G(d,p) functional. The determined results are presented in Tables S18–S21, while the main values are summarized in Table 2.
Table 2. Values of Dipole Moments (μo), Polarizability ⟨α⟩, First Hyperpolarizability (βtotal), and Second Hyperpolarizability ⟨γ⟩.
| compound | μo | ⟨α⟩ | βtotal | ⟨γ⟩ × 105 |
|---|---|---|---|---|
| 1 | 7.50 | 307.52 | 293.64 | 1.54 |
| 2 | 7.28 | 305.32 | 462.82 | 1.45 |
| 3 | 7.08 | 306.14 | 508.41 | 1.51 |
| 4 | 6.87 | 305.70 | 408.22 | 1.50 |
| 5 | 7.55 | 324.93 | 302.44 | 1.69 |
| 6 | 7.70 | 323.67 | 387.60 | 1.57 |
| 7 | 7.74 | 324.49 | 269.44 | 1.60 |
Among all compounds, a higher value of dipole moment is examined in 7 to be 7.74 D, while 4 showed the least value of 6.87D. However, the extent of dipole moments of all synthesized compounds is found to be larger as compared to the urea molecule (1.3732 D).56 Similarly, compound 5 expressed the greater values of polarizabilities ⟨α⟩ and second hyperpolarizability ⟨γ⟩ of 324.93 and 1.69 × 105 a.u., respectively. Moreover, 3 has the highest value of βtotal (508.41 a.u.), while the lowest value is investigated in 7 (269.44 a.u.). Overall, the order of the first hyperpolarizability was observed to be 3 > 2 > 4 > 6 > 5 > 1 > 7. Nevertheless, all the prepared molecules expressed a significantly higher value of hyperpolarizability than urea (βtotal= 43 a.u.), the standard molecule to analyze the NLO behavior.57 All these compounds (1–7) have 6.82, 10.76, 11.82, 9.49, 7.03, 9.01, and 6.26 times greater values of βtotal as compared to the urea molecule. This urea-relative investigation indicated that all synthesis compounds are appropriate NLO candidates.
3. Experimental Studies
3.1. Preparation of Thiosemicarbazones
Thiosemicarbazones were synthesized by mixing 5-bromo-salicylaldehyde (4.2 mmol) and substituted thiosemicarbazide (4.2 mmol) in ethanol (20 mL). A few drops of catalytic acetic acid were added through constant stirring. The precipitates formed after refluxing the mixture for 3 h. The products were then filtered and washed many times with ethanol, dried, and weighted (Scheme 1).
Scheme 1. Synthesis of Thiosemicarbazones Derivatives (1–7).
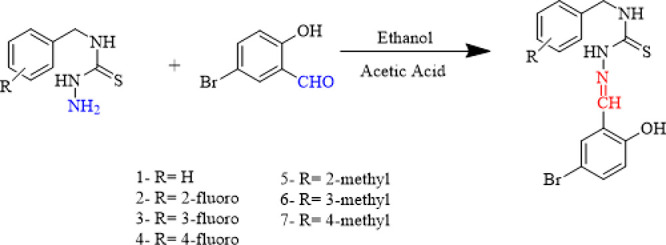
3.1.1. N-Benzyl-2-(5-bromo-2-hydroxybenzylidene)hydrazinecarbothioamide (1)58
Yield, 72%; white crystalline; mp 213 °C; IR (KBr), ν (cm–1): 3650 (O–H), 3114, 3002 (N–H), 3077 (C–H), 1524 (C=N), 1229 (C=S). UV–vis (ethanol): λmax, 296 nm. 1H NMR (4), δ (ppm): 4.834 (s, 2H, CH2–NH), 10.286 (s, 1H, N=CH), 9.157 (s, 3H, CH2–NH), 11.590 (s, 1H, NHN), 6.820 (d, J = 2.5 Hz, 1H, OH), 8.334 (s, 2H, 2 × CH phenyl), 8.161 (d, J = 3.5 Hz, 1H, CH phenyl), 7.222 (s, 4H, 4 × CH phenyl).
3.1.2. 2-(5-Bromo-2-hydroxybenzylidene)-N-(2-fluorobenzyl)hydrazinecarbothioamide (2)
Yield, 78%; white crystalline; mp 225 °C; IR (KBr), ν (cm–1): 3750 (O–H), 3114, 3002 (N–H), 2977 (CH), 1548 (C=N), 1221(C=S). UV–vis (ethanol): λmax 222 nm. 1H NMR δ (ppm): 4.847 (d, J = 8.5 Hz, 2H, CH2), 10.277 (s, 1H, N=CH), 9.111 (t, J = 8.5 Hz, 1H, CH2=NH), 11.661 (s, 1H, NHN), 6.815 (d, J = 11.0 Hz 1H, OH), 8.160 (s, 2H, 2 × CH phenyl), 8.153 (d, J = 3.5 Hz, 1H, CH phenyl), 7.291–7.274 (m, 4H, 4 × CH phenyl). 13C NMR δ (ppm): 123.334 (C1), 140.738 (C2), 76.422 (C3), 111.589 (C4), 102.522 (C5), 111.589 (C6), 70.022 (C7), 178.383 (C8), 126.837 (C9), 100.465 (C10), 156.201 (C11), 99.997 (C12), 126.744 (C13), 85.536 (C14), 126.603 (C15).
3.1.3. 2-(5-Bromo-2-hydroxybenzylidene)-N-(3-fluorobenzyl)hydrazinecarbothioamide (3)
Yield, 72%; white crystalline; mp 218 °C; IR (KBr), ν (cm–1): 3770 (OH), 3118, 3095 (N–H), 3007 (C–H), 1539 (C=N), 1264 (C=S). UV–vis (ethanol): λmax 222 nm. 1H NMR (6), δ (ppm): 4.828 (d, J = 8.5 Hz, 2H, CH2–NH), 10.271 (s, 1H, N=CH), 9.176 (t, J = 8.5 Hz, 3H, CH2=NH), 11.624 (s, 1H, NHN), 6.810 (d, J = 11.0 Hz, 1H, OH), 8.147 (s, 2H, 2 × CH phenyl), 8.140 (d, J = 3.5 Hz, 1H, CH phenyl), 7.328–7.321 (m, 4H, 2 × CH phenyl). 13C NMR δ (ppm): 175.708 (C1), 111.583 (C2), 190.705 (C3), 101.982 (C4), 156.200 (C5), 147.934 (C6), 90.898 (C7), 194.683 (C8), 178.182 (C9), 143.084 (C10), 186.841 (C11), 123.382 (C12), 160.732 (C13), 115.219 (C14), 163.737 (C15).
3.1.4. 2-(5-Bromo-2-hydroxybenzylidene)-N-(4-fluorobenzyl) hydrazinecarbothioamide (4)
Yield, 70%; white crystalline; mp 223 °C; IR (KBr), ν (cm–1): 3680 (O–H), 3150, 3050 (N–H), 2970 (C–H), 1650 (C=N), 1275 (C=S). UV–vis (ethanol): λmax 219 nm. 1H NMR (7), δ (ppm): 4.792 (d, J = 9 Hz, 2H, CH2–NH), 10.264 (s, 1H, N=CH), 9.155 (t, J = 8.5 Hz, 3H, CH2–NH), 11.624 (s, 1H, NHN), 6.805 (d, J = 11.0 Hz, 1H, OH), 8.139 (s, 2H, 2 × CH phenyl), 8.132 (d, J = 3.5 Hz, 1H, CH phenyl), 7.351–7.373 (m, 4H, 4 × CH phenyl). 13C NMR, δ (ppm): 156.162 (C1), 123.345 (C2), 100.263 (C3), 168.200 (C4), 100.263 (C5), 123.34 (C6), 99.99 (C7), 177.995 (C8), 162.850 (C9), 120.964 (C10), 165.609 (C11), 114.030 (C12), 1366.211 (C13), 111.577 (C14), 136.178 (C15).
3.1.5. 2-(5-Bromo-2-hydroxybenzylidene)-N-(2-methylbenzyl)hydrazinecarbothioamide (5)
Yield, 72%; white crystalline; mp 220 °C; IR (KBr), ν (cm–1): 3150, 3050 (N–H), 1544 (C=N), 1250 (C=S). UV–vis (ethanol): λmax 296 nm. 1H NMR (8), δ (ppm): 4.776 (d, J = 8.5 Hz, 2H, CH2–NH), 10.266 (s, 1H, N=CH), 9.061 (t, J = 8.5 Hz, 3H, CH2=NH), 11.695 (s, 1H, NHN), 6.812 (d, J = 11.0 Hz, 1H, OH), 8.327 (s, 2H, 2 × CH phenyl), 8.148 (d, J = 3.5 Hz, 1H, CH phenyl), 7.077–7.324 (m, 4H, 4 × CH phenyl), 2.285 (s, CH3). 13C NMR, δ (ppm): 170.917 (C1), 145.865 (C2), 137.200 (C3), 122.912 (C4), 121.227 (C5), 134.760 (C6), 40.379 (C7), 88.776 (C8), 206.626 (C9), 177.646 (C10), 115.587 (C11), 192.476 (C12), 111.095 (C13), 155.667 (C14), 99.509 (C15), 150.726 (C16).
3.1.6. 2-(5-Bromo-2-hydroxybenzylidene)-N-(3-methylbenzyl)hydrazinecarbothioamide (6)
Yield, 70%; white crystalline; mp 215 °C; IR (KBr), ν (cm–1): 3640 (O–H), 3250, 3067 (N–H), 3050 (C–H), 1519 (C=N), 1224 (C=S). UV–vis (ethanol): λmax 223 nm. 1H NMR (9), δ (ppm): 4.762 (d, J = 8.0 Hz, 2H, CH2–NH), 10.261 (s, 1H, N=CH), 9.106 (t, J = 8.0 Hz, 3H, CH2–NH), 11.540 (s, 1H, NHN), 6.798 (d, J = 11.0 Hz, 1H, OH), 8.296 (s, 2H, 2 × CH phenyl), 8.135 (d, J = 3.5 Hz, 1H, CH phenyl), 7.285–7.314 (m, 4H, 4 × CH phenyl), 2.231 (s, 3H, CH3). 13C NMR, δ (ppm): 136.947 (C1), 129.043 (C2), 136.204 (C3), 127.735 (C4), 128.691 (C5), 123.371 (C6), 32.165 (C7), 99.997 (C8), 177.914 (C9), 156.139 (C10), 112.701 (C11), 170.513 (C12), 111.569 (C13), 129.423 (C14), 106.774 (C15), 129.138 (C16).
3.1.7. 2-(5-Bromo-2-hydroxybenzylidene)-N-(4-methylbenzyl)hydrazinecarbothioamide (7)
Yield, 72%; white; mp 200 °C; IR (KBr), ν (cm–1): 3750 (O–H), 3152, 3000 (N–H), 3062 (C–H), 1594 (C=N), 1196 (C=S). UV–vis (ethanol): λmax 244 nm. 1H NMR (10), δ (ppm): 4.778 (d, J = 8.0 Hz, 2H, CH2–NH), 10.274 (s, 1H, N=CH), 9.113 (t, J = 7.5 Hz, 3H, CH2–NH), 11.551 (s, 4H, NHN), 6.799 (d, J = 11.0 Hz, 1H, OH), 8.301 (s, 2H, 2 × CH phenyl), 8.136 (d, J = 3.0 Hz, 1H, CH phenyl), 7.017–7.315 (m, 4H, 4 × CH phenyl), 2.247 (s, 3H, CH3). 13C NMR, δ (ppm): 139.934 (C1), 111.580 (C2), 119.537 (C3), 137.685 (C4), 121.799 (C5), 111.698 (C6), 18.902 (C7), 78.019 (C8), 177.977 (C9), 152.49 (C10), 85.31 (C11), 156.13 (C12), 81.343 (C13), 130.814 (C14), 79.171 (C15), 123.361 (C16).
3.2. Computational Studies
Quantum chemical computations at the M06/6-311G(d,p) level59 were accomplished for novel thiosemicarbazone derivatives (1–7) with aid of the Gaussian 09 program package60 to find out the vibrational wavenumbers and NBO and NLO properties. Further, GaussView 5.061 was used for creating input files and Chemcraft,62 GaussSum,63 and Avogadro64 programs were used to interpret results of 1–7 from output files. Moreover, time-dependent density functional theory (TD-DFT)65,66 calculations at the aforesaid level with the same basis set were adopted for UV–visible and frontier molecular orbital (FMO) analyses. The energy gap of HOMO/LUMO was used to investigate the global reactivity parameters of compounds. Equation 2 was used to determine the dipole moment.67
| 2 |
The average polarizability ⟨α⟩ was determined with eq 3.68
| 3 |
Analysis of the Gaussian output file yields 10 hyperpolarizability tensors oriented along x, y, and z directions: βxxx, βxyy, βxzz, βyyy, βxxy, βyzz, βzzz, βxxz, βyyz, and βxyz. The extent of the total first hyperpolarizability (βtotal) was calculated with the help of eq 4.68
| 4 |
Equation 5 was employed to calculate the second hyperpolarizability.67
| 5 |
4. Conclusions
A series of novel derivatives of thiosemicarbazones of salicylaldehyde (1–7) was prepared and characterized by 1H NMR, 13C NMR, UV–vis, and IR spectroscopies. Along with the experimental study, DFT investigations were also carried out for 1–7. A comparative study between the computational and experimental findings of the vibrational spectra and UV–vis revealed that both studies were found to be in excellent agreement. Additionally, the NBO study evaluated noncovalent interactions that were proved to offer significant stability for compounds. FMO findings explored the HOMO/LUMO bandgap of 1–7 in the range of 4.13–4.22 eV. GRPs explicated that compounds (1–7) showed greater ionization potential (IP) (5.84–5.71 eV) than the electron affinity (EA) (1.65–1.55 eV); hence, 1–7 have greater electron-accepting ability. Nevertheless, compounds 1–7 also expressed higher values of hardness (η = 2.11–2.06 eV) and a smaller value of softness (σ = 0.25 eV). These data revealed that 1–7 were found to be kinetically more stable and less reactive. NLO investigation described that compound 5 exhibited higher values of linear polarizability ⟨α⟩ and third-order polarizability ⟨γ⟩ of 324.93 and 1.69 × 105 a.u., respectively, while compound 3 exhibited a larger value of second-order polarizability (βtotal) of 508.41 a.u. A relative study with a standard molecule (urea) revealed that compounds 1–7 might be considered as a good NLO material.
Acknowledgments
M.I. expresses appreciation to the Deanship of Scientific Research at King Khalid University Saudi Arabia for funding through the research group program under grant number R.G.P. 1/318/42. A.A.C.B. (grants 2015/01491-3 and 2014/25770-6) is thankful to Fundacão de Amparo à Pesquisa do Estado de Sao Paulo for financial support. A.A.C.B. (grant 312550/2020-0) also thanks the Brazilian National Research Council (CNPq) for financial support and fellowships. This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior do Brasil (CAPES) Finance Code 001.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c01938.
Computation details and IR, 1H, and 13C NMR spectra exhibited (PDF)
Author Contributions
∇ M.K. and R.J. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Casas J.; García-Tasende M. S.; Sordo J. Main group metal complexes of semicarbazones and thiosemicarbazones. A structural review. Coord. Chem. Rev. 2000, 209, 197–261. 10.1016/S0010-8545(00)00363-5. [DOI] [Google Scholar]
- Kasuga N. C.; Sekino K.; Ishikawa M.; Honda A.; Yokoyama M.; Nakano S.; Shimada N.; Koumo C.; Nomiya K. Synthesis, structural characterization and antimicrobial activities of 12 zinc(II) complexes with four thiosemicarbazone and two semicarbazone ligands. J. Inorg. Biochem. 2003, 96, 298–310. 10.1016/S0162-0134(03)00156-9. [DOI] [PubMed] [Google Scholar]
- Taroua M.; Ribuot C.; Péra M. H.; Taillandier G.; Fatome M.; Laval J. D.; Demenge P.; Leclerc G. New α, β and y semicarbazone and thiosemicarbazone 1,3-dithiolanes as radioprotectors. Anticonvulsant activity. Eur. J. Med. Chem. 1996, 31, 589–595. 10.1016/0223-5234(96)89554-7. [DOI] [Google Scholar]
- Mishra V.; Pandeya S. N.; DeClercq E.; Pannecouque C.; Witvrouw M. Synthesis of aryl semicarbazone of 4-aminoacetophenone and their anti-HIV activity. Pharm. Acta Helv. 1998, 73, 215–218. 10.1016/S0031-6865(98)00028-4. [DOI] [PubMed] [Google Scholar]
- Turner S.; Myers M.; Gadie B.; Hale S. A.; Horsley A.; Nelson A. J.; Pape R.; Saville J. F.; Doxey J. C.; Berridge T. L. Antihypertensive thiadiazoles. 2. Vasodilator activity of some 2-aryl-5-guanidino-1,3,4-thiadiazoles. J. Med. Chem. 1988, 31, 906–913. 10.1021/jm00400a004. [DOI] [PubMed] [Google Scholar]
- Hanna M. A.; Girges M. M.; Rasała D.; Gawinecki R. Synthesis and pharmacological evaluation of some novel 5-(pyrazol-3-yl)thiadiazole and oxadiazole derivatives as potential hypoglycemic agents. Arzneim. Forsch. 1996, 27, 1074–1078. 10.1002/CHIN.199607149. [DOI] [PubMed] [Google Scholar]
- Pagano M.; Demoro B.; Toloza J.; Boiani L.; González M.; Cerecetto H.; Olea-Azar C.; Norambuena E.; Gambino D.; Otero L. Effect of ruthenium complexation on trypanocidal activity of 5-nitrofuryl containing thiosemicarbazones. Eur. J. Med. Chem. 2009, 44, 4937–4943. 10.1016/j.ejmech.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Mazzone G.; Pignatello R.; Mazzone S.; Panico A.; Pennisi G.; Castana R.; Mazzone P. Synthesis and local anesthetic activity of alkylaminoacyl derivatives of 2-amino-1,3,4-thiadiazole. Farmaco 1994, 48, 1207–1224. 10.1002/CHIN.199412195. [DOI] [PubMed] [Google Scholar]
- Andreani A.; Granaiola M.; Leoni A.; Locatelli A.; Morigi R.; Rambaldi M. Synthesis and antitubercular activity of imidazo[2,1-b]thiazoles. Eur. J. Med. Chem. 2001, 36, 743–746. 10.1016/S0223-5234(01)01266-1. [DOI] [PubMed] [Google Scholar]
- Holla B. S.; Malini K. V.; Rao B. S.; Sarojini B. K.; Kumari N. S. Synthesis of some new 2,4-disubstituted thiazoles as possible antibacterial and anti-inflammatory agents. Eur. J. Med. Chem. 2003, 38, 313–318. 10.1016/S0223-5234(02)01447-2. [DOI] [PubMed] [Google Scholar]
- Tadros A. B.; El-Batouti M. Spectral study and antifouling assessment of some thiosemicarbazone derivatives. Anti-Corros. Methods Mater. 2004, 51, 406–413. 10.1108/00035590410560958. [DOI] [Google Scholar]
- Basak P.; Gangopadhyay S.; De S.; Drew M. G. B.; Gangopadhyay P. K. Cobalt(III) complexes of some aromatic thiohydrazides–Synthesis, characterization and structure. Inorg. Chim. Acta 2010, 363, 1495–1499. 10.1016/j.ica.2010.01.011. [DOI] [Google Scholar]
- Ganjali M. R.; Hosseini M.; Salavati-Niasari M.; Poursaberi T.; Shamsipur M.; Javanbakht M.; Hashemi O. R. Nickel ion-selective coated graphite PVC-membrane electrode based on benzylbis(thiosemicarbazone). Electroanalysis 2002, 14, 526–531. . [DOI] [Google Scholar]
- Rubčić M.; Đilović I.; Cindrić M.; Matković-Čalogović D. Two thiosemicarbazones derived from salicylaldehyde: very specific hydrogen-bonding interactions of the N—H··· S= C type. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2008, 64, o570–o573. 10.1107/S0108270108029016. [DOI] [PubMed] [Google Scholar]
- Pelosi G. Thiosemicarbazone Metal Complexes: From Structure to Activity. Open Crystallogr. J. 2010, 3, 16–28. 10.2174/1874846501003010016. [DOI] [Google Scholar]
- Bal T. R.; Anand B.; Yogeeswari P.; Sriram D. Synthesis and evaluation of anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4451–4455. 10.1016/j.bmcl.2005.07.046. [DOI] [PubMed] [Google Scholar]
- Krishnakumar V.; Nagalakshmi R. Crystal growth and vibrational spectroscopic studies of the semiorganic non-linear optical crystal—bisthiourea zinc chloride. Spectrochim. Acta, Part A 2005, 61, 499–507. 10.1016/j.saa.2004.04.014. [DOI] [PubMed] [Google Scholar]
- Yousef T. A.; El-Reash G. M. A.; El-Gammal O. A.; Bedier R. A. Co(II), Cu(II), Cd(II), Fe(III) and U(VI) complexes containing a NSNO donor ligand: synthesis, characterization, optical band gap, in vitro antimicrobial and DNA cleavage studies. J. Mol. Struct. 2012, 1029, 149–160. 10.1016/j.molstruc.2012.06.050. [DOI] [Google Scholar]
- Maiman T. H. Stimulated optical radiation in ruby. Nature 1960, 187, 493. 10.1038/187493a0. [DOI] [Google Scholar]
- Franken P. A.; Hill A. E.; Peters C. W.; Weinreich G. Generation of optical harmonics. Phys. Rev. Lett. 1961, 7, 118. 10.1103/PhysRevLett.7.118. [DOI] [Google Scholar]
- Burland D. M.; Miller R. D.; Walsh C. A. Second-order nonlinearity in poled-polymer systems. Chem. Rev. 1994, 94, 31–75. 10.1021/cr00025a002S. [DOI] [Google Scholar]
- Gao L.; Huang J.; Guo S.; Yang Z.; Pan S. Structure-property survey and computer-assisted screening of mid-infrared nonlinear optical chalcohalides. Coord. Chem. Rev. 2020, 421, 213379. 10.1016/j.ccr.2020.213379. [DOI] [Google Scholar]
- Petrov V. Frequency down-conversion of solid-state laser sources to the mid-infrared spectral range using non-oxide nonlinear crystals. Prog. Quantum Electron. 2015, 42, 1–106. 10.1016/j.pquantelec.2015.04.001. [DOI] [Google Scholar]
- Delaire J. A.; Nakatani K. Linear and nonlinear optical properties of photochromic molecules and materials. Chem. Rev. 2000, 100, 1817–1846. 10.1021/cr980078m. [DOI] [PubMed] [Google Scholar]
- Evans O. R.; Lin W. Crystal engineering of NLO materials based on metal–organic coordination networks. Acc. Chem. Res. 2002, 35, 511–522. 10.1021/ar0001012. [DOI] [PubMed] [Google Scholar]
- Han S.; Mutailipu M.; Tudi A.; Yang Z.; Pan S. PbB5O7F3: A high-performing short-wavelength nonlinear optical material. Chem. Mater. 2020, 32, 2172–2179. 10.1021/acs.chemmater.0c00150. [DOI] [Google Scholar]
- Jawaria R.; Hussain M.; Khalid M.; Khan M. U.; Tahir M. N.; Shafiq Z.; Naseer M. M.; Braga A. A. C. Synthesis, crystal structure analysis, spectral characterization and nonlinear optical exploration of potent thiosemicarbazones based compounds: A DFT refine experimental study. Inorg. Chim. Acta 2019, 486, 162–171. 10.1016/j.ica.2018.10.035. [DOI] [Google Scholar]
- Cariati E.; Dragonetti C.; Lucenti E.; Nisic F.; Righetto S.; Roberto D.; Tordin E. An acido-triggered reversible luminescent and nonlinear optical switch based on a substituted styrylpyridine: EFISH measurements as an unusual method to reveal a protonation–deprotonation NLO contrast. Chem. Commun. 2014, 50, 1608–1610. 10.1039/C3CC48149B. [DOI] [PubMed] [Google Scholar]
- Tykwinski R. R.; Gubler U.; Martin R. E.; Diederich F.; Bosshard C.; Günter P. Structure–property relationships in third-order nonlinear optical chromophores. J. Phys. Chem. B 1998, 102, 4451–4465. 10.1021/jp980829o. [DOI] [Google Scholar]
- Shi Y.; Zhang C.; Zhang H.; Bechtel J. H.; Dalton L. R.; Robinson B. H.; Steier W. H. Low (sub-1-volt) halfwave voltage polymeric electro-optic modulators achieved by controlling chromophore shape. Science 2000, 288, 119–122. 10.1126/science.288.5463.119. [DOI] [PubMed] [Google Scholar]
- Lee M.; Katz H. E.; Erben C.; Gill D. M.; Gopalan P.; Heber J. D.; McGee D. J. Broadband modulation of light by using an electro-optic polymer. Science 2002, 298, 1401–1403. 10.1126/science.1077446. [DOI] [PubMed] [Google Scholar]
- Low P. J. Metal complexes in molecular electronics: progress and possibilities. Dalton Trans. 2005, 2821–2824. 10.1039/B506017F. [DOI] [PubMed] [Google Scholar]
- Adeel M.; Braga A. A. C.; Tahir M. N.; Haq F.; Khalid M.; Halim M. A. Synthesis, X-ray crystallographic, spectroscopic and computational studies of aminothiazole derivatives. J. Mol. Struct. 2017, 1131, 136–148. 10.1016/j.molstruc.2016.11.046. [DOI] [Google Scholar]
- Khalid M.; Ali A.; Adeel M.; Din Z. U.; Tahir M. N.; Rodrigues-Filho E.; Iqbal J.; Khan M. U. Facile preparation, characterization, SC-XRD and DFT/DTDFT study of diversely functionalized unsymmetrical bis-aryl-α, β-unsaturated ketone derivatives. J. Mol. Struct. 2020, 1206, 127755. 10.1016/j.molstruc.2020.127755. [DOI] [Google Scholar]
- Andersson M. P.; Uvdal P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-ζ basis set 6-311+G(d,p). J. Phys. Chem. A. 2005, 109, 2937–2941. 10.1021/jp045733a. [DOI] [PubMed] [Google Scholar]
- Muthu S.; Paulraj E. I. Molecular structure, vibrational spectra, first order hyper polarizability, NBO and HOMO–LUMO analysis of 4-amino-3(4-chlorophenyl) butanoic acid. Solid State Sci. 2012, 14, 476. 10.1016/j.solidstatesciences.2012.01.028. [DOI] [Google Scholar]
- Singh P. K.; Koacher J. K.; Tandon J. P. Boron Complexes Of Some Oxygen-Nitrogen And Sulphur-Nitrogen Containing Chelating Agents. J. Inorg. Nucl. Chem. 1981, 43, 1755–1758. 10.1016/0022-1902(81)80378-8. [DOI] [Google Scholar]
- Rittner R.; Ducati L. C.; Tormena C. F.; Fiorin B. C.; Braga C. B. Conformational preferences for some 5-substituted 2-acetylthiophenes through infrared spectroscopy and theoretical calculations. Spectrochim. Acta, Part A 2011, 79, 1071–1076. 10.1016/j.saa.2011.04.021. [DOI] [PubMed] [Google Scholar]
- Ali A.; Khalid M.; Rehman M. F. U.; Haq S.; Ali A.; Tahir M. N.; Ashfaq M.; Rasool F.; Braga A. A. C. Efficient Synthesis, SC-XRD, and Theoretical Studies of O-Benzenesulfonylated Pyrimidines: Role of Noncovalent Interaction Influence in Their Supramolecular Network. ACS Omega 2020, 5, 15115–15128. 10.1021/acsomega.0c00975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teimouri A.; Chermahini A. N.; Taban K.; Dabbagh H. A. Experimental and CIS, TD-DFT, ab initio calculations of visible spectra and the vibrational frequencies of sulfonyl azide-azoic dyes. Spectrochim. Acta, Part A 2009, 72, 369–377. 10.1016/j.saa.2008.10.006. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; Hermans J. Interaction models for water in relation to protein hydration. Intermol. Forces 1981, 331–342. 10.1007/978-94-015-7658-1_21. [DOI] [Google Scholar]
- Balachandran V.; Lakshmi A.; Janaki A. Vibrational spectroscopic studies and natural bond orbital analysis of 4,6-dichloro-2-(methylthio)pyrimidine based on density functional theory. Spectrochim. Acta, Part A 2011, 81, 1–7. 10.1016/j.saa.2011.02.011. [DOI] [PubMed] [Google Scholar]
- Irfan A.; Al-Sehemi A. G.; Chaudhry A. R.; Muhammad S. The structural, electro-optical, charge transport and nonlinear optical properties of oxazole (4Z)-4-benzylidene-2-(4-methylphenyl)-1,3-oxazol-5(4H)-one derivative. J. King Saud Univ., Sci. 2018, 30, 75–82. 10.1016/j.jksus.2016.10.004. [DOI] [Google Scholar]
- Shahid M.; Salim M.; Khalid M.; Tahir M. N.; Khan M. U.; Braga A. A. C. Synthetic, XRD, non-covalent interactions and solvent dependent nonlinear optical studies of Sulfadiazine-Ortho-Vanillin Schiff base: (E)-4-((2-hydroxy-3-methoxy-benzylidene) amino)-N-(pyrimidin-2-yl)benzene-sulfonamide. J. Mol. Struct. 2018, 1161, 66–75. 10.1016/j.molstruc.2018.02.043. [DOI] [Google Scholar]
- Srnec M.; Solomon E. I. Frontier molecular orbital contributions to chlorination versus hydroxylation selectivity in the non-heme iron halogenase SyrB2. J. Am. Chem. Soc. 2017, 139, 2396–2407. 10.1021/jacs.6b11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopmans T. Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den einzelnen Elektronen eines Atoms. Physica 1934, 1, 104–113. 10.1016/S0031-8914(34)90011-2. [DOI] [Google Scholar]
- Parr R. G.; Szentpály L. v.; Liu S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. 10.1021/ja983494x. [DOI] [Google Scholar]
- Fukui K. The Role of frontier orbitals in chemical reactions (Nobel Lecture). Angew. Chem., Int. Ed. Engl. 1982, 21, 801–809. 10.1002/anie.198208013. [DOI] [Google Scholar]
- Khalid M.; Arshad M. N.; Tahir M. N.; Asiri A. M.; Naseer M. M.; Ishaq M.; Khan M. U.; Shafiq Z. An efficient synthesis, structural (SC-XRD) and spectroscopic (FTIR, 1HNMR, MS spectroscopic) characterization of novel benzofuran-based hydrazones: An experimental and theoretical studies. J. Mol. Struct. 2020, 1216, 128318. 10.1016/j.molstruc.2020.128318. [DOI] [Google Scholar]
- Khalid M.; Ali A.; Jawaria R.; Asghar M. A.; Asim S.; Khan M. U.; Hussain R.; ur Rehman M. F.; Ennis C. J.; Akram M. S. First principles study of electronic and nonlinear optical properties of A–D−π–A and D–A–D−π–A configured compounds containing novel quinoline–carbazole derivatives. RSC Adv. 2020, 10, 22273–22283. 10.1039/D0RA02857F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhammad S.; Xu H.-L.; Zhong R.-L.; Su Z.-M.; Al-Sehemi A. G.; Irfan A. Quantum chemical design of nonlinear optical materials by sp2-hybridized carbon nanomaterials: issues and opportunities. J. Mater. Chem. C 2013, 1, 5439–5449. 10.1039/C3TC31183J. [DOI] [Google Scholar]
- Muhammad S.; Janjua M. R. S. A.; Su Z. Investigation of dibenzoboroles having π-electrons: toward a new type of two-dimensional NLO molecular switch?. J. Phys. Chem. C 2009, 113, 12551–12557. 10.1021/jp903075s. [DOI] [Google Scholar]
- Bi W.; Louvain N.; Mercier N.; Luc J.; Rau I.; Kajzar F.; Sahraoui B. A Switchable NLO Organic-Inorganic Compound Based on Conformationally Chiral Disulfide Molecules and Bi(III)I5 Iodobismuthate Networks. Adv. Mater. 2008, 20, 1013–1017. 10.1002/adma.200701753. [DOI] [Google Scholar]
- Crasta V.; Ravindrachary V.; Bhajantri R. F.; Gonsalves R. Growth and characterization of an organic NLO crystal: 1-(4-methylphenyl)-3-(4-methoxyphenyl)-2-propen-1-one. J. Cryst. Growth 2004, 267, 129–133. 10.1016/j.jcrysgro.2004.03.037. [DOI] [Google Scholar]
- Khan M. U.; Khalid M.; Ibrahim M.; Braga A. A. C.; Safdar M.; Al-Saadi A. A.; Janjua M. R. S. A. First theoretical framework of triphenylamine–dicyanovinylene-based nonlinear optical dyes: structural modification of π-linkers. J. Phys. Chem. C 2018, 122, 4009–4018. 10.1021/acs.jpcc.7b12293. [DOI] [Google Scholar]
- Reis H.; Papadopoulos M. G.; Munn R. W. Calculation of macroscopic first-, second-, and third-order optical susceptibilities for the urea crystal. J. Chem. Phys. 1998, 109, 6828–6838. 10.1063/1.477330. [DOI] [Google Scholar]
- Amiri S. S.; Makarem S.; Ahmar H.; Ashenagar S. Theoretical studies and spectroscopic characterization of novel 4-methyl-5-((5-phenyl-1,3,4-oxadiazol-2-yl)thio) benzene-1,2-diol. J. Mol. Struct. 2016, 1119, 18–24. 10.1016/j.molstruc.2016.04.053. [DOI] [Google Scholar]
- Gürsoy A.; Karali N. Synthesis, characterization and primary antituberculosis activity evaluation of 4-(3-coumarinyl)-3-benzyl-4-thiazolin-2-one benzylidenehydrazones. Turk. J. Chem. 2003, 27, 545–552. [Google Scholar]
- Poirier R.; Kari R.; Csizmadia I. G.. Handbook of Gaussian basis sets; Elsevier, 1985. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.. Gaussian 01; Gaussian, Inc.: Wallingford, CT: 2009. [Google Scholar]
- Bader R. F. W. Bond paths are not chemical bonds. J. Phys. Chem. A 2009, 113, 10391–10396. 10.1021/jp906341r. [DOI] [PubMed] [Google Scholar]
- Zhurko G.; Zhurko D.. ChemCraft, version 1.6. URL: http://www. chemcraftprog. com 2009.
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. Cclib: a library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- Mackenzie C. F.; Spackman P. R.; Jayatilaka D.; Spackman M. A. CrystalExplorer model energies and energy frameworks: extension to metal coordination compounds, organic salts, solvates and open-shell systems. IUCrJ 2017, 4, 575–587. 10.1107/S205225251700848X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross E. K. U.; Kohn W. Time-dependent density-functional theory. Adv. Quantum Chem. 1990, 255–291. 10.1016/S0065-3276(08)60600-0. [DOI] [Google Scholar]
- Burke K.; Werschnik J.; Gross E. K. U. Time-dependent density functional theory: Past, present, and future. J. Chem. Phys. 2005, 123, 062206 10.1063/1.1904586. [DOI] [PubMed] [Google Scholar]
- Valverde C.; de Lima e Castro S. A.; Vaz G. R.; de Almeida Ferreira J. L.; Baseia B.; Osório F. A. P.; et al. Acta Chim. Slov. 2018, 65, 739–749. 10.17344/ACSI.2018.4462. [DOI] [PubMed] [Google Scholar]
- Barone V.; Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J. Phys. Chem. A 1998, 102, 1995–2001. 10.1021/jp9716997. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.