

ABSTRACT
Foamy viruses (FVs) are complex retroviruses that can infect humans and other animals. In this study, by integrating transcriptomic and genomic data, we discovered 412 FVs from 6 lineages in amphibians, which significantly increased the known set of FVs in amphibians. Among these lineages, salamander FVs maintained a coevolutionary pattern with their hosts that could be dated back to the Paleozoic era, while in contrast, frog FVs were much more likely acquired from cross-species (class-level) transmission in the Cenozoic era. In addition, we found that three distinct FV lineages had integrated into the genome of a salamander. Unexpectedly, we identified a lineage of endogenous FVs in caecilians that expressed all complete major genes, demonstrating the potential existence of an exogenous form of FV outside of mammals. Our discovery of rare phenomena in amphibian FVs has significantly increased our understanding of the macroevolution of the complex retrovirus.
IMPORTANCE Foamy viruses (FVs) represent, more so than other viruses, the best model of coevolution between a virus and a host. This study represents the largest investigation so far of amphibian FVs and reveals 412 FVs of 6 distinct lineages from three major orders of amphibians. Besides a coevolutionary pattern, cross-species and repeated infections were also observed during the evolution of amphibian FVs. Remarkably, expressed FVs including a potential exogenous form were discovered, suggesting that active FVs might be underestimated in nature. These findings revealed that the multiple origins and complex evolution of amphibian FVs started from the Paleozoic era.
KEYWORDS: foamy virus, endogenous foamy virus, expressed foamy virus, amphibian, evolution, cross-species transmission, multiple origin, repeated infection
INTRODUCTION
Retroviruses (family Retroviridae) have great medical and economic significance, as some are associated with severe infectious disease or are oncogenic (1). Retroviruses are notable, as they occasionally integrate into the germ line of a host and become endogenous retroviruses (ERVs), which can be vertically inherited (2, 3). ERVs generated by simple retroviruses are widely distributed in vertebrates (4–11). However, complex retroviruses, such as lentiviruses, foamy viruses, and deltaretroviruses, have rarely appeared as endogenous forms.
Foamy viruses (FVs) (subfamily Spumavirinae) are complex retroviruses that exhibit typical codivergence with their hosts, providing an ideal framework for understanding the long-term evolutionary relationship between viruses and vertebrate hosts (12–15). Exogenous foamy viruses are prevalent in mammals, including primates (12, 16), bovines (17, 18), equines (19), bats (20), and felines (21). Vertebrate genomic analysis first identified endogenous foamy viruses (EFVs) in sloths (22), and then they were found in several primates (23–25). The subsequent discovery of EFVs and EFV-like elements in fish and amphibian genomes indicated that foamy viruses, along with their vertebrate hosts, have ancient origins (26, 27). Recently, the discovery of four novel reptile EFVs and two avian EFVs demonstrated that FVs can infect all five major classes of vertebrates (28–31).
Although a substantial number of EFVs have been found across the evolutionary history of vertebrates (22, 24, 32), to date, only 6 EFV lineages have been found in amphibians, and these were mainly found in salamanders (26). Most importantly, no potential exogenous foamy viruses have been found outside of their mammalian hosts (26, 31). In this study, by combining traditional ERV data mining with virome analysis, we found one potential exogenous foamy virus and several novel lineages of foamy viruses, which doubles the set of known foamy viruses in amphibians. Our work also provides novel insight into the early evolution of foamy viruses from the Paleozoic to the Cenozoic era.
RESULTS
Discovery and confirmation of foamy viral elements and expressed foamy viruses in amphibians.
We screened 19 amphibian genomes using a homology-based stepwise manner as described in Materials and Methods. This led to the discovery of 18 foamy-like ERVs in Spea multiplicate (Mexican spadefoot toad), 64 in Rhinatrema bivittatum (two-lined caecilian), and 131 in Ambystoma mexicanum (axolotl) (see Table S1 in the supplemental material). The foamy-like elements in S. multiplicate showed high similarity to each other. Such high similarity between copies has also been observed in foamy-like elements in R. bivittatum. However, there were two distinct groups of foamy-like ERVs in A. mexicanum that showed an average 64% identity with 99% coverage.
In order to discover any potential exogenous foamy viruses, we also searched all available transcriptome sequencing data for 61 amphibians in the transcriptome sequence assembly (TSA) database. Notably, we found 58 foamy virus-like RNA copies in Tylototriton wenxianensis (Wenxian knobby newt), 131 in Taricha granulosa (rough-skinned newt), and 10 in R. bivittatum. Homologous comparison with NviFLERV-1 (26) showed that all three groups of foamy-like viral copies harbored typical major coding regions, i.e., GAG, POL, and ENV, with an average similarity of 22% to 76%.
To examine whether these viral elements belonged to the clade of foamy viruses, a phylogenetic tree was inferred using the reverse transcriptase (RT) protein of representative viruses from all of the genera of Retroviridae (Fig. 1; Table S2). Our RT phylogenetic tree revealed that novel viral elements found in amphibians were grouped within the foamy virus clade (bootstrap value, >90%), which confirmed that they were indeed foamy viruses. In addition, these foamy-like viral elements could be divided into three major clades, where the viral elements found in different newts, including previously identified NviFLERV, clustered together, while the viral elements found in R. bivittatum and S. multiplicate formed respective monophyletic groups. Also, of note was that foamy-like ERVs in A. mexicanum divided into two clusters, indicating the existence of two lineages of EFVs in A. mexicanum.
FIG 1.
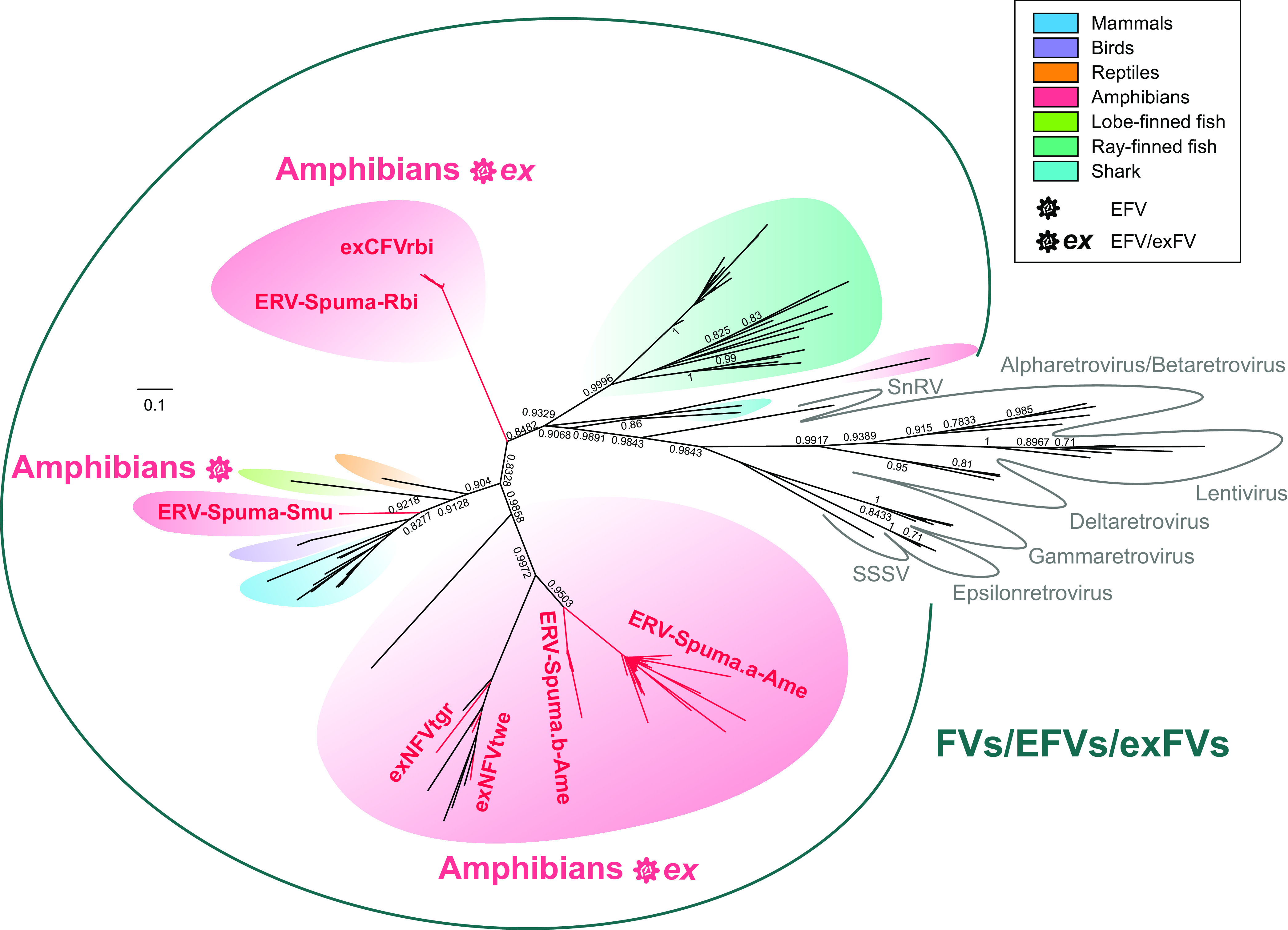
Unrooted phylogeny of retroviruses and foamy-like elements. The tree was inferred from reverse transcriptase (RT) protein alignment. The host information for FVs, EFVs, and exFVs is indicated using colors indicated in the key. The newly identified viral elements are labeled in red. The scale bar indicates the number of amino acid changes per site. Bootstrap values of <70% are not shown.
Thus, in accordance with the nomenclature proposed for ERVs (33), the foamy-like ERVs found in S. multiplicate and R. bivittatum were named ERVs-Spuma.n-Smu (n ≈ 1 to 18) and ERVs-Spuma.n-Rbi (n ≈ 1 to 64), respectively. Additionally, two lineages in A. mexicanum were designated ERVs-Spuma.an-Ame (n ≈ 1 to 104) and ERVs-Spuma.bn-Ame (n ≈ 1 to 18). Moreover, the expressed viral elements found in T. wenxianensis, T. granulosa, and R. bivittatum were then named expressed NFVtwe.n (exNFVtwe) (n ≈ 1 to 58), expressed NFVtgr.n (exNFVtgr) (n ≈ 1 to 131), and expressed CFVrbi.n (exCFVrbi) (n ≈ 1 to 10), respectively.
EFV and exFV genome characterization.
Fourteen ERVs-Spuma-Smu, 54 ERVs-Spuma-Rbi, 57 ERVs-Spuma.a-Ame, and 15 ERVs-Spuma.b-Ame, of which >5 kb was retrieved from each of their respective genomes, were used to construct a consensus sequence for each EFV lineage (Fig. 2A; Data Set S1). The consensus genomes of novel EFVs all harbored pairwise long terminal repeats (LTRs) and exhibited typical foamy virus structure, containing three major genes, gag, pol, and env, and one (ERV-Spuma-Rbi) or two (ERVs-Spuma-Smu, ERVs-Spuma.a-Ame, and ERVs-Spuma.b-Ame) putative accessory genes. It is noteworthy that none of these accessory genes showed similarity to any genes coding for known proteins. By searching against the Conserved Domain Database (CDD) using CD-Search, we were able to identify two typical foamy virus conserved domains in all four consensus EFVs: (i) the gag_spuma superfamily domain (cl26624) (34, 35) and (ii) the Foamy_virus_env superfamily domain (cl04051) (32). However, ERV-Spuma-Smu exclusively contained the Spuma_A9PTase superfamily domain (cl08397) (26) that is present in all mammalian foamy virus Pol proteins. The existence of these domains gave additional support to their classification as foamy viruses. Other regions or domains, such as the RT_like superfamily (RT) domain (cl02808), the RNase_H_like superfamily and RT_RNaseH_2 superfamily (RH) domains (cl14782, cl39038), and the rve and Integrase_H2C2 and SH3_11 superfamily domains (INT) (pfam00665, pfam17921, cl39492) were also identified in all EFVs (Fig. 2A).
FIG 2.
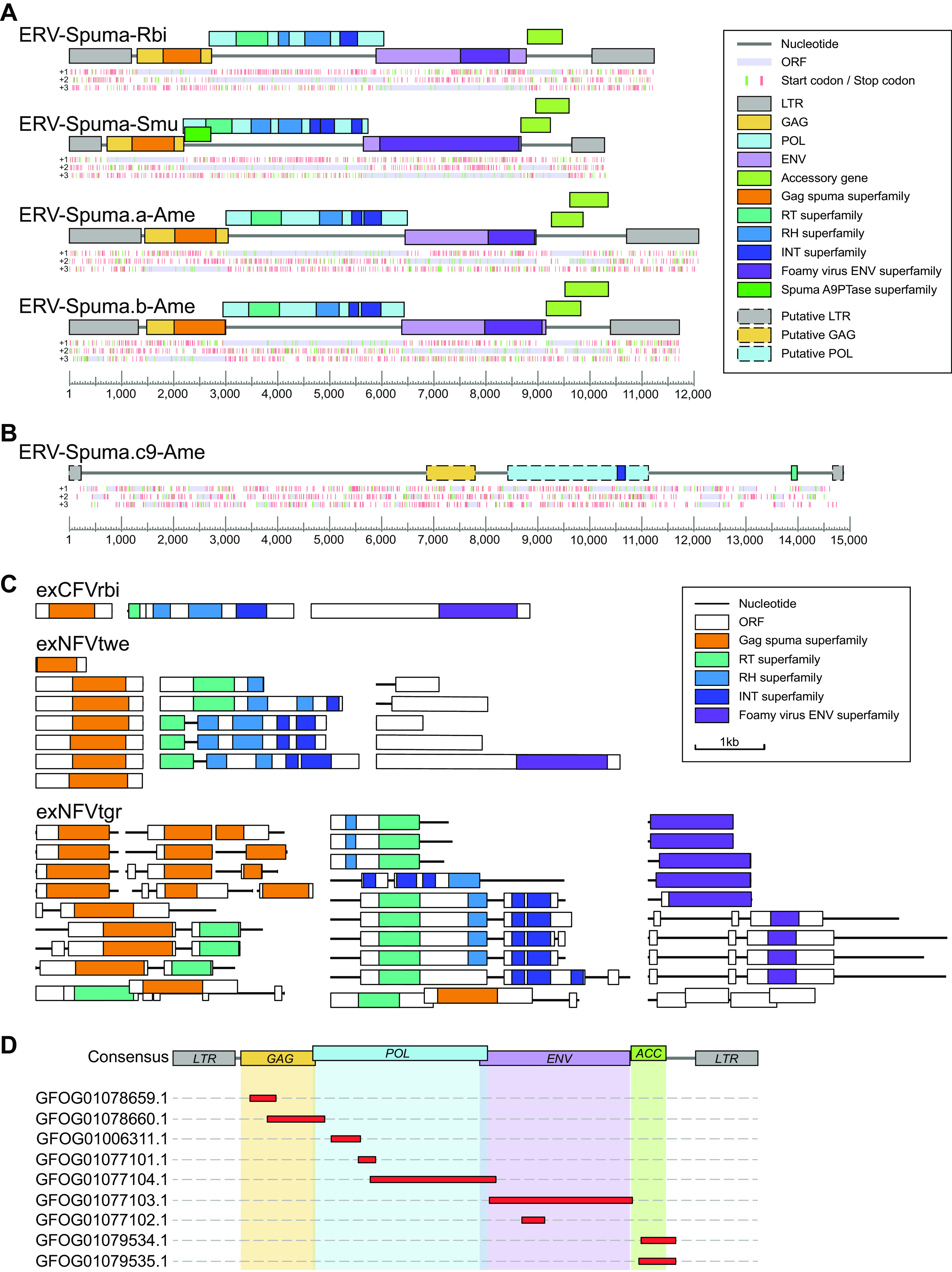
Genomic organization of novel EFVs and exFVs. (A) Consensus genomic organizations of four novel EFVs. The consensus genomes of EFVs are drawn to scale using lines and boxes. The distributions of stop (red) and start (green) codons in three forward frames (+1, +2, +3; from top to bottom) are shown under a genomic schematic diagram for each consensus genome. Putative open reading frames (ORFs) are shown in light purple and were used to determine viral coding regions. The predicted domain or regions that encode conserved proteins are represented by colored boxes. (B) Genomic organization of ERV-Spuma.c9-Ame. The only full-length ERV-Spuma.c9-Ame genome is drawn to scale using lines and boxes. The predicted putative domain or regions that encode conserved proteins are represented by colored dashed boxes. (C) Representative genomic structures of exFVs. The contigs and ORFs of exFVs are drawn to scale using lines and boxes. The predicted domain or regions that encode conserved proteins are represented by colored boxes. (D) Mapping result of the exCFVrbi genome against the consensus ERV-Spuma-Rbi genome. LTR, long terminal repeat; GAG, group-specific antigen gene; POL, polymerase gene; ENV, envelope gene; RT, reverse transcriptase; RH, RNase H.
In fact, a third lineage has also been found in A. mexicanum. However, we identified only one full-length ERV-Spuma.c-Ame among multiple copies (9 copies). Accordingly, the genome structure of this full-length ERV-Spuma.c9-Ame is presented in Fig. 2B, but its predicted pairwise LTR was too short (215 bp) in length and it harbored only a partially conserved domain (GAG, RT, and IN), which made it relatively difficult to align with other EFV lineages.
The genomes of exFVs were then separately annotated, and we found that all lineages of exFVs harbored major genes, including gag, pol, and env, and they were distributed in different contigs in most cases (Fig. 2C). Accordingly, the foamy virus conserved domains were also identified in each lineage, including (i) the gag_spuma superfamily domain (cl26624) (34, 35) and (ii) the Foamy_virus_env superfamily domain (cl04051) (32) and other retroviral domains (RT, RH, and IN). We also found that most copies of exNFVtgr contained premature stop codons or harbored only partial genes, which indicated that they might be ERV-derived RNA. It is worth noting that exCFVrbi-1 (containing gag), exCFVrbi-2 (containing pol), exCFVrbi-3 (containing env), exNFVtwe -1,2,4,5 (containing gag), exNFVtwe-4 (containing pol), and exNFVtwe-5 (containing env) harbored major genes separately without any stop codon or indels, indicating the possible existence of complete functional genomes for exCFVrbi and exNFVtwe.
Since both exCFVrbi and EFV have been found in R. bivittatum, we then further checked the homology of these two viruses (Fig. 2D). By mapping the assembled exFV contigs to a consensus ERV-Spuma-Rbi genome, we found that all exFV contigs showed high (98% to 100%) similarity to it, indicating that they were the same foamy virus. This suggests that all the major genes (gag, pol, env, and accessory genes) of exCFVrbi have been completely expressed without any stop codons or indels, indicating the high probability that exCFVrbi is the first potential exogenous form of FV outside of mammalian hosts. Also worth noting is that such phenomena were observed in koala retrovirus, where active endogenous koala retrovirus can be expressed to form exogenous viral particles (36–38).
Phylogenetic analysis.
To elucidate the relationship between novel exFVs and EFVs identified here with other vertebrate FVs and EFVs, both long POL (>500 amino acids [aa] residues in length) and ENV (>320 aa in length) protein phylogenetic trees were generated to accommodate for their different evolutionary histories (Fig. 3). The phylogenies of pol and env were slightly different, supporting the theory that different genes of FVs indeed had different evolutionary histories (26, 31, 39). However, both phylogenies indicated that the six lineages of EFVs discovered in amphibians could be divided into three clades, giving support to the idea that amphibian FVs had multiple origins. Two lineages of ERV-Spuma-Ame and two exNFVs found in salamanders clustered together with the previously identified NviFLERV, which was consistent with our codivergence theory. Also, ERV-Spuma.a-Ame and ERV-Spuma.b-Ame robustly clustered together with a long branch, reconfirming that they independently resulted from two different foamy virus infections. In addition, in the pol phylogeny, the novel Gymnophiona exCFVrbi and ERV-Spuma-Rbi sequences formed a sister clade closely related to salamander FVs, and they formed a monophyletic group with robust support in the env phylogeny, indicating the different evolutionary histories of genes from Gymnophiona FVs. Notably, frog ERV-Spuma-Smu formed a well-supported monophyletic group that was equally closely related to both avian and mammalian EFVs, indicating that ERVs-Spuma-Smu were possible acquired from cross-species transmission rather than through virus-host divergence.
FIG 3.
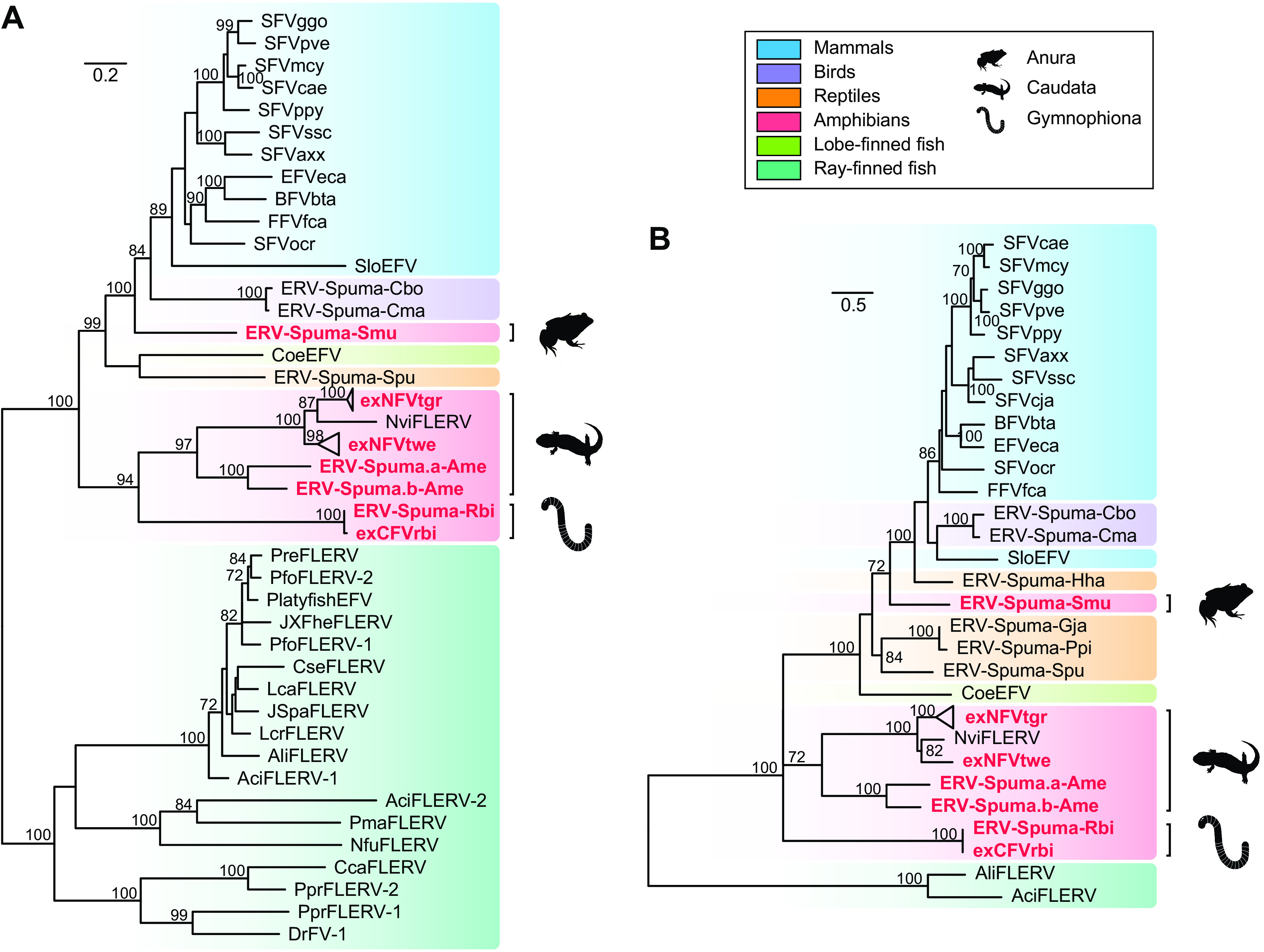
Phylogenetic trees of FVs, exFVs, and EFVs. The trees were inferred using amino acid sequences of the POL (A) and ENV (B) genes. The trees are midpoint rooted for clarity only. The newly identified exFVs and EFVs are labeled in red. The scale bar indicates the number of amino acid changes per site. Bootstrap values of <70% are not shown.
Relationship of foamy viruses with their hosts.
Previous research has provided strong evidence for the codivergence of foamy viruses and their hosts, and some cross-species transmission events have also been observed in EFVs (12, 23, 28–30). Here, to further investigate the deep histories and evolutionary relationships between FVs and their vertebrate hosts, we generated a phylogenetic tree for FVs, EFVs, and exFVs (Fig. 4). This tree showed that most FVs maintained a stable codivergence pattern with their hosts (Fig. 4A). However, the outcomes of different settings in codivergence analysis all indicated that cross-species transmission played an important role in the early evolution of foamy viruses (Fig. 4B), specifically in amphibians, where frog ERV-Spuma-Smu formed a single clade and was equally closely related to both avian and mammalian EFVs rather than other amphibian FVs (Fig. 4A). In addition, exNFVtgr clustered with exNFVtwe, which was also inconsistent with their host phylogeny. We also noted that Gymnophiona ERV-Spuma-Rbi and exCFVrbi formed a well-supported (bootstrap value, 88%) monophyletic clade, giving additional credence to the idea that amphibian FVs had multiple origins and contained several paraphyletic groups.
FIG 4.
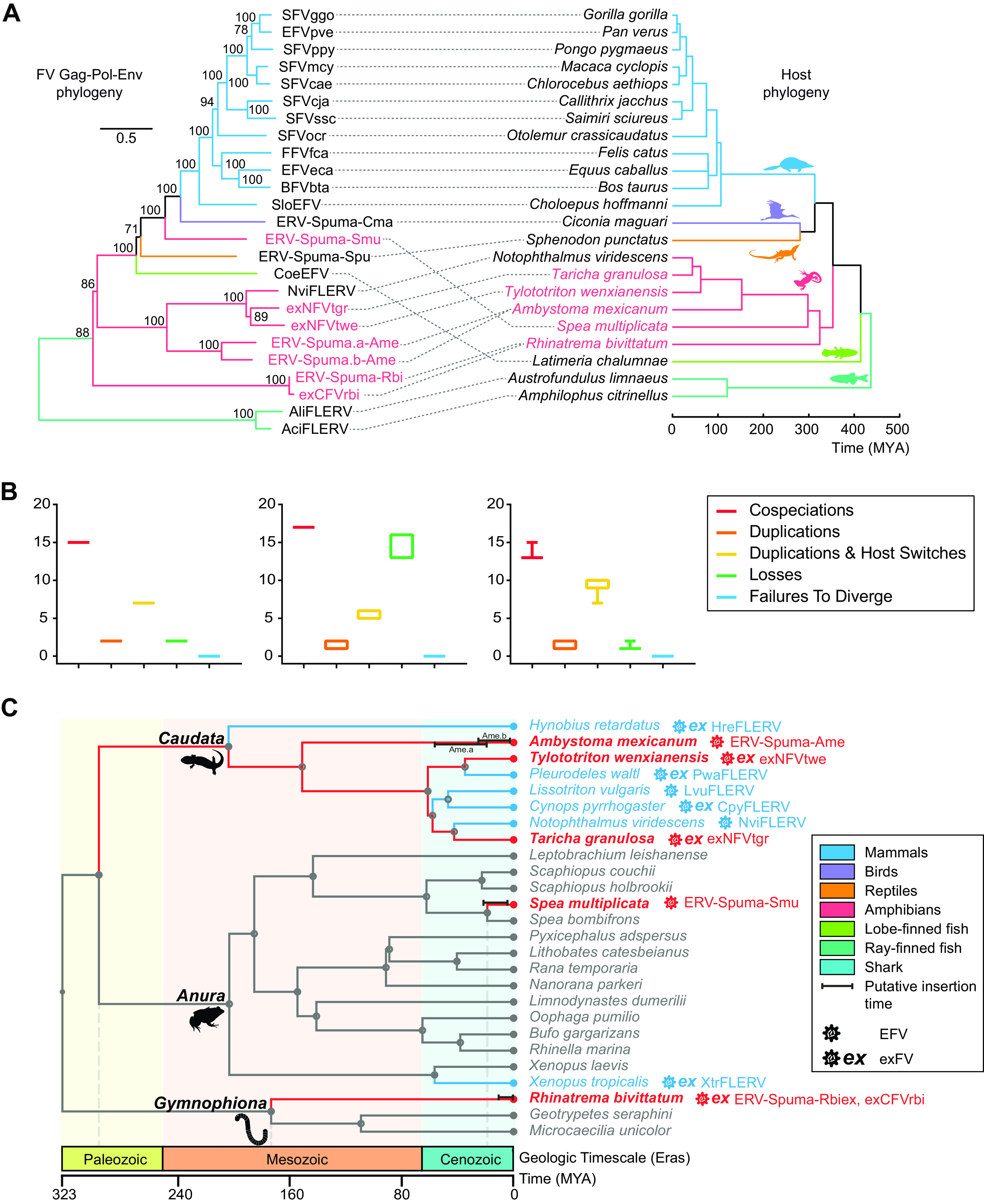
Macroevolutionary history of foamy viruses and their vertebrate hosts. (A) Association between foamy viruses (left) and their hosts (right). Associations between foamy viruses and their hosts are indicated by connecting lines. The newly identified exFVs and EFVs and their hosts are indicated in red. Scale bars indicate the number of amino acid changes per site in the viruses and the host divergence times (million years ago [MYA]). Bootstrap values of <70% are not shown. (B) Reconciliation analysis of foamy virus using Jane. Cospeciations (red), duplications (orange), host-switching (yellow), losses (green), and failure-to-diverge (blue) events are shown. The cost parameters (cospeciation, duplication, duplication and host switch, loss, and failure to diverge, respectively) used in each test were as follows: −1, 0, 0, 0, 0 (left); 0, 1, 2, 1, 1 (middle); and 0, 1, 1, 2, 0 (right). (C) Calibration of foamy virus infection timeline. A time-calibrated phylogeny of screened amphibians was obtained from TimeTree (http://www.timetree.org/). Previous estimated endogenization intervals are indicated by blue lines, while our predictions are indicated by red lines. Closed circles on nodes indicate the existence of taxon rank names. The time range of LTR dating estimation for each foamy virus is indicated by a black bar.
To roughly estimate the insertion time of amphibian EFVs, an LTR divergence-based dating method was used (40). In total, 3 ERVs-Spuma-Smu, 43 ERVs-Spuma.n-Rbi, 5 ERVs-Spuma.a-Ame, and 3 ERVs-Spuma.b-Ame were included in our dating estimation (Table 1 and Fig. 4C). This analysis revealed that ERVs-Spuma-Smu were relatively young, and their insertions could be dated back to 3.7 to 20.9 million years ago (MYA), close to the estimated divergence time of S. multiplicate (18.9 MYA). However, the insertion of ERVs-Spuma-Rbi could be traced back to as recently as 12.2 MYA, which is much more recent than the estimated divergence time for R. bivittatum (135 to 213 MYA). The insertion of ERVs-Spuma.b-Ame could date back to 2.4 to 24.9 MYA. In contrast, another lineage in A. mexicanum, ERVs-Spuma.a-Ame, could be traced back to 18.9 to 56 MYA, which is more ancient. Nevertheless, as LTR dating might severely underestimate ERV ages, these estimates should be treated with caution (41).
TABLE 1.
Dating the EFV insertion based on LTR-LTR divergence
| ERV name | Divergence | Integration time (MYA) | |
|---|---|---|---|
| ERV-Spuma.10-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.11-Rbi | 0.0018 | 0.59 | 0.97 |
| ERV-Spuma.12-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.13-Rbi | 0.0072 | 2.35 | 3.90 |
| ERV-Spuma.14-Rbi | 0.0018 | 0.59 | 0.97 |
| ERV-Spuma.15-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.1-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.21-Rbi | 0.0018 | 0.59 | 0.97 |
| ERV-Spuma.22-Rbi | 0.0027 | 0.88 | 1.46 |
| ERV-Spuma.23-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.24-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.25-Rbi | 0.0188 | 6.14 | 10.17 |
| ERV-Spuma.26-Rbi | 0.0143 | 4.67 | 7.74 |
| ERV-Spuma.27-Rbi | 0.0017 | 0.56 | 0.92 |
| ERV-Spuma.29-Rbi | 0.0022 | 0.72 | 1.19 |
| ERV-Spuma.2-Rbi | 0.0058 | 1.90 | 3.14 |
| ERV-Spuma.30-Rbi | 0.0058 | 1.90 | 3.14 |
| ERV-Spuma.31-Rbi | 0.0016 | 0.52 | 0.87 |
| ERV-Spuma.32-Rbi | 0.0063 | 2.06 | 3.41 |
| ERV-Spuma.34-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.35-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.36-Rbi | 0.0063 | 2.06 | 3.41 |
| ERV-Spuma.37-Rbi | 0.0036 | 1.18 | 1.95 |
| ERV-Spuma.3-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.41-Rbi | 0.0059 | 1.93 | 3.19 |
| ERV-Spuma.42-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.43-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.45-Rbi | 0.0059 | 1.93 | 3.19 |
| ERV-Spuma.46-Rbi | 0.0072 | 2.35 | 3.90 |
| ERV-Spuma.47-Rbi | 0.0018 | 0.59 | 0.97 |
| ERV-Spuma.4-Rbi | 0.0072 | 2.35 | 3.90 |
| ERV-Spuma.52-Rbi | 0.0018 | 0.59 | 0.97 |
| ERV-Spuma.55-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.56-Rbi | 0.0054 | 1.76 | 2.92 |
| ERV-Spuma.57-Rbi | 0.0027 | 0.88 | 1.46 |
| ERV-Spuma.58-Rbi | 0.0116 | 3.79 | 6.28 |
| ERV-Spuma.60-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.63-Rbi | 0.0075 | 2.45 | 4.06 |
| ERV-Spuma.64-Rbi | 0.0225 | 7.35 | 12.18 |
| ERV-Spuma.65-Rbi | 0.0027 | 0.88 | 1.46 |
| ERV-Spuma.67-Rbi | 0 | 0.00 | 0.00 |
| ERV-Spuma.6-Rbi | 0.0009 | 0.29 | 0.49 |
| ERV-Spuma.9-Rbi | 0.0089 | 2.91 | 4.82 |
| ERV-Spuma.4-Smu | 0.0386 | 12.61 | 20.89 |
| ERV-Spuma.5-Smu | 0.016 | 5.23 | 8.66 |
| ERV-Spuma.1-Smu | 0.0113 | 3.69 | 6.11 |
| ERV-Spuma.a10-Ame | 0.0713 | 23.30 | 38.58 |
| ERV-Spuma.a37-Ame | 0.1034 | 33.79 | 55.95 |
| ERV-Spuma.a61-Ame | 0.0758 | 24.77 | 41.02 |
| ERV-Spuma.a7-Ame | 0.0695 | 22.71 | 37.61 |
| ERV-Spuma.a9-Ame | 0.0579 | 18.92 | 31.33 |
| ERV-Spuma.b11-Ame | 0.0281 | 9.18 | 15.21 |
| ERV-Spuma.b4-Ame | 0.046 | 15.03 | 24.89 |
| ERV-Spuma.b6-Ame | 0.0129 | 4.22 | 6.98 |
| ERV-Spuma.b7-Ame | 0.0073 | 2.39 | 3.95 |
Combining our coevolution analyses with our dating estimations allowed us to further calibrate the foamy virus infection timeline in amphibians (Fig. 4C). Including previous research, it seemed that most salamander EFVs followed a codivergent pattern (Fig. 4A). To date, all screened salamander species harbored foamy viruses, five-eighths of which were exFVs, indicating the possibility that circulation of foamy virus in Caudata might date back to the Paleozoic era, and these viruses, together with their hosts, appeared to have an ancient origin. In contrast, frog EFVs and exFVs (ERV-Spuma-Smu and XtrFLERV) appeared to be the result of cross-species transmission events (Fig. 4), and other species in the same genus of their host did not harbor such FVs (26). Thus, they were relatively young compared to other amphibian FVs, which emerged in the Cenozoic era. However, this was the first time we found Gymnophiona EFV and exFV, and other related species did not harbor such FVs. Thus, infection with these viruses could date back to sometime between the Mesozoic and Cenozoic eras. Taking these facts into consideration, it seems that infection with foamy viruses in amphibians began sometime between the Paleozoic and Cenozoic eras.
DISCUSSION
In this study, we report a distinct lineage of FVs in the two-lined caecilian (order Gymnophiona) at both the DNA and RNA level, which added Gymnophiona to the list of currently known hosts of FVs. To date, in amphibians, 8 salamanders, 2 frogs, and 1 caecilian have been confirmed to carry FVs (Fig. 4A) (26). However, the evolutionary histories of FVs in different hosts appear to be extremely different (Fig. 4). We found that all screened salamanders harbored FVs and that they maintained a codivergence pattern with their host within the clade of salamanders (26). It seems that FVs might have been circulating in salamanders starting from the Paleozoic era (283 to 311 MYA). In contrast, only 2 of 15 frogs carried FVs, indicating the rarity of FVs in frogs, and they were largely not a reservoir for FVs. Notably, ERV-Spuma-Smu and the previously identified XtrFLERV in frogs were acquired from cross-class transmission recently in the Cenozoic era, where the former was from a potential unknown land retrovirus and the latter was from a marine retrovirus (26). This reconfirmed that the water-land interface was not a strict barrier to viral transmission and that cross-class transmission occurred occasionally (5). Although only three genomes were available for caecilians, we were able to identify a lineage in the two-lined caecilian. Phylogenetic analysis revealed that ERV-Spuma-Rbi was basal to all amphibians FVs, indicating the ancient origin of caecilian FVs (Fig. 4A). Taking all these data into consideration, it seemed that amphibian FVs had multiple origins and complex evolutionary histories. However, it is possible that this pattern will change with a larger sampling of taxa such that the EFV phylogeny expands (28).
Previous research identified one salamander (Japanese fire belly newt) and four fishes that harbored two EFV lineages (26). Here, we identified a novel salamander (axolotl) which harbored multiple lineages of FVs, and they were characterized at the genomic level. In fact, we found three lineages of FVs in axolotl (Fig. 2A and B), including ERV-Spuma.a-Ame, ERV-Spuma.b-Ame, and ERV-Spuma.c-Ame. However, we identified only one full-length ERV-Spuma-c-Ame among multiple copies (9 copies). Accordingly, the genome structure of this full-length ERV-Spuma.c9-Ame is presented in Fig. 2B, but its predicted pairwise LTR was too short (215 bp) in length, and it harbored only a partially conserved domain (GAG, RT, and IN), which made it relatively difficult to align with other EFV lineages. Thus, we did not include this lineage in our major analyses. But, taking this lineage into consideration, we confirmed that amphibians could be infected with several different FVs. These FVs, however, were more likely to integrate into host genomes at different periods of time (Fig. 4C). By comparing their genomes, we found that their envelope proteins showed limited similarity (46%). As the envelope gene determines the host range and binding receptor for a virus (42–45), this observation suggested that these two lineages might bind to different receptors when they infect their host.
Transcriptome data provided us with additional resources for discovering novel exogenous and endogenous viruses (46). Previous research has identified an enormous number of viruses that have shown limited similarity to well-defined viruses (47), and foamy viral contigs were also discovered in several salamanders and one caecilian (Fig. 4C) (20, 26). In our study, we improved the method by incorporating the method into virome analyses. This led to the discovery of three lineages of exFVs in two salamanders and one caecilian, and these viruses showed limited similarity to known exogenous mammal FVs (26% to 39% conservation of the POL protein). We characterized these viruses in detail and found that all lineages harbored major proteins of retroviruses in different segments (Fig. 2C), which could not be directly assembled at the genome level. Overall, the efficacy of virus discovery using transcriptome analysis depends on sequencing depth and assembly quality. However, the workflow presented here provides a new refinement for discovering novel retroviruses.
Importantly, among all exFVs, we found that exCFVrbi contained no stop codons or any indel in our contigs, which included complete major genes (Fig. 2C and D). As both genome and transcriptome data were available for R. bivittatum, we made a comparison between exCFVrbi and ERV-Spuma-Rbi and found that exCFVrbi could be the result of expression of ERV-Spuma-rbi. Thus, considering the fact that completely expressed ERV could be assembled into viral particles, which has also been reported in koala retrovirus, exCFVrbi might have been the first potential exogenous foamy virus in amphibians. Although we could not directly examine such viral particles, this research still supports the assumption that exogenous foamy viruses could exist in species other than mammals.
Recently, another study identified the exaptation of foamy virus in gecko (30). This indicated that foamy virus-derived genes could also be coopted. In our study, we annotated exFV contigs in detail and found that 7 contigs contained gag and 3 contigs contained env in NFVtwe with no stop codon, all of which had the capacity to be translated into proteins. In other words, they had potential as retrovirus-derived candidates for cooption identification. Also worth noting was that most exFVtgr-related sequences contained stop codons and their open reading frames (ORFs) were incomplete, which might indicate that they were likely to be ERV-derived long noncoding RNAs (lncRNAs) rather than exogenous retroviruses. Moreover, they might also function as lncRNAs to participate in genomic regulatory processes (48). However, this speculation should be verified by further study.
In conclusion, by integrating genomic and transcriptomic data and performing a phylogenomic analysis, we discovered six lineages of FVs with different evolutionary histories, doubling the known set of foamy viruses in amphibians. We also confirmed that amphibians could be infected by multiple FVs at different periods of time. Interestingly, we identified the first potential expressed form of FV (exCFVrbi) in caecilians, which could be the first exogenous form of foamy virus existing in species other than mammals. This research demonstrates repeated infections and multiple origins for amphibian FVs and reveals a complex macroevolution of foamy viruses with their hosts.
MATERIALS AND METHODS
Genome and transcriptome screening and EFV/exFV identification.
As most EFVs showed limited similarity to exogenous FVs and previously found EFVs, a stepwise method was used for amphibian EFV mining. First, all 19 amphibian genome assemblies (GCA_000004195.4, GCA_001663975.1, GCA_002915635.3, GCA_901001135.2, GCA_014858855.1, GCA_009364415.1, GCA_900303285.1, GCA_902459505.2, GCA_002284835.2, GCA_901765095.2, GCA_000935625.1, GCA_009667805.1, GCA_009364455.1, GCA_004786255.1, GCA_009802015.1, GCA_009364435.1, GCA_009364475.1, GCA_009801035.1, and GCA_011038615.1), excluding genomes which had previously been found for any EFV in GenBank as of November 2020, were screened for foamy-like viruses using tblastn (49), and conserved Pol proteins of foamy viruses, including EFVs, were used as probes (see Table S2 in the supplemental material). A 25% sequence identity over a 40% region with an E value set to 1E−5 was used to filter significant hits. Second, potential foamy-like elements were included in phylogenetic analysis. Hits that clustered with EFVs and FVs were considered EFVs. The flanking sequences of these EFVs were then extended to identify viral pairwise LTRs using BLASTN (49), LTR_Finder, and LTR_harvest. In total, we were able to identify 5 full-length (containing pairwise LTRs) EFVs in S. multiplicate, 46 in R. bivittatum, and 26 in A. mexicanum. These full-length EFVs were then used as a query to search for EFV copies using blastn. Sequences longer than 4 kb with 85% identity were regarded as copies of each EFV lineage (Table S1). In accordance with the nomenclature proposed for ERVs, EFVs found in S. multiplicate, R. bivittatum, and A. mexicanum were named ERVs-Spuma.n-Smu, ERVs-Spuma.n-Rbi, and ERVs-Spuma.n-Ame, respectively. As there were three lineages of EFVs in A. mexicanum, they were separately designated as ERVs-Spuma.an-Ame, ERVs-Spuma.bn-Ame, and ERVs-Spuma.cn-Ame, respectively.
To identify potential exFVs, all 61 transcriptome sequencing assemblies (TSA) (GenBank accession no. GAEG00000000, GAEI00000000, GAQK00000000, GEBK00000000, GEGF00000000, GEGG00000000, GEGH00000000, GEGI00000000, GEGJ00000000, GEGK00000000, GBET00000000, GDRL00000000, GEGL00000000, GFBS00000000, HADQ00000000, HADR00000000, HADS00000000, HADT00000000, HADU00000000, HADV00000000, GECV00000000, GESS00000000, GFBM000000000, GFLD00000000, GFLI00000000, GFLJ00000000, GFLO00000000, GFMT00000000, GFMY00000000, GFNJ00000000, GENE00000000, GFOD00000000, GFOE00000000, GFOF00000000, GFOG00000000, GFOH00000000, GFZP00000000, GGLB00000000, GGNS00000000, GGTL00000000, GDDO00000000, GGUQ00000000, GGUR00000000, GGUS00000000, GHBH00000000, GHBO00000000, GHDZ00000000, GHKF00000000, GHME00000000, HAML00000000, GHCG00000000, GHWT00000000, GICS00000000, GIKK00000000, GIKS00000000, GINY000000000, GIPO00000000, GISC00000000, ICLD00000000, ICPN00000000, and GHMZ00000000), excluding assemblies which had previously been found for any FV, were screened using tblastn, and all three major proteins of amphibian EFVs, including the newly identified ERVs-Spuma-Smu, ERVs-Spuma-Rbi, and ERVs-Spuma-Ame, were used as probes. A 25% sequence identity over a 40% region with an E value set to 1E−5 was used to filter significant hits. Then, the calling hit contigs were included in the phylogenetic analysis. The viral contigs within a clade of FVs were considered. In total, three exFVs were found, and exFVs in T. granulosa, T. wenxianensis, and R. bivittatum were named exNFVtgr.n, exNFVtwe.n, and exCFVrbi.n, respectively.
Consensus genome construction and genome annotation.
EFVs longer than 5 kb in each EFV lineage were aligned using MAFFT 7.222 (50) and then used to construct consensus sequences for each EFV lineage. The distributions of open reading frames (ORFs) in copies of EFV and exFV contigs were determined using ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) at NCBI and confirmed by BLASTP (49). Conserved domains for each sequence were found by using CD-Search against the Conserved Domain Database (CDD) (https://www.ncbi.nlm.nih.gov/cdd/) (51).
To construct putative full-length coding regions for exFVs, we used the method described below (52, 53). If a contig mapped to the same coding gene (e.g., the pol gene) with high similarity (>85%), then these contigs were used to construct consensus sequences for each gene. Otherwise, to decide which contig was used to construct genomes for exFVs, we (i) compared the phylogenetic positions of related viral proteins, (ii) compared the similarities with related proteins from a reference foamy virus, and (iii) checked the completeness of our conserved domain. Then, the selected contigs were used to construct putative genomes for exFVs. However, the putative genomes for exFVs were used only in concatenated Gag-Pol-Env phylogeny.
Molecular dating.
ERV integration time can be approximately estimated using the relationship T = (D/R)/2, in which T is the integration time (in million years [MY]), D is the number of nucleotide differences per site between a set of pairwise LTRs, and R is the genomic substitution rate (in nucleotide substitutions per site per year) (40, 54). We used a previously estimated neutral nucleotide substitution rate for frogs (9.24 × 10−10 to 1.53 × 10−9 nucleotide substitutions per site per year) (55) to estimate the evolution of amphibians. LTRs less than 300 bp in length were excluded from this analysis (29, 56). In total, 3 ERVs-Spuma -Smu, 43 ERVs-Spuma.n-Rbi, 5 ERVs-Spuma.a-Ame, and 3 ERVs-Spuma.b-Ame containing a pairwise intact LTR were used to estimate integration time in this manner (Table 1).
Phylogenetic analysis.
To investigate the evolutionary relationship between FVs, exFVs, and EFVs, protein sequences for RT, POL, ENV, and concatenated Gag-Pol-Env were aligned using MAFFT 7.222 (50) (Data Sets S2 to S5). The regions in the alignment that aligned poorly were removed using TrimAL (57) and confirmed manually in MEGA X (58). A sequence was excluded if its length was less than 75% of the alignments. The best-fit models (RT, VT+G; POL, LG+F+I+G; ENV, LG+F+I+G; and Gag-Pol-Env, LG+F+I+G) were selected using ProtTest (49), and the phylogenetic trees for these protein sequences were inferred using the maximum likelihood (ML) method in PhyML (59) or IQ-Tree (60), incorporating 100 bootstrap replicates to assess node robustness. Phylogenetic trees were viewed and annotated in FigTree v1.4.3 (https://github.com/rambaut/figtree/).
Coevolution analysis.
To assess the macroevolution of foamy viruses and their hosts, event-based Jane 4 (61) was used. We set the cost parameters (cospeciation, duplication, duplication and host switch, loss, and failure to diverge, respectively), based on previous research, as follows: (i) −1, 0, 0, 0, 0 (26); (ii) 0, 1, 2, 1, 1; and (iii) 0, 1, 1, 2, 0 (61). Then, statistical analyses were performed using Jane to assess robustness by generating random parasite trees with a sample size of 500.
Data availability.
All the data needed to generate the conclusions in the article are present in the article itself and the supplementary data.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (grant no. 31970176) and the CAS Pioneer Hundred Talents Program.
We declare no conflicts of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Jie Cui, Email: jcui@ips.ac.cn.
Frank Kirchhoff, Ulm University Medical Center.
REFERENCES
- 1.Hahn BH, Shaw GM, De Cock KM, Sharp PM. 2000. AIDS as a zoonosis: scientific and public health implications. Science 287:607–614. 10.1126/science.287.5453.607. [DOI] [PubMed] [Google Scholar]
- 2.Stoye JP. 2012. Studies of endogenous retroviruses reveal a continuing evolutionary saga. Nat Rev Microbiol 10:395–406. 10.1038/nrmicro2783. [DOI] [PubMed] [Google Scholar]
- 3.Johnson WE. 2019. Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol 17:355–370. 10.1038/s41579-019-0189-2. [DOI] [PubMed] [Google Scholar]
- 4.Hayward A, Grabherr M, Jern P. 2013. Broad-scale phylogenomics provides insights into retrovirus-host evolution. Proc Natl Acad Sci U S A 110:20146–20151. 10.1073/pnas.1315419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu X, Zhao H, Gong Z, Han GZ. 2018. Endogenous retroviruses of non-avian/mammalian vertebrates illuminate diversity and deep history of retroviruses. PLoS Pathog 14:e1007072. 10.1371/journal.ppat.1007072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayward A, Cornwallis CK, Jern P. 2015. Pan-vertebrate comparative genomics unmasks retrovirus macroevolution. Proc Natl Acad Sci U S A 112:464–469. 10.1073/pnas.1414980112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen M, Cui J. 2019. Discovery of endogenous retroviruses with mammalian envelopes in avian genomes uncovers long-term bird-mammal interaction. Virology 530:27–31. 10.1016/j.virol.2019.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Henzy JE, Gifford RJ, Johnson WE, Coffin JM. 2014. A novel recombinant retrovirus in the genomes of modern birds combines features of avian and mammalian retroviruses. J Virol 88:2398–2405. 10.1128/JVI.02863-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui J, Zhao W, Huang Z, Jarvis ED, Gilbert MT, Walker PJ, Holmes EC, Zhang G. 2014. Low frequency of paleoviral infiltration across the avian phylogeny. Genome Biol 15:539. 10.1186/s13059-014-0539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johnson WE. 2015. Endogenous retroviruses in the genomics era. Annu Rev Virol 2:135–159. 10.1146/annurev-virology-100114-054945. [DOI] [PubMed] [Google Scholar]
- 11.Chen M, Guo X, Zhang L. 2020. Unexpected discovery and expression of amphibian class II endogenous retroviruses. J Virol 95:e01806-20. 10.1128/JVI.01806-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Switzer WM, Salemi M, Shanmugam V, Gao F, Cong ME, Kuiken C, Bhullar V, Beer BE, Vallet D, Gautier-Hion A, Tooze Z, Villinger F, Holmes EC, Heneine W. 2005. Ancient co-speciation of simian foamy viruses and primates. Nature 434:376–380. 10.1038/nature03341. [DOI] [PubMed] [Google Scholar]
- 13.Lee GE, Mauro E, Parissi V, Shin CG, Lesbats P. 2019. Structural insights on retroviral DNA integration: learning from foamy viruses. Viruses 11:770. 10.3390/v11090770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rethwilm A, Bodem J. 2013. Evolution of foamy viruses: the most ancient of all retroviruses. Viruses 5:2349–2374. 10.3390/v5102349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan AS, Bodem J, Buseyne F, Gessain A, Johnson W, Kuhn JH, Kuzmak J, Lindemann D, Linial ML, Löchelt M, Materniak-Kornas M, Soares MA, Switzer WM. 2018. Spumaretroviruses: updated taxonomy and nomenclature. Virology 516:158–164. 10.1016/j.virol.2017.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muniz CP, Jia H, Shankar A, Troncoso LL, Augusto AM, Farias E, Pissinatti A, Fedullo LP, Santos AF, Soares MA, Switzer WM. 2015. An expanded search for simian foamy viruses (SFV) in Brazilian New World primates identifies novel SFV lineages and host age-related infections. Retrovirology 12:94. 10.1186/s12977-015-0217-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Malmquist WA, Van der Maaten MJ, Boothe AD. 1969. Isolation, immunodiffusion, immunofluorescence, and electron microscopy of a syncytial virus of lymphosarcomatous and apparently normal cattle. Cancer Res 29:188–200. [PubMed] [Google Scholar]
- 18.Renshaw RW, Casey JW. 1994. Transcriptional mapping of the 3' end of the bovine syncytial virus genome. J Virol 68:1021–1028. 10.1128/JVI.68.2.1021-1028.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tobaly-Tapiero J, Bittoun P, Neves M, Guillemin MC, Lecellier CH, Puvion-Dutilleul F, Gicquel B, Zientara S, Giron ML, de Thé H, Saïb A. 2000. Isolation and characterization of an equine foamy virus. J Virol 74:4064–4073. 10.1128/JVI.74.9.4064-4073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Z, Ren X, Yang L, Hu Y, Yang J, He G, Zhang J, Dong J, Sun L, Du J, Liu L, Xue Y, Wang J, Yang F, Zhang S, Jin Q. 2012. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J Virol 86:10999–11012. 10.1128/JVI.01394-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Riggs JL, Oshirls Taylor DO, Lennette EH. 1969. Syncytium-forming agent isolated from domestic cats. Nature 222:1190–1191. 10.1038/2221190a0. [DOI] [PubMed] [Google Scholar]
- 22.Katzourakis A, Gifford RJ, Tristem M, Gilbert MT, Pybus OG. 2009. Macroevolution of complex retroviruses. Science 325:1512. 10.1126/science.1174149. [DOI] [PubMed] [Google Scholar]
- 23.Katzourakis A, Aiewsakun P, Jia H, Wolfe ND, LeBreton M, Yoder AD, Switzer WM. 2014. Discovery of prosimian and afrotherian foamy viruses and potential cross species transmissions amidst stable and ancient mammalian co-evolution. Retrovirology 11:61. 10.1186/1742-4690-11-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han GZ, Worobey M. 2012. An endogenous foamy virus in the aye-aye (Daubentonia madagascariensis). J Virol 86:7696–7698. 10.1128/JVI.00650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han GZ, Worobey M. 2014. Endogenous viral sequences from the Cape golden mole (Chrysochloris asiatica) reveal the presence of foamy viruses in all major placental mammal clades. PLoS One 9:e97931. 10.1371/journal.pone.0097931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aiewsakun P, Katzourakis A. 2017. Marine origin of retroviruses in the early Palaeozoic era. Nat Commun 8:13954. 10.1038/ncomms13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ruboyianes R, Worobey M. 2016. Foamy-like endogenous retroviruses are extensive and abundant in teleosts. Virus Evol 2:vew032. 10.1093/ve/vew032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Y, Wei X, Zhang G, Holmes EC, Cui J. 2019. Identification and evolution of avian endogenous foamy viruses. Virus Evol 5:vez049. 10.1093/ve/vez049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wei X, Chen Y, Duan G, Holmes EC, Cui J. 2019. A reptilian endogenous foamy virus sheds light on the early evolution of retroviruses. Virus Evol 5:vez001. 10.1093/ve/vez001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aiewsakun P, Simmonds P, Katzourakis A. 2019. The first co-opted endogenous foamy viruses and the evolutionary history of reptilian foamy viruses. Viruses 11:641. 10.3390/v11070641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aiewsakun P. 2020. Avian and serpentine endogenous foamy viruses, and new insights into the macroevolutionary history of foamy viruses. Virus Evol 6:vez057. 10.1093/ve/vez057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han GZ, Worobey M. 2012. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathog 8:e1002790. 10.1371/journal.ppat.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gifford RJ, Blomberg J, Coffin JM, Fan H, Heidmann T, Mayer J, Stoye J, Tristem M, Johnson WE. 2018. Nomenclature for endogenous retrovirus (ERV) loci. Retrovirology 15:59. 10.1186/s12977-018-0442-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patton GS, Morris SA, Chung W, Bieniasz PD, McClure MO. 2005. Identification of domains in gag important for prototypic foamy virus egress. J Virol 79:6392–6399. 10.1128/JVI.79.10.6392-6399.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler I, Bodem J, Haas L, Zemba M, Delius H, Flower R, Flugel RM, Lochelt M. 1997. Characterization of the genome of feline foamy virus and its proteins shows distinct features different from those of primate spumaviruses. J Virol 71:6727–6741. 10.1128/JVI.71.9.6727-6741.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tarlinton RE, Meers J, Young PR. 2006. Retroviral invasion of the koala genome. Nature 442:79–81. 10.1038/nature04841. [DOI] [PubMed] [Google Scholar]
- 37.Herniou E, Martin J, Miller K, Cook J, Wilkinson M, Tristem M. 1998. Retroviral diversity and distribution in vertebrates. J Virol 72:5955–5966. 10.1128/JVI.72.7.5955-5966.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tarlinton R, Meers J, Hanger J, Young P. 2005. Real-time reverse transcriptase PCR for the endogenous koala retrovirus reveals an association between plasma viral load and neoplastic disease in koalas. J Gen Virol 86:783–787. 10.1099/vir.0.80547-0. [DOI] [PubMed] [Google Scholar]
- 39.Liu W, Worobey M, Li Y, Keele BF, Bibollet-Ruche F, Guo Y, Goepfert PA, Santiago ML, Ndjango JB, Neel C, Clifford SL, Sanz C, Kamenya S, Wilson ML, Pusey AE, Gross-Camp N, Boesch C, Smith V, Zamma K, Huffman MA, Mitani JC, Watts DP, Peeters M, Shaw GM, Switzer WM, Sharp PM, Hahn BH. 2008. Molecular ecology and natural history of simian foamy virus infection in wild-living chimpanzees. PLoS Pathog 4:e1000097. 10.1371/journal.ppat.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dangel AW, Baker BJ, Mendoza AR, Yu CY. 1995. Complement component C4 gene intron 9 as a phylogenetic marker for primates: long terminal repeats of the endogenous retrovirus ERV-K(C4) are a molecular clock of evolution. Immunogenetics 42:41–52. 10.1007/BF00164986. [DOI] [PubMed] [Google Scholar]
- 41.Kijima TE, Innan H. 2010. On the estimation of the insertion time of LTR retrotransposable elements. Mol Biol Evol 27:896–904. 10.1093/molbev/msp295. [DOI] [PubMed] [Google Scholar]
- 42.Rey MA, Prasad R, Tailor CS. 2008. The C domain in the surface envelope glycoprotein of subgroup C feline leukemia virus is a second receptor-binding domain. Virology 370:273–284. 10.1016/j.virol.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 43.Battini JL, Heard JM, Danos O. 1992. Receptor choice determinants in the envelope glycoproteins of amphotropic, xenotropic, and polytropic murine leukemia viruses. J Virol 66:1468–1475. 10.1128/JVI.66.3.1468-1475.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Konstantoulas CJ, Lamp B, Rumenapf TH, Indik S. 2015. Single amino acid substitution (G42E) in the receptor binding domain of mouse mammary tumour virus envelope protein facilitates infection of non-murine cells in a transferrin receptor 1-independent manner. Retrovirology 12:43. 10.1186/s12977-015-0168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen Y, Chen M, Duan X, Cui J. 2020. Ancient origin and complex evolution of porcine endogenous retroviruses. Biosaf Health 2:142–151. 10.1016/j.bsheal.2020.03.003. [DOI] [Google Scholar]
- 46.Zhang YZ, Chen YM, Wang W, Qin XC, Holmes EC. 2019. Expanding the RNA virosphere by unbiased metagenomics. Annu Rev Virol 6:119–139. 10.1146/annurev-virology-092818-015851. [DOI] [PubMed] [Google Scholar]
- 47.Zhang YZ, Shi M, Holmes EC. 2018. Using Metagenomics to Characterize an Expanding Virosphere. Cell 172:1168–1172. 10.1016/j.cell.2018.02.043. [DOI] [PubMed] [Google Scholar]
- 48.Wilson KD, Ameen M, Guo H, Abilez OJ, Tian L, Mumbach MR, Diecke S, Qin X, Liu Y, Yang H, Ma N, Gaddam S, Cunningham NJ, Gu M, Neofytou E, Prado M, Hildebrandt TB, Karakikes I, Chang HY, Wu JC. 2020. Endogenous retrovirus-derived lncRNA BANCR promotes cardiomyocyte migration in humans and non-human primates. Dev Cell 54:694–709.e9. 10.1016/j.devcel.2020.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 50.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, Thanki N, Yamashita RA, Yang M, Zhang D, Zheng C, Lanczycki CJ, Marchler-Bauer A. 2020. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res 48:D265–D268. 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, Qin XC, Li J, Cao JP, Eden JS, Buchmann J, Wang W, Xu J, Holmes EC, Zhang YZ. 2016. Redefining the invertebrate RNA virosphere. Nature 540:539–543. 10.1038/nature20167. [DOI] [PubMed] [Google Scholar]
- 53.Shi M, Lin XD, Chen X, Tian JH, Chen LJ, Li K, Wang W, Eden JS, Shen JJ, Liu L, Holmes EC, Zhang YZ. 2018. The evolutionary history of vertebrate RNA viruses. Nature 556:197–202. 10.1038/s41586-018-0012-7. [DOI] [PubMed] [Google Scholar]
- 54.Johnson WE, Coffin JM. 1999. Constructing primate phylogenies from ancient retrovirus sequences. Proc Natl Acad Sci U S A 96:10254–10260. 10.1073/pnas.96.18.10254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crawford AJ. 2003. Relative rates of nucleotide substitution in frogs. J Mol Evol 57:636–641. 10.1007/s00239-003-2513-7. [DOI] [PubMed] [Google Scholar]
- 56.Abascal F, Zardoya R, Posada D. 2005. ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105. 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 57.Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973. 10.1093/bioinformatics/btp348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 60.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32:268–274. 10.1093/molbev/msu300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conow C, Fielder D, Ovadia Y, Libeskind-Hadas R. 2010. Jane: a new tool for the cophylogeny reconstruction problem. Algorithms Mol Biol 5:16. 10.1186/1748-7188-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1, Tables S1 to S2, and Data Sets S1 to S5. Download JVI.00484-21-s0001.pdf, PDF file, 2.33 MB (2.3MB, pdf)
Data Availability Statement
All the data needed to generate the conclusions in the article are present in the article itself and the supplementary data.