

Abstract
Tridentate aroyl hydrazones are effective metal chelators in biological settings, and their activity has been investigated extensively for medicinal applications in metal overload, cancer, and neurodegenerative diseases. The aroyl hydrazone motif is found in the recently reported prochelator (AH1-S)2, which has shown antiproliferative pro-apoptotic activity in mammalian cancer cell lines. Intracellular reduction of this disulfide prochelator leads to the formation of mercaptobenzaldehyde benzoylhydrazone chelator AH1 and to iron sequestration, which in turn impacts cell growth. Herein, we investigate the iron coordination chemistry of AH1 to determine the structural and spectroscopic properties of the iron complexes in the solid state and in liquid solution. A neutral Fe(III) complex of 2:1 ligand-to-metal stoichiometry was isolated and characterized fully to reveal two different binding modes for the tridentate AH1 ligand. Specifically, one ligand binds in the monoanionic keto form, whereas the other ligand coordinates as a dianionic enolate. Continuous-wave EPR experiments in frozen solutions indicated that this neutral complex is one of three low-spin Fe(III) complexes observed depending on the concentrations of the metal ion, the ligand, and their ratio. Electron spin echo envelope modulation (ESEEM) experiments allowed the assignment of the three species to different protonation states of the coordinated ligands. Our ESEEM analysis provides a method to distinguish the coordination of aroyl hydrazones in the keto and enolate form, which influences both the ligand field and overall charge of the complex. As such, this type of analysis could provide valuable information in a variety of studies of iron complexes of aroyl hydrazones, ranging from investigation of spin-crossover behavior to tracking of their distribution in biological samples.
Graphical Abstract
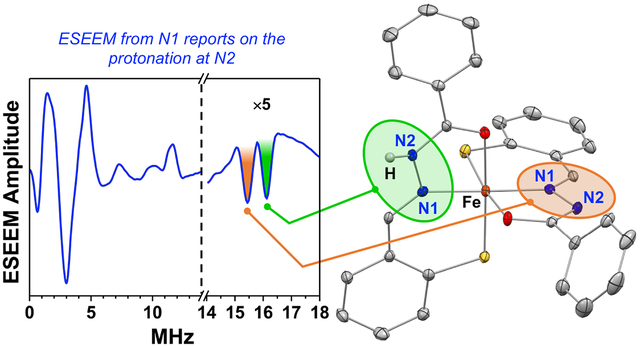
Reduction of the antiproliferative prochelator (AH1-S)2 leads to tridentate aroyl hydrazone AH1, which stabilizes low-spin ferric complexes of 2:1 binding stoichiometry. Experiments in solution by EPR/ESEEM methods detect three complexes in different protonation states. In particular, the keto and enolate binding modes of AH1 result in different quadrupole coupling constants of the nitrogen donor on the hydrazone ligand system.
INTRODUCTION
Tridentate ligands of the aroyl hydrazone family have been employed extensively to sequester iron and other metals for biological and medicinal applications.1 Early studies focused on the iron mobilization ability of pyridoxal isonicotinoyl hydrazone (PIH) in vivo and on its potential as an orally administered chelator for the treatment of iron overload disorders.2–3 The more lipophilic salicylaldehyde isonicotinoyl hydrazone (SIH) and several analogs were found to exclude intracellular iron from redox cycling, thereby protecting cardiomyocites and other cells from iron-mediated oxidative damage.4–6 In a prochelator version (BSIH) of the salicylaldehyde hydrazone SIH, the phenolic oxygen donor is masked by a boronic ester that reacts selectively in the presence of hydrogen peroxide, thus leading to the release of the iron-binding chelator as a cytoprotectant against oxidative damage.7–8 More recently, several aroyl hydrazone analogs were investigated for their ability to concurrently reduce oxidative stress and inhibit the metal-mediated aggregation of β-amyloid relevant to the pathology of Alzheimer’s disease.9
Several aroyl hydrazones, including 2-hydroxy-1-naphthaldehyde isonicotinoyl hydrazone NIH10 and 2-dipyridylketone analog PKIH,11 exhibit antiproliferative activity owing, at least in part, to their ability to sequester intracellular labile iron essential for cell cycle progression. Because rapidly proliferating malignant cells require higher iron levels when compared to normal cells,12–14 antiproliferative iron chelators, including numerous aroyl hydrazones and thiosemicarbazones, have been investigated as potential anticancer agents,1, 15–16 and several compounds have been assessed in clinical trials for cancer indications.17
Tridentate aroyl hydrazone ligands typically form octahedral iron complexes of 2:1 ligand-to-metal stoichiometry,9–10, 18–19 although 1:1 complexes have been isolated as well.20–21 Potentiometric studies indicate that these ligands (e.g., SIH, PIH) strongly stabilize the Fe(III) ion relative to Fe(II) and that the overall formation constants for the 2:1 Fe(III) complexes are extremely high (i.e., exceeding 1025).22–23 Indeed, these compounds rival many siderophores in terms of their thermodynamic iron-binding affinity. Overall, the low molecular weight, intermediate lipophilicity, and high binding affinity of aroyl hydrazones contribute to their ability to permeate cellular membranes, mobilize iron, prevent iron uptake from transferrin, and inhibit iron-dependent processes such as DNA synthesis.
For the development of a prochelator strategy, we have previously reported the incorporation of a disulfide switch in the metal-binding unit of thiosemicarbazone and hydrazone ligands.24–26 Upon cellular uptake, these disulfide-masked prochelators are converted to thiol-based tridentate chelators and reductively activated for iron sequestration.27 These compounds affect tumor progression by impacting the iron phenotype of both malignant cells and tumor-associated macrophages.28–29 The aroyl hydrazone system (AH1-S)2 (Scheme 1) exhibits antiproliferative activity in the low micromolar range in two breast cancer cell lines (i.e., MCF-7 and MDA-MB-231) and leads to cell cycle arrest at the G1/S interface, ultimately resulting in apoptotic cell death.25 In cell-based assays of cell proliferation and toxicity, (AH1-S)2 is one of the most effective compounds in this class of disulfide-based prochelators. Although several iron complexes of the thiosemicarbazone analogs have been characterized fully,24 the iron coordination of AH1 has not been investigated.
Scheme 1.
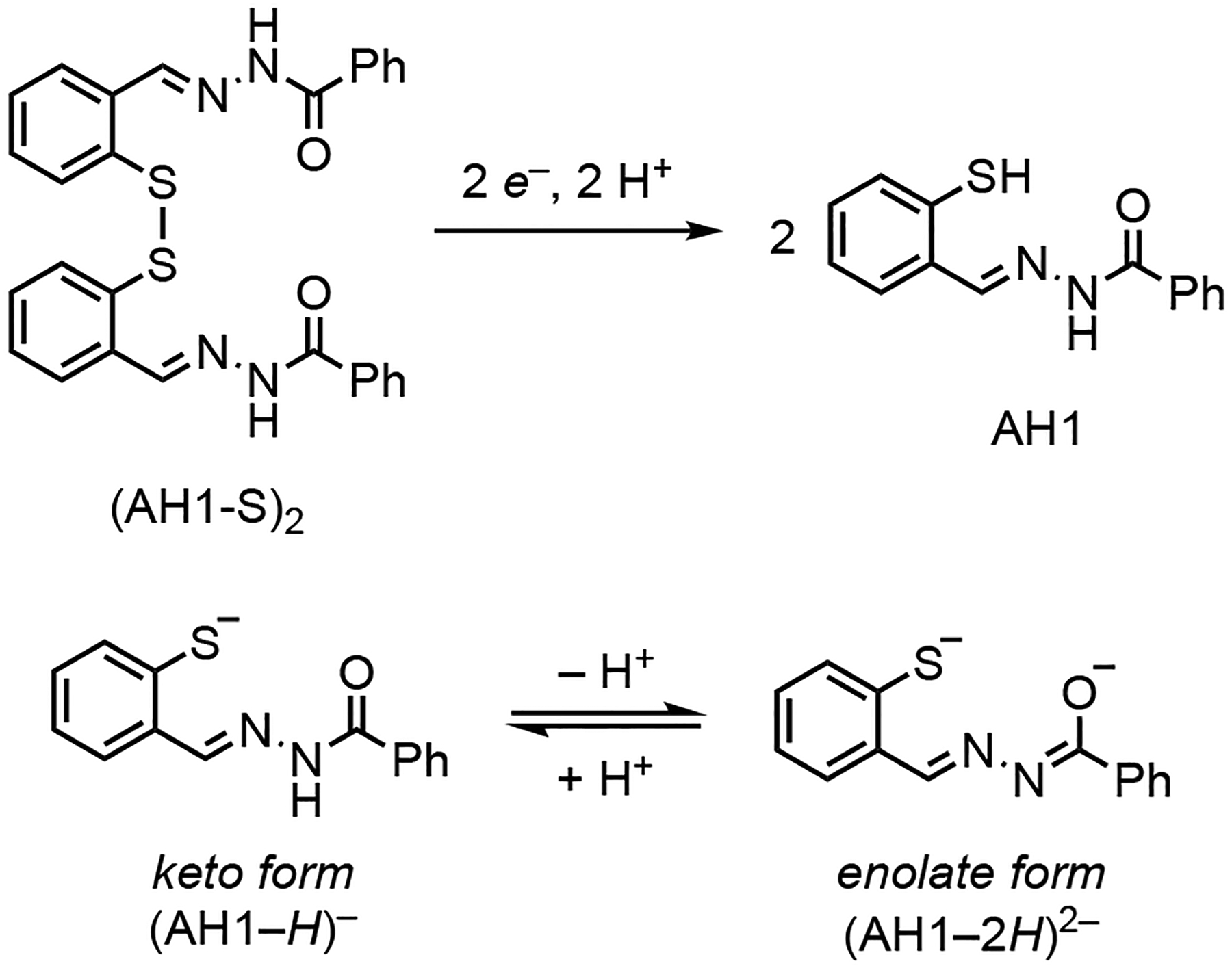
Reduction/activation of thiol chelator AH1 and protonation equilibrium of its thiolate iron-binding forms.
Herein, we sought to characterize the complex(es) formed upon reduction of (AH1-S)2 and iron coordination. In particular, we aimed to elucidate the coordination mode of the AH1 ligand, which can bind upon thiol deprotonation in the monoanionic keto form or with an additional deprotonation in the dianionic enolate form (Scheme 1). Our analysis by electron paramagnetic resonance (EPR) methods allows to identify these different ligand protonation states in iron(III) complexes in solution thus providing information that is relevant to their chemistry in a variety of contexts. For biologically active compounds like AH1 and other aroyl hydrazones, the coordination as monoanonic or dianionic ligands impacts the overall charge of the resulting iron species, an important parameter for the diffusion of the complexes through biological membranes, their cellular distribution, and ultimately for their fate in vivo. In addition, studies of magnetic properties have shown that the differences between keto and enolate coordination modes affect the spin crossover behavior observed in iron complexes of certain aroyl hydrazones,30 and this effect has been employed to achieve proton transfer coupled spin transitions (PCST).31
RESULTS AND DISCUSSION
Synthesis and Crystallographic Characterization.
The thiosalicylaldehyde aroyl hydrazone chelator AH1 was prepared by reduction of the disulfide prochelator (AH1-S)2 (Scheme 1) using dithiothreitol (DTT) in dimethylformamide. As expected for a tridentate aroyl hydrazone ligand, metal coordination occurred promptly upon addition of iron(II) tetrafluoroborate to a THF solution at room temperature in the absence of any base. The pale yellow solution of the ligand quickly turned dark brown, and the resulting complex was isolated by precipitation or crystallization. Mass spectrometry confirmed the formation of an iron complex with 2:1 ligand/metal stoichiometry, and the absence of a tetrafluoroborate counterion in the elemental analysis data and in the crystal structure (see below) indicated a neutral overall charge for the complex (see details in Experimental Section). The effective magnetic moment of the isolated complex measured at room temperature (Evans method) is 1.6±0.1 μB, and thus indicates a low-spin (S = ½) Fe(III) complex. As documented for other aroyl hydrazone ligands,18, 22, 32 AH1 stabilizes the ferric oxidation state therefore iron binding in the presence of Fe(II) leads to the isolation of a Fe(III) complex in the presence of trace oxygen. The stabilization of a low-spin configuration is in line with our previous reports on iron(III) complexes of thiol-containing chelators of the thiosemicarbazone family.24 As observed in other tridentate hydrazone ligands, the presence of a sulfur donor favors low-spin ground states.33–34
Single crystals suitable for X-ray diffraction were obtained following multiple recrystallizations of the isolated complex in THF and pentane. The crystal structure of the complex (Figures 1 and S1–S2) revealed the expected octahedral geometry with two tridentate ligands bound in meridional fashion. Unlike the Fe(III) complexes of thiol-containing tridentate chelators of the thiosemicarbazone family,24 however, the overall complex is a neutral species, and therefore the aroyl hydrazone ligands must complement the charge of the iron center in the absence of the [BF4]− ion. Indeed, the hydrogen atom at the N2 nitrogen was directly observed only for one of the two AH1 ligands in this crystal structure. This finding confirms that one of the AH1 ligands binds in the dianionic enolate form (indicated with atom labels A in Figure 1), whereas the other ligand binds in the monoanionic keto form (atom labels B in Figure 1). Accordingly, the distances to the iron center (Table 1) display a slight but convincing contraction in the case of the dianionic ligand relative to the monoanionic one (e.g., 1.9869(18) vs 2.0371(18) Å for the oxygen donor). Furthermore, on the AH1 scaffold, the O1–C8 bond is longer (1.295(2) vs 1.240(2) Å) and the N2–C8 bond is shorter (1.294(2) vs 1.333(2) Å) on the enolate ligand relative to the keto ligand. Both the keto form33 and the deprotonated enolate form19 have been previously observed in iron complexes of aroyl hydrazone ligands. More rarely, the two coordination modes have been found in the same species,32, 35 as is the case for this ferric complex of AH1. The bond distances in the primary coordination sphere are similar to those in other low-spin Fe(III) complexes of tridentate hydrazone ligands30, 33, 36 and shorter than those in analogous high-spin Fe(III) species.10, 18, 20
Figure 1.
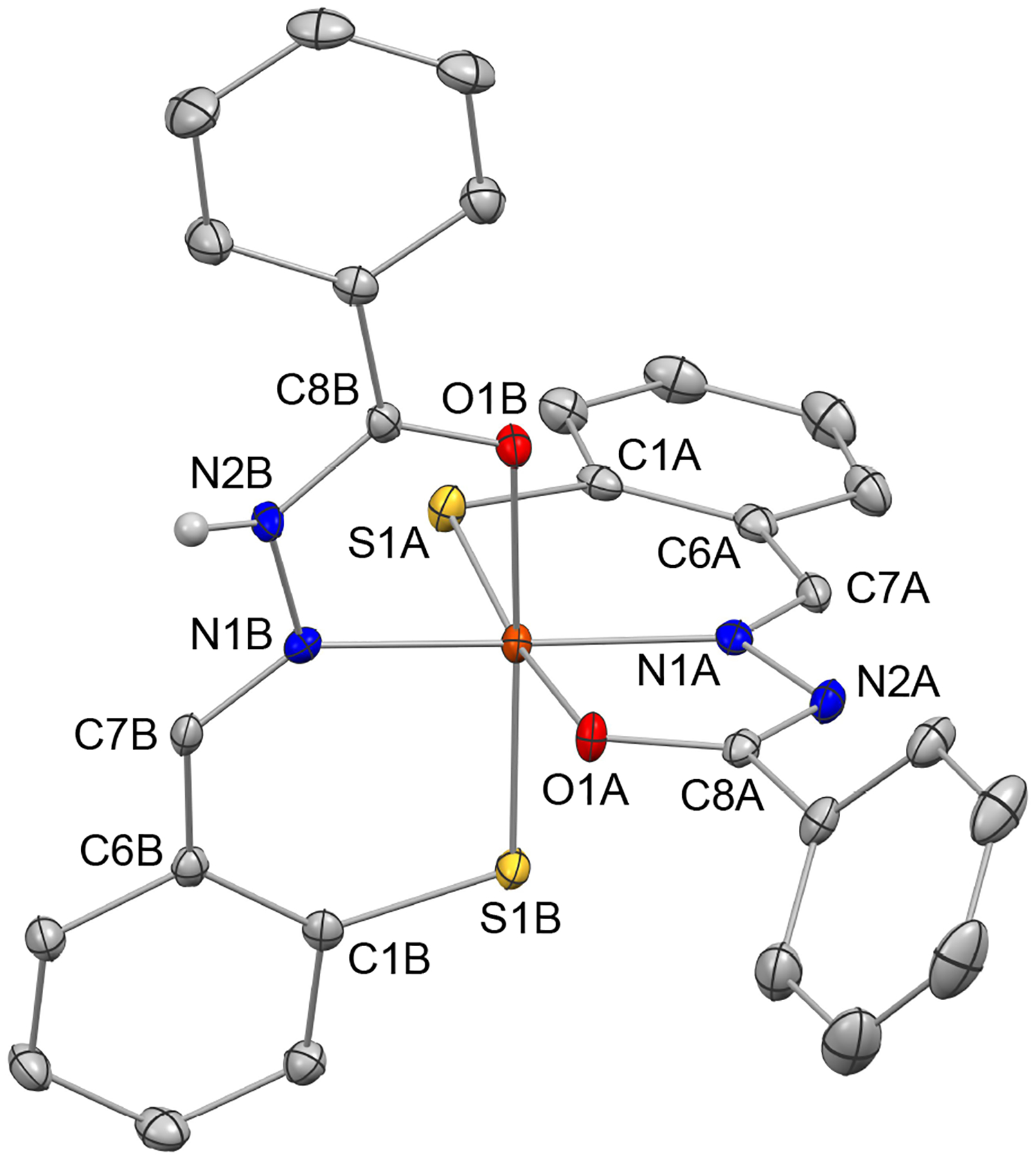
Crystal structure of [Fe(AH1−H)(AH1−2H)] showing a partial atom labeling scheme. Thermal ellipsoids are at the 50% probability level. Carbon-bound hydrogen atoms in calculated positions are not shown. The hydrogen atom bound to N2B was directly observed in the electron density map and refined explicitly (CCDC 1995248).
Table 1.
Selected bond lengths (Å) for the crystal structure of [Fe(AH1−H)(AH1−2H)].
| enolate ligand A | keto ligand B | ||
|---|---|---|---|
| bond | distance (Å) | bond | distance (Å) |
| Fe1A−S1A | 2.1622(15) | Fe1A−S1B | 2.1732(14) |
| Fe1A−N1A | 1.9014(18) | Fe1A−N1B | 1.9156(19) |
| Fe1A−O1A | 1.9869(18) | Fe1A−O1B | 2.0371(18) |
| O1A−C8A | 1.295(2) | O1B−C8B | 1.240(2) |
| N2A−C8A | 1.294(2) | N2B−C8B | 1.333(2) |
| N1A−N2A | 1.408(2) | N1B−N2B | 1.397(2) |
| N1A−C7A | 1.293(2) | N1B−C7B | 1.288(2) |
Spectroscopic Characterization in Solution.
The optical absorption changes (Figure 2) associated with the iron-binding reaction are reminiscent of those observed for other tridentate aroyl hydrazones and their ferric complexes.23–24, 37 The free ligand is pale yellow in solution and presents little absorption in the visible region, whereas the iron complex is darker and brown, exhibiting a maximum at 280 nm and very broad overlapping bands up to approximately 450 nm. Saturation was reached at approximately 0.5 equiv of added iron and the spectrum of the resulting species is consistent with that of the structurally characterized complex [Fe(AH1−H)(AH1−2H)] (vide infra). These experiments therefore confirmed the formation of a ferric complex of 2:1 ligand/metal stoichiometry in neutral aqueous solution. The strong preference of aroyl hydrazones for Fe(III) relative to Fe(II)22 explains the facile oxidation of the iron center from the divalent to the trivalent state that we observed both during the isolation of the complex and the titration experiments. In addition, we found that the ferric complex has a limited solubility (i.e., low micromolar range) in aqueous solutions and, further complicating these experiments, the unwanted oxidation of the thiol ligand AH1 led in some cases to the formation of the practically insoluble disulfide prochelator.
Figure 2.
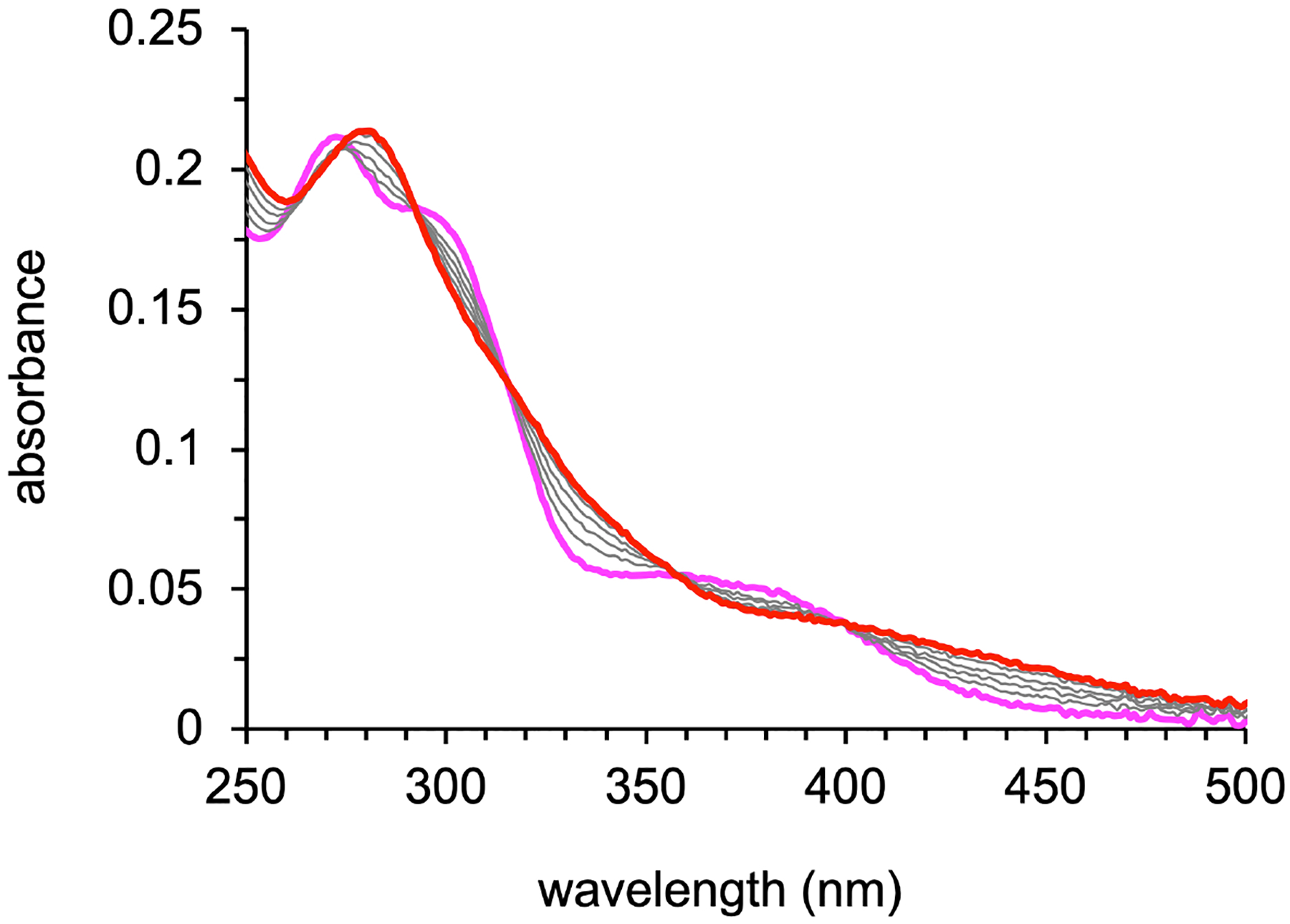
Optical absorption changes observed during the conversion of AH1 (15 μM, magenta trace) to the iron complex (red trace) in a buffered aqueous solution (10 mM HEPES, pH 7.4). Each addition corresponded to 0.1 equiv Fe(II), and saturation was reached at approximately 0.5 equiv.
To further characterize the iron AH1 complex(es) formed in solution, we continued the investigation by continuous-wave (CW) and pulsed EPR methods. The experiments were conducted with frozen solutions in 70/30% (v/v) DMSO/water mixtures, which ensured sufficient solubility as well as glass formation upon freezing. Initial measurements indicated the formation of three distinct low-spin Fe(III) complexes (or mixtures thereof) depending on the concentrations of the ligand and metal salt, as well as their ratio. Based on previous reports on iron binding by aroyl hydrazone ligands,22, 30, 35 we hypothesized that the three slightly different spectra could be attributed to three protonation states of the isolated 2:1 Fe(III) AH1 complex and sought to observe their speciation at different pH values. Buffer systems were omitted because their buffering capacity is significantly altered in 70% DMSO solutions in which the autoprotolysis of water is markedly suppressed (i.e., pKw is 18.4 in aqueous mixtures containing 80% DMSO).38 The sample solutions were prepared by dissolving the isolated complex [Fe(AH1−H)(AH1−2H)] in 70/30% DMSO/water mixtures and the pH was adjusted by addition of diluted HCl or NaHCO3 solutions. Because high percentages of organic solvents are incompatible with standard pH-meters, the pH of the sample solutions was estimated using colorimetric indicator pH strips with a sensitivity of 0.5 pH units. Under these conditions, we reasoned that the change of pH upon freezing in liquid nitrogen for EPR measurements would be within our experimental error.
The pH study confirmed the formation of three different low-spin Fe(III) complexes (Figure 3). At pH 8.5, we detected a species (complex I) characterized by a rhombic spectrum with principal g-values 2.163, 2.107, and 1.997. The narrower spectrum of a different species (g-values: 2.146, 2.093, 2.005) appeared as the pH was decreased and this was practically the only species observed at pH 5.5 (complex II). At lower pH, a third species (complex III) was detected presenting an even narrower spectrum (g-values: 2.134, 2.080, 2.013) and we could observe it as the dominant species in solution at very low pH (<2.0) in spite of partial demetalation.
Figure 3.
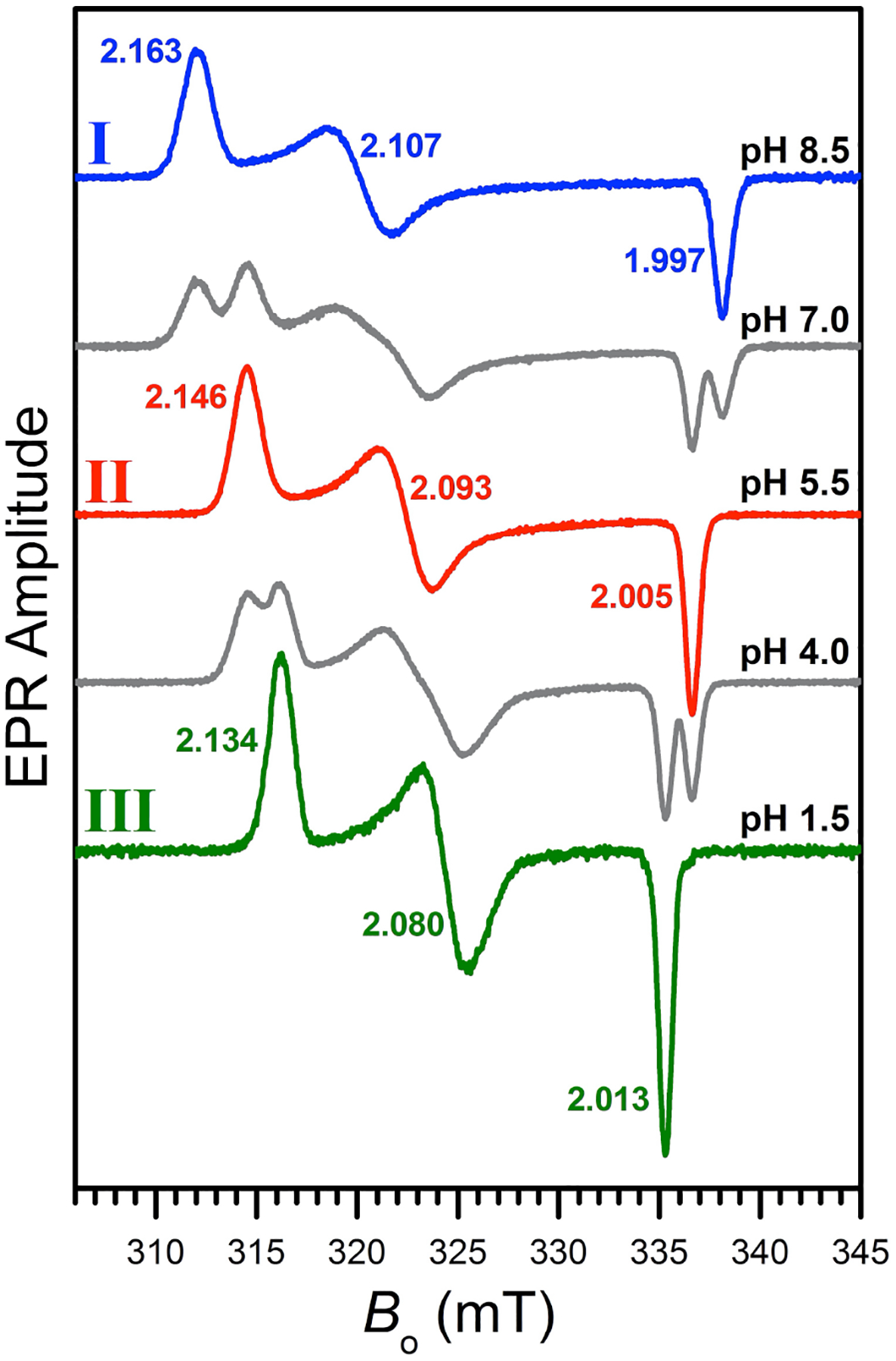
EPR spectra of frozen solutions of [Fe(AH1−H)(AH1−2H)] (200 μM) in DMSO/water (70/30% v/v) at various pH values. The spectra corresponding to three distinct low-spin Fe(III) species are indicated as complexes I, II, and III. The spectra of mixtures are shown in grey. Experimental conditions: mw frequency, 9.444 GHz; mw power, 2 mW; magnetic field modulation amplitude, 0.2 mT; temperature, 77 K.
To further characterize the three species observed in the pH study, we performed Ka-band (~34 GHz) electron spin echo envelope modulation (ESEEM) measurements on complexes I and II, which could be generated as single detectable species in solution. The ESEEM spectra of complex III could not be obtained because it appears as a single species in solution only at very low pH (below 2) and partial demetalation/precipitation in those conditions led to a signal amplitude that was not suitable for pulsed EPR measurements. Figure 4 shows the field-integrated spectra of two-pulse ESEEM obtained for complexes I and II as explained in the SI. The field-sweep spectra and the original orientation-selective ESEEM spectra are also shown in the SI (Figures S3–S5).
Figure 4.
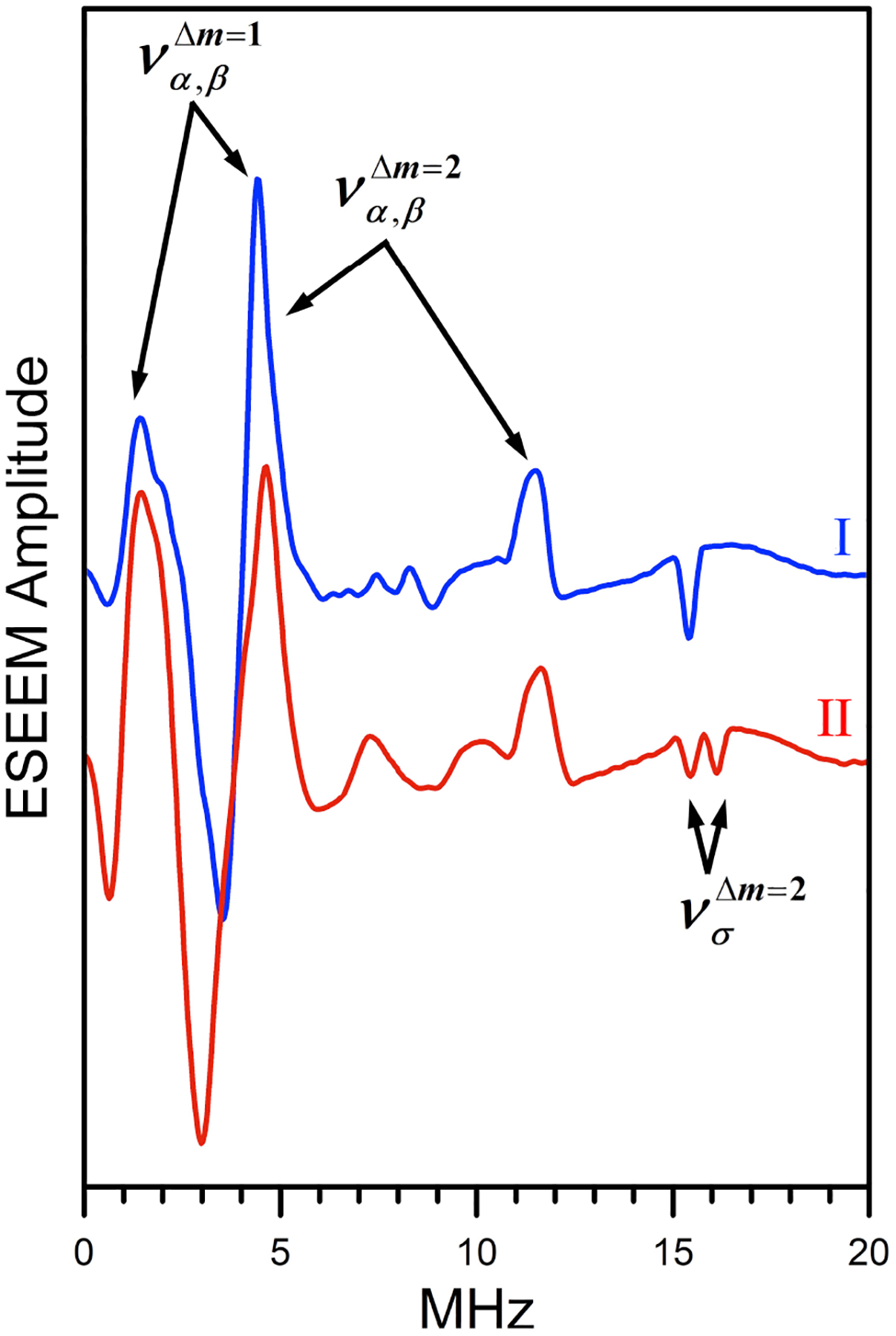
Field-integrated spectra of Ka-band 2-pulse ESEEM for complexes I and II in DMSO/water solutions (70/30% v/v). Experimental conditions: mw frequency, 34.268 GHz; effective magnetic field, 1163 and 1171 mT for I and II, respectively; temperature, 15 K.
The main isotopes of carbon, oxygen, and sulfur nuclei are not magnetic (spin I = 0), and the protons in our experimental conditions (Bo ~ 1.17 T) have a Zeeman frequency of about 50 MHz and an extremely low ESEEM amplitude. Thus, all lines/features observed in the spectra in Figure 4 originate from the 14N nuclei of the ligands. The observed 14N ESEEM spectra are characteristic of weak hyperfine interaction (hfi): νN > |AN|/2, where νN is the nitrogen Zeeman frequency and AN is the (effective) diagonal component of the nitrogen hfi. Such an assignment of the spectroscopic situation is supported by the hyperfine sublevel correlation (HYSCORE) spectra showing the prominent lines in the (++) quadrant (see Figure 5 showing a Ka-band HYSCORE spectrum obtained at g2). The assignment of the most essential lines observed in the primary ESEEM and HYSCORE spectra of complexes I and II to fundamental (να,β) and sum combination (νσ = να+νβ) harmonics is indicated in the figures. The hfi constant can be estimated as: ~ 4 MHz for both complexes I and II.
Figure 5.
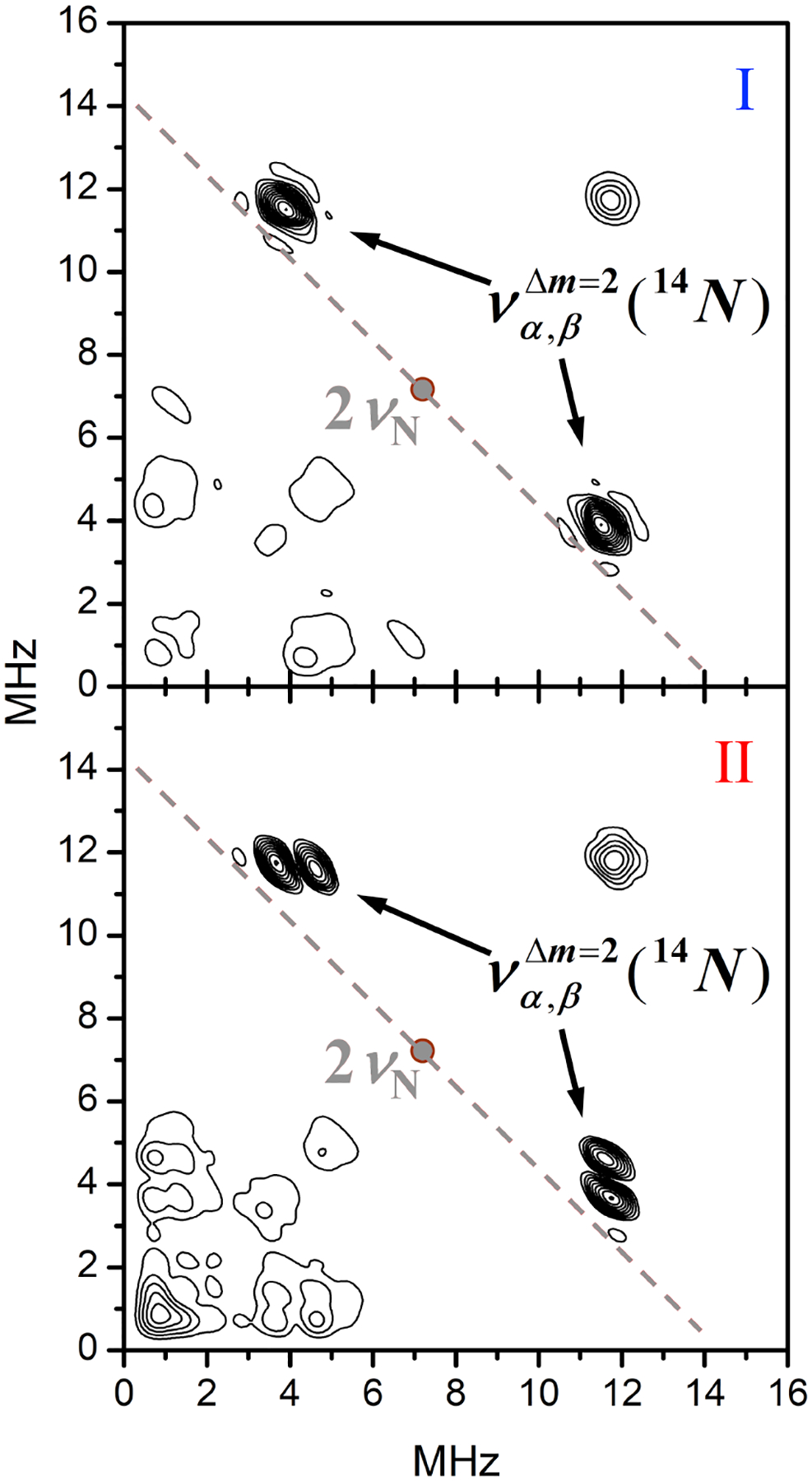
The (++) quadrants of the HYSCORE spectra at g2 for complexes I and II in DMSO/water solutions (70/30% v/v). Experimental conditions: mw frequency, 34.268 GHz; magnetic field, 1163 and 1171 mT for I and II, respectively; temperature, 15 K.
In the primary ESEEM spectrum of complex I (Figure 4), a single sum combination line is observed at the frequency ≈ 15.4 MHz. The shift of this frequency from 4νN ≈ 14.4 MHz is mostly explained by the second order effect of the nuclear quadrupole interaction (nqi):39
| (1) |
where k = e2Qq/4h is the reduced quadrupole coupling constant (e is the electron charge, Q is the 14N quadrupole moment, q is the largest component of the electron field gradient tensor at the position of the 14N nucleus, and h is the Planck constant) and η ∈[0, 1] is the asymmetry parameter of the nqi tensor. This equation is readily obtained from the expression for the frequencies of the ΔmI = 2 transitions:40
Using Eq. 1, the quadrupole coupling constant can be estimated as e2Qq/h = 2.2–2.6 MHz. This estimated range of e2Qq/h corresponds to the range of possible η values between 0 and 1. Because a single set of 14N lines (including the νσ line) is observed for complex I, we conclude that the coordinated nitrogen atoms in these species are equivalent.
The fundamental lines in the primary ESEEM spectrum of complex II (Figure 4) remain at approximately the same frequencies and have the same amplitudes as those observed for complex I. The HYSCORE spectrum of II (Figure 5), however, shows two closely located sets of crosspeaks instead of one set observed for complex I (Figure 5) and thus indicates a slight inequivalence between the coordinated nitrogen atoms in complex II. In the primary ESEEM spectrum of this complex in Figure 4, this inequivalence mostly results in the presence of two sum combination lines instead of one. The amplitudes of the νσ lines in the spectrum of II are similar and approximately equal to one half of the νσ line amplitude in the spectrum of I. Additionally, one of the νσ lines for II is at the same frequency of 15.4 MHz as the νσ line for I, indicating that the corresponding coordinated nitrogen in complex II is spectroscopically and electronically similar to those in complex I. For the second coordinated nitrogen in complex II, the sum combination line is located at the frequency of 16.1 MHz. The quadrupole coupling constant of this 14N estimated using Eq. (1) is within the range e2Qq/h = 2.9–3.4 MHz (depending on the assumed η value).
Based on the ESEEM analysis, complex II, which presents two spectroscopically distinct iron-bound nitrogen donors, is fully consistent with the complex isolated by crystallization. In [Fe(AH1−H)(AH1−2H)] (Figure 1), the nitrogen donor (N1A) in the enolate ligand is adjacent to a deprotonated nitrogen (N2A), whereas the nitrogen donor on the keto ligand (N1B) is adjacent to a protonated nitrogen (N2B). Our experiments demonstrate that these different binding modes of the AH1 ligand can be discriminated by ESEEM methods.
A qualitative analysis based on the Townes-Dailey approximation41–42 and literature examples43–44 supports our explanation of the changes in the nqi of the nitrogen donor N1 upon protonation of the nearby nitrogen N2. The quadrupole coupling constant of N1 is determined by the electronic populations of its valence orbitals, with the largest contribution being made by the lone pair electronic orbital directed toward the iron center. The second largest contribution comes from the p-type electronic orbital included in the ligand π-system. Importantly, these two contributions are of opposite signs. For the experimental geometry of the C7-N1-N2 fragment, the contributions of the two remaining orbitals are negligible. The partial positive charge at N2 caused by protonation will result in a withdrawal of electronic density from the N1 p-orbital toward N2 and an increase of the N1 nqi in full agreement with the ESEEM experimental observations.
The broader spectrum observed at higher pH can therefore be assigned to anionic complex [Fe(AH1−2H)2]− (Chart 1), which features two dianionic enolate ligands and hence two spectroscopically equivalent nitrogen donors. In other words, the neutral complex [Fe(AH1−H)(AH1−2H)] is the prevalent species at pH ~5.5 but is deprotonated as the pH of the solution increases to form the anionic complex [Fe(AH1−2H)2]−. The narrowest EPR spectrum in this series, which is observed at low pH, is then assigned to the cationic complex [Fe(AH1−H)2]+ featuring two monoanionic keto ligands (shown in gray on Chart 1).
Chart 1.
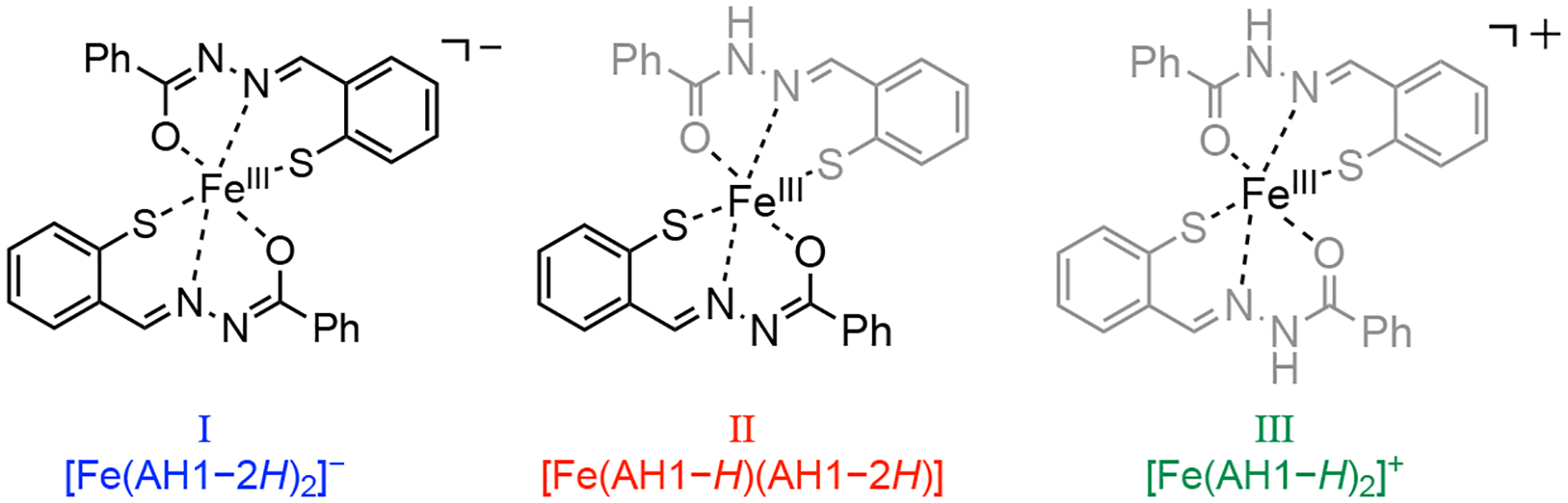
Low-spin iron(III) complexes of aroyl hydrazone AH1 identified by EPR/ESEEM.
The deconvolution and integration of CW EPR spectra for Fe(III) AH1 solutions (200 μM) provided an estimate of the distribution of the three complexes in the range from pH 1.5 to 8.0 (Figure S6). The apparent pKa values in 70/30% DMSO/water estimated based on the relative amounts of each complex were 3.7±0.5 and 7.0 ±0.5, with the neutral complex II observed as the prevalent species in the (4.5–6.5) pH range. To reproduce these findings in conditions more relevant to biological settings, optical absorption measurements were conducted at lower complex concentrations (15 μM) in buffered aqueous solutions with 10% DMSO. Only slight changes in the absorption of the complex were observed, and solubility limitations prevented reliable data acquisition below pH 6.5; nevertheless, the conversion between complexes I and II (Figure S7) could be observed in the same pH range (6.5−8.0) observed for the EPR experiments. Taken collectively, these results on the speciation of the Fe(III) AH1 complexes are consistent with those obtained via potentiometric methods for the aroyl hydrazone SIH (with pKa values of 4.6 and 7.8)22 and indicate that the neutral complex II is the prevalent species in the slightly acidic pH conditions of many biological environments.
CONCLUSIONS
In summary, we have investigated the iron coordination chemistry of mercaptobenzaldehyde benzoylhydrazone AH1, which results from the reduction of antiproliferative prochelator (AH1-S)2. AH1 stabilizes ferric centers in the low-spin configuration and both the keto and enolate coordination modes were observed in a neutral complex that was isolated and characterized by crystallography (Figure 1). In solution, three protonation states of the 2:1 AH1/Fe(III) complex (Chart 1) are associated with three distinct EPR spectra (Figure 3). In particular, the keto and enolate binding modes result in different quadrupole coupling constants of the donor nitrogen atoms as evidenced by our ESEEM study (Figures 4 and 5). This analysis indicates that the anionic and neutral complexes are prevalent at biologically relevant pH values and is therefore consistent with the acid dissociation constants determined by potentiometry for Fe(III) complexes of similar tridentate aroyl hydrazones (e.g., PIH, SIH). These findings provide valuable information for the detection of iron complexes of aroyl hydrazones in frozen intact cells by EPR methods, which we have previously employed to demonstrate the activation and iron binding of thiosemicarbazone prochelator analogs.27 More in general, this study contributes to the rich chemistry of iron aroyl hydrazones, which ranges from the engineering of new spin-crossover systems to medicinal chemistry.
EXPERIMENTAL SECTION
Materials and methods.
The prochelator disulfide (AH1-S)2 was prepared as previously described.25 N,N-Dimethylformamide (DMF), tetrahydrofuran (THF) and pentane were dried by passage through a solvent purifier. All other reagents were obtained commercially and used as received.
UV/visible spectra were recorded on an Agilent 8453 UV/visible spectrophotometer. The pH of buffered aqueous solutions (10% DMSO, 50 mM HEPES or PIPES) was measured on a Thermo Scientific Orion™ 3-Star pH-meter with a Ross™ Ultra pH/ATC Epoxy probe. The pH of 70/30% DMSO/water solutions was estimated using Whatman® Panpeha™ pH indicator strips with a sensitivity of 0.5 pH units. Solution magnetic moments were measured by the Evans method45–46 using reported diamagnetic corrections.47 A solution of the paramagnetic complex in THF-d8 was transferred into a 5-mm NMR tube, and a Wilmad® coaxial insert filled with the deuterated solvent was employed as an internal reference. Solution magnetic susceptibilities were calculated based on the difference in chemical shift for the 1H NMR resonance of the residual solvent protons in neat THF-d8 and in the solution containing the paramagnetic species (1.0 or 10.0 mM). 1H NMR data were recorded at the University of Arizona NMR Facility on a Bruker DRX−500 instrument. High-resolution mass spectrometry data were acquired at the University of Arizona Mass Spectrometry Facility. Elemental analyses were performed by Numega Resonance Labs, San Diego, CA.
The EPR experiments were carried out at the University of Arizona EPR Facility. In CW EPR experiments, the X-band EPR spectrometer Elexsys E500 (Bruker Biospin) was used. The pulsed EPR experiments were performed on the homebuilt Ka-band spectrometer described elsewhere.48
Synthesis of AH1.
The disulfide prochelator (AH1-S)2 (100 mg, 0.196 mmol) and dithiothreitol (DTT, 75.5 mg, 0.490 mmol) were dissolved in DMF (3 mL). The reaction mixture was allowed to stir for 3 hours under nitrogen and then evaporated in the presence of toluene (multiple additions) until the final volume was about 0.5 mL. Water was added to precipitate the desired product as a pale yellow solid, which was filtered and washed with water. The final product was dried under vacuum (88.3 mg, 88%). 1H NMR (500 MHz, DMSO-d6) δ 12.1 (s, 1H), 8.8 (s, 1H), 8.0 (d, 2H), 7.8 (d, 1H), 7.6 (d, 1H) 7.5 (t, 1H), 7.4 (d, 2H), 7.3 (dt, 2H), 6.8 (s, 1H). 13C NMR (126 MHz, DMSO-d6) δ 163.6, 147.1, 134.1, 133.6, 132.4, 131.4, 131.1, 130.3, 129.4, 129.0, 128.2, 125.7. HRMS m/z [M+H]+ calculated for C14H13N2OS, 257.0744; found 257.0745; m/z [M+Na]+ calculated for C14H12N2OSNa, 279.0563; found 279.0563.
Synthesis of [Fe(AH1−H)(AH1−2H)].
Chelator AH1 (20 mg) was combined with Fe(BF4)2•6H2O (13 mg, 0.5 equiv) in THF (2 mL) and the reaction mixture was allowed to stir for 30 min at room temperature. Pentane was added dropwise until a brown precipitate formed. This solid was collected by filtration and dried under high vacuum to afford the desired complex (14 mg, 64%). HRMS-ESI (m/z): [M+H]+ calculated for C28H22N4O2S2Fe 566.0529, found 566.0524. Magnetic moment (Evans): μeff = 1.6±0.1 μB. Anal. Calcd. for C33H33FeN4O2S2: C, 62.2; H, 5.2; N, 8.8; S 10.1%; found: C, 61.7; H, 5.2; N, 8.4; S, 9.6%.
Crystal structure determination of [Fe(AH1−H)(AH1−2H)].
The X-ray quality crystals of Fe(AH1)2 as dark brown plates were grown from a THF solution by slow vapor diffusion of pentane. The single crystal XRD data were collected on a Bruker Kappa APEX-II diffractometer at the XRD Facility of the University of Arizona, using the Mo Kα irradiation (λ ≈ 0.7107 Å). The data collection temperature was 100 K. The experiment was controlled by the APEX2 software (Bruker AXS Inc.), which also served as a user interface for calling the data reduction/integration (SAINT V6.28A), absorption correction (SADABS version 2.10), and space group determination (XPREP version 2008/2) programs. The crystal system was identified as triclinic, with the space group P-1. The structure solution and refinement were performed by ShelXT49 and ShelXL50 programs, respectively, called from the Olex2 interface.51 The carbon-bound hydrogen atoms were placed at predicted ideal locations stemming from the molecular geometry and refined using a riding model with Uiso = 1.2Ueq of the attached carbon atom. The nitrogen-bound hydrogen atoms were directly observed in the electron density map and refined explicitly.
The unit cell contains two identical complexes cocrystallized with two THF molecules. One of these solvent molecules had a well-defined orientation but presented two stable ring conformations. This disorder was modeled explicitly by splitting the positions of the ring carbons C2 and C3. To stabilize the refinement and ensure sensible bond lengths, the involved THF carbons were restrained using six DFIX and two ISOR statements. The second THF molecule, in which both the orientation and conformation were disordered, was excluded from refinement by using a solvent mask. The excluded region had a volume of 153.7 Å. The crystallographic information is summarized in Table S1.
Supplementary Material
Funding Sources
This work was supported by the US National Institutes of Health (GM127646).
Footnotes
Supporting Information. The following files are available free of charge:
X-ray diffraction and ESEEM data acquisition details (PDF)
REFERENCES
- 1.Kalinowski DS; Richardson DR, The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev 2005, 57, 547–583. [DOI] [PubMed] [Google Scholar]
- 2.Ponka P; Borova J; Neuwirt J; Fuchs O; Necas E, Study of intracellular iron-metabolism using pyridoxal isonicotinoyl hydrazone and other synthetic chelating agents. Biochim. Biophys. Acta 1979, 586, 278–297. [DOI] [PubMed] [Google Scholar]
- 3.Richardson DR; Ponka P, Pyridoxal isonicotinoyl hydrazone and its analogs: Potential orally effective iron-chelating agents for the treatment of iron overload disease. J. Lab. Clin. Med 1998, 131, 306–315. [DOI] [PubMed] [Google Scholar]
- 4.Horackova M; Ponka P; Byczko Z, The antioxidant effects of a novel iron chelator salicylaldehyde isonicotinoyl hydrazone in the prevention of H2O2 injury in adult cardiomyocytes. Cardiovasc. Res 2000, 47, 529–536. [DOI] [PubMed] [Google Scholar]
- 5.Simunek T; Boer C; Bouwman RA; Vlasblom R; Versteilen AMG; Sterba M; Gersl V; Hrdina R; Ponka P; de Lange JJ; Paulus WJ; Musters RJP, SIH - a novel lipophilic iron chelator - protects H9c2 cardiomyoblasts from oxidative stress-induced mitochondrial injury and cell death. J. Mol. Cell. Cardiol 2005, 39, 345–354. [DOI] [PubMed] [Google Scholar]
- 6.Haskova P; Kovarikova P; Koubkova L; Vavrova A; Mackova E; Simunek T, Iron chelation with salicylaldehyde isonicotinoyl hydrazone protects against catecholamine autoxidation and cardiotoxicity. Free Rad. Biol. Med 2011, 50, 537–549. [DOI] [PubMed] [Google Scholar]
- 7.Charkoudian LK; Pham DM; Franz KJ, A pro-chelator triggered by hydrogen peroxide inhibits iron-promoted hydroxyl radical formation. J. Am. Chem. Soc 2006, 128, 12424–12425. [DOI] [PubMed] [Google Scholar]
- 8.Charkoudian LK; Dentchev T; Lukinova N; Wolkow N; Dunaief JL; Franz KJ, Iron prochelator BSIH protects retinal pigment epithelial cells against cell death induced by hydrogen peroxide. J. Inorg. Biochem 2008, 102, 2130–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Palanimuthu D; Wu ZX; Jansson PJ; Braidy N; Bernhardt PV; Richardson DR; Kalinowski DS, Novel chelators based on adamantane-derived semicarbazones and hydrazones that target multiple hallmarks of Alzheimer’s disease. Dalton Trans. 2018, 47, 7190–7205. [DOI] [PubMed] [Google Scholar]
- 10.Richardson DR; Bernhardt PV, Crystal and molecular structure of 2-hydroxy-1-naphthaldehyde isonicotinoyl hydrazone (NIH) and its iron(III) complex: an iron chelator with anti-tumour activity. J. Biol. Inorg. Chem 1999, 4, 266–273. [DOI] [PubMed] [Google Scholar]
- 11.Becker EM; Lovejoy DB; Greer JM; Watts R; Richardson DR, Identification of the di-pyridyl ketone isonicotinoyl hydrazone (PKIH) analogues as potent iron chelators and anti-tumour agents. Br. J. Pharmacol 2003, 138, 819–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torti SV; Torti FM, Iron and cancer: more ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Torti SV; Manz DH; Paul BT; Blanchette-Farra N; Torti FM, Iron and Cancer. Annu. Rev. Nutr 2018, 38, 97–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jung M; Mertens C; Tomat E; Brune B, Iron as a Central Player and Promising Target in Cancer Progression. Int. J. Mol. Sci 2019, 20, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crielaard BJ; Lammers T; Rivella S, Targeting iron metabolism in drug discovery and delivery. Nat. Rev. Drug Discov 2017, 16, 400–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Utterback RD; Tomat E, Developing Ligands to Target Transition Metals in Cancer. In Encyclopedia of Inorganic and Bioinorganic Chemistry, Scott RA, Ed. Wiley: 2020; p eibc2694. [Google Scholar]
- 17.Yu Y; Kalinowski DS; Kovacevic Z; Siafakas AR; Jansson PJ; Stefani C; Lovejoy DB; Sharpe PC; Bernhardt PV; Richardson DR, Thiosemicarbazones from the Old to New: Iron Chelators That Are More Than Just Ribonucleotide Reductase Inhibitors. J. Med. Chem 2009, 52, 5271–5294. [DOI] [PubMed] [Google Scholar]
- 18.Avramovicigrisaru S; Sarel S; Cohen S; Bauminger RE, The synthesis, crystal and molecular structure, and oxidation state of iron complex from pyridoxal isonicotinoyl hydrazone and ferrous sulfate. Isr. J. Chem 1985, 25, 288–292. [Google Scholar]
- 19.Bernhardt PV; Wilson GJ; Sharpe PC; Kalinowski DS; Richardson DR, Tuning the antiproliferative activity of biologically active iron chelators: characterization of the coordination chemistry and biological efficacy of 2-acetylpyridine and 2-benzoylpyridine hydrazone ligands. J. Biol. Inorg. Chem 2008, 13, 107–119. [DOI] [PubMed] [Google Scholar]
- 20.Murphy TB; Rose NJ; Schomaker V; Aruffo A, Syntheses of iron(III) aroyl hydrazones containing pyridoxal and salicylaldehyde – The crystal and molecular structure of two iron(III) pyridoxal isonicotinoyl hydrazone complexes. Inorg. Chim. Acta 1985, 108, 183–194. [Google Scholar]
- 21.Aruffo AA; Murphy TB; Johnson DK; Rose NJ; Schomaker V, Structural studies of Fe(III) and Cu(II) complexes of salicylaldehyde benzoyl hydrazone, a synthetic chelating agent exhibiting diverse biological properties. Inorg. Chim. Acta 1982, 67, L25–L27. [Google Scholar]
- 22.Wis Vitolo LM; Hefter GT; Clare BW; Webb J, Iron chelators of the pyridoxal isonicotinoyl hydrazone class Part II. Formation constants with iron(III) and iron(II). Inorg. Chim. Acta 1990, 170, 171–176. [Google Scholar]
- 23.Chen YL; Kong XL; Xie YY; Hider RC, The interaction of pyridoxal isonicotinoyl hydrazone (PIH) and salicylaldehyde isonicotinoyl hydrazone (SIH) with iron. J. Inorg. Biochem 2018, 180, 194–203. [DOI] [PubMed] [Google Scholar]
- 24.Chang TM; Tomat E, Disulfide/thiol switches in thiosemicarbazone ligands for redox-directed iron chelation. Dalton Trans. 2013, 42, 7846–7849. [DOI] [PubMed] [Google Scholar]
- 25.Akam EA; Utterback RD; Marcero JR; Dailey HA; Tomat E, Disulfide-masked iron prochelators: Effects on cell death, proliferation, and hemoglobin production. J. Inorg. Biochem 2018, 180, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akam EA; Tomat E, Targeting Iron in Colon Cancer via Glycoconjugation of Thiosemicarbazone Prochelators. Bioconjugate Chem. 2016, 27, 1807–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akam EA; Chang TM; Astashkin AV; Tomat E, Intracellular reduction/activation of a disulfide switch in thiosemicarbazone iron chelators. Metallomics 2014, 6, 1905–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mertens C; Akam EA; Rehwald C; Brüne B; Tomat E; Jung M, Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS ONE 2016, 11, e0166164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnetz M; Meier KJ; Rehwald C; Mertens C; Urbschat A; Tomat E; Akam AE; Baer P; Roos CF; Brüne B; Jung M, The Disturbed Iron Phenotype of Tumor Cells and Macrophages in Renal Cell Carcinoma Influences Tumor Growth. Cancers 2020, 12, 530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shongwe MS; Al-Rahbi SH; Al-Azani MA; Al-Muharbi AA; Al-Mjeni F; Matoga D; Gismelseed A; Al-Omari IA; Yousif A; Adams H; Morris MJ; Mikuriya M, Coordination versatility of tridentate pyridyl aroylhydrazones towards iron: tracking down the elusive aroylhydrazono-based ferric spin-crossover molecular materials. Dalton Trans. 2012, 41, 2500–2514. [DOI] [PubMed] [Google Scholar]
- 31.Nakanishi T; Hori Y; Sato H; Wu S-Q; Okazawa A; Kojima N; Yamamoto T; Einaga Y; Hayami S; Horie Y; Okajima H; Sakamoto A; Shiota Y; Yoshizawa K; Sato O, Observation of Proton Transfer Coupled Spin Transition and Trapping of Photoinduced Metastable Proton Transfer State in an Fe(II) Complex. J. Am. Chem. Soc 2019, 141, 14384–14393. [DOI] [PubMed] [Google Scholar]
- 32.Sutradhar M; Alegria ECBA; Mahmudov KT; da Silva MFCG; Pombeiro AJL, Iron(III) and cobalt(III) complexes with both tautomeric (keto and enol) forms of aroylhydrazone ligands: catalysts for the microwave assisted oxidation of alcohols. RSC Adv. 2016, 6, 8079–8088. [Google Scholar]
- 33.Kalinowski DS; Sharpe PC; Bernhardt PV; Richardson DR, Design, synthesis, and characterization of new iron chelators with anti-proliferative activity: Structure-activity relationships of novel thiohydrazone analogues. J. Med. Chem 2007, 50, 6212–6225. [DOI] [PubMed] [Google Scholar]
- 34.Basha MT; Chartres JD; Pantarat N; Ali MA; Mirza AH; Kalinowski DS; Richardson DR; Bernhardt PV, Heterocyclic dithiocarbazate iron chelators: Fe coordination chemistry and biological activity. Dalton Trans. 2012, 41, 6536–6548. [DOI] [PubMed] [Google Scholar]
- 35.Matoga D; Szklarzewicz J; Stadnicka K; Shongwe MS, Iron(III) complexes with a biologically relevant aroylhydrazone: Crystallographic evidence for coordination versatility. Inorg. Chem 2007, 46, 9042–9044. [DOI] [PubMed] [Google Scholar]
- 36.Richardson DR; Kalinowski DS; Richardson V; Sharpe PC; Lovejoy DB; Islam M; Bernhardt PV, 2-Acetylpyridine Thiosemicarbazones are Potent Iron Chelators and Antiproliferative Agents: Redox Activity, Iron Complexation and Characterization of their Antitumor Activity. J. Med. Chem 2009, 52, 1459–1470. [DOI] [PubMed] [Google Scholar]
- 37.Ponka P; Borova J; Neuwirt J; Fuchs O, Mobilization of iron from reticulocytes – Identification of pyridoxal isonicotinoyl hydrazone as a new iron chelating agent. FEBS Lett. 1979, 97, 317–321. [DOI] [PubMed] [Google Scholar]
- 38.Baughman EH; Kreevoy MM, Determination of Acidity in 80% Dimethyl Sulfoxide − 20% Water. J. Phys. Chem 1974, 78, 421–423. [Google Scholar]
- 39.Astashkin AV; Nesmelov YE, Mn2+-Nucleotide Coordination at the Myosin Active Site As Detected by Pulsed Electron Paramagnetic Resonance. J. Phys. Chem. B 2012, 116, 13655–13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dikanov SA; Tsvetkov YD; Bowman MK; Astashkin AV, Parameters of quadrupole coupling of 14N nuclei in chlorophyll a cations determined by the electron spin echo method. Chem. Phys. Lett 1982, 90, 149–153. [Google Scholar]
- 41.Townes CH; Dailey BP, Determination of electronic structure of molecules from nuclear quadrupole effects. J. Chem. Phys 1949, 17, 782–796. [Google Scholar]
- 42.Stoll S; Goldfarb D, EPR Interactions - Nuclear quadrupole couplings. In EPR Spectroscopy: Fundamentals and methods, Stoll S; Goldfarb D, Eds. Wiley: 2018; pp 95–114. [Google Scholar]
- 43.Hsieh YN; Rubenacker GV; Cheng CP; Brown TL, N-14 Nuclear quadrupole resonance spectra of coordinated pyridine. J. Am. Chem. Soc 1977, 99, 1384–1389. [Google Scholar]
- 44.Ashby CIH; Cheng CP; Brown TL, N-14 Nuclear quadrupole resonance spectra of coordinated imidazole. J. Am. Chem. Soc 1978, 100, 6057–6063. [Google Scholar]
- 45.Evans DF, The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. J. Chem. Soc 1959, 2003–2005. [Google Scholar]
- 46.Schubert EM, Utilizing the Evans method with a super-conducting NMR spectrometer in the undergraduate laboratory. J. Chem. Ed 1992, 69, 62. [Google Scholar]
- 47.Bain GA; Berry JF, Diamagnetic corrections and Pascal’s constants. J. Chem. Ed 2008, 85, 532–536. [Google Scholar]
- 48.Astashkin AV; Enemark JH; Raitsimring A, 26.5–40 GHz Ka-band pulsed EPR spectrometer. Concepts Magn. Reson. Part B: Magn. Reson. Eng 2006, 29B, 125–136. [Google Scholar]
- 49.Sheldrick GM, SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sheldrick GM, Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dolomanov OV; Bourhis LJ; Gildea RJ; Howard JAK; Puschmann H, OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr 2009, 42, 339–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.