

ABSTRACT
Emerging evidence has revealed the crucial role of transcriptional RNA methyladenosine modification in immune response. However, the potential role of RNA N1-methyladenosine (m1A) modification of immune cells in the tumor microenvironment (TME) still remains unclear. In this study, we identified three distinct m1A modification patterns based on the integrated analyses of nine m1A regulators, which are significantly related to Relapse-free survival (RFS), Overall survival (OS), and TME infiltration cells in colon cancer patients. Furthermore, the m1AScore was generated by using principal components analysis (PCA) of expression of the 71 m1A-related genes to further demonstrate the characteristics of m1A patterns in colon cancer. In summary, a low m1AScore could be characterized by lower EMT, pan-F TBRS, and TNM stages, as well as less presence of lymphatic invasion, and, hence, good prognosis. At the same time, a low m1AScore could also be linked to CD8 + T effector proliferation, in addition to high microsatellite instability (MSI), neoantigen burden and PD-L1 expression, showing prolonged survival and better response after undergoing an anti-PD-L1 immunotherapy regimen in the public immunotherapy cohort. Our work reveals that m1A modification patterns play a key role in the formation of TME complexity and diversity in the context of immune cell infiltration. Accordingly, this m1AScore system provides an efficient method by which to identify and characterize TME immune cell infiltration, thereby allowing for more personalized and effective antitumor immunotherapy strategies.
KEYWORDS: m1A, tumor microenvironment, immunotherapy, colon cancer
Introduction
Over 160 post-transcriptional RNA chemical modifications have been identified in different cellular RNAs of all living organisms, including non-coding RNAs (primarily rRNAs, tRNAs and snRNAs) and mRNAs.1 Among these chemical modifications, RNA modifications include, but are not limited to, N6-methyladenosine (m6A), N1-methyladenosine (m1A), 5-methylcytosine (m5C) and Pseudouridine in eukaryotic cells.2–5 Similar to the modification of m6A, m1A is a kind of dynamic reversible modification process in mammalian cells that is regulated by methyltransferases, demethylases and binding proteins, which are known respectively as “Writers”, “Erasers” and “Readers”. The modification process of m1A methylation is catalyzed by methyltransferases consisting of TRMT6, TRMT61A, Trmt61B and TRMT10C, whereas the removal process is conducted by demethylases composed of FTO, ALKBH1 and ALKBH3. Furthermore, four special RNA-binding proteins, including YTHDF1, YTHDF2, YTHDF3, and YTHDC1, are required to complete the process.6–9 Previous study revealed that m1A regulators were dysregulated in gastrointestinal cancers and correlated with the ErbB and mTOR pathway.10 In addition, Shi et al. found that m1A-related regulatory genes play a crucial role in regulating hepatocellular carcinoma progression.11 However, the function of m1A modification still remains largely unclear. A deeper understanding of these regulators would help reveal the potential role of m1A modification in various physiological and pathological processes.
Nowadays, emerging evidence has proven that the tumor microenvironment (TME) plays a critical role in the growth and survival of tumor cells. Providing a novel insight into tumorigenesis, the TME is composed of cancer cells, stromal cells (cancer-associated fibroblast, mesenchymal stem cells and endothelial cells), infiltrating immune cells (macrophages, myeloid cells and lymphocytes), as well as secreted factors, such as chemokines, cytokines and growth factors. Among the infiltrating myeloid cells, five distinct myeloid cell groups are found, including tumor-associated neutrophils (TANs), tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), dendritic cells (DCs) and Tie2-expressing monocytes consisting of the tumor-associated myeloid cells (TAMCs).12 Greater understanding has accompanied the ever-broadening of the diverse and complex TME landscape. Accordingly, considerable evidence has shown its crucial role in the immune escape process of tumorigenesis and the effect on response to immunotherapy. Unlike traditional chemotherapy and targeted therapy, immunotherapy is a method of reactivating antitumor immune response by inhibiting co-suppressive molecules at the surface of tumor cells or immune cells. Unfortunately, only a small percentage of patients have experienced clinical benefit, far from meeting clinical goals.13 This allows for the possibility of identifying novel tumor-related immune phenotypes, which could prove efficient in recognizing immunotherapeutic response and prove valuable in the search for new therapeutic targets through a more comprehensive understanding of the heterogeneity and complexity of the TME.14,15
Recently, several studies have revealed the correlation between m6A modifications and TME-infiltrating immune cells. Han et al. reported that the loss of m6A-binding protein YTHDF1 of DCs plays a crucial role in enhancing the cross-presentation of tumor antigen and the cross-priming of CD8 + T cells, indicating YTHDF1 as a new potential anticancer immunotherapeutic target.16 In addition, Wang et al. revealed that depletion of RNA methyltransferase Mettl3 of m6A in DCs resulted in impaired functional activation and DCs-based T cell response, implicating a new role for Mettl3-mediated m6A modification in innate immunity.17 However, as another important form of post-transcriptional modification, the correlation between m1A modification and TME-infiltrating immune cells still remains unclear. This calls for a comprehensive identification and characterization of TME-infiltrating immune cells, as mediated by multiple m1A regulators.
Therefore, we herein integrated the genomic information of 794 colon cancer samples to systematically evaluate putative m1A modification patterns, correlating, at the same time, these m1A-modified patterns with our characterization of TME-infiltrating immune cells. From three distinct modification patterns, we found that 71 out of 233 differentially expressed genes (DEGs) had significant prognosis in colon cancer. By using the method of principal components analysis (PCA) for the expression of the 71 m1A-related prognostic genes, we generated the m1AScore to further demonstrate the characteristics of m1A patterns in colon cancer. In a word, our novel m1AScore system provides an efficient method by which to identify and characterize TME infiltration, thereby allowing for more personalized and effective antitumor immunotherapy strategies.
Materials and methods
Study Design and Collection of Patient Dataset
The flowchart of the present study was shown in Figure 1, which was produced in BioRender (https://app.biorender.com/). Notably, to fully assess types of m1A methylation of colon cancer, we searched the public datasets in the Gene-Expression Omnibus (GEO) and The Cancer Genome Atlas (TCGA) databases. Patients without prognostic data were removed from our study. In summary, 8 eligible colon cancer patient cohorts (GSE39582, GSE17538, TCGA-COAD, GSE41258, GSE33113, GSE37892, GSE38832 and GSE39084) were collected for further analysis. The baseline information on current colon cancer patients was listed in Supplemental Table S1. For microarray data, we used the present log scale matrix files directly downloaded from GEO. For TCGA-COAD, we used the FPKM value of RNA transcriptome obtained from the UCSC Public Hub (https://xenabrowser.net/). The somatic mutation data of TCGA-COAD patients were also gathered for further analysis in this study. As to datasets in pan-cancer, the RNA sequencing data were also directly downloaded from the UCSC Public Hub.
Figure 1.
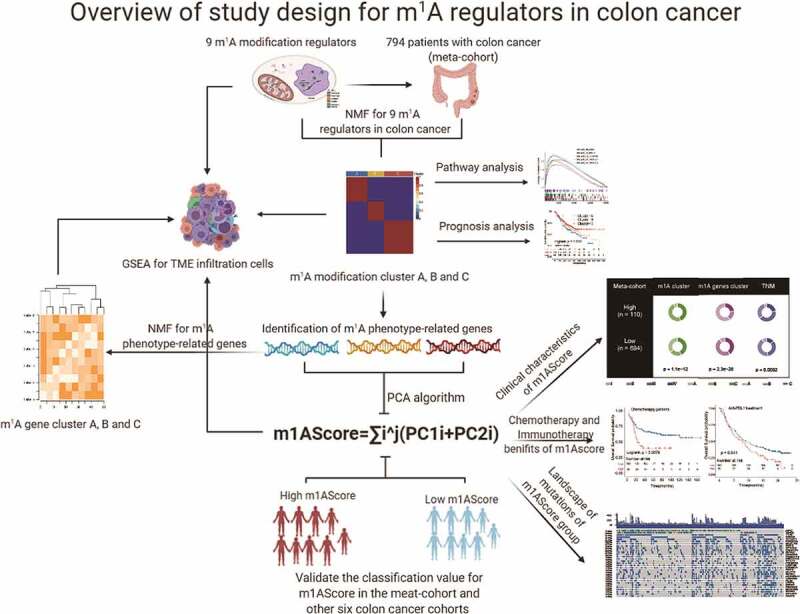
Diagram of analytic workflow. The drawing of the syringe in the figure from the BioRender (https://biorender.com/)
Nonnegative Matrix Factorization for nine m1A regulators
First, to eliminate the batch effects of different cohorts, we used the “ComBat” algorithm of the “sva” R package to obtain a meta-dataset for the incorporated patients (GSE39582 and GSE17538).18 Then, nine previously reported m1A regulators were achieved for the subsequent Nonnegative Matrix Factorization (NMF) clustering19 and used to determine the m1A modification patterns for a total of 794 colon cancer patients. The k values where the magnitude of the cophenetic correlation coefficient began to fall were chosen as the best number of clusters.19 The heatmap of m1A regulators, basis components and the connectivity matrix of NMF in different clusters were also estimated by the “NMF” R package.
Generation of tumor-infiltrating immune cells
The ssGSEA (single-sample gene-set enrichment analysis) algorithm was used to quantify the relative abundance of each tumor-infiltrating immune cell by using the 782 meta-genes (Supplemental Table S2)20 in the “GSVA” R package21 for colon cancer. We also used the scale algorithm to normalize the values of each immune cell in the meta-cohort for further analysis.
Identification of differentially expressed genes among distinct m1A modification patterns
To identify the m1A-related genes based on the three m1A modification NMF patterns, we applied the “limma” R package to determine the differentially expressed genes (DEGs). The adjusted P values were set as < 0.05. Veen and chart plot directly showed the number of DEGs among these m1A modification patterns by using the “UpSetR” and “VennDiagram” R packages. Also, the T-distributed stochastic neighbor embedding (t-SNE)-based approach in the “Rtsne” R package was used to visualize the distribution of colon cancer patients with the mRNA expression data of DEGs.22
Generation of m1A gene signature
To quantify the m1A modification patterns of each patient, we aimed to construct a scoring system to assess all individuals with colon cancer, and it was termed as m1AScore. We determined the prognostic value for DEGs of the distinct m1A modification patterns by using Cox proportional hazards regression analysis. Next, the DEGs with significant prognostic value were included in calculating the m1AScore. Given the prognostic DEGs in colon cancer, we then performed principal component analysis (PCA) to establish the m1AScore. Similar to a previous study,23 we added the (principal components) PC1 and PC2 as the last gene signature scores. Concomitantly, the NMF clustering algorithm was used to generate gene clusters and their stability. The m1AScore is expressed as
m1AScore =,
where i and j are the order and the total number of the m1A-related prognostic genes in colon cancer. Here, the Z-score of the m1AScore was used for further analysis.
Correlation between m1A gene signature and previous biological processes
To reveal the distinct TME between two m1AScore groups, we collected some biological processes demonstrated by Mariathasan et al.24 These biological processes included the following gene sets: 1) epithelial-mesenchymal transition (EMT) markers, including EMT1, EMT2 and EMT3; 2) immune-checkpoint; 3) antigen processing machinery (APM); 4) CD8 + T-effector signature; 5) Angiogenesis signature; and (7) pan-fibroblast TGFb response signature (Pan-FTBRS), all of which were listed in Supplemental Table S3. Many immune co-inhibitors and -stimulators were also extracted to prove their relationship to the m1AScore. Using the “xCell” R package, Dvir Aran et al. showed that a microenvironment score could be calculated for colon cancer by integrating such TME-infiltrating cells as B-cells, CD4 + T-cells, CD8 + T-cells, DCs, ‘Eosinophils’, ‘Macrophages, ‘Monocytes, ‘Mast cells, Neutrophils, NK cells, Adipocytes, Endothelial cells, and Fibroblasts,25 thus supporting our proposition about the relationship of m1Ascore with TME.
Collection of Clinical Treatment and Transcriptomic Information
The information on GSE39582 patients was complete; therefore, we were able to reorganize their chemotherapeutic outcomes and, as a result, discover 232 colon cancer patients who had undergone chemotherapy. This allowed us to use our m1AScore to predict the prognosis of these patients. Concomitantly, given the relationship between m1AScore and TME, two other immunotherapeutic cohorts were included: advanced urothelial cancer with atezolizumab, an anti-PD-L1 antibody (IMvigor210 n = 348) from the “IMvigor210” R package,24 and metastatic melanoma treated with pembrolizumab, an anti-PD-1 antibody from the GEO database (GSE78220, n = 27).26
PPI string network and Functional and Pathway Enrichment Analysis
To explore the distinct pathways among the m1A modification patterns, we downloaded the classical hallmark gene sets in the MSigDB (https://www.gsea-msigdb.org/). We conducted GSEA for hallmark pathways in different groups. With the prognostic m1A-related genes and nine m1A regulators, we constructed the PPI network by using STRING (http://string-db.org/) with R < 0.15. Gene Ontology (GO) of prognostic m1A-related genes was performed by the “clusterProfiler” R package, and the barplot and heatplot of related terms were used in the enrichplot R package. Moreover, the biological processes of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways among different m1A modification subtypes were also investigated in the combat dataset (GSE17538 and GSE59382). The enrichment P value was also adjusted by the Benjamini–Hochberg procedure.27
Statistical analysis
All statistical analysis was performed in R 4.0, and all reported P-values were 2-sided. Statistical significance was set at 0.05 or 0.001. In summary, standard tests included the Student’s t-test, Wilcoxon rank-sum test, and Fisher exact test to determine the differences of variables among different modification patterns. Also, Benjamini–Hochberg (BH-FDR) was used to adjust the P-values for multiple comparisons of the multiple variables.27 For prognosis analysis, log-rank test and Cox regression analysis were used to test the prognostic value of these special variables, including m1A-related genes, m1A modification patterns, m1A-related gene clusters and m1AScore. The best cutoff of these genes and signatures was determined by the cut point value of the “survminer” package, according to prognostic status, Overall survival (OS) and Relapse-free survival (RFS) for colon patients of these cohorts. The area under the receiver operating characteristic curve (AUC) of time-dependent ROC analysis was performed to detect the prediction values of current signatures for different therapies applied at distinct times by the “timeROC” package.
Results
The landscape of m1A regulators in colon cancer
In summary, among the regulators mentioned above, a total of nine m1A regulators, including three writers (TRMT6, TRMT61A and TRMT10C), two erasers (ALKBH1 and ALKBH3) and four readers (YTHDF1, YTHDF2, YTHDF3 and YTHDC1), were identified in the present study. Figure 2a demonstrated the dynamic reversible process mediated by m1A regulators for RNA modification. To be complete, we also gave the landscape of the incidence of somatic mutations of nine m1A regulators in the TCGA-COAD patients (Figure 2b). In the available samples, 45 out of 399 patients experienced mutations of nine m1A regulators, with frequency ranging from 4% to 1%. YTHDC1 had the highest number of mutations of all m1A regulators (4%). Then, we calculated the relationships of the m1A regulators in the metadata set (GSE39582 and GSE17538). Weak correlations among these m1A regulators were proved and shown in Figure 2c. In addition, to investigate the difference of m1A regulators between normal and tumor tissue RNA expression, a comparison of results showed that 7 out of the 9 m1A regulators were higher in TCGA-COAD patients (Figure 2d), but not ALKBH1 and YTHDC1. Through the application of the GSEA algorithm for immune-infiltration cells, we found a strong relationship of the nine m1A regulators with the current TME-infiltrating immune cells using Spearman’s correlation analyses in the metadata set. Most of these regulators were negatively related to the immune cells in varying degrees. However, the regulator of ALKBH1 was positively related to almost all of these immune infiltration cells (Figure 2e). The above analysis indicated that m1A regulators play a crucial role in the ever-changing immune microenvironment as colon cancer develops.
Figure 2.
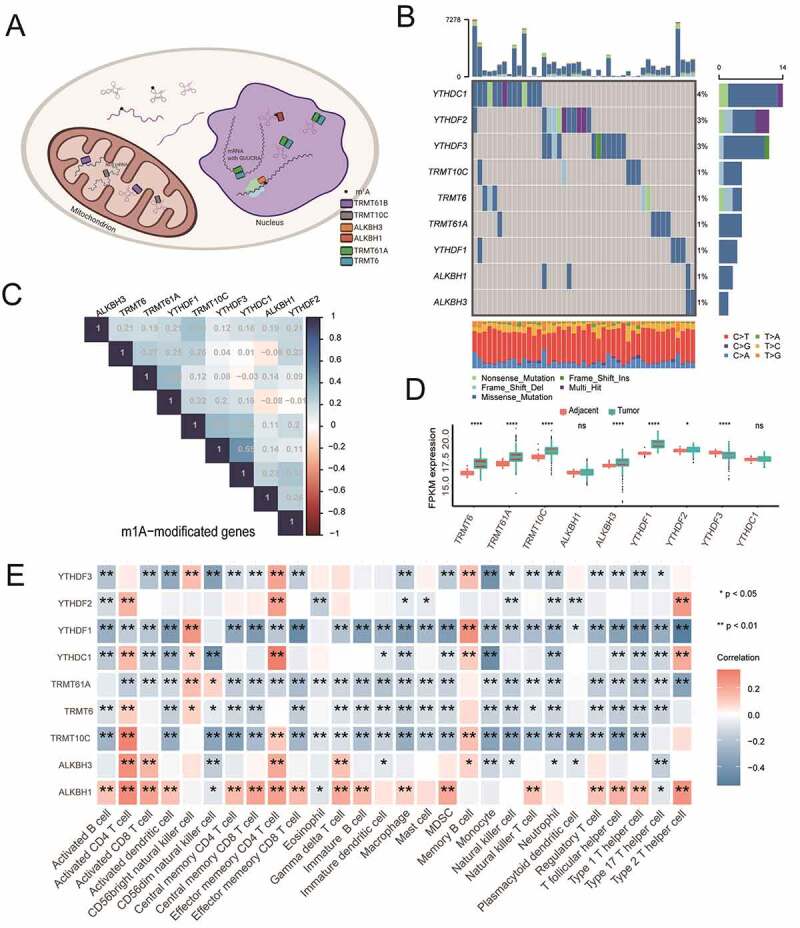
The landscape of m1A regulators in colon cancer. A). Methylation process for the m1A regulators in erasers, writers and readers in cancers. B) The mutation of nine m1A regulators in the TCGA-COAD cohort. C) Relationships of the nine m1A regulators in the meta-cohort. D) Comparisons of the nine m1A regulators between normal and tumor tissue in the TCGA-COAD cohorts. E) Relationships of the nine m1A regulators with immune infiltration cells in the meta-cohort (GSE39582, GSE17538)
Distinct modification patterns of m1A regulators and hallmark pathway analysis
In the GSE39582 and GSE17538 cohorts, we found no significant differences in the prognosis of OS and RFS (Supplemental Figure S1A and S1B). In the metadata set, we applied the NMF method to achieve 3 m1A modification patterns with significant features of m1A regulators for the colon cancer patients, termed as Cluster A, Cluster B, and Cluster C, based on the choice of k = 3 as the optimal k value after calculating the cophenetic correlation coefficients of NMF (Supplemental Figure2A, S2B, Figure 3a and Figure 3b). Not surprisingly, data revealed in Figure 3c also uncovered the significant differences of the relative expression of immune infiltration cells among three m1A modification patterns. More specifically, the colon cancer patients in cluster B showed lower enrichment score in the most of the immune infiltration cells than that in other clusters (P < .05). In addition, Cluster A was relatively rich in innate immune cell infiltration, including NK cells, macrophages and MDSCs, while Cluster A and Cluster C were both characterized by adaptive immune cell infiltration. Prognostic analysis for the three m1A modification subtypes also revealed that Cluster B had poor OS or RFS probability in the colon cancer patients of the metadata set (Figure 3d and Figure 3e). Further analysis indicated that significantly different pathways were activated among the main m1A modification patterns by conducting the GSEA method of hallmark gene sets. The top rank of the hallmarks in Cluster B with poor prognosis was occupied by “HEME_METABOLISM” and “KRAS_SIGNALING_DN”, which revealed unusual development when compared to other m1A modification patterns (Cluster A and Cluster C) in the metadata set (Figure 3g). Other active hallmark pathways of the three patterns were listed in figure 3f, Figure 3h and Supplemental Table S4.
Figure 3.
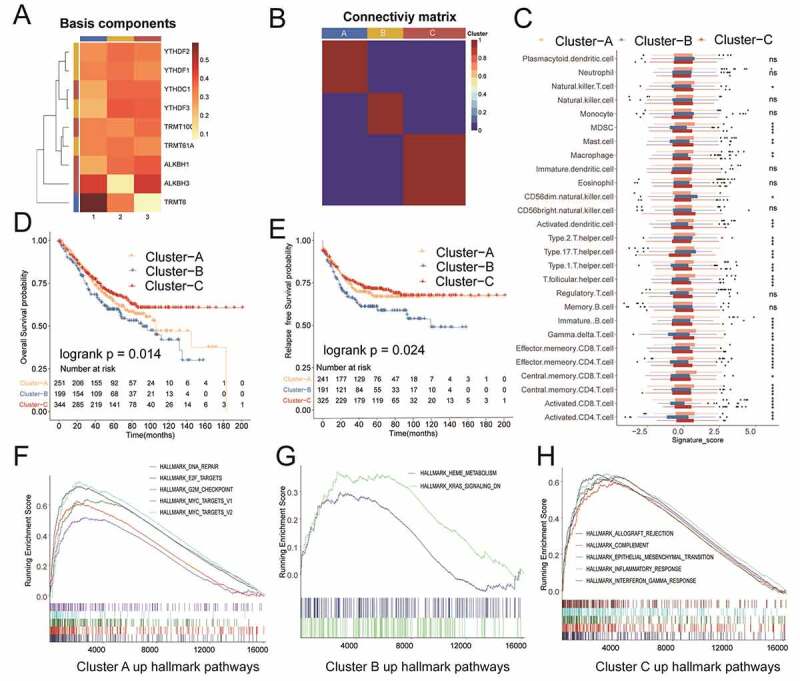
NMF for m1A modification patterns, characteristics of immune infiltration cells, and biological processes of each pattern. A) Heat map of basic components of m1A regulator expression in three m1A modification patterns by NMF. B) Connectivity matrix for patients with colon cancer in the meta-cohort by NMF. C) Different expressions of immune infiltration cells in each pattern. D) and E) OS and RFS analysis for patterns in the meta-cohort. F)-H) GSEA method for the activation of KEGG pathways in each pattern in the meta-cohort
Roles of DEGs of m1A modification patterns
A total of 233 DEGs were identified from three m1A modification patterns using the “limma” package based on RNA expression in the metadata set (Supplemental Figure S3A and S3B and Table S5). We then used Cox regression analysis to detect their relationship with the RFS status of the colon cancer patients, and 71 m1A-related genes with significant prognostic value were recognized with FDR <0.05 to further establish the m1AScore signature (Figure 4a and Supplemental Table S6). Their detailed hazard ratio (HR) with 95% confidence interval (CI) was listed in Supplemental Table S6. Such functional analysis for these genes indicated that some m1A-related genes with some equally important annotations played a crucial role in the m1A modification subtypes (Figure 4b, Supplemental Figure S3C and Table S7). To validate this conclusion, we conducted another NMF step for 71 m1A-related genes to reveal three distinct m1A-related gene phenotypes, named gene-clusterA, gene-clusterB and gene-clusterC. Obviously, the RNA expression of m1A regulators was significantly different among the gene-clusters in the metadata set (Supplemental Figure S4A). Using the current prognostic DEGs, the plot of t-SNE exhibited the distinct distance of the three gene-clusters (Supplemental Figure S4B). Similar to m1A modification patterns, the gene-clusters could also be used to distinguish the immune-infiltration cells, RFS and OS in the metadata set (Supplemental Figure S4C, S4D and S4E).
Figure 4.
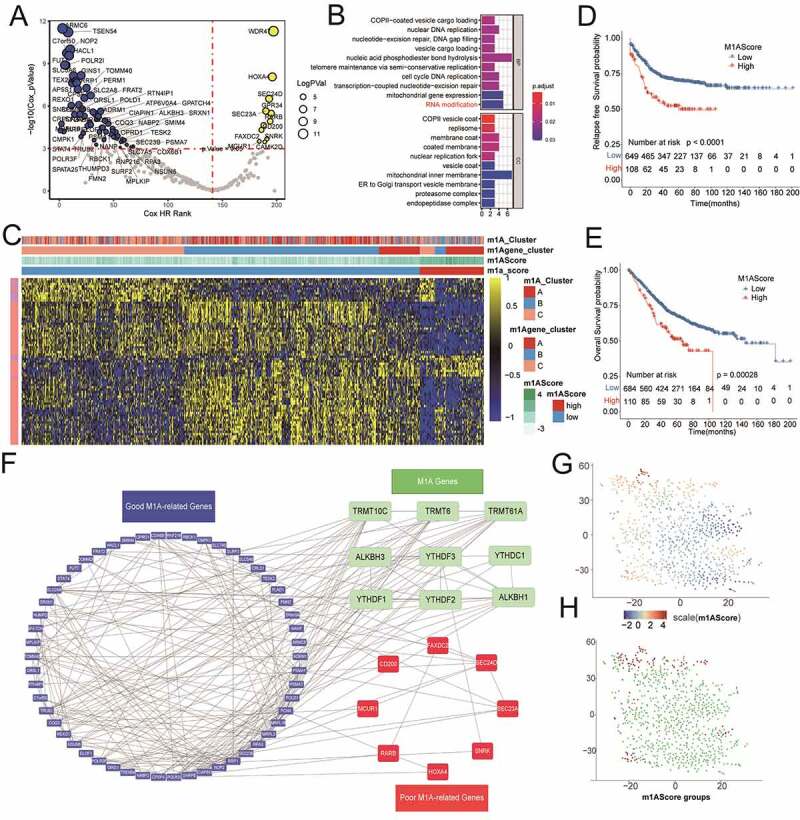
Construction of m1AScore for patients with colon cancer. The figures referred to meta-cohort. A) The significant prognostic value of selecting m1A-related genes from DEGs in each m1A modification pattern. B) Biological process for the 71 prognostic genes. C) Heat map for the relationship between the expression of 71 prognostic genes and m1AScore, m1A modification patterns and m1A gene-clusters. D) and E) The significant differences between two m1AScore groups based on RFS and OS analysis. F) PPI string proteins network for the 71 m1A-related genes and 9 m1A regulators. G) and H) The t-SNE distribution of m1AScore and m1AScore groups for all the meta-patients with colon cancer
Generation of m1A gene signature
The complexity of m1A modification could restrict the application of m1A modification patterns in the real world. Therefore, we established our m1AScore based on the 71 prognostic DEGs and the algorithm mentioned above to further analyze the potential biological processes related to m1A modification patterns. The detailed coefficients of 71 m1A-related prognostic genes and all m1AScores of the metadata patients were also listed in Supplemental Table S8 and Table S9. According to the optimal truncation of m1AScore, patients with colon cancer could be divided into two groups: high m1AScore groups (n = 110) and low m1AScore groups (n = 684), which were largely in accordance with the gene-clusters (Figure 4c). A high m1AScore had poor OS and RFS in the metadata set (Figure 4d and Figure 4e, all log-rank P < .001). In addition, the PPI string network plot for the m1A regulators and m1A-related genes uncovered the complex relationships among them (figure 4f). Similar to the m1A gene-cluster classification, the t-NSE could also be used to visualize the distribution of the continuous m1AScore and associated categories (Figure 4g and Figure 4h).
Correlation of m1A-related phenotypes with immune cell infiltration and previous biological processes
To better demonstrate the characteristics of m1AScore, we further analyzed the relationship of m1AScore to m1A modification patterns and m1A-related gene-clusters. All significant differences of m1AScore among these subtypes could be found in the two cohorts shown in Figure 5a and Figure 5b. The alluvial diagram showed the changes among the m1A modification cluster, m1A gene-cluster, m1AScore, RFS and OS in the meta-data set (Figure 5c). Considering the heterogeneity and complexity of the TME in colon cancer patients, we compared the different immune cells among the different m1AScore groups in detail. We found that the m1AScore was closely linked to at least 12 out of 28 TME immune cells, albeit not always consistent with the TME score (Figure 5d). As a whole index for the microenvironment, we found that the m1AScore was significantly related to the microenvironment Score in the metadata set (r = 0.259, p < .001) (Figure 5e). By comparing interferons and receptors, interleukins, as well as receptor co-stimulators and co-inhibitors between two m1AScore groups (Supplemental Figure S6A and S6B), we also found that significantly different genes could be in distinctly different m1AScore groups in the meta-cohort (GSE17538 and GSE39582). This discovery inspired us to further investigate the biological processes linked to m1AScores. We found that our m1AScore was positively related to EMT1, EMT2, EMT3 and pan-F TBRS, but negatively related to CD8 + T effector and APM in colon cancer patients (figure 5f). Figure 5g revealed that some significant differential pathways could be activated in the low m1AScore range, such as Mismatch repair (MMR), RNA degradation, RNA polymerase, RNA transport, DNA replication and cell cycle. The details of these pathways can be seen in Supplemental Table S10.
Figure 5.
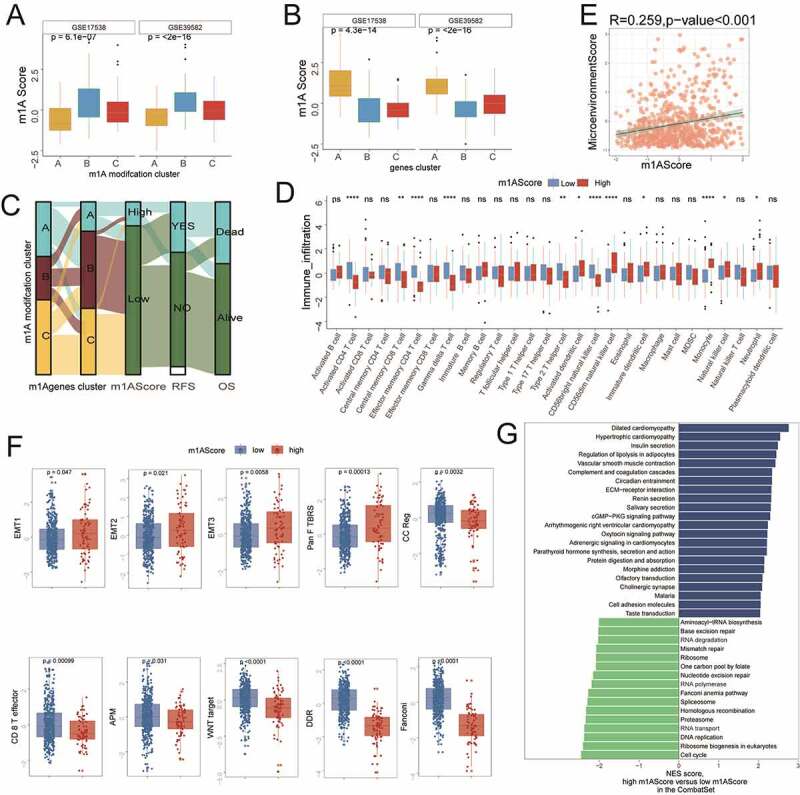
Immune-related characteristics of m1AScore. These figures referred to the meta-cohort. A) and B) The significant differences of m1AScore in m1A patterns and m1A-related gene-clusters. C) Sankey plot for the change of patients in different subgroups. D) Comparisons of immune cells between two m1AScore groups (i.e., high and low). E) The significant relationship between Microenvironment Score and m1AScore. F) Boxplot for the significant differences of the current immune-related signatures between two m1AScore groups (i.e., high and low). G) Significant KEGG pathways for the high vs. low m1AScore group
Characteristics of clinical traits in m1A-related phenotypes and tumor somatic mutation
To ensure the completion of expansibility of m1AScore and generate m1AScore for the available cohorts, we matched all 71 m1A pattern-related genes from different platforms, including GPL570 (71/71, 100%), GPL96 (52/71, 72%), and Illumina-sequence (64/72, 90.1%). Then, to validate it for clinical applicability of the m1AScore, we collected the clinical traits of colon cancer patients in all cohorts. We found that the patients with high m1AScore had higher TNM stages in the variables of both m1AScore Group and m1AScore (Figure 6a and Figure 6b). In the GSE41258 cohort (n = 182), the high m1AScore could still predict poor prognosis of colon cancer (Figure 6c, log rank test P = .039). In addition, the patients with low m1AScore had more MSI-high status (Figure 6d, p = .011). In the TCGA-COAD cohort, significant differences of RFS could be detected between two m1AScore groups (Figure 6e, log-rank test, p = .015). figure 6f also showed that the m1AScore could be linked to lymphatic invasion (P = .0061). The oncoplot of tumor somatic mutation in the TCGA-COAD cohort showed that APC and TP53 gene mutation in the high m1AScore group was approximately ten percent higher than that in the low m1AScore (Figure 6g). Meanwhile, to validate the m1AScore prognostic values, we found that colon cancer patients could be distinguished by m1AScore in most of the available cohorts, including GSE14333 (RFS, p = .0053; OS, p = .0026), GSE37892 (RFS, P = .17), GSE38832 (RFS, p = .013; OS, p = .00056), GSE39084 (OS, p = .0055), and GSE39084 (RFS, p = .042) (Supplemental Figure S7). Given its excellent performance in identifying the prognosis of colon cancer, we also conducted the m1AScore algorithm for all pan-cancer patients. Significant relationships between m1AScore and OS were found in 18 out of 28 cancers (Supplemental Figure S8A and Table S11). However, high m1AScore had poor prognosis in 6 out of 18, and the remaining were well. Among them, a high m1AScore was strongly related to poor OS in both Rectum Adenocarcinoma (READ) and COAD (Supplemental Figure S8B and S8C, P < .05).
Figure 6.
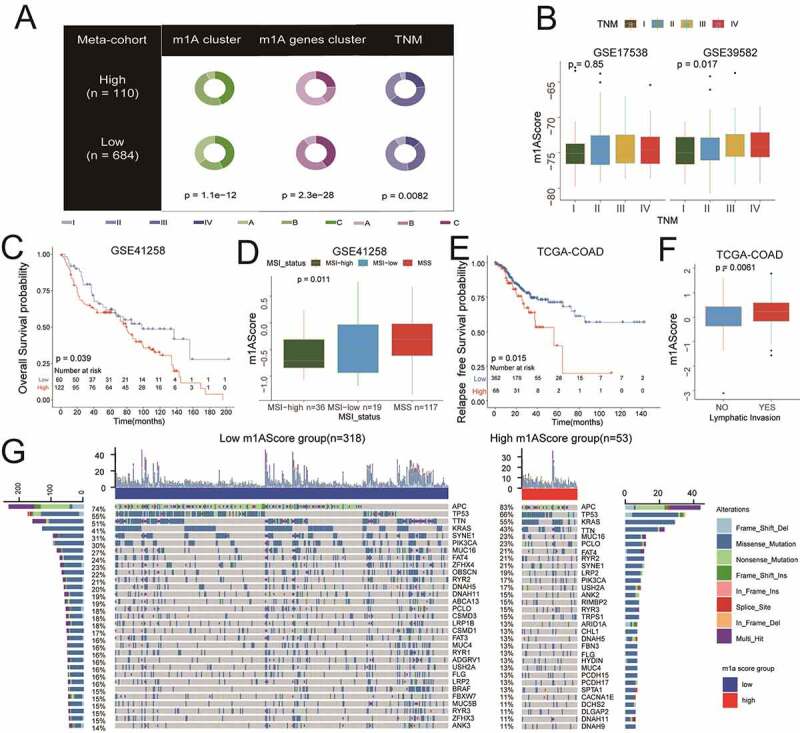
Clinical characteristics and tumor somatic mutation for m1AScore. A) Pie plots for the different distributions of m1AScore groups in different subgroups, such as m1A patterns, m1A gene-clusters and TNM (meta-cohort). B) The higher the TNM stage, the higher the m1AScore (meta-cohort). C) The m1AScore indicating significantly distinct OS in the GSE41258 cohort. D) Patients with MSI-high status showing significantly low m1AScore. E) and F) m1AScore indicating significantly distinct RFS and lymphatic invasion of TCGC-COAD patients. G) The landscape of tumor somatic mutation between the two m1AScore groups
Clinical benefit of m1A gene signature for chemotherapy and anti-PD-1/L1 immunotherapy
In the GSE39582 cohort, 232 colon cancer patients who had undergone chemotherapy showed higher m1AScore than that in the remaining patients without chemotherapy (Figure 7a, p = .011). Based on this evidence, we went further to identify the clinical benefits of our m1AScore for the chemotherapy. Accordingly, we also conducted Cox regression for the m1AScore in these patients and found that it could still distinguish OS (Figure 7b, log-rank test, p = .0076). This result indicated that low m1AScore patients generally had better response to chemotherapy. Time-dependent ROC showed that the AUCs of the m1AScore were 0.55, 0.64, 0.63 and 0.67 in prediction of 1-, 3-, 5- and 10-year OS (Figure 7c).
Figure 7.
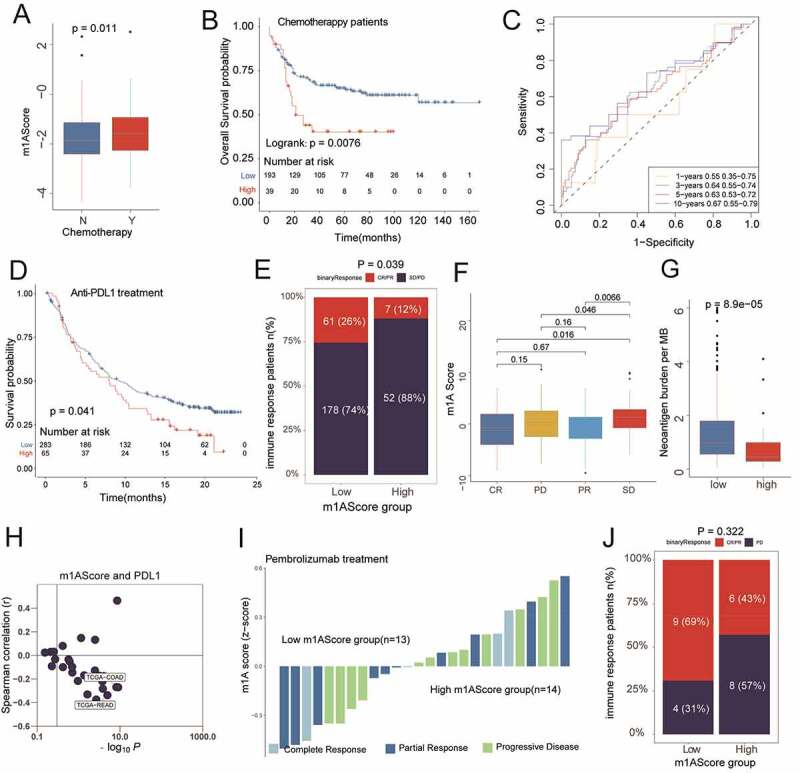
Clinical benefits of m1AScore in revealing the better prognosis for patients who underwent chemotherapy and immunotherapy. A) The patients with chemotherapy had a high m1AScore, P = .011 (patients selected from GSE39582). B) The significant difference between high m1AScore group and low m1AScore groups (GSE39582, P = .0076). C) The time-ROC analysis for m1AScore for predicting OS rate (GSE39582, range: 0.55–0.67). D) High m1AScore had a worse OS rate in patients who underwent anti-PDL1 treatments (IMvigor210cohort, P = .041). E) High m1AScore group had significantly lower CR/PR rate (IMvigor210cohort, P = .039). F) The significantly different expressions of m1AScore among four immune responses (IMvigor210cohort). G) High m1AScore had a significantly lower neoantigen burden (IMvigor210cohort, P < .001). H) Relationship between m1AScore and PDL1 in the pan-cancer cohorts. I) and J) Bar plots of the immune response in each patient who underwent PD1 treatment (KEYTRUDA® pembrolizumab) and two m1AScore groups (not significant)
Similarly, in terms of immunotherapy, we found that the m1AScore could predict the OS for patients who had undergone anti-PD-L1 treatment (IMvigor210, Figure 7d, log-rank test, p = .041). Also, significant differences of m1AScore among patients with different main responses were observed and shown in Figure 7e and 7f (all p < .05). Patients with low m1AScore also had a higher neoantigen burden (Figure 7g, p < .001). Pan-cancer analysis showed that the m1AScore was negatively related to PD-L1 expression in most cancer patients, such as the TCGA-COAD and TCGA-READ cohorts (Figure 7h, all r > 0.3, P < .001), indicating that patients with low m1AScore may have had a potential response to anti-PD-L1 immunotherapy similar to that of the IMvigor210 cohort. However, we could not find any significant relationship between m1AScore and the responses of patients who had undergone anti-PD1 treatment (GSE78220, Figure 7i and 7j, P = .322). The correlations of m1AScore with CTLA4, PDCD1 (PD1), CD274 (PD-L1) and PDCD1LG2 (PD-L2) and their detail results in all pan-cancers were also listed in Supplemental Figure S9 and TableS12. In summary, our work indicated that the construction of m1A gene signatures could help in predicting the response to chemotherapy and anti-PDL1 immunotherapy.
Discussion
The m1A methylation modification, as another form of post-transcriptional chemical modifications of RNA, likely plays an indispensable role in tumorigenesis. Nonetheless, little research has been reported on m1A modification in tumor development. In this study, we, for the first time, systematically characterized the infiltration of TME cells, as mediated by the integrated roles of multiple m1A regulators and corresponding modification patterns. In fact, three distinct m1A modification patterns were identified as playing a key role in the identified and characterized TME-infiltrating (immune) cells. Furthermore, a scoring system, named m1AScore, was established to correlate m1A modification patterns in individual patients and their response to immunotherapy, providing a clinical tool for more individualized and effective antitumor immunotherapy strategies.
Here, we first globally evaluated somatic mutations and RNA expression of nine m1A regulators in TCGA-COAD patients and found that 45 out of 399 patients experienced mutations and that 7 m1A regulators expression were higher in TCGA-COAD patients. Furthermore, the frequency of mutations ranged from 4% to 1%, and YTHDC1 had the highest mutations of all m1A regulators. The alteration of m1A regulators could contribute to the dysregulation of m1A-related genes, thus indicating their involvement in tumorigenesis. Woo et al. reported that ALKBH3-induced m1A demethylation increased CSF-1 expression and the degree of cancer cell invasiveness in breast and ovarian cancer.28 As described above, dysregulation of m1A regulators was correlated with the development of gastrointestinal cancers and hepatocellular carcinoma progression.10,11 However, little was known about the correlation between m1A regulators and TME-infiltrating cells. Therefore, by the application of the GSEA algorithm for immune-infiltration cells, we found a strong relationship between the nine identified m1A regulators and the current TME immune-infiltration cells, indicating the potential role of m1A regulators in the immune TME and subsequent development of colon cancer.
We then revealed three distinct m1A modification patterns based on the nine m1A regulators in the cohort of colon cancer patients and named them as Cluster A, Cluster B and Cluster C, respectively. Compared with Cluster A and C, Cluster B exhibited relatively lower infiltration of immune cells, including both innate and adaptive immune cells. Not surprisingly, Cluster B had the poorest OS and RFS probabilities in the colon cancer patients owing to the suppression of innate and adaptive immunity. To our surprise, analyses of TME cell infiltration indicated that Cluster A was relatively rich in innate immune cell infiltration, including NK cells, macrophages, and MDSC, simultaneously characterized by adaptive immune cell infiltration. However, patients with this m1A modification pattern did not show prolonged OS compared with other patterns. As reported in a previous study, the presence of abundant immune cells retained in the stroma surrounding tumor cell nests could activate the stroma of TME, thus suppressing adaptive immunity.29 Therefore, we speculated that the activation of stroma in Cluster A inhibited the antitumor effect of immune cells. Furthermore, GSEA analysis revealed significantly different pathways among the three m1A modification patterns. Cluster C with prolonged overall survival mainly exhibited EPITHELIAL_MESENCHYMAL_TRANSITI-ON and INFLAMMATORY_RESPO- NSE, cluster A with the poorer prognosis was mainly DNA_REPAIR and E2F_TARGETS, while cluster B with the poorest prognosis was HEME_METABOLISM and KRAS_SIGNALING_DN.
Further, in our study, 233 differentially expressed mRNA transcriptomes were identified among the distinct m1A modification patterns. The differentially expressed mRNA may have been a product of post-transcriptional modifications, which could be mediated by the m1A regulators. Among them, 71 genes were related to the RFS status of colon cancer patients, and these were named as m1A-related signature genes. Similar to the clustering results of m1A modification patterns, three gene clusters, named as gene-clusterA, gene-clusterB and gene-clusterC, were also correlated with distinct immune infiltration cells, RFS and OS. The complexity of m1A modification in individuals called for a new method to quantify the m1A modification patterns of individual cancer patients. With this in mind, we established a scoring system, named as m1AScore, based on the 71 m1A-related signature genes, to evaluate the m1A modification patterns of colon cancer patients. Furthermore, two m1AScore groups exhibited distinct TME infiltration characterizations. That is, a high m1AScore appeared to have a relatively lower amount of infiltrating immune cells, while a low m1AScore had more adaptive immune cells. Survival analysis revealed that a low m1AScore had prolonged OS and RFS compared with OS and RFS of high m1AScore. These results suggest that the m1AScore was a robust and reliable tool for comprehensive clinical assessment of m1A modification patterns in individual patients and that it could be used to evaluate the characterized TME-infiltrating cells to indicate the level of immunotherapy response in colon cancer patients.
Our study also found a positive correlation between m1AScore and EMT1, EMT2, EMT3 and pan-F TBRS, but a negative relationship with the CD8 + T effector and DNA damage response (DDR) in colon cancer patients. As reported in a previous study, inhibition of EMT- and TGFβ-related pathways could increase the trafficking of cytotoxic T-cells against tumor cells, thus preventing metastasis.30,31 In addition, we found that patients with high m1AScore had higher TNM stages and lymphatic invasion, which indicated a poor prognosis.
The initial success of target immune checkpoint blockade (ICB) for the treatment of tumor patients has aroused great interest in utilizing the antitumor potential of our own immune system across many cancer types. Frustratingly, however, only a few patients can benefit from ICB. Recent research has revealed that alterations in the DDR pathway may influence response to ICB.32,33 In addition, microsatellite instability (MSI) and elevated mutational load derived from MMR deficiency can elevate the sensitivity to ICB.34 Our study revealed that patients with a low m1AScore had better response in undergoing anti-PDL1 immunotherapy and chemotherapy in two cohorts. In accordance with previous studies, we found that patients with low m1AScore had more MSI-high status and neoantigen burden. However, we could not find a significant relationship between m1AScore and the responses of patients who had undergone anti-PD1 treatment. Pan-cancer analysis showed that the m1AScore was negatively related to PDL1 and PDL2 expression in most cancer patients, including TCGA-COAD and TCGA-READ. In contrast, m1AScore was not significantly related to PD1 expression in the TCGA-COAD and TCGA-READ cohorts. Unfortunately, we also could not detect the significant relationship between m1AScore and CTLA4 in colon cancer. Further analysis suggested that some significant differential pathways could be activated in those patients presenting with a low m1AScore, such as Mismatch repair, DNA replication and cell cycle. We could predict not only patients’ clinical responses to anti-PDL1 immunotherapy through our m1AScore, but also the efficiency of adjuvant chemotherapy. Our findings provide both foundation and framework for a better understanding of patients’ antitumor immune response and a better tool in the novel m1AScore for guiding more individualized and effective immunotherapy strategies.
However, there still exists some controversies for detecting the m1A methylation in mRNA at present. The research group of Prof. S Schwartz claimed that m1A in mRNA was low when using the method of single-base resolution.9 In contrast, many high-quality articles revealed that m1A methylation sites present more in mRNA by using the method of transcriptome-wide mapping and reverse transcriptase.35–37 However, Prof. S Schwartz still published a refute paper about a single-nucleotide resolution to argue with the published articles.38 Due to the insufficiency of detection methods, we do not verify the m1A modification levels in mRNA of colon cancer patients. In the near future, more effective and accurate methods would be established to detect the m1A modification sites in mRNA to further explore the key role in tumorigenesis.
Conclusions
For the first time, our study systematically demonstrated how the regulatory mechanisms of m1A modification patterns behave in the tumor microenvironment. Simply stated, distinct m1A modification patterns play a key role in the formation of individual TME complexity and diversity. The comprehensive evaluation of m1A modification pattern of individual tumors will, therefore, increase our understanding of TME immune cell infiltration and guide more effective and personalized antitumor immunotherapy strategies.
Supplementary Material
Funding Statement
This work was supported by the Natural Science Foundation of China (No: 81972012) and Medical and Health Science and Technology project of Zhejiang Province (2017KY418); Medical and Health Science and Technology project of Zhejiang Province [2017KY418].
Disclosure of potential conflicts of interest
The authors declare that they have no conflicts of interest for this work.
Author contributions
Conception and Design: J.Z., L.W., Y.G. and H.W.; Financial Support: J.Z. and J.L.; Provision of Study Materials: Y.G. and H.W.; Collection and Assembly of Data: All authors; Data Analysis and Interpretation: YG and H.W.; Manuscript Writing: H.W. and Y.G.; Manuscript Supervised: J.Z., L.W. and Y.X.;
Data availability statement
All data generated or analyzed during this study are included in this published article from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/), and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/).
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Boccaletto P, Machnicka MA, Purta E, Piatkowski P, Baginski B, Wirecki TK, de Crecy-Lagard V, Ross R, Limbach PA, Kotter A, et al. MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018;46(D1):D303–13. doi: 10.1093/nar/gkx1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Desrosiers R, Friderici K, Rottman F (1974). Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proceedings of the National Academy of Sciences of the United States of America 71: 3971–3975. doi: 10.1073/pnas.71.10.3971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams JM, Cory S.. Modified nucleosides and bizarre 5′-termini in mouse myeloma mRNA. Nature. 1975;255(5503):28–33. doi: 10.1038/255028a0. [DOI] [PubMed] [Google Scholar]
- 4.Wei CM, Moss B (1975). Methylated nucleotides block 5ʹ-terminus of vaccinia virus messenger RNA. Proceedings of the National Academy of Sciences of the United States of America 72: 318–322. doi: 10.1073/pnas.72.1.318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei CM, Gershowitz A, Moss B.. Methylated nucleotides block 5ʹ terminus of HeLa cell messenger RNA. Cell. 1975;4(4):379–386. doi: 10.1016/0092-8674(75)90158-0. [DOI] [PubMed] [Google Scholar]
- 6.Wiener D, Schwartz S. The epitranscriptome beyond m(6)A. Nat Rev Genet. 2020;22(2):119–131. doi: 10.1038/s41576-020-00295-8. [DOI] [PubMed] [Google Scholar]
- 7.Dai X, Wang T, Gonzalez G, Wang Y. Identification of YTH Domain-Containing Proteins as the Readers for N1-Methyladenosine in RNA. Anal Chem. 2018;366(11):6380–6384. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Z, Qi M, Shen B, Luo G, Wu Y, Li J, Lu Z, Zheng Z, Dai Q, Wang H. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019;47(5):2533–2545. doi: 10.1093/nar/gky1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Safra M, Sas-Chen A, Nir R, Winkler R, Nachshon A, Bar-Yaacov D, Erlacher M, Rossmanith W, Stern-Ginossar N, Schwartz S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature. 2017;551(7679):251–255. doi: 10.1038/nature24456. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Zhao Q, Kaboli PJ, Shen J, Li M, Wu X, Yin J, Zhang H, Wu Y, Lin L, et al. m1A Regulated Genes Modulate PI3K/AKT/mTOR and ErbB Pathways in Gastrointestinal Cancer. Transl Oncol. 2019;12(10):1323–1333. doi: 10.1016/j.tranon.2019.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Q, Xue C, Yuan X, He Y, Yu Z. Gene signatures and prognostic values of m1A-related regulatory genes in hepatocellular carcinoma. Sci Rep. 2020;10:15083. doi: 10.1038/s41598-020-72178-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Annals of Oncology: Official Journal of the European Society for Medical Oncology. 2016;27(8):1482–1492. doi: 10.1093/annonc/mdw168. [DOI] [PubMed] [Google Scholar]
- 13.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fang H, Declerck YA. Targeting the tumor microenvironment: from understanding pathways to effective clinical trials. Cancer Res. 2013;73(16):4965–4977. doi: 10.1158/0008-5472.CAN-13-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han D, Liu J, Chen C, Dong L, Liu Y, Chang R, Huang X, Liu Y, Wang J, Dougherty U, et al. Anti-tumour immunity controlled through mRNA m6A methylation and YTHDF1 in dendritic cells. Nature. 2019;566(7743):270–274. doi: 10.1038/s41586-019-0916-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, Zhou Q, Cao X. Mettl3-mediated mRNA m6A methylation promotes dendritic cell activation. Nat Commun. 2019;10(1):1898. doi: 10.1038/s41467-019-09903-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teschendorff AE, Zhuang J, Widschwendter M. Independent surrogate variable analysis to deconvolve confounding factors in large-scale microarray profiling studies. Bioinformatics. 2011;27(11):1496. doi: 10.1093/bioinformatics/btr171. [DOI] [PubMed] [Google Scholar]
- 19.Gaujoux R, Seoighe C. A flexible R package for nonnegative matrix factorization. BMC Bioinform. 2010;11(1):367. doi: 10.1186/1471-2105-11-367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Charoentong P, Finotello F, Angelova M, Mayer C, Efremova M, Rieder D, Hackl H, Trajanoski Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017;18(1):248–262. doi: 10.1016/j.celrep.2016.12.019. [DOI] [PubMed] [Google Scholar]
- 21.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14(1):7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Belkina AC, Ciccolella CO, Anno R, Halpert R, Spidlen J, Snyder-Cappione JE. Automated optimized parameters for T-distributed stochastic neighbor embedding improve visualization and analysis of large datasets. Nat Commun. 2019;10(1):5415. doi: 10.1038/s41467-019-13055-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B, Wu Q, Li B, Wang D, Zhou YL. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer. 2020;19(1). doi: 10.1186/s12943-020-01170-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18(1):220. doi: 10.1186/s13059-017-1349-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165(1):35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thissen D, Steinberg L, Kuang D. Quick and Easy Implementation of the Benjamini-Hochberg Procedure for Controlling the False Positive Rate in Multiple Comparisons. Journal of Educational and Behavioral Statistics. 2002;27(1):77–83. doi: 10.3102/10769986027001077. [DOI] [Google Scholar]
- 28.Woo HH, Chambers SK. Human ALKBH3-induced m(1)A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochimica Et Biophysica Acta Gene Regulatory Mechanisms. 2019;1862(1):35–46. doi: 10.1016/j.bbagrm.2018.10.008. [DOI] [PubMed] [Google Scholar]
- 29.Chen DS, Mellman I. Elements of cancer immunity and the cancer–immune set point. Nature. 2017;541(7637):321–330. doi: 10.1038/nature21349. [DOI] [PubMed] [Google Scholar]
- 30.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554(7693):538–543. doi: 10.1038/nature25492. [DOI] [PubMed] [Google Scholar]
- 31.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554(7693):544–548. doi: 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, NY). 2015;348(6230):124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouw KW, Goldberg MS, Konstantinopoulos PA, D’Andrea AD. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017;7(7):675–693. doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, NY). 2017;357(6349):409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, Ben-Haim MS, Dai Q, Di Segni A, Salmon-Divon M, Clark WC, et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature. 2016;530(7591):441–446. doi: 10.1038/nature16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Xiong X, Wang K, Wang L, Shu X, Ma S, Yi C. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311–316. doi: 10.1038/nchembio.2040. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, Rauch S, Dai Q, Cui X, Zhang Z, Nachtergaele S, Sepich C, He C, Dickinson BC. Evolution of a reverse transcriptase to map N(1)-methyladenosine in human messenger RNA. Nat Methods. 2019;16(12):1281–1288. doi: 10.1038/s41592-019-0550-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwartz S. m1 A within cytoplasmic mRNAs at single nucleotide resolution: a reconciled transcriptome-wide map. Rna. 2018;24(11):1427–1436. doi: 10.1261/rna.067348.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article from the Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/), and Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/).