

Abstract
BACKGROUND:
Fibrolamellar carcinoma (FLC) is a rare primary liver cancer of young adults. A functional chimeric transcript resulting from the in-frame fusion of the DNAJ homolog, subfamily B, member 1 (DNAJB1), and the catalytic subunit of protein kinase A (PRKACA) genes on chromosome 19 is believed to be unique in FLC, with a possible role in pathogenesis, yet with no established therapeutic value. The objective of the current study was to understand the molecular landscape of FLC and to identify potential novel therapeutic targets.
METHODS:
Archival fresh, formalin-fixed, paraffin-embedded samples from patients with FLC who prospectively consented to an institutional review board-approved protocol were analyzed using Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a next-generation sequencing assay encompassing up to 468 key cancer genes. Custom targeted RNA-Seq was performed in selected patients. Demographics, treatment, and outcome data were collected prospectively. Survival outcomes were estimated and correlated with mutation and/or copy number alterations.
RESULTS:
A total of 33 tumor samples from 31 patients with FLC were analyzed. The median age of the patients at the time of diagnosis was 18 years and approximately 53% were women. The DNAJB1-PRKACA fusion transcript was detected in 100% of patients. In 10 of 31 patients in which MSK-IMPACT did not detect the fusion, its presence was confirmed by targeted RNA-Seq. TERT promoter mutation was the second most common, and was detected in 7 patients. The median follow up was 30 months (range, 6-153 months). The 3-year overall survival rate was 84% (95% CI, 61%-93%).
CONCLUSIONS:
The DNAJB1-PRKACA fusion transcript is nonspecific and nonsensitive to FLC. Its potential therapeutic value currently is under evaluation. Opportunities currently are under development for therapy that may be driven or related to the DNAJB1-PRKACA fusion transcript or any therapeutic target identified from next-generation sequencing in patients with FLC.
Keywords: catalytic subunit of protein kinase A (PRKACA), DNAJ homolog, subfamily B, member 1 (DNAJB1), fibrolamellar carcinoma (FLC), Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), TERT
INTRODUCTION
Fibrolamellar carcinoma (FLC) is an extremely rare form of liver cancer that is estimated to comprise <1% of all primary liver cancers and is distinct from conventional hepatocellular carcinoma.1,2 FLC comprises up to 13.4% of all liver cancer patients occurring in patients aged <40 years.1 Although FLC cells histologically have been characterized by well-differentiated neoplastic hepatocytes and thick fibrous bands in a noncirrhotic background,3 to our knowledge the cell of origin in FLC has yet to be demonstrated. FLC occurs particularly in adolescents and young adults without evidence of underlying liver disease. 4–6 Survival is impacted mainly by lymph node and distant organ metastases.7 FLC has a poor outcome due to its resistance to chemotherapy and limited therapeutic options. Although to our knowledge surgical resection remains the only curative approach, it also is performed in patients with recurrent and/or metastatic disease.8–10 This is especially pertinent in the absence of alternative therapeutic approaches. FLC recently has gained more attention based on the discovery of an approximately 400-kilobase deletion in chromosome 19, which resulted in a fusion of the heat shock protein genes DNAJ homolog, subfamily B, member 1 (DNAJB1) and the catalytic subunit of protein kinase A (PRKACA).11,12 It is interesting to note that the DNAJB1-PRKACA fusion transcript is not detected in adjacent normal tissue. Analysis of the RNA sequencing data from The Cancer Genome Atlas for >9100 tumors across approximately 30 cancer types suggested the DNAJB1-PRKACA fusion transcript to be specific to FLC.13 However, recent data have demonstrated that the same fusion can occur in intraductal neoplasms of the pancreas and bile ducts,14 and also is not present in all patients of FLC. Biallelic protein kinase CAMP-dependent type I regulatory subunit alpha (PRKAR1A) mutations with loss of PRKAR1A expression have been reported in FLC without PRKACA gene rearrangement15 and another case of FLC characterized by PRKACA amplification without PRKACA rearrangement.16 To further understand FLC and identifying therapeutic targets, recent efforts at Memorial Sloan Kettering Cancer Center (MSKCC) using clustered regularly interspaced short palindromic repeats (CRISPR)–mediated mouse models demonstrated that the DNAJB1-PRKACA gene fusion actually drives tumorigenesis in mice, and that fusion to DNAJB1 leads to the initiation of FLC more effectively than wild-type PRKACA overexpression.15 The fact that PRKACA kinase domain appears to be conditional in tumor initiation suggests the potential value of developing kinase inhibitors of the fusion. Similarly, other investigators delivered CRISPR/Cas9 vectors designed to juxtapose exon 1 of DNAJB1 with exon 2 of PRKACA to create the DNAJB1-PRKACA fusion, or control Cas9 vector, to livers of 8-week-old female mice. Twelve of 15 mice who were given the CRISPR/Cas9 vectors and none of the 11 control mice developed neoplasms in the liver parenchyma. The oncogenic lesions exhibited cytologic and histologic features of human FLC.15 It is interesting to note that the generated mice tumors contained the DNAJB1-PRKACA gene fusion. Despite that, to our knowledge the predictive and targetable value of this fusion transcript has yet to be identified. The objective of the current study was to understand the molecular landscape of FLC past the DNAJB1-PRKACA fusion and to help to identify potential novel therapeutic targets.
MATERIALS AND METHODS
Over a 3-year period between July 2014 and June 2017, patients with a confirmed histologic diagnosis of FLC were identified and included. Informed consent was obtained from all patients under ClinicalTrials.gov identifier NCT01775072 for tumor genomic profiling in patients evaluated for targeted cancer therapy. The study protocol was reviewed by the institutional review board at MSKCC. The current study was conducted in accordance with the principles of the Declaration of Helsinki.
Results from 31 patients with FLC were available at the time of analysis. Clinical data were collected including demographics (age, sex, and race), family and personal history of malignancy, treatment details, and survival outcomes including progression-free survival (PFS) and 3-year overall survival (OS).
Sample Preparation
Histopathological review and microdissection of tissue from formalin-fixed, paraffin-embedded tumor samples were performed as needed to ensure adequate cellularity. Archival tissue (tissue obtained from prior surgical resection or biopsy) was used. Matched normal blood DNA from prospectively collected blood samples was analyzed in all patients to verify the somatic nature of the alterations detected. Germline mutation analysis also was performed if patients provided consent.
Genetic Analysis
Tumors were profiled for genomic alterations using Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a US Food and Drug Administration–approved hybridization capture-based clinical sequencing assay.16,17 Custom DNA probes were designed to capture all exons and selected introns (depending on the version of the test in use at the time the individual case was sequenced) of 341 (used for 8 patients), 410 (used for 12 patients), or 468 (used for 11 patients) oncogenes, tumor suppressor genes, and members of pathways deemed potentially actionable by targeted therapies (see Supporting Tables 1–3).
Genomic DNA from tumor and patient-matched normal samples was analyzed as previously described.16,18 Somatic copy number alterations were identified by comparing sequence coverage of targeted regions in the tumor sample relative to standard diploid normal. The resulting high-confidence single-nucleotide variant, indels, somatic copy number alterations, and structural variants as detected by MSK-IMPACT were used to produce a binary alteration matrix across all altered genes and samples. Genetic alterations were classified as actionable using a scale of 1 to 4, with level 1 to 2A alterations indicating standard therapeutic interventions and levels 2B to 4 suggesting investigational therapeutic alterations.19 Classification was performed using the OncoKB19 database, which integrates biologic, clinical, and therapeutic information curated from multiple resources, including recommendations derived from US Food and Drug Administration labeling, National Comprehensive Cancer Network guidelines, and the medical literature. The tumor mutational burden per sample was calculated as the total number of nonsynonymous mutations divided by the actual number of bases analyzed.16
Targeted RNA Sequencing
Targeted RNA sequencing using Archer DX platform18 was performed in patients that had undergone analysis using the previous MSK-IMPACT (version v3; 341 genes), which did not include the genes DNAJB1 or PRKACA. Archer targeted RNA sequencing also was performed in patients in which the fusion was not detected through version 5 of MSK-IMPACT (468 genes) due to low tumor content. cDNA libraries were created using the Archer FusionPlex standard protocol (Illimunia Inc, San Diego, California), Archer MBC adapters (Illimunia Inc; catalog no. #SA0040-45), and our custom-designed gene-specific primer (GSP) pool kit. Fusion unidirectional GSPs were designed to target specific exons in 62 genes known to be involved in chromosomal rearrangements based on the current literature. GSPs, in combination with adapter-specific primers, enriched for known and novel fusion transcripts, were sequenced using an Illumina MiSeq sequencer (Illumina Inc). At the end of MiSeq sequencing, FASTQ files were generated automatically using the MiSeq reporter software (version 2.6.2.3; Illumina Inc) and analyzed using the Archer analysis software (version 5.0.4; Illumina Inc).18
Statistical Analysis
PFS was calculated from the date of diagnosis of FLC to the time of first disease progression or death, whichever occurred first. OS was calculated from the date of diagnosis to the date of death or last follow-up. OS and PFS were estimated using Kaplan-Meier methods20. For genes with a frequency of mutation of at least 5%, an association between patients with and without mutations and/or copy number alterations and survival outcomes was computed using the permuted log-rank test.21 Among the subset of patients who underwent surgery (28 patients), PFS then was calculated from the date of surgery until the time of first disease progression or death and the log-rank test was used to compare PFS between patients who underwent an R0 versus R1 surgical resection. Patients who did not undergo surgery (3 patients) were excluded from this subgroup analysis. All statistical analyses were performed using R statistical software (version 3.3.2; R Foundation for Statistical Computing, Vienna, Austria).22 All P values were 2-sided and P < .05 was considered to indicate statistical significance.
RESULTS
Demographics
A total of 33 samples from 31 individual patients with histologically confirmed FLC were analyzed. Clinical characteristics are summarized in Table 1. All patients were white, with a slight female predominance (52%) noted. The majority of patients underwent upfront surgical resection (28 patients; 90%), 14 of whom underwent R0 resections.
Table 1.
Patient Characteristics (N = 31)
| Variable | Value |
|---|---|
| Age (range), y | 18 (17-24.5) |
| Sex, no.(%) | |
| Female | 16 (51.6) |
| Male | 15 (48.4) |
| Race, no. (96) | |
| White | 31 (100.0) |
| Stage at of disease at the time of diagrams, no. (%) (AJCC 8th edition) | |
| I | 1 (3.2) |
| II | 9 (29.0) |
| II/III | 1 (3.2) |
| III | 7 (22.6) |
| IV | 13 (41.9) |
| OCP use, no. (%) | |
| No | 25 (80.6) |
| PCOS | 1 (3.2) |
| Unknown | 2 (6.5) |
| Yes | 3 (9.7) |
| Family history of cancer | 12 (38.7%) |
Abbreviations: OCP, oral contraceptive pill; PCOS, polycystic ovary syndrome.
Landscape of Genomic Alterations in FLC
Genomic alterations were observed in all the examined samples. The DNAJB1-PRKACA fusion transcript was identified in all 31 patients (100%). In 12 patients, the DNAJB1-PRKACA fusion was not captured through MSK-IMPACT but rather through Archer targeted RNA sequencing: 8 samples tested negative when testing was performed using MSK-IMPACT version 3 (341 genes), which did not include the genes DNAJB1 or PRKACA, and in 4 samples the fusion was not detected by MSK-IMPACT version 5 (468 genes) due to low tumor content. Genetic sequencing of brain metastasis samples from 3 patients demonstrated the DNAJB-PRKACA fusion transcript in all metastatic samples.
In 6 tumor samples, there was the suggestion of a different splice variant. In addition to the typical fusion that reads from the end of exon 1 of DNAJB1 to the start of exon 2 of PRKACA, there were reads that started in the middle of exon 2 of DNAJB1. These findings were reported previously and suggested the presence of a predominant chimera and a second, minority chimera, incorporating the first exon and a part of the second exon of DNAJB1.12 Samples with the minority chimera, as identified by MSK-IMPACT, all had a breakpoint within exon 2 of DNAJB1. We were unable to predict whether this was in-frame or not. Of these samples, only 2 had Archer targeted RNA sequencing results and 1 sample demonstrated evidence of the “minority chimera.” That single sample suggested retention of intron 2 following exon 2. Both chimeras continued with exons 2 to 10 of PRKACA (Fig. 1).
FIGURE 1.
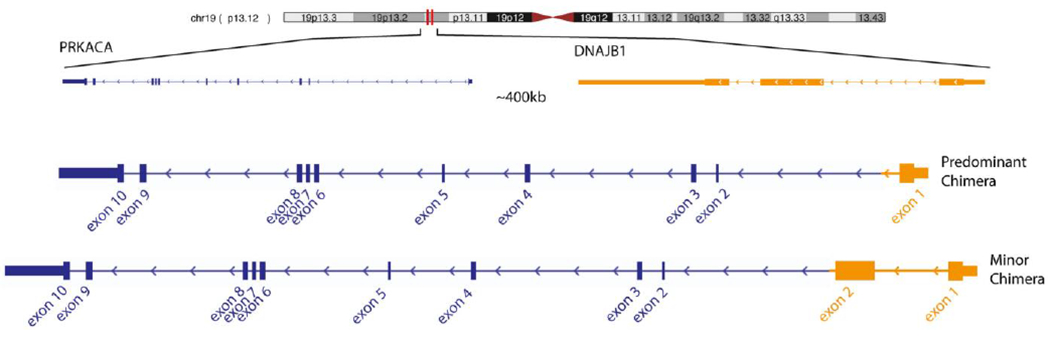
Predominant chimera incorporating only the first exon of DNAJ homolog, subfamily B, member 1 (DNAJB1) and a second, minority chimera, incorporating both the first and a part of the second exon of DNAJB1. Both chimeras continue with exons 2 to 10 of catalytic subunit of protein kinase A (PRKACA).
The second most frequently observed alterations were mutations in the TERT promoter and TERT amplifications (7 patients; 23%): 4 were promoter mutations, 2 were amplifications, and 1 was fusion. This was followed by mutations in SF3B1, ZFHX3, and CTNNB1 (2 patients each), as illustrated in the oncoprint shown in Figure 2 and Table 2. Both patients harboring CTNNB1 mutations were examined using immunohistochemistry, and neither was found to have abnormal nuclear β-catenin staining. Forty-three additional gene alterations were detected in 1 sample each (Table 2). No mTOR genetic alterations were identified. The tumor mutational burden score ranged from 0.9 to 5.3. Because to our knowledge there is no standard-of-care targeted therapy for patients with FLC, we looked for genetic alterations for which potentially active drugs were under development or drugs that have been used for other cancer types. To our knowledge, there currently is no specific agent to therapeutically target the DNAJB1-PRKACA fusion, and it remains unclear whether the DNAJB1-PRKACA chimera harbors a potential target. The oncogenic and likely oncogenic mutations per OncoKB were KRAS p.D119G, TERT promoter mutations, CTNNB1 p.K335I, the truncating mutations ARID1A p.Y1734*, and NCOR p.S1519 (Fig. 2) (Table 2). Several telomere-targeting strategies currently are in various stages of development.23
FIGURE 2.
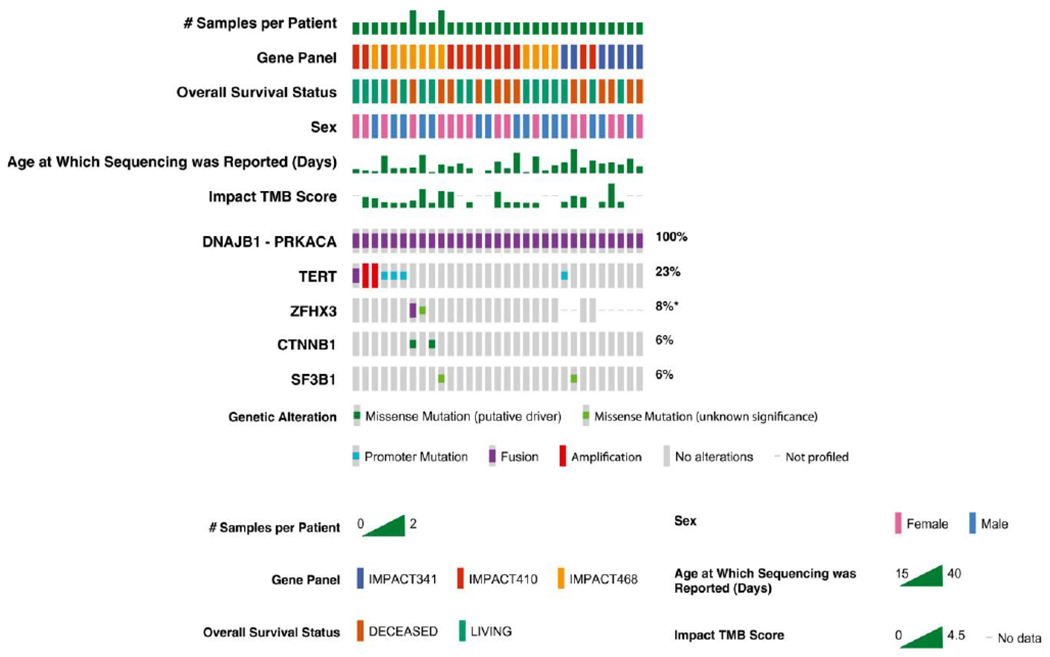
Oncoprint per sample evaluated. DNAJB1 indicates DNAJ homolog, subfamily B, member 1; IMPACT, Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets; PRKACA, catalytic subunit of protein kinase A; TMB, tumor mutational burden.
Table 2.
Oncogenic Mutations
| Gene | cDNA | Amino Acid |
|---|---|---|
| APC | c.1585C>T | p.L529F |
| ARID1A | c.5202T>G | p.Y1734* |
| ATR | c.4430C>G | p.P1477R |
| BCOR | c.2360C>T | p.P787L |
| BRD4 | c.886A>C | p.T296P |
| CHEK1 | c.1235T>C | p.V412A |
| CHEK2 | c.680G>A | p.G227E |
| CIC | c.89G>A | p.R30H |
| CTNNB1 | c.1004A>T | p.K335I |
| CTNNB1 | c.122C>T | p.T41I |
| DIS3 | c.2417A>G | p.D806G |
| EIF4A2 | c.881A>G | p.H294R |
| EPHA5 | c.2382G>C | p.M794I |
| ERBB3 | c.621_622delinsCC | p.K207_T208delinsNP |
| FAT1 | c.12791C>T | p.S4264L |
| GRIN2A | c.3211C>A | p.H1071N |
| HGF | c.1504C>G | p.R502G |
| INPP4B | c.7delA | p.I3fs |
| KMT2C | c.8539G>A | p.D2847N |
| KRAS | c.356A>G | p.D119G |
| MED12 | c.6118G>T | p.A2040S |
| MPL | c.1586G>A | p.W529* |
| NCOR1 | c.4556delC | p.S1519* |
| NKX3-1 | c.225G>C | p.Q75H |
| NOTCH1 | c.3340C>T | p.R1114C |
| NOTCH3 | c.6743C>T | p.S2248F |
| NSD1 | c.6531G>A | p.M2177I |
| KTRK2 | c.1784A>G | p.D595G |
| PAK7 | c.996T>A | p.D332E |
| RASA1 | c.143C>A | p.P48H |
| ROS1 | c.377G>A | p.G126E |
| SF3B1 | c.1447G>T | p.D483Y |
| SF3B1 | c.674G>T | p.G225V |
| SYK | c.1850C>A | p.P617H |
| TERT | g.1295228C>T | |
| TET1 | c.5973T>A | p.D1991E |
| WT1 | c.811C>A | p.H271N |
| YAP1 | c.370C>G | p.R124G |
| ZFHX3 | c.3195G>C | p.E1065D |
Abbreviation: cDNA, complementary DNA.
Means Substitution - Nonsense.
Germline Mutations
Fourteen patients provided consent for germline sequencing. Of these, 3 patients (21%) had pathogenic germline mutations detected: MUTYH: c.1187G>A in 2 patients and NBN c.657_661delACAAA in one patient. In one of the patients with a MUTYH germline mutation, histology demonstrated characteristic foci of FLC features with certain regions in which such features are less well developed. Reportedly, the tumor cells were positive for CK7 in a diffuse pattern, and positive for HepPar-1. The patient with NBN germline mutation histology was found to be positive for HepPar-1, CK7, and CK8/18. Family history was negative for both patients with MUTYH germline mutations. The father of the patient with the NBN germline mutation had prostate cancer. Based on the transition and/or transversion rate ratio using the Single Nucleotide Polymorphism database (dbSNP), no increased transversions or double-strand breaks were noted.
Clinical Outcomes and Genetic Correlation
The median follow-up among survivors was 30 months (range, 6-153 months). The most common sites of distant metastasis included the lungs (14 patients) and peritoneal cavity (10 patients). Three patients had bone metastases, 3 patients developed brain metastases, 1 patient had a urinary bladder lesion, and 1 patient had an intragastric metastasis.
Fifteen patients received cytotoxic therapy during their clinical course (5-fluorouracil, oxaliplatin, cisplatin, doxorubicin, temozolomide, and gemcitabine); the median number of therapies was 7, including local and systemic therapies. The 3-year OS rate was 83.7% (95% CI, 61%-93%) in the overall group (Fig. 3). The median PFS for the study population was 12 months (95% CI, 5-14 months) (Fig. 4). Four patients remained free of disease recurrence at the time of last follow-up. One may speculate a borderline association between PFS and surgical resection margin status (R0 vs R1). PFS from the time of surgery was 14 months (95% CI, 6-31 months) for patients who achieved an R0 surgical resection versus 5 months (95% CI, 0.98-12 months) for patients who underwent R1 surgical resections (P = .10) (Fig. 5). The median OS for the patients with R0 surgical resections was not reached, whereas the median OS for the R1 surgical resection group was 112 months (P = .093).
FIGURE 3.
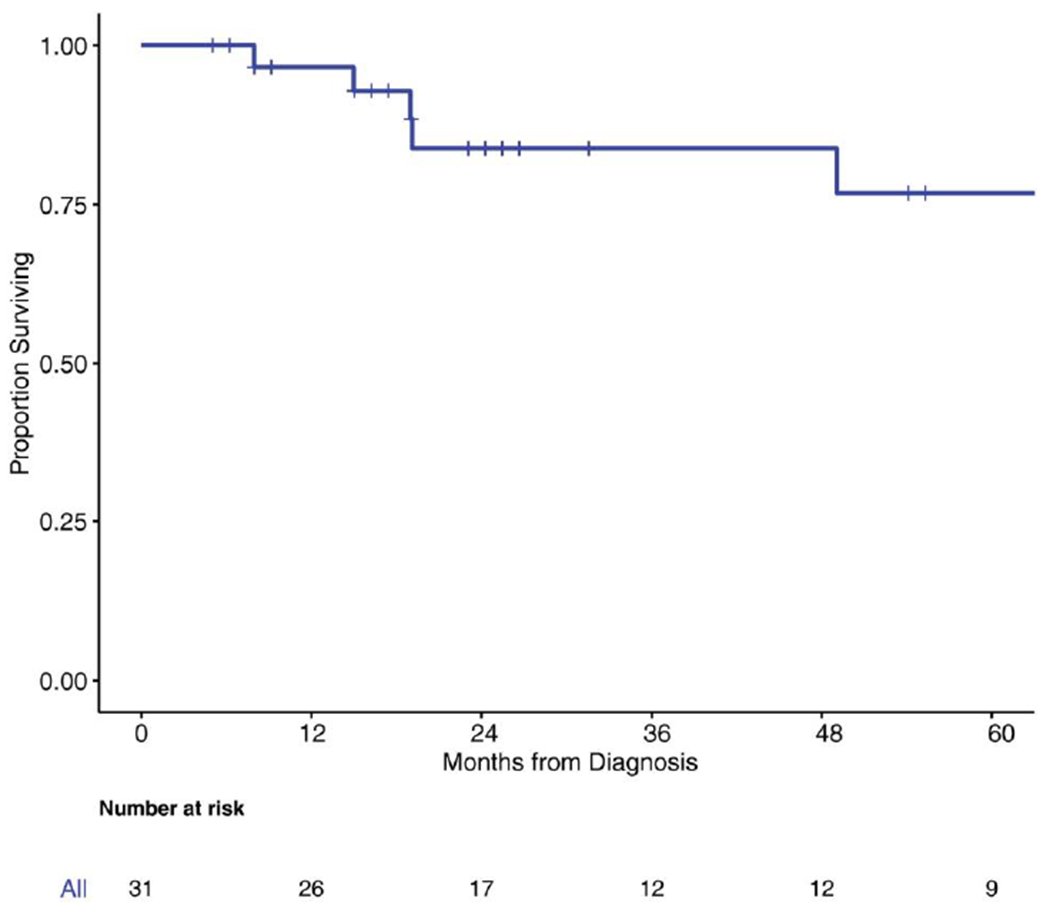
Kaplan-Meier curve for overall survival.
FIGURE 4.
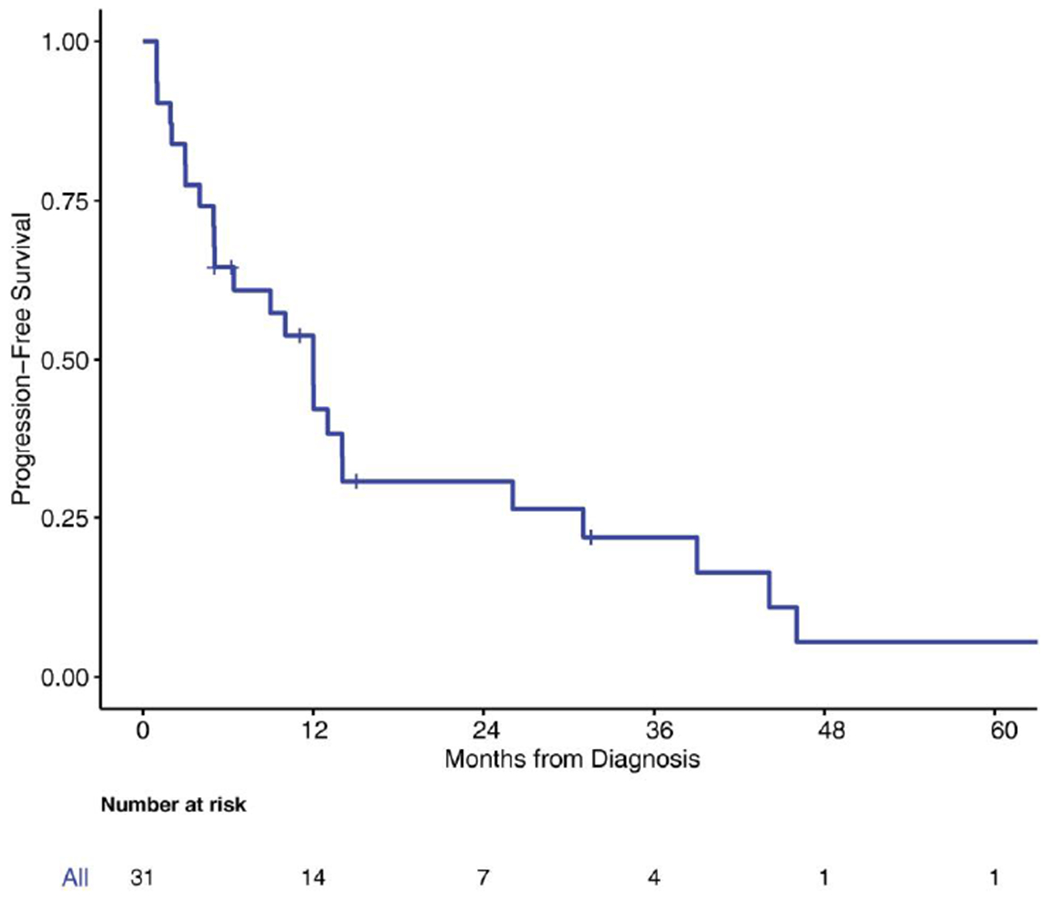
Kaplan-Meier curve for progression-free survival.
FIGURE 5.
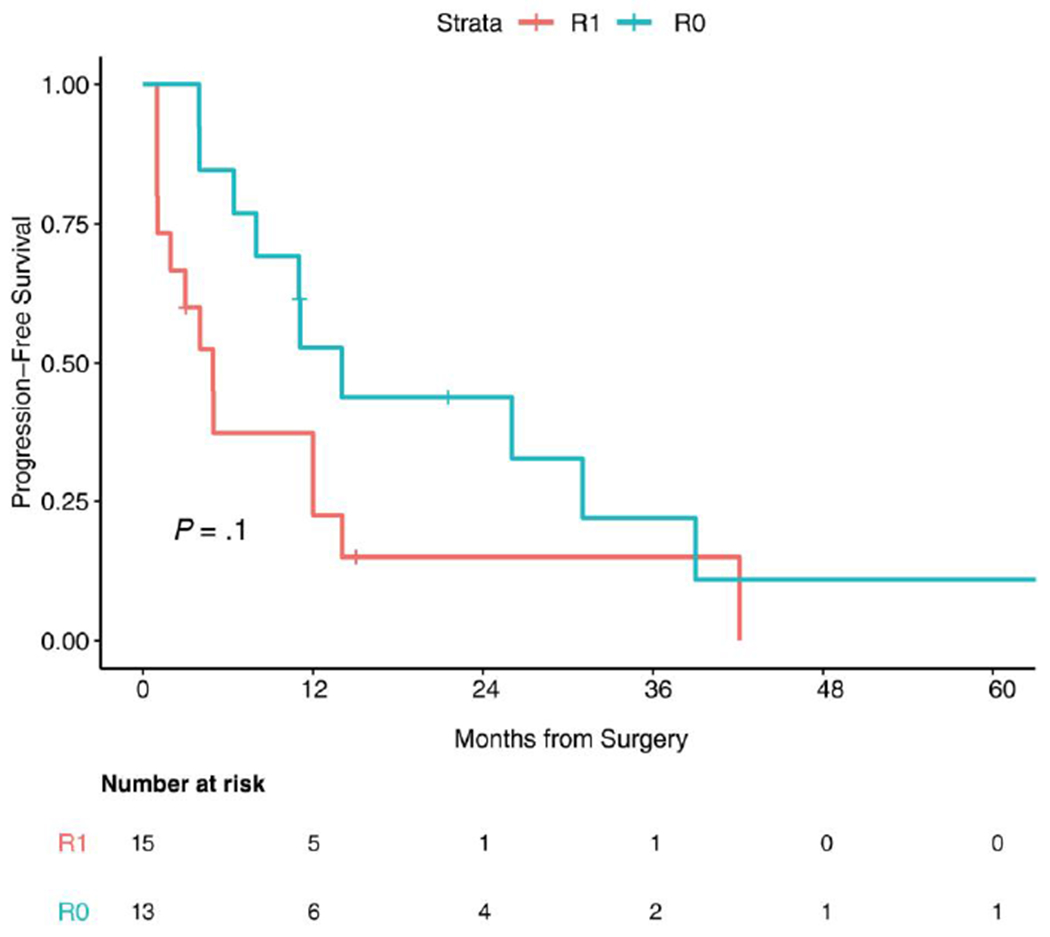
Kaplan-Meier curve for progression-free survival based on surgical resection margin status (R0 vs R1).
Sixteen patients underwent subsequent surgery. Among this group, the median time from first treatment until subsequent surgery was approximately 8.98 months (range, 2.03-117.138 months). To examine the association between subsequent surgery and OS, subsequent surgery was treated as time-dependent in the Cox proportional hazards model. No significant association between subsequent surgery and OS was noted in this population (hazard ratio, 1.28; 95% CI, 0.29-5.52 [P = .73]).
No other clinical features were found to be associated with outcome. No association between OS and PFS and recurrently detected genetic alterations was noted (Table 3).
Table 3.
Study of the Association Between Recurrently Mutated Genes and Survival Outcomes
| Characteristics | No. at Risk | No. Dead | 36-Month OS (95% CI) | P |
|---|---|---|---|---|
| TERT | .105 | |||
| 0 | 24 | 8 | 0.79 (0.63-1) | |
| 1 | 7 | 0 | 1 (1-1) | |
| CTNNB1 | .604 | |||
| 0 | 28 | 8 | 0.83 (0.68-1) | |
| 1 | 2 | 0 | NA | |
| ZFHX3 | .668 | |||
| 0 | 28 | 8 | 0.83 (0.69-1) | |
| 1 | 2 | 0 | NA |
Abbrevations: NA, not applicable; OS, overall survival.
DISCUSSION
Similar to previous efforts, the results of the current study helped to establish the presence of the chimeric DNAJB1-PRKACA transcript across examined FLC samples despite it not being universal and neither specific14 nor sensitive.15,16 The data from the current study are consistent with a previous report of transcriptome and whole-genome sequencing.12
In another report, whole-genome sequencing was performed on 10 paired tumor and adjacent normal liver tissues from an FLC cohort to identify recurrent oncogenic mutations24. There were relatively few somatic mutations, thereby placing FLC on the low end of the mutational spectrum. There were a few recurrent mutations, with the most frequently mutated gene being MUC4 in 4 patients. Aside from the heterozygous deletion on chromosome 19 that encodes for the functional chimeric protein, there were no other recurrent structural variations that contributed to the tumor genotype. In the current study, we observed a different splicing pattern with breakpoints outside of the canonical region in 6 patients (19%), 2 of which were run on Archer RNA sequencing, which supported the presence of a minority chimera in 1 sample (Fig. 1). However, the small number of samples limited our ability to make conclusions.
In another report, a similar observation was made based on DNA sequencing. This finding then was validated at the RNA level using Sanger sequencing and at the protein level by Western blot analysis, with sodium dodecyl sulfate–polyacrylamide gel electrophoresis demonstrating the chimeric protein.12
The DNAJB1-PRKACA fusion previously was reported in a study that examined 26 samples of FLC. All neoplastic cells harbored the gene fusion, as demonstrated by fluorescence in situ hybridization analysis. The fusion was not detected in the background liver, suggesting that it is a rather early step in FLC carcinogenesis25.
To our knowledge, the cell of origin in FLC is an area of controversy26; previous studies suggested a neuroendocrine signature. In the current study, we did not detect genes that are considered hallmarks of neuroendocrine differentiation. In one study evaluating genetic pathways in FLC tumors, among the genes that were found to be the most significantly overexpressed were prohormone convertase 1 (PCSK1), neurotensin, delta/notch-like EGF, and calcitonin, all of which were considered neuroendocrine genes27. Genetic studies have indicated that patients with FLC do not have genetic lesions that are common to other forms of liver cancer, such as loss-of-function mutations in TP53 or PTEN28. Of the alterations observed in FLC, the most frequent are gains in 1q and 8q (which also frequently are noted in other hepatic tumors) and loss of 18q. In the MSKCC cohort in the current study, the second most common genetic alteration was TERT promoter mutations, amplifications, and fusions, which were detected in 22% of patients.
The chimeric transcript is predicted to code for a protein that includes the catalytic domain of protein kinase A. Upregulation of aurora kinase A (AURKA) may be responsible for subsequent tumor formation in FLC.29 Targeting AURKA currently is underway in a recent study evaluating ENMD-2076 with selective activities against Aurora kinases30 that recently was reported, disappointingly, as negative.31
Riehle et al examined mTOR pathway activation in 23 samples of FLC. Phosphorylated S6 ribosomal protein (P-S6), a downstream target of mTORC1, and fibroblast growth factor receptor 1 (FGFR1) demonstrated strong staining activity on the tissue microarray.32 The results of the current study did not demonstrate expression of mTOR or FGFR pathway genes. Immunohistochemical staining for P-S6 and FGFR1 was not conducted. The mTOR inhibitor everolimus was evaluated alone and in combination with the aromatase inhibitor letrozole in 28 patients with FLC. Stable disease was reported in 6 patients (21%). The study was halted due to the low probability of extending PFS and the lack of efficacy.33
The FGFR pathway also was explored in another effort to understand the downstream targets of protein kinase A. Nineteen patients were evaluated: immunohistochemistry for FGFR1 was negative in all examined patients.34 There was modest FGFR1 messenger RNA expression and weak or absent protein expression noted. Furthermore, the FGFR2 rearrangement was not detected. To the best of our knowledge, this controversial and limited understanding of any potential role of the FGFR pathway in FLC has yet to be analyzed further, and would not exclude the exploration of FGFR inhibitors for the treatment of patients with FLC.
Whole-genome and RNA sequencing11 revealed an FLC subset associated with the EGF/ErbB2 gene signaling pathway, suggesting a potential therapeutic target. Neratinib, an irreversible pan-HER tyrosine kinase inhibitor, was evaluated as a single agent in patients with FLC in an open-label, multinational, phase 2 study (ClinicalTrials.gov identifier NCT01953926) that recently was discontinued because of a lack of efficacy.
This disappointing experience may suggest that future efforts should be directed toward targeting multiple pathways in FLC instead of using a single-target approach. Using immunohistochemistry to look for immune checkpoint targetable pathways in 32 tumor samples of FLC, PD-L1 was found to be expressed in tumor cells in approximately 63% of patients, and significant expression of B7-H3, PD-1, and IDO was noted both in tumor cells and in the tumor microenvironment.35 Checkpoint inhibitors have been used anecdotally with no significant results to our knowledge. Potential combination therapy as used for patients with hepatocellular carcinoma would be worth exploring.
The limitations of the current study included the small number of patients; nonetheless, its results have established the feasibility and potential clinical value for next-generation sequencing in patients with this rare disease. The use of multiple next-generation sequencing platforms (older versions of MSK-IMPACT) was a possible constraint to the study because some alterations may have been missed; however, the DNAJB1-PRKACA fusion was confirmed using Archer targeted RNA sequencing. Last, to the best of our knowledge, the etiology of FLC remains largely unknown, and the association between risk factors and a genetic signature could not be made; large-scale epidemiological studies are needed for this purpose.
In the current study, we were unable to identify mutations with known clinical actionability or an association between clinical outcome and genetic signature. Beyond the chimera, the TERT pathway alternations appeared frequently in the current study cohort, therefore justifying future therapeutic exploration. The results of the current study indicated DNAJB1-PRKACA as an ubiquitous fusion that can be detected by both targeted DNA and RNA sequencing in a clinical setting, despite its nonsecificity14 and nonsensitivity.15,16 Beyond the presence of DNAJB1-PRKACA, FLC tumorigenesis remains poorly understood, and ongoing efforts currently are underway to clarify oncogenesis and identify novel therapeutic targets.
Supplementary Material
FUNDING SUPPORT
Funded in part by Cycle for Survival, the Marie-Josée and Henry R. Kravis Center for Molecular Oncology, and National Cancer Institute Cancer Center Core Grant P30-CA008748.
CONFLICT OF INTEREST DISCLOSURES
Michael F. Berger has received personal fees from Roche and grants from Grail for work performed outside of the current study. Ahmet Zehir has received speaking honoraria from Illumina for work performed outside of the current study. Eileen M. O’Reilly has received grants from ActaBiologica, Agios, Astra Zeneca, Bayer, Beigene, Berry Genomics, BMS, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Mabvax, Puma, QED, Roche, Sillajen, Yiviva and has acted as a paid consultant for Agios, Astra Zeneca, Autem, Bayer, Beigene, Berry Genomics, Celgene, CytomX, Debio, Eisai, Eli Lilly, Flatiron, Genentech, Gilead, Incyte, Ipsen, LAM, Loxo, Merck, MINA, Polaris, QED, Redhill, Roche, Silenseed, Sillajen, Sobi, Therabionics, TWoxar, Vector, Yiviva. David B. Solit has acted as a paid member of the Scientific Advisory Boards for Pfizer, Loxo Oncology, Lilly Oncology, Illumina, and QED Therapeutics for work performed outside of the current study. Ghassan K. Abou-Alfa has received grants from ActaBiologica, Agios, Astra Zeneca, Bayer, Beigene, Berry Genomics, BMS, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Mabvax, Puma, QED, Roche, Sillajen, Yiviva and has acted as a paid consultant for Agios, Astra Zeneca, Autem, Bayer, Beigene, Berry Genomics, Celgene, CytomX, Debio, Eisai, Eli Lilly, Flatiron, Genentech, Gilead, Incyte, Ipsen, LAM, Loxo, Merck, MINA, Polaris, QED, Redhill, Roche, Silenseed, Sillajen, Sobi, Therabionics, Twoxar, Vector, Yiviva. In addition, Dr. Abou- Alfa has a patent “Articles and Methods for Preventing and Treating Dermatologic Adverse Events” identified by International Patent Application No. PCT/US2014/031545 filed on March 24, 2014 and priority application Serial No. 61/804,907 filed on March 25, 2013 issued. The other authors made no disclosures.
Footnotes
Additional supporting information may be found in the online version of this article.
REFERENCES
- 1.El-Serag HB, Davila JA. Is fibrolamellar carcinoma different from hepatocellular carcinoma? A US population-based study. Hepatology. 2004;39:798–803. [DOI] [PubMed] [Google Scholar]
- 2.Sooklim K, Sriplung H, Piratvisuth T. Histologic subtypes of hepatocellular carcinoma in the southern Thai population. Asian Pac J Cancer Prev. 2003;4:302–306. [PubMed] [Google Scholar]
- 3.Craig JR, Peters RL, Edmondson HA, Omata M. Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer. 1980;46:372–379. [DOI] [PubMed] [Google Scholar]
- 4.Moreno-Luna LE, Arrieta O, Garcia-Leiva J, et al. Clinical and pathologic factors associated with survival in young adult patients with fibrolamellar hepatocarcinoma. BMC Cancer. 2005;5:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Okuda K Natural history of hepatocellular carcinoma including fibrolamellar and hepato-cholangiocarcinoma variants. J Gastroenterol Hepatol. 2002;17:401–405. [DOI] [PubMed] [Google Scholar]
- 6.Pinna AD, Iwatsuki S, Lee RG, et al. Treatment of fibrolamellar hepatoma with subtotal hepatectomy or transplantation. Hepatology. 1997;26:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ang CS, Kelley RK, Choti MA, et al. Clinicopathologic characteristics and survival outcomes of patients with fibrolamellar carcinoma: data from the fibrolamellar carcinoma consortium. Gastrointest Cancer Res. 2013;6:3–9. [PMC free article] [PubMed] [Google Scholar]
- 8.Epstein BE, Pajak TF, Haulk TL, Herpst JM, Order SE, Abrams RA. Metastatic nonresectable fibrolamellar hepatoma: prognostic features and natural history. Am J Clin Oncol. 1999;22:22–28. [DOI] [PubMed] [Google Scholar]
- 9.Kakar S, Burgart LJ, Batts KP, Garcia J, Jain D, Ferrell LD. Clinicopathologic features and survival in fibrolamellar carcinoma: comparison with conventional hepatocellular carcinoma with and without cirrhosis. Mod Pathol. 2005;18:1417–1423. [DOI] [PubMed] [Google Scholar]
- 10.Yen JB, Chang KW Fibrolamellar hepatocellular carcinoma–report of a case. Chang Gung Med J. 2009;32:336–339. [PubMed] [Google Scholar]
- 11.Simon EP, Freije CA, Farber BA, et al. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2015;112:E5916–E5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Honeyman JN, Simon EP, Robine N, et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343:1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dinh TA, Vitucci EC, Wauthier E, et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci Rep. 2017;7:44653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vyas M, Hechtman JF, Zhang Y, et al. DNAJB1-PRKACA fusions occur in oncocytic pancreatic and biliary neoplasms and are not specific for fibrolamellar hepatocellular carcinoma. Mod Pathol. 2020;33:648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kastenhuber ER, Lalazar G, Houlihan SL, et al. DNAJB1-PRKACA fusion kinase interacts with beta-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2017;114:13076–13084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zehir A, Benayed R, Shah RH, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benayed R, Offin M, Mullaney K, et al. High yield of RNA sequencing for targetable kinase fusions in lung adenocarcinomas with no mitogenic driver alteration detected by DNA sequencing and low tumor mutation burden. Clin Cancer Res. 2019;25:4712–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. JCO Precis Oncol. 2017;2017. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 21.Heller G, Venkatraman ES. Resampling procedures to compare two survival distributions in the presence of right-censored data. Biometrics. 1996;52:1204–1213. [Google Scholar]
- 22.R Foundation for Statistical Computing. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2014. Accessed March 23, 2020. http://wwwR-projectorg/ [Google Scholar]
- 23.Xu Y, Goldkorn A. Telomere and telomerase therapeutics in cancer. Genes (Basel). 2016;7(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Darcy DG, Chiaroni-Clarke R, Murphy JM, et al. The genomic landscape of fibrolamellar hepatocellular carcinoma: whole genome sequencing of ten patients. Oncotarget. 2015;6:755–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graham RP, Jin L, Knutson DL, et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod Pathol. 2015;28:822–829. [DOI] [PubMed] [Google Scholar]
- 26.Ward SC, Huang J, Tickoo SK, Thung SN, Ladanyi M, Klimstra DS. Fibrolamellar carcinoma of the liver exhibits immunohistochemical evidence of both hepatocyte and bile duct differentiation. Mod Pathol. 2010;23:1180–1190. [DOI] [PubMed] [Google Scholar]
- 27.Malouf GG, Job S, Paradis V, et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology. 2014;59:2228–2237. [DOI] [PubMed] [Google Scholar]
- 28.Ward SC, Waxman S. Fibrolamellar carcinoma: a review with focus on genetics and comparison to other malignant primary liver tumors. Semin Liver Dis. 2011;31:61–70. [DOI] [PubMed] [Google Scholar]
- 29.Lim IIP, Greene-Colozzi EA, Murphy JM, Heaton TE, Simon SM, LaQuaglia MP. DNAJB1-PRKACA chimera increases Aurora kinase A expression in fibrolamellar hepatocellular carcinoma [abstract]. Cancer Res. 2015;75(15 suppl):LB-214-LB-. Abstract LB-214. [Google Scholar]
- 30.Fletcher GC, Brokx RD, Denny TA, et al. ENMD-2076 is an orally active kinase inhibitor with antiangiogenic and antiproliferative mechanisms of action. Mol Cancer Ther. 2011;10:126–137. [DOI] [PubMed] [Google Scholar]
- 31.Abou-Alfa GK, Mayer R, Venook AP, et al. Phase II multicenter, open-label study of oral ENMD-2076 for the treatment of patients with advanced fibrolamellar carcinoma. Oncologist. Published online March 10, 2020. doi: 10.1634/theoncologist.2020-0093 [DOI] [PMC free article] [PubMed]
- 32.Riehle KJ, Yeh MM, Yu JJ, et al. mTORC1 and FGFR1 signaling in fibrolamellar hepatocellular carcinoma. Mod Pathol. 2015;28: 103–110. [DOI] [PubMed] [Google Scholar]
- 33.El Dika I, Mayer RJ, Venook AP, et al. A multicenter randomized three-arm phase II study of (1) everolimus, (2) estrogen deprivation therapy (EDT) with leuprolide + letrozole, and (3) everolimus + EDT in patients with unresectable fibrolamellar carcinoma. Oncologist. 2020. doi: 10.1634/theoncologist.2020-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graham RP, Garcia JJ, Greipp PT, Barr Fritcher EG, Kipp BR, Torbenson MS. FGFR1 and FGFR2 in fibrolamellar carcinoma. Histopathology. 2016;68:686–692. [DOI] [PubMed] [Google Scholar]
- 35.Kim AK, Gani F, Layman AJ, et al. Multiple immune-suppressive mechanisms in fibrolamellar carcinoma. Cancer Immunol Res. 2019;7:805–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.