

Abstract
Oximes have been studied for decades because of their significant roles as acetylcholinesterase reactivators. Over the last twenty years, a large number of oximes have been reported with useful pharmaceutical properties, including compounds with antibacterial, anticancer, anti-arthritis, and anti-stroke activities. Many oximes are kinase inhibitors and have been shown to inhibit over 40 different kinases, including AMP-activated protein kinase (AMPK), phosphatidylinositol 3-kinase (PI3K), cyclin-dependent kinase (CDK), serine/threonine kinases glycogen synthase kinase 3 α/β (GSK-3α/β), Aurora A, B-Raf, Chk1, death-associated protein-kinase-related 2 (DRAK2), phosphorylase kinase (PhK), serum and glucocorticoid-regulated kinase (SGK), Janus tyrosine kinase (JAK), and multiple receptor and non-receptor tyrosine kinases. Some oximes are inhibitors of lipoxygenase 5, human neutrophil elastase, and proteinase 3. The oxime group contains two H-bond acceptors (nitrogen and oxygen atoms) and one H-bond donor (OH group), versus only one H-bond acceptor present in carbonyl groups. This feature, together with the high polarity of oxime groups, may lead to a significantly different mode of interaction with receptor binding sites compared to corresponding carbonyl compounds, despite small changes in the total size and shape of the compound. In addition, oximes can generate nitric oxide. This review is focused on oximes as kinase inhibitors with anticancer and anti-inflammatory activities. Oximes with non-kinase targets or mechanisms of anti-inflammatory activity are also discussed.
Keywords: oxime, kinase inhibitor, indirubin, nitric oxide, molecular modeling, inflammation, cancer
1. Introduction
Oxime compounds have been investigated for decades because of their significant roles as acetylcholinesterase reactivators and their use as therapeutics for a number of diseases [1,2,3]. Metabolites of various oximes have also been identified in plants as intermediates in biosynthesis and can facilitate a range of processes important for plant growth and development (for review [4]). Since amidoximes were found to be synthetic antimicrobial agents [5], oximes with different scaffolds have been developed for the treatment of bacterial infections, including tuberculosis [6,7,8,9,10]. Oximes have also been reported to exhibit a wide range of biological activities, such as anti-inflammatory [11,12,13,14,15] and anti-human immunodeficiency (HIV) agents that can inhibit HIV protease [16,17]. Indeed, the anti-inflammatory activity of some oximes has been reported to be comparable to standard anti-inflammatory drugs, such as indomethacin, diclofenac, and dexamethasone [18,19,20]. On the other hand, the introduction of an oxime group into an appropriate chemical backbone is a reasonable approach for the preparation of cytotoxic agents, and many oxime derivatives have been reported to have therapeutic activity for cancer [2,21,22,23,24,25,26,27] and neurodegenerative disorders [28,29,30].
The introduction of oxime groups has been reported to increase the biological activity of several natural compounds (Figure 1). For example, oxime derivatives of gossypol, a natural phenol derived from the cotton plant, exhibit antiviral, insecticidal, and fungicidal activity [31]. Another example is psammaplin A analog, the free oxime group which was responsible for high anticancer activity [32]. Moreover, oxime derivatives of radicicol, a macrocyclic antifungal antibiotic, showed higher inhibitory activity toward Src tyrosine kinase and anticancer activity in comparison with the parent compound [33,34]. Similarly, the oxime modifications made on the biflorin structure led to an increase in antibacterial potential [7]. Acylated oximes derived from triterpenes have shown cytotoxic or antiproliferative activity against many lines of cancer cells [35]. The biological activity of several indirubin oxime derivatives is much higher than that of the plant alkaloid indirubin [36,37]. Finally, we recently reported that the oxime derivative of the natural alkaloid tryptanthrin is a c-Jun N-terminal kinase (JNK) inhibitor [38] (Figure 1).
Figure 1.

Introduction of oxime groups increases kinase inhibitory activity of natural compounds.
Oximes have been used in the design of various kinase inhibitors, including phosphatidyl inositol 3-kinase (PI3K) inhibitors [39], phosphorylase kinase (PhK) [40], and JNK [38,41] (see Table 1 and Table 2). For example, indirubin oximes are of interest because of their high affinity binding to the ATP-binding site of protein kinases involved in tumorigenesis, e.g., cyclin-dependent kinases (CDK), glycogen synthase kinase (GSK) 3β, vascular endothelial growth factor receptor 2 (VEGFR-2), c-Src, and casein kinase 2 (CK2) [42,43,44,45,46,47,48]. Many of these kinases are molecular targets for compounds with anticancer activity.
Table 1.
Indirubin oxime-based kinase inhibitors and their kinase targets.
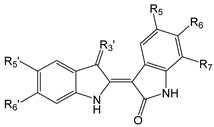
| Compound | R5 | R6 | R7 | R3′ | R5′ | R6′ |
|---|---|---|---|---|---|---|
| 1 | H | H | H | =N-OH | H | H |
| 2 | H | H | H | =N-OAc | H | H |
| 3 | H | H | H | =N-OCH2CHOHCH2OH | H | H |
| 4 | OCH3 | H | H | =N-O-CHOH-CH2OH | H | H |
| 5 | OCH3 | H | H | =N-O-(CH2)2OH | F | F |
| 6 |
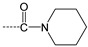
|
H | H | =N-OH | H | H |
| 7 | NO2 | H | H | =N-OH | OH | H |
| 8 | NHC(O)Bu | H | H | =N-OH | H | H |
| 9 | C(O)OCH3 | H | H |
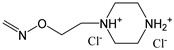
|
H | H |
| 10 | I | H | H | =N-OH | H | H |
| 11 | H | Br | H | =N-OH | H | H |
| 12 | H | Br | H | =N-OAc | H | H |
| 13 | H | Br | H |
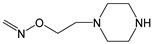
|
H | H |
| 14 | H | H | Br | =N-OH | H | H |
| 15 | H | H | Br | =N-OH | COOH | H |
| 16 | F | H | H |
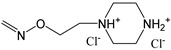
|
H | H |
| Compound | Kinase target (IC50, μM) | Ref. | ||||
| 1 | GSK-3α/β (0.022), CDK1 (0.18), CDK2 (0.7), CDK5 (0.1), CDK9 (2.4), PhK (0.21), FLT3 (0.033), AMPK (0.22), Lck (0.3), SGK (0.38), DRAK2 (0.71) | [37,40,43,44,49,50,51] | ||||
| 2 | CDK1 (1.2), CD5 (0.7), PhK (0.17), GSK-3α/β (0.2) | [40] | ||||
| 3 | CDK2 (0.23), Src (0.43), CDK6, CDK16, GSK-3β | [47,52] | ||||
| 4 | CDK2 (0.043), JAK1 (0.01), JAK2 (0.074), Tyk2 (0.001), c-Src (0.011), Lyn (0.03), Hck (0.264), Aurora A, c-Kit, GSK-3β, IGF1R, VEGFR2, ABL | [47,52] | ||||
| 5 | CDK2 (0.4), CDK9 (0.3) | [53] | ||||
| 6 | Aurora A (0.37) | [54] | ||||
| 7 | CDK2 (0.002) | [55] | ||||
| 8 | DRAK2 | [51] | ||||
| 9 | FLT3 (0.003), JAK2 (0.52), JAK3 (0.69), cMET (0.24), IRAK4 (0.3) | [56] | ||||
| 10 | GSK-3α/β, CDK1, CDK5 | [37] | ||||
| 11 | GSK-3β (0.005), CDK1 (0.32), CDK5 (0.083), PhK, Aurora A (0.6), Aurora B (0.9), Aurora C (0.2), DYRK1a (1.7), DYRK2 (2.1) | [40,44,57,58] | ||||
| 12 | CDK5 (2.4), GSK-3α/β (0.01), PhK (0.33) | [40,44] | ||||
| 13 | c-Src (0.0002), JAK1 (0.6), JAK2 (0.03), TYK2 (0.05), GSK-3β (0.003) | [59,60] | ||||
| 14 | Aurora B (4.6), Aurora C (0.7), DYRK1a (1.9), DYRK2 (1.3) | [57,58] | ||||
| 15 | DYRK1a (0.21), DYRK2 (0.13) | [58] | ||||
| 16 | FLT3 (0.001) | [61] | ||||
Table 2.
Miscellaneous oxime-based kinase inhibitors and their kinase targets.
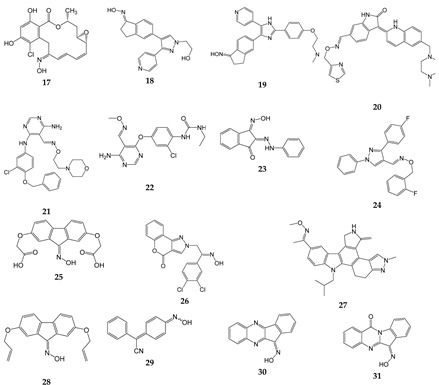
| Compound | Kinase target (IC50, µM) | Ref. |
|---|---|---|
| 17 | Src (0.056) | [34] |
| 18 | B-Raf (0.0001) | [62] |
| 19 | B-Raf (Ki = 0.0002), c-Raf (Ki = 0.0017) | [63] |
| 20 | VEGFR2 (0.009) | [64] |
| 21 | ErbB1 (0.022), ErbB2 (0.038), ErbB4 (0.021) | [65] |
| 22 | VEGFR-2 (0.04), Ret (0.18), Kit (0.5 | [66] |
| 23 | EGFR (50.3% at 100 µM) | [67] |
| 24 | PI3Kγ (1.3) | [68] |
| 25 | Chk1 (13.4) | [69] |
| 26 | PI3Kα (0.012), PI3Kβ (0.187), PI3Kγ (0.293), PI3Kσ (0.219) | [39] |
| 27 | VEGF-R1 (0.008), VEGF-R3 (0.01), TIE-2 (0.03) | [70] |
| 28 | EGFR (55.3% at 100 µM) | [67] |
| 29 | CK2 | [71] |
| 30 | JNK1/2/3 | [41,72] |
| 31 | JNK1/2/3 | [38] |
2. Chemical Characterization of Oximes
Oxime groups can be easily introduced into organic molecules by reaction of a carbonyl compound (ketone or aldehyde) with hydroxylamine (NH2OH) or a hydroxylammonium salt. This chemical modification leads to the appearance of a new pharmacophoric feature, since the oxime moiety contains two H-bond acceptors (nitrogen and oxygen atoms) and one H-bond donor (OH group), instead of the single H-bond acceptor of the C=O group present in the carbonyl precursor. These features, together with the high polarity of oxime groups, can lead to significantly different modes of interaction with receptor binding sites versus the corresponding carbonyl compound, although only small changes occur in the total size and shape of the ligand.
Oximes of aldehydes and non-symmetrical ketones can be obtained in two geometrical isomeric forms that are assigned either E or Z configurations with respect to the C=N bond (Figure 2). For many oximes, the energy barrier for Z,E-isomerization is high, i.e., the isomers exist as individual compounds at room temperature and under physiological conditions, as demonstrated by NMR spectroscopy [73]. Oxime stereochemistry can be important for pharmacological properties, as demonstrated by the antidepressant fluvoxamine, where only the E isomer is active [74]. It should also be noted that nitric oxide (NO) can catalyze E/Z isomerization of some oximes, most likely by a spin catalytic mechanism [75].
Figure 2.

E/Z isomerism of aldoximes.
Major plant oximes are amino acid-derived metabolites. It should be noted that the E isomers but not the Z isomers of plant oximes have high biological activity, including growth regulation, plant defense, pollinator attraction, and plant communication [4].
The hydrogen atom of the oxime OH group can be replaced with alkyl, acyl, or other substituents, and the general synthetic paths for O-substituted derivatives (oxime ethers or esters) include direct alkylation or acylation [76]. In addition, oximation of a corresponding aldehyde or ketone precursor by an appropriate O-substituted hydroxylamine is widely used (see, e.g., [38,77]). Many biologically active compounds discussed in the present review are actually oxime ethers or esters. For example, compounds 2 and 12 (Table 1) are O-acylated oximes, whereas 3–5, 9, 13, 16, 20–22, 24, and 27 (Table 1 and Table 2) contain an O-alkyloxime fragment. The high reactivity of the oxime OH group makes it possible to obtain corresponding salts (oximates). Typical pKa values for aryloximes in aqueous solutions are ~11 [78], but may decrease to 7–8 in some special cases of oximes with stabilized conjugated bases, e.g., bearing pyridinium moieties [79]. Hence, it is expected that oximates are prone to hydrolysis in an organism. Thus, oxime salts can be regarded as pro-drugs with better bioavailability than the parent oximes.
3. Anticancer Activity of Oximes
Several kinases shown in Table 1 and Table 2 are potential targets for anticancer therapy, and the anticancer activities of selected oximes in various in vitro and in vivo models are summarized in Table 3 and Table 4.
Table 3.
In vitro anticancer activity of selected oximes.
| Compound | Cells | Concentration range (µM) | Effect/Mechanism a | Ref. |
|---|---|---|---|---|
| 1 | Pancreatic ductal adenocarcinoma cells | 1–10 | ↓ p-CDK1/cyclinB1 | [80] |
| MG63 and U2-OS osteosarcoma | 1–10 | ↓ CDK2/4, FAK | [81] | |
| Cholangiocarcinoma linesNOZ, HuCCT1, OCUG-1, and OZ | 1–60 | [82] | ||
| 11, 14 | MDA-MB-231-TXSA breast cancer | 10–50 | ↑ Caspase-3 | [83] |
| 14 | Thyroid carcinoma | 1–10 | ↑ Caspase-3 | [84] |
| Neuroblastoma SH-SY5Y | 10–100 | [85] | ||
| 16 | MG63 and Saos-2 osteosarcoma | 1–30 | ↑ AMPK | [86] |
| MV4-11 and FLT3/D835Y expressed MOLM14 | IC50 = 0.001 (toward FLT3) | ↓ FLT3 | [61] | |
| 17 | 3Y1-B, SR-3Y1, NRK,KNRK5.2 cells | IC50 = 0.025 (toward v-Src) | ↓ v-Src activity; ↓ Raf-1 expression |
[34] |
| 26 | Human colorectal carcinoma HCT-116, human lung cancer A549, human liver carcinoma Huh7, human leukemia HL60 | 0.1–1 | Inhibitor of PI3Kα, PI3Kβ, PI3Kγ and PI3Kδ | [39] |
a ↓ and ↑ indicate decreasing or increasing enzyme activity or protein expression after treatment with compound, respectively.
Table 4.
In vivo anticancer activity of selected oximes.
| Compound | Model | Treatment | Ref. |
|---|---|---|---|
| 1 | Pancreatic ductal adenocarcinoma cells, inoculated s.c. | 10–40 mg/kg, i.p., daily for 4 days | [80] |
| 16 | MG63 osteosarcoma cells, inoculated s.c. | 5 mg/kg, i.p. daily for 45 days | [86] |
| MV-4-11 B-myelomonocytic leukemia cells, inoculated s.c. | 20 mg/kg, orally, daily for 21 days | [61] | |
| 20 | Lung cancer A549 cells, inoculated s.c. | 4 mg/kg, orally, daily for 14 day | [64] |
| 22 | A431 epidermoid carcinoma cells, HCT116 colorectal carcinoma cells, A375 skin melanoma cells; all cells inoculated s.c. |
10, 50, 100 and 200 mg/kg, intragastically, daily for 35 days, | [66] |
| 27 | A375 skin melanoma cells, inoculated s.c. | 10 mg/kg, orally, for 22 days | [70] |
s.c., subcutaneous; i.p., intraperitoneal; i.g., intragastic.
For example, CDKs are serine/threonine kinases that represent promising therapeutic oxime targets for treating various types of cancer [87]. Likewise, GSK-3 performs critical functions in many cellular processes, such as tumor growth, cell invasion, metastasis, and apoptosis (reviewed in [88,89]). Additional oxime targets include Aurora kinases, which are a group of serine/threonine kinases responsible for the regulation of mitosis. Aurora A and Aurora B are validated anticancer targets, and the development of Aurora kinase inhibitors has progressed from preclinical to clinical studies [90,91]. Oximes can also inhibit FMS-like tyrosine kinase-3 (FLT3), which is recognized as a drug target for the treatment of acute myeloid leukemia (AML), as activating mutations of FLT3 have been found in ~30% of AML patients. Targeted inhibition of FLT3 has shown promising results in the treatment of FLT3 mutation-dependent AML (for review [92]). Janus kinase 1 and 2 (JAK1/2) inhibitors represent an emerging and promising pharmacological class of anticancer drugs used notably for the treatment of some myeloproliferative neoplasms [93]. Finally, dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A) is overexpressed in a variety of diseases, including a number of human malignancies such as hematological and brain cancers (for review [94]), and could be an ideal oxime target.
Most reported oximes are multitargeted kinase inhibitors (see Table 1 and Table 2 and Figure 3) and can inhibit multiple intracellular signal transduction pathways. Therefore, they have therapeutic advantages over single-targeted inhibitors and have become a focus of antitumor drug research in recent years [95,96]. Recent reviews summarize the mechanisms of action of multitarget therapies and results of the latest clinical trials (e.g., [97,98]). On the other hand, these compounds may exhibit adverse events involving several biological systems due to their mechanism of multitargeted inhibition [99].
Figure 3.

Compound 11 is a multitargeted kinase inhibitor.
4. Anti-Inflammatory Activity of Oximes
Several of the kinases targeted by oximes represent potential targets for anti-inflammatory therapy, and the activities of selected oximes in various in vitro and in vivo models of inflammation are summarized in Table 5 and Table 6, respectively. For example, CDKs are potential oxime targets that play regulatory roles in influencing the pro-inflammatory functions of various cytokines during inflammation. CDKs initiate inflammatory responses by triggering the activity of prominent pro-inflammatory transcription factors, such as nuclear factor κB (NF-κB), signal transducer and activator of transcription 3 (STAT3), and activator protein 1 (AP-1) [100]. Likewise, the serine/threonine protein kinase GSK-3β has been implicated as an important regulator of the inflammatory response [101], and contributes to NF-κB activation, as well as to the induction of NF-κB-targeted pro-inflammatory molecules [102]. Indeed, GSK-3β inhibitors have potent anti-inflammatory activity and have been shown to be useful in treating neurodegenerative diseases, including Alzheimer’s disease (reviewed in [103,104]). Finally, recent findings from a Phase 3 clinical trial showed that patients with rheumatoid arthritis who were treated with a JAK1/2 inhibitor had significantly greater improvements in pain reduction [93,105]. Thus, oximes targeting JAK1/2 also have anti-inflammatory potential.
Table 5.
In vitro anti-inflammatory activity of selected oximes.
| Compound | Cell Culture | Model | Concentration Range (µM) | Effect/Mechanism a | Ref. |
|---|---|---|---|---|---|
| 1 | Adipocytes | Saturated free fatty acid-induced inflammation | 2–10 | ↑ Cell viability; ↑ mRNA for IL-4, IL-10, IL-13, TGF-β; ↓ mRNA for TNF, IL-1β, IL-6 | [106] |
| H9C2 rat cardiac myocyte cells | Incubation of cells with high glucose | 3–30 | ↓ PKR protein and mRNA; ↓ JNK and NF-κB mRNA; ↓ Caspase-3 mRNA; ↓ ROS |
[107] | |
| Cultured rat brain microglia, hippocampal slice cultures | LPS stimulation | 0.5–4 | ↓ NF-κB activation; ↓ TNF, IL-1β, PGE2, ROS; ↓ Hippocampal cell death | [108] | |
| Mouse microglia BV-2 cells, hippocampal slice cultures | LPS stimulation | 10 | ↓ Migration; ↓ iNOS expression; ↓ IL-6 and NO production | [30] | |
| Human neutrophils, monocytes, VSMCs | LTB4, CysLT and LT-enriched medium | 0.3–10 | ↓ LT-induced VSMC migration; ↑ HO-1 induction; ↓ 5-LO in monocytes and neutrophils |
[109] | |
| Human macrophages, primary type-I like pneumocytes | Influenza virus H5N1 infection | 10 | ↓ IP-10, IL-1β, RANTES, IFN-β, TNF; ↑ Delay of virus replication | [110] | |
| SH-SY5Y cells, primary cerebellar granule neurons | H2O2-induced apoptosis | 0.1–3 | ↑ Cell viability; ↓ p-Akt and p-GSK-3β | [111] | |
| 11 | Human FLS | TNF stimulation | 0.050 | ↓ mRNA for IL-1, IL-6, CCL-2, CCL-7, COX-2, MMP-9; ↓ IL-1, IL-6, CCL-2, CCL-7, COX-2, MMP-9; ↓ NF-κB, p-JNK, p-c-Jun, p-ATF-2, p-p38 | [13] |
| RAW264.7 macrophages | LPS stimulation | 2.5–20 | ↓ NO, PGE2; ↓ iNOS mRNA, COX-2;↓ IL-1β, IL-6; ↓ p-JNK, p-IκB-α; ↑ IκB-α |
[112] | |
| Neutrophils, RAW264.7 macrophages |
LPS stimulation | 5 | ↓ TNF; ↑ IκB-α | [113] | |
| Mouse mammary epithelial cells | LPS stimulation | 5–50 | ↓ mRNA for IL-1β, IL-6, IL-10, TNF; ↓ IL-1β, IL-6, TNF; ↑ IL-10; ↓ TLR4/NF-κB and TLR4/MAPK expression and phosphorylation |
[12] | |
| 30 | PBMCs, MonoMac-6, J774.A1 cells |
LPS stimulation | 0.2–30 | ↓ IL-1α, IL-1β, IL-6, TNF, IFN-γ, GM-CSF, NO production by human and murine monocyte/macrophages. | [41] |
| HUVECs | 0.3–10 | ↓ Endothelin-1 secretion | [114] | ||
| Macrophages, T-cells | LPS stimulation | 1 | ↓ TNF, IL-6, IL-1β; ↓ p-JNK2, p-p38, p-IκBα, p-IKKβ; ↓ IL-6 mRNA, TNF, iNOS | [115] | |
| 31 | Human FLS, synovial SW982 cells, HUVECs, monocytic THP-1 cells |
IL-1β stimulation | 1–25 | ↓MMP-3 gene expression; ↓ MMP-1/3 and IL-6 secretion | [116] |
| 32 | Human neutrophils | fMLF stimulation | 0.03–20 | ↓ HNE and Pr3 activities; ↓ ROS generation, HNE release | [15] |
a ↓ and ↑ indicate decreasing or increasing enzyme activity or protein/mRNA expression, or functional activity after treatment with compound, respectively.
Table 6.
In vivo anti-inflammatory activity of selected oximes.
| Compound | Animal | Model | Dose | Effect/Mechanism a | Ref. |
|---|---|---|---|---|---|
| 1 | Swiss albino mice | High fat-high fructose diet-induced neuropathological changes | 0.4 mg/kg for 7 days | ↓ Area occupied by dark neurons; ↓ Amyloid spots in hippocampus ↓ NF-κB; ↓ TNF, IL-6 ↓ Bax and caspase-3; ↑ Bcl-2 |
[29] |
| 11 | C57BL/6 mice | TPA-induced ear skin inflammation | 1.5 µg/ear | ↓ GSK-3β activity; ↓ IFN-γ production; ↓ Ear skin edema, epidermis hyperproliferation and dermis angiogenesis | [117] |
| Rats | Intracerebral hemorrhage | 10, 20, 40, 60, 80, & 100 µg/kg | ↓ NF-κB, COX-2, GSK-3β phosphorylation; ↑ Brain-derived neurotrophic factor; ↓ IL-1β and IL-6, ↑ IL-10; ↓ Microglia activation and cell apoptosis | [118] | |
| C57BL6/J mice | Transient occlusion of the MCA | 1 mg/kg i.p., 3 and 6 h after occlusion | ↑ Wnt/β-catenin pathway activation ;↓ Brain edema, IgG extravasation, perivascular petechial bleeding; ↓ Hemorrhagic transformation after ischemic stroke |
[119] | |
| C57BL/6 mice | Ligature + LPS-induced periodontitis | 0.5−5 μg in 1 mL hydrogel | ↓ Inflammatory cell infiltration; ↑ Expression of ALP, and Runx2 | [120] | |
| Mice | Aging | 1 mg/kg, i.p. during 2 weeks | ↓ IL-6 in liver and serum; ↑ SOD and GSH in liver; ↓ Total cholesterol and triglycerides in liver & serum | [121] | |
| Mice | Arthritis (collagen + complete Freund’s adjuvant) | 1 and 10 mg/kg | ↓ Synovial hyperplasia, infiltration of inflammatory cells, cartilage destruction, and bone erosion; ↓ TNF, IL-1, IL-6, and IFN-γ in serum | [13] | |
| 30 | Mice | Ovalbumin-specific DTH response | Every 12 h with 12.5 mg/kg, i.p., 5 injections | ↓Ear thickness | [41] |
| Mice | Acute lung inflammation (LPS plus D-galactosamine) |
200 µg/mouse, i.p. | ↓ Lethality and lung inflammation; ↓ TNF, IL-6 and IL-1β; ↓ p-JNK2, p-p38, p-IκBα & p-IKKβ; ↓ mRNA for IL-6, TNF and iNOS |
[115] | |
| Mice | CIA | 5, 20, 30 and 50 mg/kg, daily, i.p. | ↓CIA and CAIA severity; ↓Cartilage erosion; ↓ Collagen II-specific antibody | [122] | |
| Rats | Focal cerebral ischemia/reperfusion | 5 and 25 mg/kg, i.p. | ↓ p-c-Jun | [123] | |
| 31 | Mice | CIA and CAIA | 30 mg/kg i.p., daily, 34 days | ↓ CIA and CAIA severity; ↓ Cartilage erosion; ↓ IL-17A, GM-CSF, RANKL | [116] |
| 32 | Mice | HNE-induced paw edema |
50–100 mg/kg, i.p. | ↓ Paw edema | [15] |
| LPS-induced acute lung injury | 100 mg/kg, i.p. | ↓ MPO; ↓ Edematous changes, alveolar thickening, leukocyte infiltration, and lung tissue destruction |
a ↓ and ↑ indicate decreasing or increasing enzyme activity or protein/mRNA expression, or functional activity after treatment with compound, respectively.
5. Indirubin Oxime-Based Kinase Inhibitors
Indirubin, a bis-indole alkaloid found in some terrestrial plants and sea shells, is the active ingredient of Danggui Longhui Wan, a traditional Chinese herbal medicine used in the treatment of chronic myelocytic leukemia [25]. Indirubin inhibits various kinases in vitro and is thought to exert its action in vivo by this mechanism [43]. There have been a number of attempts to improve the biological activity and selectivity of indirubin through analog synthesis. Most efforts have focused on modifying this natural product structure by adding substituents around the fused phenyl rings or by converting its 3′-carbonyl group into an oxime [37] (Figure 1). The latter modification was shown to increase the potency of indirubin and its halogenated derivatives toward CDK1, CDK2, CDK5, GSK-3α/β, Aurora kinases A-C, FLT3, and JAK1/2 (reviewed in [36]).
Among the synthetic indirubin analogs, indirubin-3′-monoxime (compound 1) has been reported to inhibit growth of human MCF-7 breast cancer cells [124,125], osteosarcoma [82], and cholangiocarcinoma cells [81]. Compound 1 also suppressed the production of interleukin (IL)-1β, IL-6, NO, inducible nitric oxide synthase (iNOS), and cyclooxygenase 2 (COX-2) expression via downregulation of NF-κB and JNK signaling pathways in lipopolysaccharide (LPS)-treated murine RAW264.7 macrophage cells [112].
Using a combination of in silico virtual screening of potential anti-diabetic candidates and an in vitro study using an insulin-resistant model of 3T3-L1 adipocytes, Choudhary et al. [106] showed that 1 prevented lipid-induced impairment of the insulin signaling pathway in adipocytes via A2A adenosine receptor activation. While compound 1 reduced lipid-induced adipocyte inflammation by inhibiting NF-κB dependent pro-inflammatory cytokine expression, it also augmented cAMP responsive element binding protein (CREB) activation, favoring an overall anti-inflammatory state [106].
The pathogenesis of atherosclerosis is complex and depends on altered cholesterol metabolism and inflammation [126]. During the onset of atherosclerosis, monocytes adhere to sites of endothelial damage and migrate into the subendothelial layer, where they contribute to early lesion development by accumulating lipids and by secreting cytokines, growth factors, and leukotrienes. Those mediators facilitate further recruitment of immune cells and stimulate migration of vascular smooth muscle cells (VSMCs) from the medial to the intimal layer and, finally, to their proliferation [127,128]. Interestingly, Blazevic et al. [109] found that 1 inhibited leukotriene-mediated VSMC migration. Moreover, 1 inhibited 5-lipoxygenase (5-LO) in cell-based and cell-free assays [109].
Microglia are the resident immune cells of the brain and play a role in the pathogenesis of various central nervous system diseases [129,130,131]. Activated microglia promote neuronal injury through the release of proinflammatory and cytotoxic factors, including tumor necrosis factor (TNF), IL-1β, NO and reactive oxygen species (ROS) [132]. In LPS-induced models of inflammation in cultured rat brain microglia and in organotypic hippocampal slice cultures, compound 1 was shown to inhibit LPS-related hippocampal cell death, decrease the production of TNF, IL-1β, prostaglandin E2 (PGE2), and ROS, and also reduce LPS-induced NF-κB activation [108]. In addition, compound 1 was also reported to prevent neuronal apoptosis via inhibition of GSK-3β and extracellular signal-regulated kinase (ERK) [111,133]. These results suggest that compound 1 provides neuroprotection by reducing the production of various inflammatory mediators by activated microglia. Likewise, Sathiya Priya et al. [29] reported that compound 1 reduced the levels of NF-κB in nuclear extracts and decreased expression of TNF and IL-6 in a model of neuroinflammation (high fat/high fructose diet in mice). In addition, compound 1 may suppress aberrant NF-κB signaling via inactivation of IκB kinase β (IKK-β), an enzyme that is involved in propagating the cellular response to inflammation. Treatment with 1 significantly decreased the formation of dark neurons, which clearly indicates its recuperative effects towards neuronal apoptosis. Among the anti-apoptotic effects reported for compound 1 are the attenuation of pro-apoptotic Bax and caspase-3 expression, along with an increase in anti-apoptotic Bcl-2 [29].
Compound 1 can inhibit several different kinases, including CDK1, CDK2, CDK5, CDK9, GSK-3α/β, PhK, FLT3, AMP-activated protein kinase (AMPK), Lck, and serum- and glucocorticoid-induced kinase (SGK) [17,37,40,43,44,49,50] (Figure 3). The interaction mode of 1 is similar for both CDK2 and CDK9 [53]. According to reported docking studies, compound 1 forms H-bonds with hinge residue Cys106 via N1′ and O2 atoms. In addition, the oxime moiety is H-bonded via the OH group to the backbone carbonyl group of Ile25 [53]. There is an important link between activation of GSK-3β, amyloid deposition, and neuroinflammation. Indeed, treatment of murine microglial BV-2 cells with compound 1 greatly reduced LPS-stimulated migration, IL-6, and the expression of iNOS and NO production [30]. Likewise, 1 effectively prevented neuronal apoptosis via inhibition of GSK-3β [111,133] and suppression of inflammation, as GSK-3β has been shown to activate NF-κB in LPS-stimulated RAW264.7 macrophages [134] and increase expression of pro-inflammatory genes in LPS-stimulated human monocytic cells and mouse hippocampal slice cultures [30].
RNA-dependent protein kinase R (PKR) plays an important role in inflammation, insulin sensitivity, and glucose homeostasis [135]. For example, treatment of cultured rat cardiomyocytes with high glucose induced a significant increase in PKR, JNK, caspase-3, NF-ĸB, and ROS generation. Notably, all of these inflammatory responses were attenuated by pretreatment with compound 1 [107].
Compounds 1 and 2 (indirubin-3′-acetoxime) have also been shown to be relatively moderate inhibitors of PhK [40,136], which coordinates hormonal and neuronal signals to initiate the breakdown of glycogen. In comparison with indirubin, the parent non-oxime analog, compound 1 forms additional H-bond interactions with Glu110 of the γ catalytic subunit of PhK [136]. Compound 2 has higher PhK inhibitory activity and, in docking experiments, the acetoxime methyl group of 2 partially occupies a space of negative electrostatic potential created by the Glu153 oxygen and the PhK Glu110 and Asp167 side chain carboxylates [40]. These authors performed a thorough quantum mechanics/molecular mechanics (QM/MM) study of PhK-inhibitor interactions and found that the introduction of an oxime or acetoxime moiety in place of the 3′-carbonyl group in the indirubin molecule led to significantly more negative ΔEQM/MM, indicating more effective binding to PhK due to strong anchoring of the oxime or acetoxime group within a subpocket between Glu110, Glu153, and Asp167 [40].
In the context of the current coronavirus COVID-19 pandemic, the antiviral and anti-inflammatory properties of indirubin oxime derivatives should also be considered. Notably, compound 1 can suppress pro-inflammatory factors associated with viral infection, including chemokine CXCL10 (one of the key factors contributing to lung inflammation during H5N1 influenza virus infection), interferon (IFN)-β, and monocyte chemoattractant protein 1 (MCP-1) [110]. In addition, compound 1 delayed H5N1 virus replication in primary cell culture models [110].
Several different indirubin analogs have been synthesized to improve water solubility and bioavailability. A variety of side chains were introduced at the 3′-position, leading to the synthesis of compounds 3–5 (Figure 1) [46,47,53,124]. Likewise, compound 3, which contains a dihydroxypropyl 3′-oxime substituent together with an OCH3 group, is a potent inhibitor of Src kinase, and it downregulated the constitutively activated signal transducer and activator of transcription 3 (STAT3) or STAT5 in human breast cancer CML cells [46]. Compound 3 also inhibited CDK2, CDK6, CDK16, and GSK-3β [46,52,53]. High-grade gliomas can secrete large amounts of inflammatory cytokines and growth factors that promote autocrine tumor growth. Interestingly, 3 was able to suppress pro-inflammatory genes, including IL-1α, IL-1β, IL-12, prostaglandin endoperoxide synthase 2 (PTGS-2), and Toll-like receptor 4 (TLR4), as well as the secretion of the pro-inflammatory cytokine IL-6 in LN-18 and T98G glioblastoma cells [137]. Similarly, 6-bromoindirubin-3′-glycerol-oxime ether suppressed LPS-induced secretion of IL-1β and PGE2 via the inhibition of GSK-3β [138]. Unexpectedly, compound 4 appears to be a strong dual inhibitor of JAK/signal transducer and activator of transcription 3 (STAT3) and Src family of protein tyrosine kinases (SFKs)/STAT3 signaling that is associated with the induction of apoptosis in human pancreatic cancer cells [47,139]. It was also found that 4 is a potent inhibitor of a broad spectrum of serine/threonine and tyrosine kinases, including CDK2, JAK1/2, Tyk2, c-Src, Lyn, Hck, Aurora A, c-Kit, GSK-3β, IGF1R, VEGFR2, and ABL [47,52]. Another important family of synthetic indirubins are the 5-substituted analogs of compound 1, such as compounds 6–8 and 10 (Figure 1). Compound 6 was found to inhibit Aurora kinase A but had no effect on the kinase activities of c-Met, ALK, and JAK2 [55]. Likewise, compound 7 was reported to inhibit CDK2 and induce apoptosis of lung cancer cells [140,141]. The presence of an oxime group was also found to be essential for increasing the inhibitory activity of compound 8 against death-associated protein kinase-related apoptosis-inducing protein kinase (DRAK) 1/2, a serine/threonine kinase belonging to the death-associated protein kinase (DAPK) family [51]. According to docking results, the oxime OH group of compound 8 acts as an H-bond donor with respect to the Glu117 carboxyl oxygen of DRAK. The authors suggested an important role for this interaction in the binding of 8 to DRAK, along with other H-bonds formed by 8 with Glu111 and Ala113 via NH and C=O in the indolin-2-one moiety [51]. Finally, compound 9 and 5-fluoro-indirubin-3′-oxime have been recognized as potent inhibitors of FLT3, which is involved in cancer development, especially leukemia [49,56].
Halogenated indirubins are among the most important subcategories of indirubins, with the main representatives being 6-bromoindirubin and 6-bromoindirubin-3′-oxime (11). Notably, the affinity of compound 11 for GSK-3β (IC50 = 5 nM) is 100-fold greater than that of 6-bromoindirubin [44]. Indeed, the oxime analogs generally exhibit 5–10 times greater inhibitory activity toward GSK-3 β compared to the corresponding non-oxime halogenated indirubin derivatives [142]. Docking of 11 into GSK-3β was reported by Nisha et al. [143], who found that an oxime group forms H-bonds with Val135.
Compound 11 appears to have significant therapeutic potential due to its anti-inflammatory properties. For example, Liu et al. [12] investigated the effects of 11 on inflammatory signaling in mouse mammary epithelial cells (MMECs) and on LPS-induced mastitis in mice [12] and reported that it inhibited the TLR4/NF-κB and TLR4/mitogen-activated protein kinase (MAPK) pathways. This resulted in inhibition of JNK, ERK, and p38 phosphorylation, downregulation of IL-6, IL-1β, TNF, and myeloperoxidase (MPO) expression, and upregulation of IL-10 expression in MMECs. Consequently, compound 11 pretreatment downregulated the expression of the proinflammatory factors IL-1β, IL-6, TNF, and MPO in mammary glands and reduced inflammatory lesions in breast tissue of LPS-injected mice [12]. Similarly, Park et al. [113] showed that the inhibition of GSK-3β activity by 11 delayed the inhibitor of nuclear factor κB (IκBα) degradation and diminished expression of TNF in LPS-stimulated neutrophils and macrophages. In addition, compound 11 blocked GSK-3β phosphorylation/activation, decreased the levels of the proinflammatory cytokines TNF, IL-1β, and IL-6, elevated the level of anti-inflammatory cytokine IL-10, inhibited microglia activation and cell apoptosis, and improved the sensorimotor deficits of rats after intracerebral hemorrhage [118].
Kwon et al. [13] showed that compound 11 inhibited the NF-κB, JNK, c-Jun, activating transcription factor (ATF)-2 and p38 pathways in fibroblast-like synoviocytes (FLS). Consequently, 11 treatment also diminished the production of proinflammatory mediators IL-1, IL-6, MCP-1, MCP-3, COX-2, and matrix metalloproteinase (MMP)-9 by these FLS. The anti-inflammatory effects of compound 11 were also evaluated in vivo in a mouse model of collagen-induced arthritis (CIA). Treatment of CIA mice with 11 attenuated clinical and histological signs of arthritis. For example, infiltration of T cells, macrophages, and tartrate-resistant acid phosphatase positive cells was decreased in joint sections of mice with arthritis. Likewise, serum levels of IL-1β, IL-6, TNF, and IFN-γ were inhibited by compound 11 treatment [13]. Similarly, 11 inhibited production of IFN-γ and nuclear translocation of T-box (Tbx21), a transcription factor of IFN-γ, in CD3+ T cells in mouse model of skin inflammation [117]. In addition, this treatment attenuated epidermal hyperproliferation and dermal angiogenesis [117]. Compound 11 has also been shown to inhibit periodontal inflammation, promote bone regeneration, and induce the expression of bone-forming markers in a mouse periodontitis model [120].
Ischemic stroke triggers blood–brain barrier (BBB) breakdown via destabilization of the tight junctions and deregulation of the transport mechanisms [144]. Subsequently, BBB breakdown can contribute to the progression of secondary brain injury by causing edema formation, increasing the accumulation of toxic metabolites, and exacerbating the inflammatory response [145]. Another consequence of BBB disruption can be hemorrhagic transformation, which is a major complication of ischemic stroke, causing significant morbidity and mortality in patients [146]. The formation and maintenance of the BBB is ensured by correct functioning of the Wnt/β-catenin pathway [147]. Interestingly, compound 11 can induce Wnt/β-catenin pathway activation and reduce the incidence of hemorrhagic transformation associated with delayed recombinant tissue plasminogen activator (rtPA) administration [119]. Specifically, compound 11 treatment was shown to limit BBB breakdown via the promotion of tight junction formation and repression of endothelial basal permeability, independently of rtPA proteolytic activity. The effects of 11 on tight junctions was apparently due to is ability to stabilize β-catenin in the cytosol and stimulate its subsequent translocation to the nucleus. As a consequence, compound 11 treatment decreased brain edema, reduced IgG extravasation, and diminished the incidence of perivascular petechial bleeding 24 h after middle cerebral artery occlusion [119].
A newer area of compound 11 investigation is focused on aging. Liver aging is associated with age-related histopathological and functional changes that significantly enhance the risk of numerous diseases or disorders developing in elderly populations. Studies have demonstrated that 11 can reduce oxidative stress, improve lipid metabolism, enhance autophagy, and significantly reduce liver aging via modulation of the GSK-3β and mTOR pathways [121].
Compounds 12–15 are brominated indirubin derivatives, and 12 has been reported to be a potent inhibitor of GSK-3α/β and PhK [40,44]. Likewise, compound 13 exhibited inhibitory activity toward c-Src, JAK1, JAK2, and TYK2 [60]. In contrast, the 7-bromoindirubin-3′-oxime (14) was found to be a selective inhibitor of Aurora C [57]. 6-Bromoindirubin-3′-[O-(2-piperazine-1-ylethyl)] oxime has also been reported to inhibit proinflammatory pathways, including GSK-3α/β [59]. Finally, 5′-carboxylate derivative 15 was reported to inhibit DYRK1a and DYRK2 with enhanced selectivity [58].
6. Miscellaneous Oxime Group-Containing Kinase Inhibitors
The structures of oxime kinase inhibitors with non-indirubin scaffolds are shown in Table 2. These inhibitors were designed to inhibit various kinases, including vascular endothelial growth factor receptor 2 (VEGFR-1/2/3), B-Raf, ErbB1/2/3, PI3K isoforms α, β, γ, σ, and γ. Radicicol is a naturally occurring macrocyclic antifungal agent. Interestingly, oximation of radicicol increases its inhibitory activity towards Src [34], a tyrosine kinase that can regulate a number of signaling pathways impacting tumor cell behavior, including proliferation, survival, migration, invasion, and angiogenesis [148]. Similarly, the radicicol oxime derivative 17 had even higher anticancer activity than radicicol [33]. Raf isoforms are activated by phosphorylation via downstream regulation from the MAPK pathway. For example, B-Raf kinase plays a significant role in healthy cell growth by regulating B-Raf activity, and B-Raf mutations can lead to the development of cancer and other diseases [149]. Indeed, the oxime 18 is a highly selective, potent, and orally bioavailable B-Raf inhibitor with anticancer activity [62,150]. The major ketone metabolite of compound 18 is inactive [151], strongly suggesting that that the oxime group is responsible for kinase inhibitory activity. Likewise, oxime 19 was identified by Takle et al. [63] as another potent inhibitor of B-Raf.
To evaluate the role of the oxime group in binding, we conducted additional molecular docking of compounds 18 and 19 towards B-Raf (PDB: 1UWH). Our modeling experiments showed that the best docking pose of 18 with B-Raf structure forms a strong H-bond to Cys531 with participation of the pyridine nitrogen atom (Figure 4). In addition, a weaker H-bond is formed between the OH group of the 2-hydroxyethyl moiety and Phe594, while the oxime group has non-valent attractive interactions with Leu504, Ile526, and Thr528. The partial docking score for the =N-OH moiety of compound 18 is −8.04 kcal/mol. We found that the inactive ketone metabolite of 18 has a similar docking pose, with the pyridine nitrogen atom H-bonded to Cys531, while the dihydroindene moiety is rotated about the exocyclic C-C bond. In this conformation, the ketone oxygen atom has much weaker Van der Waals interactions with Lys482, Ile526, and Thr528 (partial docking score is −2.52 kcal/mol). Compound 19 has a bulky imidazole ring, and is bound to B-Raf, with the dimethylamino tail directed outwards from the kinase cavity (Figure 4). However, strong H-bonding is present between the oxime nitrogen atom and Cys531, and the oxime OH group forms an H-bond with Gln529.
Figure 4.

Docking pose of compound 19 (Z-isomer) in B-Raf (PDB: 1UWH). Amino acid residues within 3 Å from the pose are visible. H-bonds are shown in dashed blue lines. H-bond lengths with Gln529 and Cys531 are equal to 1.73 and 1.86 Å. They are formed with participation of oxime OH group and nitrogen atom, respectively.
Vasculature development is believed to be dependent on VEGF and its receptor tyrosine kinases, mainly VEGFR-2 and the angiopoietins (Ang-1 and Ang-2) and their receptor tyrosine kinase (primarily TIE-2). Thus, optimal antiangiogenic kinase therapy may require concurrently blocking both TIE-2 and VEGFR-2 signaling to inhibit tumor growth and metastasis. Compound 20 was reported to be a potent VEGFR-2 tyrosine kinase inhibitor [64] and, according to our results of docking into VEGFR-2 (PDB: 1YWN), is H-bonded to the NH group of Cys917 via its carbonyl moiety. The substituted oxime group =N-O- does not form H-bonds with the enzyme, although it has attractive Van der Waals interactions, mainly with Cys1043 and Asp1044. The partial docking score for the oxygen and nitrogen atoms of the oxime group is −9.11 kcal/mol. Compound 20 inhibited VEGF-dependent proliferation of human vascular endothelial cells and markedly regressed tumors in an A549 lung cancer xenograft model [64]. Compound 22 was also identified as a potent and selective inhibitor of VEGFR-2. This oxime also inhabited the closely related tyrosine kinases, Ret and Kit, but had no significant activity against VEGFR-1 or VEGFR-3 [66]. Notably, treatment of nude mice bearing human A431, HCT116, and A375 tumors with compound 22 resulted in up to 90% tumor growth inhibition [66].
Fused dihydroindazolopyrrolocarbazole oximes have been identified as low nanomolar dual TIE-2 and VEGFR-2 receptor tyrosine kinase inhibitors, with the most potent being compound 27. This compound inhibited VEGF-induced human umbilical vein endothelial cell (HUVEC) capillary-tube formation and was orally active in an A375 human tumor xenograft melanoma model with no observed toxicity [70].
Checkpoint kinase 1 (Chk1) and epidermal growth factor receptor (EGFR) are therapeutic targets for treatment of acute and chronic leukemias [152] and high-grade serous ovarian cancer [153,154]. Thus, it is significant that compounds 23, 25, and 27 have been reported as Chk1 and EGFR tyrosine kinase inhibitors [67,69].
Signaling pathways regulated by PI3Ks have been shown to play a role in cancer development and progression. Thus, therapeutic targeting of PI3K has been considered as a possible strategy for treating several types of cancer, including gastrointestinal cancer [155]. For example, compound 24 inhibited PI3Kγ and IL-6 release by concanavalin A-simulated mouse lymph node cells [68]. Similarly, several chromeno [4,3-c]pyrazol-4(2H)-one oxime derivatives have been shown to target PI3Ks, including PI3Kα, which is inhibited by compound 26. This compound also exhibited the most potent antiproliferative activity against human colorectal carcinoma HCT-116 cells [39].
CK2 is a ubiquitously expressed and highly conserved serine/threonine or tyrosine kinase that regulates diverse signaling pathways responsible for cell proliferation and apoptosis via interactions with over 500 known substrates. CK2 also plays an extrinsic role in cancer stroma or in the tumor microenvironment [156]. Thus, it is significant that compound 29 can inhibit CK2 kinase with moderate potency [71].
JNKs play important roles in many pathological processes, including autoimmune inflammatory disorders such as rheumatoid arthritis [157]. A number of JNK inhibitors with anti-inflammatory properties have been developed [158], yet few have been developed for the treatment of rheumatoid arthritis. Recently, we reported that 11H-indeno[1,2-b]quinoxalin-11-one oxime (compound 30), its sodium salt IQ-1S, and tryptanthrin-6-oxime (compound 31) were JNK inhibitors [41,116]. We found that the side chain oxime substituent was critical for JNK binding and biological activity of these compounds [38,41].
Molecular modeling studies suggested that H-bonding interactions with participation of the oxime group play an important role in the JNK inhibitory activity of compounds 30 and 31. In support of this conclusion, the inactive ketone of 30 (IQ-18) formed one weak H-bond with Gln37 of JNK1, whereas the oxime group of 30 formed two stronger H-bonds with Lys55 and Glu73 (Figure 5). Similarly, the high JNK inhibitory activity of compound 30 could be modulated by H-bonding interactions with Asn152, Gln155, or Met149 in the JNK3 binding site [41]. Compound 30 inhibited matrix metalloproteinase 1 and 3 (MMP1/3) gene expression induced by IL-1β in human FLS, and significantly attenuated development of CIA [122]. Treatment with 30 either before or after induction of CIA resulted in decreased clinical scores, and joint sections from compound 30-treated CIA mice exhibited only mild signs of inflammation and minimal cartilage loss compared with those from control mice. Collagen II-specific antibody responses were also reduced. Compound 30 treatment also suppressed proinflammatory cytokine and chemokine levels in joints and lymph node cells [122].
Figure 5.

Modeling H-bond interactions of oxime 30 (A) and its inactive ketone derivative IQ-18 (B) in the JNK1 binding site (PDB code 1UKI). Residues within 3 Å from the pose are visible. H-bonds are shown as dashed blue lines. Compound 30 forms H-bonds with Lys55 and Glu73. These H-bonds have lengths of 2.08 and 2.58 Å and are formed with the participation of oxime oxygen and hydrogen atoms, respectively. In contrast, IQ-18 forms one very weak H-bond with Gln37 (calculated length of 2.76 Å).
The docking pose of compound 31 was also characterized by strong H-bonding between the oxygen atom of the amide group and the Met111 of JNK1. This compound was H-bonded with JNK2 through its oxime group with Gly171. Finally, 31 was anchored in the JNK3 cavity via H-bonding of the oxime group with Asp207 [38]. Compound 31 demonstrated high binding activity toward all three JNK isoforms (JNK 1-3) [38], inhibited MMP-3 gene expression in IL-1β-stimulated human FLS, and inhibited IL-1β-induced secretion of MMP-1/3 by FLS and synovial SW982 cells and IL-6 by FLS, SW982 cells, HUVECs, and monocytic THP-1 cells [116]. Evaluation of the therapeutic potential of compound 31 in vivo in murine arthritis models showed that it attenuated the development of CIA and collagen-antibody-induced arthritis (CAIA). Collagen II-specific antibody levels were reduced in compound 31-treated CIA mice. This compound also suppressed the production of proinflammatory cytokines IL-17A, granulocyte-macrophage colony-stimulating factor (GM-CSF), and receptor activator of nuclear factor-κB ligand (RANKL) by lymph node cells from CIA mice [116].
JNK-mediated signaling pathways also play an essential role in cerebral and myocardial ischemia/reperfusion injury [159], and the neuroprotective activity of oxime 30 has been demonstrated in models of focal cerebral ischemia in mice [160] and rats [123], as well as in a model of total cerebral ischemia in rats [161]. Compound 30 inhibited JNK activity in the hippocampus and protected against stroke injury, reduced the infarct size, and limited the neurological deficit of rats after focal ischemia/reperfusion. After global ischemia/reperfusion, 30 decreased the number of animals with severe neurological deficit, increased density of the pyramidal neurons in the hippocampal CA1 area, improved the cerebral microcirculation, and attenuated the endothelial dysfunction. In addition, compound 30 treatment resulted in decreased systolic blood pressure, mean arterial blood pressure, and total peripheral resistance in spontaneously hypertensive rats [114]. Overall, the antihypertensive effects of compound 30 may be due to a combination of the inhibition of myocardial and aorta remodeling, attenuation of blood viscosity due to hematocrit decrease, vasodilatory effects, and decreased endothelin-1 production by the endothelial cells.
7. Oximes with Non-kinase Targets
While most of the identified oxime targets have been various kinases, there are some oximes that also have non-kinase targets of action. These targets include 5-lipoxygenase (5-LO), proteases, phosphodiesterase, chemokine receptors, growth factor receptors, and various channels (Table 7). For example, several indirubin oximes, such as compounds 1 and 11, have been reported to inhibit 5-LO [162], which is required for leukotriene synthesis. Replacement of the 3′-oxime in 1 by a keto group, 3′-methoxime or acetoxime resulted in loss of 5-LO inhibitory activity, indicating that a free oxime moiety in the 3′-position and a hydrogen in position N1 are required for effective inhibitory activity [162]. Additionally, newer derivatives of oleanolic acid oxime, and particularly their conjugates with acetylsalicylic acid, have been shown to downregulate the expression of cyclooxygenase 2 (COX-2) in human hepatoma HepG2 cells by modulating NF-κB signaling [163]. A reduction in COX-2 leads to reduced prostaglandin synthesis, which also inhibits inflammation in a similar fashion to other nonsteroidal anti-inflammatory drugs (NSAIDs).
Table 7.
Chemical structures of oximes with non-kinase targets and mechanisms of action.
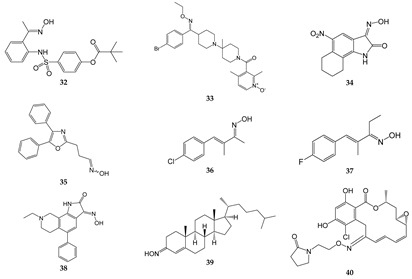
| Compound | Molecular Target/Mechanism | Ref. |
|---|---|---|
| 32 | Dual inhibitor of HNE and Pr3 | [15] |
| 33 | CCR5 antagonist | [169,170] |
| 34 | GluR6 antagonist, amelioration of inflammatory hyperalgesia | [171,172] |
| 35 | TRPA1 and TRPV1 antagonist | [14] |
| 36 | TRPA1 antagonist | [173,174] |
| 37 | TRPA1 antagonist | [173,174] |
| 38 | ASIC blocker, attenuation of pathophysiological nociceptive behaviors in CFA-inflamed and CCI rats | [175] |
| 39 | Binds directly to two components of the mitochondrial permeability pore, the VDAC, and translocator protein; inhibits MPTP opening | [176] |
| 40 | Binds to Hsp90 and provides a significant decrease in HIF-1α expression | [177] |
16α,17β-Epoxypregnenolone-20-oxime was reported to inhibit LPS-induced JNK phosphorylation, iNOS expression, and NO production in BV-2 microglial cells and RAW264.7 macrophages [164,165]. Likewise, the introduction of an oxime at position 12 of dehydroabietic acid, an aromatic abietane-type diterpenoid, increased its anti-proliferative and anti-inflammatory activities in pancreatic cancer Aspc-1 cells [166]. Moreover, a kinase profiling study showed that dehydroabietic oxime had modest inhibitory activity for p90 ribosomal S6 kinase 2 (RSK2) [166], a kinase that has been implicated in cellular invasion and metastasis [166,167,168]. In addition, Chen et al. [167,168] found that oxime derivatives of furo[2,3-b]quinolines were more potent than their respective ketone precursors for their ability to inhibit mast cell and neutrophil degranulation, as well as neutrophil ROS production. The precise targets of these oximes have not been identified.
Pillai et al. [178] synthesized a series of tetra-substituted thiophenes and reported that they had anti-inflammatory activity in a carrageenin-induced rat paw edema model [178]. They also found that compounds with aliphatic oxime esters attached with a ketone bridge to the thiophene had higher anti-inflammatory activity than the aromatic oximes. These oxime analogs were also weak to moderate free radical scavengers; however, a direct correlation between anti-inflammatory activity and free radical scavenging activity was not seen [178]. Nevertheless, the authors suggested that these oximes could have potential as anti-inflammatory agents. Likewise, 2-phenylindole-3-carboxaldehyde oxime was reported to inhibit NO production in RAW 264.7 macrophage cells, as well as NF-κB inhibition in human embryonic kidney cells 293 [179]. In addition, oxime derivatives of β-acetoxy-17β-hydroxy-androst-5-ene, such as 3β-acetoxy-androst-5-ene-17 oxime, were shown to have anti-inflammatory activity in a mouse model of ear inflammation [18]. Other steroidal oximes, such as 22-oxocholestane oximes, that were also evaluated as anti-inflammatory agents in the acute ear inflammation model exhibited anti-inflammatory activity [20]. The most active oximes downregulated NF-κB and inhibited expression of pro-inflammatory genes TNF, COX-2, and IL-6, and reduced ear-induced inflammation and edema. Notably, the activity of these oximes was comparable to the potent anti-inflammatory agent dexamethasone [20]. Similarly, (Z)-(2-carbethoxyamino-4-methyl-1,3-thiazol-5-yl)-(4-methylphenyl)methanone oxime exhibited anti-inflammatory activity in acute and chronic inflammatory models of rat paw edema [180]. Likewise, the adamantane-containing molecules O-(α-acetoxy-benzeneacetyl)-2-tricyclo[3.3.1.13,7]decan-2-one oxime and O-(α-propoxy-benzeneacetyl)-2-tricyclo[3.3.1.13,7]decan-2-one oxime) had anti-inflammatory activity comparable to that of diclofenac in a mouse paw edema model [19]. Finally, oral dosing with (E)-1-(4-((1R,2S,3R)-1,2,3,4-tetrahydroxybutyl)-1H-imidazol-2-yl)ethanone oxime resulted in a decrease in circulating lymphocytes, decreased hind limb swelling, and reduced circulating anti-type II collagen antibodies in a CIA mouse model of rheumatoid arthritis [181].
Human neutrophil elastase (HNE) and proteinase 3 (Pr3) also represent potential oxime targets for the development of anti-inflammatory therapeutics to treat adult respiratory distress syndrome, autoimmune disorders, and hypersensitivity reactions [182,183]. For example, 2-aminobenzaldehyde oxime analogs such as compound 32 were found to have dual inhibitory effects on HNE and Pr3 [15]. This compound was slightly more potent than the commercial HNE inhibitor Sivelestat, which is used in Japan and Korea for the treatment of acute lung injury associated with systemic inflammation [184]. In mouse models of inflammation, treatment with 32 reduced paw edema and acute lung injury [15].
Oxime-based phosphodiesterase (PDE) 4 inhibitors are also being evaluated as potential anti-inflammatory agents, as they have the ability to inhibit the production of inflammatory mediators and cytokines [185]. Several oxime derivatives of rolipram, an inhibitor of PDE4, have been reported to inhibit TNF production in LPS-stimulated RAW264.7 macrophages with higher potency than rolipram [186]. Interestingly, the E/Z-geometry of oxime was important for activity of these compounds, with cis-isomers being more active than the corresponding trans-isomers [186].
Several oximes target receptors or ion channels. Among the oxime receptor targets are chemokine receptors, kainate receptors, and growth factor receptors. For example, compound 33 has been reported to be an orally bioavailable, small molecule antagonist of CCR5. Indeed, this compound exhibited potent antiviral activity against HIV-1 infection in vitro and in vivo [169,170]. Another oxime derivative, compound 34 was reported to be a low-affinity inhibitor of the ionotropic kainite receptor GluR6, and treatment with 34 was reported to attenuate inflammation-induced thermal hyperalgesia [171,172]. Compound 34 has also been proposed to inhibit neurotoxic effects of kainate receptor agonists in murine cultured cortical neurons [187]. Finally, El-Sherief et al. [188] synthesized a series of oximes with a 1,2,4-triazole scaffold. Some of these oxime hybrids had higher anti-proliferative activity than their corresponding ketones [188], and were determined to be epidermal growth factor receptor (EGFR) inhibitors, as well as moderate inhibitors of B-Raf and tubulin.
Among the oxime channel or transporter targets are transient receptor potential (TRP) channels, acid-sensing channels, and mitochondrial transition pores. For example, compound 35 has been reported to be a potent transient receptor potential ankyrin 1 and vanilloid 1 (TRPA1 and V1) channel antagonist [14]. Similarly, compounds 36 and 37 were found to be selective TRPA1 channel blockers [173,174]. These compounds represent promising new candidates for drug development focusing on neuropathic pain, migraine, and arthritis. Compound 38 is an acid-sensing ion channel (ASIC) blocker with specificity for ASIC1a and ASIC3. This oxime compound reduced pathophysiological nociceptive behaviors in complete Freund’s adjuvant-inflamed and reversed mechanical hypersensitivity in a rat chronic constriction injury model [175]. Interestingly, 38 had no adverse effects on motor function, which are major problems with morphine-based analgesics.
Compound 39 (cholest-4-en-3-one, oxime) is a neuroprotective and neuroregenerative compound that has been reported to rescue motor neurons from axotomy-induced cell death and promote nerve regeneration following sciatic nerve crush in vivo [176]. This compound is thought to bind to two components of the mitochondrial permeability transition pore, the voltage-dependent anion channel (VDAC) and translocator protein, and inhibit pore opening and reduce neuronal apoptosis [176]. The authors suggested that 39 may have therapeutic potential for amyotrophic lateral sclerosis (ALS).
Although most of the known oximes exhibit anti-inflammatory activity, oxime IMR-23 has been reported to exhibit pro-inflammatory activity in J774A.1 cells and in a mouse model [189], and has been suggested to have potential in the development of adjuvants. Specifically, treatment with IMR-23 induced the release of pro-inflammatory cytokines IL-1β, IL-6, and TNF, induced the production of antibodies, and led to the generation of antigen-specific T cells [189].
8. Metabolism of Oximes and NO Production
Metabolism of oximes catalyzed by cytochrome P450 can lead to release of NO [190,191,192,193]. For example, oxidative breaks of the oxime C=N bond and the formation of a C=O bond lead to the transfer of one oxygen atom from O2 to the compound and simultaneous release of NO [194]. For various oximes, it has been reported that this reaction proceeds in liver microsomes with the participation of cytochromes P450, NADPH, and O2 [190,191,195,196]. The participation of cytochromes P450 is confirmed by the fact that inducers and inhibitors of microsomal oxidation can activate or inhibit oxidative metabolism of oximes, respectively [190,195]. For acetoxime, it was shown that ROS play a key role in oxidation of the compound to NO by liver microsomes [191]. Jousserandot et al. [196] described a mechanism for such oxidative cleavages of oximes with formation of nitrogen oxides by cytochrome P450, with the involvement of O2•− and its Fe-complexes [(FeIII-O2−), or (FeII-O2)] as the main reactive species. Amidoximes oxidized together with NO also release NO-related products, such as NO2− and NO3− [190,191]. For example, the rate of arylamidoxime microsomal oxidation of para-hexyloxy-benzamidoxime rapidly decreases with time, which is related to the inactivation of cytochromes by the formation of P450-Fe(II)-NO and P420-Fe(II)-NO complexes [190].
Microsomal oxidation of amidoximes to the corresponding nitriles, and of ketoximes to the corresponding nitroalkanes, are not inhibited by superoxide dismutase (SOD), and are performed by a cytochrome P450 active species, presumably the high-valent P450-Fe-oxo complex. In contrast, microsomal oxidation of amidoximes to the corresponding ureas and amides was also found to be mainly performed by O2•−, as shown by the inhibitory effect of SOD and the ability of the xanthine-xanthine oxidase system to give similar oxidation products [196]. Further steps in the metabolism of keto-derivatives and their excretion from the organism will depend on the specific structure of the aryl ring. For example, Figure 6 shows the pharmacokinetic curves of compound 30 and its keto-derivative.
Figure 6.

Pharmacokinetic profile of compound 30 (A) and its keto-derivative IQ-18 (B) in blood plasma of SD rats after a single intragastric administration of compound 30 at a dose of 50 mg/kg. Mass spectrometric analyses were performed using a Shimadzu LC-20 (Kyoto, Japan), coupled with an ABSCIEX API 3200 triple quadrupole mass spectrometer (USA).
The vasodilator effects of oximes on isolated vessels with denuded endothelium and endothelium [161,190,197,198] substantiated the existence of other (non-microsomal) pathways of oxime biotransformation and the production of NO. Treatment with formamidoxime, acetaldoxime, acetone oxime, acetohydroxamic acid, or formaldoxime resulted in a relaxation of rat endothelium-denuded rings [198]. Neither inhibitors of NO synthases nor inhibitors of cytochrome P450 reduced the vasodilator effect of oxime derivatives. Furthermore, inhibition of the vasodilatory effects of these oximes under the influence of 7-ethoxyresorufin suggests the possibility of the participation of NAD(P)H-dependent reductases in the NO-donating properties of oximes [190,197,198]. For a broader review on the biological pathways of amidoximes, see [193]. NO is involved in many physiological processes, such as neurotransmission, blood pressure regulation, and immune modulation. However, in some diseases, such as hypertension and diabetes, the ability of endothelial NO synthase (eNOS) to generate NO is impaired [6,199]. For this reason, compounds capable of being oxidized to release NO in pathways other than NOS are of high interest. Indeed, NO donors have been reported to exhibit anti-inflammatory and anticancer activities [200,201]. For example, NO-donating NSAIDs, which are safer than their NSAID counterparts, inhibit the growth of colon cancer cells with greater potency than traditional NSAIDs [202]. Due to their NO-donating capacities, some oxime derivatives have also been shown to offer therapeutic potential for the treatment of erectile dysfunction, as well as cardiovascular diseases [203,204]. Likewise, a number of oxime derivatives have been shown to exhibit antithrombogenic, hypotensive, and cardiotonic activity [198,205,206]. For example, amidoximes and oximes have been shown to inhibit platelet aggregation, decrease thrombus formation, induce vasodilation, and lower intraocular pressure [192,197,199,207,208,209]. NO-donating oxime hybrids also have been reported to have gastroprotective activity versus their corresponding ketone precursors, which also may be attributed to the release of NO [210].
9. Conclusions and Perspectives
Oxime groups have been successfully introduced into a large number of therapeutic leads for the development of kinase inhibitors with anticancer and anti-inflammatory activities. The kinase selectivity of oximes does not appear to be due to the oxime group. Rather, selectivity seems to be due to the scaffold of the molecule, since some oximes are highly selective (e.g., JNK inhibitors 30 and 31 [38,41]), while others, such as indirubin, have a wide spectrum of kinase targets. In this regard, compounds 30 and 31 are of particular interest as candidates for the development of new anti-inflammatory drugs, since they are highly selective for JNKs.
While the presence of a terminal oxime group is necessary for the activity of these compounds, the oxime group also offers a significant advantage in drug design versus carbonyl groups because of the presence of two H-bond acceptors (N and O atoms) and one donor (OH group). Additionally, the metabolism of oximes can lead to the release of NO, which may also be therapeutically beneficial [56]. The important role of the oxime group is supported by docking results revealing direct participation of oxime moiety in interactions with kinase binding sites. On the other hand, there has been some concern regarding the development of new drugs based on oxime derivatives. For example, a disadvantage of compound 11 and other indirubin derivatives is the high affinity of indirubin for ATP-binding pockets and the high degree of similarity between ATP cavities within the serine/threonine and tyrosine kinases, leading to multi-targeting. However, single molecules targeting two (or three) kinases is considered less problematic for current pharmaceutical development, and 11 is considered to have significant potential as a therapeutic for treatment of inflammatory and degenerative diseases. One major unsolved issue related to oxime derivatives is their unfavorable physicochemical properties, including poor solubility and membrane permeability, which results in low plasma bioavailability and a short half-life that limits their suitability as drugs [211,212]. However, compounds 1 and 30 can apparently cross the BBB easily, suggesting that these oximes might be useful for treating brain disorders. New approaches are being developed to improve oxime PK/PD parameters [213,214,215]. For example, complexing oxime molecules into a dendrimer carrier has been proposed as a strategy to extend their plasma duration through a mechanism of release kinetics, so that loaded drug molecules are released over a longer half-life. Choi et al. [215] demonstrated that drug-dendrimer complexes form in a specific manner, wherein each oxime molecule interacts through electrostatic attraction with the primary amine terminated at the peripheral branch of the dendrimer [215]. The importance of the oxime group in kinase binding suggests that additional introduction of this group in the structures of known kinase inhibitors could improve their potency. In addition, oximes with non-kinase targets could be screening toward a broad kinase panel for identification of novel kinase inhibitors.
It is important to note that most of the oximes reviewed here were discovered during compound optimization and not high-throughput screening (HTS). In addition, most of these compounds were characterized in cell-free enzymatic systems and supported in independent test systems. Although compound 30 was originally discovered using HTS in a cell-based assay, the target of this compound was verified using multiple enzymatic assays, cell-based assays, structure–activity relationship (SAR) analysis, and animal experiments. Based on this compound and the absolute requirement for the oxime group in JNK inhibitory activity, we also developed compound 31, which was also validated in cell- and enzyme-based assays and in animal experiments. Thus, it is unlikely that these compounds or the oximes reviewed here are pan assay interference compounds (PAINS) [216,217]. Nevertheless, this is an important consideration in small molecule screening and will need to be addressed as oximes are developed for new therapeutics.
Abbreviations
| Kinase Abbreviations | |
| AMPK | AMP-activated protein kinase |
| CDK1/2/5/6/9 | cyclin-dependent kinases |
| Chk1, | checkpoint kinase 1 |
| CK2 | casein kinase 2 |
| DRAK2 | death-associated protein-related apoptotic kinase 2 |
| DYRK | dual-specificity tyrosine-phosphorylated and regulated kinase |
| EGFR | epidermal growth factor receptor tyrosine kinase |
| ERK | extracellular signal-regulated kinase |
| FLT3 | FMS-related receptor tyrosine kinase 3 |
| GSK-3α/β | glycogen synthase kinase 3 |
| IGF1R | receptor of insulin-like growth factor type 1 |
| IRAK4 | interleukin-1 receptor-associated kinase 4 |
| JAK1/2/3 | Janus kinases 1/2/3, tyrosine kinases |
| JNK | c-Jun N-terminal kinase |
| Lck | lymphocyte-specific protein tyrosine kinase |
| Lyn | non-receptor tyrosine-protein kinase |
| PhK | serine/threonine-specific phosphorylase kinase |
| PI3K | phosphatidylinositol 3-kinase |
| PKR | RNA-dependent protein kinase R |
| RSK2 | ribosomal S6 kinase 2 |
| SGK | serine/threonine-protein kinase Sgk1 (serum and glucocorticoid-regulated kinase 1) |
| VEGFR1/2 | vascular endothelial growth factor receptor tyrosine kinase |
| Other Abbreviations | |
| ALP | alkaline phosphatase |
| AP-1 | activator protein 1 |
| ASIC | acid-sensing ion channel |
| ATF-2 | activating transcription factor 2 |
| CAIA | collagen-antibody-induced arthritis |
| CCI | chronic constriction injury |
| CCL | chemokine ligand |
| CCR5 | chemokine receptor 5 |
| CFA | complete Freund’s adjuvant |
| CIA | collagen-induced arthritis |
| COX-2 | cyclooxygenase 2 |
| CysLT | cysteinyl leukotriene |
| DTH | delayed-type hypersensitivity |
| eNOS | endothelial NO synthase |
| FLS | fibroblast-like synoviocytes |
| GluR6 | glutamate receptor 6 |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| HIV | human immunodeficiency virus |
| HNE | human neutrophil elastase |
| HO-1 | heme oxygenase 1 |
| Hsp90 | heat shock protein 90 |
| HUVECs | human umbilical vein endothelial cells |
| IFN | interferon |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| IP-10 | interferon γ-induced protein 10 |
| LO | lipoxygenase |
| LPS | lipopolysaccharide |
| LTB4 | leukotriene B4 |
| MAPK | mitogen-activated protein kinase |
| MCA | middle cerebral artery |
| MCP | monocyte chemoattractant protein |
| MMECs | mouse mammary epithelial cells |
| MMP | matrix metalloproteinase |
| MPO | myeloperoxidase |
| NF-κB | nuclear factor κB |
| NO | nitric oxide |
| NSAIDs | nonsteroidal anti-inflammatory drugs |
| OCN | osteocalcin |
| PBMCs | peripheral blood mononuclear cells |
| PDE | phosphodiesterase |
| PGE2 | prostaglandin E2 |
| Pr3 | proteinase 3 |
| PTGS-2 | prostaglandin endoperoxide synthase 2 |
| RANKL | receptor activator of NF-κB ligand |
| RANTES | regulated on activation, normal T cell expressed and secreted |
| ROS | reactive oxygen species |
| Runx2 | runt-related transcription factor 2 |
| S.c. | subcutaneous |
| SOD | superoxide dismutase |
| STAT | signal transducer and activator of transcription |
| TGF-β | transforming growth factor β |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| TRPA1 | transient receptor potential ankyrin 1 |
| TRPV1 | transient receptor potential vanilloid 1 |
| VDAC, | voltage-dependent anion channel |
| VEGFA | vascular endothelial growth factor A |
| VSMCs | vascular smooth muscle cells |
| Chemical Names | |
| Compound 1 (E231) | indirubin-3′-oxime |
| Compound 2 | indirubin-3′-acetoxime |
| Compound 3 (E804) | indirubin-3′-oxime 2,3-dihydroxypropyl ether |
| Compound 4 (E738) | 5-methoxyindirubin-3′-oxime 1,2-dihydroxyethyl ether |
| Compound 5 | 5′,6′-difluoro-5-methoxy-indirubin-3′-oxime 2-hydroxyethyl ether |
| Compound 6 (LDD970) | 5-[(1-morpholino)carbonyl]indirubin-3′-oxime |
| Compound 7 (AGM130) | 5-nitro-5′-hydroxyindirubin-3′-oxime |
| Compound 8 | 5-(pentanamido)indirubin-3′-oxime |
| Compound 9 (LDD1937) | 5-(methoxycarbonyl)indirubin-3′-oxime 2-(piperazin-1-yl)ethyl ether dihydrochloride |
| Compound 10 | 5-iodoindirubin-3′-oxime |
| Compound 11 | 6-bromoindirubin-3′-oxime |
| Compound 12 | 6-bromoindirubin-3′-acetoxime |
| Compound 13 (MLS-2384) | 6-bromoindirubin-3′-oxime 2-(piperazin-1-yl)ethyl ether |
| Compound 14 | 7-bromoindirubin-3′-oxime |
| Compound 15 | 7-bromo-5′-carboxyindirubin-3′-oxime |
| Compound 16 | 5-fluoroindirubin-3′-oxime 2-(piperazin-1-yl)ethyl ether dihydrochloride |
| Compound 17 | radicicol 6-oxime |
| Compound 18 (GDC 0879) | 2,3-dihydro-5-[1-(2-hydroxyethyl)-3-(4-pyridinyl)-1H-pyrazol-4-yl]-1H-inden-1-one oxime |
| Compound 19 (SB 590885) | 5-[2-[4-[2-(dimethylamino)ethoxy]phenyl]-5-(4-pyridinyl)-1H-imidazol-4-yl]-2,3-dihydro-1H-inden-1-one oxime |
| Compound 20 (YM-359445) | (3Z)-3-[6-[(4-methylpiperazin-1-yl)methyl]quinolin-2(1H)-ylidene]-2-oxoindoline-6-carbaldehyde O-(1,3-thiazol-4-ylmethyl)oxime. |
| Compound 21 (JNJ-28871063) | 5E-4-amino-6-(4-benzyloxy-3-chlorophenylamino)pyrimidine-5-carboxaldehyde N-(2-morpholin-4-ylethyl) oxime |
| Compound 22 (JNJ-38158471) | (E)-1-(4-((6-amino-5-((methoxyimino)methyl)pyrimidin-4-yl)oxy)-2-chlorophenyl)-3-ethylurea |
| Compound 23 | 1H-indene-1,2,3-trione-2-(phenylhydrazone) 1-oxime |
| Compound 24 | (E)-3-(4-fluorophenyl)-1-phenyl-1H-pyrazole-4-carbaldehyde O-(2-fluorobenzyl) oxime |
| Compound 25 | 2,2′-((9-(hydroxyimino)-9H-fluorene-2,7-diyl)bis(oxy))diacetic acid |
| Compound 26 | ((E)-2-(2-(3,4-dichlorophenyl)-2-(hydroxyimino)ethyl)chromeno[4,3-c]pyrazol-4(2H)-one) |
| Compound 27 | (E)-1-(13-isobutyl-4-methyl-6-methylene-2,4,6,7,8,13-hexahydro-1H-indazolo[5,4-a]pyrrolo[3,4-c]carbazol-10-yl)ethan-1-one O-methyl oxime |
| Compound 28 | 2,7-bis(allyloxy)-9H-fluoren-9-one oxime |
| Compound 29 (4-AN) | phenylcyanomethylenequinone oxime-4-(hydroxyimino) cyclohexa-2,5-dien-1-ylidene](phenyl)ethanenitrile |
| Compound 30 (IQ-1) | 11H-indeno[1,2-b]quinoxalin-11-one oxime |
| Compound 31 | tryptanthrin-6-oxime |
| Compound 32 | (E)-4-(N-(2-(1-(hydroxyimino)ethyl)phenyl)sulfamoyl)phenyl pivalate |
| Compound 34 (NS 102) | 6,7,8,9-tetrahydro-5-nitro-1H-benz[g]indole-2,3-dione 3-oxime |
| Compound 35 (SZV-1287) | 3-(4,5-diphenyl-1,3-oxazol-2-yl)propanal oxime |
| Compound 36 (AP 18) | 4-(4-chlorophenyl)-3-methyl-3-buten-2-one oxime |
| Compound 37 (A 967079) | (1E,3E)-1-(4-fluorophenyl)-2-methyl-1-pentene-3-one oxime |
| Compound 38 (NS 383) | 8-ethyl-6,7,8,9-tetrahydro-5-phenyl-1H-pyrrolo[3,2-h]isoquinoline-2,3-dione-3-oxime |
| fMLF | formyl-l-methionyl-l-leucyl-l-phenylalanine |
| MPTP | 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine |
| TPA | 12-O-tetradecanoylphorbol-13-acetate |
Author Contributions
I.A.S., M.B.P. and T.M.P. carried out the literature review and drafted the manuscript. I.A.S. and M.B.P. proposed the concept and edited the manuscript. A.I.K. conducted molecular docking, prepared the section on chemical characterization of oxime groups and provided a revision of the chemical structures and chemical names. M.B.P. evaluated the pharmacokinetic profile of compound 30. M.T.Q. and A.I.K. extensively revised the manuscript, provided critical revision and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by National Institutes of Health NIGMS IDeA P01 Program Grants GM115371 and GM103474; the Tomsk Polytechnic University Development Program; USDA National Institute of Food and Agriculture Hatch project 1009546; and the Montana State University Agricultural Experiment Station. The docking results were obtained with the support of the Russian Science Foundation grant No. 17-15-01111.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support our specific findings in this review are available from the authors upon reasonable request.
Conflicts of Interest
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Musilek K., Dolezal M., Gunn-Moore F., Kuca K. Design, evaluation and structure-activity relationship studies of the AChE reactivators against organophosphorus pesticides. Med. Res. Rev. 2011;31:548–575. doi: 10.1002/med.20192. [DOI] [PubMed] [Google Scholar]
- 2.Canario C., Silvestre S., Falcao A., Alves G. Steroidal oximes: Useful compounds with antitumor activities. Curr. Med. Chem. 2018;25:660–686. doi: 10.2174/0929867324666171003115400. [DOI] [PubMed] [Google Scholar]
- 3.Franjesevic A.J., Sillart S.B., Beck J.M., Vyas S., Callam C.S., Hadad C.M. Resurrection and reactivation of acetylcholinesterase and butyrylcholinesterase. Chemistry. 2019;25:5337–5371. doi: 10.1002/chem.201805075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sorensen M., Neilson E.H.J., Moller B.L. Oximes: Unrecognized chameleons in general and specialized plant metabolism. Mol. Plant. 2018;11:95–117. doi: 10.1016/j.molp.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 5.Fuller A.T. Antibacterial action of some aromatic amines, amidines, amidoximes, guanidines and diguanides. Biochem. J. 1947;41:403–408. doi: 10.1042/bj0410403. [DOI] [PubMed] [Google Scholar]
- 6.Fylaktakidou K.C., Hadjipavlou-Litina D.J., Litinas K.E., Varella E.A., Nicolaides D.N. Recent developments in the chemistry and in the biological applications of amidoximes. Curr. Pharm. Des. 2008;14:1001–1047. doi: 10.2174/138161208784139675. [DOI] [PubMed] [Google Scholar]
- 7.Souza L.G.D., Almeida M.C.S., Lemos T.L.G., Ribeiro P.R.V., de Brito E.S., Silva V.L.M., Silva A.M.S., Braz R., Costa J.G.M., Rodrigues F.F.G., et al. Synthesis, antibacterial and cytotoxic activities of new biflorin-based hydrazones and oximes. Bioorg. Med. Chem. Lett. 2016;26:435–439. doi: 10.1016/j.bmcl.2015.11.095. [DOI] [PubMed] [Google Scholar]
- 8.Reddy D.S., Kongot M., Netalkar S.P., Kurjogi M.M., Kumar R., Avecilla F., Kumar A. Synthesis and evaluation of novel coumarin-oxime ethers as potential anti-tubercular agents: Their DNA cleavage ability and BSA interaction study. Eur. J. Med. Chem. 2018;150:864–875. doi: 10.1016/j.ejmech.2018.03.042. [DOI] [PubMed] [Google Scholar]
- 9.Hall J.E., Kerrigan J.E., Ramachandran K., Bender B.C., Stanko J.P., Jones S.K., Patrick D.A., Tidwell R.R. Anti-pneumocystis activities of aromatic diamidoxime prodrugs. Antimicrob. Agents Chemother. 1998;42:666–674. doi: 10.1128/AAC.42.3.666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clement B., Burenheide A., Rieckert W., Schwarz J. Diacetyldiamidoximeester of pentamidine, a prodrug for treatment of protozoal diseases: Synthesis, in vitro and in vivo biotransformation. ChemMedChem. 2006;1:1260–1267. doi: 10.1002/cmdc.200600079. [DOI] [PubMed] [Google Scholar]
- 11.Li Q., Zhang J.P., Chen L.Z., Wang J.Q., Zhou H.P., Tang W.J., Xue W., Liu X.H. New pentadienone oxime ester derivatives: Synthesis and anti-inflammatory activity. J. Enzym. Inhib. Med. Chem. 2017;33:130–138. doi: 10.1080/14756366.2017.1396455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu C., Tang X., Zhang W., Li G., Chen Y., Guo A., Hu C. 6-bromoindirubin-3′-oxime suppresses LPS-induced inflammation via inhibition of the TLR4/NF-κB and TLR4/MAPK signaling pathways. Inflammation. 2019;42:2192–2204. doi: 10.1007/s10753-019-01083-1. [DOI] [PubMed] [Google Scholar]
- 13.Kwon Y.J., Yoon C.H., Lee S.W., Park Y.B., Lee S.K., Park M.C. Inhibition of glycogen synthase kinase-3β suppresses inflammatory responses in rheumatoid arthritis fibroblast-like synoviocytes and collagen-induced arthritis. Jt. Bone Spine. 2014;81:240–246. doi: 10.1016/j.jbspin.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Payrits M., Saghy E., Matyus P., Czompa A., Ludmerczki R., Deme R., Sandor Z., Helyes Z., Szoke E. A novel 3-(4,5-diphenyl-1,3-oxazol-2-yl)propanal oxime compound is a potent transient receptor potential ankyrin 1 and vanilloid 1 (TRPA1 and V1) receptor antagonist. Neuroscience. 2016;324:151–162. doi: 10.1016/j.neuroscience.2016.02.049. [DOI] [PubMed] [Google Scholar]
- 15.Hwang T.L., Wang W.H., Wang T.Y., Yu H.P., Hsieh P.W. Synthesis and pharmacological characterization of 2-aminobenzaldehyde oxime analogs as dual inhibitors of neutrophil elastase and proteinase 3. Bioorg. Med. Chem. 2015;23:1123–1134. doi: 10.1016/j.bmc.2014.12.056. [DOI] [PubMed] [Google Scholar]
- 16.Komai T., Yagi R., Suzuki-Sunagawa H., Ishikawa Y., Kasuya A., Miyamoto S., Handa H., Nishigaki T. Inhibition of HIV-1 protease by oxim derivatives. Biochem. Biophys. Res. Commun. 1997;230:557–561. doi: 10.1006/bbrc.1996.5907. [DOI] [PubMed] [Google Scholar]
- 17.Heredia A., Davis C., Bamba D., Le N., Gwarzo M.Y., Sadowska M., Gallo R.C., Redfield R.R. Indirubin-3 ‘-monoxime, a derivative of a chinese antileukemia medicine, inhibits P-TEFb function and HIV-1 replication. AIDS. 2005;19:2087–2095. doi: 10.1097/01.aids.0000194805.74293.11. [DOI] [PubMed] [Google Scholar]
- 18.Chaubal R., Mujumdar A.M., Misar A., Deshpande V.H., Deshpande N.R. Structure-activity relationship study of androstene steroids with respect to local anti-inflammatory activity. Arzneimittelforschung. 2006;56:394–398. doi: 10.1055/s-0031-1296740. [DOI] [PubMed] [Google Scholar]
- 19.Antoniadou-Vyza E., Avramidis N., Kourounakis A., Hadjipetrou L. Anti-inflammatory properties of new adamantane derivatives. Design, synthesis, and biological evaluation. Arch. Pharm. 1998;331:72–78. doi: 10.1002/(SICI)1521-4184(199802)331:2<72::AID-ARDP72>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 20.Zeferino-Diaz R., Olivera-Castillo L., Davalos A., Grant G., Kantun-Moreno N., Rodriguez-Canul R., Bernes S., Sandoval-Ramirez J., Fernandez-Herrera M.A. 22-oxocholestane oximes as potential anti-inflammatory drug candidates. Eur. J. Med. Chem. 2019;168:78–86. doi: 10.1016/j.ejmech.2019.02.035. [DOI] [PubMed] [Google Scholar]
- 21.Shen S., Xu N., Klamer G., Ko K.H., Khoo M., Ma D., Moore J., O’Brien T.A., Dolnikov A. Small-molecule inhibitor of glycogen synthase kinase 3β 6-bromoindirubin-3-oxime inhibits hematopoietic regeneration in stem cell recipient mice. Stem. Cells Dev. 2015;24:724–736. doi: 10.1089/scd.2014.0230. [DOI] [PubMed] [Google Scholar]
- 22.Zhang X., Castanotto D., Nam S., Horne D., Stein C. 6bio enhances oligonucleotide activity in cells: A potential combinatorial anti-androgen receptor therapy in prostate cancer cells. Mol. Ther. 2017;25:79–91. doi: 10.1016/j.ymthe.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qu H.E., Huang R.Z., Yao G.Y., Li J.L., Ye M.Y., Wang H.S., Liu L. Synthesis and pharmacological evaluation of novel bisindole derivatives bearing oximes moiety: Identification of novel proapoptotic agents. Eur. J. Med. Chem. 2015;95:400–415. doi: 10.1016/j.ejmech.2015.03.058. [DOI] [PubMed] [Google Scholar]
- 24.Chiou C.T., Lee W.C., Liao J.H., Cheng J.J., Lin L.C., Chen C.Y., Song J.S., Wu M.H., Shia K.S., Li W.T. Synthesis and evaluation of 3-ylideneoxindole acetamides as potent anticancer agents. Eur. J. Med. Chem. 2015;98:1–12. doi: 10.1016/j.ejmech.2015.04.062. [DOI] [PubMed] [Google Scholar]
- 25.Blazevic T., Heiss E.H., Atanasov A.G., Breuss J.M., Dirsch V.M., Uhrin P. Indirubin and indirubin derivatives for counteracting proliferative diseases. Evid. Based Complement. Alternat. Med. 2015;2015:654098. doi: 10.1155/2015/654098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiong B., Chen S., Zhu P., Huang M., Gao W., Zhu R., Qian J., Peng Y., Zhang Y., Dai H., et al. Design, synthesis, and biological evaluation of novel thiazolyl substituted bis-pyrazole oxime derivatives with potent antitumor activities by selectively inducing apoptosis and ROS in cancer cells. Med. Chem. 2019;15:743–754. doi: 10.2174/1573406414666180827112724. [DOI] [PubMed] [Google Scholar]
- 27.Galmozzi E., Facchetti F., La Porta C.A. Cancer stem cells and therapeutic perspectives. Curr. Med. Chem. 2006;13:603–607. doi: 10.2174/092986706776055661. [DOI] [PubMed] [Google Scholar]
- 28.Avrahami L., Farfara D., Shaham-Kol M., Vassar R., Frenkel D., Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the alzheimer disease mouse model: In vivo and in vitro studies. J. Biol. Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sathiya Priya C., Vidhya R., Kalpana K., Anuradha C.V. Indirubin-3′-monoxime prevents aberrant activation of gsk-3beta/nf-kappab and alleviates high fat-high fructose induced abeta-aggregation, gliosis and apoptosis in mice brain. Int. Immunopharmacol. 2019;70:396–407. doi: 10.1016/j.intimp.2019.02.053. [DOI] [PubMed] [Google Scholar]
- 30.Yuskaitis C.J., Jope R.S. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell. Signal. 2009;21:264–273. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L., Li Z., Wang K.L., Liu Y.X., Li Y.Q., Wang Q.M. Synthesis and antiviral, insecticidal, and fungicidal activities of gossypol derivatives containing alkylimine, oxime or hydrazine moiety. Bioorg. Med. Chem. 2016;24:474–483. doi: 10.1016/j.bmc.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 32.Hong S., Shin Y., Jung M., Ha M.W., Park Y., Lee Y.J., Shin J., Oh K.B., Lee S.K., Park H.G. Efficient synthesis and biological activity of psammaplin a and its analogues as antitumor agents. Eur. J. Med. Chem. 2015;96:218–230. doi: 10.1016/j.ejmech.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 33.Soga S., Neckers L.M., Schulte T.W., Shiotsu Y., Akasaka K., Narumi H., Agatsuma T., Ikuina Y., Murakata C., Tamaoki T., et al. KF25706, a novel oxime derivative of radicicol, exhibits in vivo antitumor activity via selective depletion of Hsp90 binding signaling molecules. Cancer Res. 1999;59:2931–2938. [PubMed] [Google Scholar]
- 34.Ikuina Y., Amishiro N., Miyata M., Narumi H., Ogawa H., Akiyama T., Shiotsu Y., Akinaga S., Murakata C. Synthesis and antitumor activity of novel O-carbamoylmethyloxime derivatives of radicicol. J. Med. Chem. 2003;46:2534–2541. doi: 10.1021/jm030110r. [DOI] [PubMed] [Google Scholar]
- 35.Bednarczyk-Cwynar B., Zaprutko L. Recent advances in synthesis and biological activity of triterpenic acylated oximes. Phytochem. Rev. 2015;14:203–231. doi: 10.1007/s11101-014-9353-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vougogiannopoulou K., Skaltsounis A.L. From tyrian purple to kinase modulators: Naturally halogenated indirubins and synthetic analogues. Planta Med. 2012;78:1515–1528. doi: 10.1055/s-0032-1315261. [DOI] [PubMed] [Google Scholar]
- 37.Leclerc S., Garnier M., Hoessel R., Marko D., Bibb J.A., Snyder G.L., Greengard P., Biernat J., Wu Y.Z., Mandelkow E.M., et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/p25, two protein kinases involved in abnormal tau phosphorylation in alzheimer’s disease—A property common to most cycline-dependent kinase inhibitors? J. Biol. Chem. 2001;276:251–260. doi: 10.1074/jbc.M002466200. [DOI] [PubMed] [Google Scholar]
- 38.Schepetkin I.A., Khlebnikov A.I., Potapov A.S., Kovrizhina A.R., Matveevskaya V.V., Belyanin M.L., Atochin D.N., Zanoza S.O., Gaidarzhy N.M., Lyakhov S.A., et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno[1,2-b]quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019;161:179–191. doi: 10.1016/j.ejmech.2018.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu L., Sha S., Wang K., Zhang Y.H., Liu Y.D., Ju G.D., Wang B., Zhu H.L. Discovery of chromeno[4,3-c]pyrazol-4(2H)-one containing carbonyl or oxime derivatives as potential, selective inhibitors PI3Kα. Chem. Pharm. Bull. 2016;64:1576–1581. doi: 10.1248/cpb.c16-00388. [DOI] [PubMed] [Google Scholar]
- 40.Begum J., Skamnaki V.T., Moffatt C., Bischler N., Sarrou J., Skaltsounis A.L., Leonidas D.D., Oikonomakos N.G., Hayes J.M. An evaluation of indirubin analogues as phosphorylase kinase inhibitors. J. Mol. Graph. Model. 2015;61:231–242. doi: 10.1016/j.jmgm.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 41.Schepetkin I.A., Kirpotina L.N., Khlebnikov A.I., Hanks T.S., Kochetkova I., Pascual D.W., Jutila M.A., Quinn M.T. Identification and characterization of a novel class of c-Jun N-terminal kinase inhibitors. Mol. Pharmacol. 2012;81:832–845. doi: 10.1124/mol.111.077446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nam S., Scuto A., Yang F., Chen W., Park S., Yoo H.S., Konig H., Bhatia R., Cheng X., Merz K.H., et al. Indirubin derivatives induce apoptosis of chronic myelogenous leukemia cells involving inhibition of STAT5 signaling. Mol. Oncol. 2012;6:276–283. doi: 10.1016/j.molonc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoessel R., Leclerc S., Endicott J.A., Nobel M.E., Lawrie A., Tunnah P., Leost M., Damiens E., Marie D., Marko D., et al. Indirubin, the active constituent of a chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999;1:60–67. doi: 10.1038/9035. [DOI] [PubMed] [Google Scholar]
- 44.Meijer L., Skaltsounis A.L., Magiatis P., Polychronopoulos P., Knockaert M., Leost M., Ryan X.P., Vonica C.A., Brivanlou A., Dajani R., et al. GSK-3-selective inhibitors derived from tyrian purple indirubins. Chem. Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 45.Chan Y.K., Kwok H.H., Chan L.S., Leung K.S., Shi J., Mak N.K., Wong R.N., Yue P.Y. An indirubin derivative, E804, exhibits potent angiosuppressive activity. Biochem. Pharmacol. 2012;83:598–607. doi: 10.1016/j.bcp.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Nam S., Buettner R., Turkson J., Kim D., Cheng J.Q., Muehlbeyer S., Hippe F., Vatter S., Merz K.H., Eisenbrand G., et al. Indirubin derivatives inhibit STAT3 signaling and induce apoptosis in human cancer cells. Proc. Natl. Acad. Sci. USA. 2005;102:5998–6003. doi: 10.1073/pnas.0409467102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nam S., Wen W., Schroeder A., Herrmann A., Yu H., Cheng X., Merz K.H., Eisenbrand G., Li H., Yuan Y.C., et al. Dual inhibition of janus and src family kinases by novel indirubin derivative blocks constitutively-activated STAT3 signaling associated with apoptosis of human pancreatic cancer cells. Mol. Oncol. 2013;7:369–378. doi: 10.1016/j.molonc.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng X., Merz K.H., Vatter S., Christ J., Wolfl S., Eisenbrand G. 7,7′-diazaindirubin--a small molecule inhibitor of casein kinase 2 in vitro and in cells. Bioorg. Med. Chem. 2014;22:247–255. doi: 10.1016/j.bmc.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 49.Choi S.J., Moon M.J., Lee S.D., Choi S.U., Han S.Y., Kim Y.C. Indirubin derivatives as potent FLT3 inhibitors with anti-proliferative activity of acute myeloid leukemic cells. Bioorg. Med. Chem. Lett. 2010;20:2033–2037. doi: 10.1016/j.bmcl.2010.01.039. [DOI] [PubMed] [Google Scholar]
- 50.Bain J., McLauchlan H., Elliott M., Cohen P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003;371:199–204. doi: 10.1042/bj20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung M.E., Byun B.J., Kim H.M., Lee J.Y., Park J.H., Lee N., Son Y.H., Choi S.U., Yang K.M., Kim S.J., et al. Discovery of indirubin derivatives as new class of DRAK2 inhibitors from high throughput screening. Bioorg. Med. Chem. Lett. 2016;26:2719–2723. doi: 10.1016/j.bmcl.2016.03.111. [DOI] [PubMed] [Google Scholar]
- 52.Cheng X.L., Merz K.H., Vatter S., Zeller J., Muehlbeyer S., Thommet A., Christ J., Wolfl S., Eisenbrand G. Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. J. Med. Chem. 2017;60:4949–4962. doi: 10.1021/acs.jmedchem.7b00324. [DOI] [PubMed] [Google Scholar]
- 53.Yan L., Lai F.F., Chen X.G., Xiao Z.Y. Discovery of novel indirubin-3 ‘-monoxime derivatives as potent inhibitors against CDK2 and CDK9. Bioorg. Med. Chem. Lett. 2015;25:2447–2451. doi: 10.1016/j.bmcl.2015.03.066. [DOI] [PubMed] [Google Scholar]
- 54.Ndolo K.M., Park K.R., Lee H.J., Bin Yoon K., Kim Y.C., Han S.Y. Characterization of the indirubin derivative LDD970 as a small molecule aurora kinase a inhibitor in human colorectal cancer cells. Immune. Netw. 2017;17:110–115. doi: 10.4110/in.2017.17.2.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi S.J., Lee J.E., Jeong S.Y., Im I., Lee S.D., Lee E.J., Lee S.K., Kwon S.M., Ahn S.G., Yoon J.H., et al. 5,5′-Substituted indirubin-3′-oxime derivatives as potent cyclin-dependent kinase inhibitors with anticancer activity. J. Med. Chem. 2010;53:3696–3706. doi: 10.1021/jm100080z. [DOI] [PubMed] [Google Scholar]
- 56.Lee H.J., Lee J., Jeong P., Choi J., Baek J., Ahn S.J., Moon Y., Heo J.D., Choi Y.H., Chin Y.W., et al. Discovery of a FLT3 inhibitor LDD1937 as an anti-leukemic agent for acute myeloid leukemia. Oncotarget. 2018;9:924–936. doi: 10.18632/oncotarget.23221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Myrianthopoulos V., Magiatis P., Ferandin Y., Skaltsounis A.L., Meijer L., Mikros E. An integrated computational approach to the phenomenon of potent and selective inhibition of aurora kinases B and C by a series of 7-substituted indirubins. J. Med. Chem. 2007;50:4027–4037. doi: 10.1021/jm070077z. [DOI] [PubMed] [Google Scholar]
- 58.Myrianthopoulos V., Kritsanida M., Gaboriaud-Kolar N., Magiatis P., Ferandin Y., Durieu E., Lozach O., Cappel D., Soundararajan M., Filippakopoulos P., et al. Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. ACS Med. Chem. Lett. 2013;4:22–26. doi: 10.1021/ml300207a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vougogiannopoulou K., Ferandin Y., Bettayeb K., Myrianthopoulos V., Lozach O., Fan Y., Johnson C.H., Magiatis P., Skaltsounis A.L., Mikros E., et al. Soluble 3‘,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J. Med. Chem. 2008;51:6421–6431. doi: 10.1021/jm800648y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu L., Gaboriaud N., Vougogianopoulou K., Tian Y., Wu J., Wen W., Skaltsounis L., Jove R. MLS-2384, a new 6-bromoindirubin derivative with dual JAK/Src kinase inhibitory activity, suppresses growth of diverse cancer cells. Cancer Biol. Ther. 2014;15:178–184. doi: 10.4161/cbt.26721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jeong P., Moon Y., Lee J.H., Lee S.D., Park J., Lee J., Kim J., Lee H.J., Kim N.Y., Choi J., et al. Discovery of orally active indirubin-3 ‘-oxime derivatives as potent type 1 FLT3 inhibitors for acute myeloid leukemia. Eur. J. Med. Chem. 2020;195:112205. doi: 10.1016/j.ejmech.2020.112205. [DOI] [PubMed] [Google Scholar]
- 62.Hansen J.D., Grina J., Newhouse B., Welch M., Topalov G., Littman N., Callejo M., Gloor S., Martinson M., Laird E., et al. Potent and selective pyrazole-based inhibitors of b-raf kinase. Bioorg. Med. Chem. Lett. 2008;18:4692–4695. doi: 10.1016/j.bmcl.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 63.Takle A.K., Brown M.J.B., Davies S., Dean D.K., Francis G., Gaiba A., Hird A.W., King F.D., Lovell P.J., Naylor A., et al. The identification of potent and selective imidazole-based inhibitors of B-Raf kinase. Bioorg. Med. Chem. Lett. 2006;16:378–381. doi: 10.1016/j.bmcl.2005.09.072. [DOI] [PubMed] [Google Scholar]
- 64.Amino N., Ideyama Y., Yamano M., Kuromitsu S., Tajinda K., Samizu K., Hisamichi H., Matsuhisa A., Shirasuna K., Kudoh M., et al. YM-359445, an orally bioavailable vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor, has highly potent antitumor activity against established tumors. Clin. Cancer Res. 2006;12:1630–1638. doi: 10.1158/1078-0432.CCR-05-2028. [DOI] [PubMed] [Google Scholar]
- 65.Emanuel S.L., Hughes T.V., Adams M., Rugg C.A., Fuentes-Pesquera A., Connolly P.J., Pandey N., Moreno-Mazza S., Butler J., Borowski V., et al. Cellular and in vivo activity of JNJ-28871063, a nonquinazoline pan-ErbB kinase inhibitor that crosses the blood-brain barrier and displays efficacy against intracranial tumors. Mol. Pharmacol. 2008;73:338–348. doi: 10.1124/mol.107.041236. [DOI] [PubMed] [Google Scholar]
- 66.LaMontagne K.R., Butler J., Borowski V.B., Fuentes-Pesquera A.R., Blevitt J.M., Huang S.L., Li R.H., Connolly P.J., Greenberger L.M. A highly selective, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor has potent activity in vitro and in vivo. Angiogenesis. 2009;12:287–296. doi: 10.1007/s10456-009-9151-7. [DOI] [PubMed] [Google Scholar]
- 67.Cavasotto C.N., Ortiz M.A., Abagyan R.A., Piedrafita F.J. In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg. Med. Chem. Lett. 2006;16:1969–1974. doi: 10.1016/j.bmcl.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 68.Lv X.H., Li Q.S., Ren Z.L., Chu M.J., Sun J., Zhang X., Xing M., Zhu H.L., Cao H.Q. (E)-1,3-diphenyl-1H-pyrazole derivatives containing O-benzyl oxime moiety as potential immunosuppressive agents: Design, synthesis, molecular docking and biological evaluation. Eur. J. Med. Chem. 2016;108:586–593. doi: 10.1016/j.ejmech.2015.12.020. [DOI] [PubMed] [Google Scholar]
- 69.Foloppe N., Fisher L.M., Howes R., Potter A., Robertson A.G.S., Surgenor A.E. Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening. Biorg. Med. Chem. 2006;14:4792–4802. doi: 10.1016/j.bmc.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 70.Dandu R., Zulli A.L., Bacon E.R., Underiner T., Robinson C., Chang H., Miknyoczki S., Grobelny J., Ruggeri B.A., Yang S., et al. Design and synthesis of dihydroindazolo[5,4-a] pyrrolo[3,4-c] carbazole oximes as potent dual inhibitors of TIE-2 and VEGF-R2 receptor tyrosine kinases. Bioorg. Med. Chem. Lett. 2008;18:1916–1921. doi: 10.1016/j.bmcl.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 71.Maslyk M., Janeczko M., Demchuk O.M., Boguszewska-Czubara A., Golczyk H., Sieroslawska A., Rymuszka A., Martyna A., Kubinski K. A representative of arylcyanomethylenequinone oximes effectively inhibits growth and formation of hyphae in Candida albicans and influences the activity of protein kinases in vitro. Saudi Pharm. J. 2018;26:244–252. doi: 10.1016/j.jsps.2017.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ansideri F., Dammann M., Boeckler F.M., Koch P. Fluorescence polarization-based competition binding assay for c-Jun N-terminal kinases 1 and 2. Anal. Biochem. 2017;532:26–28. doi: 10.1016/j.ab.2017.05.022. [DOI] [PubMed] [Google Scholar]
- 73.Karabatsos G.J., Taller R.A. Structural studies by nuclear magnetic resonance XV. Conformations and configurations of oximes. Tetrahedron. 1968;24:3347–3360. doi: 10.1016/S0040-4020(01)92633-X. [DOI] [Google Scholar]
- 74.Claassen V., Davies J.E., Hertting G., Placheta P. Fluvoxamine, a specific 5-hydroxytryptamine uptake inhibitor. Br. J. Pharmacol. 1977;60:505–516. doi: 10.1111/j.1476-5381.1977.tb07528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bohle D.S., Chua Z., Perepichka I., Rosadiuk K. E/Z oxime isomerism in PhC(NOH)CN. Chem. Eur. J. 2013;19:4223–4229. doi: 10.1002/chem.201203357. [DOI] [PubMed] [Google Scholar]
- 76.Wylie B.B., Isaacson E.I., Delgado J.N. Synthesis of oxime esters and ethers as potential psychotropic aegents. J. Pharmaceut. Sci. 1965;54:1373–1376. doi: 10.1002/jps.2600540932. [DOI] [PubMed] [Google Scholar]
- 77.Hatem J., Henriet-Bernard C., Grimaldi J., Maurin R. Radical Cyclization of β-allenic oxime ethers. Tetrahedron Lett. 1992;3:1057–1058. doi: 10.1016/S0040-4039(00)91859-8. [DOI] [Google Scholar]
- 78.Kurtz A.P., D’Silva T.D. Estimation of dissociation constants (pKa’s) of oximes from proton chemical shifts in dimethyl sulfoxide solution. J. Pharm. Sci. 1987;76:599–610. doi: 10.1002/jps.2600760805. [DOI] [PubMed] [Google Scholar]
- 79.Musil K., Florianova V., Bucek P., Dohnal V., Kuca K., Musilek K. Development and validation of a FIA/UV-vis method for pK(a) determination of oxime based acetylcholinesterase reactivators. J. Pharmaceut. Biomed. Anal. 2016;117:240–246. doi: 10.1016/j.jpba.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 80.Sano M., Ichimaru Y., Kurita M., Hayashi E., Homma T., Saito H., Masuda S., Nemoto N., Hemmi A., Suzuki T., et al. Induction of cell death in pancreatic ductal adenocarcinoma by indirubin 3 ‘-oxime and 5-methoxyindirubin 3 ‘-oxime in vitro and in vivo. Cancer Lett. 2017;397:72–82. doi: 10.1016/j.canlet.2017.03.031. [DOI] [PubMed] [Google Scholar]
- 81.Zhang Y., Song L., Li J., Zhang Y., Lu X., Zhang B. Inhibitory effects of indirubin-3′-monoxime against human osteosarcoma. IUBMB Life. 2019;71:1465–1474. doi: 10.1002/iub.2058. [DOI] [PubMed] [Google Scholar]
- 82.Lee M.Y., Li Y.Z., Huang K.J., Huang H.C., Lin C.Y., Lee Y.R. Indirubin-3′-oxime suppresses human cholangiocarcinoma through cell-cycle arrest and apoptosis. Eur. J. Pharmacol. 2018;839:57–65. doi: 10.1016/j.ejphar.2018.09.023. [DOI] [PubMed] [Google Scholar]
- 83.Nicolaou K.A., Liapis V., Evdokiou A., Constantinou C., Magiatis P., Skaltsounis A.L., Koumas L., Costeas P.A., Constantinou A.I. Induction of discrete apoptotic pathways by bromo-substituted indirubin derivatives in invasive breast cancer cells. Biochem. Bioph. Res. Commun. 2012;425:76–82. doi: 10.1016/j.bbrc.2012.07.053. [DOI] [PubMed] [Google Scholar]
- 84.Broecker-Preuss M., Becher-Boveleth N., Gall S., Rehmann K., Schenke S., Mann K. Induction of atypical cell death in thyroid carcinoma cells by the indirubin derivative 7-bromoindirubin-3′-oxime (7BIO) Cancer Cell Int. 2015;15:97. doi: 10.1186/s12935-015-0251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ribas J., Bettayeb K., Ferandin Y., Knockaert M., Garrofe-Ochoa X., Totzke F., Schachtele C., Mester J., Polychronopoulos P., Magiatis P., et al. 7-bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene. 2006;25:6304–6318. doi: 10.1038/sj.onc.1209648. [DOI] [PubMed] [Google Scholar]
- 86.Fu B., Yin G., Song K., Mu X., Xu B., Zhang X. Indirubin-3′-oxime (IDR3O) inhibits proliferation of osteosarcoma cells in vitro and tumor growth in vivo through AMPK-activation and PGC-1α/TFAM up-regulation. Dokl. Biochem. Biophys. 2020;495:354–360. doi: 10.1134/S1607672920060022. [DOI] [PubMed] [Google Scholar]
- 87.Brighi N., Conteduca V., Lolli C., Gurioli G., Schepisi G., Palleschi M., Mariotti M., Casadei C., De Giorgi U. The cyclin-dependent kinases pathway as a target for prostate cancer treatment: Rationale and future perspectives. Crit. Rev. Oncol. Hematol. 2021;157:103199. doi: 10.1016/j.critrevonc.2020.103199. [DOI] [PubMed] [Google Scholar]
- 88.Augello G., Emma M.R., Cusimano A., Azzolina A., Montalto G., McCubrey J.A., Cervello M. The role of GSK-3 in cancer immunotherapy: GSK-3 inhibitors as a new frontier in cancer treatment. Cells. 2020;9:1427. doi: 10.3390/cells9061427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sahin I., Eturi A., De Souza A., Pamarthy S., Tavora F., Giles F.J., Carneiro B.A. Glycogen synthase kinase-3β inhibitors as novel cancer treatments and modulators of antitumor immune responses. Cancer Biol. Ther. 2019;20:1047–1056. doi: 10.1080/15384047.2019.1595283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qi G., Liu J., Mi S., Tsunematsu T., Jin S., Shao W., Liu T., Ishimaru N., Tang B., Kudo Y. Aurora kinase inhibitors in head and neck cancer. Curr. Top. Med. Chem. 2018;18:199–213. doi: 10.2174/1568026618666180112163741. [DOI] [PubMed] [Google Scholar]
- 91.Falchook G.S., Bastida C.C., Kurzrock R. Aurora kinase inhibitors in oncology clinical trials: Current state of the progress. Semin. Oncol. 2015;42:832–848. doi: 10.1053/j.seminoncol.2015.09.022. [DOI] [PubMed] [Google Scholar]
- 92.Yuan T., Qi B., Jiang Z., Dong W., Zhong L., Bai L., Tong R., Yu J., Shi J. Dual FLT3 inhibitors: Against the drug resistance of acute myeloid leukemia in recent decade. Eur. J. Med. Chem. 2019;178:468–483. doi: 10.1016/j.ejmech.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 93.Alim K., Bruyere A., Lescoat A., Jouan E., Lecureur V., Le Vee M., Fardel O. Interactions of janus kinase inhibitors with drug transporters and consequences for pharmacokinetics and toxicity. Expert Opin. Drug Metab. Toxicol. 2021:1–13. doi: 10.1080/17425255.2021.1862084. [DOI] [PubMed] [Google Scholar]
- 94.Abbassi R., Johns T.G., Kassiou M., Munoz L. DYRK1A in neurodegeneration and cancer: Molecular basis and clinical implications. Pharmacol. Therapeut. 2015;151:87–98. doi: 10.1016/j.pharmthera.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 95.Guo T., Ma S. Recent advances in the discovery of multitargeted tyrosine kinase inhibitors as anticancer agents. ChemMedChem. 2021;16:600–620. doi: 10.1002/cmdc.202000658. [DOI] [PubMed] [Google Scholar]
- 96.Gerritse S.L., Janssen J.B.E., Labots M., de Vries R., Rudek M., Carducci M., van Erp N.P., Verheul H.M.W. High-dose administration of tyrosine kinase inhibitors to improve clinical benefit: A systematic review. Cancer Treat. Rev. 2021;97:102171. doi: 10.1016/j.ctrv.2021.102171. [DOI] [PubMed] [Google Scholar]
- 97.Yumura M., Nagano T., Nishimura Y. Novel multitarget therapies for lung cancer and respiratory disease. Molecules. 2020;25:3987. doi: 10.3390/molecules25173987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sola A.M., Johnson D.E., Grandis J.R. Investigational multitargeted kinase inhibitors in development for head and neck neoplasms. Expert Opin. Investig. Drugs. 2019;28:351–363. doi: 10.1080/13543784.2019.1581172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Basolo A., Matrone A., Elisei R., Santini F. Effects of tyrosine kinase inhibitors on thyroid function and thyroid hormone metabolism. Semin. Cancer Biol. 2021 doi: 10.1016/j.semcancer.2020.12.008. [DOI] [PubMed] [Google Scholar]
- 100.Sundar V., Vimal S., Mithlesh M.S.S., Dutta A., Tamizhselvi R., Manickam V. Transcriptional cyclin-dependent kinases as the mediators of inflammation—A review. Gene. 2021;769:145200. doi: 10.1016/j.gene.2020.145200. [DOI] [PubMed] [Google Scholar]
- 101.Martin M., Rehani K., Jope R.S., Michalek S.M. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Beurel E., Michalek S.M., Jope R.S. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maity A., Sen D., Kandar C.C. Anti-inflammatory potential of GSK-3 inhibitors. Curr. Drug Targets. 2021 doi: 10.2174/1389450122666210118150313. [DOI] [PubMed] [Google Scholar]
- 104.Wadhwa P., Jain P., Jadhav H.R. Glycogen synthase kinase 3 (GSK3): Its role and inhibitors. Curr. Top. Med. Chem. 2020;20:1522–1534. doi: 10.2174/1568026620666200516153136. [DOI] [PubMed] [Google Scholar]
- 105.Simon L.S., Taylor P.C., Choy E.H., Sebba A., Quebe A., Knopp K.L., Porreca F. The JAK/STAT pathway: A focus on pain in rheumatoid arthritis. Semin. Arthritis. Rheum. 2021;51:278–284. doi: 10.1016/j.semarthrit.2020.10.008. [DOI] [PubMed] [Google Scholar]
- 106.Choudhary S.A., Bora N., Banerjee D., Arora L., Das A.S., Yadav R., Klotz K.N., Pal D., Jha A.N., Dasgupta S. A novel small molecule A2A adenosine receptor agonist, indirubin-3′-monoxime, alleviates lipid-induced inflammation and insulin resistance in 3T3-l1 adipocytes. Biochem. J. 2019;476:2371–2391. doi: 10.1042/BCJ20190251. [DOI] [PubMed] [Google Scholar]
- 107.Udumula M.P., Medapi B., Dhar I., Bhat A., Desai K., Sriram D., Dhar A. The small molecule indirubin-3′-oxime inhibits protein kinase R: Antiapoptotic and antioxidant effect in rat cardiac myocytes. Pharmacology. 2016;97:25–30. doi: 10.1159/000441727. [DOI] [PubMed] [Google Scholar]
- 108.Jung H.J., Nam K.N., Son M.S., Kang H., Hong J.W., Kim J.W., Lee E.H. Indirubin-3′-oxime inhibits inflammatory activation of rat brain microglia. Neurosci. Lett. 2011;487:139–143. doi: 10.1016/j.neulet.2010.10.009. [DOI] [PubMed] [Google Scholar]
- 109.Blazevic T., Schaible A.M., Weinhaupl K., Schachner D., Nikels F., Weinigel C., Barz D., Atanasov A.G., Pergola C., Werz O., et al. Indirubin-3′-monoxime exerts a dual mode of inhibition towards leukotriene-mediated vascular smooth muscle cell migration. Cardiovasc. Res. 2014;101:522–532. doi: 10.1093/cvr/cvt339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mok C.K., Kang S.S., Chan R.W., Yue P.Y., Mak N.K., Poon L.L., Wong R.N., Peiris J.S., Chan M.C. Anti-inflammatory and antiviral effects of indirubin derivatives in influenza a (H5N1) virus infected primary human peripheral blood-derived macrophages and alveolar epithelial cells. Antiviral. Res. 2014;106:95–104. doi: 10.1016/j.antiviral.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 111.Yu J., Zheng J., Lin J., Jin L., Yu R., Mak S., Hu S., Sun H., Wu X., Zhang Z., et al. Indirubin-3-oxime prevents H2O2-induced neuronal apoptosis via concurrently inhibiting GSK3β and the ERK pathway. Cell. Mol. Neurobiol. 2017;37:655–664. doi: 10.1007/s10571-016-0402-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim J.K., Park G.M. Indirubin-3-monoxime exhibits anti-inflammatory properties by down-regulating NF-κB and JNK signaling pathways in lipopolysaccharide-treated RAW264.7 cells. Inflamm. Res. 2012;61:319–325. doi: 10.1007/s00011-011-0413-7. [DOI] [PubMed] [Google Scholar]
- 113.Park D.W., Jiang S., Liu Y., Siegal G.P., Inoki K., Abraham E., Zmijewski J.W. GSK3β-dependent inhibition of AMPK potentiates activation of neutrophils and macrophages and enhances severity of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;307:L735–L745. doi: 10.1152/ajplung.00165.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plotnikov M.B., Aliev O.I., Shamanaev A.Y., Sidekhmenova A.V., Anishchenko A.M., Fomina T.I., Rydchenko V.S., Khlebnikov A.I., Anfinogenova Y.J., Schepetkin I.A., et al. Antihypertensive activity of a new c-Jun N-terminal kinase inhibitor in spontaneously hypertensive rats. Hypertens. Res. 2020 doi: 10.1038/s41440-020-0446-9. [DOI] [PubMed] [Google Scholar]
- 115.Nie Z., Xia X., Zhao Y., Zhang S., Zhang Y., Wang J. JNK selective inhibitor, IQ-1S, protects the mice against lipopolysaccharides-induced sepsis. Bioorg. Med. Chem. 2021;30:115945. doi: 10.1016/j.bmc.2020.115945. [DOI] [PubMed] [Google Scholar]
- 116.Kirpotina L.N., Schepetkin I.A., Hammaker D., Kuhs A., Khlebnikov A.I., Quinn M.T. Therapeutic effects of tryptanthrin and tryptanthrin-6-oxime in models of rheumatoid arthritis. Front. Pharmacol. 2020;11:1145. doi: 10.3389/fphar.2020.01145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hsieh C.Y., Chen C.L., Tsai C.C., Huang W.C., Tseng P.C., Lin Y.S., Chen S.H., Wong T.W., Choi P.C., Lin C.F. Inhibiting glycogen synthase kinase-3 decreases 12-o-tetradecanoylphorbol-13-acetate-induced interferon-γ-mediated skin inflammation. J. Pharmacol. Exp. Ther. 2012;343:125–133. doi: 10.1124/jpet.112.194100. [DOI] [PubMed] [Google Scholar]
- 118.Zhao S., Liu Z., Yu Z., Wu X., Li R., Tang X. Bio alleviates inflammation through inhibition of GSK-3β in a rat model of intracerebral hemorrhage. J. Neurosurg. 2019:1–9. doi: 10.3171/2019.4.JNS183501. [DOI] [PubMed] [Google Scholar]
- 119.Jean LeBlanc N., Menet R., Picard K., Parent G., Tremblay M.E., ElAli A. Canonical wnt pathway maintains blood-brain barrier integrity upon ischemic stroke and its activation ameliorates tissue plasminogen activator therapy. Mol. Neurobiol. 2019;56:6521–6538. doi: 10.1007/s12035-019-1539-9. [DOI] [PubMed] [Google Scholar]
- 120.Shen S., Zhang Y., Zhang S., Wang B., Shang L., Shao J., Lin M., Cui Y., Sun S., Ge S. 6-bromoindirubin-3′-oxime promotes osteogenic differentiation of periodontal ligament stem cells and facilitates bone regeneration in a mouse periodontitis model. ACS Biomater. Sci. Eng. 2021;7:232–241. doi: 10.1021/acsbiomaterials.0c01078. [DOI] [PubMed] [Google Scholar]
- 121.Guo D., Shen Y., Li W., Li Q., Zhao Y., Pan C., Chen B., Zhong Y., Miao Y. 6-bromoindirubin-3′-oxime (6BIO) suppresses the mtor pathway, promotes autophagy, and exerts anti-aging effects in rodent liver. Front. Pharmacol. 2019;10:320. doi: 10.3389/fphar.2019.00320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schepetkin I.A., Kirpotina L.N., Hammaker D., Kochetkova I., Khlebnikov A.I., Lyakhov S.A., Firestein G.S., Quinn M.T. Anti-inflammatory effects and joint protection in collagen-induced arthritis after treatment with IQ-1S, a selective c-Jun N-terminal kinase inhibitor. J. Pharmacol. Exp. Ther. 2015;353:505–516. doi: 10.1124/jpet.114.220251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Plotnikov M.B., Chernysheva G.A., Smolyakova V.I., Aliev O.I., Trofimova E.S., Sherstoboev E.Y., Osipenko A.N., Khlebnikov A.I., Anfinogenova Y.J., Schepetkin I.A., et al. Neuroprotective effects of a novel inhibitor of c-Jun N-terminal kinase in the rat model of transient focal cerebral ischemia. Cells. 2020;9:1860. doi: 10.3390/cells9081860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marko D., Schatzle S., Friedel A., Genzlinger A., Zankl H., Meijer L., Eisenbrand G. Inhibition of cyclin-dependent kinase 1 (CDK1) by indirubin derivatives in human tumour cells. Br. J. Cancer. 2001;84:283–289. doi: 10.1054/bjoc.2000.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Xiao Z., Hao Y., Liu B., Qian L. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in china. Leuk. Lymphoma. 2002;43:1763–1768. doi: 10.1080/1042819021000006295. [DOI] [PubMed] [Google Scholar]
- 126.Orekhov A.N., Oishi Y., Nikiforov N.G., Zhelankin A.V., Dubrovsky L., Sobenin I.A., Kel A., Stelmashenko D., Makeev V.J., Foxx K., et al. Modified LDL particles activate inflammatory pathways in monocyte-derived macrophages: Transcriptome analysis. Curr. Pharm. Des. 2018;24:3143–3151. doi: 10.2174/1381612824666180911120039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Back M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc. Drugs Ther. 2009;23:41–48. doi: 10.1007/s10557-008-6140-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hlawaty H., Jacob M.P., Louedec L., Letourneur D., Brink C., Michel J.B., Feldman L., Back M. Leukotriene receptor antagonism and the prevention of extracellular matrix degradation during atherosclerosis and in-stent stenosis. Arterioscler. Thromb. Vasc. Biol. 2009;29:518–524. doi: 10.1161/ATVBAHA.108.181750. [DOI] [PubMed] [Google Scholar]
- 129.Davalos D., Grutzendler J., Yang G., Kim J.V., Zuo Y., Jung S., Littman D.R., Dustin M.L., Gan W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005;8:752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 130.Nimmerjahn A., Kirchhoff F., Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 131.Hanisch U.K., Kettenmann H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- 132.Walter L., Neumann H. Role of microglia in neuronal degeneration and regeneration. Semin. Immunopathol. 2009;31:513–525. doi: 10.1007/s00281-009-0180-5. [DOI] [PubMed] [Google Scholar]
- 133.Zhang S.G., Wang X.S., Zhang Y.D., Di Q., Shi J.P., Qian M., Xu L.G., Lin X.J., Lu J. Indirubin-3′-monoxime suppresses amyloid-β-induced apoptosis by inhibiting tau hyperphosphorylation. Neural. Regen. Res. 2016;11:988–993. doi: 10.4103/1673-5374.184500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Konda V.R., Desai A., Darland G., Bland J.S., Tripp M.L. Rho iso-alpha acids from hops inhibit the GSK-3/NF-κB pathway and reduce inflammatory markers associated with bone and cartilage degradation. J. Inflamm. 2009;6:26. doi: 10.1186/1476-9255-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hotamisligil G.S. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 136.Hayes J.M., Skamnaki V.T., Archontis G., Lamprakis C., Sarrou J., Bischler N., Skaltsounis A.L., Zographos S.E., Oikonomakos N.G. Kinetics, in silico docking, molecular dynamics, and MM-GBSA binding studies on prototype indirubins, KT5720, and staurosporine as phosphorylase kinase ATP-binding site inhibitors: The role of water molecules examined. Proteins. 2011;79:703–719. doi: 10.1002/prot.22890. [DOI] [PubMed] [Google Scholar]
- 137.Scobie M.R., Houke H.R., Rice C.D. Modulation of glioma-inflammation crosstalk profiles in human glioblastoma cells by indirubin-3′-(2,3 dihydroxypropyl)-oximether (E804) and 7-bromoindirubin-3′-oxime (7BIO) Chem. Biol. Interact. 2019;312:108816. doi: 10.1016/j.cbi.2019.108816. [DOI] [PubMed] [Google Scholar]
- 138.Czapka A., Konig S., Pergola C., Grune C., Vougogiannopoulou K., Skaltsounis A.L., Fischer D., Werz O. The indirubin derivative 6-bromoindirubin-3′-glycerol-oxime ether (6BIGOE) potently modulates inflammatory cytokine and prostaglandin release from human monocytes through GSK-3 interference. Biochem. Pharmacol. 2020;180:114170. doi: 10.1016/j.bcp.2020.114170. [DOI] [PubMed] [Google Scholar]
- 139.Freyberg Z., Ferrando S.J., Javitch J.A. Roles of the AKT/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am. J. Psychiatry. 2010;167:388–396. doi: 10.1176/appi.ajp.2009.08121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ahn M.Y., Kim T.H., Kwon S.M., Yoon H.E., Kim H.S., Kim J.I., Kim Y.C., Kang K.W., Ahn S.G., Yoon J.H. 5-nitro-5′-hydroxy-indirubin-3′-oxime (AGM130), an indirubin-3′-oxime derivative, inhibits tumor growth by inducing apoptosis against non-small cell lung cancer in vitro and in vivo. Eur. J. Pharm. Sci. 2015;79:122–131. doi: 10.1016/j.ejps.2015.08.015. [DOI] [PubMed] [Google Scholar]
- 141.Moon M.J., Lee S.K., Lee J.W., Song W.K., Kim S.W., Kim J.I., Cho C., Choi S.J., Kim Y.C. Synthesis and structure-activity relationships of novel indirubin derivatives as potent anti-proliferative agents with CDK2 inhibitory activities. Biorg. Med. Chem. 2006;14:237–246. doi: 10.1016/j.bmc.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 142.Polychronopoulos P., Magiatis P., Skaltsounis A.L., Myrianthopoulos V., Mikros E., Tarricone A., Musacchio A., Roe S.M., Pearl L., Leost M., et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J. Med. Chem. 2004;47:935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- 143.Nisha C.M., Kumar A., Vimal A., Bai B.M., Pal D., Kumar A. Docking and ADMET prediction of few GSK-3 inhibitors divulges 6-bromoindirubin-3-oxime as a potential inhibitor. J. Mol. Graph. Model. 2016;65:100–107. doi: 10.1016/j.jmgm.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 144.Sandoval K.E., Witt K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008;32:200–219. doi: 10.1016/j.nbd.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 145.Brouns R., Wauters A., De Surgeloose D., Marien P., De Deyn P.P. Biochemical markers for blood-brain barrier dysfunction in acute ischemic stroke correlate with evolution and outcome. Eur. Neurol. 2011;65:23–31. doi: 10.1159/000321965. [DOI] [PubMed] [Google Scholar]
- 146.Sussman E.S., Connolly E.S., Jr. Hemorrhagic transformation: A review of the rate of hemorrhage in the major clinical trials of acute ischemic stroke. Front. Neurol. 2013;4:69. doi: 10.3389/fneur.2013.00069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liebner S., Corada M., Bangsow T., Babbage J., Taddei A., Czupalla C.J., Reis M., Felici A., Wolburg H., Fruttiger M., et al. Wnt/β-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008;183:409–417. doi: 10.1083/jcb.200806024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Creedon H., Brunton V.G. Src kinase inhibitors: Promising cancer therapeutics? Crit. Rev. Oncog. 2012;17:145–159. doi: 10.1615/CritRevOncog.v17.i2.20. [DOI] [PubMed] [Google Scholar]
- 149.Chavda J., Bhatt H. Systemic review on B-RafV600E mutation as potential therapeutic target for the treatment of cancer. Eur. J. Med. Chem. 2020;206 doi: 10.1016/j.ejmech.2020.112675. [DOI] [PubMed] [Google Scholar]
- 150.Hoeflich K.P., Herter S., Tien J., Wong L., Berry L., Chan J., O’Brien C., Modrusan Z., Seshagiri S., Lackner M., et al. Antitumor efficacy of the novel RAF inhibitor GDC-0879 is predicted by BRAFV600E mutational status and sustained extracellular signal-regulated kinase/mitogen-activated protein kinase pathway suppression. Cancer Res. 2009;69:3042–3051. doi: 10.1158/0008-5472.CAN-08-3563. [DOI] [PubMed] [Google Scholar]
- 151.Choo E.F., Driscoll J.P., Feng J., Liederer B., Plise E., Randolph N., Shin Y., Wong S., Ran Y. Disposition of GDC-0879, a B-Raf kinase inhibitor in preclinical species. Xenobiotica. 2009;39:700–709. doi: 10.1080/00498250902991827. [DOI] [PubMed] [Google Scholar]
- 152.Boudny M., Trbusek M. ATR-CHK1 pathway as a therapeutic target for acute and chronic leukemias. Cancer Treat. Rev. 2020;88 doi: 10.1016/j.ctrv.2020.102026. [DOI] [PubMed] [Google Scholar]
- 153.Evangelisti G., Barra F., Moioli M., Sala P., Stigliani S., Gustavino C., Costantini S., Ferrero S. Prexasertib: An investigational checkpoint kinase inhibitor for the treatment of high-grade serous ovarian cancer. Expert Opin. Investig. Drug. 2020;29:779–792. doi: 10.1080/13543784.2020.1783238. [DOI] [PubMed] [Google Scholar]
- 154.Bonello M., Sims A.H., Langdon S.P. Human epidermal growth factor receptor targeted inhibitors for the treatment of ovarian cancer. Cancer Biol. Med. 2018;15:375–388. doi: 10.20892/j.issn.2095-3941.2018.0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hashemzadeh K., Jokar M.H., Sedighi S., Moradzadeh M. Therapeutic potency of PI3K pharmacological inhibitors of gastrointestinal cancer. Middle East J. Dig. Dis. 2019;11:5–16. doi: 10.15171/mejdd.2018.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Husain K., Williamson T.T., Nelson N., Ghansah T. Protein kinase 2 (CK2): A potential regulator of immune cell development and function in cancer. Immunol. Med. 2020:1–16. doi: 10.1080/25785826.2020.1843267. [DOI] [PubMed] [Google Scholar]
- 157.Bogoyevitch M.A., Boehm I., Oakley A., Ketterman A.J., Barr R.K. Targeting the JNK MAPK cascade for inhibition: Basic science and therapeutic potential. Biochim. Biophys. Acta. 2004;1697:89–101. doi: 10.1016/j.bbapap.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 158.Bhagwat S.S. Map kinase inhibitors in inflammation and autoimmune disorders. Annu. Rep. Med. Chem. 2007;42:265–278. doi: 10.1016/S0065-7743(07)42017-6. [DOI] [Google Scholar]
- 159.Shvedova M., Anfinogenova Y., Atochina-Vasserman E.N., Schepetkin I.A., Atochin D.N. C-Jun N-terminal kinases (JNKs) in myocardial and cerebral ischemia/reperfusion injury. Front. Pharmacol. 2018;9:715. doi: 10.3389/fphar.2018.00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Atochin D.N., Schepetkin I.A., Khlebnikov A.I., Seledtsov V.I., Swanson H., Quinn M.T., Huang P.L. A novel dual no-donating oxime and c-Jun N-terminal kinase inhibitor protects against cerebral ischemia-reperfusion injury in mice. Neurosci. Lett. 2016;618:45–49. doi: 10.1016/j.neulet.2016.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Plotnikov M.B., Chernysheva G.A., Aliev O.I., Smol’iakova V.I., Fomina T.I., Osipenko A.N., Rydchenko V.S., Anfinogenova Y.J., Khlebnikov A.I., Schepetkin I.A., et al. Protective effects of a new c-Jun N-terminal kinase inhibitor in the model of global cerebral ischemia in rats. Molecules. 2019;24:1722. doi: 10.3390/molecules24091722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Pergola C., Gaboriaud-Kolar N., Jestadt N., Konig S., Kritsanida M., Schaible A.M., Li H.K., Garscha U., Weinigel C., Barz D., et al. Indirubin core structure of glycogen synthase kinase-3 inhibitors as novel chemotype for intervention with 5-lipoxygenase. J. Med. Chem. 2014;57:3715–3723. doi: 10.1021/jm401740w. [DOI] [PubMed] [Google Scholar]
- 163.Krajka-Kuzniak V., Bednarczyk-Cwynar B., Paluszczak J., Szaefer H., Narozna M., Zaprutko L., Baer-Dubowska W. Oleanolic acid oxime derivatives and their conjugates with aspirin modulate the NF-κB-mediated transcription in HEPG2 hepatoma cells. Bioorg. Chem. 2019;93:103326. doi: 10.1016/j.bioorg.2019.103326. [DOI] [PubMed] [Google Scholar]
- 164.Sun H.N., Jin M.H., Han B., Feng L., Han Y.H., Shen G.N., Yu Y.Z., Jin C.H., Lian Z.X., Lee D.S., et al. 16α,17α-epoxypregnenolone-20-oxime prevent LPS-induced NO production and iNOS expression in BV-2 microglial cells by inhibiting JNK phosphorylation. Biol. Pharm. Bull. 2014;37:1096–1102. doi: 10.1248/bpb.b13-00706. [DOI] [PubMed] [Google Scholar]
- 165.Sun H.N., Han Y.H., Feng L., Jin C.H., Han B., Liu L., Lee D.S., Kwon T.H., Li L.G., Ge W.Z., et al. 16α,17α-epoxypregnenolone-20-oxime inhibits NO and IL-6 production in LPS-treated RAW264.7 cells. Mol. Med. Rep. 2016;13:4927–4933. doi: 10.3892/mmr.2016.5125. [DOI] [PubMed] [Google Scholar]
- 166.Kolsi L.E., Leal A.S., Yli-Kauhaluoma J., Liby K.T., Moreira V.M. Dehydroabietic oximes halt pancreatic cancer cell growth in the G1 phase through induction of p27 and downregulation of cyclin D1. Sci. Rep. 2018;8:15923. doi: 10.1038/s41598-018-34131-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chen Y.L., Zhao Y.L., Lu C.M., Tzeng C.C., Wang J.P. Synthesis, cytotoxicity, and anti-inflammatory evaluation of 2-(furan-2-yl)-4-(phenoxy)quinoline derivatives. Part 4. Bioorg. Med. Chem. 2006;14:4373–4378. doi: 10.1016/j.bmc.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 168.Chen Y.L., Chen I.L., Lu C.M., Tzeng C.C., Tsao L.T., Wang J.P. Synthesis and anti-inflammatory evaluation of 4-anilinofuro[2,3-b]quinoline and 4-phenoxyfuro[2,3-b]quinoline derivatives. Part 3. Bioorg. Med. Chem. 2004;12:387–392. doi: 10.1016/j.bmc.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 169.Strizki J.M., Xu S., Wagner N.E., Wojcik L., Liu J., Hou Y., Endres M., Palani A., Shapiro S., Clader J.W., et al. SCH-C (SCH 351125), an orally bioavailable, small molecule antagonist of the chemokine receptor CCR5, is a potent inhibitor of HIV-1 infection in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2001;98:12718–12723. doi: 10.1073/pnas.221375398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Tsamis F., Gavrilov S., Kajumo F., Seibert C., Kuhmann S., Ketas T., Trkola A., Palani A., Clader J.W., Tagat J.R., et al. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581 inhibit human immunodeficiency virus type 1 entry. J. Virol. 2003;77:5201–5208. doi: 10.1128/JVI.77.9.5201-5208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Johansen T.H., Drejer J., Watjen F., Nielsen E.O. A novel non-NMDA receptor antagonist shows selective displacement of low-affinity [H-3] kainate binding. Eur. J. Pharm. Molec. Pharmacol. 1993;246:195–204. doi: 10.1016/0922-4106(93)90031-4. [DOI] [PubMed] [Google Scholar]
- 172.Guo W., Zou S.P., Tal M., Ren K. Activation of spinal kainate receptors after inflammation: Behavioral hyperalgesia and subunit gene expression. Eur. J. Pharmacol. 2002;452:309–318. doi: 10.1016/S0014-2999(02)02333-6. [DOI] [PubMed] [Google Scholar]
- 173.Petrus M., Peier A.M., Bandell M., Hwang S.W., Huynh T., Olney N., Jegla T., Patapoutian A. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol. Pain. 2007;3:40. doi: 10.1186/1744-8069-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.McGaraughty S., Chu K.L., Perner R.J., Didomenico S., Kort M.E., Kym P.R. TRPA1 modulation of spontaneous and mechanically evoked firing of spinal neurons in uninjured, osteoarthritic, and inflamed rats. Mol. Pain. 2010;6:14. doi: 10.1186/1744-8069-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Munro G., Christensen J.K., Erichsen H.K., Dyhring T., Demnitz J., Dam E., Ahring P.K. NS383 selectively inhibits acid-sensing ion channels containing 1a and 3 subunits to reverse inflammatory and neuropathic hyperalgesia in rats. CNS Neurosci. Ther. 2016;22:135–145. doi: 10.1111/cns.12487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Bordet T., Buisson B., Michaud M., Drouot C., Galea P., Delaage P., Akentieva N.P., Evers A.S., Covey D.F., Ostuni M.A., et al. Identification and characterization of cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007;322:709–720. doi: 10.1124/jpet.107.123000. [DOI] [PubMed] [Google Scholar]
- 177.Kurebayashi J., Otsuki T., Kurosumi M., Soga S., Akinaga S., Sonoo H. A radicicol derivative, KF58333, inhibits expression of hypoxia-inducible factor-1α and vascular endothelial growth factor, angiogenesis and growth of human breast cancer xenografts. Jpn. J. Cancer Res. 2001;92:1342–1351. doi: 10.1111/j.1349-7006.2001.tb02159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pillai A.D., Rathod P.D., Franklin P.X., Padh H., Vasu K.K., Sudarsanam V. Design, synthesis, and sar studies of some 5-aliphatic oximino esters of thiophene as potential anti-inflammatory leads: Comparative biological activity profile of aliphatic oximes vs aromatic oximes. Biochem. Biophys. Res. Commun. 2004;317:1067–1074. doi: 10.1016/j.bbrc.2004.03.148. [DOI] [PubMed] [Google Scholar]
- 179.Yu X., Park E.J., Kondratyuk T.P., Pezzuto J.M., Sun D. Synthesis of 2-arylindole derivatives and evaluation as nitric oxide synthase and NF-κB inhibitors. Org. Biomol. Chem. 2012;10:8835–8847. doi: 10.1039/c2ob26456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Franklin P.X., Pillai A.D., Rathod P.D., Yerande S., Nivsarkar M., Padh H., Vasu K.K., Sudarsanam V. 2-amino-5-thiazolyl motif: A novel scaffold for designing anti-inflammatory agents of diverse structures. Eur. J. Med. Chem. 2008;43:129–134. doi: 10.1016/j.ejmech.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 181.Bagdanoff J.T., Donoviel M.S., Nouraldeen A., Carlsen M., Jessop T.C., Tarver J., Aleem S., Dong L., Zhang H., Boteju L., et al. Inhibition of sphingosine 1-phosphate lyase for the treatment of rheumatoid arthritis: Discovery of (E)-1-(4-((1R,2S,3R)-1,2,3,4-tetrahydroxybutyl)-1H-imidazol-2-yl)ethanone oxime (LX2931) and (1R,2S,3R)-1-(2-(isoxazol-3-yl)-1H-imidazol-4-yl)butane-1,2,3,4-tetraol (LX2932) J. Med. Chem. 2010;53:8650–8662. doi: 10.1021/jm101183p. [DOI] [PubMed] [Google Scholar]
- 182.Kessenbrock K., Frohlich L., Sixt M., Lammermann T., Pfister H., Bateman A., Belaaouaj A., Ring J., Ollert M., Fassler R., et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J. Clin. Investig. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Crocetti L., Quinn M.T., Schepetkin I.A., Giovannoni M.P. A patenting perspective on human neutrophil elastase (HNE) inhibitors (2014–2018) and their therapeutic applications. Expert Opin. Ther. Pat. 2019;29:555–578. doi: 10.1080/13543776.2019.1630379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hayakawa M., Katabami K., Wada T., Sugano M., Hoshino H., Sawamura A., Gando S. Sivelestat (selective neutrophil elastase inhibitor) improves the mortality rate of sepsis associated with both acute respiratory distress syndrome and disseminated intravascular coagulation patients. Shock. 2010;33:14–18. doi: 10.1097/SHK.0b013e3181aa95c4. [DOI] [PubMed] [Google Scholar]
- 185.Essayan D.M. Cyclic nucleotide phosphodiesterase (PDE) inhibitors and immunomodulation. Biochem. Pharmacol. 1999;57:965–973. doi: 10.1016/S0006-2952(98)00331-1. [DOI] [PubMed] [Google Scholar]
- 186.Yoo E.S., Son H.J., Park J.S., Kim A.R., Baik K.U., Park M.H., Cho J.Y. Effects of dialkoxylphenyl compounds with oxime group on macrophage function and the proliferation of lymphocytes. J. Pharm. Pharmacol. 2004;56:503–512. doi: 10.1211/0022357023042. [DOI] [PubMed] [Google Scholar]
- 187.Larm J.A., Cheung N.S., Beart P.M. (S)-5-fluorowillardiine-mediated neurotoxicity in cultured murine cortical neurones occurs via AMPA and kainate receptors. Eur. J. Pharmacol. 1996;314:249–254. doi: 10.1016/S0014-2999(96)00633-4. [DOI] [PubMed] [Google Scholar]
- 188.El-Sherief H.A.M., Youssif B.G.M., Bukhari S.N.A., Abdelazeem A.H., Abdel-Aziz M., Abdel-Rahman H.M. Synthesis, anticancer activity and molecular modeling studies of 1,2,4-triazole derivatives as EGFR inhibitors. Eur. J. Med. Chem. 2018;156:774–789. doi: 10.1016/j.ejmech.2018.07.024. [DOI] [PubMed] [Google Scholar]
- 189.Sanchez-Pavon E., Lopez-Monteon A., Hernandez-Romero D., de la Soledad Lagunes-Castro M., Zanatta-Garcia D.Y., Ramos-Ligonio A. Design and synthesis of IMR-23, an oxime derived from nitroimidazole as an immunomodulatory molecule. Curr. Drug Deliv. 2020;17:324–332. doi: 10.2174/1567201817666200214110442. [DOI] [PubMed] [Google Scholar]
- 190.Androniklion V., Boucher J.L., Delaforge M., Henry Y., Mansuy D. Formation of nitric-oxide by cytochrome P450-catalyzed oxidation of aromatic amidoximes. Biochem. Biophys. Res. Commun. 1992;185:452–458. doi: 10.1016/S0006-291X(05)81006-X. [DOI] [PubMed] [Google Scholar]
- 191.Caro A.A., Cederbaum A.L., Stoyanovsky D.A. Oxidation of the ketoxime acetoxime to nitric oxide by oxygen radical-generating systems. Nitric Oxide Biol. Chem. 2001;5:413–424. doi: 10.1006/niox.2001.0362. [DOI] [PubMed] [Google Scholar]
- 192.Veras R.C., Rodrigues K.G., Alustau M.D., Araujo I.G.A., de Barros A.L.B., Alves R.J., Nakao L.S., Braga V.A., Silva D.F., de Medeiros I.A. Participation of nitric oxide pathway in the relaxation response induced by E-cinnamaldehyde oxime in superior mesenteric artery isolated from rats. J. Cardiovasc. Pharmacol. 2013;62:58–66. doi: 10.1097/FJC.0b013e31829013ff. [DOI] [PubMed] [Google Scholar]
- 193.Sahyoun T., Arrault A., Schneider R. Amidoximes and oximes: Synthesis, structure, and their key role as NO donors. Molecules. 2019;24:2470. doi: 10.3390/molecules24132470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Mansuy D., Boucher J.L., Clement B. On the mechanism of nitric oxide formation upon oxidative cleavage of C=N(OH) bonds by no-synthases and cytochromes P450. Biochimie. 1995;77:661–667. doi: 10.1016/0300-9084(96)88181-8. [DOI] [PubMed] [Google Scholar]
- 195.Volkel W., Wolf N., Derelanko M., Dekant W. Slow oxidation of acetoxime and methylethyl ketoxime to the corresponding nitronates and hydroxy nitronates by liver microsomes from rats, mice, and humans. Toxicol. Sci. 1999;47:144–150. doi: 10.1093/toxsci/47.2.144. [DOI] [PubMed] [Google Scholar]
- 196.Jousserandot A., Boucher J.L., Henry Y., Niklaus B., Clement B., Mansuy D. Microsomal cytochrome p450 dependent oxidation of N-hydroxyguanidines, amidoximes, and ketoximes: Mechanism of the oxidative cleavage of their C=N(OH) bond with formation of nitrogen oxides. Biochemistry. 1998;37:17179–17191. doi: 10.1021/bi981175c. [DOI] [PubMed] [Google Scholar]
- 197.Vetrovsky P., Boucher J.L., Schott C., Beranova P., Chalupsky K., Callizot N., Muller B., Entlicher G., Mansuy D., Stoclet J.C. Involvement of NO in the endothelium-independent relaxing effects of N-omega-hydroxy-L-arginine and other compounds bearing a C=NOH function in the rat aorta. J. Pharmacol. Exp. Ther. 2002;303:823–830. doi: 10.1124/jpet.102.038612. [DOI] [PubMed] [Google Scholar]
- 198.Chalupsky K.I.L., Nepveu F.I.G., Beranova P., Entlicher G., Stoclet J.C., Muller B. Relaxant effect of oxime derivatives in isolated rat aorta: Role of nitric oxide (NO) formation in smooth muscle. Biochem. Pharmacol. 2004;67:1203–1214. doi: 10.1016/j.bcp.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 199.Jaros F., Straka T., Dobesova Z., Pinterova M., Chalupsky K., Kunes J., Entlicher G., Zicha J. Vasorelaxant activity of some oxime derivatives. Eur. J. Pharmacol. 2007;575:122–126. doi: 10.1016/j.ejphar.2007.07.040. [DOI] [PubMed] [Google Scholar]
- 200.Hassan G.S., Hegazy G.H., Ibrahim N.M., Fahim S.H. New ibuprofen derivatives as H2S and NO donors as safer anti-inflammatory agents. Future Med. Chem. 2019;11:3029–3045. doi: 10.4155/fmc-2018-0467. [DOI] [PubMed] [Google Scholar]
- 201.Mauge L., Fotopoulou T., Deemasure S., Dutartre P., Koufaki M., Connat J.L. In vitro inflammatory/anti-inflammatory effects of nitrate esters of purines. Eur. J. Pharmacol. 2014;730:148–156. doi: 10.1016/j.ejphar.2014.02.022. [DOI] [PubMed] [Google Scholar]
- 202.Kashfi K., Ryann Y., Qiao L.L., Williams J.L., Chen J., Del Soldato P., Traganos F., Rigas B. Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: Evidence of a tissue type-independent effect. J. Pharmacol. Exp. Ther. 2002;303:1273–1282. doi: 10.1124/jpet.102.042754. [DOI] [PubMed] [Google Scholar]
- 203.Pauwels B., Boydens C., Brouckaert P., Van de Voorde J. Oximes induce erection and are resistant to oxidative stress. J. Sex. Med. 2015;12:906–915. doi: 10.1111/jsm.12846. [DOI] [PubMed] [Google Scholar]
- 204.Pauwels B., Boydens C., Decaluwe K., Van de Voorde J. NO-donating oximes relax corpora cavernosa through mechanisms other than those involved in arterial relaxation. J. Sex. Med. 2014;11:1664–1674. doi: 10.1111/jsm.12564. [DOI] [PubMed] [Google Scholar]
- 205.Rehse K., Bade S., Harsdorf A., Clement B. New NO-donors with antithrombotic and vasodilating activities, Part 17—Arylazomidoximes and 3-arylazo-1,2,4-oxadiazol-5-ones. Arch. Pharm. 1997;330:392–398. doi: 10.1002/ardp.19973301207. [DOI] [PubMed] [Google Scholar]
- 206.Shahid M., Martorana M.G., Cottney J.E., Marshall R.J. Pharmacological and biochemical effects of the cardiotonic agent ORG10325 in isolated cardiac and vascular tissue preparations. Br. J. Pharmacol. 1990;100:735–742. doi: 10.1111/j.1476-5381.1990.tb14084.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Rehse K., Brehme F. New NO donors with antithrombotic and vasodilating activities, Part 27: Azide oximes and 1-hydroxytetrazoles. Arch. Pharm. 2000;333:157–161. doi: 10.1002/1521-4184(20006)333:6<157::AID-ARDP157>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 208.Dantas B.P.V., Ribeiro T.P., Assis V.L., Furtado F.F., Assis K.S., Alves J.S., Silva T.M.S., Camara C.A., Franca-Silva M.S., Veras R.C., et al. Vasorelaxation induced by a new naphthoquinone-oxime is mediated by NO-SGC-CGMP pathway. Molecules. 2014;19:9773–9785. doi: 10.3390/molecules19079773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Oresmaa L., Kotikoski H., Haukka M., Oksala O., Pohjala E., Vapaatalo H., Moilanen E., Vainiotalo P., Aulaskari P. Synthesis, ocular effects, and nitric oxide donation of imidazole amidoximes. Eur. J. Med. Chem. 2006;41:1073–1079. doi: 10.1016/j.ejmech.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 210.Abuo-Rahma G.E.D.A.A., Abdel-Aziz M., Beshr E.A.M., Ali T.F.S. 1,2,4-triazole/oxime hybrids as new strategy for nitric oxide donors: Synthesis, anti-inflammatory, ulceroginicity and antiproliferative activities. Eur. J. Med. Chem. 2014;71:185–198. doi: 10.1016/j.ejmech.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 211.Gaboriaud-Kolar N., Vougogiannopoulou K., Skaltsounis A.L. Indirubin derivatives: A patent review (2010-present) Expert Opin. Ther. Pat. 2015;25:583–593. doi: 10.1517/13543776.2015.1019865. [DOI] [PubMed] [Google Scholar]
- 212.Tchoumtchoua J., Halabalaki M., Gikas E., Tsarbopoulos A., Fotaki N., Liu L., Nam S., Jove R., Skaltsounis L.A. Preliminary pharmacokinetic study of the anticancer 6BIO in mice using an UHPLC-MS/MS approach. J. Pharm. Biomed. Anal. 2019;164:317–325. doi: 10.1016/j.jpba.2018.10.039. [DOI] [PubMed] [Google Scholar]
- 213.Lorke D.E., Kalasz H., Petroianu G.A., Tekes K. Entry of oximes into the brain: A review. Curr. Med. Chem. 2008;15:743–753. doi: 10.2174/092986708783955563. [DOI] [PubMed] [Google Scholar]
- 214.Kobrlova T., Korabecny J., Soukup O. Current approaches to enhancing oxime reactivator delivery into the brain. Toxicology. 2019;423:75–83. doi: 10.1016/j.tox.2019.05.006. [DOI] [PubMed] [Google Scholar]
- 215.Choi S.K., Thomas T.P., Leroueil P., Kotlyar A., Van Der Spek A.F., Baker J.R., Jr. Specific and cooperative interactions between oximes and pamam dendrimers as demonstrated by 1H NMR study. J. Phys. Chem. B. 2012;116:10387–10397. doi: 10.1021/jp305867v. [DOI] [PubMed] [Google Scholar]
- 216.Baell J.B. Screening-based translation of public research encounters painful problems. ACS Med. Chem. Lett. 2015;6:229–234. doi: 10.1021/acsmedchemlett.5b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Dahlin J.L., Walters M.A. How to triage PAINS-full research. Assay Drug Dev. Technol. 2016;14:168–174. doi: 10.1089/adt.2015.674. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support our specific findings in this review are available from the authors upon reasonable request.