

Abstract
Advances in synthetic biology, nanotechnology, and genetic engineering are allowing parallel advances in areas such as drug delivery and rapid diagnostics. Although our current visions of nanobots may be far off, a generation of nanobots synthesized by engineering viruses is approaching. Such tools can be used to solve complex problems where current methods do not meet current demands. Assuring safe drinking water is crucial for minimizing the spread of waterborne illnesses. Although extremely low levels of fecal contamination in drinking water are sufficient to cause a public health risk, it remains challenging to rapidly detect Escherichia coli, the standard fecal indicator organism. Current methods sensitive enough to meet regulatory standards suffer from either prohibitively long incubation times or requirement of expensive, impractical equipment. Bacteriophages, tuned by billions of years of evolution to bind viable bacteria and readily engineered to produce custom proteins, are uniquely suited to bacterial detection. We have developed a biosensor platform based on magnetized phages encoding luminescent reporter enzymes. This system utilizes bio-orthogonally functionalized phages to enable site-specific conjugation to magnetic nanoparticles. The resulting phage-based nanobots, when combined with standard, portable field equipment, allow for detection of <10 cfu/100 mL of viable E. coli within 7 h, faster than any methods published to date.
Keywords: bacteriophages, pathogen detection, water safety, magnetic nanoparticles, nanobot, nanoprobe
Graphical Abstract

INTRODUCTION
The concept of nanobots has long intrigued scientists and engineers. Self-replicating nanoscale robots which can be used to repair, detect, deliver, or destroy bacteria, cancer cells, toxins, or the like would be a technological revolution in science and medicine. Although the popular depictions of mechanical nanobots are still in the distant future, scientists have proposed many versions of nanobots which can perform tasks such as deliver drugs,1,2 detect cancer,3 and move within cells.4 The nanobots to date have typically been biological in nature, such as DNA structures, or inorganic such as nanocoils. The next generation of nanobots is possibly a hybrid of organic and inorganic components which can be put together to perform custom functions. These technologies can be used to provide tools for real-world problems for which the current technologies are limited.
The United Nations has declared safe drinking water a fundamental human right,5 yet fecal contamination affects the primary drinking water sources of over 2 billion people worldwide6 and the associated pathogenic bacteria caused 502,000 deaths in 2012 alone.7 Regulations mandate zero-detectable colony-forming units (cfu) of generic Escherichia coli (an indicator of fecal contamination8) in 100 mL of drinking water.9 Because fecal contamination may be undetectable by taste or smell, illnesses often spread via inadvertent consumption of contaminated water. Indeed, the World Health Organization estimates that assuring drinking water safety can reduce deaths from waterborne fecal bacterial pathogens by up to 73%.7 Diagnostic assays giving observable signal for low levels (<10 cfu/100 mL) of E. coli are therefore crucial in limiting the public health impact of contaminated drinking water.
Standard methods detect E. coli by culturing on selective media until visible colonies form (generally 24–48 h).10,11 Although extremely sensitive [limit of detection (LOD) < 10 cfu/100 mL], the required multiday incubation time translates to a slow response time and thus additional illnesses. This prolonged time to detection is due to the rate-limiting step of colony formation. More recently developed biosensors promise faster detection12–18 by shifting signal away from colony formation but often require equipment unsuitable for the low-resource settings where demand for these tests is the highest.7 Bacteriophage-based detection offers an unparalleled combination of sensitivity and speed,19–28 leveraging the specificity of bacteriophages (“phages”, bacteria-specific viruses) to their target bacteria.19–21,23,24,26,27,29–31 Phage replication, which takes place on host cell machinery, involves precise regulation of viral gene expression levels. Phage-based detection takes advantage of their remarkable biorecognition and directed protein expression properties to create a unique platform for rapid, sensitive diagnostic assays.20–22,27,30,32–35
Introducing genes with an observable product (e.g., fluorescent proteins) into the phage genome21,22,27,32,33,35 creates a reporter phage that gives a detectable signal during infection. Reporter phage-based assays have been reported to detect 1 cfu/100 mL in 10 h.19 The limiting step in reporter gene expression is phage infection, although this can take <30 min. Therefore, increasing the likelihood of infection by engineering concentration steps into reporter phage assays can reduce detection time further.
Immobilizing phages to solid supports (filters, nanoparticles, etc.) creates bacterial affinity materials that capture cells from solution for detection in a concentrated volume.23,28,29 Phage orientation is crucial in these systems, although challenging to control. Although phage capsids and tails have similar chemistry, only phages conjugated via their capsids retain infectivity. Conjugation techniques reliant on native phage chemistry cannot distinguish between capsids and tails, limiting system potency. Although capsid biotinylation orients phages on avidin-decorated surfaces,17,22,28 the required reagents are cost-prohibitive for water testing in low-resource areas. Phages represent an ideal nanobot scaffold because of their ability to bind specific bacteria, replicate, and be genetically engineered.32,36,37 The remarkable tunability of phage properties allows for precise control over the structure and function.
In this paper, we present a novel water test integrating sensitive reporter phage technology with low-cost, site-specific immobilization. We use amber suppression and directed protein self-assembly to alkynylate a luminescent reporter phage38 that covalently conjugates to azide-decorated magnetic nanoparticles (MNPs)39 to afford a phage-based bioaffinity nanobot that, in combination with two concentration steps, detects <10 cfu E. coli in a 100 mL sample within 7 h. By incorporating unnatural amino acids with unique conjugation abilities on phages, additional functionalities can be incorporated by conjugating enzymes or other moieties in the future.
RESULTS AND DISCUSSION
Assay Design.
E. coli detection in drinking water requires a highly sensitive detection platform to meet the 1 cfu/100 mL regulatory requirement, and to be practical for use in the low-resource areas most affected by poor water safety, an inexpensive, portable apparatus must be accomplished. The assay outlined in Figure 1 is engineered to achieve these disparate goals by employing a robust luminescent reporter phage [NRGp17, a variant of T4 expressing the reporter enzyme nanoluciferase40 fused to a cellulose-binding module27 under the native small outer capsid protein (SOC) promoter30 and multiple concentration steps with equipment already common in field testing. Luminescent intensity is inversely proportional to the surface area, so we maximized sensitivity by concentrating the signal from the entire 100 mL into a single 0.05 cm2 filter. To achieve this concentration using a standard, portable field equipment, we employ two separate steps: (1) magnetic separation of target bacteria from solution and (2) vacuum filtration to immobilize the reporter enzyme fusion [Nluc cellulose-binding module (CBM)] in the cellulose filter. Although we quantified luminescence in the lab using a spectrophotometer, previous research has demonstrated that luminescent signals can be readily captured using portable methods such as long-exposure photography using either a digital camera or smartphone.19,38 Similarly, vacuum filtration is the current standard for portable water testing in developing countries.
Figure 1.
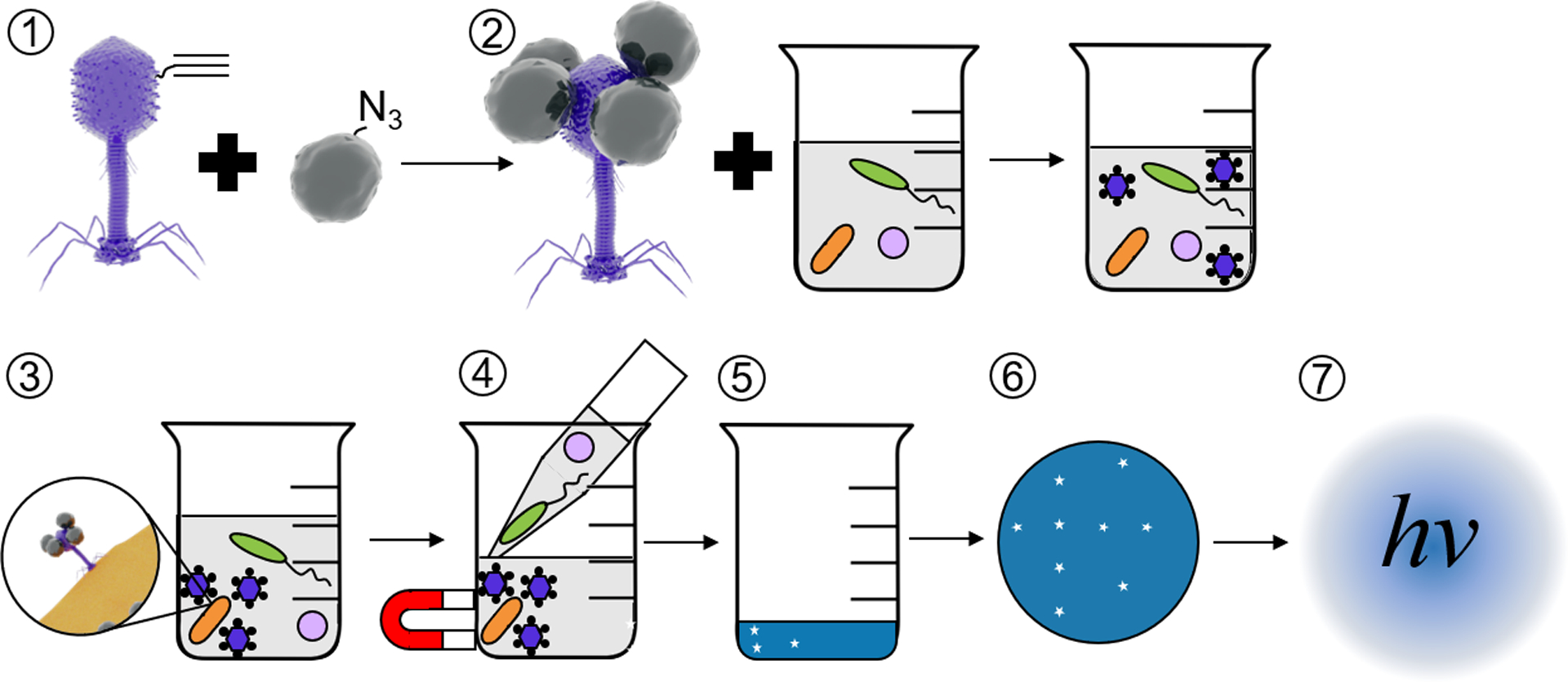
Schematic of E. coli detection assay. Self-assembled alkynylated T4 Nluc CBM phages are conjugated to azide-coated MNPs (1) using click chemistry. The resulting magnetic phage-based bioaffinity nanobot is added to samples of environmental water (2), where the phages bind specifically to E. coli (orange rods, 3). Beads, along with phages and E. coli, are collected with a magnet and excess volume is aspirated (4). Infection cycle completes (5), releasing Nluc CBM into solution. Samples are filtered (6) on cellulose to immobilize Nluc CBM and luminescence is measured (7) to detect E. coli. Please note that the schematics in this figure are not to scale. T4 Nluc CBM is approximately 200 nm long and 900 nm in diameter. MNPs are approximately 15 nm in diameter (Table S9).
Click T4 Nluc CBM Design.
The described assay uses luminescent bacteriophages conjugated to superparamagnetic MNPs as the primary detection and concentration agent (Figure 1). To avoid nonspecific interactions that minimize potency, we used Huisgen cycloaddition (click chemistry)39 to form covalent bonds between alkynylated phages and azide-decorated MNPs. We functionalized the modified T4 phage NRGp17 with alkyne groups (Click NRGp17) by developing an alkynylated variant of the SOC. This 9 kDa nonessential T4 protein self-assembles in a honeycomb pattern to form a homomeric 870-unit cage on the capsid41–43 (Figure 2a). Previous research has shown that recombinant SOC can self-assemble on phage scaffolds with its native SOC deleted.41,42 Phage display experiments have shown that SOC tolerates modification at both the N and C termini,43 and structural studies demonstrate that both termini are exposed to solution in the fully assembled cage.42 Because NRGp17 is a SOC knockout, we therefore hypothesized that by incubating it with recombinant SOC alkynylated at the N-terminus, we could build phages that display up to 870 alkyne groups on their capsids (Figure 2b).
Figure 2.
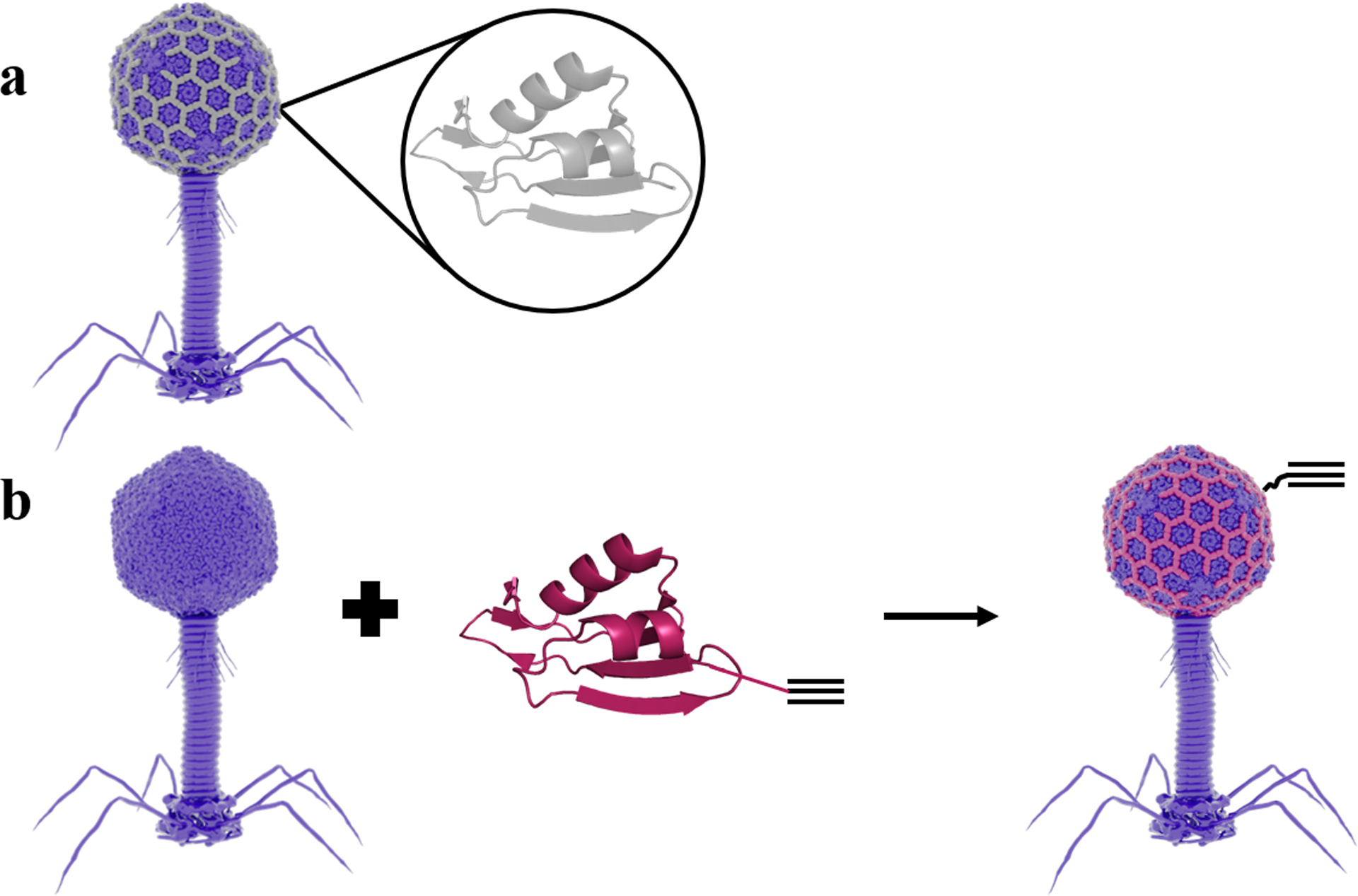
T4 phage capsid alkyne decoration strategy and click SOC construct design. (a) Schematic of WT SOC (gray, PDB ID 3IGE) assembly on the T4 capsid (PDB ID 5VF3). (b) SOC with an alkyne-containing unnatural amino acid is incorporated into the phage capsid to allow for click chemistry conjugation. Each phage capsid can accommodate 870 SOC proteins. Click SOC self-assembled on T4 Nluc CBM (Δsoc). Click SOC is shown in red with an alkyne schematic.
Click SOC Design, Expression, and Analysis.
Because no native amino acids contain alkyne functionality, we used the pEvol system44 to engineer click SOC, an SOC derivative containing propargylglyoxy phenylalanine (prp), an alkynyl derivative of tyrosine, at its N-terminus in addition to a C-terminal 6× polyhistidine tag for purification (Figure S1a,b). For the protein scaffold, we chose SOC from RB69, a phage closely related to T4. This protein is homologous to T4 SOC but expresses with higher yield in recombinant systems.42 Previous research has shown that RB69 SOC has high affinity for the T4 capsid as the native T4 SOC.42 After recombinantly expressing click SOC in BL21 (DE3) E. coli and purifying using immobilized metal affinity chromatography (Figure S2), we verified alkyne insertion by reacting the protein with 3-azido-7-hydroxycoumarin, a molecule that becomes fluorescent upon undergoing click cycloaddition45 (Figure 3a). Click SOC was significantly more fluorescent in this experiment than WT SOC [649 ± 2.2 relative fluorescence units (rfu) vs 329 ± 34.5 rfu, p < 0.05], indicating the presence of free alkyne groups. WT SOC was not significantly more fluorescent than a protein-free control, indicating that the alkyne group on click SOC is specifically reacting with the azido fluorophore.
Figure 3.
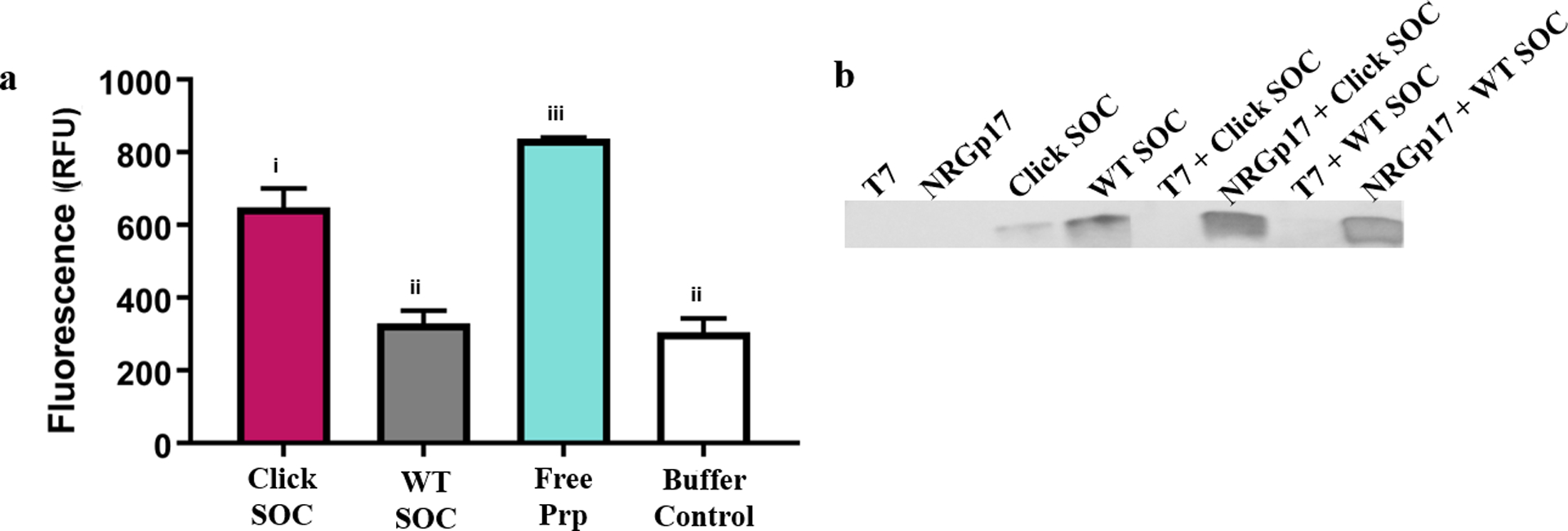
Click SOC has alkyne functionality and retains self-assembly properties. (a) Fluorescence of 3-azido-7-hydroxycoumarin after the click reaction. Click SOC has significantly higher fluorescence than WT SOC or the buffer control, indicating the successful introduction of available alkyne groups. Error bars indicate standard deviation of four technical replicates. Letters indicate significance by one-way ANOVA and Tukey’s multiple comparison test (p < 0.05). (b) Western blot against the C-terminal 6× His tag on WT SOC and click SOC before and after self-assembly on phages confirms click SOC has the same T4 self-assembly properties as WT SOC. T7 was used as a negative control for self-assembly. The raw data used to construct this figure are reported in Table S3.
Directed Self-Assembly of Click SOC on NRGp17.
The alkynylation of NRGp17 relies on previously demonstrated self-assembly of SOC on the phage capsid. To show that click SOC has these same self-assembly properties, we incubated the purified protein with NRGp17, which was engineered as a soc deletion mutant. After centrifugation to pellet the phages, we performed a western blot against the C-terminal 6× polyhistidine tag (Figure 3b). Both WT SOC and click SOC pelleted along with T4, showing self-assembly. Importantly, this pulldown behavior was not observed in samples of WT SOC or click SOC incubated with T7, another icosahedral phage distantly related to T4, indicating that assembly is specific to the T4 capsid and not merely nonspecific adsorption to protein surfaces.
Synthesis of Phage-Based Bioaffinity Nanobots.
Having shown that click SOC is both alkynylated and able to self-assemble onto the T4 capsid, we next incorporated the magnetic component of the nanobot using the click reaction39 to conjugate click NRGp17 to azide-decorated MNPs. After the reaction was complete, we magnetically captured the resulting phage-based bioaffinity nanobots and washed the resulting pellet with excess buffer. We found that upon resuspension, the nanobots had an infectivity of 5.10 ± 0.17 log plaques per mL of suspension (pfu/mL), while the final washes were only infective at 3.42 ± 0.10 log pfu/mL (Figure 4a). This significant difference in infectivity indicates oriented, immobilized phages, critical for the bacterial capture step of the assay in Figure 1. Furthermore, WT SOC gave a significantly lower yield of immobilized phages than click SOC (4.00 ± 0.13 log pfu/mL) (Figure 4b). This click SOC-dependent activity retention demonstrates the stronger conjugation and site specificity of the click conjugation as compared to any potential random adsorption of the WT SOC.
Figure 4.
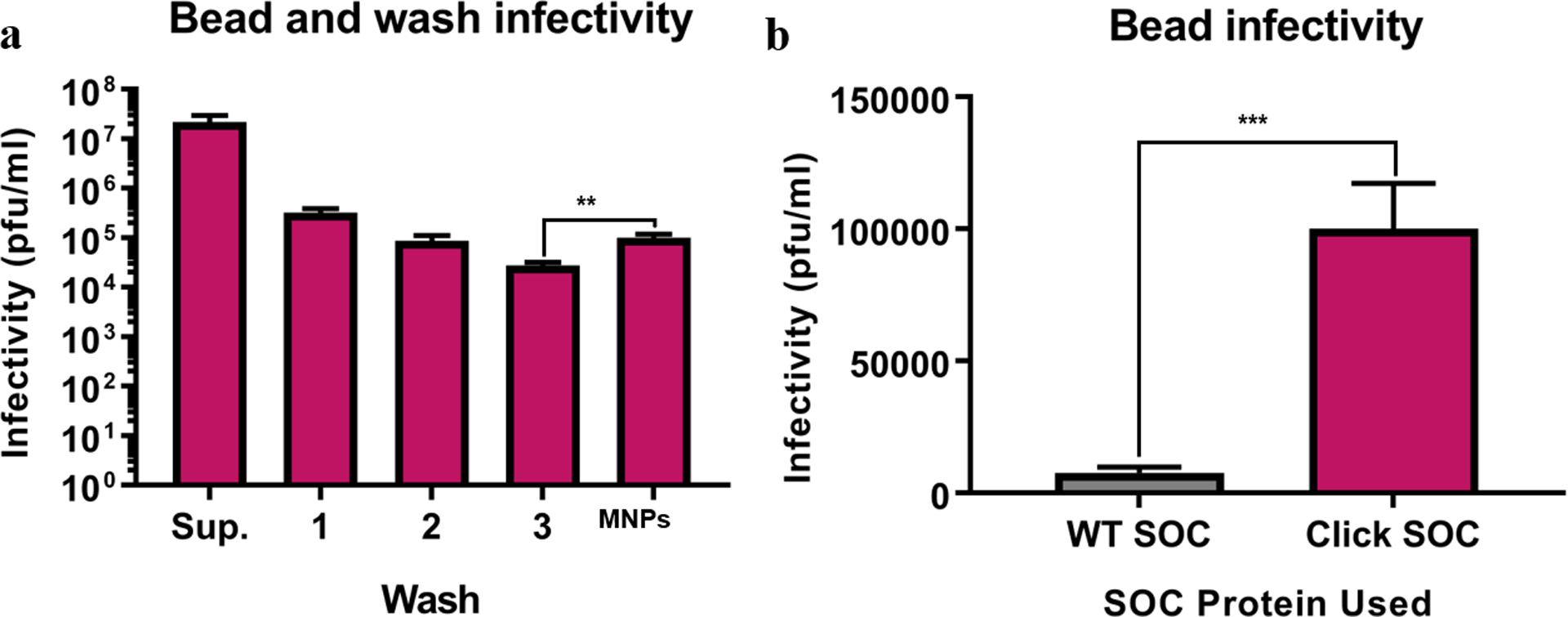
Immobilized phages retain infective properties after magnetic capture. (a) Resuspended nanobots are significantly more infective than the final wash, indicating that phages have been magnetically captured and resuspended during the washing process. (b) Infectivity of MNPs conjugated to NRGP17 by either WT SOC or click SOC. Click SOC conjugation results in significantly higher infectivity, indicating a stronger conjugation between the phage and the bead. Error bars indicate the standard deviation of three technical replicates. Asterisks indicate significance (** = p < 0.01, *** = p < 0.001) by unpaired two-tailed t-test. The raw data used to construct this figure are reported in Table S4.
Click Conjugation of Phages to MNPs Creates Sensitive Nanobots.
Although the plaque assays showed that click NRGp17 nanobots have significantly more infectivity than their WT SOC counterparts (Figure 4b), real-world challenge studies are necessary to demonstrate their high sensitivity and potential in a true assay setting. Therefore, we set up small (100 μL) samples containing 3.64 ± 0.32 log cfu/mL of E. coli (ECOR#13), an environmental E. coli isolate previously used in detection studies. The four detection reagents we tested (MNPs on their own, free NRGp17, WT NRGp17 nanobots, and click NRGp17 nanobots) gave luminescent signals of 3.5 ± 2.4, 74 ± 59, 75 ± 31, and 170 ± 100 relative luminescence units (rlu), respectively (Figure 5a). Importantly, only click NRGp17 nanobots were sensitive enough to give luminescence above the baseline sterile water signal (Figure 5a). These data, in combination with the plaque infectivity data, suggest that every component (phages, MNPs, and click SOC) is crucial to the sensitivity, supporting the scheme in Figure 1.
Figure 5.
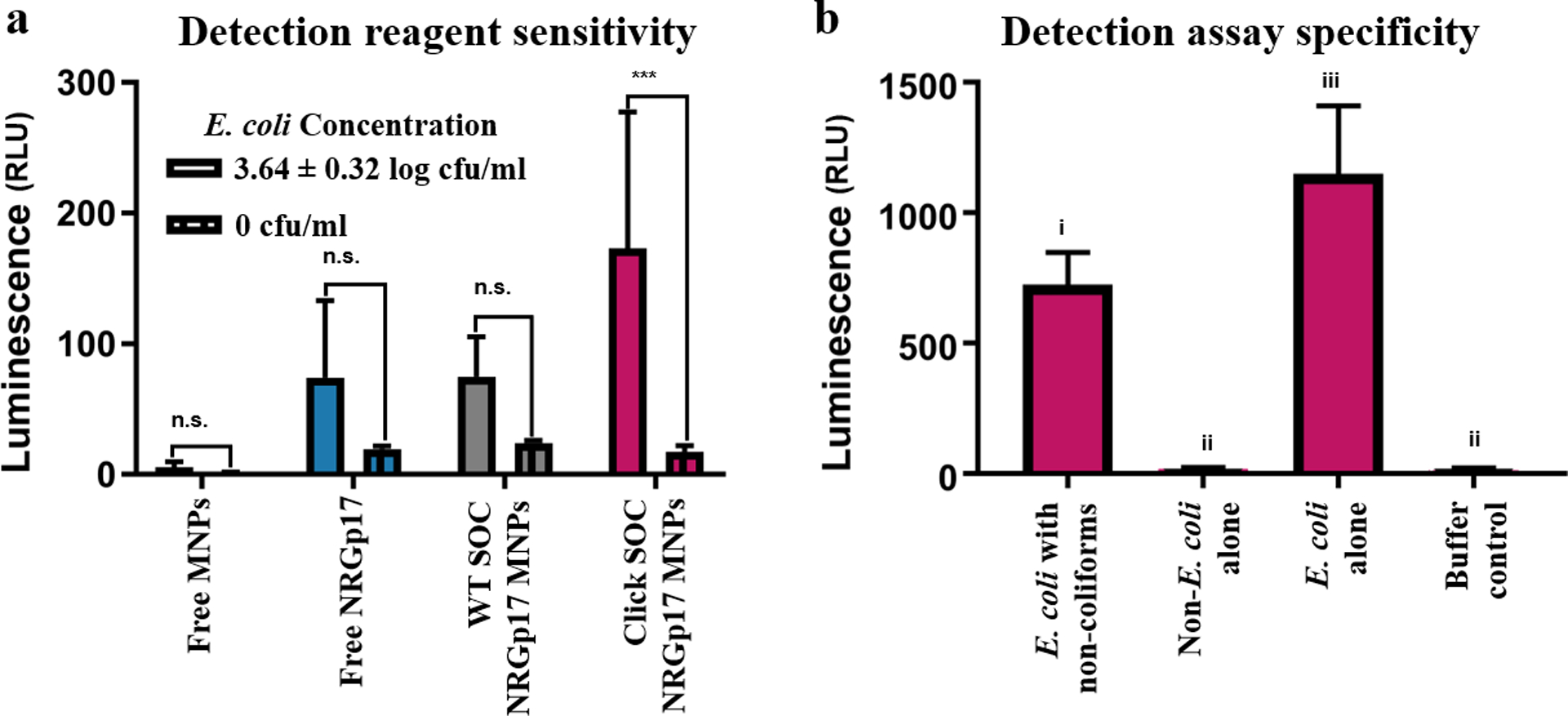
MNPs with click-immobilized NRGp17 are a specific, sensitive detection tool. (a) Small-scale E. coli (ECOR#13) detection with no preenrichment. 100 μL samples were treated with 5 log mL−1 unconjugated azide-coated MNPs, 5 log pfu/mL free NRGp17, NRGp17 conjugated to MNPs via WT SOC, or click NRGp17 conjugated to MNPs. Only the latter gives a signal significantly higher than the baseline luminescence. (b) Small-scale detection of 4.01 ± 0.15 log cfu/mL E. coli with no pre-enrichment in pure and mixed culture. Non-coliform bacteria included 3.27 ± 0.33 log cfu/mL P. aeruginosa, 3.82 ± 0.22 log cfu/mL L. monocytogenes, and 3.23 ± 0.07 log cfu/mL S. alaskensis. Error bars indicate the standard deviation of four technical replicates. Bacterial concentration reported as mean ± standard deviation of 12 plate counts. Letters and stars indicate significance (p < 0.05; n.s. = p > 0.05; *** = p < 0.001) by one-way ANOVA and Tukey’s multiple comparison test. Raw data used to construct this figure are reported in Tables S5 and S6.
Phage-Based Nanobots are Specific to E. coli.
To show that the click NRGp17 nanobots are specific to E. coli, we prepared a cocktail containing 3–4 log cfu/mL of each of three non-E. coli bacterial strains (Pseudomonas aeruginosa, Listeria monocytogenes, and Sphingopyxis alaskensis), all water-isolated or otherwise common in fresh water. The click NRGp17 nanobots gave negligible (19 ± 4.5 rlu against a background of 19 ± 2.9 rlu) luminescent signal for these nontarget strains, demonstrating their specificity to E. coli (Figure 5b). Furthermore, the nanobots gave a significantly more luminescent signal (724 ± 124 rlu) for E. coli in mixed culture, demonstrating the high specificity of this assay to its target organism, with little interference by nontarget environmental strains.
Magnetic Nanobots Detect <10 cfu E. coli in 100 mL within 7 h.
Drinking water regulations require testing 100 mL of sample46 for E. coli, so we scaled up the detection platform to demonstrate performance at this volume. We prepared sterile tap water samples in 100 mL collection vials similar to those used in the field and inoculated them with known concentrations of E. coli (ECOR#13). To maximize the sensitivity of the assay, we pre-enriched the samples by adding concentrated media and incubating for 3 h to allow injured bacteria to recover and allow all bacteria to approach the log growth phase. Because maximizing the magnetic surface area minimizes collection time, we taped bar magnets to 10 cm2 supports in the pattern shown in Figure S1c. These magnetic rigs have the same footprint as 10 cm2 sterile Petri dishes, which hold ~100 mL, thus making them ideal magnetic incubation vessels. We incubated the water samples for 10 min with the click NRGp17 nanobots, captured in square Petri dishes on the magnetic rigs for 10 min longer, and concentrated the samples 200-fold by resuspending captured bacteria in 500 μL of LB media. The capture media was spiked with additional NRGp17 to ensure that all cells present were included in the luminescent signal. After allowing the infections to progress during a further 3 h incubation, we vacuum-filtered the samples through single wells of a 384-well filter plate, which immobilized the Nluc CBM expressed by the phages into a 0.05 cm2 area for detection using the NanoGlo kit. This assay gave a luminescent signal of 29 ± 3 rlu for 7 ± 2 cfu of E. coli in 100 mL against a background luminescence of 7 ± 3 rlu. The entire assay, from collecting water samples to reading luminescence, required less than 7 h, which is less than 1/3 the time required for standard culture methods.11 Although it is impossible to reproducibly make samples containing exactly 1 cfu in 100 mL, we consistently detected <10 cfu in the same volume and the 16 rlu LOD (3 standard deviations above the baseline luminescence) was well below the 29 rlu observed luminescence for 7 ± 2 cfu, the lowest inoculated samples tested (Figure 6).
Figure 6.
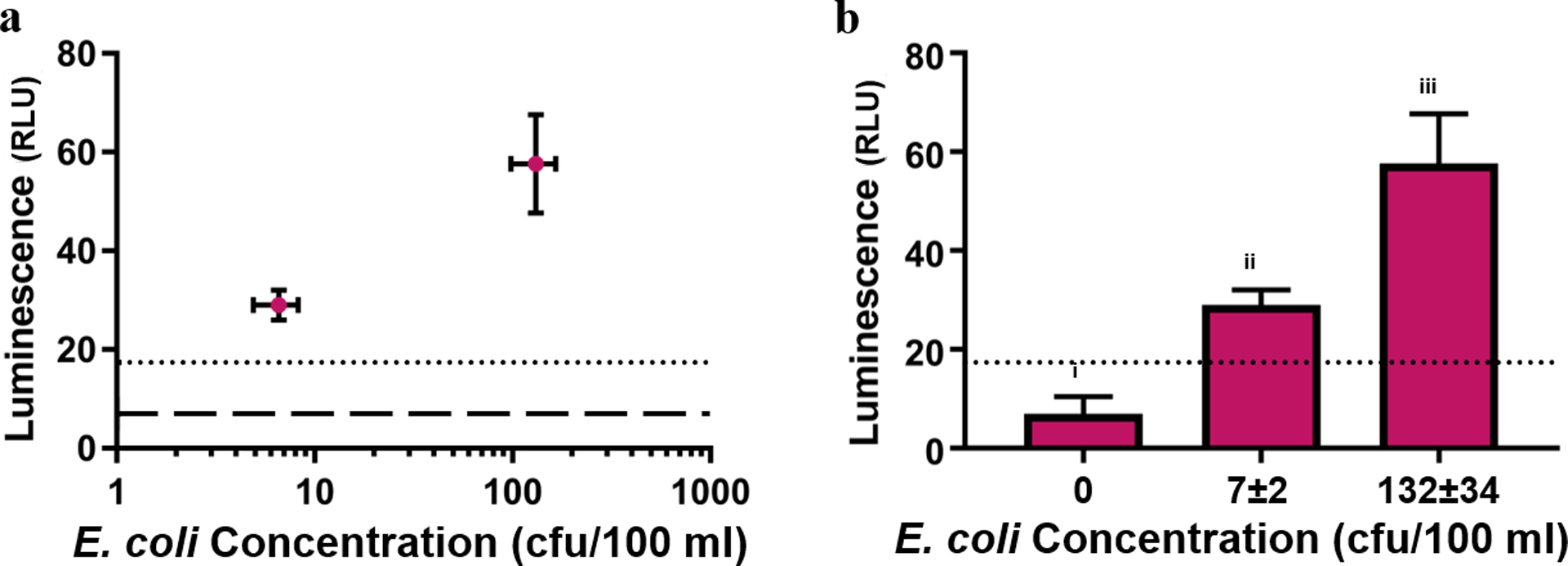
Phage-based bioaffinity nanobots can detect <10 cfu E. coli in 100 mL of tap water within 7 h. Luminescence error bars indicate the standard deviation of three technical replicates. Dotted lines indicate LOD (3 standard deviations above mean baseline luminescence). (a) E. coli (ECOR#13) concentration plotted against luminescence. Dashed line indicates baseline luminescence. Concentration error bars indicate the standard deviation of 12 plate counts. (b) Luminescence values obtained from different ECOR#13 concentrations. Letters indicate significance (p < 0.05) by one-way ANOVA and Tukey’s multiple comparison test. Bacterial concentration reported as mean ± standard deviation of 12 plate counts. Raw data used to construct this figure are reported in Table S7.
CONCLUSIONS
Testing drinking water for the presence of E. coli is an essential step in mitigating the spread of waterborne illnesses in the developing world. To comply with international and national regulatory standards, water tests must be able to detect 1 cfu of generic E. coli in 100 mL of drinking water.46 We developed an assay for E. coli contamination that detects <10 cfu in 100 mL of drinking water within 7 h, more than threefold faster than the standard EPA method11 and 30% faster than previously published phage-based detection methods,19 and with demonstrated specificity in a cocktail of nontarget organisms. It remains unknown if the assay can detect a single cfu given the practical limitations of preparing a sample containing a single cfu in 100 mL of water. This assay uses a combination of standard, portable field equipment (magnets, vacuum filtration, and long-exposure photography) in combination with novel phage-based magnetic nanobots. Using amber suppression and directed self-assembly, we created a luminescent reporter bacteriophage with alkyne functionality. By conjugating this alkynylated phage to MNPs via the click reaction, we achieved spatial control of phage orientation on a solid surface without the need for expensive reagents. The resulting phage-based magnetic nanobots are capable of binding bacteria, separating them from solution via magnetic capture and detecting them via luminescence. By engineering a reagent with this level of polyfunctionality, we are able to achieve high sensitivity through multiple concentration steps.
Phage functionalization expands the potential utility of phages in bioengineering. Amber suppression technology is widely used for real-time visualization of biological processes, development of biocompatible materials, and spatial resolution in living systems.47 The technique described in this study: production of recombinant phage-display proteins site-specifically tagged with artificial amino acids followed by directed self-assembly is widely applicable beyond just the alkyne-tagged SOC reported. Given the range of artificial amino acids previously described in the literature,47 it is possible to use this technique to build phages with a wide range of functionality (e.g., photosensitivity, fluorescence, incorporation in polymers, etc.). Although amber suppression has been previously used in phage display,48 the method we used offers over 100-fold higher density of the artificial amino acid per capsid and allows for functionalization without the need for genetically modifying a potentially complex phage genome. Phages are widely used in biotechnology research and are an emerging antimicrobial and biocontrol agent,49 so the potential impact of facile introduction of novel functionality extends far beyond the water testing presented in this study. Although this paper demonstrates the ability to detect low concentrations of E. coli (ECOR#13) from a large volume sample, the selection of different phage cocktails can allow the detection of numerous other bacterial indicators of pathogens.
MATERIALS AND METHODS
Materials.
All reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted. pEvol-pPrpRS,44 encoding propargylglyoxy prp RNA synthetase under an araBAD promoter, was a gift from Peter Schultz at the Scripps Research Institute (San Diego, CA, USA) and used unaltered. Sterile coliform test bottles were purchased from Corning Inc. (Corning, NY, USA). Multiwell filter plates (0.2 μm pore-size, nitrocellulose membranes) were purchased from Pall (Cortland, NY, USA). 4–20% gradient sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE) gels and nitrocellulose western blot membranes were purchased from BioRad (Hercules, CA, USA). Custom DNA oligonucleotides were synthesized by Integrated DNA Technologies (IDT, Coralville, IA, USA).
Bacterial and Phage Strains and Growth Conditions.
All strains are described in Table S1. Unless otherwise noted, E. coli strains were grown aerobically at 37 °C with shaking in an LB medium (solid or liquid as needed) supplemented with antibiotics (100 μg/mL ampicillin and/or 50 μg/mL chloramphenicol) as necessary. Non-coliform bacteria were grown aerobically at 32 °C with shaking in tryptic soy medium (solid or liquid as needed). Bacterial strains were stored as glycerol stocks at −80 °C and streaked out on solid media as needed. All phages were stored at 4 °C until needed.
Cloning.
Expression plasmids pSOC6H and pClickSOC6H were constructed from the pET 11a backbone (Invitrogen, Burlington, MA, USA) using HiFi DNA assembly (NEB, Ipswich, MA, USA). RB69 SOC was amplified from genomic DNA and modified to include a C-terminal 6×-His tag and an N-terminal amber codon using Q5 PCR master mix (NEB) and the primers in Table S2 (designed with the NEBuilder tool). Plasmids were amplified in E. cloni 10G Electro-competent Cells (Lucigen, Middleton, WI, USA), purified with the QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA, USA) and validated by Sanger DNA sequencing.
Wild-Type SOC Expression and Purification.
pSOC6H was transformed into BL21(DE3) E. coli for expression. Cultures were grown to an OD600 of 0.5 in 2xYT medium supplemented with 10 μg/mL ampicillin and induced with 400 μM isopropyl-β-D-1 thiogalactopyranoside (IPTG, Thermo Fisher Scientific, Waltham, MA, USA) for 15–16 h at 30 °C. Cells were pelleted by centrifugation (3260g, 15 min), flash-frozen in an ethanol/dry ice bath, and lysed by incubating with 5 mL of B-PER complete (Thermo Fisher, Waltham, MA, USA) per gram of wet cell pellet for 15 min. Lysates were diluted 10-fold in wash buffer [500 mM NaCl, 25 mM imidazole ([pH 8.0), and 50 mM sodium phosphate (pH 8.0)]. After clarifying the lysate by centrifugation (3260g, 1 h), the supernatant was batch-bound for 1 h to 500 μL HisPur Cobalt Resin (Thermo Fisher) equilibrated in 1 mL of wash buffer. Resin was collected by centrifugation (700g, 2 min) and washed six times in excess wash buffer, with resin pelleted after each wash. Protein was eluted in 2 mL of elution buffer (wash buffer with 150 mM imidazole) and dialyzed against excess dialysis buffer [150 mM NaCl and 50 mM sodium phosphate (pH 8.0)] for 3 h, after which the buffer was changed and dialysis continued for 18 h more. The resulting purified protein was analyzed by SDS-PAGE, quantified using bicinchoninic acid assay (Thermo Fisher Scientific, Waltham, MA, USA), and stored at 4 °C until further use.
Click SOC Expression.
pClickSOC6H and pEvol pPrpRS were cotransformed into BL21(DE3) E. coli for expression. Cultures were grown from single colonies in ampicillin- and chloramphenicol-supplemented minimal glycerol medium as previously described50 in a fed-batch fermentor (BioFlo 2000, New Brunswick Scientific, Edison, NJ, USA). When initial carbon was exhausted (indicated by reduced agitation speed), glycerol feeding proceeded exponentially for 18 h with a fixed growth rate of 0.1 h−1 calculated as previously described.51 After 14 h feeding, 1 mM of the artificial amino acid prp (Chem-Impex International, Wood Dale, IL, USA) was added to the culture. Expression of prp tRNA synthetase was induced by addition of 0.02% L-(+)-arabinose after 16 h of feeding. Expression of click SOC was induced by addition of 1 mM IPTG and 0.2% yeast extract after 18 h of feeding. After IPTG induction, expression continued for 21 h at 30 °C with pH-stat feeding as previously described.51 Cells were pelleted by centrifugation (4000g, 30 min), flash-frozen in an ethanol/dry ice bath, and stored at −80 °C until further use. For purification, pellets were thawed overnight at 4 °C and mixed with 3 mL of lysis buffer [50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 25 mM imidazole (pH 8.0), 25% sucrose, 0.5 mg/mL lysozyme, 40 μg/mL DNAse I, and 0.5% IGEPAL] for 30 min at room temperature. Lysates were clarified by centrifugation (3260g, 1 h) and purification proceeded as described above.
Fluorescent Cycloaddition Assays.
Approximately, 90 ng of SOC was mixed to a final volume of 200 μL with the following: 37.5 mM NaCl, 12.5 mM sodium phosphate (pH 8.0), 6 M guanidinium, 250 μM CuSO4, 200 μM 3-azido-7-hydroxycoumarin (Marker Gene, Eugene, OR, USA), 250 μM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine, and 1 mg of copper wire as a reducing agent. SOC was replaced with 50 nM prp for a positive control. The cycloaddition reaction proceeded at 37 °C for 4 h. Fluorescence (ex: 404 nm/em: 477 nm) was measured with a spectrophotometer (BioTek, Winooski, VT, USA).
Phage Propagation.
Phage particles were added to logarithmically growing (OD600 ≈ 0.5) DH5α E. coli with a 0.1 multiplicity of infection. Infection proceeded at 37 °C with shaking (150 rpm) for 4 h, at which point there was visible clearance of the culture. Cell debris was pelleted by centrifugation (3260g, 15 min) and the resulting clarified lysates were incubated at room temperature for 16–18 h. Phages were pelleted by centrifugation (30,000g, 3 h), resuspended in 15 mL of sterile phosphate-buffered saline (PBS), and enumerated using standard plaque-counting techniques.
Directed Self-Assembly of SOC on Phages and Western Blots.
Approximately, 200 ng of SOC was mixed with 10 log pfu of phages in 1 mL of assembly buffer [75 mM NaCl, 50 mM sodium phosphate (pH 7.0), and 1 mM MgSO4] and incubated for 90 min at 4 °C. Phages were pelleted by centrifugation at 16,000g for 40 min. Pellets were washed twice in excess buffer, centrifuging under the same conditions after each wash, and then resuspended to a final volume of 16 μL. Reducing SDS-PAGE sample buffer (BioRad) was added to a final concentration of 1× and samples were boiled for 5 min before being run on 4–20% gradient SDS-PAGE gels. Protein was transferred to nitrocellulose membranes with a semidry process and blocked in 5% nonfat dry milk powder. SOC was detected by alkaline phosphatase-conjugated antibody against the 6× His tag (Invitrogen).
Synthesis of Phage-Based Bioaffinity Nanobots.
Azide-coated MNPs (TurboBeads, Zurich, SUI) were stored in PBS and protected from light. To make 1 mL of click-immobilized phages, 9 log pfu of NRGp17 was mixed with 1010 MNPs, SOC (20 ng Click-SOC or 2 μg WT SOC), 1 mM CuSO4, and 1 mg of copper wire in 1 mL of assembly buffer supplemented with 0.1% Tween 20. Reactions were incubated 16–18 h at 4 °C, after which beads were collected by incubation against a magnet for 15 min. Although still on the magnet, beads were washed three times in excess buffer and resuspended to a final volume of 1 mL in fresh buffer. The resulting nanobots were stored at 4 °C.
Small-Scale Detection of E. coli.
Single colonies of E. coli (ECOR#13) were grown for 16–18 h in 10 mL of the total culture volume. Serial dilutions were performed in sterile PBS and cultures were enumerated using standard plate-counting techniques. Aliquots (100 μL) of 10−6 (3–4 log cfu/mL) and 10−8 (1–2 log cfu/mL) dilutions were placed in separate tubes and spiked with 2 μL of phage-decorated beads. A total of 100 μL of sterile PBS was used as a 0 cfu control. Other controls included 2 μL of free NRGp1738 at 5 log pfu/mL, 2 μL of unconjugated MNPs at 105 mL−1, and 2 μL of phage-decorated MNPs prepared as mentioned above but with click SOC replaced with additional assembly buffer. Samples were statically incubated for 15 min to allow phages to bind to bacteria and then moved to a magnetic rack and incubated for 15 min more to collect bacteria. The supernatant was aspirated and beads were resuspended in 100 μL of LB medium containing 7 log pfu/mL NRGp17 to ensure lysis of all captured cells. Resuspended samples were incubated with shaking for 6 h at 37 °C and then vacuumed through single wells of a 384-well filter plate. A total of 50 μL of the freshly prepared NanoGlo luciferase assay substrate (Promega, Madison, WI, USA) was added to each well and luminescence was read on the BioTek spectrophotometer.
Large-Scale Detection of E. coli.
Starter culture and dilutions were prepared as described above. Samples (100 mL) of autoclaved tap water were prepared in disposable coliform test bottles (Corning Inc.) and spiked with either 20 μL of the 10−6 E. coli (ECOR#13) dilution (1.3–2.3 log cfu), or 100 μL of the 10−8 E. coli dilution (0–1 log cfu), or else left sterile. A total of 5 mL of 20× LB broth was added to each sample and bottles were incubated with shaking at 37 °C for 3 h. A total of 2 mL of phage-decorated beads, prepared as described above, was added to each bottle followed by a 10 min incubation. Samples were poured into square Petri dishes to maximize the surface area and incubated against a custom-built magnetic rig (bar magnets adhered to 10 cm2 supports) for 10 min. Supernatants were aspirated and discarded, dishes were removed from magnets, and beads were resuspended in 500 μL of LB broth containing 107 pfu/mL NRGp17. Resuspended beads were incubated with shaking for 3 h at 37 °C followed by filtration and detection as described above.
Competitive Detection of E. coli.
Single colonies of non-coliform, water-associated bacteria (L. monocytogenes, P. aeruginosa, and S. alaskensis) were grown in 10 mL of liquid culture for 24 h. Serial dilutions were performed in sterile PBS and cultures were enumerated using standard plate-counting techniques. E. coli (ECOR#13) dilutions were prepared as described above. Cocktail cultures were prepared by combining ~3 log cfu/mL of each non-coliform strain. Detection samples included non-coliform cocktail spiked with E. coli, E. coli with no competitors, cocktail with no added E. coli, and sterile PBS. 100 μL samples were processed as described above for small-scale detection samples.
Magnetic Nanoparticle Size Determination.
MNPs in aqueous suspension at a concentration of ~109 mL−1 were analyzed using a Zetasizer (Malvern Pananalytical, Malvern, UK). Dynamic light-scattering data are shown in Table S9.
Statistics and Data Processing.
All quantitative assays were performed with 3–4 technical replicates and at least 1 biological replicate. Not all biological replicates included technical replicates; these were not included in the statistical analyses and are reported with the raw data in Tables S3–S8. Pair-wise comparisons were done using two-tailed, unpaired t-tests. Small-scale E. coli (ECOR#13) tests, including competitor assays, were analyzed in JMP Pro 14.0.0 (Cary, NC, USA) using a standard least-squares model. Large-scale E. coli (ECOR#13) detection and fluorescent cycloaddition were analyzed in GraphPad Prism (La Jolla, CA, USA) using standard one-way ANOVA. Significance was determined with a cutoff of p < 0.05.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation (award #1705797). Research reported in this article was also supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under award number R01EB027895. DNA sequencing was performed by Biotechnology Resource Center genomics facility at Cornell University. The authors thank Peter G. Schultz of the Scripps Research Institute for providing the vector pEvol pPrpRS. We also thank Carl Batt and Mariely Medina Rivera for allowing us to use the BioFlo 2000 bioreactor and providing technical support. Finally, we thank the members of both the Goddard and Nugen research groups for their insight on this project.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsabm.0c00546.
Phage and bacteria strains used in this study and their sources; all primers used in this study, their sequences, and their purposes; raw data displayed in Figure 3a; raw data displayed in Figure 4; raw data displayed in Figure 5a; raw data displayed in Figure 5b; raw data displayed in Figure 6; additional biological replicates of large-scale detection; overview of the genetic system used to construct click SOC; and SOC purification data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Daljit Singh JK; Luu MT; Abbas A; Wickham SFJ Switchable DNA-Origami Nanostructures That Respond to Their Environment and Their Applications. Biophys. Rev 2018, 10, 1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fu J; Yan H Controlled Drug Release by a Nanorobot. Nat. Biotechnol 2012, 30, 407–408. [DOI] [PubMed] [Google Scholar]
- (3).Maheswari R; Sheeba Rani S; Gomathy V; Sharmila P Cancer Detecting Nanobot Using Positron Emission Tomography. Procedia Comput. Sci 2018, 133, 315–322. [Google Scholar]
- (4).Ogawa K; Uesugi K; Morishima K On-Chip Internalization Process of an Intracellular Nanobot into a Single Cell. In Proceedings of the IEEE International Conference on Micro Electro Mechanical Systems (MEMS), 2017; pp 581–584. [Google Scholar]
- (5).United Nations. 64/292. The Human Right to Water and Sanitation, 2010; Vol. 64, p 3. [Google Scholar]
- (6).World Health Organisation. Drinking-water fact sheet https://www.who.int/news-room/fact-sheets/detail/drinking-water (accessed Dec 12, 2019).
- (7).World Health Organisation. Preventing Diarrhoea through Better Water; Sanitation and Hygiene: Geneva, 2014. [Google Scholar]
- (8).Edberg SC; Rice EW; Karlin RJ; Allen MJ Escherichia coli: The Best Biological Drinking Water Indicator for Public Health Protection. J. Appl. Microbiol. Symp. Suppl 2000, 88, 106S–116S. [DOI] [PubMed] [Google Scholar]
- (9).United States Environmental Protection Agency. Revised Total Coliform Rule. Code of Federal Regulations, 2013; Vol. 78 (30), p 10269. [Google Scholar]
- (10).Edberg SC; Allen MJ; Smith DB Rapid, Specific, Defined Substrate Technology for the Simultaneous Detection of Total Coliforms and Escherichia coli. Toxic. Assess 1988, 3, 565–580. [Google Scholar]
- (11).United States Environmental Protection Agency. Method 1603: Escherichia coli (E. coli) in Water by Membrane Filtration Using Modified Membrane-Thermotolerant Escherichia coli Agar (Modified MTEC); United States Environmental Protection Agency, 2002, No. EPA-821-R-R-09–007. [Google Scholar]
- (12).Bej AK; DiCesare JL; Haff L; Atlas RM Detection of Escherichia coli and Shigella Spp. in Water by Using the Polymerase Chain Reaction and Gene Probes for Uid. Appl. Environ. Microbiol 1991, 57, 1013–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fratamico PM; Strobaugh TP; Medina MB; Gehring AG Detection of Escherichia coli O157:H7 Using a Surface Plasmon Resonance Biosensor. Biotechnol. Tech 1998, 12, 571–576. [Google Scholar]
- (14).Hammes F; Broger T; Weilenmann H-U; Vital M; Helbing J; Bosshart U; Huber P; Peter Odermatt R; Sonnleitner B Development and Laboratory-Scale Testing of a Fully Automated Online Flow Cytometer for Drinking Water Analysis. Cytometry, Part A 2012, 81, 508–516. [DOI] [PubMed] [Google Scholar]
- (15).Van Poucke SO; Nelis HJA 210-min solid phase cytometry test for the enumeration of Escherichia coli in drinking water. J. Appl. Microbiol 2000, 89, 390–396. [DOI] [PubMed] [Google Scholar]
- (16).Temur E; Boyacı IH; Tamer U; Unsal H; Aydogan N A Highly Sensitive Detection Platform Based on Surface-Enhanced Raman Scattering for Escherichia coli Enumeration. Anal. Bioanal. Chem 2010, 397, 1595–1604. [DOI] [PubMed] [Google Scholar]
- (17).Lee J; Deininger RA Detection of E. coli in Beach Water within 1 Hour Using Immunomagnetic Separation and ATP Bioluminescence. Luminescence 2004, 19, 31–36. [DOI] [PubMed] [Google Scholar]
- (18).Vikesland PJ Nanosensors for Water Quality Monitoring. Nat. Nanotechnol 2018, 13, 651–660. [DOI] [PubMed] [Google Scholar]
- (19).Hinkley TC; Singh S; Garing S; Le Ny A-LM; Nichols KP; Peters JE; Talbert JN; Nugen SR A Phage-Based Assay for the Rapid, Quantitative, and Single CFU Visualization of E. coli (ECOR #13) in Drinking Water. Sci. Rep 2018, 8, 14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Kim J; Kim M; Kim S; Ryu S Sensitive Detection of Viable Escherichia coli O157:H7 from Foods Using a Luciferase-Reporter Phage PhiV10lux. Int. J. Food Microbiol 2017, 254, 11–17. [DOI] [PubMed] [Google Scholar]
- (21).Born Y; Fieseler L; Thöny V; Leimer N; Duffy B; Loessner MJ Engineering of Bacteriophages Y2::DpoL1-C and Y2::LuxAB for Efficient Control and Rapid Detection of the Fire Blight Pathogen, Erwinia Amylovora. Appl. Environ. Microbiol 2017, 83, No. e00341–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Singh S; Hinkley T; Nugen SR; Talbert JN Colorimetric Detection of Escherichia coli Using Engineered Bacteriophage and an Affinity Reporter System. Anal. Bioanal. Chem 2019, 411, 7273–7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen J; Alcaine SD; Jiang Z; Rotello VM; Nugen SR Detection of Escherichia coli in Drinking Water Using T7 Bacteriophage-Conjugated Magnetic Probe. Anal. Chem 2015, 87, 8977–8984. [DOI] [PubMed] [Google Scholar]
- (24).Alcaine SD; Pacitto D; Sela DA; Nugen SR Phage & Phosphatase: A Novel Phage-Based Probe for Rapid, Multi-Platform Detection of Bacteria. Analyst 2015, 140, 7629–7636. [DOI] [PubMed] [Google Scholar]
- (25).Alcaine SD; Tilton L; Serrano MAC; Wang M; Vachet RW; Nugen SR Phage-Protease-Peptide: A Novel Trifecta Enabling Multiplex Detection of Viable Bacterial Pathogens. Appl. Microbiol. Biotechnol 2015, 99, 8177–8185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wang D; Hinkley T; Chen J; Talbert JN; Nugen SR Phage Based Electrochemical Detection of: Escherichia coli in Drinking Water Using Affinity Reporter Probes. Analyst 2019, 144, 1345–1352. [DOI] [PubMed] [Google Scholar]
- (27).Zhang D; Coronel-Aguilera CP; Romero PL; Perry L; Minocha U; Rosenfield C; Gehring AG; Paoli GC; Bhunia AK; Applegate B The Use of a Novel NanoLuc-Based Reporter Phage for the Detection of Escherichia coli O157:H7. Sci. Rep 2016, 6, 33235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Wang Z; Wang D; Chen J; Sela DA; Nugen SR Development of a Novel Bacteriophage Based Biomagnetic Separation Method as an Aid for Sensitive Detection of Viable Escherichia coli. Analyst 2016, 141, 1009–1016. [DOI] [PubMed] [Google Scholar]
- (29).Minikh O; Tolba M; Brovko LY; Griffiths MW Bacteriophage-Based Biosorbents Coupled with Bioluminescent ATP Assay for Rapid Concentration and Detection of Escherichia coli. J. Microbiol. Methods 2010, 82, 177–183. [DOI] [PubMed] [Google Scholar]
- (30).Vinay M; Franche N; Grégori G; Fantino J-R; Pouillot F; Ansaldi M Phage-Based Fluorescent Biosensor Prototypes to Specifically Detect Enteric Bacteria Such as E. coli and Salmonella enterica Typhimurium. PLoS One 2015, 10, No. e0131466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Burnham S; Hu J; Anany H; Brovko L; Deiss F; Derda R; Griffiths MW Towards Rapid On-Site Phage-Mediated Detection of Generic Escherichia coli in Water Using Luminescent and Visual Readout. Anal. Bioanal. Chem 2014, 406, 5685–5693. [DOI] [PubMed] [Google Scholar]
- (32).Pulkkinen EM; Hinkley TC; Nugen SR Utilizing in Vitro DNA Assembly to Engineer a Synthetic T7 Nanoluc Reporter Phage for Escherichia coli Detection. Integr. Biol 2019, 11, 63–68. [DOI] [PubMed] [Google Scholar]
- (33).Hinkley TC; Garing S; Singh S; Le Ny A-LM; Nichols KP; Peters JE; Talbert JN; Nugen SR Reporter Bacteriophage T7NLC Utilizes a Novel NanoLuc::CBM Fusion for the Ultra-sensitive Detection of: Escherichia coli in Water. Analyst 2018, 143, 4074–4082. [DOI] [PubMed] [Google Scholar]
- (34).Pires DP; Cleto S; Sillankorva S; Azeredo J; Lu TK Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev 2016, 80, 523–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jackson AA; Hinkley TC; Talbert JN; Nugen SR; Sela DA Genetic Optimization of a Bacteriophage-Delivered Alkaline Phosphatase Reporter to Detect: Escherichia coli. Analyst 2016, 141, 5543–5548. [DOI] [PubMed] [Google Scholar]
- (36).Ando H; Lemire S; Pires DP; Lu TK Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tao P; Wu X; Tang W-C; Zhu J; Rao V Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol 2017, 6, 1952–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Duong M; Carmody C; Nugen S Report of T4 Nluc-CBM.
- (39).Rostovtsev VV; Green LG; Fokin VV; Sharpless KB A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem., Int. Ed 2002, 41, 2596–2599. [DOI] [PubMed] [Google Scholar]
- (40).Hall MP; Unch J; Binkowski BF; Valley MP; Butler BL; Wood MG; Otto P; Zimmerman K; Vidugiris G; MacHleidt T; Robers MB; Benink HA; Eggers CT; Slater MR; Meisenheimer PL; Klaubert DH; Fan F; Encell LP; Wood KV Engineered Luciferase Reporter from a Deep Sea Shrimp Utilizing a Novel Imidazopyrazinone Substrate. ACS Chem. Biol 2012, 7, 1848–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li Q; Shivachandra SB; Zhang Z; Rao VB Assembly of the Small Outer Capsid Protein, Soc, on Bacteriophage T4: A Novel System for High Density Display of Multiple Large Anthrax Toxins and Foreign Proteins on Phage Capsid. J. Mol. Biol 2007, 370, 1006–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Qin L; Fokine A; O’Donnell E; Rao VB; Rossmann MG Structure of the Small Outer Capsid Protein, Soc: A Clamp for Stabilizing Capsids of T4-like Phages. J. Mol. Biol 2010, 395, 728–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ren Z.-j.; Black LW Phage T4 SOC and HOC Display of Biologically Active, Full-Length Proteins on the Viral Capsid. Gene 1998, 215, 439–444. [DOI] [PubMed] [Google Scholar]
- (44).Young TS; Ahmad I; Yin JA; Schultz PG An Enhanced System for Unnatural Amino Acid Mutagenesis in E. coli. J. Mol. Biol 2010, 395, 361–374. [DOI] [PubMed] [Google Scholar]
- (45).Zhou Z; Fahrni CJ A Fluorogenic Probe for the Copper(I)-Catalyzed Azide-Alkyne Ligation Reaction: Modulation of the Fluorescence Emission via 3(n, Π*)-1(π,Π*) Inversion. J. Am. Chem. Soc 2004, 126, 8862–8863. [DOI] [PubMed] [Google Scholar]
- (46).World Health Organisation. Guidelines for Drinking-Water Quality, 4th ed. Incorporating the First Addendum, 2014. [PubMed] [Google Scholar]
- (47).Liu CC; Schultz PG Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem 2010, 79, 413–444. [DOI] [PubMed] [Google Scholar]
- (48).Tian F; Tsao M-L; Schultz PG A Phage Display System with Unnatural Amino Acids. J. Am. Chem. Soc 2004, 126, 15962–15963. [DOI] [PubMed] [Google Scholar]
- (49).Ul Haq I; Chaudhry WN; Akhtar MN; Andleeb S; Qadri I Bacteriophages and Their Implications on Future Biotechnology: A Review. Virol. J 2012, 9, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Korz DJ; Rinas U; Hellmuth K; Sanders EA; Deckwer W-D Simple Fed-Batch Technique for High Cell Density Cultivation of Escherichia coli. J. Biotechnol 1995, 39, 59–65. [DOI] [PubMed] [Google Scholar]
- (51).Kim BS; Lee SC; Lee SY; Chang YK; Chang HN High Cell Density Fed-Batch Cultivation of Escherichia ColiEscherichia coli Using Exponential Feeding Combined with PH-Stat . Bioprocess Biosyst. Eng 2004, 26, 147–150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.