

Abstract
Objective
Radiation therapy is a mainstay in the treatment of numerous neoplasms. Numerous publications have reported good clinical outcomes for primary radiation therapy for Vestibular Schwannomas (VS). However, there are relatively few pathologic specimens of VSs available to evaluate post‐radiation, which has led to a relative dearth in research on the cellular mechanisms underlying the effects of radiation therapy on VSs.
Methods
Here we review the latest literature on the complex biological effects of radiation therapy on these benign tumors—including resistance to oxidative stress, mechanisms of DNA damage repair, alterations in normal growth factor pathways, changes in surrounding vasculature, and alterations in immune responses following radiation.
Results
Although VSs are highly radioresistant, radiotherapy is often successful in arresting their growth.
Conclusion
By better understanding the mechanisms underlying these effects, we could potentially harness such mechanisms in the future to potentiate the clinical effects of radiotherapy on VSs.
Level of Evidence
N/A.
Keywords: radiation therapy, radiobiology, radiosurgery, vestibular schwannoma
Although Vestibular Schwannomas (VS) are highly radioresistant, radiotherapy is often successful in arresting their growth. Here we review the latest literature on the complex biological effects of radiation therapy on these benign tumors—including resistance to oxidative stress, mechanisms of DNA damage repair, alterations in normal growth factor pathways, changes in surrounding vasculature, and alterations in immune responses following radiation. By better understanding the mechanisms underlying these effects, we could potentially harness such mechanisms in the future to potentiate the clinical effects of radiotherapy on VSs.
1. INTRODUCTION
Radiation therapy is a mainstay in the treatment of numerous neoplasms, whether as an adjuvant therapy to surgical resection or as an alternative to surgery. In the case of vestibular schwannomas (VSs), there are numerous publications reporting clinical outcomes for each therapeutic strategy. However, given that VSs are benign tumors and thus do not typically result in a patient's death, there are few pathologic specimens available to evaluate post‐radiation, which has led to a relative dearth in research on the cellular mechanisms underlying the effectiveness of radiation therapy for VSs.
Sporadic cases of VS constitute approximately 95% of cases. 1 The remainder are accounted for by neurofibromatosis type 2 (NF2), a syndrome of autosomal dominant inheritance secondary to mutation in NF2 gene, which encodes the tumor suppressor protein merlin. 2 The hallmark of NF2 is bilateral VS, which poses a particular challenge towards managing and treating this condition.
Broadly speaking, one can divide radiation therapy into two primary groups: conventional fractionated radiotherapy (FRT) and stereotactic radiosurgery (SRS). This distinction is based on the number of sessions over which the target dose is delivered, with FRT administered in small doses over many sessions—hence, fractionated—and SRS given as larger doses over one to five sessions. 3 FRT is commonly delivered with linear accelerator, or LINAC, technology, of which there are multiple commercial manufacturers. The name SRS was first coined by the famous Swedish neurosurgeon Lars Leksell in 1951, and it was Leksell who later developed the first GammaKnife device in 1967 (Elekta AB, Stockholm, Sweden). 4 GammaKnife has undergone several iterations after becoming commercially available in the United States in 1987, and is perhaps the most commonly used SRS device in the United States today. 5 Some LINAC devices are also capable of delivering SRS.
The efficacy of FRT compared to SRS depends greatly on the characteristics of the pathology being treated. Historically, the decision to use one or the other in a particular clinical scenario was almost entirely empirical—that is, clinical outcomes were used to guide treatment decisions with little to no knowledge of the underlying biological response to the therapy. 6 With time, the study of radiobiology has advanced significantly, but in many ways the field remains in its infancy. Here we will first review the basic biological effects of ionizing radiation, the classic “5 R's” of radiobiology, and how they apply to SRS. Next, we will review the latest literature specifically focused on the radiobiological effects of SRS (and, to a lesser extent, FRT) on vestibular schwannomas. Finally, we will review the latest work on specific cell signaling pathways and molecules which are thought to take part in these effects.
1.1. Cellular effects of ionizing radiation
The anti‐tumor effect of ionizing radiation delivered as FRT or SRS is complex and multifactorial. Ionizing radiation has both direct and indirect effects on tumor cells. 6 , 7 At the most basic level, radiation acts upon tissues by depositing excess energy into the molecules of said tissue, which results in ionization and subsequent formation of free radicals. Although the ionizing radiation can affect any molecule in its path—including proteins, nucleic acids, lipids, or water—the most common molecule in human tissue is water, and thus water is the most frequently ionized substance. The ionization of water produces free radicals such as OH‐ (hydroxyl radical) which can then damage cellular DNA; this indirect damage via hydroxyl radicals may cause as much as two‐thirds of the DNA damage observed after ionizing radiation, with direct DNA ionization accounting for only one‐third. 6 , 8 , 9 The production of these hydroxyl radicals also leads to the formation of additional reactive oxygen species by inducing activity of mitochondrial oxidase, nitric oxide synthase, and cytoplasmic NADPH synthase. 10 , 11 , 12 , 13 These indirect effects can last well beyond the immediate radiation exposure and even spread to other nearby cells. 14 , 15 , 16 , 17 Ultimately, these different pathways all lead to damage of key cellular components including DNA and cell membranes. DNA can be damaged in multiple ways following radiation, but the most important are double‐strand breaks. When cellular machinery recognizes DNA damage, the cell cycle is arrested to repair the error prior to proceeding with reproduction. Checkpoints are key stoppage points between cell cycle phases to ensure that damaged DNA is not replicated. Double‐strand breaks (DSB) are repaired in one of two ways: homologous recombination and nonhomologous end joining (NHEJ). Homologous recombination uses the replicated sister chromatid as a template to replace the damaged or missing DNA sequence following DSB; hence, the repair is highly accurate. However, because it relies on the sister chromatid, it can only be done in the late S or the G2 phase, after replication has occurred. Otherwise, the cell must use NHEJ, which removes bases from the 3′ end of each strand prior to reattaching them, a process that is highly error‐prone. Other erroneous changes such as translocations can occur. Ultimately, severe changes to DNA will frequently result in cell death, of which there are multiple mechanisms. 6
Four key cellular processes leading to tumor control post‐radiation are mitotic catastrophe, apoptosis, necrosis, and senescence. Mitotic catastrophe occurs when a neoplastic cell with damaged or misrepaired DNA passes a checkpoint to enter mitosis when it otherwise should not, typically due to dysfunctional checkpoint control; subsequent death occurs via apoptosis or necrosis. 18 The programmed cell death of apoptosis is brought on by one of three pathways—intrinsic, extrinsic, and the ceramide pathway. In the intrinsic, or mitochondrial, pathway, excessive DNA damage triggers mitochondrial release of cytochrome c, which leads to a cascade of events resulting in apoptosis; p53 is a key component of this process. 19 The extrinsic pathway occurs when cell membrane death receptors are activated by extracellular tumor necrosis factor (TNF); cells often upregulate these death receptors following radiation. 20 , 21 , 22 Finally, in the ceramide pathway, radiation activates acid sphingomyelinase, which catalyzes the production of ceramide, which itself serves to initiate apoptosis via a distinct mechanism. 23 , 24 Necrosis is a histological description of unregulated cell death resulting from severe changes to the cellular microenvironment, such as energy loss or extreme pH changes, making the cell incapable of survival. Finally, senescence refers to permanent arrest of the cell cycle without true death, meaning that while the cell remains alive, it cannot contribute to tumor growth.
1.2. Indirect effects of stereotactic radiosurgery
In addition to directly killing tumor cells, damage to surrounding vasculature and induction of immunological responses have also been proposed to take part in the anti‐tumor effects of SRS (Figure 1). Traditional models for FRT, such as the linear quadratic model, do not always accurately reflect the clinical outcomes seen in SRS. 6 Mouse models of SRS have shown that highly radioresistant tumors including fibosarcomas and melanomas were nonetheless significantly impacted by SRS due to destruction of tumor‐associated vasculature. 25 , 26 , 27 Others have shown that there can be an immune‐mediated antitumor response following SRS, which is proposed to result from increased tumor antigen display following SRS. 28 , 29
FIGURE 1.
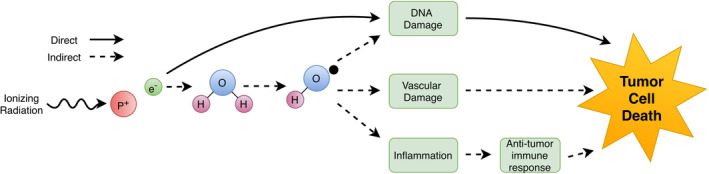
The direct and indirect effects of ionizing radiation leading to tumor cell death. Aside from directly damaging cellular DNA and other machinery and thereby inducing cell death, there are indirect manners of cell death—both acute and chronic—via damage to peritumoral endothelial cells and via induction of a systemic immune response
1.3. The five R's of radiobiology
While originally conceived in the context of conventional FRT, the five R's of radiobiology can also be applied to SRS. Given that our focus is SRS, we will only briefly discuss this here. The five R's are as follows: Repair, Redistribution, Reoxygenation, Repopulation, and Radiosensitivity. Repair refers to repair of DNA strand breaks and chromosomal alterations, which as discussed before are a key component of the cell's response to radiation and, when overwhelmed, will result in cell death. SRS and FRT work similarly with respect to repair. Redistribution refers to the stage of cell cycle a given cell is in when the treatment is given; certain stages are far more sensitive to radiation than others, so it is beneficial to hit the target multiple times so as to increase the chance that any given cell will be maximally susceptible to IR damage at some point during treatment. SRS does not benefit from redistribution due to the small number of treatments; at the same time, unlike malignant tumors with high proliferation indices, VS would not likely benefit from redistribution with FRT due to the small percentage of their cells that are dividing at any given time. Reoxygenation refers to the phenomenon that tumors often have regions of relative hypoxia, and since formation of ROS is a key mediator of IR damage, lacking oxygen can make cells relatively more radioresistant. FRT is thought to counteract this with changes in the tumor microenvironment over time during the multiple fractions of treatment; SRS does not address this problem. Repopulation refers to tumor cell regrowth between treatments; SRS is presumed to be superior to FRT in this respect given that there are few or no gaps between multiple fractions. Finally, radiosensitivity is a characteristic of the tumor type being treated. Contemporary knowledge of tumor radiosensitivity is derived primarily from clinical data. Multiple mathematical models—such as the Target Theory and the Linear Quadratic Theory—have been proposed to explain this response at the biophysical level, but this remains a topic of much controversy and further discussion is beyond the scope of this article. 6 , 30 , 31 , 32 , 33
2. VESTIBULAR SCHWANNOMA RADIOBIOLOGY
Vestibular schwannomas (VS) are benign tumors of the CN VIII nerve sheath. They represent about 80% of all tumors in the cerebellopontine angle but fewer than 6% of all intracranial tumors. 34 While VS are benign tumors, they can lead to various cranial nerve deficits, such as hearing loss, tinnitus, imbalance, hypoesthesia (due to compression of the trigeminal nerve), and hydrocephalus.
The three pillars of VS treatment are close observation, surgical resection, and irradiation. Traditionally, surgical eradication of the tumor was the first choice of treatment regardless of tumor size, however over the past three decades less invasive treatment options have become increasingly more prevalent. 35 , 36 Aided by the benign nature of VS and advances in noninvasive neuroimaging, close observation or a “wait‐and‐scan” approach has become the first choice of treatment for many cerebellopontine angle tumors under 1.5 cm in size. 37 , 38 Interventions such as microsurgery and irradiation are considered only in cases of continued tumor growth.
Stereotactic Radiosurgery (SRS) has become a well‐accepted noninvasive treatment alternative to surgery. In this case, tumors are not eliminated, rather the goal is to arrest further growth. SRS controls tumor growth and offers cranial nerve preservation rates comparable to surgical resection. 39 , 40 , 41 , 42 While some tumors shrink following radiation, all exhibit tumor viability and hold the possibility of eventual tumor growth. Furthermore, not all tumors are controlled with radiation alone, which suggests that radiosensitivity is variable among VS. 43 , 44 Tumor control rate (stable or decreased size) is 91%, while approximately 9% of the tumors are radioresistant; some tumors continue to grow despite high doses of radiation. 45 All this suggests that unlike malignant tumors, VS are particularly radioresistant, so clinical surveillance is required indefinitely in irradiated VS. 46 This relative resistance to radiation is not surprising given the low proliferative capacity of VS cells reflected in the slow clinical growth rate of VSs. Understanding the cellular mechanisms that render VS cells resistant to radiation provides an opportunity to target these mechanisms in an effort to enhance VS cell sensitivity to radiation and, perhaps, expand the effectiveness of this treatment strategy. Here we first provide a brief overview of the effects of ionizing radiation on cells generally. We then discuss recent data that informs the radiobiology of VSs.
In NF2 patients, VS seem particularly radioresistant with a high escape rate. 47 , 48 In this review, we consider the radiobiology of both sporadic and NF2‐associated VS. Given their somewhat distinct behavior, further discussion of the radiobiologic features that are specific to NF2‐associated VS is highly warranted; however, this is not well described in the current literature.
To date, research has revealed several factors linked to the biology of radioresistance in VS, including histological features, resistance to oxidative stress, effects of cell cycle and proliferation on radiosensitivity, alteration of cell‐cycle checkpoint and apoptotic pathways, key growth factor pathways, and angiogenesis. Unfortunately, clinical studies which report histopathological changes following SRS inherently select for non‐responding tumors because tumors with successful SRS do not typically undergo surgical resection. Although such selection bias may limit the generalizability of pathologic features reported in some clinical case series, this bias is unavoidable. In addition, the difficulty of replicating the tumor microenvironment in the laboratory is an inherent hurdle to investigating the radiobiology of VS. Nonetheless, a body of research both in vitro and in vivo has provided the framework to deepen our understanding of radiobiology of VS. In this section, we will review these factors contributing to the radiobiology of VS.
2.1. Histopathologic features of irradiated VSs
Typical VS morphology demonstrates bipolar spindle cells with moderate cellularity, interspersed Antoni A and Antoni B pattern regions with Verocay bodies and hyalinized blood vessels.
The in vivo radiobiology of human VS was first assessed in immunocompromised mice‐xenograft models. 49 Mice implanted with VS harvested from patients were irradiated with varying doses and tissue was harvested for histological assessment 3 months later. Increasing doses of radiation up to 40 Gy significantly reduced tumor volume and vascularity, while at 10 Gy there was no change in tumor vascularity. In 2003, Lee et al assessed the histological features of VS that failed radiation and underwent salvage microsurgical resection. 50 Light microscopy demonstrated varying degrees of nuclear pleomorphism with hyperchromasia, vascular hyalinization with surrounding hemosiderin deposition, and hypercellular areas similar to normal, non‐irradiated VS tissue. Others have described partial necrotic, fibrotic and vascular changes following radiation of the tumor. 39 , 43 Although histological features of irradiated VSs vary, overall features unique to radiation‐induced changes in VS have not been identified. 10 , 39 , 43 , 51 , 52
2.2. Resistance to oxidative stress
Recently, Robinett et al assessed the histopathological features in four VS patients who recurred after initial microsurgical resection, were then treated with SRS, and later underwent re‐resection due to failure of salvage SRS. 10 Tyrosine nitrosylation, a marker of oxidative stress following radiation in malignant tumors, was used to assess whether VS treated with SRS show signs of oxidative stress, despite being benign tumors treated with significantly lower radiation doses than malignant tumors. In three of four tumors, nitrotyrosine immunostaining was significantly higher post‐radiation, even when several years had passed since radiation treatment. The authors concluded that these irradiated VS persistently grew despite the presence of oxidative stress. These results indicate that irradiated VSs are able to grow despite the cells being under long‐term oxidative stress, implying that VS cells have mechanisms to mitigate oxidative stress and continue to proliferate.
Taken together, these observations suggest that the effect of radiation on VS may be indirect, perhaps involving damage to the surrounding vasculature and/or induction of immunological responses.
2.3. Effects of cell cycle and proliferation on radiosensitivity
It is well established that rapidly dividing cells are more sensitive to radiation than slowly dividing cells. 6 In cell culture conditions, VS cells proliferate very slowly and are less sensitive to radiation than malignant tumor cells, and they require higher doses of radiation to prevent growth. 43 , 53 , 54 Cultured primary human VS cells require over 20 Gy radiation to induce cell death and cell cycle arrest. 55 , 56 , 57 The low proliferation rate is thought to correlate to low radiosensitivity of VS cells. Consistent with the notion that the relative radioresistance of VS cells is due, at least in part, to their limited proliferative capacity, augmenting cell proliferation by application of exogenous mitogens that increase cell proliferation enhances radiation‐induced cell death, while reducing proliferation with ErbB2 inhibitors limits radiation‐induced cell death in VS. 56
Lee et al assessed proliferation potential of VS regrowth following SRS with Gamma Knife vs microsurgery using the immunohistochemical marker PCNA (proliferating cell nuclear antigen) to assess the tumor proliferation capacity. 53 Fifteen patients underwent microsurgical resection and eight patients underwent SRS. The nuclei of schwannoma cells in all tumors were labeled with PCNA. In tumors that underwent SRS the PCNA index was significantly lower than the microsurgery group, suggesting that radiation‐induced apoptosis may reduce proliferation. However, two of the eight patients that underwent SRS had increased proliferation levels, which highlight the variable response of VSs to radiation; the authors did not specify whether those two tumors demonstrated a clinically significant difference in growth compared to the other six tumors. 53
2.4. Alteration of cell‐cycle checkpoint and apoptotic pathways
As previously mentioned, radiation activates cell‐cycle checkpoints leading to tumor growth arrest or necrosis. Cells are most sensitive to radiation during mitosis (M) and the G2 phase, less sensitive in G1, and least sensitive during the latter part of the S phase. 58 Radiation‐induced cell death typically requires re‐entry into the cell cycle. Jacob et al reported the histological results from a non‐growing VS based on MRI that underwent biopsy 3 years after radiation. The tumor section was immunostained for S‐100 and Ki67, a marker of proliferating cells. Ki‐67 is expressed during the active phases of the cell cycle (G1, S, G2, and mitosis), and is absent during quiescent phases (G0). 59 Interestingly, the irradiated tumor expressed Ki67 protein suggesting that while VSs remain grossly stable in size, at the molecular level, the cell cycle was active in some portion of the tumor. 60 This also suggests that cells are capable of repairing DNA damage prior to re‐entering the cell cycle and cells that do so can bypass apoptosis. Within a single tumor, proliferation rates of different sub‐populations can vary. 61
2.5. Key growth factor pathways
2.5.1. Merlin
In both sporadic VS and NF2‐associated VSs, inactivation of the tumor suppressor gene NF2 plays a central role (Figure 2). 62 , 63 , 64 The NF2 gene resides on chromosome 22q12 and encodes the schwannomin/merlin protein, which shares homology to ezrin‐radixin‐moesin (ERM) family of membrane–cytoskeleton‐linking proteins. Merlin regulates transmembrane and signaling molecules' interaction with cytoskeletal actin, thereby affecting cell–cell attachments, cell motility, and subcellular localization in response to cell‐to‐cell contact inhibition. 65 , 66 , 67 Thus, merlin is an important mediator of contact inhibition. Lack of merlin leads to disruption of cell‐to‐cell contact inhibition promoting cell proliferation and tumorigenesis. Recent evidence supports that merlin may directly or indirectly interact with several proteins leading to the suppression of mitogenic activity at both the cellular membrane and nucleus levels. 68 , 69 At the cell membrane level, merlin blocks signaling by integrins and tyrosine receptor kinases (RTKs) such as ErB2 and platelet derived growth factor receptor and regulates multiple downstream pathways, including the Ras/Raf/MEK/ERK, FAK/Src, PI3K/AKT, Rac/PAK/ JNK, mTORC1, and Wnt/β‐catenin pathways. 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 (p200), 81 , 82 , 83 , 84 , 85 , 86 , 87 In addition, the Hippo pathway is inhibited upstream by merlin to suppress the function of Yes‐associated protein 1 (YAP1), an oncogene associated with meningioma proliferation. 88 , 89 , 90 , 91 At the nuclear level, merlin downregulates the E3 ubiquitin ligase CRL4 (DCAF1) to inhibit proliferation. 92
FIGURE 2.
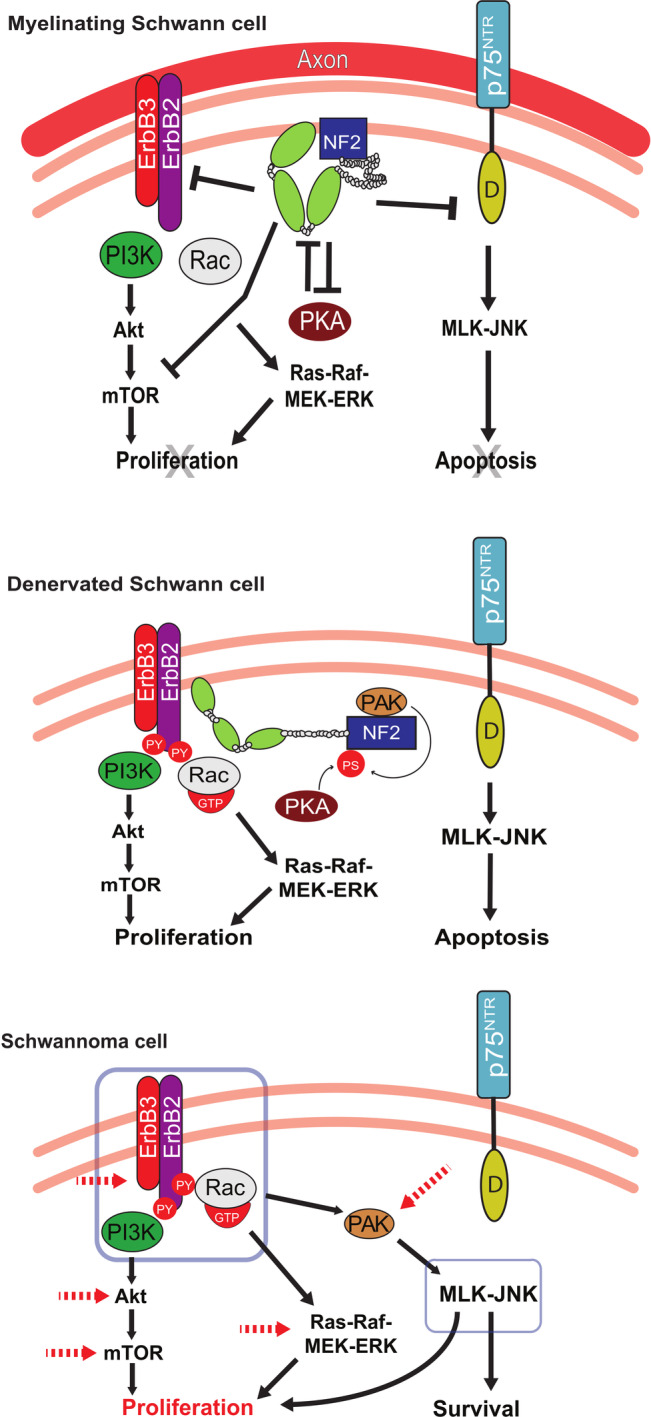
Overview of signal transduction pathways in myelinated Schwann cells vs denervated Schwann cells vs Schwannoma cells. In myelinated Schwann cells, NF2/merlin is dephosphorylated and serves as an “active” tumor suppressor, which promotes cellular quiescence. NF2/merlin inhibits expression of cell membrane receptors p75NTR and ErbB2/3, regulating multiple downstream pathways, including the Ras/Raf/MEK/ERK, PI3K/AKT, Rac/PAK, MLK/JNK, and mTOR pathways. In injured nerves, denervated Schwann cells and NF2/merlin become phosphorylated and “inactive” as tumor suppressors. An increase in p75NTR and ErbB2/3 levels lead to Schwann cell proliferation or apoptosis. In the absence of NF2/Merlin as demonstrated in schwannoma cells, p75NTR and ErbB2/3 are activated similar to denervated Schwann cells, but schwannoma cells can survive in the presence of the high‐affinity p75NTR ligand, proNGF, unlike the denervated Schwann cells. Signal transduction pathways downstream of ErbB2/3 signaling are elevated (red dotted arrows) leading to proliferation of schwannoma cells. PS, phosphorylated serine; PY, phosphorylated tyrosine
2.5.2. Mitogen activated protein kinase (MAP‐kinase) pathway
The mitogen activated protein kinase (MAP‐K) superfamily has specific and overlapping roles in normal SC plasticity regulation. In response to radiation and nerve injury, multiple MAP‐K pathways are activated, including extracellular signal regulated kinase (ERK), c‐jun N‐terminal kinase (JNK), P13‐K/Alt signaling, NF‐ĸB, and p38 MAP kinases. The activation of Ras/Raf‐ERK and Rac‐JNK pathways regulates cell motility, axonal growth, cell death, and cell proliferation. Merlin dephosphorylation (activation) suppresses both Ras/Raf‐ERK1/2 and Rac‐JNK signaling, whereas Merlin phosphorylation (inactivation) enhances Rac‐JNK signaling. 86 The effects of irradiation on VS cells and the specific pathways that are turned on and off are largely unknown.
2.5.3. Mammalian target of rapamycin (mTOR)
NF2/Merlin deactivation is associated with increased signaling of mTOR complex 1 (mTORC1), and increased mTORC1 signaling has been shown to increase growth of schwannomas and meningiomas. 80 , 87 , 93 These findings have led to clinical trials testing mTORC1 inhibitors, such as everolimus, in NF2 patients with progressive vestibular schwannomas. 94 In a recent microarray and pathway analysis by Gugel et al, mTOR signaling was found to be upregulated in VSs that recurred after radiation therapy when compared to VSs that were resected without prior radiation therapy. This was the case for both sporadic tumors and tumors in NF2 patients. Furthermore, the same tumors demonstrated downregulation of phosphate and tensin homolog (PTEN) signaling; PTEN downregulation leads to increased mTORC1 signaling via overactivation of the AKT/PKB pathway. Together, these findings suggest that increased mTORC1 activity plays a key role in VS radioresistance. 95 Mutations affecting the mTOR and PTEN pathway may therefore play a role in tumor transformation leading to recurrence or treatment escape following radiation therapy.
2.5.4. c‐Jun N‐terminal kinases (JNK)
Merlin suppresses JNK activity. JNK is activated by dual phosphorylation of threonine and tyrosine residues by two MAPK kinases—MKK4 and MKK7—in response to cellular stress. In normal SCs, JNK activity promotes apoptosis (eg, following nerve injury). However, in human VS cells (where merlin protein expression is reduced), JNK is persistently phosphorylated and activated; providing functional merlin to VS cells reduces JNK activity. Further, in human VS cells this persistent JNK activation promotes cell survival by suppressing oxidative stress, particularly in the mitochondria. 96 Thus while JNK activation leads to normal SC apoptosis, JNK is persistently active in VS cells and enhances cell survival. Given that JNK suppresses oxidative stress, Yue et al investigated the extent to which JNK signaling contributes to VS cell radiosensitivity. Primary human VS cultures were utilized; the tissue received single doses of radiation (5‐10 Gy) in the presence or absence of JNK inhibitors. Histone 2AX (HDAX) phosphorylation, a marker of radiation‐induced DNA damage, reactive oxygen species (ROS) levels, and cell death were analyzed. The results demonstrated that JNK activity in VS cells suppressed radiation‐induced oxidative stress, DNA damage (as measured by H2AX phosphorylation), and cell death. This suggests that concurrent use of JNK inhibitors with radiosurgery may increase VS radiosensitivity and may be useful in tumors that are refractory to current radiation protocols. 57
2.5.5. Low affinity neurotrophin receptor—p75NTR
p75NTR is the one of the founding members of the TNF receptor superfamily. 97 It binds with low affinity to mature neurotrophins (eg, nerve growth factor, NGF) but with high affinity to precursor forms of neurotrophins (eg, proNGF). 98 Activation of p75NTR leads to apoptosis or cell survival depending on the cellular context. In the absence of Trk receptors, p75NTR activates the sphingomyelin cycle, JNK, and NF‐κB. 99 , 100 , 101 , 102 p75NTR activation of JNK is necessary for pro‐death signal, whereas activation of NF‐κB is thought to promote survival. 100 , 103 , 104 , 105 In injured nerves with axonal degeneration, p75NTR is upregulated in the denervated SCs, which in turn leads to p75NTR‐mediated apoptosis without reinnervation. 106 , 107 , 108 However, VS cells exhibit survival long‐term without the neighboring axonal contact. Ahmad et al found that VSs express p75NTR levels similar to those of denervated SCs in nerves following axotomy. 109 Expression of p75NTR in SCs and VS cells appears to be regulated by merlin status. 110 Interestingly, VS cells are able to survive in the presence of the high‐affinity p75NTR ligand, proNGF, unlike the non‐neoplastic SC counterpart. Furthermore, proNGF rescues VS cells from cell death due to JNK inhibition by activating NF‐ĸB, suggesting a paradoxical anti‐apoptotic role of p75NTR leading to VS. Interestingly, upregulation of NF‐ĸB can enhance the survival of cells treated with chemotherapeutic drugs and SRS, while down regulation may inhibit the effect of SRS. Therefore, targeting the p75NTR pathway along with or without JNK may provide a therapeutic target that acts specifically to impair VS growth while sparing normal SCs.
2.5.6. ErbB2
ErbB2 (erb‐b2 receptor tyrosine kinase 2) and ErbB3 are members of the epidermal growth factor receptor family of receptor tyrosine kinases; both are essential for SC growth and survival. ErbB2/ErbB3 function as heterodimeric receptors for neuregulin‐1 (NRG1), which is a glial growth factor expressed on the axonal surface essential for normal SC proliferation, development, and survival. 111 , 112 , 113 In VS cells, NRG1 and ErbB2/3 are constitutively expressed and activated. 112 , 114 Further, in VS cells ErbB2 appears to constitutively reside in lipid raft regions of the cell membrane where it promotes VS proliferation and survival. 112 , 114 , 115 In contrast, in normal myelinating SCs ErbB2 expression is relatively low and excluded from lipid rafts, perhaps under control by merlin. 115 Following denervation, the growth suppressive function of merlin becomes inactivated by phosphorylation in SCs and ErbB2 expression is elevated with movement into lipid rafts akin to VS cells that lack functional merlin and axonal contact. 110 , 115 These observations raise the possibility that constitutive ErbB2 signaling in VS cells could modulate the effects of radiation. In cultured human VS cells, radiation doses over 20 Gy induce VS cell apoptosis and cell cycle arrest. Inhibition of ErbB2 signaling with PD158780, a small molecule ErbB2 inhibitor, or trastuzumab, an inhibitory anti‐ErbB2 monoclonal antibody, protected VS cells from radiation by reducing the proliferation rate. In contrast, treatment with NRG1 promoted mitosis and enhanced radiation‐induced cell apoptosis. 56 These observations suggest that the relative radioresistance of VS reflects their low proliferation rate and suggests that the effects of SRS on VSs could be largely indirect. 56
2.5.7. p53
In multiple tissues, radiation activates tumor suppressor genes such as p53 thereby inducing activation of pro‐apoptotic Bax protein and cytochrome C/caspase, thus leading to apoptosis. Molecular genetic analysis of blood‐tumor DNA has demonstrated that p53 is not critical for the tumorigenesis of VS. 116 Likewise, no mutation, deletion, or loss of heterozygosity in p53 was found in VS tissue. 117 These studies support the hypothesis that p53 contribution in VS proliferation is likely minimal. However, although not well described for VS, it is possible that mutations in p53 or its signaling pathways could contribute to treatment failure or recurrence following radiation, as these mutations are known to prevent post‐radiation apoptosis in various other neoplasms.
The expression pattern of other apoptotic markers has also been investigated. In Mawrin et al, the expression levels of the Fas‐Fas‐L system were quantitatively analyzed with immunohistochemistry in 14 sporadic VS samples. This system regulates apoptosis, pro‐apoptotic factor Bax, and anti‐apoptotic factor Bcl‐2. The results of the study demonstrated that while most VS cells express Fas‐L, Bax, and Bcl‐2, levels of Fas were limited, suggesting that Fas‐Fas‐L system may not be critical to apoptosis in VS. However, the expression of Bax and Bcl‐2 suggest that theses apoptotic markers can be expressed independent of p53 expression. 118
Neurod1 is a basic helix‐loop‐helix transcription factor that is critical for neuronal development and maturation. 119 It is highly expressed in a variety of tumors including neuroblastoma, glioblastoma, and colorectal cancer. 120 Neurod1 affects cell‐cycle progression and overexpression leads to exit of cell‐cycle in part by increasing p21 in a p53‐dependent manner. In contrast, absence of Neurod1 induces proliferation. 121 Recently, Kersigo et al demonstrated that Neurod1 overexpression reduces SC proliferation in primary human VS culture and axotomized sciatic nerves. 122 However, the impact of Neurod1 in genetic mouse models of schwannoma was highly variable, suggesting that a tightly regulated Neurod1 expression level may be necessary to drive VS cells out of the cell cycle. Adjuvant irradiation may potentiate this therapeutic approach.
2.5.8. Angiogenesis: VEGF and radiation
Vascular supply is essential for tumor growth and cell proliferation. Indirect effects of radiation may lead to an inadequate blood supply for VS and result in tumor shrinkage. 123 However, because oxygen is a potent radiosensitizer, hypoxia may also lead to radioresistance. At the same time, hypoxia in tumor cells may induce angiogenesis, and tumor cells may self‐repair in a state of hypoxia. Vascular endothelial growth factor (VEGF) is a signaling protein that induces neo‐angiogenesis, contributes to vasodilation, and increases vascular permeability. In VSs, expression levels of pro‐angiogenic factors such as VEGF‐A and corresponding receptors VEGFR correlate positively with VS growth rate. 124 , 125 , 126 NF2 patients treated with bevacizumab—a humanized monoclonal antibody that neutralizes VEGF‐A—have demonstrated VS tumor control and improved hearing in some cases. 126 , 127 , 128 , 129 , 130 However, this treatment effect is not durable and long‐term side effects have been reported. 131 In a mouse model of NF2, the efficacy of bevacizumab in combination with radiation was investigated. The researchers demonstrated that anti‐VEGF treatment led to normalization of VS vasculature thereby improving vascular supply and oxygenation. When anti‐VEGF was combined with low dose IR during the window of normalized VS vasculature, tumor control rates were superior compared to either alone. 132 Treatment with lower dose IR in combination with anti‐VEGF was comparable to higher dose of IR without anti‐VEGF, suggesting that combination therapy may contribute to lowering the total dose of IR in NF2 patients.
3. CONCLUSIONS
The anti‐tumor effect of radiotherapy is complex and multifactorial, with both direct and indirect effects on tumor cells. VSs are relatively radioresistant tumors, which one expects given their low proliferative capacity and slow growth rates. Despite this, radiotherapy is often successful in arresting VS growth. Several key factors in the radiobiology of VSs have been described. VSs can grow despite long‐term oxidative stress, implying that their cells have mechanisms to mitigate oxidative stress. VS cells appear to be capable of repairing DNA damage prior to re‐entering the cell cycle and thus bypass apoptosis. Several growth factor pathways regulate VS cell growth and appear to be altered in the setting of radiotherapy. Finally, damage to surrounding vasculature and/or induction of immunological responses also seem to play an important indirect role in the response of VS to radiotherapy. By better understanding the mechanisms underlying these effects, in the future we could potentially harness these mechanisms to potentiate the clinical effects of radiotherapy on VSs.
CONFLICT OF INTEREST
The authors report no conflicts of interest relating to the current work.
ACKNOWLEDGMENTS
The authors would like to thank the editorial board of Laryngoscope Invesitgative Otolaryngology for their kind invitation for a manuscript submission. The authors would like to thank Danika Simonson for her assistance with figure creation. M. R. H. is supported by National Institutes of Health grant NCATS U54 TR001013. M. C. D. and S. B. S. report no external funding.
Dougherty MC, Shibata SB, Hansen MR. The biological underpinnings of radiation therapy for vestibular schwannomas: Review of the literature. Laryngoscope Investigative Otolaryngology. 2021;6:458–468. 10.1002/lio2.553
Funding information National Institutes of Health, Grant/Award Number: NCATS U54 TR001013
BIBLIOGRAPHY
- 1. Fong B, Barkhoudarian G, Pezeshkian P, Parsa AT, Gopen Q, Yang I. The molecular biology and novel treatments of vestibular schwannomas. J Neurosurg. 2011;115(5):906‐914. 10.3171/2011.6.JNS11131. [DOI] [PubMed] [Google Scholar]
- 2. Schulz A, Büttner R, Hagel C, et al. The importance of nerve microenvironment for schwannoma development. Acta Neuropathol. 2016;132(2):289‐307. 10.1007/s00401-016-1583-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Linskey M, Kuo J. General and historical considerations of radiotherapy and radiosurgery. Youmans and Winn Neurological Surgery. Vol 3. 7th ed. Philadelphia, PA: Elsevier; 2017:2135‐2143. [Google Scholar]
- 4. Leksell L. The stereotaxic method and radiosurgery of the brain. Acta Chir Scand. 1951;102(4):316‐319. [PubMed] [Google Scholar]
- 5. Lunsford LD, Flickinger J, Lindner G, Maitz A. Stereotactic radiosurgery of the brain using the first United States 201 cobalt‐60 source gamma knife. Neurosurgery. 1989;24(2):151‐159. 10.1227/00006123-198902000-00001. [DOI] [PubMed] [Google Scholar]
- 6. Yu J, Brown M, Suh J, Ma L, Saghal A. Radiobiology of radiotherapy and radiosurgery. Youmans & Winn Neurological Surgery. Vol 3. 7th ed. Philadelphia, PA: Elsevier; 2017:2144‐2154. [Google Scholar]
- 7. Sia J, Szmyd R, Hau E, Gee HE. Molecular mechanisms of radiation‐induced cancer cell death: a primer. Front Cell Dev Biol. 2020;8:41. 10.3389/fcell.2020.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Biaglow JE, Mitchell JB, Held K. The importance of peroxide and superoxide in the X‐ray response. Int J Radiat Oncol Biol Phys. 1992;22(4):665‐669. 10.1016/0360-3016(92)90499-8. [DOI] [PubMed] [Google Scholar]
- 9. Michaels HB, Hunt JW. A model for radiation damage in cells by direct effect and by indirect effect: a radiation chemistry approach. Radiat Res. 1978;74(1):23‐34. [PubMed] [Google Scholar]
- 10. Robinett ZN, Bathla G, Wu A, et al. Persistent oxidative stress in vestibular schwannomas after stereotactic radiation therapy. Otol Neurotol. 2018;39(9):1184‐1190. 10.1097/MAO.0000000000001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gao Z, Sarsour EH, Kalen AL, Li L, Kumar MG, Goswami PC. Late ROS accumulation and radiosensitivity in SOD1‐overexpressing human glioma cells. Free Radic Biol Med. 2008;45(11):1501‐1509. 10.1016/j.freeradbiomed.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Robbins MEC, Zhao W. Chronic oxidative stress and radiation‐induced late normal tissue injury: a review. Int J Radiat Biol. 2004;80(4):251‐259. 10.1080/09553000410001692726. [DOI] [PubMed] [Google Scholar]
- 13. Szumiel I. Ionizing radiation‐induced oxidative stress, epigenetic changes and genomic instability: the pivotal role of mitochondria. Int J Radiat Biol. 2015;91(1):1‐12. 10.3109/09553002.2014.934929. [DOI] [PubMed] [Google Scholar]
- 14. Hei TK, Zhou H, Chai Y, Ponnaiya B, Ivanov VN. Radiation induced non‐targeted response: mechanism and potential clinical implications. Curr Mol Pharmacol. 2011;4(2):96‐105. 10.2174/1874467211104020096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Du C, Gao Z, Venkatesha VA, et al. Mitochondrial ROS and radiation induced transformation in mouse embryonic fibroblasts. Cancer Biol Ther. 2009;8(20):1962‐1971. 10.4161/cbt.8.20.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Azzam EI, Jay‐Gerin J‐P, Pain D. Ionizing radiation‐induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327(1–2):48‐60. 10.1016/j.canlet.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dayal D, Martin SM, Owens KM, et al. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat Res. 2009;172(6):737‐745. 10.1667/RR1617.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vakifahmetoglu H, Olsson M, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death Differ. 2008;15(7):1153‐1162. 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 19. Eriksson D, Stigbrand T. Radiation‐induced cell death mechanisms. Tumour Biol. 2010;31(4):363‐372. 10.1007/s13277-010-0042-8. [DOI] [PubMed] [Google Scholar]
- 20. Wu GS, Burns TF, McDonald ER, et al. KILLER/DR5 is a DNA damage‐inducible p53‐regulated death receptor gene. Nat Genet. 1997;17(2):141‐143. 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 21. Chakraborty M, Abrams SI, Camphausen K, et al. Irradiation of tumor cells up‐regulates Fas and enhances CTL lytic activity and CTL adoptive immunotherapy. J Immunol. 2003;170(12):6338‐6347. 10.4049/jimmunol.170.12.6338. [DOI] [PubMed] [Google Scholar]
- 22. Sheard MA, Uldrijan S, Vojtesek B. Role of p53 in regulating constitutive and X‐radiation‐inducible CD95 expression and function in carcinoma cells. Cancer Res. 2003;63(21):7176‐7184. [PubMed] [Google Scholar]
- 23. Pettus BJ, Chalfant CE, Hannun YA. Ceramide in apoptosis: an overview and current perspectives. Biochim Biophys Acta. 2002;1585(2–3):114‐125. 10.1016/s1388-1981(02)00331-1. [DOI] [PubMed] [Google Scholar]
- 24. Kolesnick R, Fuks Z. Radiation and ceramide‐induced apoptosis. Oncogene. 2003;22(37):5897‐5906. 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- 25. Garcia‐Barros M, Paris F, Cordon‐Cardo C, et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science. 2003;300(5622):1155‐1159. 10.1126/science.1082504. [DOI] [PubMed] [Google Scholar]
- 26. Park HJ, Griffin RJ, Hui S, Levitt SH, Song CW. Radiation‐induced vascular damage in tumors: implications of vascular damage in ablative hypofractionated radiotherapy (SBRT and SRS). Radiat Res. 2012;177(3):311‐327. 10.1667/rr2773.1. [DOI] [PubMed] [Google Scholar]
- 27. Garcia‐Barros M, Lacorazza D, Petrie H, et al. Host acid sphingomyelinase regulates microvascular function not tumor immunity. Cancer Res. 2004;64(22):8285‐8291. 10.1158/0008-5472.CAN-04-2715. [DOI] [PubMed] [Google Scholar]
- 28. Brown JM, Carlson DJ, Brenner DJ. The tumor radiobiology of SRS and SBRT: are more than the 5 Rs involved? Int J Radiat Oncol Biol Phys. 2014;88(2):254‐262. 10.1016/j.ijrobp.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown JM, Brenner DJ, Carlson DJ. Dose escalation, not “new biology,” can account for the efficacy of stereotactic body radiation therapy with non‐small cell lung cancer. Int J Radiat Oncol Biol Phys. 2013;85(5):1159‐1160. 10.1016/j.ijrobp.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bedford JS. Sublethal damage, potentially lethal damage, and chromosomal aberrations in mammalian cells exposed to ionizing radiations. Int J Radiat Oncol Biol Phys. 1991;21(6):1457‐1469. 10.1016/0360-3016(91)90320-4. [DOI] [PubMed] [Google Scholar]
- 31. Tomé WA. Universal survival curve and single fraction equivalent dose: useful tools in understanding potency of ablative radiotherapy: In Regard to Parks et al. (Int J Radiat Oncol Biol Phys 2008;70:847–852). International Journal of Radiation Oncology, Biology, Physics. 2008;72(5):1620. 10.1016/j.ijrobp.2008.08.032. [DOI] [PubMed] [Google Scholar]
- 32. Kirkpatrick JP, Meyer JJ, Marks LB. The linear‐quadratic model is inappropriate to model high dose per fraction effects in radiosurgery. Semin Radiat Oncol. 2008;18(4):240‐243. 10.1016/j.semradonc.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 33. Song CW, Cho LC, Yuan J, Dusenbery KE, Griffin RJ, Levitt SH. Radiobiology of stereotactic body radiation therapy/stereotactic radiosurgery and the linear‐quadratic model. Int J Radiat Oncol Biol Phys. 2013;87(1):18‐19. 10.1016/j.ijrobp.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 34. Lanser MJ, Sussman SA, Frazer K. Epidemiology, pathogenesis, and genetics of acoustic tumors. Otolaryngol Clin North Am. 1992;25(3):499‐520. [PubMed] [Google Scholar]
- 35. Ramsden RT. The bloody angle: 100 years of acoustic neuroma surgery. J R Soc Med. 1995;88(8):464P‐468P. [PMC free article] [PubMed] [Google Scholar]
- 36. Strasnick B, Glasscock ME, Haynes D, McMenomey SO, Minor LB. The natural history of untreated acoustic neuromas. Laryngoscope. 1994;104(9):1115‐1119. 10.1288/00005537-199409000-00011. [DOI] [PubMed] [Google Scholar]
- 37. Carlson ML, Habermann EB, Wagie AE, et al. The changing landscape of vestibular schwannoma management in the United States—a shift toward conservatism. Otolaryngol Head Neck Surg. 2015;153(3):440‐446. 10.1177/0194599815590105. [DOI] [PubMed] [Google Scholar]
- 38. Carlson ML, Van Gompel JJ, Wiet RM, et al. A cross‐sectional survey of the North American Skull Base Society: current practice patterns of vestibular schwannoma evaluation and management in North America. J Neurol Surg B Skull Base. 2018;79(3):289‐296. 10.1055/s-0037-1607319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hirato M, Inoue H, Zama A, Ohye C, Shibazaki T, Andou Y. Gamma knife radiosurgery for acoustic schwannoma: effects of low radiation dose and functional prognosis. Stereotact Funct Neurosurg. 1996;66(Suppl 1):134‐141. 10.1159/000099803. [DOI] [PubMed] [Google Scholar]
- 40. Hasegawa T, Kida Y, Kobayashi T, Yoshimoto M, Mori Y, Yoshida J. Long‐term outcomes in patients with vestibular schwannomas treated using gamma knife surgery: 10‐year follow up. J Neurosurg. 2005;102(1):10‐16. 10.3171/jns.2005.102.1.0010. [DOI] [PubMed] [Google Scholar]
- 41. Lunsford LD, Niranjan A, Flickinger JC, Maitz A, Kondziolka D. Radiosurgery of vestibular schwannomas: summary of experience in 829 cases. J Neurosurg. 2005;102(Suppl):195‐199. [PubMed] [Google Scholar]
- 42. Friedman RA, Brackmann DE, Hitselberger WE, Schwartz MS, Iqbal Z, Berliner KI. Surgical salvage after failed irradiation for vestibular schwannoma. Laryngoscope. 2005;115(10):1827‐1832. 10.1097/01.mlg.0000175063.76945.75. [DOI] [PubMed] [Google Scholar]
- 43. Yeung AH, Sughrue ME, Kane AJ, Tihan T, Cheung SW, Parsa AT. Radiobiology of vestibular schwannomas: mechanisms of radioresistance and potential targets for therapeutic sensitization. Neurosurg Focus. 2009;27(6):E2. 10.3171/2009.9.FOCUS09185. [DOI] [PubMed] [Google Scholar]
- 44. Yomo S, Arkha Y, Delsanti C, Roche P‐H, Thomassin J‐M, Régis J. Repeat gamma knife surgery for regrowth of vestibular schwannomas. Neurosurgery. 2009;64(1):48‐54; discussion 54‐55. 10.1227/01.NEU.0000327692.74477.D5. [DOI] [PubMed] [Google Scholar]
- 45. Kaylie DM, Horgan MJ, Delashaw JB, McMenomey SO. A meta‐analysis comparing outcomes of microsurgery and gamma knife radiosurgery. Laryngoscope. 2000;110(11):1850‐1856. 10.1097/00005537-200011000-00016. [DOI] [PubMed] [Google Scholar]
- 46. Macielak RJ, Patel NS, Lees KA, et al. Delayed tumor growth in vestibular schwannoma: an argument for lifelong surveillance. Otol Neurotol. 2019;40(9):1224‐1229. 10.1097/MAO.0000000000002337. [DOI] [PubMed] [Google Scholar]
- 47. Rowe JG, Radatz M, Walton L, Kemeny AA. Stereotactic radiosurgery for type 2 neurofibromatosis acoustic neuromas: patient selection and tumour size. Stereotact Funct Neurosurg. 2002;79(2):107‐116. 10.1159/000070106. [DOI] [PubMed] [Google Scholar]
- 48. Wowra B, Muacevic A, Jess‐Hempen A, Hempel J‐M, Müller‐Schunk S, Tonn J‐C. Outpatient gamma knife surgery for vestibular schwannoma: definition of the therapeutic profile based on a 10‐year experience. J Neurosurg. 2005;102(Suppl):114‐118. [PubMed] [Google Scholar]
- 49. Linskey ME, Martinez AJ, Kondziolka D, et al. The radiobiology of human acoustic schwannoma xenografts after stereotactic radiosurgery evaluated in the subrenal capsule of athymic mice. J Neurosurg. 1993;78(4):645‐653. 10.3171/jns.1993.78.4.0645. [DOI] [PubMed] [Google Scholar]
- 50. Lee DJ, Westra WH, Staecker H, Long D, Niparko JK, Slattery WH. Clinical and histopathologic features of recurrent vestibular schwannoma (acoustic neuroma) after stereotactic radiosurgery. Otol Neurotol. 2003;24(4):650‐660; discussion 660. 10.1097/00129492-200307000-00020. [DOI] [PubMed] [Google Scholar]
- 51. Kwon Y, Khang SK, Kim CJ, Lee DJ, Lee JK, Kwun BD. Radiologic and histopathologic changes after gamma knife radiosurgery for acoustic schwannoma. Stereotact Funct Neurosurg. 1999;72(Suppl 1):2‐10. 10.1159/000056433. [DOI] [PubMed] [Google Scholar]
- 52. Slattery WH, Brackmann DE. Results of surgery following stereotactic irradiation for acoustic neuromas. Am J Otol. 1995;16(3):315‐319. discussion 319‐321. [PubMed] [Google Scholar]
- 53. Lee F, Linthicum F, Hung G. Proliferation potential in recurrent acoustic schwannoma following gamma knife radiosurgery versus microsurgery. Laryngoscope. 2002;112(6):948‐950. 10.1097/00005537-200206000-00002. [DOI] [PubMed] [Google Scholar]
- 54. Anniko M, Arndt J, Norén G. The human acoustic neurinoma in organ culture. II. Tissue changes after gamma irradiation. Acta Otolaryngol. 1981;91(3–4):223‐235. 10.3109/00016488109138503. [DOI] [PubMed] [Google Scholar]
- 55. Anniko M. Early morphological changes following gamma irradiation. A comparison of human pituitary tumours and human acoustic neurinomas (schwannomas). Acta Pathol Microbiol Scand A. 1981;89(2):113‐124. [PubMed] [Google Scholar]
- 56. Hansen MR, Clark JJ, Gantz BJ, Goswami PC. Effects of ErbB2 signaling on the response of vestibular schwannoma cells to gamma‐irradiation. Laryngoscope. 2008;118(6):1023‐1030. 10.1097/MLG.0b013e318163f920. [DOI] [PubMed] [Google Scholar]
- 57. Yue WY, Clark JJ, Telisak M, Hansen MR. Inhibition of c‐Jun N‐terminal kinase activity enhances vestibular schwannoma cell sensitivity to gamma irradiation. Neurosurgery. 2013;73(3):506‐516. 10.1227/01.neu.0000431483.10031.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys. 2004;59(4):928‐942. 10.1016/j.ijrobp.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 59. Bruno S, Darzynkiewicz Z. Cell cycle dependent expression and stability of the nuclear protein detected by Ki‐67 antibody in HL‐60 cells. Cell Prolif. 1992;25(1):31‐40. 10.1111/j.1365-2184.1992.tb01435.x. [DOI] [PubMed] [Google Scholar]
- 60. Jacob A, Igarashi S, Platto T, Khan R, Jain R. The solid component of radiographically non‐growing, post‐radiated vestibular schwannoma retains proliferative capacity: implications for patient counseling. Ann Otol Rhinol Laryngol. 2015;124(10):834‐840. 10.1177/0003489415588128. [DOI] [PubMed] [Google Scholar]
- 61. Deacon J, Peckham MJ, Steel GG. The radioresponsiveness of human tumours and the initial slope of the cell survival curve. Radiother Oncol. 1984;2(4):317‐323. 10.1016/s0167-8140(84)80074-2. [DOI] [PubMed] [Google Scholar]
- 62. Seizinger BR, Martuza RL, Gusella JF. Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature. 1986;322(6080):644‐647. 10.1038/322644a0. [DOI] [PubMed] [Google Scholar]
- 63. Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin‐, ezrin‐, radixin‐like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72(5):791‐800. 10.1016/0092-8674(93)90406-g. [DOI] [PubMed] [Google Scholar]
- 64. Rouleau GA, Merel P, Lutchman M, et al. Alteration in a new gene encoding a putative membrane‐organizing protein causes neuro‐fibromatosis type 2. Nature. 1993;363(6429):515‐521. 10.1038/363515a0. [DOI] [PubMed] [Google Scholar]
- 65. Welling DB, Packer MD, Chang L‐S. Molecular studies of vestibular schwannomas: a review. Curr Opin Otolaryngol Head Neck Surg. 2007;15(5):341‐346. 10.1097/MOO.0b013e3282b97310. [DOI] [PubMed] [Google Scholar]
- 66. McClatchey AI, Giovannini M. Membrane organization and tumorigenesis—the NF2 tumor suppressor, Merlin. Genes Dev. 2005;19(19):2265‐2277. 10.1101/gad.1335605. [DOI] [PubMed] [Google Scholar]
- 67. Xiao G‐H, Chernoff J, Testa JR. NF2: the wizardry of merlin. Genes Chromosomes Cancer. 2003;38(4):389‐399. 10.1002/gcc.10282. [DOI] [PubMed] [Google Scholar]
- 68. Li W, Cooper J, Karajannis MA, Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012;13(3):204‐215. 10.1038/embor.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhou L, Hanemann CO. Merlin, a multi‐suppressor from cell membrane to the nucleus. FEBS Lett. 2012;586(10):1403‐1408. 10.1016/j.febslet.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 70. Bosco EE, Nakai Y, Hennigan RF, Ratner N, Zheng Y. NF2‐deficient cells depend on the Rac1‐canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. Oncogene. 2010;29(17):2540‐2549. 10.1038/onc.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chadee DN, Kyriakis JM. MLK3 is required for mitogen activation of B‐Raf, ERK and cell proliferation. Nat Cell Biol. 2004;6(8):770‐776. 10.1038/ncb1152. [DOI] [PubMed] [Google Scholar]
- 72. Chadee DN, Xu D, Hung G, et al. Mixed‐lineage kinase 3 regulates B‐Raf through maintenance of the B‐Raf/Raf‐1 complex and inhibition by the NF2 tumor suppressor protein. Proc Natl Acad Sci U S A. 2006;103(12):4463‐4468. 10.1073/pnas.0510651103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Flaiz C, Chernoff J, Ammoun S, Peterson JR, Hanemann CO. PAK kinase regulates Rac GTPase and is a potential target in human schwannomas. Exp Neurol. 2009;218(1):137‐144. 10.1016/j.expneurol.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Fraenzer J‐T, Pan H, Minimo L, Smith GM, Knauer D, Hung G. Overexpression of the NF2 gene inhibits schwannoma cell proliferation through promoting PDGFR degradation. Int J Oncol. 2003;23(6):1493‐1500. [PubMed] [Google Scholar]
- 75. Houshmandi SS, Emnett RJ, Giovannini M, Gutmann DH. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2‐ and Src‐dependent manner. Mol Cell Biol. 2009;29(6):1472‐1486. 10.1128/MCB.01392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. James MF, Stivison E, Beauchamp R, et al. Regulation of mTOR complex 2 signaling in neurofibromatosis 2‐deficient target cell types. Mol Cancer Res. 2012;10(5):649‐659. 10.1158/1541-7786.MCR-11-0425-T. [DOI] [PubMed] [Google Scholar]
- 77. Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Hum Mol Genet. 2003;12(11):1211‐1221. 10.1093/hmg/ddg146. [DOI] [PubMed] [Google Scholar]
- 78. Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21‐activated kinase, Pak1. Mol Cell. 2003;12(4):841‐849. 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 79. Lim JY, Kim H, Kim YH, et al. Merlin suppresses the SRE‐dependent transcription by inhibiting the activation of Ras‐ERK pathway. Biochem Biophys Res Commun. 2003;302(2):238‐245. 10.1016/s0006-291x(03)00124-4. [DOI] [PubMed] [Google Scholar]
- 80. López‐Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin‐dependent mTORC1 signaling. Mol Cell Biol. 2009;29(15):4235‐4249. 10.1128/MCB.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3‐kinase through binding to PIKE‐L. Proc Natl Acad Sci U S A. 2004;101(52):18200‐18205. 10.1073/pnas.0405971102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Yi C, Wilker EW, Yaffe MB, Stemmer‐Rachamimov A, Kissil JL. Validation of the p21‐activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68(19):7932‐7937. 10.1158/0008-5472.CAN-08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Zhou L, Ercolano E, Ammoun S, Schmid MC, Barczyk MA, Hanemann CO. Merlin‐deficient human tumors show loss of contact inhibition and activation of Wnt/β‐catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia. 2011;13(12):1101‐1112. 10.1593/neo.111060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rangwala R, Banine F, Borg J‐P, Sherman LS. Erbin regulates mitogen‐activated protein (MAP) kinase activation and MAP kinase‐dependent interactions between Merlin and adherens junction protein complexes in Schwann cells. J Biol Chem. 2005;280(12):11790‐11797. 10.1074/jbc.M414154200. [DOI] [PubMed] [Google Scholar]
- 85. Fernandez‐Valle C, Tang Y, Ricard J, et al. Paxillin binds schwannomin and regulates its density‐dependent localization and effect on cell morphology. Nat Genet. 2002;31(4):354‐362. 10.1038/ng930. [DOI] [PubMed] [Google Scholar]
- 86. Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer Res. 2007;67(2):520‐527. 10.1158/0008-5472.CAN-06-1608. [DOI] [PubMed] [Google Scholar]
- 87. James MF, Han S, Polizzano C, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Mol Cell Biol. 2009;29(15):4250‐4261. 10.1128/MCB.01581-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baia GS, Caballero OL, Orr BA, et al. Yes‐associated protein 1 is activated and functions as an oncogene in meningiomas. Mol Cancer Res. 2012;10(7):904‐913. 10.1158/1541-7786.MCR-12-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hamaratoglu F, Willecke M, Kango‐Singh M, et al. The tumour‐suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat Cell Biol. 2006;8(1):27‐36. 10.1038/ncb1339. [DOI] [PubMed] [Google Scholar]
- 90. Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia. 2008;10(11):1204‐1212. 10.1593/neo.08642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang N, Bai H, David KK, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Dev Cell. 2010;19(1):27‐38. 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li W, You L, Cooper J, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140(4):477‐490. 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Giovannini M, Bonne N‐X, Vitte J, et al. mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro Oncol. 2014;16(4):493‐504. 10.1093/neuonc/not242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Goutagny S, Raymond E, Esposito‐Farese M, et al. Phase II study of mTORC1 inhibition by everolimus in neurofibromatosis type 2 patients with growing vestibular schwannomas. J Neurooncol. 2015;122(2):313‐320. 10.1007/s11060-014-1710-0. [DOI] [PubMed] [Google Scholar]
- 95. Gugel I, Ebner FH, Grimm F, et al. Contribution of mTOR and PTEN to radioresistance in sporadic and NF2‐associated vestibular schwannomas: a microarray and pathway analysis. Cancers (Basel). 2020;12(1):177. 10.3390/cancers12010177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Yue WY, Clark JJ, Fernando A, Domann F, Hansen MR. Contribution of persistent C‐Jun N‐terminal kinase activity to the survival of human vestibular schwannoma cells by suppression of accumulation of mitochondrial superoxides. Neuro Oncol. 2011;13(9):961‐973. 10.1093/neuonc/nor068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Bothwell M. Functional interactions of neurotrophins and neurotrophin receptors. Annu Rev Neurosci. 1995;18:223‐253. 10.1146/annurev.ne.18.030195.001255. [DOI] [PubMed] [Google Scholar]
- 98. Feng D, Kim T, Ozkan E, et al. Molecular and structural insight into proNGF engagement of p75NTR and sortilin. J Mol Biol. 2010;396(4):967‐984. 10.1016/j.jmb.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low‐affinity neurotrophin receptor. Science. 1994;265(5178):1596‐1599. 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- 100. Gentry JJ, Casaccia‐Bonnefil P, Carter BD. Nerve growth factor activation of nuclear factor kappaB through its p75 receptor is an anti‐apoptotic signal in RN22 schwannoma cells. J Biol Chem. 2000;275(11):7558‐7565. 10.1074/jbc.275.11.7558. [DOI] [PubMed] [Google Scholar]
- 101. Harrington AW, Kim JY, Yoon SO. Activation of Rac GTPase by p75 is necessary for c‐jun N‐terminal kinase‐mediated apoptosis. J Neurosci. 2002;22(1):156‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Roux PP, Barker PA. Neurotrophin signaling through the p75 neurotrophin receptor. Prog Neurobiol. 2002;67(3):203‐233. 10.1016/s0301-0082(02)00016-3. [DOI] [PubMed] [Google Scholar]
- 103. Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20(17):6340‐6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Koshimizu H, Hazama S, Hara T, Ogura A, Kojima M. Distinct signaling pathways of precursor BDNF and mature BDNF in cultured cerebellar granule neurons. Neurosci Lett. 2010;473(3):229‐232. 10.1016/j.neulet.2010.02.055. [DOI] [PubMed] [Google Scholar]
- 105. Lu B, Pang PT, Woo NH. The yin and yang of neurotrophin action. Nat Rev Neurosci. 2005;6(8):603‐614. 10.1038/nrn1726. [DOI] [PubMed] [Google Scholar]
- 106. Petratos S, Butzkueven H, Shipham K, et al. Schwann cell apoptosis in the postnatal axotomized sciatic nerve is mediated via NGF through the low‐affinity neurotrophin receptor. J Neuropathol Exp Neurol. 2003;62(4):398‐411. 10.1093/jnen/62.4.398. [DOI] [PubMed] [Google Scholar]
- 107. Provenzano MJ, Minner SA, Zander K, et al. p75(NTR) expression and nuclear localization of p75(NTR) intracellular domain in spiral ganglion Schwann cells following deafness correlate with cell proliferation. Mol Cell Neurosci. 2011;47(4):306‐315. 10.1016/j.mcn.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Provenzano MJ, Xu N, Ver Meer MR, Clark JJ, Hansen MR. p75NTR and sortilin increase after facial nerve injury. Laryngoscope. 2008;118(1):87‐93. 10.1097/MLG.0b013e31814b8d9f. [DOI] [PubMed] [Google Scholar]
- 109. Ahmad I, Yue WY, Fernando A, Clark JJ, Woodson EA, Hansen MR. p75NTR is highly expressed in vestibular schwannomas and promotes cell survival by activating nuclear transcription factor κB. Glia. 2014;62(10):1699‐1712. 10.1002/glia.22709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ahmad I, Fernando A, Gurgel R, Jason Clark J, Xu L, Hansen MR. Merlin status regulates p75(NTR) expression and apoptotic signaling in Schwann cells following nerve injury. Neurobiol Dis. 2015;82:114‐122. 10.1016/j.nbd.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Adlkofer K, Lai C. Role of neuregulins in glial cell development. Glia. 2000;29(2):104‐111. . [DOI] [PubMed] [Google Scholar]
- 112. Hansen MR, Linthicum FH. Expression of neuregulin and activation of erbB receptors in vestibular schwannomas: possible autocrine loop stimulation. Otol Neurotol. 2004;25(2):155‐159. 10.1097/00129492-200403000-00013. [DOI] [PubMed] [Google Scholar]
- 113. Hansen MR, Vijapurkar U, Koland JG, Green SH. Reciprocal signaling between spiral ganglion neurons and Schwann cells involves neuregulin and neurotrophins. Hear Res. 2001;161(1–2):87‐98. 10.1016/s0378-5955(01)00360-4. [DOI] [PubMed] [Google Scholar]
- 114. Hansen MR, Roehm PC, Chatterjee P, Green SH. Constitutive neuregulin‐1/ErbB signaling contributes to human vestibular schwannoma proliferation. Glia. 2006;53(6):593‐600. 10.1002/glia.20316. [DOI] [PubMed] [Google Scholar]
- 115. Brown KD, Hansen MR. Lipid raft localization of ErbB2 in vestibular schwannoma and schwann cells. Otol Neurotol. 2008;29(1):79‐85. 10.1097/mao.0b013e31815dbb11. [DOI] [PubMed] [Google Scholar]
- 116. Irving RM, Moffat DA, Hardy DG, Barton DE, Xuereb JH, Maher ER. Molecular genetic analysis of the mechanism of tumorigenesis in acoustic neuroma. Arch Otolaryngol Head Neck Surg. 1993;119(11):1222‐1228. 10.1001/archotol.1993.01880230066011. [DOI] [PubMed] [Google Scholar]
- 117. Monoh K, Ishikawa K, Yasui N, Mineura K, Andoh H, Togawa K. p53 tumor suppressor gene in acoustic neuromas. Acta Otolaryngol Suppl. 1998;537:11‐15. 10.1080/00016489850182288. [DOI] [PubMed] [Google Scholar]
- 118. Mawrin C, Kirches E, Dietzmann K, Roessner A, Boltze C. Expression pattern of apoptotic markers in vestibular schwannomas. Pathol Res Pract. 2002;198(12):813‐819. 10.1078/0344-0338-00340. [DOI] [PubMed] [Google Scholar]
- 119. Lee JE, Hollenberg SM, Snider L, Turner DL, Lipnick N, Weintraub H. Conversion of Xenopus ectoderm into neurons by NeuroD, a basic helix‐loop‐helix protein. Science. 1995;268(5212):836‐844. 10.1126/science.7754368. [DOI] [PubMed] [Google Scholar]
- 120. Huang P, Kishida S, Cao D, et al. The neuronal differentiation factor NeuroD1 downregulates the neuronal repellent factor Slit2 expression and promotes cell motility and tumor formation of neuroblastoma. Cancer Res. 2011;71(8):2938‐2948. 10.1158/0008-5472.CAN-10-3524. [DOI] [PubMed] [Google Scholar]
- 121. Lei K, Li W, Huang C, et al. Neurogenic differentiation factor 1 promotes colorectal cancer cell proliferation and tumorigenesis by suppressing the p53/p21 axis. Cancer Sci. 2020;111(1):175‐185. 10.1111/cas.14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kersigo J, Gu L, Xu L, et al. Effects of Neurod1 expression on mouse and human schwannoma cells. Laryngoscope. 2020;21:E259‐E270. 10.1002/lary.28671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Arvold ND, Guha N, Wang D, et al. Hypoxia‐induced radioresistance is independent of hypoxia‐inducible factor‐1A in vitro. Int J Radiat Oncol Biol Phys. 2005;62(1):207‐212. 10.1016/j.ijrobp.2005.01.019. [DOI] [PubMed] [Google Scholar]
- 124. Cayé‐Thomasen P, Baandrup L, Jacobsen GK, Thomsen J, Stangerup S‐E. Immunohistochemical demonstration of vascular endothelial growth factor in vestibular schwannomas correlates to tumor growth rate. Laryngoscope. 2003;113(12):2129‐2134. 10.1097/00005537-200312000-00014. [DOI] [PubMed] [Google Scholar]
- 125. Brieger J, Bedavanija A, Lehr H‐A, Maurer J, Mann WJ. Expression of angiogenic growth factors in acoustic neurinoma. Acta Otolaryngol. 2003;123(9):1040‐1045. 10.1080/00016480310005101. [DOI] [PubMed] [Google Scholar]
- 126. Plotkin SR, Stemmer‐Rachamimov AO, Barker FG, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358‐367. 10.1056/NEJMoa0902579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Mautner V‐F, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14‐18. 10.1093/neuonc/nop010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Plotkin SR, Merker VL, Halpin C, et al. Bevacizumab for progressive vestibular schwannoma in neurofibromatosis type 2: a retrospective review of 31 patients. Otol Neurotol. 2012;33(6):1046‐1052. 10.1097/MAO.0b013e31825e73f5. [DOI] [PubMed] [Google Scholar]
- 129. Alanin MC, Klausen C, Caye‐Thomasen P, et al. The effect of bevacizumab on vestibular schwannoma tumour size and hearing in patients with neurofibromatosis type 2. Eur Arch Otorhinolaryngol. 2015;272(12):3627‐3633. 10.1007/s00405-014-3398-3. [DOI] [PubMed] [Google Scholar]
- 130. Hochart A, Gaillard V, Baroncini M, et al. Bevacizumab decreases vestibular schwannomas growth rate in children and teenagers with neurofibromatosis type 2. J Neurooncol. 2015;124(2):229‐236. 10.1007/s11060-015-1828-8. [DOI] [PubMed] [Google Scholar]
- 131. Slusarz KM, Merker VL, Muzikansky A, Francis SA, Plotkin SR. Long‐term toxicity of bevacizumab therapy in neurofibromatosis 2 patients. Cancer Chemother Pharmacol. 2014;73(6):1197‐1204. 10.1007/s00280-014-2456-2. [DOI] [PubMed] [Google Scholar]
- 132. Gao X, Zhao Y, Stemmer‐Rachamimov AO, et al. Anti‐VEGF treatment improves neurological function and augments radiation response in NF2 schwannoma model. Proc Natl Acad Sci U S A. 2015;112(47):14676‐14681. 10.1073/pnas.1512570112. [DOI] [PMC free article] [PubMed] [Google Scholar]