

ABSTRACT
Mouse mammary tumor virus (MMTV) encodes a Rem precursor protein that specifies both regulatory and accessory functions. Rem is cleaved at the endoplasmic reticulum (ER) membrane into a functional N-terminal signal peptide (SP) and the C terminus (Rem-CT). Rem-CT lacks a membrane-spanning domain and a known ER retention signal, and yet it was not detectably secreted into cell supernatants. Inhibition of intracellular trafficking by the drug brefeldin A (BFA), which interferes with the ER-to-Golgi secretory pathway, resulted in dramatically reduced intracellular Rem-CT levels that were not rescued by proteasomal or lysosomal inhibitors. A Rem mutant lacking glycosylation was cleaved into SP and Rem-CT but was insensitive to BFA, suggesting that unglycosylated Rem-CT does not reach this BFA-dependent compartment. Treatment with endoglycosidase H indicated that Rem-CT does not traffic through the Golgi apparatus. Analysis of wild-type Rem-CT and its glycosylation mutant by confocal microscopy revealed that both were primarily localized to the ER lumen. A small fraction of wild-type Rem-CT, but not the unglycosylated mutant, was colocalized with Rab5-positive (Rab5+) early endosomes. The expression of a dominant-negative (DN) form of ADP ribosylation factor 1 (Arf1) (containing a mutation of threonine to asparagine at position 31 [T31N]) mimicked the effects of BFA by reducing Rem-CT levels and increased Rem-CT association with early and late endosomes. Inhibition of the AAA ATPase p97/VCP rescued Rem-CT in the presence of BFA or DN Arf1 and prevented localization to Rab5+ endosomes. Thus, Rem-CT uses an unconventional p97-mediated scheme for trafficking to early endosomes.
IMPORTANCE Mouse mammary tumor virus is a complex retrovirus that encodes a regulatory/accessory protein, Rem. Rem is a precursor protein that is processed at the endoplasmic reticulum (ER) membrane by signal peptidase. The N-terminal SP uses the p97/VCP ATPase to elude ER-associated degradation to traffic to the nucleus and serve a human immunodeficiency virus Rev-like function. In contrast, the function of the C-terminal glycosylated cleavage product (Rem-CT) is unknown. Since localization is critical for protein function, we used mutants, inhibitors, and confocal microscopy to localize Rem-CT. Surprisingly, Rem-CT, which lacks a transmembrane domain or an ER retention signal, was detected primarily within the ER and required glycosylation and the p97 ATPase for early endosome trafficking without passage through the Golgi apparatus. Thus, Rem-CT uses a novel intracellular trafficking pathway, potentially impacting host antiviral immunity.
KEYWORDS: Rem, mouse mammary tumor virus, endoplasmic reticulum, endosomal trafficking, brefeldin A sensitivity, p97/VCP, Arf1
INTRODUCTION
Mouse mammary tumor virus (MMTV) is a murine complex retrovirus with organizational features similar to those of human-pathogenic retroviruses, such as human immunodeficiency virus type 1 (HIV-1) (1–4). Unlike HIV-1, which is difficult to study in its native host, MMTV provides a suitable animal model to elucidate viral interactions within the context of a natural host immune system. Like human complex retroviruses, MMTV also encodes accessory and regulatory genes (4–6). Our previous studies have shown that MMTV specifies both regulatory and accessory proteins from a precursor protein, Rem (2–4). Rem is generated from the doubly spliced MMTV mRNA, which is translated on the endoplasmic reticulum (ER) membrane and then cleaved by signal peptidase into a 98-amino-acid N-terminal signal peptide (SP) and a 203-amino-acid C-terminal glycosylated protein (Rem-CT). Since Rem is translated in the same reading frame and from the same start codon as the MMTV envelope protein (Env), SP is synthesized from both rem and env mRNAs, although env mRNA is much more abundant (7). After Rem or Env cleavage, MMTV-encoded SP is retrotranslocated from the ER to the cytosol in a p97/VCP ATPase-dependent manner (7, 8). SP then traffics to the nucleus to bind to unspliced MMTV RNA for nuclear export and subsequent steps of virus replication (7–9). Thus, SP has a function similar to that of the HIV-1-encoded Rev (4). Our recent experiments indicate that Rem and/or Rem-CT act as accessory factors in vivo to counteract Apobec-mediated restriction of MMTV replication, particularly to antagonize the mutagenic activity of activation-induced cytidine deaminase (AID) (10). Since protein localization and function are tightly linked, the exact localization and trafficking of Rem-CT within host cells will contribute toward understanding its activity.
Proteins synthesized in the ER usually traffic from the ER to the ER-Golgi intermediate compartment (ERGIC) and then the cis-Golgi network in coatomer II (COPII) vesicles. Proteins are further transported through the medial- and trans-Golgi or secreted depending on the presence of transmembrane domains, where they may be processed by cleavage or modification of the simple sugars added in the ER (11). Integral membrane proteins require a sufficiently long transmembrane sequence, which forms an α-helix to span the lipid bilayer (12). Proteins with or without transmembrane domains translated on the ER membrane may be retained within intracellular membrane compartments if they bear retrograde transport sequences, such as a C-terminal KDEL/KKXX for ER retention or, alternatively, an LPYS or KXE/D motif for localization within the Golgi compartment (11, 13).
ADP-ribosylation factor 1 (Arf1) is a small GTP-binding protein that mediates the retrograde transport of cargo from the cis-Golgi or ERGIC to the ER (14). The activated GTP-bound Arf1 recruits the COPI coatomer complexes to the Golgi/ERGIC membrane to form retrograde transport vesicles (15–17). Arf1 recruits the clathrin adaptor proteins (APs) to form clathrin-coated vesicles on the trans-Golgi for trafficking between this compartment and endosomes (18–20). Arf1 also functions in a clathrin-independent pathway for endocytosis of the morphogen Wingless during Drosophila wing development (21, 22), during chemotaxis of migrating neutrophils (23), and in delivery of HIV-1 viral particles from dendritic cells to CD4+ T cells through virological synapses (24). Thus, Arf1 plays different roles to mediate viral protein trafficking, as well as immune responses.
As noted above, p97/VCP plays an integral role in the trafficking of the Rem precursor protein, as well as cleaved SP (8, 9). Recent studies indicate that the p97 ATPase is necessary for ER-associated protein quality control and interacts with other membranous organelles associated with mitochondria-associated degradation and Golgi apparatus-related degradation (MAD and GARD, respectively) (25). During these degradation processes, p97 extracts membrane-associated polyubiquitinated proteins recognized by multiple adapter proteins for delivery to the proteasome. Furthermore, the p97 ATPase provides a chaperone-like activity for the assembly and disassembly of multiprotein complexes, both in the nucleus and the cytosol (26). This ATPase is required for coronavirus escape from the early endosomes during viral entry (27) and the relocation of antigen cross-presentation machinery from the ER to the endosomal compartment (28). ER-to-endosome membrane contacts are needed for regulation of receptor tyrosine kinases and bidirectional cholesterol transfer, as well as endosomal trafficking, positioning, and fission (29), but an exact role for p97/VCP has not been described.
Here, we have examined the trafficking of the MMTV Env-related protein, Rem-CT. Our results indicated that Rem-CT is a glycoprotein lacking a transmembrane domain and a canonical ER retention signal and, yet, that it is localized primarily within the ER. In addition, Rem-CT has a unique trafficking scheme from the ER to early endosomes without passage through the Golgi or detectable secretion. We observed that Rem-CT intracellular levels were reduced both by brefeldin A (BFA), a fungal metabolite that disrupts protein secretion and collapses the Golgi compartment into the ER (30), and by dominant-negative (DN) Arf1. Complete rescue of Rem-CT levels after BFA treatment was achieved by the expression of a DN p97/VCP ATPase or loss of N-linked glycosylation, and p97/VCP inhibitors blocked the trafficking of Rem-CT to early endosomes. Our data reveal the first virus or host protein that employs such a pathway, providing new insights into intracellular protein trafficking.
RESULTS
Rem-CT is not a secreted protein.
Rem precursor is cleaved by signal peptidase in the ER to generate an N-terminal SP and Rem-CT (Fig. 1A) (7–9). Since Rem-CT is glycosylated and lacks a transmembrane domain or any known canonical KDEL or KKXX sequence at the C terminus for ER retention (Fig. 1A) (31, 32), we predicted that Rem-CT would be secreted into the extracellular medium. To test this idea initially, we transfected 293T cells with a plasmid expressing Rem with an N-terminal green fluorescent protein (GFP) tag and a C-terminal T7 tag (GFP-Rem-T7). This approach allowed us to measure SP and Rem-CT levels independently after Rem cleavage by signal peptidase (4). As a positive control, we also transfected cells with an expression plasmid for a secreted form of embryonic alkaline phosphatase (SEAP), which was tagged at the C terminus with T7 (SEAP-T7). SEAP is known to be synthesized in the ER and secreted through the classical Golgi pathway (33). As expected, both Rem-CT-T7 and SEAP-T7 were detected in cell lysates by Western blotting with T7-specific antibody (Fig. 1B, lanes 1 and 3). Rem precursor also was detectable with T7 antibody, and cleaved SP was observed using GFP-specific antibody (Fig. 1B, lane 1). To determine if both SEAP and Rem-CT were secreted, supernatants from the transfected cells were collected. Proteins with a mass of >15 kDa, which included T7-tagged Rem-CT and SEAP, were concentrated through a centrifugal filtration unit prior to Western blotting using T7-specific antibody. The secretory protein SEAP was easily detectable in cell supernatants, whereas Rem-CT was not (Fig. 1B, compare lanes 5 and 7). These results indicated that Rem-CT is not a secreted protein.
FIG 1.
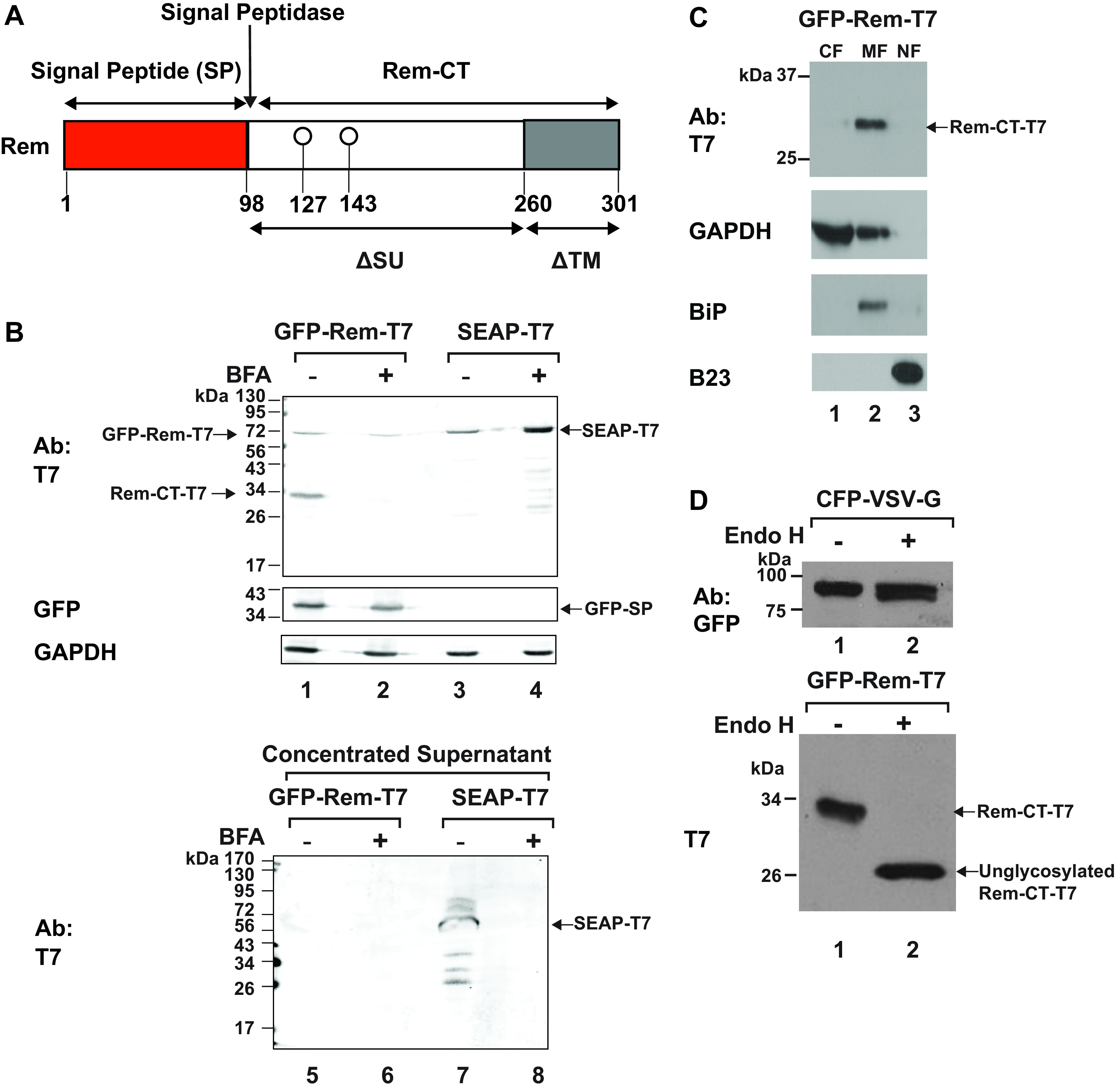
Rem-CT is localized to cytosolic membranes and contains only simple glycans. (A) Diagram of Rem and its cleavage products, SP and Rem-CT. Rem is directed to the ER for translation by the signal peptide sequence. The Rem precursor is then cleaved by signal peptidase at the ER membrane into SP (red box) and Rem-CT (white and gray boxes). The ΔSU and ΔTM designations indicate that Rem represents an in-frame deletion of the Env protein due to a second splicing event within the env gene. (B) Rem-CT is not secreted either with or without BFA treatment. Cells (293T) were transfected with GFP-Rem-T7 (lanes 1, 2, 5, and 6) or CMV-SEAP-T7 (lanes 3, 4, 7, and 8). Top, cells were treated with (lanes 2 and 4) or without BFA (lanes 1 and 3). Cells were lysed and subjected to Western blotting with T7, GFP, or GAPDH-specific antibodies. GFP-Rem-T7 is the uncleaved tagged precursor, whereas Rem-CT-T7 is the cleaved C terminus tagged with T7. The cleaved N terminus was detected with GFP-specific antibody. Supernatants from transfected cells were collected and concentrated using Amicon ultra-15 centrifugal filter units. Bottom, concentrated CMV-SEAP-T7 supernatant (50 μg) was loaded as a positive control (lane 7) for secretion, and the concentrated SEAP treated with BFA (50 μg) was loaded as a BFA secretion inhibition control. In the same experiment, concentrated supernatants (500 μg) from cells transfected with the Rem expression plasmid with or without BFA (lanes 5 and 6) were subjected to Western blotting using the T7-specific antibody. Only SEAP-T7 in untreated cells was detected in culture supernatants (lane 7). (C) Rem-CT localizes to the cytosolic membrane fraction. Cells (293T) were transfected with GFP-Rem-T7 expression plasmid and then lysed and fractionated as described in Materials and Methods. Different fractions were then subjected to Western blotting with T7, BiP, B23, or GAPDH-specific antibodies. (D) Rem-CT glycan is cleavable by Endo H. Cells (293T) were transfected with vectors expressing either CFP-VSV-G (lanes 1 and 2, top) or GFP-Rem-T7 (lanes 1 and 2, bottom). The indicated cell extracts (20 μg) were exposed to Endo H for 1 h at 37°C prior to Western blotting with GFP- or T7-specific antibodies.
To confirm that Rem-CT is primarily expressed intracellularly, we transfected 293T cells with the GFP-Rem-T7 expression vector and, after 48 h, lysed the cells to obtain cytosolic, membrane, and nuclear fractions (Fig. 1C). As expected, the nuclear fraction was enriched for the nucleolar protein B23 (34), whereas the ER protein BiP (35) was enriched in the membrane fraction. The majority of the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was found in the cytosolic fraction, but it also was associated with ER membranes, as previously described (36, 37). Rem-CT showed the same distribution as BiP (lane 2), whereas we previously have shown that the highest steady-state level of SP occurs in nucleoli (4, 9). Therefore, Rem-CT is localized primarily within the intracellular membrane fraction containing the ER protein BiP.
Brefeldin A reduces intracellular levels of Rem-CT.
To confirm the lack of Rem-CT secretion, we used a strategy previously designed to promote the intracellular retention of secreted cytokines (38). This strategy takes advantage of the drug BFA, a fungal metabolite that disrupts intracellular vesicular trafficking between the ER and Golgi networks (39–44). BFA causes dissociation of guanine nucleotide exchange factors (GEFs) specific for Arfs from the Golgi membrane. Furthermore, BFA inhibits the retrograde transport of proteins from the Golgi to the ER (45, 46) and subsequently results in the fusion of ER and Golgi membranes (39, 41, 42). Therefore, BFA inhibits the early steps of protein secretion to intracellularly trap proteins that traffic through the Golgi compartment (30).
To determine the effect of BFA on Rem-CT trafficking, we transfected 293T cells with GFP-Rem-T7. Transfected cells then were treated with 3 μg/ml BFA for 6 h, and lysates from treated and untreated cells were subjected to Western blotting. We anticipated that the presence of BFA would increase the amount of Rem-CT in cell lysates. Instead, Rem-CT levels were dramatically reduced in lysates from BFA-treated cells, whereas SP and Rem precursor levels were not affected (Fig. 1B, compare lanes 1 and 2). The addition of BFA increased the intracellular levels of SEAP-T7 (Fig. 1B, compare lanes 3 and 4). The levels of GAPDH confirmed the integrity of lysates and equal loading of protein in each lane of the gel. In agreement with our failure to detect Rem-CT in concentrated supernatants (Fig. 1B, lanes 5 and 6), the results with BFA were consistent with a lack of Rem-CT secretion. BFA blocked the production of SEAP-T7 in the supernatants (Fig. 1B, compare lanes 7 and 8), confirming that BFA worked as expected. Together, our data indicate that Rem-CT does not traffic through the classical secretory pathway involving the Golgi network.
Protein glycosylation is known to be modified after trafficking through the ER and Golgi, due to the presence of specific glycosyltransferases (47). Following the addition of simple glycans in the ER, these polysaccharides are modified to a more complex structure by glycosyltransferases within the Golgi compartment (48). Therefore, lysates from 293T cells transfected with GFP-Rem-T7 were treated with endoglycosidase H (Endo H). Endo H removes simple glycans, which are present on proteins that fail to traffic to the Golgi compartment (49). As a control for proteins that traffic through the Golgi, we also transfected cells with a cyan fluorescent protein (CFP)-tagged form of the vesicular stomatitis virus glycoprotein (CFP-VSV-G) (50). As expected, most glycans of the surface-localized VSV-G protein were not cleavable by Endo H (Fig. 1D, top, compare lanes 1 and 2). Surprisingly, the glycans on Rem-CT were cleaved by Endo H (Fig. 1D, bottom, compare lanes 1 and 2). Since Endo H only cleaves high-mannose or hybrid oligosaccharides that have not been processed by Golgi enzymes (49), Rem-CT does not traverse the medial-Golgi where the conversion to complex glycans occurs (47, 48).
Lack of glycosylation reduces Rem-CT susceptibility to BFA, but not proteasomal or lysosomal inhibition.
Our previous results have shown that Rem-CT has two glycosylation sites, at positions 127 and 143 relative to the Rem N terminus (Fig. 1A) (9, 51). Trafficking of viral and host glycoproteins and exit from the ER are known to be affected by glycosylation (47, 52). Therefore, we used expression vectors for GFP-Rem-T7 or its glycosylation-null variant (N127Q/N143Q), which contains mutations of both asparagines to glutamines, for transfection of 293T cells. Lysates of transfected cells were analyzed by Western blotting for Rem-CT and SP. As expected, N127Q/N143Q migrated faster than wild-type Rem-CT. However, unlike the wild-type protein, the level of the Rem glycosylation mutant did not change in the presence of BFA (Fig. 2A, top, compare lanes 1 and 2 to lanes 5 and 6). Furthermore, with the same amounts of transfected plasmids, the expression of unglycosylated Rem-CT was more stable than that of glycosylated Rem-CT. Incubation of Western blots with GFP-specific antibody indicated that both the glycosylated and unglycosylated Rem proteins were cleaved appropriately by signal peptidase (Fig. 2A, middle). Similar results were observed with HC11 mouse mammary epithelial cells, which are targets of MMTV infection (1) (Fig. 2B). Therefore, BFA’s effects on Rem-CT are not cell type specific, but glycosylation is required for the drug to decrease Rem-CT levels.
FIG 2.

BFA treatment reduces Rem-CT levels, and the reduction is not rescued by a proteasome inhibitor. (A) Proteasomal inhibitor MG132 did not reverse BFA-mediated reduction of Rem-CT levels. Cells (293T) were transfected with equal amounts of expression vectors for GFP-Rem-T7 wild type (WT) (lanes 1 to 4) or the N127Q/N143Q mutant (lanes 5 to 8) and pretreated with MG132 (10 μM) for 3 h prior to the addition of BFA (3 μg/ml) for another 6 h. Cell lysates were subjected to Western blotting with T7-, GFP-, or GAPDH-specific antibodies and then LI-COR imaging. Quantitation of the Rem-CT-T7 signal or GFP-SP is indicated relative to that of GAPDH. (B) Reduced levels of Rem-CT after BFA treatment are prevented by lack of glycosylation in mouse mammary epithelial cells. HC11 cells were transfected with equal amounts of expression vectors for GFP-Rem-T7 (lanes 1 and 2) or dually tagged N127Q/N143Q mutant (lanes 3 and 4) and treated with or without 3 μg/ml BFA. Cells were lysed and subjected to Western blotting with T7, GFP, or GAPDH-specific antibodies. LI-COR imaging was performed after incubation of Western blots with appropriate secondary antibodies. The asterisk indicates the unglycosylated Rem-CT protein produced by the N127Q/N143Q mutant (lanes 3 and 4).
Since the Rem precursor is highly susceptible to endoplasmic reticulum-associated degradation (ERAD) but SP is not, we also tested whether the diminished levels of Rem-CT observed in the presence of BFA were rescued in the presence of the proteasomal inhibitor MG132 (8, 9). MG132 was added for 3 h prior to the addition of BFA for another 6 h, and then cell extracts were obtained for Western blotting. As expected, tagged-Rem precursor (GFP-Rem-T7) levels increased after MG132 treatment (Fig. 2A, top, compare lanes 2 and 4). SP levels produced by cleavage of the glycosylated or unglycosylated Rem precursor did not change after proteasomal inhibition (Fig. 2A, middle). Extracts also were prepared from cells treated with both MG132 and BFA. Treatment with MG132 primarily increased the levels of unglycosylated Rem-CT (Fig. 2A, top, compare the unglycosylated Rem-CT-T7 bands in lanes 3 and 4 with the bands in lanes 1 and 2), consistent with the removal of carbohydrates by glycosidases in the cytosol prior to ERAD (53). The proteasomal inhibitor showed minimal rescue of glycosylated Rem-CT levels in the presence of BFA (Fig. 2A, top, lanes 3 and 4) but did not affect the levels of unglycosylated Rem-CT (Fig. 2A, lanes 7 and 8). These results suggest that decreased Rem-CT levels resulting from BFA-induced reorganization of the secretory pathway are not due primarily to ERAD.
Another possibility is that Rem-CT leaves the ER for an atypical secretory compartment and that BFA treatment leads to fusion with lysosomes, which contain large amounts of degradative enzymes (54). Therefore, we transfected 293T cells with expression vectors for either wild-type Rem or the N127Q/N143Q glycosylation mutant. A portion of the transfected cells was treated with BFA or BFA plus the inhibitor bafilomycin A1 (Fig. 3A). This inhibitor blocks the activity of the vacuolar H+ ATPase, thus preventing the low pH needed for proteolytic enzyme activation within endosomes and lysosomes (55). As anticipated, BFA reduced the intracellular levels of Rem-CT but not the unglycosylated protein, as detected by Western blots of transfected cell lysates (Fig. 3A, compare lanes 1 and 2 with lanes 5 and 6). Bafilomycin A1 did not rescue wild-type Rem-CT levels in the presence of BFA (Fig. 3A, compare lanes 3 and 4) or change the levels of the N127Q/N143Q mutant (Fig. 3A, lanes 7 and 8). As a control for the activity of bafilomycin A1, we confirmed the conversion of LC3-I to the lipidated form, LC3-II (56, 57). We also tested whether a different inhibitor, chloroquine, had an effect on Rem-CT levels in the presence of BFA. Chloroquine has recently been shown to primarily inhibit autophagy by reducing the fusion of lysosomes with autophagosomes, as well as by promoting disorganization of the Golgi network and endolysosomes (58). Treatment of transiently transfected 293 cells with chloroquine did not rescue the levels of Rem-CT in the presence of BFA (Fig. 3B, compare lanes 2 and 5). Again, the detection of LC3-II served as a control for chloroquine activity. Therefore, MG132, bafilomycin A1, and chloroquine all failed to restore Rem-CT levels in the presence of BFA.
FIG 3.

BFA-induced reduction of Rem-CT levels was not rescued by lysosomal or protease inhibitors. (A) Bafilomycin A1 does not reverse BFA-induced reduction of Rem-CT levels. 293T cells transfected with equal amounts of expression vectors for GFP-Rem-T7 wild type (WT) (lanes 1 to 4) or the N127Q/N143Q mutant (lanes 5 to 8) were treated with BFA (3 μg/ml) and/or bafilomycin A1 (1 μM). Cell lysates were subjected to Western blotting with T7-, GFP-, LC3B-, or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used. The appearance of LC3-II served as a positive control for bafilomycin A1 activity. (B) Chloroquine and MG132 do not reverse BFA-induced reduction of Rem-CT levels. Transfected 293T cells were treated with BFA (3 μg/ml) and/or chloroquine (200 μM) and/or MG132 (10 μM) for 6 h. Cell lysates were subjected to Western blotting with T7-, LC3B-, or actin-specific antibodies. The appearance of LC3-II served as a positive control for chloroquine activity. (C) A pancaspase inhibitor does not reverse BFA-induced reduction of Rem-CT levels. Transfected 293T cells were treated with BFA (1 μg/ml) and/or Z-VAD(OMe)-FMK (20 μM) for 16 h. Cell lysates were subjected to Western blotting with T7-, GFP-, PARP1-, or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used. The asterisk indicates the expected cleaved PARP1 size. The absence of cleaved PARP1 indicates no caspase activities. (D) The inhibition of cysteine proteases, including calpains and cathepsins, by E64d does not reverse the BFA-reduced Rem-CT levels. Transfected 293T cells were treated with BFA (1 μg/ml) and/or E64d (50 μM) for 16 h. Cell lysates were subjected to Western blotting with T7-, GFP-, LC3B-, or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used. The appearance of LC3-II served as a positive control for inhibition of cathepsin activity in endosomes. (E) The inhibition of serine proteases by AEBSF does not reverse the BFA-reduced Rem-CT levels. Transfected 293T cells were treated with serine protease inhibitor AEBSF (0.1 mM) overnight before the addition of BFA (3 μg/ml) for another 6 h. As expected, protein concentrations in cell lysates from AEBSF-treated samples (lanes 3 and 4) were ∼1.5 μg/μl higher than in the non-AEBSF treated samples (lanes 1 and 2). Cell lysates were subjected to Western blotting with T7-, GFP-, or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used. (F) Rem-CT is not detectable in exosomes after BFA treatment. Cells (293T) were transfected with expression plasmids for GFP-Rem-T7 and treated with or without 3 μg/ml BFA. Supernatants were filtered prior to isolation and concentration of exosomes as described in Materials and Methods. Cell lysates (lanes 1 and 2) and concentrated exosomes (lanes 3 and 4) were subjected to Western blotting using T7 or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used.
To determine whether protease inhibitors would reverse the effects of BFA on Rem-CT levels, we treated GFP-Rem-T7-expressing cells with the cell-permeable caspase inhibitor Z-VAD-FMK (Fig. 3C) (59). Z-VAD-FMK had no effect on Rem-CT levels in the presence or absence of BFA (Fig. 3C, lanes 3 and 4). No cleavage of poly(ADP-ribose) polymerase 1 (PARP1) was detectable under any condition, indicating that caspases were not activated by BFA (60). In addition, the levels of the unglycosylated N127Q/N143Q Rem mutant were unchanged by the caspase inhibitor either with or without BFA treatment. Similarly, we observed no effect of the cysteine protease inhibitor E64d, which is effective against calpains and cathepsins (61–63), or the serine protease inhibitor AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride] (64), on BFA-mediated reduction of Rem-CT levels (Fig. 3D and E). None of the protease inhibitors blocked Rem cleavage by signal peptidase, as determined by the uniform expression of GFP-SP, and cellular toxicity was not observed by monitoring total protein and GAPDH levels.
Because we had determined that Rem-CT is localized in a membrane compartment (Fig. 1C), we also isolated exosomes from 293T cells transiently transfected with a vector expressing GFP-Rem-T7 in the presence or absence of BFA treatment. Rem-CT remained undetectable, whereas GAPDH can be detected in exosomes (65) as a positive control (Fig. 3F). Together, our data suggest that BFA neither induces proteasome, lysosome, or autophagosome-mediated degradation of Rem-CT nor promotes extracellular production of Rem-CT-containing exosomes.
Rem-CT is localized primarily within the ER.
Although our data indicated that Rem-CT is located within intracellular membranes, its exact location has not been determined. Our previous studies using fluorescence microscopy indicated that Rem with a C-terminal GFP tag was not cleaved or folded properly to allow a detectable signal (7), and therefore, live-cell imaging has not been possible using our current methods. However, Rem expressing a C-terminal T7 tag (Rem-T7) was cleaved to give functional SP in a dose-dependent manner (Fig. 4A). Therefore, we used indirect fluorescence to track Rem-CT within transfected 293 cells. Fluorescence was observed in a mottled pattern within the cytoplasm of transfected cells incubated with primary T7-specific antibody and a fluorescein isothiocyanate (FITC)-tagged secondary antibody (Fig. 4B). This pattern was similar to that obtained after transfection of a plasmid expressing the fluorescent protein mCherry with an N-terminal signal peptide and a C-terminal KDEL to promote ER entry and retention (ER-mCherry) (31, 66). Both Rem-T7 and ER-mCherry were excluded from the nucleus, whereas Rem-CT was highly colocalized with the ER-mCherry (Fig. 4B, merged image).
FIG 4.
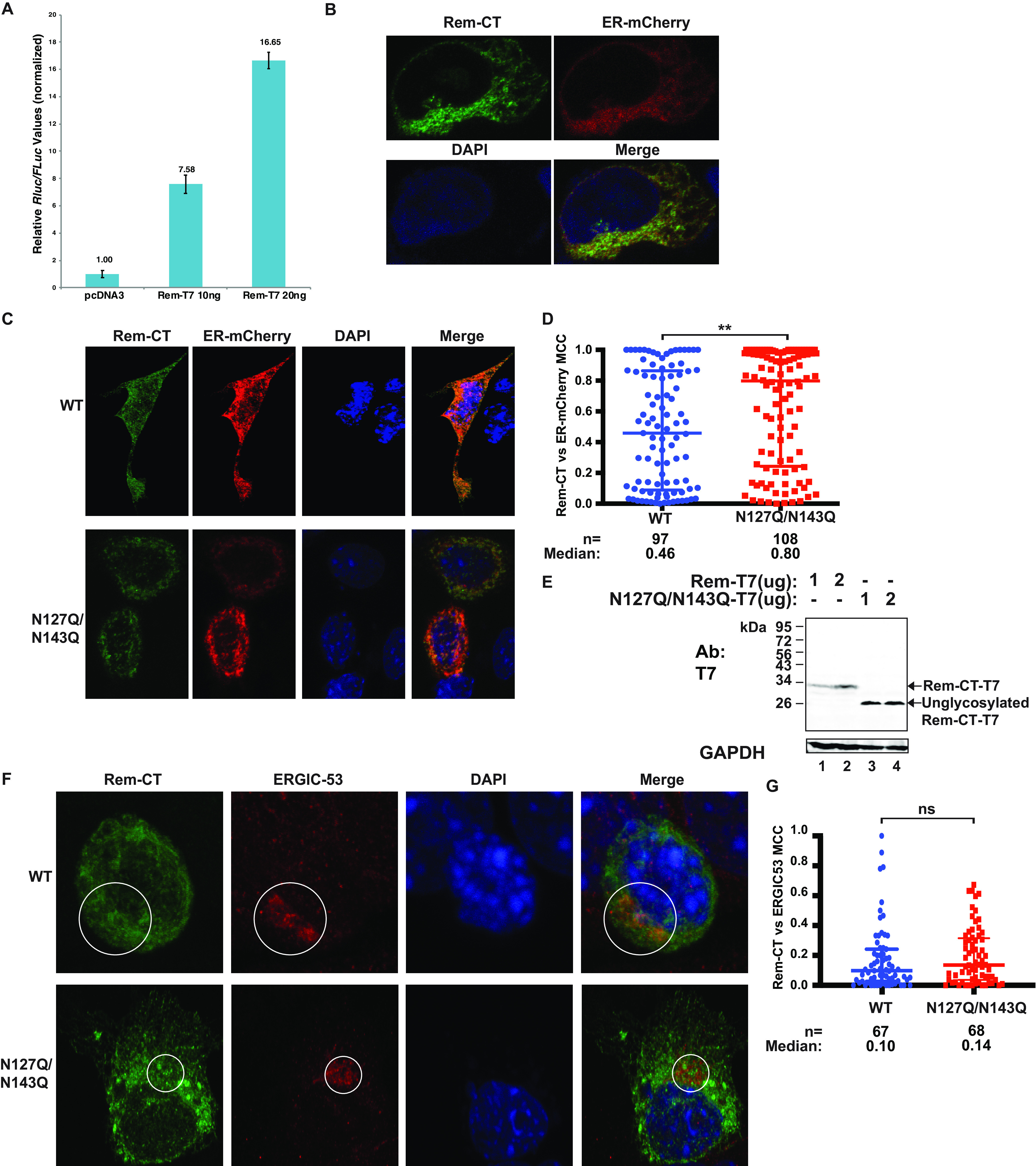
Both wild-type and unglycosylated Rem-CT localize to the ER. (A) Luciferase assays confirmed SP function from Rem-T7 constructs. Cells (293T) were transfected with either 10 or 20 ng of Rem-T7 constructs together with luciferase reporter constructs as described in Materials and Methods. Luciferase activities normalized for transfection efficiency were reported as the mean values and standard deviations from triplicate transfections. The sample lacking a Rem expression construct (pcDNA3) was assigned a relative value of 1. (B) Rem-CT colocalized with an ER marker. Cells (293) were cotransfected with expression plasmids for Rem-T7 and ER-mCherry. Rem-CT was detected using a T7-specific antibody followed by Alexa Fluor 488-tagged secondary antibody. Images were obtained by confocal microscopy (see Materials and Methods). Green shows the localization of Rem-CT (top left) as detected by a T7-specific antibody and an Alexa Fluor 488 secondary antibody. Red detects ER-mCherry (top right). DAPI (blue) was used to stain the nucleus (bottom left). The bottom right shows the merged image. (C) Representative confocal microscopy images of wild-type and N127Q/N143Q mutant Rem-CT-transfected cells. Equal amounts of wild-type or mutant expression constructs were cotransfected into mouse mammary HC11 cells and detected as described in the legend to panel A. (D) Scatterplot of Mander’s colocalization coefficients (MCCs) for Rem-CT wild type or glycosylation mutant versus ER-mCherry. Dots represent MCCs in individual cells. The middle line in each group shows the median value for the group, whereas the highest and lowest lines represent the interquartile range. The numbers (n) of cells analyzed and the median MCCs are given. Statistical analysis was performed using nonparametric Mann-Whitney tests. **, P < 0.01. (E) Rem precursor is not detectable in mouse mammary cells at the DNA levels used for confocal microscopy. HC11 cells were transfected with the indicated amounts of expression constructs for Rem-T7 WT or N127Q/N143Q mutant. Cell lysates were subjected to Western blotting with T7- or GAPDH-specific antibodies. LI-COR imaging was performed after appropriate secondary antibodies were used. Note that the unglycosylated N127Q/N143Q mutant migrated faster than WT Rem-CT (compare lanes 1 and 2 with 3 and 4). (F) Representative confocal microscopy images of WT Rem-CT and the glycosylation mutant stained with the ERGIC marker ERGIC-53. Equal amounts of WT and N127Q/N143Q Rem-T7 expression constructs were transfected into mouse mammary HC11 cells. Tagged Rem-CT was detected using a T7-specific antibody. Appropriate Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies were used for T7 and ERGIC-53, respectively. DAPI (blue) was used to stain the nucleus. Circles mark the colocalization of Rem-CT with ERGIC-53. (G) Quantitation of Rem-CT colocalization with ERGIC-53. Dots represent the MCCs in individual cells. Nonparametric Mann-Whitney test comparisons between WT and mutant were performed. ns, nonsignificant.
To further confirm Rem-CT localization, we cotransfected plasmids expressing either Rem-T7 or the N127Q/N143Q mutant together with the ER-mCherry expression vector into HC11 mouse mammary epithelial cells (Fig. 4C). Similar to the results from 293 cells, we observed Rem-CT colocalization with the ER marker in HC11 cells expressing either the wild type or the glycosylation mutant (N127Q/N143Q). To determine intracellular location more quantitatively, we measured the Mander’s colocalization coefficients (MCCs) (67), which quantified the ER-colocalized fraction of Rem-CT pixels in multiple individual cells (Fig. 4D). Because these cells are transiently transfected, a range of colocalization values were obtained. The median value of the colocalization coefficient for wild-type Rem-CT was ∼0.5, whereas the coefficient for the mutant was significantly higher (∼0.8). As previously reported, higher levels of transfected Rem-T7 expression plasmids led to abundant detection of both uncleaved Rem precursor and cleaved Rem-CT (10). Therefore, we performed Western blot analyses of cell lysates from these transfections using T7-specific antibody. The results confirmed that under these conditions, only the cleaved Rem-CT was detectable (Fig. 4E). Moreover, similar to the results in 293T cells, the unglycosylated N127Q/N143Q mutant accumulated to higher levels than the wild-type protein. These results indicated that the majority of both glycosylated and unglycosylated Rem-T7 remains associated with the ER. Nevertheless, the distribution of the signals suggests that neither the wild type nor the glycosylation mutant traffics exactly with the ER-mCherry marker, showing the potential to localize at other intracellular sites.
To determine whether Rem-CT leaves the ER to travel to the ERGIC, we transfected HC11 cells with plasmids expressing T7-tagged Rem-CT or the glycosylation mutant. After 48 h, cells were fixed, permeabilized, and incubated with antibodies specific for T7 or ERGIC-53 (Fig. 4F). We observed significant colocalization of Rem-CT, as well as the mutant, in a perinuclear compartment consistent with the ERGIC. Quantitation of multiple cells indicated that the colocalization of wild-type or mutant Rem-CT was not significantly different (Fig. 4G). Therefore, these results indicate that lack of glycosylation does not prevent the export of a substantial fraction of Rem-CT from the ER to the ERGIC, and yet, its higher median colocalization with ER-mCherry suggests that Rem-CT returns to the ER.
Since Rem-CT lacks a transmembrane domain and a C-terminal KDEL/KKXX retrograde transport signal for ER retention (68), we attempted to map the Rem-CT sequences required for its primary localization within the ER. Four deletion mutants were constructed that lacked sequential 50-amino-acid regions, starting from residue 103 (Δ103–155 mutant) and C terminal to the signal peptidase cleavage site (7, 9, 10). All mutants retained the C-terminal T7 tag. Expression plasmids for the wild type, deletion mutants, and the N127Q/N143Q glycosylation mutant were transfected independently into 293T cells, and after 48 h, cells were lysed for Western blotting with different antibodies. To ensure that the deletions did not affect Rem cleavage in the ER, we examined SP expression with GFP-specific antibody (Fig. 5A and B). Compared to the results for wild-type Rem, similar amounts of SP were produced by all mutants with the exception of a Δ103–155 mutant. Equal loading of protein extracts was confirmed by GAPDH levels. Although the deletion of residues 103 to 155 does not remove the consensus signal peptidase cleavage site, these results suggest that sequences C terminal to the consensus site affect cleavage efficiency, since the level of uncleaved Rem is similar to that observed with the wild-type expression plasmid.
FIG 5.
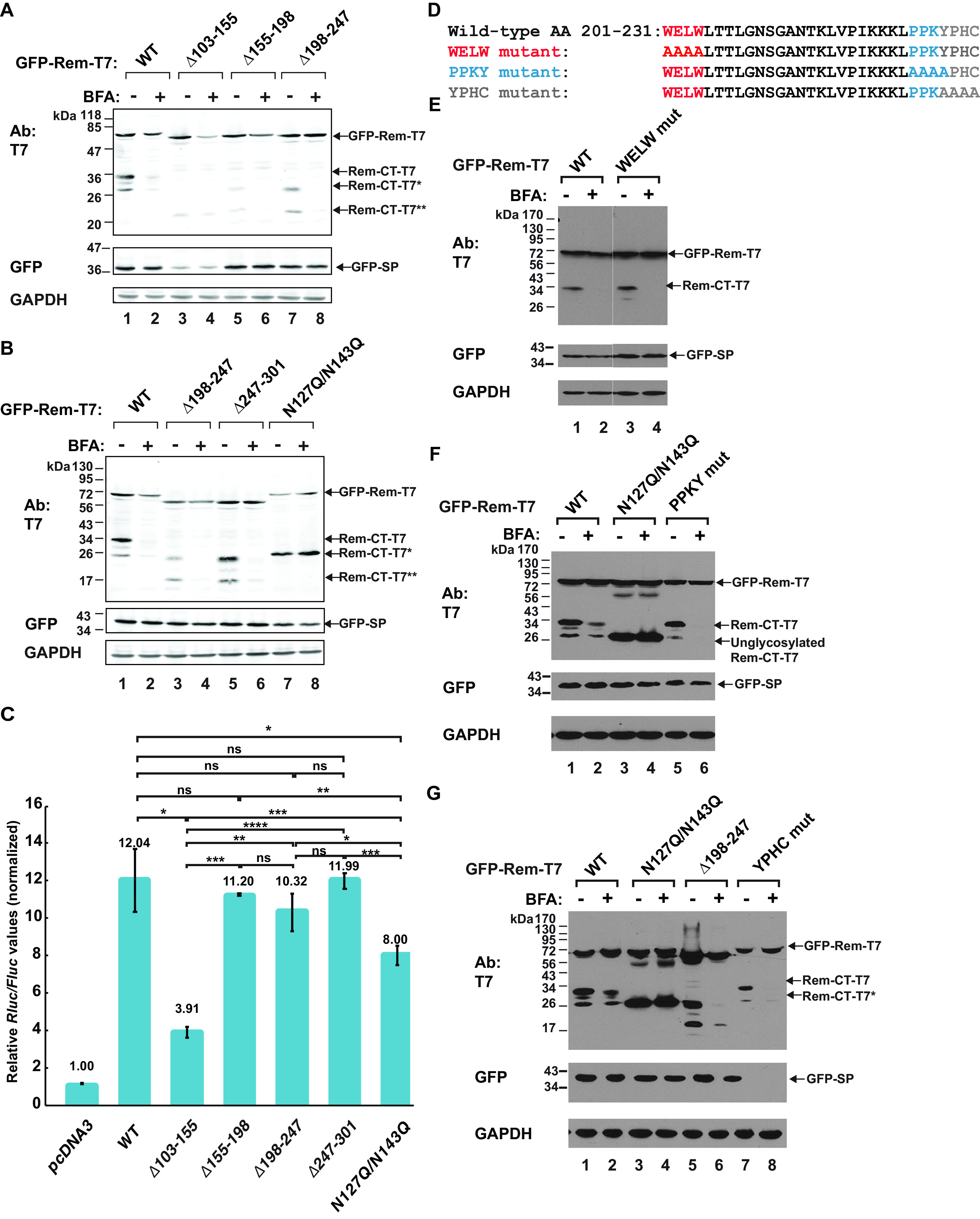
Loss of glycosylation sites rescues Rem-CT levels in the presence of BFA. (A and B) BFA sensitivity of Rem-CT deletion and substitution mutants. Expression plasmids for GFP-Rem-T7 wild type, the GFP-RemΔ103–155-T7, GFP-RemΔ155–198-T7, GFP-RemΔ198–247-T7, and GFP-RemΔ247–301-T7 deletion mutants, or the GFP-N127Q/N143Q-T7 glycosylation site substitution mutant were transfected into 293T cells and treated with 3 μg/ml BFA as indicated. Whole-cell lysates were subjected to Western blotting with T7-, GFP-, or GAPDH-specific antibodies. LI-COR imaging was performed after incubation with tagged secondary antibodies. The arrows represent different forms of Rem detected by the T7 or GFP antibodies. GFP-Rem-T7 is the uncleaved precursor, whereas Rem-CT-T7 is the wild-type C-terminal cleavage product. Arrows indicate Rem-CT-T7*, the unglycosylated form of Rem-CT (B, lanes 7 and 8), the deleted Rem-CT (A, lanes 5 and 7, and B, lanes 3 and 5), and Rem-CT-T7**, which has a deletion that spans the two glycosylation sites (A, lanes 3 and 4). (C) SP activities of Rem-CT deletion or substitution mutants. Cells (293T) were transfected with 2.5 ng of different GFP-Rem-T7 constructs together with Rem-responsive and Rem-nonresponsive luciferase reporter constructs. Luciferase activities normalized for transfection efficiency were reported as the mean values and standard deviations for triplicate transfections. The pcDNA3 control sample lacked any Rem expression vector and was set to a relative value of 1. Statistical significance is indicated as follows: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant. (D) Potential cellular trafficking motifs were identified within Rem-CT using the Eukaryotic Linear Motif (ELM) prediction tool. The motifs are identified in separate colors as WELW (clathrin binding), YPHC (an SH3 interaction motif for cytoskeletal organization and membrane trafficking), and PPKY (a WW domain recognition motif for protein interactions). The mutations introduced are shown below the wild-type sequence. (E to G) Results of transfections of 293T cells with expression constructs for wild-type GFP-Rem-T7 or the same amounts of the N127Q/N143Q glycosylation mutant or motif mutants. Transfected cells were treated with or without 3 μg/ml BFA. Cells were lysed and subjected to Western blotting with T7-, GFP-, or GAPDH-specific antibodies. The YPHC mutant produced no detectable GFP-tagged SP.
To further confirm the reduced generation of SP from the Δ103–155 mutant, we used our quantitative SP reporter assay (4, 7, 9). As expected, the wild-type Rem expression plasmid gave ∼12-fold induction of the Renilla luciferase reporter at the DNA level transfected relative to the results for the reporter vector alone (Fig. 5C). The Δ103–155 plasmid, lacking sequences just downstream from the signal peptidase cleavage site, including the two glycosylation sites (Fig. 1A), showed a significantly lower SP activity than wild-type Rem (Fig. 5C). Interestingly, our previous results showed that substitution of single glycosylation sites preserved wild-type SP activity, whereas mutation of both glycosylation sites (N127Q/N143Q) reduced SP function (9). As expected, the N127Q/N143Q mutant also showed significantly lower SP activity than wild-type Rem (Fig. 5C), but its activity was twice that of the Δ103–155 mutant. Western blotting indicated that the glycosylation mutant gave normal levels of SP and Rem-CT, whereas the Δ103–155 mutant did not (Fig. 5A and B). These results suggested that sequences between residues 103 and 155 affect signal peptidase recognition independent of glycosylation status. Nevertheless, our results indicated that the majority of the deletion mutants were sufficiently stable to assess Rem-CT levels in the presence and absence of BFA.
To test the effect of BFA on the deletion mutants, half of the transfected cells from the same experiment were treated with the drug prior to Western blotting with T7-specific antibodies. As anticipated, wild-type Rem-CT levels were greatly reduced in the presence of BFA, and some unglycosylated protein was observed, which comigrated with the protein produced from the N127Q/N143Q glycosylation mutant (Fig. 5B, compare lanes 1 and 7). Also, the levels of the glycosylation mutant were unchanged in the presence of BFA (Fig. 5B, lanes 7 and 8). The Δ103–155 mutant had low levels of Rem-CT in the presence and absence of BFA, and this protein had a mobility consistent with the deletion and loss of both glycosylation sites. Uncleaved Δ103–155 mutant Rem levels were more affected by BFA than were wild-type Rem levels. Rem-CT levels of the Δ103–155 mutant were affected minimally by BFA (Fig. 5B, lanes 3 and 4). These results were consistent with those from the N127Q/N143Q mutant, indicating that loss of glycosylation leads to resistance of Rem-CT to BFA. The other mutants, with deletions of residues 155 to 198, 198 to 247, and 247 to 301, had reduced levels of Rem-CT in the presence of BFA (Fig. 5A and B), although only the Δ247–301 mutant had wild-type levels of Rem-CT. Our previous results with N-terminally GFP-tagged Rem mutants containing sequential deletions from the C terminus also showed that the removal of more than 50 amino acids from the Rem C terminus led to destabilization of SP and uncleaved Rem (7).
To identify alternative sequences that may affect Rem-CT trafficking and response to BFA, we also performed a motif search within Rem-CT using the Eukaryotic Linear Motif (ELM) prediction tool. The sequences identified were WELW for clathrin binding, PPKY (a WW domain recognition motif), and YPHC, an Src homology 3 (SH3) domain interaction motif (Fig. 5D). These motifs were modified by alanine substitutions, and the resulting mutants were transfected into 293T cells. Half of the cells were treated with BFA as described for the internal deletion mutants. None of these mutants showed resistance to the drug (Fig. 5E, F, and G). Together, these results suggested that only the loss of glycosylation sites rescues Rem-CT levels in the presence of BFA, by promoting retention within the ER.
Rem-CT traffics to endosomes.
Our previous data indicated that Rem-CT is primarily found in the ER and does not traffic through the Golgi as is typical of other viral glycoproteins. Endosomes act as sorting stations between different membrane compartments within the cell (69). To test whether Rem-CT traffics out of the ER to endosomes, we used confocal microscopy to examine HC11 mammary epithelial cells transfected with plasmids expressing C-terminally T7-tagged wild-type Rem-CT or the N127Q/N143Q glycosylation mutant. Transfected cells were then fixed and incubated with T7-specific and Rab5-specific primary antibodies prior to detection with fluorescently tagged secondary antibodies. Rab5 is a regulatory GTPase that serves as a marker for early endosomes (70). We observed colocalization of Rem-CT with Rab5 in a small percentage of transfected cells (Fig. 6A). To quantitate these results, we determined the Rem-CT and Rab5 colocalization coefficient after observation of multiple cells (Fig. 6B). The fraction of wild-type Rem-CT colocalized with Rab5 was very small, indicating that only a small fraction of Rem-CT trafficked to the early endosomes. However, trafficking of Rem-CT out of the ER to early endosomes may occur very quickly, since our results measured only a single fixed time. We did observe a significantly higher colocalization coefficient of Rem-CT with Rab5 compared with that of the N127Q/N143Q glycosylation mutant. Our results suggest that Rem-CT traffics to the early endosomes but that trafficking to this compartment is impaired in the absence of glycosylation.
FIG 6.
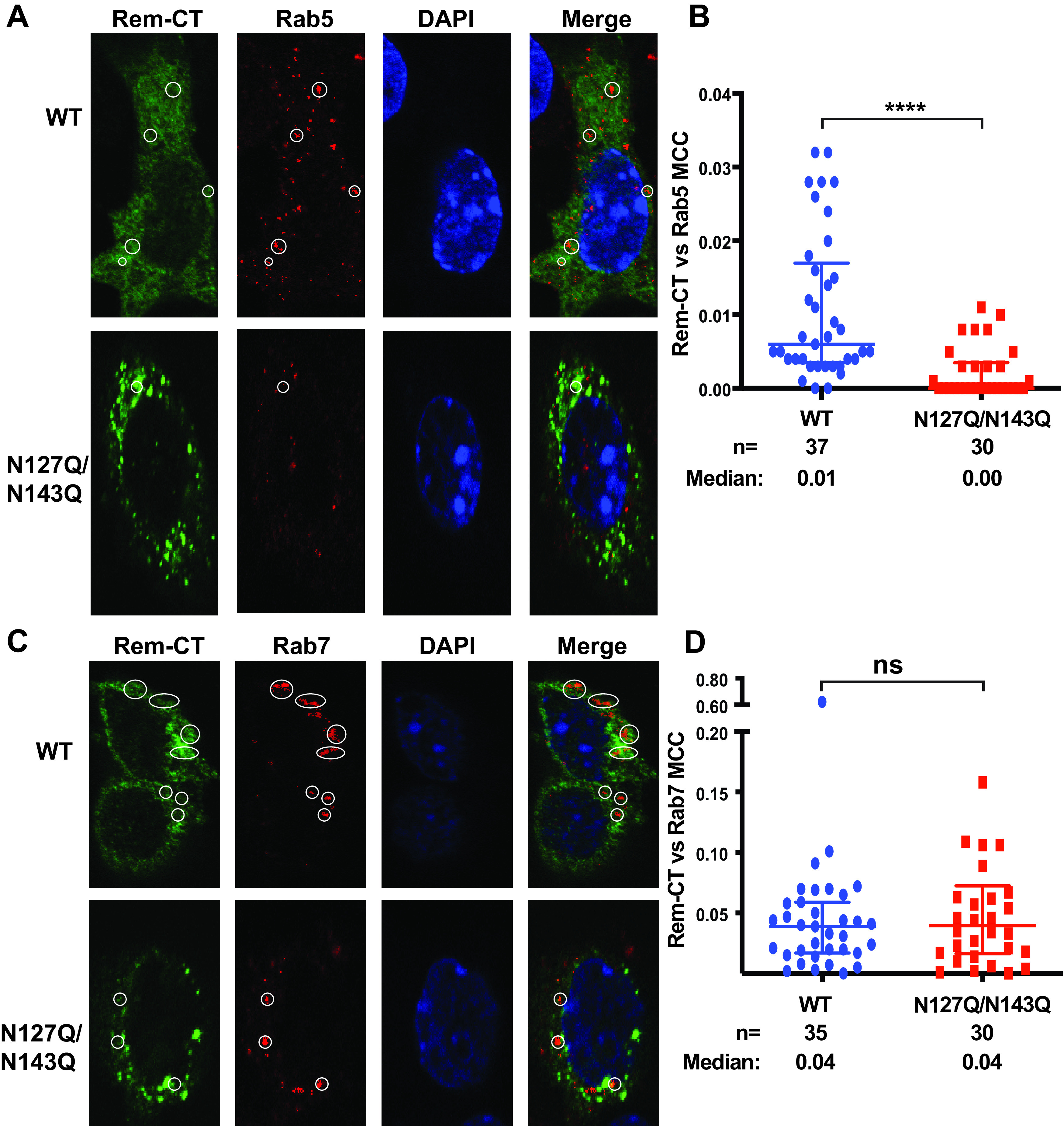
Rem-CT traffics to the endosomes. (A) Representative images of WT and N127Q/N143Q Rem-CT stained with the early endosome marker Rab5. Equal amounts of WT and N127Q/N143Q Rem-T7 expression constructs were transfected into mouse mammary HC11 cells. Rem-CT was detected using a T7-specific antibody. Appropriate Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies were used for T7 and Rab5, respectively. DAPI (blue) stained the nucleus. Circles mark the colocalization of Rem-CT with Rab5. (B) Quantitation of Rem-CT colocalization with Rab5. Dots represent the MCCs in individual cells. The middle line in each group shows the median value for the group, whereas the highest and lowest lines represent the interquartile range. The numbers (n) of cells analyzed and the median MCCs are given. Statistical analysis performed using nonparametric Mann-Whitney tests indicated a highly significant difference between WT and mutant. ****, P < 0.0001. (C) Representative images of WT and N127Q/N143Q Rem-CT stained with the late endosome marker Rab7. Equal amounts of WT (Rem-T7) or N127Q/N143Q expression constructs were transfected into mouse mammary HC11 cells. Rem-CT was detected using a T7-specific antibody. Appropriate Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies were used for T7 and Rab7, respectively. DAPI (blue) stained the nucleus. Circles mark the colocalization of Rem-CT with Rab7. (D) Quantitation of Rem-CT colocalization with Rab7. Dots represent the MCCs of individual cells. The middle line in each group represents the median value for the group, whereas the highest and lowest lines represent the interquartile range. The numbers (n) of cells analyzed and the median MCCs are given. Nonparametric Mann-Whitney test comparisons between WT and mutant were nonsignificant (ns).
Proteins found within early endosomes often traffic to late endosomes prior to lysosomal fusion and degradation (71). Therefore, we also tested Rem-CT-T7-expressing cells with antibodies specific for the late endosomal marker, Rab7, to determine if a portion of Rem-CT is localized to late endosomes. We again observed low levels of colocalization between Rem-CT and Rab7 (Fig. 6C). Quantitation of colocalization of Rem-CT with Rab7 in multiple transfected cells was performed to determine the colocalization coefficient (Fig. 6D). Unlike the results with Rab5-specific antibodies, these experiments indicated similar low levels of colocalization of wild-type Rem-CT and the N127Q/N143Q mutant with Rab7 (median coefficient of 0.04). However, these coefficients were about 4-fold higher than those in early endosomes, perhaps indicative of Rem-CT residence times. These data suggest that portions of both wild-type Rem-CT and the glycosylation mutant reach the late endosomes.
Rem-CT levels are rescued after BFA treatment in the presence of DN p97/VCP.
The Arf guanine nucleotide exchange factors (GEFs) release GDP from Arf proteins, leading to their activation (14) by GTP binding. Arf1 activation has multiple effects on the cellular vesicular network, including the formation of COPI vesicles that mediate retrograde trafficking from the Golgi network to the ER, changes in the activity of lipid-modifying enzymes, and altered composition of the Golgi compartment, such as loss of golgin-160 (72). BFA treatment of mammalian cells promotes an inactive GEF-Arf1-GDP complex (73). Therefore, we tested whether a hemagglutinin (HA)-tagged DN Arf1 (containing a mutation of threonine to asparagine at position 31 [T31N]) has the same effect as BFA on Rem-CT levels. We cotransfected expression vectors for the DN Arf1 and either wild-type Rem-T7 or the N127Q/N143Q mutant into HC11 cells. Cell lysates subjected to Western blotting with HA-specific antibody confirmed the expression of the DN Arf1 (Fig. 7A). Similar to the results for BFA treatment, we observed reduced levels of wild-type Rem-CT and no effect on the levels of the Rem-CT glycosylation mutant by coexpression of DN Arf1 (Fig. 7A, compare lanes 1 and 2 to lanes 3 and 4). Equal loading of cellular extracts was verified by Western blotting with GAPDH-specific antibodies. Together with previous data, these results suggest that reduction of Rem-CT levels by BFA is partially determined by Arf1.
FIG 7.
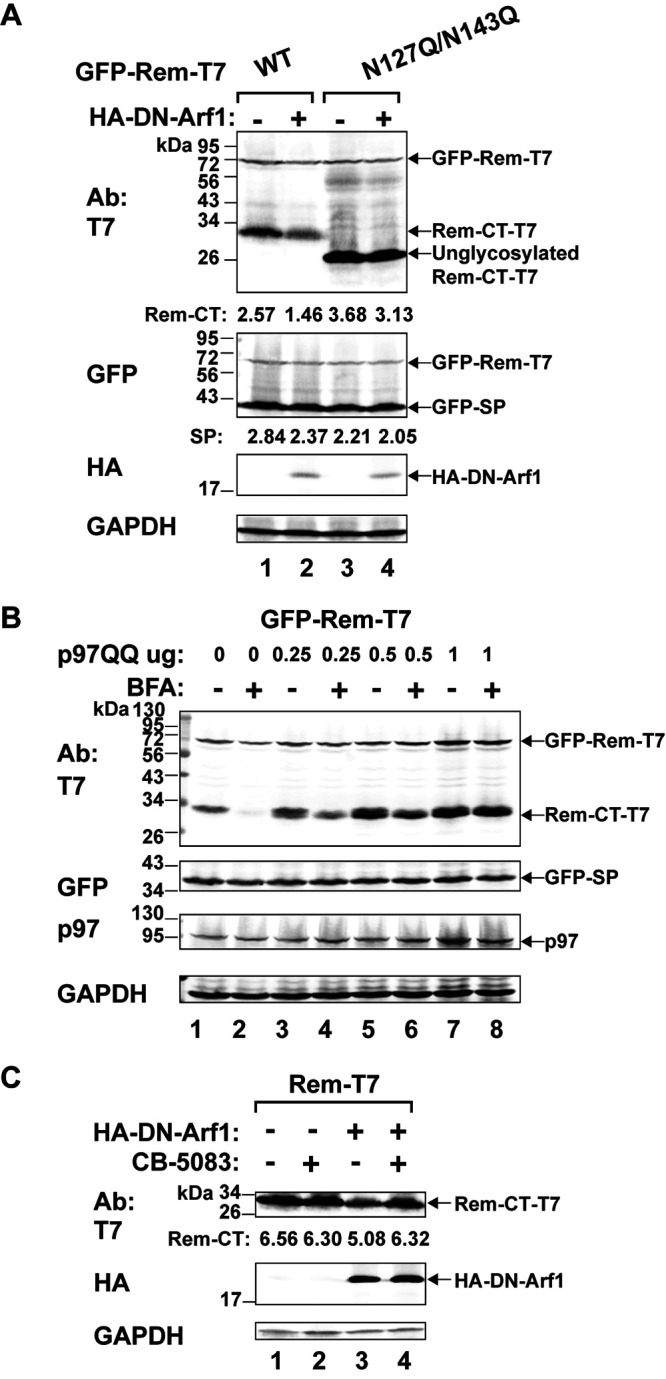
Inhibition of p97/VCP rescues Rem-CT levels in the presence of BFA or DN Arf1. (A) Expression of DN Arf1 mimics the reduced Rem-CT levels observed after BFA treatment. Cells (HC11) were cotransfected with equal amounts of expression vectors for wild-type GFP-Rem-T7 (lanes 1 and 2) or the N127Q/N143Q glycosylation mutant (lanes 3 and 4) with and without HA-tagged DN Arf1. Cells were lysed and subjected to Western blotting with T7-, GFP-, HA-, or GAPDH-specific antibodies. LI-COR imaging was performed after incubation with appropriate secondary antibodies. Arrows indicate the positions of tagged Rem precursor, glycosylated Rem-CT, or unglycosylated Rem-CT. Quantitation of Rem-CT and SP signals relative to that of GAPDH is shown. (B) A DN form of p97 rescues Rem-CT levels in the presence of BFA. Increasing amounts of an expression plasmid for DN p97 (p97QQ) were transfected together with a constant amount of the GFP-Rem-T7 expression construct into 293T cells. Cells were treated with 3 μg/ml of BFA prior to lysis. Lysates were subjected to Western blotting with T7-, GFP-, p97-, or GAPDH-specific antibodies. (C) The inhibition of p97 rescues DN Arf1-reduced Rem-CT levels. HC11 cells were transfected with expression vectors for wild-type Rem-CT or the N127Q/N143Q mutant with or without the HA-tagged DN Arf1. The p97 inhibitor CB-5083 (1 μM) was added for 9 h prior to harvest and Western blotting with antibodies as indicated above. Quantitation of Rem-CT signal relative to that of GAPDH is shown.
Our previous experiments indicated that the stability of uncleaved Rem is regulated by the AAA ATPase p97/VCP, which is required for SP extraction from the ER membrane and nuclear function (7, 8). Furthermore, p97 has a role in regulation of vesicle fusion during the reformation of the ER and Golgi network after mitosis (74, 75), as well as in receptor-mediated endocytosis (76–78). Therefore, we determined whether p97 also affected the response of Rem-CT to BFA. Cells (293T) were cotransfected with a GFP-Rem-T7 expression vector and increasing amounts of a plasmid expressing the DN p97 (p97QQ) (79) in the presence or absence of BFA. Lysates were used for Western blotting with GAPDH-specific antibodies to confirm equal loading of extracts (Fig. 7B). Incubation of blots with p97-specific antibodies revealed increased levels of the ATPase after transfection with the DN p97 expression plasmid. Examination of SP levels revealed that DN p97 did not affect Rem cleavage, consistent with our previously reported results (8). As expected, incubation of Western blots with T7-specific antibodies confirmed reduced levels of Rem-CT in the presence of BFA. However, the expression of increasing amounts of DN p97 completely reversed the effect of BFA (Fig. 7B, compare lanes 2 and 8).
To confirm the results with the DN p97, we also used a chemical inhibitor of p97 activity, CB-5083 (80), in HC11 mammary cells. An expression vector for the Rem-T7 wild type was transfected in the presence or absence of the DN Arf1 expression plasmid. Duplicate transfected samples were then treated with CB-5083, and then all samples were used for lysate preparation and Western blotting (Fig. 7C). Although not as dramatic as the results in 293T cells, quantitation showed that inhibition of p97 in HC11 mammary cells rescued wild-type Rem-CT levels in the presence of DN Arf1 (Fig. 7C, compare lanes 3 and 4). Together, our data suggest that trafficking of wild-type Rem-CT requires glycosylation and p97 to localize to early endosomes and a BFA-sensitive compartment.
Inhibition of p97 prevents Rem-CT trafficking to early endosomes.
Inhibition of the p97/VCP function reverses the effect of BFA or DN Arf1 on Rem-CT levels. Since p97 is involved in endosomal trafficking (76, 78), we tested whether inhibition of p97 affects the localization of Rem-CT to early endosomes. HC11 mammary cells were transfected with expression vectors for Rem-CT-T7 with or without a plasmid expressing DN Arf1. After 48 h, cells were fixed and incubated with antibodies specific for T7 or the early endosomal marker Rab5, followed by confocal microscopy (Fig. 8A). As expected from our previous results, quantitation of multiple cells indicated that a small percentage of wild-type Rem-CT colocalized with early endosomes, and this percentage increased slightly in the presence of DN Arf1 (Fig. 8B). Treatment of transfected cells with the p97 inhibitor CB-5083 diminished the ability of Rem-CT to localize to early endosomes regardless of DN Arf1 expression (Fig. 8A). Cells from two independent transfection experiments were used for quantitation (Fig. 8B). Rem-T7 localization following cotransfection of expression vectors for DN Arf1 and Rem-T7 for ∼48 h with p97 inhibition for the last 9 h was not significantly different from Rem-T7 localization in the absence of DN Arf1 and p97 inhibition. We could not use longer periods for p97 inhibition because of cellular toxicity. These results suggested that p97 and glycosylation are required for Rem-CT anterograde trafficking to early endosomes. In contrast, Arf1 likely functions in retrograde trafficking of Rem-CT.
FIG 8.
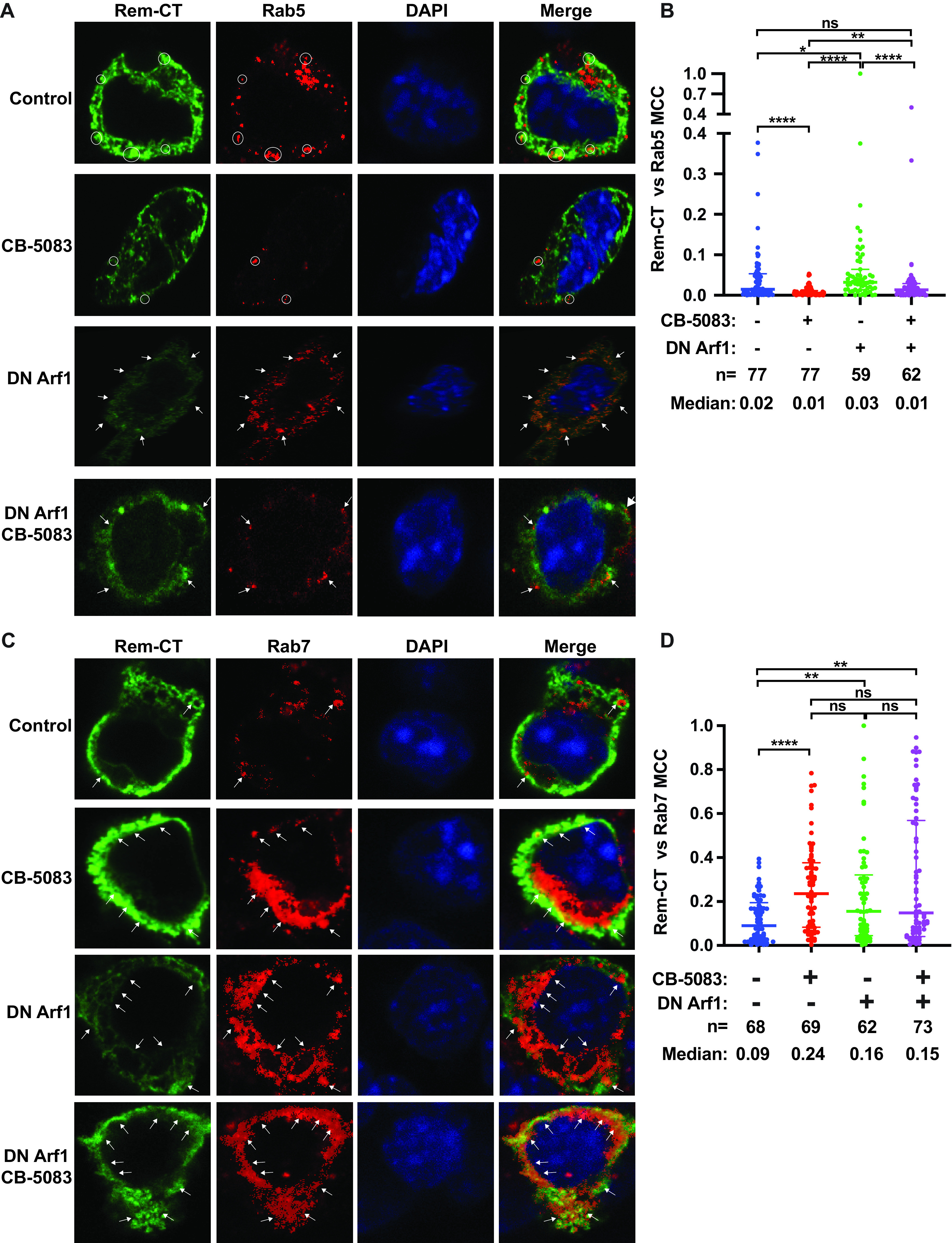
Inhibition of p97 activity blocks Rem-CT trafficking to early endosomes and allows accumulation in late endosomes. (A) Representative images of wild-type Rem-CT expression with or without p97 inhibition by CB-5083 and/or DN Arf1 stained with Rab5-specific antibody. Transfections and treatments of HC11 cells were performed using the same conditions described in the legend to Fig. 7C. Rem-CT-T7 was detected using a T7-specific antibody. Appropriate Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies were used for T7 and Rab5, respectively. DAPI (blue) was used to stain nuclei. Arrows and circles mark the colocalization of Rem-CT with Rab5. (B) Quantitation of Rem-CT colocalization with Rab5. Dots represent the Mander’s colocalization coefficients (MCCs) of individual cells. Median MCCs are given, as well as the numbers (n) of cells analyzed in two independent experiments. The middle line in each group represents the median value of the group, whereas the highest and lowest lines represent the interquartile range. Statistical analysis was performed between the treated and untreated cells. (C) Chemical inhibition of p97 increases Rem-CT colocalization with late endosomes stained with Rab7-specific antibody. Representative images of wild-type Rem-CT expression with or without p97 inhibition by CB-5083 and/or DN Arf1 stained with Rab7-specific antibody. Transfections and treatments of HC11 cells were performed using the same conditions described in the legend to Fig. 7C. Rem-CT-T7 was detected using a T7-specific antibody. Appropriate Alexa Fluor 488 (green) and Alexa Fluor 594 (red) secondary antibodies were used for T7 and Rab7, respectively. DAPI (blue) was used to stain nuclei. Arrows mark the colocalization of Rem-CT with Rab7. (D) Quantitation of Rem-CT colocalization with Rab7. Dots represent the MCCs of individual cells. The middle line in each group represents the median value of the group, whereas the highest and lowest lines represent the interquartile range. The numbers (n) of cells analyzed and the median MCCs are given. (B and D) Nonparametric Mann-Whitney test was used for comparisons between different conditions. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; ns, nonsignificant.
To test the roles of p97 and Arf1 in Rem-CT localization to late endosomes, we performed similar inhibition experiments on Rem-T7-transfected cells for analysis by confocal microscopy using Rab7-specific antibody (Fig. 8C). Inhibition of p97 or Arf1 increased the colocalization of Rem-T7 with Rab7. Quantitation of cells from two different transfections showed no significant difference in Rem-CT colocalization with Rab7-positive (Rab7+) late endosomes when only the levels of inhibition of Arf1, p97/VCP, or both proteins were compared (Fig. 8D). Our experiments indicated that p97 and/or Arf1 inhibition increases Rem-CT retention in late endosomes. Together with our observations that inhibition of p97 rescues Rem-CT levels from BFA or DN Arf1 treatment (Fig. 7B and C), our immunostaining results suggested that reduced Rem-CT in the presence of BFA or DN Arf1 occurs in early endosomes or during retrograde trafficking from endosomes to the ER.
DISCUSSION
Our recent studies show that Rem C-terminal sequences antagonize the mutagenic effects of Apobecs, particularly by proteasomal degradation of activation-induced cytidine deaminase (AID) during MMTV replication in lymphocytes (10). Since Apobec proteins function in both the cytoplasm and nucleus (81), determination of Rem-CT cellular localization was key to understanding its role in Apobec antagonism. In this paper, we showed that despite lacking a C-terminal KDEL/KKXX sequence (31, 68) and transmembrane domains, the majority of Rem-CT remains in the ER at steady state. Furthermore, our data demonstrated that Rem-CT, like SP, has an unusual trafficking scheme that allows a small portion of the protein to localize, at least transiently, to early and late endosomes. Rem-CT trafficking does not involve passage through Golgi compartments that allow the addition of complex glycans (47, 48), and it requires p97/VCP. To our knowledge, no other viral or host proteins have been described with such a trafficking pathway.
A primary indicator of this unique pathway was our observation that BFA, a fungal metabolite that disrupts intracellular membrane trafficking between the ER and Golgi (39, 41, 43), reduced Rem-CT levels. We identified several factors that affected the sensitivity of Rem-CT to BFA. First, elimination of glycosylation sites either through substitution or deletion mutation prevented the reduction of Rem-CT levels in the presence of BFA. Our results indicated that glycosylated Rem-CT contains only simple glycans, as determined by sensitivity to Endo H cleavage (Fig. 1D). Thus, Rem-CT does not traffic through the medial-Golgi where modification to complex glycans occurs (48). Second, coexpression of Rem-CT and a DN form of Arf1 mimics the effects of BFA (Fig. 7A and C). Arf1 participates in multiple parts of the secretory pathway, and the Golgi-specific BFA resistance guanine nucleotide exchange factor (GBF) and BFA-inhibited guanine nucleotide-exchange protein (BIG) subfamilies of Arf1 GEFs are affected in the presence of BFA by stabilizing the inactive GEF-Arf1-GDP complex (72). Third, Rem-CT is not detectably secreted in the presence or absence of BFA (Fig. 1B and 3F), and we cannot rescue Rem-CT in the presence of BFA using known inhibitors of degradation pathways like the proteasome or lysosome (Fig. 2 and 3). In addition, inhibitors of caspases, serine proteases, and cysteine proteases fail to rescue Rem-CT from BFA treatment (Fig. 3). Fourth, Rem transcription and translation are not reduced by BFA since the Rem precursor and SP expression are unaffected (Fig. 1, 5, and 7). Fifth, a DN form of p97/VCP (p97QQ) prevents decreased Rem-CT levels in the presence of BFA (Fig. 7B). Since p97 is involved in multiple cellular processes that involve the assembly and disassembly of large protein complexes, as well as membrane or vesicle fusions, we propose that Rem-CT has a function that requires trafficking outside the ER.
To our knowledge, Rem-CT is the only reported viral protein that displays this type of BFA sensitivity. Previous data suggested that two other cellular proteins, caspase 3 (CASP3) and Golgi-localized endoplasmic reticulum α-1,2-mannosidase (ER-ManI), have lower intracellular levels in the presence of BFA (82, 83). CASP3 levels were rescued by a proteasome inhibitor, and this effect was observed only in one ovarian cell line, W1PR (82). This situation differs from that of Rem-CT, since the proteasome inhibitor MG132 showed weak restoration of the unglycosylated Rem-CT levels even when MG132 was added prior to BFA (Fig. 2A). Moreover, the BFA sensitivity of Rem-CT appears to involve a conserved pathway, since we observed this effect in both human embryonic kidney and mouse mammary epithelial cells (Fig. 1B and 2). ER-ManI in the medial-Golgi binds the gamma subunit of COPI (84) to mediate retrieval of ERAD substrates from the Golgi to the ER (83). This study did not report a mechanism for reduced ER-ManI levels in the presence of BFA. BFA collapses the cis-Golgi network to fuse with the ER (39, 41, 42) and the trans-Golgi to fuse with endosomes to form a tubular endosomal structure (85, 86). It is possible that proteases in the Golgi degrade Rem-CT after BFA induces an ER-Golgi fusion network or that Rem-CT is degraded in the tubular endosomal structure.
The majority of the wild-type protein and the N127Q/N143Q glycosylation mutant colocalized with the ER marker (Fig. 4), whereas portions of both proteins traffic to the late endosomes (Fig. 6 and 8). In contrast, the N127Q/N143Q mutant showed significantly less colocalization with the early endosomal marker (Rab5) than did wild-type Rem-CT (Fig. 6A and B). Since inhibition of p97 rescued the Rem-CT levels following BFA treatment or the DN Arf1 reduced Rem-CT levels (Fig. 7B and C), our confocal microscopy results (Fig. 8) showed that without a functional p97, wild-type Rem-CT could not reach the early endosomes but was trapped in the late endosomes. Inhibition of Arf1 resulted in Rem-CT accumulation in early and late endosomes, suggesting that DN Arf1 or BFA effects occur after Rem-CT reaches the early endosomes or during the Rem-CT retrograde-trafficking process (Fig. 9). The ER and endosomes have numerous contact sites for cholesterol transfer, endosome positioning, and trans-Golgi-to-endosome protein transport, as well as receptor dephosphorylation and endosome fission (87–92). Trafficking through the endosomal system may allow Rem-CT to counteract cellular proteins that function in host antiviral defenses. For example, tetherin/BST-2 localizes to the cell surface and endosomes through the classical ER-to-Golgi transport system to inhibit viral release. The HIV-1-encoded Vpu protein reverses this inhibition by redirecting BST-2 from the cell surface to late endosomes (93–96). Moreover, major histocompatibility complex class I (MHC-I), which is exported through the classical ER-Golgi pathway to the cell surface, is sequestered by HIV-1-encoded Nef within early endosomes. Redirection of MHC-I by Nef into late endosomes allows HIV-1 to evade immune responses (97).
FIG 9.

Model for Rem-CT trafficking. Rem is directed to the ER membrane for translation and cleaved by signal peptidase into an N-terminal SP and Rem-CT. SP is retrotranslocated into the cytosol by the p97/VCP ATPase and traffics to the nucleus to perform a Rev-like function (4, 7–9). The Rem-CT cleavage product is glycosylated with simple glycans and maintains high steady-state levels in the ER lumen. Both glycosylated and unglycosylated Rem-CT exit the ER to the ERGIC and late endosomes. Only glycosylated Rem-CT is detectable in early endosomes (Rab5+), and inhibition of p97 activity reduces anterograde trafficking of Rem-CT from early endosomes to late endosomes, leading to accumulation in late endosomes (Rab7+). Inhibition of retrograde trafficking by the DN Arf1 also leads to Rem-CT accumulation in early endosomes and late endosomes. This unique scheme may allow Rem-CT-mediated capture or degradation of host transmembrane or secreted proteins that traffic through the traditional secretory pathway, thus thwarting the immune response. Rem (no CHO), Rem lacking glycosylation (carbohydrate).
Recent evidence from plant cell experiments indicates that BFA relocalizes a number of Golgi enzymes to the ER. Specific enzymes associated with cis-Golgi trafficking using a COPII-independent mechanism enter a compartment known as the Golgi entry core compartment (GECCO) near ER exit sites (ERES) in the presence of BFA (84). GECCO may be similar to the ERGIC, which serves as a sorting station for proteins at ERES (98, 99). Our data have shown that a portion of Rem-CT is present in the ERGIC (Fig. 4F and G). In addition, some proteins that lack a KDEL/KKXX motif are subjected to retrograde transport to the ER by binding to other KDEL-containing proteins (100). We propose that after acquisition of simple glycans in the ER, Rem-CT traffics to the ERGIC. Rem-CT anterograde sorting to early endosomes is dependent on p97/VCP, whereas Arf1 is required for retrograde trafficking (Fig. 9) (101). This model leaves many unanswered questions, and yet, the results of these experiments suggest that Rem-CT must reach the early endosomes for BFA sensitivity. In addition, our results predict that uncleaved Rem, rather than Rem-CT, may be the antagonist of Apobec family enzymes (10), since Apobecs are not known to reside in cellular membrane compartments where Rem-CT is localized.
In summary, our study identifies the first viral protein that uses p97-dependent trafficking from the ER to the early endosomes without transit through the Golgi network. Determining the benefit of this pathway for viruses and/or host cell proteins requires additional experiments. Thus, the study of viruses continues to elucidate novel cell processes that allow the development of new antiviral and cancer therapies.
MATERIALS AND METHODS
Cell culture and transfection.
HEK293 or 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Sigma-Aldrich) supplemented with 10% fetal calf serum, gentamicin sulfate (50 μg/ml), penicillin (100 units/ml), and streptomycin (50 μg/ml). HC11 cells were maintained in RPMI medium (Sigma-Aldrich) supplemented with 10% fetal calf serum, gentamicin sulfate (50 μg/ml), insulin (0.5 μg/ml), and epidermal growth factor (EGF) (1 μg/ml). Cells were routinely tested for contamination with bacteria using a PCR-based assay. Transfections of HEK293 or 293T cells were performed using polyethylenimine (PEI) (Polysciences) transfection. Briefly, 1 × 106 cells were seeded into each well of a 6-well plate on the day prior to transfection. DNA (3 μg) was mixed with 200 μl of DMEM and 9 μl of PEI (1 μg/μl) and incubated at room temperature for 20 min before adding to preseeded cells. At 7 h posttransfection, cells were washed with phosphate-buffered saline (PBS) once and replaced with fresh medium. Transfections of HC11 cells were performed using Lipofectamine 3000 (Thermo Fisher Scientific), following the protocols from the manufacturer. Transfection experiments with DN Arf1 were performed by electroporation of 7.5 × 106 HC11 cells with 40 μg of DNA at 260 V, 950 μF for ∼20 ms. Cells were harvested 48 h after transfection. For some experiments, as indicated in the figure legends, BFA (Cell Signaling Technology), MG132 (Boston Biochem), bafilomycin A1 (Sigma-Aldrich), chloroquine (Sigma-Aldrich), Z-VAD(OMe)-FMK (Cell Signaling Technology), E64d (Sigma-Aldrich), or AEBSF (Sigma-Aldrich) was added at the indicated concentrations and times before cells were harvested. Cells were treated with the p97/VCP inhibitor CB-5083 (MedChemExpress) for 9 h prior to analysis by Western blotting or confocal microscopy. Endoglycosidase H (Endo H) (catalog number P0702S; NEB) (500 U) was added to 20 μg of cell extracts for 1 h at 37°C prior to analysis by Western blotting.
Plasmid construction and sources.
GFP-Rem-T7 and Rem-T7 plasmids were constructed based on previously described N-terminally GFP-tagged (GFP-Rem) or untagged Rem constructs (3) with the addition of six glycine linkers and the T7 tag sequence (MASMTGGQQMG) at the C terminus as indicated. CMV-SEAP was a gift from Alan Cochrane (Addgene plasmid number 24595; RRID Addgene_24595). The CMV-SEAP-T7 construct was made based on plasmid CMV-SEAP with the addition of six glycine linkers and the T7 tag at the C terminus. The linkers and the T7 tag were added using PCR and CloneAmp HiFi (high-fidelity) PCR premix (TakaRa). Mutants (N127Q/N143Q mutant and mutants with motif substitutions) were obtained by mutagenesis PCR using CloneAmp HiFi PCR premix (TakaRa). The GFP-tagged N127Q/N143Q mutant was constructed by replacement of asparagines with glutamines at amino acids 127 and 143 by site-directed mutagenesis (Stratagene). The GFP-Rem-T7 partial deletion mutants (GFP-RemΔ103–155-T7, GFP-RemΔ155–198-T7, GFP-RemΔ198–247-T7, and GFP-RemΔ247–301-T7 mutants) were constructed from GFP-Rem-T7 similarly by deleting the designated 50-amino-acid segments from the Rem C terminus. These mutations were prepared by inserting SalI sites into the rem gene by site-directed mutagenesis at the designated codon sites, followed by digestion with SalI and religation of the vector. Forward and reverse primers were used for PCR in separate tubes with the following parameters: 98°C for 1 min, 5 cycles at 98°C for 10 s, 55°C for 30s, and 72°C for 3.5 min (or 0.5 min per kb, based on the final product length) with a final extension at 72°C for 30 min. Subsequently, the forward and reverse primer PCRs were mixed and used for additional cycles: 98°C for 1 min, followed by 20 cycles at 98°C for 10 s, 55°C for 30 s, and 72°C for 3.5 min (or 0.5 min per kb, based on the final product length) with a final extension at 72°C for 30 min. All constructs were sequenced to confirm the mutations, and restriction digests were used to confirm the plasmid integrity.
The ER-mCherry expression plasmid was a gift from N. Hosokawa (Kyoto University, Kyoto, Japan) (66). The HA Arf1 DN (T31N) plasmid was a gift from Thomas Roberts (Addgene plasmid number 10833; RRID Addgene_10833) (102). The CFP-VSV-G construct was a gift from Michael Davidson (Addgene plasmid number 55397; RRID Addgene_55397).
Luciferase reporter assays.
The SP-responsive reporter assays have been described previously (7–9) using the pHMRluc Rem-responsive construct (4). The pHMRluc plasmid contains the 3′ end of the C3H-MMTV genome missing the MMTV SP coding sequence. Insertion of the Renilla luciferase reporter gene between splice donor and acceptor sites within the MMTV envelope gene avoided interruption of the Rem-responsive element (4). Briefly, transfections were performed in triplicate by cotransfection of the pHMRluc Rem-responsive construct and a Rem-nonresponsive firefly luciferase reporter construct (pGL3-Control; Promega) in the presence and absence of a Rem expression plasmid. After 48 h, transfected cells were lysed in buffers provided with the Dual-Luciferase assay kit (Promega). Protein concentrations were determined (Bio-Rad protein assay system), and lysates (∼40 μg) were used to detect firefly and Renilla luciferase activities. Rem-responsive Renilla luciferase data were then normalized to Rem-nonresponsive firefly luciferase levels to control for differences in transfection efficiency. Values are reported for 100 μg of protein lysate. The mean values of normalized Renilla luciferase activities for triplicate transfections and standard deviations are shown.
Western blotting.
Western blots were performed as previously described (8). Monoclonal antibodies to the following proteins were obtained from the sources named in parentheses: T7 (Cell Signaling Technology), GFP (Clontech), GAPDH (Cell Signaling Technology), p97/VCP (Invitrogen), PARP1 (Cell Signaling Technology), and LC3B (Cell Signaling Technology). Horseradish peroxidase-conjugated secondary antibodies were obtained from Cell Signaling Technology. Fluorescently labeled IRDye secondary antibodies were purchased from LI-COR Biosciences.
Cell supernatant concentration.
After cells were treated with BFA, the medium was collected and subjected to centrifugation to remove cell debris. Supernatants were filtered through a 0.45-μm filter. Supernatants were concentrated based on the manufacturer’s instructions. Briefly, Amicon ultra-15 centrifugal filter units were rinsed with Milli-Q water before use, and filtered supernatants were added to each unit. Filter units were then subjected to centrifugation at 4,000 × g for 40 min, and the concentrated solute was recovered from the unit for Western blotting.
Exosome isolation.
After cells were treated with BFA, the medium was collected and subjected to centrifugation to remove cell debris. Supernatants were filtered through a 0.45-μm filter. The miRCURY exosome kits were purchased from Qiagen (catalog number 76743), and exosome isolation was performed according to the manufacturer’s instructions. Briefly, 0.4 volume of precipitation buffer B was added for every 1 volume of the sample and mixed thoroughly. Following an overnight precipitation at 4°C, samples were centrifuged and supernatants were removed. Resuspension buffer was used for pellets, which were processed for Western blotting.
Cellular fractionation.
Transfected cells were harvested 48 h posttransfection. Cells were washed with PBS twice to remove the residual medium and then permeabilized for 5 min on ice with 0.02% (wt/vol) digitonin using a fractionation buffer containing Roche protease inhibitor cocktail. The cytosolic fraction was isolated from the cells by centrifugation at 13,000 × g for 5 min at 4°C. The remaining pellet was incubated in 1% Triton X-100 for 5 min on ice, followed by a second centrifugation at 13,000 × g for 5 min at 4°C to separate the membrane and nuclear fractions.
Immunofluorescence and image analysis.
On the day of transfection, coverslips were washed with ethanol, placed into 6-well plates, and dried under UV light in a laminar flow hood. After 24 h, coverslips were washed with PBS once, and ∼0.6 × 106 cells were trypsinized and plated in each well. Transfected cells were incubated for 48 h and fixed with 4% paraformaldehyde in PBS for 15 min. Cells were permeabilized with 0.1% Triton X-100 and then washed three times with PBS containing 0.1% Tween 20 (PBST) for 5 min each. Cells were then blocked with blocking buffer (2% fetal bovine serum [FBS], 0.1% Tween 20, 150 mM NaCl, 10 mM Tris-HCl, pH 7.4) for 1 h at room temperature and then incubated with primary antibodies overnight at 4°C. After three PBST washes following the primary antibody incubation, Alexa Fluor secondary antibodies (Invitrogen, Thermo Fisher Scientific) were added for 2 h at room temperature. Samples were then washed thrice with PBST and stained with DAPI (4′,6-diamidino-2-phenylindole) (150 nM) for 10 min at room temperature. After three PBST washes, coverslips were mounted on slides with Vectashield antifade mounting medium (Vector Laboratories) and sealed for fluorescence imaging. Monoclonal Rab5- and Rab7-specific antibodies were purchased from Cell Signaling Technology. When two different primary antibodies were used, the first primary antibody was incubated overnight before samples were washed with PBST three times. The second primary antibody was then incubated with the sample overnight. During secondary antibody incubation, Alexa Fluor secondary antibodies with different spectral characteristics from different animal sources were used to ensure recognition of only one primary antibody.
The Zeiss LSM 710 confocal and Elyra S.1 structured-illumination (SIM) superresolution microscope was used to capture Z-stack sample images. All slices in a Z-stack across samples were kept at 0.33 μm per slice interval. Lasers (405 nm, 488 nm, and 561 nm) were used with the same power and gain across samples to obtain images. Quantitation and image processing were performed using Zeiss ZEN software, GraphPad (Prism), and ImageJ. Briefly, Mander’s colocalization coefficient (67) was used to quantify the fraction of Rem-CT colocalized with ER-mCherry or other markers (ERGIC-53, Rab5, and Rab7), using pixels from each cell in single slices and ZEN software. Mann-Whitney t tests were used to compare different samples in GraphPad. Images were processed in ImageJ.
Reproducibility and statistical analysis.
All experiments were performed two to five times with similar results. Individual statistical tests are indicated in each section or figure. A P value of <0.05 was considered to be significant.
ACKNOWLEDGMENTS
We thank members of the Dudley laboratory for helpful comments on experimental design and the manuscript. We also are grateful for advice from the Microscopy and Imaging Core facilities at The University of Texas at Austin. We thank John Digiovanni at UT Austin for his kind donation of the PARP1-specific antibody.
This work was supported by National Institutes of Health R01 grant numbers CA167053 and AI131660.
Contributor Information
Jaquelin P. Dudley, Email: jdudley@austin.utexas.edu.
Viviana Simon, Icahn School of Medicine at Mount Sinai.
REFERENCES
- 1.Dudley JP, Golovkina TV, Ross SR. 2016. Lessons learned from mouse mammary tumor virus in animal models. ILAR J 57:12–23. 10.1093/ilar/ilv044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mertz JA, Chadee AB, Byun H, Russell R, Dudley JP. 2009. Mapping of the functional boundaries and secondary structure of the mouse mammary tumor virus Rem-responsive element. J Biol Chem 284:25642–25652. 10.1074/jbc.M109.012476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mertz JA, Lozano MM, Dudley JP. 2009. Rev and Rex proteins of human complex retroviruses function with the MMTV Rem-responsive element. Retrovirology 6:10. 10.1186/1742-4690-6-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mertz JA, Simper MS, Lozano MM, Payne SM, Dudley JP. 2005. Mouse mammary tumor virus encodes a self-regulatory RNA export protein and is a complex retrovirus. J Virol 79:14737–14747. 10.1128/JVI.79.23.14737-14747.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golovkina TV, Dudley JP, Jaffe AB, Ross SR. 1995. Mouse mammary tumor viruses with functional superantigen genes are selected during in vivo infection. Proc Natl Acad Sci U S A 92:4828–4832. 10.1073/pnas.92.11.4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bergman AC, Bjornberg O, Nord J, Nyman PO, Rosengren AM. 1994. The protein p30, encoded at the gag-pro junction of mouse mammary tumor virus, is a dUTPase fused with a nucleocapsid protein. Virology 204:420–424. 10.1006/viro.1994.1547. [DOI] [PubMed] [Google Scholar]
- 7.Byun H, Halani N, Gou Y, Nash AK, Lozano MM, Dudley JP. 2012. Requirements for mouse mammary tumor virus Rem signal peptide processing and function. J Virol 86:214–225. 10.1128/JVI.06197-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byun H, Das P, Yu H, Aleman A, Lozano MM, Matouschek A, Dudley JP. 2017. Mouse mammary tumor virus signal peptide uses a novel p97-dependent and Derlin-independent retrotranslocation mechanism to escape proteasomal degradation. mBio 8:e00328-17. 10.1128/mBio.00328-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Byun H, Halani N, Mertz JA, Ali AF, Lozano MM, Dudley JP. 2010. Retroviral Rem protein requires processing by signal peptidase and retrotranslocation for nuclear function. Proc Natl Acad Sci U S A 107:12287–12292. 10.1073/pnas.1004303107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh GB, Byun H, Ali AF, Medina F, Wylie D, Shivram H, Nash AK, Lozano MM, Dudley JP. 2019. A protein antagonist of activation-induced cytidine deaminase encoded by a complex mouse retrovirus. mBio 10:e01678-19. 10.1128/mBio.01678-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. 2002. Transport from the ER through the Golgi apparatus. In Molecular Biology of the Cell, Garland Science, New York, NY. [Google Scholar]
- 12.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. 2002. Membrane proteins. In Molecular biology of the cell, Garland Science, New York, NY. [Google Scholar]
- 13.Lodish H, Berk A, Zipursky SL, Matsudaira P, Baltimore D, Darnell J. 2000. Molecular mechanisms of vesicular traffic. In Molecular cell biology, W.H. Freeman, New York, NY. [Google Scholar]
- 14.Sztul E, Chen PW, Casanova JE, Cherfils J, Dacks JB, Lambright DG, Lee FS, Randazzo PA, Santy LC, Schurmann A, Wilhelmi I, Yohe ME, Kahn RA. 2019. ARF GTPases and their GEFs and GAPs: concepts and challenges. Mol Biol Cell 30:1249–1271. 10.1091/mbc.E18-12-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu X, Breitman M, Goldberg J. 2012. A structure-based mechanism for Arf1-dependent recruitment of coatomer to membranes. Cell 148:530–542. 10.1016/j.cell.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stearns T, Willingham MC, Botstein D, Kahn RA. 1990. ADP-ribosylation factor is functionally and physically associated with the Golgi complex. Proc Natl Acad Sci U S A 87:1238–1242. 10.1073/pnas.87.3.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spang A, Matsuoka K, Hamamoto S, Schekman R, Orci L. 1998. Coatomer, Arf1p, and nucleotide are required to bud coat protein complex I-coated vesicles from large synthetic liposomes. Proc Natl Acad Sci U S A 95:11199–11204. 10.1073/pnas.95.19.11199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traub LM, Ostrom JA, Kornfeld S. 1993. Biochemical dissection of AP-1 recruitment onto Golgi membranes. J Cell Biol 123:561–573. 10.1083/jcb.123.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stamnes MA, Rothman JE. 1993. The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell 73:999–1005. 10.1016/0092-8674(93)90277-W. [DOI] [PubMed] [Google Scholar]
- 20.Sauvageau E, McCormick PJ, Lefrancois S. 2017. In vivo monitoring of the recruitment and activation of AP-1 by Arf1. Sci Rep 7:7148. 10.1038/s41598-017-07493-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta GD, Swetha MG, Kumari S, Lakshminarayan R, Dey G, Mayor S. 2009. Analysis of endocytic pathways in Drosophila cells reveals a conserved role for GBF1 in internalization via GEECs. PLoS One 4:e6768. 10.1371/journal.pone.0006768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hemalatha A, Prabhakara C, Mayor S. 2016. Endocytosis of Wingless via a dynamin-independent pathway is necessary for signaling in Drosophila wing discs. Proc Natl Acad Sci U S A 113:E6993–E7002. 10.1073/pnas.1610565113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mazaki Y, Nishimura Y, Sabe H. 2012. GBF1 bears a novel phosphatidylinositol-phosphate binding module, BP3K, to link PI3Kgamma activity with Arf1 activation involved in GPCR-mediated neutrophil chemotaxis and superoxide production. Mol Biol Cell 23:2457–2467. 10.1091/mbc.E12-01-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bayliss R, Wheeldon J, Caucheteux SM, Niessen CM, Piguet V. 2020. Identification of host trafficking genes required for HIV-1 virological synapse formation in dendritic cells. J Virol 94:e01597-19. 10.1128/JVI.01597-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg-Lerner A, Benyair R, Hizkiahou N, Nudel N, Maor R, Kramer MP, Shmueli MD, Zigdon I, Cherniavsky Lev M, Ulman A, Sagiv JY, Dayan M, Dassa B, Rosenwald M, Shachar I, Li J, Wang Y, Dezorella N, Khan S, Porat Z, Shimoni E, Avinoam O, Merbl Y. 2020. Golgi organization is regulated by proteasomal degradation. Nat Commun 11:409. 10.1038/s41467-019-14038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ye Y, Tang WK, Zhang T, Xia D. 2017. A mighty “protein extractor” of the cell: structure and function of the p97/CDC48 ATPase. Front Mol Biosci 4:39. 10.3389/fmolb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wong HH, Kumar P, Tay FP, Moreau D, Liu DX, Bard F. 2015. Genome-wide screen reveals valosin-containing protein requirement for coronavirus exit from endosomes. J Virol 89:11116–11128. 10.1128/JVI.01360-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Embgenbroich M, Burgdorf S. 2018. Current concepts of antigen cross-presentation. Front Immunol 9:1643. 10.3389/fimmu.2018.01643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wong LH, Eden ER, Futter CE. 2018. Roles for ER:endosome membrane contact sites in ligand-stimulated intraluminal vesicle formation. Biochem Soc Trans 46:1055–1062. 10.1042/BST20170432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klausner RD, Donaldson JG, Lippincott-Schwartz J. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J Cell Biol 116:1071–1080. 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vincent MJ, Martin AS, Compans RW. 1998. Function of the KKXX motif in endoplasmic reticulum retrieval of a transmembrane protein depends on the length and structure of the cytoplasmic domain. J Biol Chem 273:950–956. 10.1074/jbc.273.2.950. [DOI] [PubMed] [Google Scholar]
- 32.Gaynor EC, Te Heesen S, Graham TR, Aebi M, Emr SD. 1994. Signal-mediated retrieval of a membrane protein from the Golgi to the ER in yeast. J Cell Biol 127:653–665. 10.1083/jcb.127.3.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettan M, Darteil R, Scherman D. 1999. Secreted human placental alkaline phosphatase as a reporter gene for in vivo gene transfer. Anal Biochem 271:187–189. 10.1006/abio.1999.4144. [DOI] [PubMed] [Google Scholar]
- 34.Biggiogera M, Burki K, Kaufmann SH, Shaper JH, Gas N, Amalric F, Fakan S. 1990. Nucleolar distribution of proteins B23 and nucleolin in mouse preimplantation embryos as visualized by immunoelectron microscopy. Development 110:1263–1270. 10.1242/dev.110.4.1263. [DOI] [PubMed] [Google Scholar]
- 35.Bole DG, Dowin R, Doriaux M, Jamieson JD. 1989. Immunocytochemical localization of BiP to the rough endoplasmic reticulum: evidence for protein sorting by selective retention. J Histochem Cytochem 37:1817–1823. 10.1177/37.12.2685110. [DOI] [PubMed] [Google Scholar]
- 36.Sirover MA. 2012. Subcellular dynamics of multifunctional protein regulation: mechanisms of GAPDH intracellular translocation. J Cell Biochem 113:2193–2200. 10.1002/jcb.24113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tisdale EJ. 2002. Glyceraldehyde-3-phosphate dehydrogenase is phosphorylated by protein kinase Ciota/lambda and plays a role in microtubule dynamics in the early secretory pathway. J Biol Chem 277:3334–3341. 10.1074/jbc.M109744200. [DOI] [PubMed] [Google Scholar]
- 38.Schuerwegh AJ, Stevens WJ, Bridts CH, De Clerck LS. 2001. Evaluation of monensin and brefeldin A for flow cytometric determination of interleukin-1 beta, interleukin-6, and tumor necrosis factor-alpha in monocytes. Cytometry 46:172–176. 10.1002/cyto.1102. [DOI] [PubMed] [Google Scholar]
- 39.Doms RW, Russ G, Yewdell JW. 1989. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J Cell Biol 109:61–72. 10.1083/jcb.109.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lippincott-Schwartz J, Donaldson JG, Schweizer A, Berger EG, Hauri HP, Yuan LC, Klausner RD. 1990. Microtubule-dependent retrograde transport of proteins into the ER in the presence of brefeldin A suggests an ER recycling pathway. Cell 60:821–836. 10.1016/0092-8674(90)90096-w. [DOI] [PubMed] [Google Scholar]
- 41.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801–813. 10.1016/0092-8674(89)90685-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strous GJ, Berger EG, van Kerkhof P, Bosshart H, Berger B, Geuze HJ. 1991. Brefeldin A induces a microtubule-dependent fusion of galactosyltransferase-containing vesicles with the rough endoplasmic reticulum. Biol Cell 71:25–31. 10.1016/0248-4900(91)90048-r. [DOI] [PubMed] [Google Scholar]
- 43.Ulmer JB, Palade GE. 1991. Effects of brefeldin A on the Golgi complex, endoplasmic reticulum and viral envelope glycoproteins in murine erythroleukemia cells. Eur J Cell Biol 54:38–54. [PubMed] [Google Scholar]
- 44.Young WW, Jr, Lutz MS, Mills SE, Lechler-Osborn S. 1990. Use of brefeldin A to define sites of glycosphingolipid synthesis: GA2/GM2/GD2 synthase is trans to the brefeldin A block. Proc Natl Acad Sci U S A 87:6838–6842. 10.1073/pnas.87.17.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morinaga N, Tsai SC, Moss J, Vaughan M. 1996. Isolation of a brefeldin A-inhibited guanine nucleotide-exchange protein for ADP ribosylation factor (ARF) 1 and ARF3 that contains a Sec7-like domain. Proc Natl Acad Sci U S A 93:12856–12860. 10.1073/pnas.93.23.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helms JB, Rothman JE. 1992. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360:352–354. 10.1038/360352a0. [DOI] [PubMed] [Google Scholar]
- 47.Barlowe CK, Miller EA. 2013. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193:383–410. 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schwarz F, Aebi M. 2011. Mechanisms and principles of N-linked protein glycosylation. Curr Opin Struct Biol 21:576–582. 10.1016/j.sbi.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 49.Maley F, Trimble RB, Tarentino AL, Plummer TH, Jr.. 1989. Characterization of glycoproteins and their associated oligosaccharides through the use of endoglycosidases. Anal Biochem 180:195–204. 10.1016/0003-2697(89)90115-2. [DOI] [PubMed] [Google Scholar]
- 50.Alvarez C, Fujita H, Hubbard A, Sztul E. 1999. ER to Golgi transport: requirement for p115 at a pre-Golgi VTC stage. J Cell Biol 147:1205–1222. 10.1083/jcb.147.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dultz E, Hildenbeutel M, Martoglio B, Hochman J, Dobberstein B, Kapp K. 2008. The signal peptide of the mouse mammary tumor virus Rem protein is released from the endoplasmic reticulum membrane and accumulates in nucleoli. J Biol Chem 283:9966–9976. 10.1074/jbc.M705712200. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe Y, Bowden TA, Wilson IA, Crispin M. 2019. Exploitation of glycosylation in enveloped virus pathobiology. Biochim Biophys Acta Gen Subj 1863:1480–1497. 10.1016/j.bbagen.2019.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Byun H, Gou Y, Zook A, Lozano MM, Dudley JP. 2014. ERAD and how viruses exploit it. Front Microbiol 5:330. 10.3389/fmicb.2014.00330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Settembre C, Ballabio A. 2014. Lysosome: regulator of lipid degradation pathways. Trends Cell Biol 24:743–750. 10.1016/j.tcb.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Crider BP, Xie XS, Stone DK. 1994. Bafilomycin inhibits proton flow through the H+ channel of vacuolar proton pumps. J Biol Chem 269:17379–17381. 10.1016/S0021-9258(17)32447-X. [DOI] [PubMed] [Google Scholar]
- 56.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. 2000. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19:5720–5728. 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Redmann M, Benavides GA, Berryhill TF, Wani WY, Ouyang X, Johnson MS, Ravi S, Barnes S, Darley-Usmar VM, Zhang J. 2017. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol 11:73–81. 10.1016/j.redox.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mauthe M, Orhon I, Rocchi C, Zhou X, Luhr M, Hijlkema KJ, Coppes RP, Engedal N, Mari M, Reggiori F. 2018. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14:1435–1455. 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. 1996. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J 315:21–24. 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. 1995. Yama/CPP32 beta, a mammalian homolog of CED-3, is a CrmA-inhibitable protease that cleaves the death substrate poly(ADP-ribose) polymerase. Cell 81:801–809. 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 61.Tamai M, Matsumoto K, Omura S, Koyama I, Ozawa Y, Hanada K. 1986. In vitro and in vivo inhibition of cysteine proteinases by EST, a new analog of E-64. J Pharmacobiodyn 9:672–677. 10.1248/bpb1978.9.672. [DOI] [PubMed] [Google Scholar]
- 62.Zhang X, Yang X, Wang H, Li S, Guo K, Jiang D, Xiao J, Liang D. 2017. Design, synthesis, and structure-activity relationship study of epoxysuccinyl-peptide derivatives as cathepsin B inhibitors. Biol Pharm Bull 40:1240–1246. 10.1248/bpb.b17-00075. [DOI] [PubMed] [Google Scholar]
- 63.Tamai M, Hanada K, Adachi T, Oguma K, Kashiwagi K, Omura S, Ohzeki M. 1981. Papain inhibitions by optically active E-64 analogs. J Biochem 90:255–257. 10.1093/oxfordjournals.jbchem.a133458. [DOI] [PubMed] [Google Scholar]
- 64.Gold AM, Fahrney D. 1964. Sulfonyl fluorides as inhibitors of esterases. II. Formation and reactions of phenylmethanesulfonyl alpha-chymotrypsin. Biochemistry 3:783–791. 10.1021/bi00894a009. [DOI] [PubMed] [Google Scholar]
- 65.Jella KK, Yu L, Yue Q, Friedman D, Duke BJ, Alli AA. 2016. Exosomal GAPDH from proximal tubule cells regulate ENaC activity. PLoS One 11:e0165763. 10.1371/journal.pone.0165763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hosokawa N, Wada I, Nagasawa K, Moriyama T, Okawa K, Nagata K. 2008. Human XTP3-B forms an endoplasmic reticulum quality control scaffold with the HRD1-SEL1L ubiquitin ligase complex and BiP. J Biol Chem 283:20914–20924. 10.1074/jbc.M709336200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc 169:375–382. 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- 68.Stornaiuolo M, Lotti LV, Borgese N, Torrisi MR, Mottola G, Martire G, Bonatti S. 2003. KDEL and KKXX retrieval signals appended to the same reporter protein determine different trafficking between endoplasmic reticulum, intermediate compartment, and Golgi complex. Mol Biol Cell 14:889–902. 10.1091/mbc.e02-08-0468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tu Y, Zhao L, Billadeau DD, Jia D. 2020. Endosome-to-TGN trafficking: organelle-vesicle and organelle-organelle interactions. Front Cell Dev Biol 8:163. 10.3389/fcell.2020.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorvel JP, Chavrier P, Zerial M, Gruenberg J. 1991. rab5 controls early endosome fusion in vitro. Cell 64:915–925. 10.1016/0092-8674(91)90316-Q. [DOI] [PubMed] [Google Scholar]
- 71.Hu YB, Dammer EB, Ren RJ, Wang G. 2015. The endosomal-lysosomal system: from acidification and cargo sorting to neurodegeneration. Transl Neurodegener 4:18. 10.1186/s40035-015-0041-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jackson CL. 2018. Activators and effectors of the small G protein Arf1 in regulation of Golgi dynamics during the cell division cycle. Front Cell Dev Biol 6:29. 10.3389/fcell.2018.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Peyroche A, Antonny B, Robineau S, Acker J, Cherfils J, Jackson CL. 1999. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol Cell 3:275–285. 10.1016/s1097-2765(00)80455-4. [DOI] [PubMed] [Google Scholar]
- 74.Meyer HH. 2005. Golgi reassembly after mitosis: the AAA family meets the ubiquitin family. Biochim Biophys Acta 1744:481–492. [PubMed] [Google Scholar]
- 75.Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. 1998. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell 92:603–610. 10.1016/s0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- 76.Ritz D, Vuk M, Kirchner P, Bug M, Schutz S, Hayer A, Bremer S, Lusk C, Baloh RH, Lee H, Glatter T, Gstaiger M, Aebersold R, Weihl CC, Meyer H. 2011. Endolysosomal sorting of ubiquitylated caveolin-1 is regulated by VCP and UBXD1 and impaired by VCP disease mutations. Nat Cell Biol 13:1116–1123. 10.1038/ncb2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pleasure IT, Black MM, Keen JH. 1993. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature 365:459–462. 10.1038/365459a0. [DOI] [PubMed] [Google Scholar]
- 78.Ramanathan HN, Ye Y. 2012. The p97 ATPase associates with EEA1 to regulate the size of early endosomes. Cell Res 22:346–359. 10.1038/cr.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ye Y, Meyer HH, Rapoport TA. 2001. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414:652–656. 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 80.Anderson DJ, Le Moigne R, Djakovic S, Kumar B, Rice J, Wong S, Wang J, Yao B, Valle E, Kiss von Soly S, Madriaga A, Soriano F, Menon MK, Wu ZY, Kampmann M, Chen Y, Weissman JS, Aftab BT, Yakes FM, Shawver L, Zhou HJ, Wustrow D, Rolfe M. 2015. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell 28:653–665. 10.1016/j.ccell.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harris RS, Dudley JP. 2015. APOBECs and virus restriction. Virology 479-480:131–145. 10.1016/j.virol.2015.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wojtowicz K, Januchowski R, Sosińska P, Nowicki M, Zabel M. 2016. Effect of brefeldin A and castanospermine on resistant cell lines as supplements in anticancer therapy. Oncol Rep 35:2896–2906. 10.3892/or.2016.4656. [DOI] [PubMed] [Google Scholar]
- 83.Pan S, Cheng X, Sifers RN. 2013. Golgi-situated endoplasmic reticulum alpha-1,2-mannosidase contributes to the retrieval of ERAD substrates through a direct interaction with gamma-COP. Mol Biol Cell 24:1111–1121. 10.1091/mbc.E12-12-0886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ito Y, Uemura T, Nakano A. 2018. The Golgi entry core compartment functions as a COPII-independent scaffold for ER-to-Golgi transport in plant cells. J Cell Sci 131:jcs203893. 10.1242/jcs.203893. [DOI] [PubMed] [Google Scholar]
- 85.Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. 1991. Brefeldin A’s effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67:601–616. 10.1016/0092-8674(91)90534-6. [DOI] [PubMed] [Google Scholar]
- 86.Wood SA, Park JE, Brown WJ. 1991. Brefeldin A causes a microtubule-mediated fusion of the trans-Golgi network and early endosomes. Cell 67:591–600. 10.1016/0092-8674(91)90533-5. [DOI] [PubMed] [Google Scholar]
- 87.Dong R, Saheki Y, Swarup S, Lucast L, Harper JW, De Camilli P. 2016. Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166:408–423. 10.1016/j.cell.2016.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rocha N, Kuijl C, van der Kant R, Janssen L, Houben D, Janssen H, Zwart W, Neefjes J. 2009. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 Glued and late endosome positioning. J Cell Biol 185:1209–1225. 10.1083/jcb.200811005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eden ER, White IJ, Tsapara A, Futter CE. 2010. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat Cell Biol 12:267–272. 10.1038/ncb2026. [DOI] [PubMed] [Google Scholar]
- 90.Raiborg C, Wenzel EM, Pedersen NM, Olsvik H, Schink KO, Schultz SW, Vietri M, Nisi V, Bucci C, Brech A, Johansen T, Stenmark H. 2015. Repeated ER-endosome contacts promote endosome translocation and neurite outgrowth. Nature 520:234–238. 10.1038/nature14359. [DOI] [PubMed] [Google Scholar]
- 91.Rowland AA, Chitwood PJ, Phillips MJ, Voeltz GK. 2014. ER contact sites define the position and timing of endosome fission. Cell 159:1027–1041. 10.1016/j.cell.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Friedman JR, Dibenedetto JR, West M, Rowland AA, Voeltz GK. 2013. Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24:1030–1040. 10.1091/mbc.E12-10-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schmidt S, Fritz JV, Bitzegeio J, Fackler OT, Keppler OT. 2011. HIV-1 Vpu blocks recycling and biosynthetic transport of the intrinsic immunity factor CD317/tetherin to overcome the virion release restriction. mBio 2:e00036-11. 10.1128/mBio.00036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perez-Caballero D, Zang T, Ebrahimi A, McNatt MW, Gregory DA, Johnson MC, Bieniasz PD. 2009. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 139:499–511. 10.1016/j.cell.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Neil SJ, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–430. 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 96.Habermann A, Krijnse-Locker J, Oberwinkler H, Eckhardt M, Homann S, Andrew A, Strebel K, Krausslich HG. 2010. CD317/tetherin is enriched in the HIV-1 envelope and downregulated from the plasma membrane upon virus infection. J Virol 84:4646–4658. 10.1128/JVI.02421-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dirk BS, Pawlak EN, Johnson AL, Van Nynatten LR, Jacob RA, Heit B, Dikeakos JD. 2016. HIV-1 Nef sequesters MHC-I intracellularly by targeting early stages of endocytosis and recycling. Sci Rep 6:37021. 10.1038/srep37021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ito Y, Uemura T, Shoda K, Fujimoto M, Ueda T, Nakano A. 2012. cis-Golgi proteins accumulate near the ER exit sites and act as the scaffold for Golgi regeneration after brefeldin A treatment in tobacco BY-2 cells. Mol Biol Cell 23:3203–3214. 10.1091/mbc.E12-01-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sesso A, de Faria FP, Iwamura ES, Correa H. 1994. A three-dimensional reconstruction study of the rough ER-Golgi interface in serial thin sections of the pancreatic acinar cell of the rat. J Cell Sci 107:517–528. 10.1242/jcs.107.3.517. [DOI] [PubMed] [Google Scholar]
- 100.Kakihana T, Araki K, Vavassori S, Iemura S, Cortini M, Fagioli C, Natsume T, Sitia R, Nagata K. 2013. Dynamic regulation of Ero1alpha and peroxiredoxin 4 localization in the secretory pathway. J Biol Chem 288:29586–29594. 10.1074/jbc.M113.467845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kaczmarek B, Verbavatz JM, Jackson CL. 2017. GBF1 and Arf1 function in vesicular trafficking, lipid homoeostasis and organelle dynamics. Biol Cell 109:391–399. 10.1111/boc.201700042. [DOI] [PubMed] [Google Scholar]
- 102.Furman C, Short SM, Subramanian RR, Zetter BR, Roberts TM. 2002. DEF-1/ASAP1 is a GTPase-activating protein (GAP) for ARF1 that enhances cell motility through a GAP-dependent mechanism. J Biol Chem 277:7962–7969. 10.1074/jbc.M109149200. [DOI] [PubMed] [Google Scholar]