

Advances in amyotrophic lateral sclerosis (ALS) research: Research in ALS has gained unprecedented momentum in recent years fueled by important conceptual developments, establishment of international consortia, breakthrough genetic discoveries and relentless technological advances. The first genotype-specific pharmaceutical trials signal the paradigm shift from the notion of ’one-drug-for-all’ to precision, individualized therapies. The once arcane presymptomatic phase of the disease is gradually unraveled by seminal studies of asymptomatic mutation carriers (Geevasinga et al., 2015; Querin et al., 2019). The meticulous analysis of data from large population-based registries has contributed to the identification of etiological factors, genetic risk profiles, epigenetic and environmental modifiers. Progression patterns have been characterized in vivo by robust clinical, neurophysiology and neuroimaging studies and led to the development of clinical staging systems and biomarkers with practical utility in clinical trials (Chipika et al., 2019). While ALS was once considered a ’pure’ motor system disorder, it is now widely regarded as multisystem condition with frontotemporal, cerebellar, and subcortical grey matter involvement and a range of extrapyramidal, cognitive, and behavioral manifestations (Elamin et al., 2017). Disease-specific functional rating scales are now routinely used and screening instruments have been developed to assess the most commonly affected cognitive and behavioral domains in ALS. Advances in genetics paved the way for the first large presymptomatic studies which confirmed considerable cerebral and spinal cord alterations decades before symptom manifestation (Vucic et al., 2008; Querin et al., 2019). The characterization of genotype-associated molecular cascades, pathological signatures and clinical features were important milestones for the development of novel therapies, and the first antisense oligonucleotide trials are now underway. The datasets generated by multicenter initiatives offer unprecedented data mining opportunities; clustering patterns, prognostic determinants, and reliable diagnostic indicators were identified using machine-learning approaches that could not have previously been applied to smaller datasets. Technological advances in electrophysiology and the emergence of magnetoencephalography generated important functional insights (Bede et al., 2018). Novel imaging modalities, such as multi-voxel spectroscopy, spinal cord imaging, diffusion kurtosis imaging captured pathological changes that were previously impossible to ascertain in vivo (Bede et al., 2017; Huang et al., 2020). Advanced neurophysiology techniques, such as transcranial magnetic stimulation or motor unit number estimation are now widely used in both clinical and academic settings and contribute to diagnostic clarification and the monitoring of individual patients. In response to the inevitable sample size limitations of single-center studies (Schuster et al., 2016), ambitious international initiatives such as Project MinE established large biobanks to conduct genetic studies with sufficient statistical power. Societies such as NISALS provide pioneering frameworks to conduct large multicenter neuroimaging studies.
Barriers to drug development: Despite the coordinated work of large research centers, research consortia, patient charities, advocacy groups and pharmacological companies, relatively limited progress has been made in the development of effective disease-modifying therapies. The barriers to successful drug development in ALS include the marked clinical heterogeneity of the condition, the relatively late inclusion of patients into clinical trials, and an inadvertent selection bias to patients with limited cognitive impairment, who may live closer to research centers and who may have been diagnosed relatively early. Clinical heterogeneity in ALS is multidimensional and encompasses considerable differences in age of onset, progression rates, extra-motor manifestations, bulbar versus limb disability, lower- versus upper motor neuron predominance. The considerable differences in clinical profiles necessitate individualized management and a series of well-timed multidisciplinary interventions such as feeding-tube placement, initiation of non-invasive ventilation and ultimately palliative measures tailored to the patient’s specific medical needs and care preferences. While the benefits of individualized clinical care in contrast to a blanket strategy are widely accepted, the ill-conceived expectation that a single drug may be useful for all patients with ALS prevails. It is increasingly clear that unique genotype-associated clinical profiles exist and patients with specific mutations may have relatively distinct disease trajectories. It is also apparent that considerable phenotypic differences exist in survival, progression rates and disability profiles. It is therefore likely that patients may benefit from individualized pharmacological interventions tailored to their genotype, phenotype and disease-stage as opposed to the notion of ’one-drug-for-all’. Despite the enthusiasm generated by the first antisense oligonucleotide studies, it is noteworthy that the vast majority of patients with ALS are seemingly sporadic and test negative for large panels of mutations linked to ALS such as SOD1, ALS2, SETX, SPG11, FUS, VAPB, ANG, TARDBP, FIG4, OPTN, ATXN2, VCP, C9orf72, UBQLN2, SQSTM1, NEK1, FUS, TBK1 etc. Accordingly, the majority of patients with ALS are not candidates for genotype-specific interventions, cannot be included in presymptomatic studies and the risk of their relatives developing neurodegenerative change is unclear. Another barrier to successful clinical trial is the relatively late inclusion of patients into clinical trials due to stringent inclusion criteria. Large epidemiology studies in ALS have repeatedly demonstrated that the interval between symptom onset and diagnosis is in the range of 12–14 months which is a considerable delay with a multitude of adverse ramifications. Quantitative radiology studies have shown that by the time the diagnosis is confirmed, patients already exhibit considerable motor cortex, corticospinal tract and corpus callosum degeneration which are unlikely to be ameliorated by pharmacological intervention. The observation that significant pathological changes have already taken place by the time a patient fulfills diagnostic criteria suggests that the optimal therapeutic window is earlier. The development of novel technologies, efficient referral frameworks and revised diagnostic criteria would potentially capture ALS patients at an earlier stage, with limited disease burden, increasing the benefit of pharmacological intervention. Another shortcoming of current clinical trial designs is the exclusive reliance on respiratory measures, functional rating scales and survival as primary end-points at a time when a multitude of quantitative cerebrospinal fluid, serum, electrophysiology and imaging biomarkers show promise in tracking pathological changes in vivo. Neuroradiological end-points are firmly integrated in the clinical trials of other conditions, such as multiple sclerosis, are non-invasive and can be quantitatively interpreted in an observer-independent fashion. From a practical biomarker perspective, neurophysiology techniques have the added advantage of appraising both upper and lower motor neuron function, being relatively cost effective, and applicable to patients with considerable disability. The addition of quantitative biomarker panels to clinical outcome measures may allow a more nuanced appraisal of response to therapy.
Conceptual barriers: Academic efforts to characterize progressive changes are limited by a multitude of methodological and conceptual constraints. Clinical studies typically rely on batteries of clinical and neuropsychological instruments, the scores of which have to be carefully adjusted for fatigue, motor disability, motivation, mood, and medications. Neuroradiological studies suffer from selection bias to patients who can tolerate MR scanning, able to lie flat, comply with instructions and therefore tend to represent patients with limited motor disability, limited bulbar involvement, no orthopnea, no behavioral deficits and no sialorrhoea. Post mortem studies, by definition, only capture the terminal phase of the disease and brain banking is only available in certain centers due to a number of financial and cultural factors which limit the widespread availability of brain and spinal cord donation. Conceptual constraints also limit the interpretation of large clinical, neurophysiology, radiology and histology studies. Cross-sectional and longitudinal studies overwhelmingly focus on disease-burden and evaluate the most affected anatomical regions. These studies typically also seek correlations with clinical metrics and propose phenotype or genotype-associated anatomical patterns. This conservative approach can ascertain core disease signatures, but overlooks compensatory changes which are ill-characterized contributors to the clinical picture. Direct correlations between focal imaging metrics and clinical scores are often requested during peer-review, even though the majority of motor and cognitive functions are mediated by multi-synaptic networks, making the allocation of a specific function to a single structure contentious. In ALS, hippocampal volumes have been linked to memory scores, motor cortex metrics to ALSFRS-r, accumbens nucleus volume to apathy etc. overlooking the contribution of other anatomical structures to a specific function. One of the biggest shortcomings of existing studies however is the exclusive focus on degenerative changes and the lack of attention to putative compensatory processes which are also likely to occur. Analogous to the concept of ’cognitive reserve’ in Alzheimer’s disease, the notion of ’motor reserve’ merits consideration in ALS. Corticospinal tract alterations and spinal cord atrophy have been described in relatives of ALS patients without overt clinical signs or symptoms (Querin et al., 2019). The striking discrepancy between the severity of radiological changes and limited functional impairment, especially in the earlier phases of the disease, suggests a degree of network redundancy, functional resilience or ’motor reserve’.
Evidence for compensatory and adaptive changes: Imaging studies have consistently suggested a degree of cerebral reorganization and proposed that key functions may be taken over by structurally less affected brain regions. These processes however are poorly characterized despite their proposed role in delaying symptom manifestation and compensating for the effects of relentless neurodegeneration (Abidi et al., 2020). Adaptive processes underpin multidisciplinary rehabilitation efforts and anecdotal observations from physiotherapists, occupational therapists, and speech and language therapists suggest that functional gains can be made with individualized exercise regimes. Neural plasticity and rehabilitation has a robust literature in other neurological conditions such as stroke, multiple sclerosis, traumatic brain and spinal cord injury, yet adaptive mechanisms in ALS are woefully understudied. Functional MRI studies using motor paradigms have consistently demonstrated an activation shift from the primary motor cortex to premotor, supplementary motor, ipsilateral motor, basal ganglia, and cerebellar regions (Figure 1). Resting state functional studies often identify increased connectivity (Nasseroleslami et al., 2019), typically in the default mode and cortico-cerebellar networks. While these alterations are typically interpreted as ’compensatory’, the confounding effects of medications (Riluzole, Baclofen) and hypoxia on fMRI signal are seldom acknowledged. Aberrant network activity and increased connectivity could equally be considered pathological and the binary categorization of functional observations into ’adaptive’ and ’pathological’ is likely to be simplistic. Structural imaging studies in ALS provide limited evidence of compensatory processes (Bede et al., 2018), but increased cortical volumes in supplementary motor regions have been reported (Christidi et al., 2018). PET studies primarily report patterns of hypometabolism in brain regions susceptible to ALS pathology, and clusters of hypermetabolism are likely to represent microglial activation rather than compensatory changes. Post mortem studies in ALS have also primarily concentrated on the characterization of affected brain regions and the molecular analysis of intraneuronal inclusions. Pathological staging systems have been developed and robust post mortem imaging studies have been undertaken, but only few post mortem studies specifically investigated stem cell mobilization, adaptive or ’regenerative’ processes.
Figure 1.
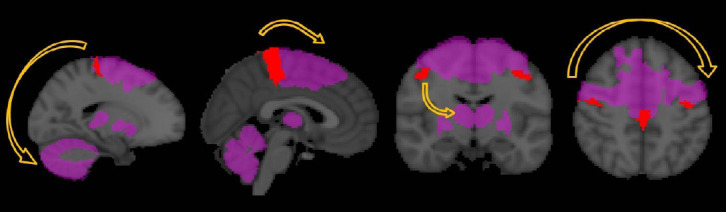
Functional reorganization in ALS based on functional MRI studies.
Motor task studies in ALS have consistently captured increased cerebellar, thalamic and basal ganglia signal and an activation shift from the primary motor cortex to premotor, supplementary motor and ipsilateral motor cortices. Cognitive paradigms have also revealed broadened and aberrant activation patterns and resting state studies captured increased cerebro-cerebellar and default mode network connectivity. ALS: Amyotrophic lateral sclerosis.
Demographic and clinical variables are likely to influence the efficacy of adaptive processes in ALS. Plasticity is thought to be more significant in younger patients and ALS phenotypes with slower progression rates are likely to permit more from efficient compensatory mechanisms. There is considerable variation in progression rates in ALS; patients with restrictive phenotypes, such as monomelic forms of the disease, or upper motor neuron predominant phenotypes typically have longer survival. Bulbar onset, respiratory compromise, older age at onset, and comorbid dementia, are established negative prognostic indicators. Pharmacological targets in ALS typically focus on ’neuroprotection’, glutamate inhibition, anti-inflammatory effects, oxidative stress mitigation, iron chelation and antisense therapy, but recent studies have also shown promise in mobilizing stem cells through granulocyte colony stimulation (Peters et al., 2018).
Conclusions: Due to the striking biological heterogeneity of ALS, the concept of ’one-drug-for-all’ should be superseded by precision, phenotype-, genotype-, and stage-specific therapeutic strategies. Regenerative and adaptive processes are poorly characterized in ALS despite offering a potential target for intervention. Robust, dedicated studies are urgently required to study compensatory mechanisms in ALS and explore therapies that facilitate stem cell mobilization and network reorganization.
PB is supported by the Spastic Paraplegia Foundation, Inc. (SPF), the Health Research Board (HRB EIA-2017-019), the EU Joint Programme – Neurodegenerative Disease Research (JPND), the Andrew Lydon scholarship, the Irish Institute of Clinical Neuroscience (IICN), and the Iris O’Brien Foundation; PB is the patron of the Irish Motor Neuron Disease Association (IMNDA). FC is supported by the EU-IKY Scholarship Program (European Social Fund-ESF), the Greek “Reinforcement of Postdoctoral Researchers” grant (5033021) of the “Human Resources Development Program, Education and Lifelong Learning” of the National Strategic Reference Framework (NSRF 2014-2020). The sponsors of the authors had no bearing on the opinions expressed herein.
Footnotes
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
References
- 1.Abidi M, de Marco G, Couillandre A, Feron M, Mseddi E, Termoz N, Querin G, Pradat PF, Bede P. Adaptive functional reorganization in amyotrophic lateral sclerosis: coexisting degenerative and compensatory changes. Eur J Neurol. 2020;27:121–128. doi: 10.1111/ene.14042. [DOI] [PubMed] [Google Scholar]
- 2.Bede P, Iyer PM, Finegan E, Omer T, Hardiman O. Virtual brain biopsies in amyotrophic lateral sclerosis: Diagnostic classification based on in vivo pathological patterns. Neuroimage Clin. 2017;15:653–658. doi: 10.1016/j.nicl.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bede P, Querin G, Pradat PF. The changing landscape of motor neuron disease imaging: the transition from descriptive studies to precision clinical tools. Curr Opin Neurol. 2018;31:431–438. doi: 10.1097/WCO.0000000000000569. [DOI] [PubMed] [Google Scholar]
- 4.Chipika RH, Finegan E, Li Hi Shing S, Hardiman O, Bede P. Tracking a fast-moving disease: longitudinal markers, monitoring, and clinical trial endpoints in ALS. Front Neurol. 2019;10:229. doi: 10.3389/fneur.2019.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christidi F, Karavasilis E, Riederer F, Zalonis I, Ferentinos P, Velonakis G, Xirou S, Rentzos M, Argiropoulos G, Zouvelou V, Zambelis T, Athanasakos A, Toulas P, Vadikolias K, Efstathopoulos E, Kollias S, Karandreas N, Kelekis N, Evdokimidis I. Gray matter and white matter changes in non-demented amyotrophic lateral sclerosis patients with or without cognitive impairment: A combined voxel-based morphometry and tract-based spatial statistics whole-brain analysis. Brain Imaging Behav. 2018;12:547–563. doi: 10.1007/s11682-017-9722-y. [DOI] [PubMed] [Google Scholar]
- 6.Elamin M, Pinto-Grau M, Burke T, Bede P, Rooney J, O’Sullivan M, Lonergan K, Kirby E, Quinlan E, Breen N, Vajda A, Heverin M, Pender N, Hardiman O. Identifying behavioural changes in ALS: Validation of the Beaumont Behavioural Inventory (BBI) Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:68–73. doi: 10.1080/21678421.2016.1248976. [DOI] [PubMed] [Google Scholar]
- 7.Geevasinga N, Menon P, Nicholson GA, Ng K, Howells J, Kril JJ, Yiannikas C, Kiernan MC, Vucic S. Cortical function in asymptomatic carriers and patients with C9orf72 amyotrophic lateral sclerosis. JAMA Neurol. 2015;72:1268–1274. doi: 10.1001/jamaneurol.2015.1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang NX, Zou ZY, Xue YJ, Chen HJ. Abnormal cerebral microstructures revealed by diffusion kurtosis imaging in amyotrophic lateral sclerosis. J Magn Reson Imaging. 2020;51:554–562. doi: 10.1002/jmri.26843. [DOI] [PubMed] [Google Scholar]
- 9.Nasseroleslami B, Dukic S, Broderick M, Mohr K, Schuster C, Gavin B, McLaughlin R, Heverin M, Vajda A, Iyer PM, Pender N, Bede P, Lalor EC, Hardiman O. Characteristic increases in EEG connectivity correlate with changes of structural MRI in amyotrophic lateral sclerosis. Cereb Cortex. 2019;29:27–41. doi: 10.1093/cercor/bhx301. [DOI] [PubMed] [Google Scholar]
- 10.Peters S, Zitzelsperger E, Kuespert S, Iberl S, Heydn R, Johannesen S, Petri S, Aigner L, Thal DR, Hermann A, Weishaupt JH, Bruun TH, Bogdahn U. Biomarker supervised G-CSF (Filgrastim) response in ALS patients. Front Neurol. 2018;9:971. doi: 10.3389/fneur.2018.00971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Querin G, Bede P, El Mendili MM, Li M, Pélégrini-Issac M, Rinaldi D, Catala M, Saracino D, Salachas F, Camuzat A, Marchand-Pauvert V, Cohen-Adad J, Colliot O, Le Ber I, Pradat PF. Predict to Prevent Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis Study Group (2019) Presymptomatic spinal cord pathology in c9orf72 mutation carriers: A longitudinal neuroimaging study. Ann Neurol. 86:158–167. doi: 10.1002/ana.25520. [DOI] [PubMed] [Google Scholar]
- 12.Schuster C, Hardiman O, Bede P. Development of an automated MRI-based diagnostic protocol for amyotrophic lateral sclerosis using disease-specific pathognomonic features: a quantitative Disease-State Classification Study. PLoS One. 2016;11:e0167331. doi: 10.1371/journal.pone.0167331. [DOI] [PMC free article] [PubMed] [Google Scholar]