

Abstract
Small molecule mitochondrial uncouplers are emerging as a new class of molecules for the treatment of nonalcoholic steatohepatitis. We utilized BAM15, a potent protonophore that uncouples the mitochondria without depolarizing the plasma membrane, as a lead compound for structure-activity profiling. Using oxygen consumption rate as an assay for determining uncoupling activity, changes on the 5- and 6-position of the oxadiazolopyrazine core were introduced. Our studies suggest that unsymmetrical aniline derivatives bearing electron withdrawing groups are preferred compared to the symmetrical counterparts. In addition, alkyl substituents are not tolerated, and the N-H proton of the aniline ring is responsible for the protonophore activity. In particular, compound 10b had an EC50 value of 190 nM in L6 myoblast cells. In an in vivo model of NASH, 10b decreased liver triglyceride levels and showed improvement in fibrosis, inflammation, and plasma ALT. Taken together, our studies indicate that mitochondrial uncouplers have potential for the treatment of NASH.
Keywords: Oxidative phosphorylation, small molecule mitochondrial uncoupler, protonophore, BAM15, NASH
Graphical Abstract
Introduction
Oxidative phosphorylation is an essential aerobic process that couples nutrient oxidation in mitochondria to adenosine triphosphate (ATP) production. Mechanistically, nutrient oxidation in the mitochondrial matrix drives efflux of protons from the matrix into the mitochondrial intermembrane space creating potential energy in the form of a proton motive force (pmf).1 Protons then re-enter the matrix along their concentration gradient via ATP synthase to produce ATP and fuel cellular function and signaling pathways.2 Mitochondrial uncoupling refers to processes that uncouple nutrient oxidation from ATP production; an example is proton leak from the intermembrane space into the mitochondrial matrix independent of ATP synthase.3–4
Mitochondrial uncoupling occurs naturally in mammals as both basal and inducible proton leak, with estimates that approximately 20–30% of resting metabolic rate in rats is due to basal proton leak.5 Inducible proton leak is a process activated by cold exposure and other stresses that increase metabolic rate in part through activation of uncoupling proteins (UCPs).6–10 Mammals have five known UCPs, UCP1-5. Of the five UCPs, UCP1-3 are the most studied and better understood than either UCP4 or UCP5. UCP1 has long been established as a promoter of non-shivering thermogenesis11–12 while UCP2-3 are thought to play a protective role within the cell by helping to prevent oxidative damage from radical oxygen species.13–14
Mitochondrial uncoupling can also be induced with small molecule protonophores (Figure 1).4, 15–17 Chemical protonophores are typically lipophilic weak acids that transport protons from the mitochondrial intermembrane space into the mitochondrial matrix. Their potential for the treatment of various disease states has been explored, particularly for obesity and type 2 diabetes (T2D), due to their ability to increase nutrient metabolism and energy expenditure.6, 18–25 Multiple reports have shown that mice on a high fat diet supplemented with small molecule mitochondrial uncouplers gained less weight than controls and/or had decreased blood glucose and lipid levels with improved insulin sensitivity.19, 22–24, 26–29 Additionally, treatment with uncouplers slowed down or even prevented hepatic steatosis and nonalcoholic steatohepatitis (NASH) phenotypes in mice and rats fed high fat diet or methionine/choline deficient diet, highlighting their therapeutic benefit for fatty liver disease.22–24, 26–27, 29 Furthermore, protonophores decrease the generation of reactive oxygen species30–32 and recent studies explored their use as anticancer,33–36 anti-ageing,37–39 and antibacterial agents.40
Figure 1.
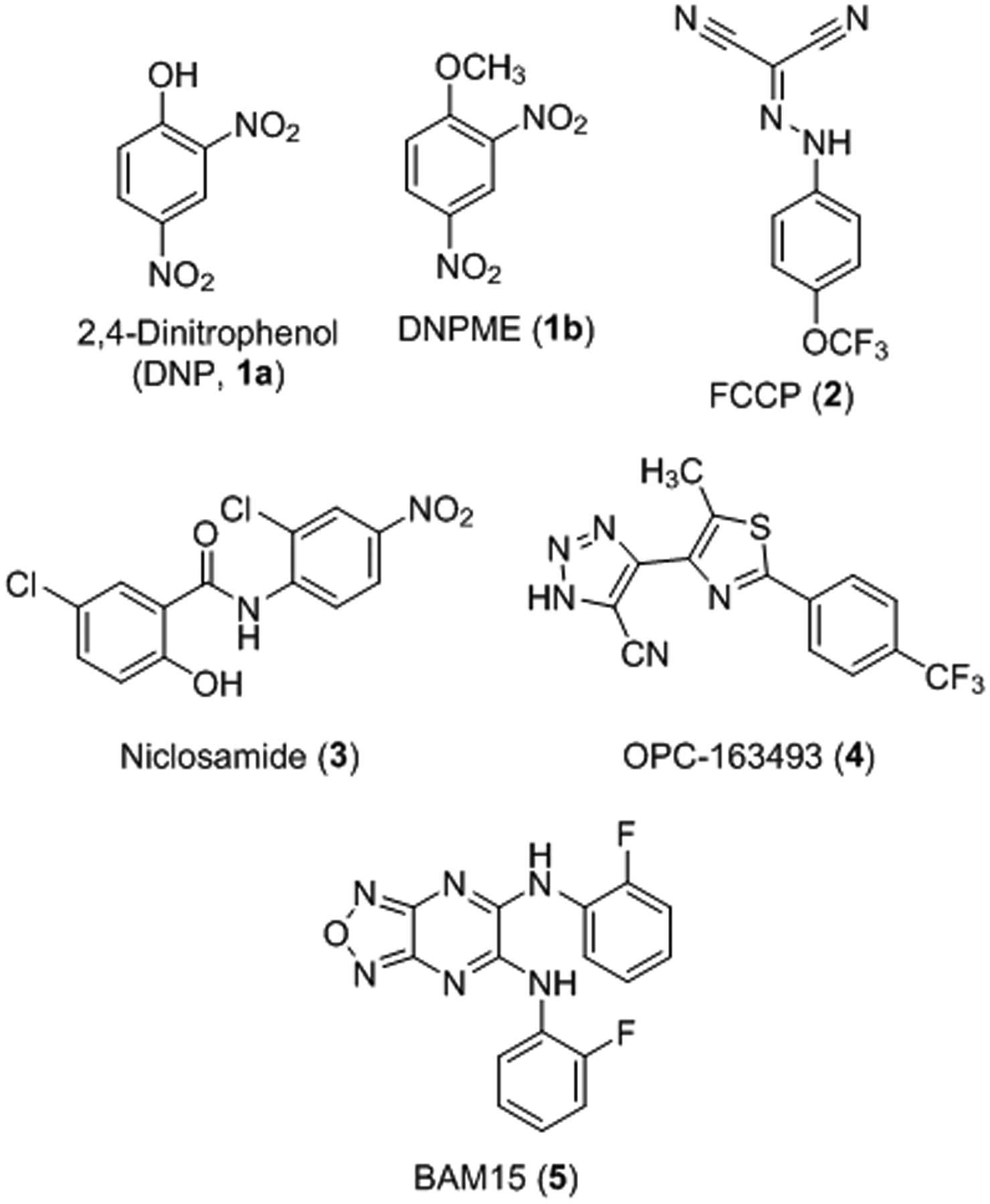
Chemical structures of select protonophore mitochondrial uncouplers.
The great promise of mitochondrial uncouplers in preclinical disease models has had limited translation to the clinic. The main challenge for preclinical development is finding uncouplers that are selective for the mitochondria without additional mechanisms of action. In fact, any lipophilic weak acid, within a certain lipophilicity and acidity window, has the potential to uncouple mitochondria, albeit unselectively.41 Some of the most well-studied examples of mitochondrial protonophore uncouplers were discussed in a recent review and are illustrated in Figure 1.17 2,4-Dinitrophenol (DNP, 1a) is the most pertinent example as it was found to have weight-loss-inducing effects in humans, promoting upwards of 50% increase in metabolism.42–45 However, DNP caused adverse effects in some patients including increased body temperature,43, 46–48 cataracts,49–52 and blindness,51 which led the U. S. Food and Drug Administration (FDA) to remove this drug from the market in 1938. Studies now indicate that the toxicity issues associated with DNP are due to a narrow therapeutic window, which may result from unwanted depolarization of non-mitochondrial membranes such as the plasma membrane.53–56 In an effort to control DNP toxicity, Perry et al. reported a controlled-release version that is orally bioavailable with an increased therapeutic range, reduced toxicity, improved insulin sensitivity in diabetic rats, and improvements in nonalcoholic fatty liver disease (NAFLD) phenotypes.24 Further, a prodrug version of DNP, DNPME (1b), was found to be non-toxic and liver-targeted while improving glucose tolerance and insulin sensitivity in diabetic rats.22 Despite these advancements, the active compound in both cases is DNP and, therefore, off-target actions may still occur. Another notable potent mitochondrial uncoupler is FCCP (2), which is limited by an extremely narrow therapeutic window and off-target effects at the plasma membrane.57–61 FCCP is commonly used as a control in biochemical assays to measure maximal mitochondrial capacity, but it has a narrow maximally effective concentration range. The well-known anthelmintic drug niclosamide (3) has uncoupling activity and is widely pursued for a number of diseases, most notably cancer.62–63 However, the niclosamide mechanism of action is complex and extends beyond uncoupling the mitochondria to other cancer targets including STAT3, Wnt, and PKA signaling. More recently, a liver- and kidney-targeted compound OPC-163493 (4) is reported to have efficacy in multiple animal models of type I and type II diabetes via insulin-independent antidiabetic effects, in addition to antisteatotic effects in fatty liver models.25 Finally, nitazoxanide is a FDA-approved anti-parasitic compound that inhibits pyruvate:ferredoxin oxidoreductase but was recently found to also have uncoupling activity64 and is currently in phase 2 clinical trials for NASH-associated fibrosis (ClinicalTrials.gov Identifier: NCT03656068).
Mitochondrial uncouplers have the potential for the treatment of metabolic diseases such as NAFLD, which affects 25% of the world’s population.65 NAFLD is a chronic liver disease that progresses to more severe conditions such as nonalcoholic steatohepatitis (NASH) and eventually liver cancer. NASH is associated with hepatocyte injury with ballooning, lobular inflammation, and often fibrosis. The number of people affected with NASH is unknown because biopsy is required for definitive diagnosis: however, NASH incidence is growing and is expected to be the major cause of liver transplantation.66 Unfortunately, there are no FDA-approved drugs for the treatment of NASH. A recent review details current strategies and ongoing clinical trials for the treatment of NASH.67 As an alternative to these drug discovery campaigns, mitochondrial uncouplers have a mechanism of action that increases energy expenditure and decreases reactive oxygen species formation to directly target the key underlying drivers of NASH pathophysiology.
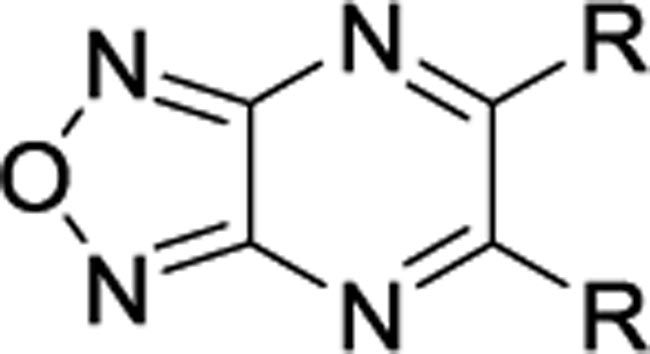
Recently, we reported the identification of a novel mitochondrial uncoupler, BAM15 (5), which selectively depolarizes the inner mitochondrial membrane but not the plasma membrane.68 Preliminary structure–activity relationship (SAR) studies revealed that the oxadiazole, pyrazine, and aniline moieties on 5 are all necessary for mitochondrial uncoupling activity.69 Replacing the aniline moiety with phenol caused a complete loss of activity, suggesting that the aniline N-H was the source of the acidic proton. While neutral anilines are typically basic moieties under physiological conditions, the aniline of BAM15 is acidic due to the strongly electron withdrawing effect of the [1,2,5]oxadiazolo[3,4-b]pyrazine core.70 In this study, we performed an SAR investigation to explore the electronic effects of substituents on the oxadiazolopyrazine ring of BAM15. In particular, compound 10b is efficacious in a streptozotocin (STZ)-induced mouse model of NASH demonstrating improvements in hepatocyte liver triglyceride content, inflammation, and fibrosis.
Results and Discussion
In this study, we focused our investigations on understanding the effect of the aniline moiety on the 5- and 6-position of the pyrazine ring. The synthesis of symmetrical and unsymmetrical derivatives is shown in Scheme 1. The requisite precursor 5,6-dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (7) was synthesized from the reaction of diaminofurazan 6 and oxalic acid in an aqueous 10% HCl solution, followed by chlorination with PCl5/POCl3.71–73 Intermediate 7 was then directly substituted with various alkyl or aryl amines in tetrahydrofuran (THF) to afford the desired symmetrical derivatives 8. To synthesize the unsymmetrical derivatives, compound 7 was reacted with an equivalent of 2-fluoroaniline and triethylamine in THF followed by the addition of a different amine to yield the unsymmetrical derivatives 10. Intermediate 9 may be isolated and purified after the first addition or both substitutions may occur sequentially in a one-pot reaction. Lastly, to confirm that BAM15 is a proton shuttle, the acidic N-Hs were replaced with a methyl groups on both anilines (8am) (Table 2).
Scheme 1.
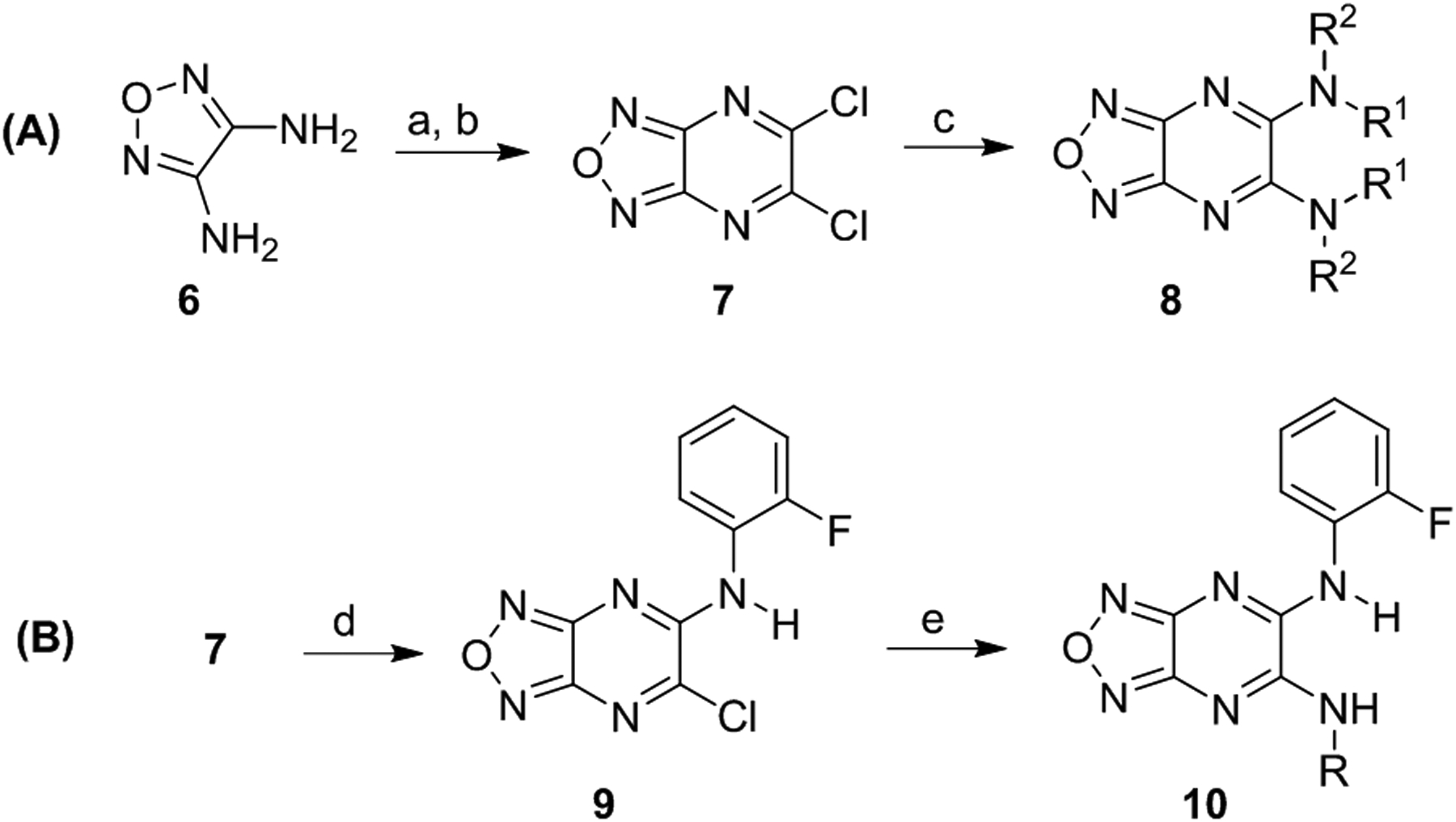
Synthesis of symmetrical (A) and unsymmetrical (B) derivatives. Reagents and conditions: (a) oxalic acid, 10% HCl, reflux, 3 h, quantitative; (b) PCl5, POCl3, reflux, 2 h, 30%; (c) aniline, THF, reflux, 19 h, 19 – 95%; (d) ortho-fluoroaniline, Et3N, THF, 0 °C, 0.25 to 1 h; (e) amine, Et3N, THF, 0 °C to rt, 19 h, 13 – 59%.
Table 2.
Oxygen consumption rates of alkyl and cycloalkyl derivatives in L6 rat myoblast cells.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | % of BAM15 OCRa | EC50, μM | Entry | R | % of BAM15 OCRa | EC50, μM |
| 8ah | 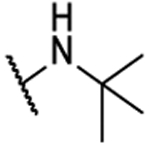 |
NA | -- | 8ak | 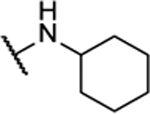 |
NA | -- |
| 8ai | 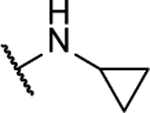 |
NA | -- | 8al | 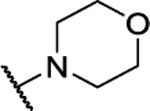 |
NA | -- |
| 8aj | 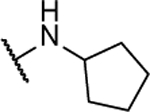 |
20%b | 45.0 ± 21.3 | 8am | 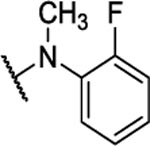 |
NA | -- |
Integrated area under OCR dose curve above baseline relative to that of BAM15 expressed as percent. NA = No Activity.
Highest tested concentration is 50 μM. BAM15 was tested up to 50 μM.
Three classes of oxadiazolopyrazine derivatives were synthesized: (1) symmetrical anilines, (2) unsymmetrical anilines, and (3) symmetrical and unsymmetrical alkyl amines. All compounds were tested for mitochondrial uncoupling activity as a function of oxygen consumption rate (OCR) in L6 rat myoblast cells using the Agilent Seahorse XF analyzer.74 Briefly, in this assay, L6 myoblasts were treated with increasing concentrations of the compounds over a 90 minute time period. As mitochondrial uncouplers promote proton leak, effective compounds increased OCR. The impact on cellular respiration in compound-treated cells was compared to BAM15 as a positive control, which induced a maximum increase in respiration. Since the increase in cellular respiration can be variable from one plate to another due to the confluency of the cells, each experiment contained a fresh batch of BAM15 as a positive control. To allow for direct comparison of the analogs across several experiments, analog activity is normalized to BAM15 control in each experiment and reported as the area under the curve relative to BAM15. Low relative activity may be due to poor uncoupling activity or due to treatment-induced crash of cellular respiration. The half maximal effective concentration (EC50) is measured based on the maximum OCR% of each individual compound. Both parameters are considered in conjunction when evaluating the analogs as a measure of potency and efficacy, and are reported in Tables 1–4. Using these criteria, good mitochondrial uncouplers will have OCR close to 100% of BAM15 activity and low EC50 values.
Table 1.
Oxygen consumption rates of symmetrical derivatives in L6 rat myoblast cells.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | % of BAM15 OCRa | EC50, μM | Entry | R | % of BAM15 OCRa | EC50, μM |
| BAM15 | 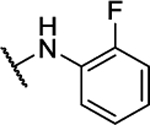 |
100%b | 0.49 ± 0.06 | 8q | 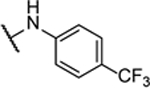 |
NA | -- |
| 8a | 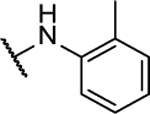 |
33%b | 2.3 ± 0.7 | 8r | 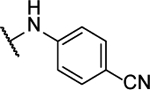 |
NA | -- |
| 8b | 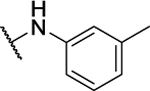 |
22%c | 2.1 ± 1.3 | 8s | 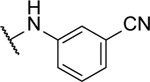 |
NA | -- |
| 8c | 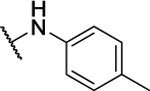 |
59%c | >20 | 8t | 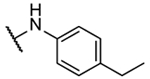 |
21%b | 3.9 ± 1.1 |
| 8d | 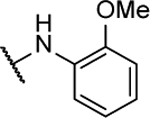 |
NA | -- | 8u | 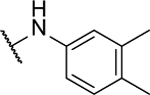 |
18%b | >100 |
| 8e | 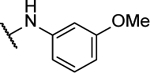 |
43%c | >100 | 8v | 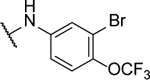 |
8%b | 2.2 ± 0.4 |
| 8f | 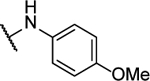 |
14%c | >100 | 8w | 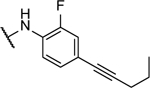 |
NA | -- |
| 8g | 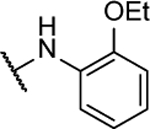 |
NA | -- | 8x | 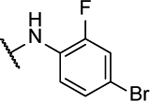 |
5%b | <3 |
| 8h | 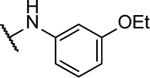 |
45%c | 10.1 ± 0.7 | 8y | 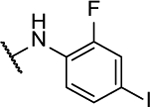 |
27%b | 8.8 ± 3.2 |
| 8i | 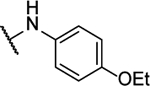 |
NA | -- | 8z | 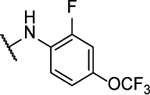 |
NA | -- |
| 8j | 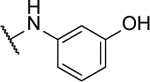 |
NA | -- | 8aa | 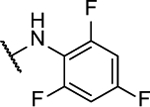 |
17%b | 1.1 ± 0.4 |
| 8k | 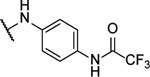 |
NA | -- | 8ab | 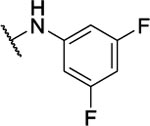 |
7%b | <1 μM |
| 8l | 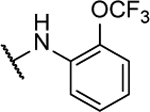 |
NA | -- | 8ac | 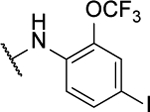 |
NA | -- |
| 8m | 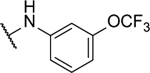 |
53%c | 1.1 ± 0.2 | 8ad | 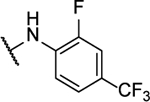 |
0.5%b | <1 μM |
| 8n | 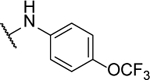 |
14%c | <5 | 8ae | 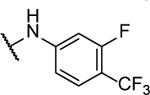 |
1%b | 1.7 ± 0.3 |
| 8o | 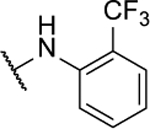 |
39%c | 1.0 ± 0.3 | 8af | 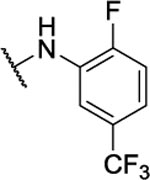 |
0.3%b | <1 μM |
| 8p | 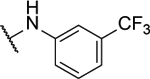 |
28%c | 0.7 ± 0.1 | 8ag | 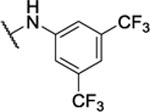 |
5%b | 6.1 ± 2.6 |
Integrated area under OCR dose curve above baseline relative to that of BAM15 expressed as percent. NA = No Activity.
Highest tested concentration is 200 μM. BAM15 was tested up to 200 μM.
Highest tested concentration is 50 μM. BAM15 was tested up to 50 μM.
Table 4.
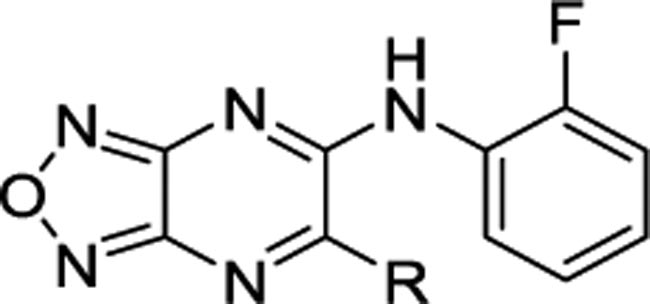
Oxygen consumption rates (OCR) of unsymmetrical derivatives in L6 rat myoblast cells.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | % of BAM15 OCRa | EC50, μM | Entry | R | % of BAM15 OCRa | EC50, μM |
| 10aa |  |
NA | -- | 10ad | 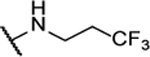 |
46%b | >100 |
| 10ab | 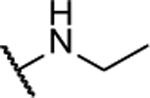 |
NA | -- | 10ae |  |
54%b | 13.4 ± 3.2 |
| 10ac | 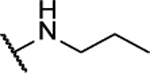 |
NA | -- | 10af | 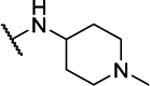 |
NA | -- |
Integrated area under OCR dose curve above baseline relative to that of BAM15 expressed as percent. NA = No Activity.
Highest tested concentration is 200 μM. BAM15 was tested up to 200 μM.
Starting with the symmetrical series (Tables 1–2), electron-donating groups such as alkyl (8a–8c; 8t–8u) and alkoxy (8d–8i) groups in various positions on the aniline ring were tolerated and resulted in a moderate increase in respiration over basal levels, up to 45% of BAM15 OCR; however, these compounds were significantly less potent than BAM15. Interestingly, polar groups such as hydroxy in 8j, trifluoroacetamide in 8k, and cyano in 8r–8s were not tolerated and showed no activity. The position of the substituent on the ring plays a role in the activity, but it is dependent on the substituent. For example, ortho-OCF3 in 8l was inactive, but meta- and para-OCF3 in 8m and 8n respectively showed low to moderate efficacy and good potency at 1 μM. In contrast, ortho- and meta-CF3 in 8o and 8p respectively were well tolerated, but para-CF3 in 8q was inactive. Combinations of two or more halogenated groups resulted in diminished efficacy and potency (8v, 8x-8ag) and an alkynyl substituent is not tolerated (8w). As shown in Table 2, replacement of the aniline with alkyl (8ah) and cycloalkyl (8ai-8al) groups resulted in complete loss of activity. Therefore, these types of structure were not pursued. In addition, the methylated version of BAM15, 8am, was inactive, supporting the notion that the N-H proton as the acidic proton.
Next, the importance of the symmetry was evaluated (Tables 3 and 4). For the unsymmetrical analogs, one ortho-fluoroaniline was maintained and the second aniline was substituted with various functional groups. In stark contrast to the symmetrical series, most of these analogs showed high efficacy (>50% of BAM15 OCR) and good potency (<5 μM). Interestingly, diversity among the aniline substituents was more tolerated in the unsymmetrical analogs than the symmetrical counterparts. Notable examples include the activity of the para-trifluoromethyl aniline in the unsymmetrical analog 10v with 45% OCR and EC50 of 0.27 μM as compared to the inactive symmetrical analog 8q in Table 1. Furthermore, ortho-OCF3 was not tolerated in the symmetrical analog 8l, but highly active in the unsymmetrical 10q with 67% of BAM15 OCR and EC50 of 1.7 μM. As seen with the symmetrical analogs (Table 2), replacing one aniline with an alkyl or cycloalkyl group resulted in no or poor activity (Table 4). Among unsymmetrical analogs (10b, 10g, 10h and 10p) with OCR >70%, meta-fluoroaniline 10b was the most potent with EC50 of 191 nM (Figure 2). Therefore, 10b was selected for validation of efficacy in rat primary hepatocytes where we observed a 2-fold increase in OCR, similar to BAM15 (Figure S1). Consequently, 10b was identified as the candidate for in vivo anti-NASH efficacy studies.
Table 3.
Oxygen consumption rates (OCR) of unsymmetrical derivatives in L6 rat myoblast cells.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | R | % of BAM15 OCRa | EC50, μM | Entry | R | % of BAM15 OCRa | EC50, μM |
| BAM15 | 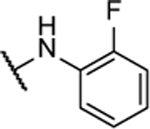 |
100% | 0.49 ± 0.06 | 10n | 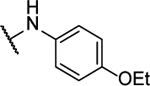 |
16%b | 3.8 ± 0.6 |
| 10a | 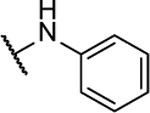 |
65%c | 2.2 ± 1.7 | 10o | 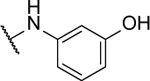 |
NA | -- |
| 10b | 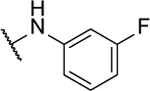 |
71%b | 0.19 ± 0.01 | 10p | 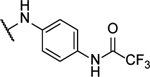 |
77%b | 4.1 ± 0.3 |
| 10c | 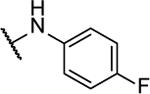 |
65%c | 2.3 ± 0.3 | 10q | 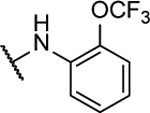 |
67%c | 1.7 ± 0.7 |
| 10d | 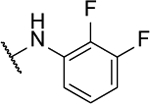 |
15%c | 0.5 ± 0.1 | 10r | 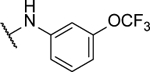 |
8%c | 0.6 ± 0.03 |
| 10e | 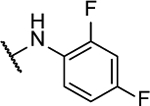 |
22%c | 0.3 ± 0.01 | 10s | 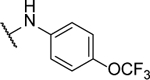 |
23%b | 1.3 ± 0.4 |
| 10f | 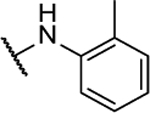 |
58%c | 1.1 ± 0.3 | 10t | 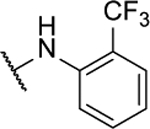 |
57%c | 1.4 ± 0.2 |
| 10g | 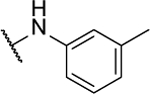 |
77%b | 1.5 ± 0.3 | 10u | 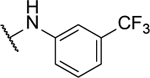 |
21%b | <1 |
| 10h | 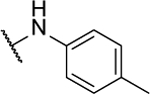 |
71%b | 2.1 ± 0.4 | 10v | 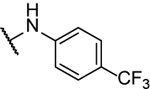 |
45%b | 0.27 ± 0.08 |
| 10i | 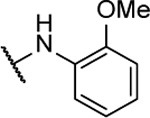 |
NA | -- | 10w | 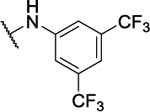 |
1%b | <0.37 |
| 10j | 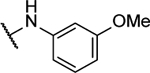 |
18%b | 0.83 ± 0.09 | 10x | 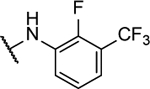 |
32%b | <0.37 |
| 10k | 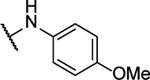 |
52%c | 1.6 ± 1.2 | 10y | 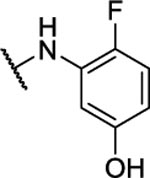 |
45%c | 30 ± 17 |
| 10l | 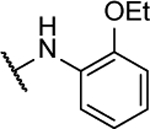 |
15%c | 5.6 ± 2.2 | 10z | 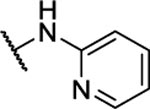 |
20%c | 9.0 ± 4.9 |
| 10m | 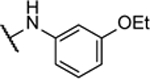 |
28%c | 2.2 ± 1.1 | ||||
Integrated area under OCR dose curve above baseline relative to that of BAM15 expressed as percent. NA = No Activity.
Highest tested concentration is 200 μM. BAM15 was tested up to 200 μM.
Highest tested concentration is 50 μM. BAM15 was tested up to 50 μM.
Figure 2.
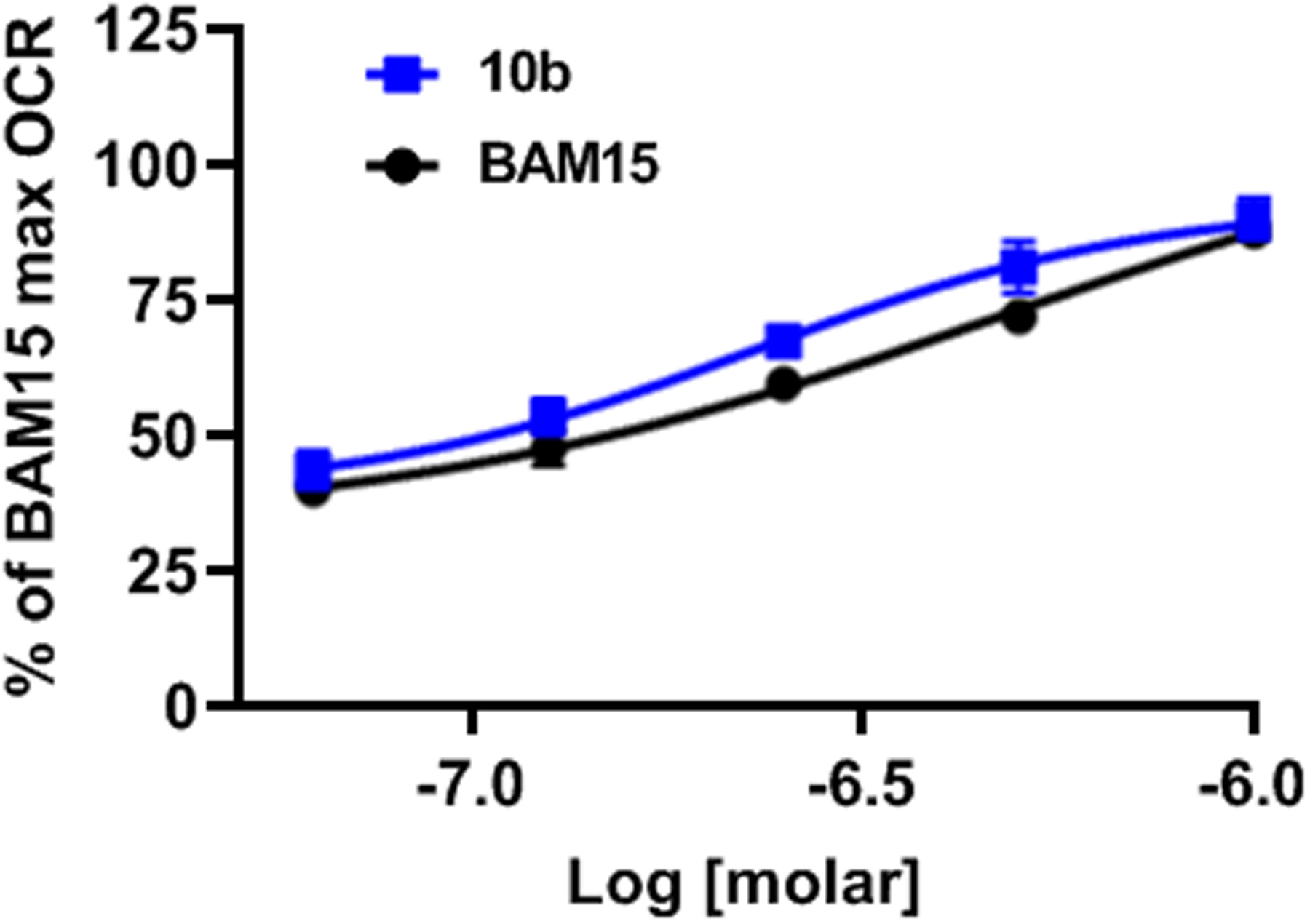
Oxygen consumption rate comparing 10b to BAM15 in L6 myoblasts. BAM15 EC50 = 0.486 ± 0.057 μM and Compound 10b EC50 = 0.191 ± 0.014 μM. Data are averaged over 3 separate experiments. Error bars represent standard error of mean.
The effect 10b in vivo was first assessed with acute dosing of male C57BL/6J mice. To determine whether 10b had adverse effects on core body temperature, mice were treated with 10b at doses of 25, 50, or 100 mg/kg or vehicle control by oral gavage. As shown in Figure 3A, no alterations in core temperature were observed with 10b treatment as measured by rectal probe thermometer at 1, 2, 4, and 24 hours following drug exposure. To determine tissue distribution, mice were given 50 mg/kg 10b by oral gavage. After 1 hour post treatment, the animals were sacrificed and levels of 10b determined by LC/MS. Whereas low levels were detected in white adipose tissue (WAT) (2.85 μM), quadricep muscle (0.26 μM), heart (2.09 μM) and brain (0.85 μM), the highest concentration of 10b was found in the liver (13.4 μM) followed by the kidney (4.83 μM) (Figure 3B).
Figure 3.
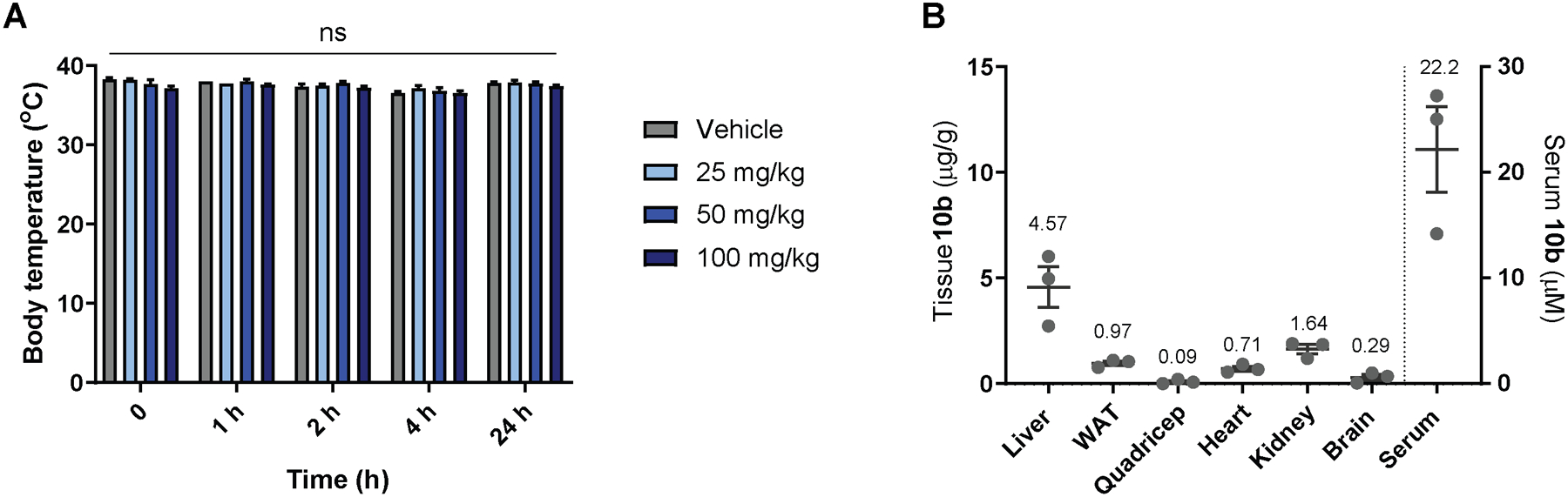
Body temperature (A) was not altered by acute oral dose of up to 100 mg/kg 10b. Tissue distribution of 10b was determined 1 h after acute oral dose of 50 mg/kg body weight (B). n=5–6 male mice per group for body temperature (A) and n=3 male mice per group for tissue distribution (B). Statistical significance was assessed by Two-Way Repeated Measures ANOVA with Dunnett’s correction.
Because 10b localized primarily in the liver, we determined its efficacy in vivo using the Stelic animal model (STAM) of NASH. Briefly, two-day old neonatal C57BL/6 male mice were administered low-dose 200 μg STZ followed by a high fat diet (HFD32, CLEA Japan) from 4 weeks of age. By 6 weeks of age, mice develop fatty change, increased serum markers of liver damage including alanine aminotransferase (ALT), and increased NAFLD activity score (NAS). NAS is defined as the sum of the histological scores for steatosis, lobular inflammation and hepatocyte ballooning. Mice were randomized at 6 weeks of age into 3 groups of 8 mice the day before the start of treatment with 10b or telmisartan, an angiotensin II receptor blocker shown to improve NAS in human NASH patients and commonly used as positive control in this model.75–76 10b was administered in food at 25 mg/kg/d over 21 days. Telmisartan was administered by oral gavage in water once daily at a dose of 10 mg/kg.77 Food intake was recorded during the treatment period; however, mice were grouped 4 per cage with 2 cages per treatment group and statistics could not be performed. Nevertheless, the data shows that telmisartan groups ate 23% less food while 10b had negligible effect on food intake (Figure S2). As shown in Figure 4A–B, 10b markedly improved fibrosis score as measured by picrosirius red-stained area (0.49 ± 0.23) compared to vehicle control (0.91 ± 0.25) and outperformed the telmisartan (0.60 ± 0.19) positive control. 10b also showed some improvement of the NAS score (3.9 ± 0.8) compared to vehicle control (4.6 ± 0.7), but telmisartan had the greatest protective effect (2.9 ± 0.8). The improvement in NAS by 10b was primarily driven by a statistically significant improvement in inflammation score (Figure 4C–F). No effect on ballooning was seen in this study with either compound.
Figure 4.
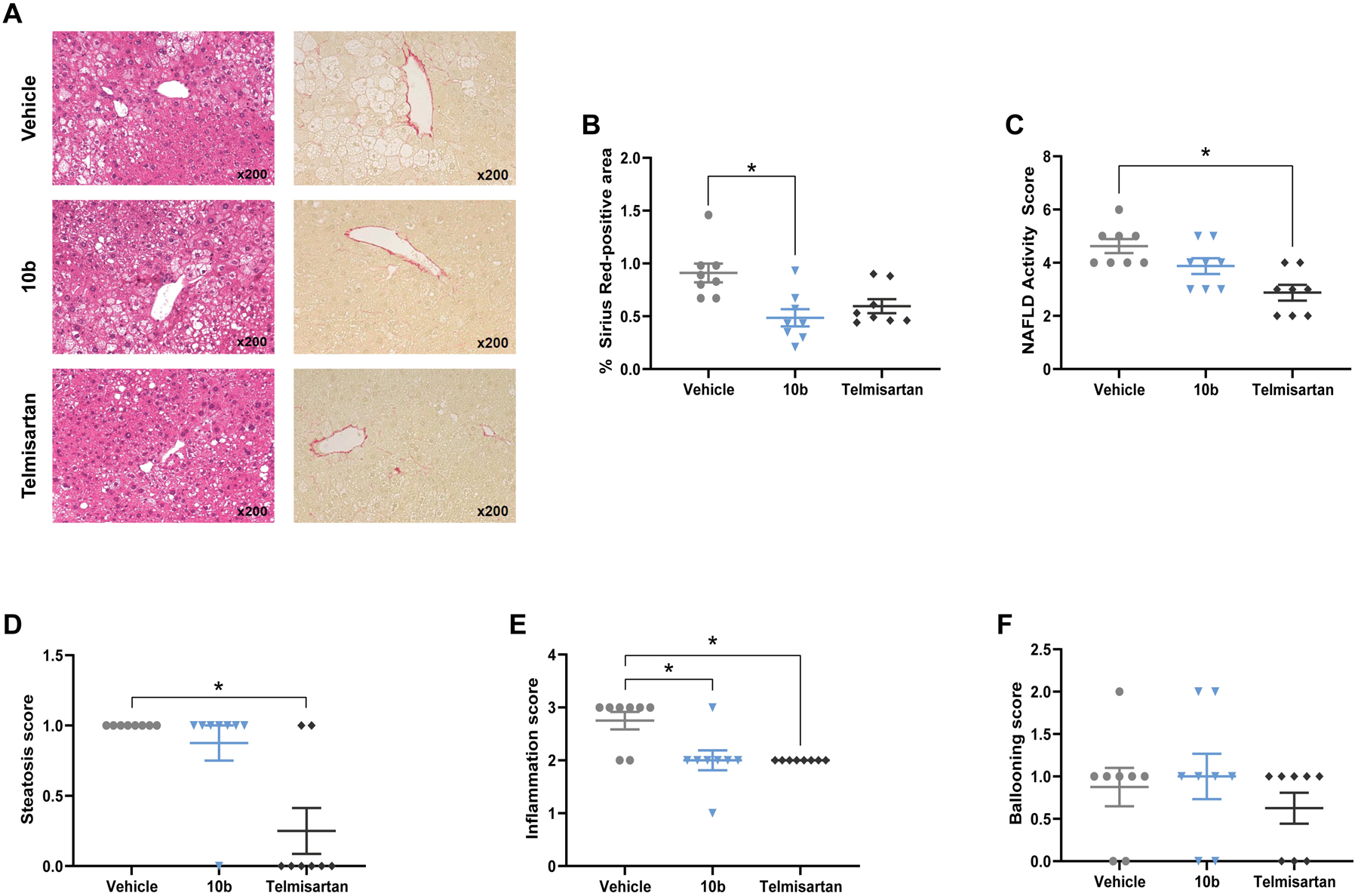
Effect of compound 10b on liver histopathologic and biochemical phenotypes in the STAM mouse model. Representative images of Hematoxylin and Eosin staining and Sirius Red staining in liver sections (A), Sirius Red staining quantified as a measure of fibrosis (B). NAFLD Activity Score (C) is the sum of individual parameters: steatosis (D), inflammation (E), and ballooning (F). Graphs show mean ± SEM. * indicates p < 0.05, by Kruskal-Wallis test with Dunn’s correction for multiple comparisons for non-parametric data. n=8 male mice per group.
To investigate the effect of 10b on whole body physiology, we measured several parameters of body composition and biochemistry. In contrast to telmisartan-treated animals, no changes in body weight or liver weight were observed with 10b treatment (Figure 5A–C). Curiously, while 10b did not affect the steatosis score determined by a pathologist (Figure 4D), it did markedly decrease liver triglyceride content measured from biochemical assay of liver tissue (Figure 5E). Blood biochemical analysis demonstrated that while telmisartan mildly decreased plasma triglyceride and increased plasma cholesterol, 10b had no effect on either of these parameters (Figure 5 E–F). Telmisartan treatment lead to hyperglycemia; however, blood glucose levels were unaffected by 10b (Figure 5G). Importantly, 10b improved the primary serum marker of liver damage, alanine aminotransferase (ALT) (Figure 5H). Plasma levels of the aspartate aminotransferase (AST), a marker of tissue damage, were not altered by 10b or telmisartan (Figure 5I). We assessed liver tissue for markers of energy status by western blot of activated AMPK (pAMPK/AMPK) and its downstream substrate acetyl-CoA carboxylase (pACC/ACC). No difference in AMPK activity was detected, suggesting that 10b treatment at this dose did not trigger energetic stress (Figure S3). Finally, we assessed liver tissue for mRNA of genes involved in fibrosis including collagen 1A1 (Col1a1); transforming growth factor β1 (Tgfb1), alpha-smooth muscle actin (a-SMA), tissue inhibitor matrix metalloproteinase 1 (Timp1), and matrix metallopeptidase 2 (Mmp2); however, compound 10b had minimal effects on the expression of these genes that did not reach statistical significance (Figure S4). Hepatic stellate cells transition from a quiescent cell phenotype to a myofibroblast proliferative, migratory, and fibrogenic phenotype that drives liver fibrosis; therefore, we investigated cluster of differentiation 146 (Cd146) as a marker of activated stellate cells and glial fibrillary acidic protein (Gfap) as a marker of quiescent stellate cells and found that neither marker was affected by 10b or telmisartan treatment (Figure S4). Despite no major changes in fibrogenic response gene expression, the physiological data shows that compound 10b reduced fibrosis and improved NAS without evidence of toxicity suggesting that mitochondrial uncouplers have potential as NASH therapeutics.
Figure 5.
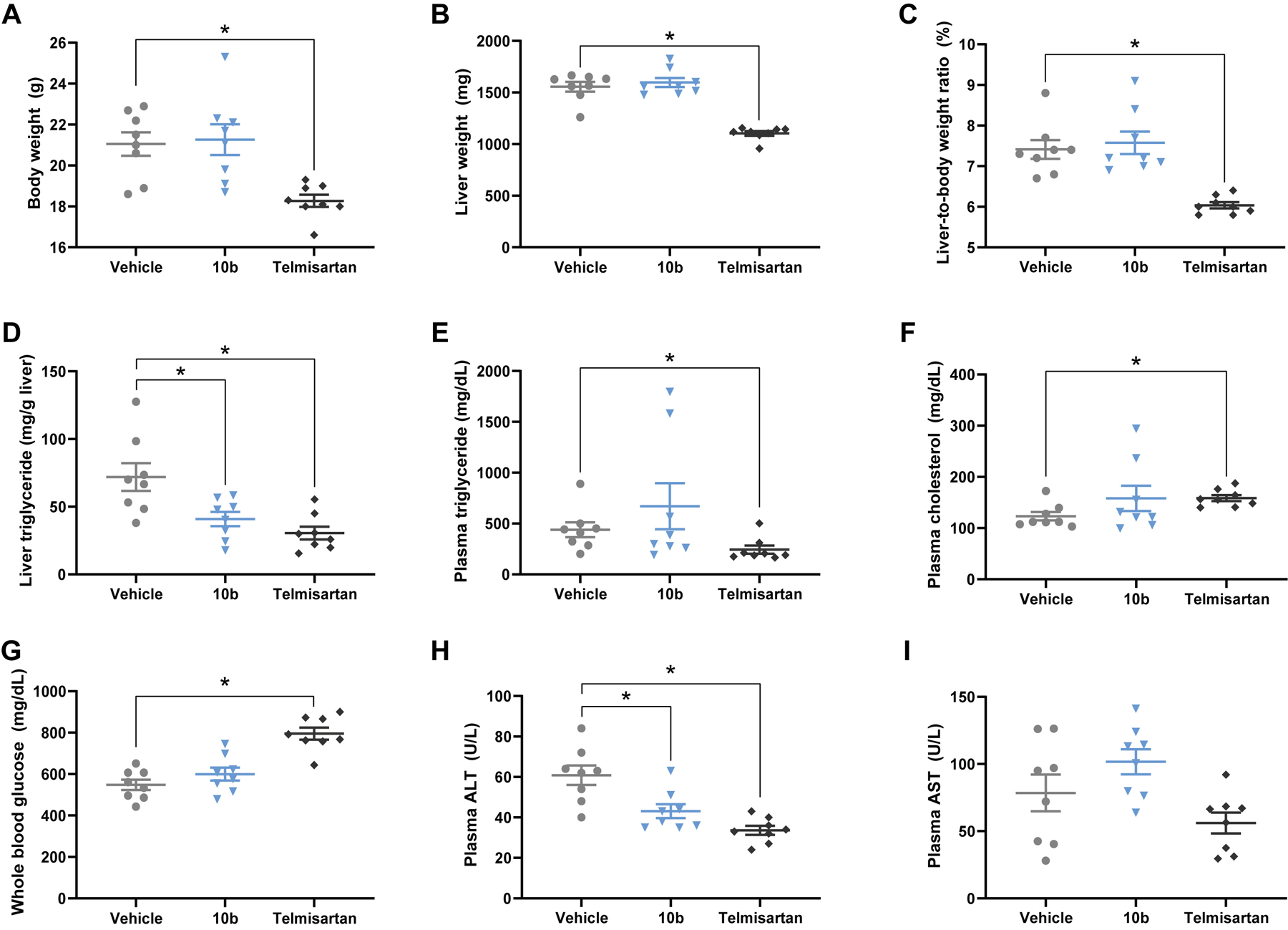
Effect of compound 10b and telmisartan on body composition and biochemistry in the STAM mouse model. Telmisartan, but not 10b, significantly decreased body weight (A), liver weight (B) and liver-to-body weight ratio (C). Both 10b and telmisartan significantly decreased liver triglyceride (D), but only telmisartan affected plasma triglyceride (E) and cholesterol (F) Telmisartan, but not 10b, exacerbated hyperglycemia (G). Plasma ALT levels were reduced by both 10b and telmisartan (H) but plasma AST levels were unaffected (I). Graphs show mean ± SEM. * indicates p < 0.05, by One-Way ANOVA with Dunnett’s correction for multiple comparisons for normally distributed data (A, D,G–I) or Kruskal-Wallis test with Dunn’s correction for multiple comparisons for non-parametric data (B–C,E–F). n=8 male mice per group.
Conclusion
Mitochondrial uncouplers can modulate the bioenergetic profile of cells by increasing respiration rates. This effect has proven useful in a number of preclinical disease models including obesity and fatty liver disease. However, drug development remains challenging due to toxicity or complex mechanism of action. The current state-of-the-art includes the acutely toxic DNP molecule that has been formulated to control Cmax-mediated toxicity24, 78 and improve liver targeting via a liver-targeted methyl ether prodrug of DNP (DNPME),22 and the repurposed drug niclosamide that have efficacy in fatty liver models despite a number of non-mitochondrial off-target effects.17 Specifically, the controlled-release DNP molecule had efficacy against NASH and fibrosis in a rat model of NASH induced by methionine/choline-deficient diet and a mouse model of lipodystrophy (fatless AZIP/F-1 mice).
Our discovery of BAM15 as a new class of mitochondria-selective uncoupler68 prompted a medicinal chemistry campaign to garner a better understanding of the compound’s structural properties and determine in vivo efficacy for this class of molecule in the STAM mouse model of NASH. In this study, we focused profiling structural changes on the 5- and 6-positions of the oxadiazolopyrazine core of BAM15. Our investigations revealed that the aniline rings with electron withdrawing groups are preferred. Compared to symmetrical derivatives, unsymmetrical aniline analogs were significantly more potent, with 10b bearing a 2-fluoro and 3-fluoro aniline being the best. In addition, we found that alkyl substituents were completely inactive and removal of the N-H proton was deleterious for the protonophore activity as measured by the OCR. In a mouse model of NASH, mitochondrial uncoupler 10b decreased liver triglyceride levels and improved liver enzymes (ALT and AST), NAFLD activity score as well as fibrosis without any alterations on body temperature and food intake. Overall, our studies support the development of mitochondrial uncouplers towards the treatment of NASH.
Experimental Section
General Material and Synthetic Procedures
All solvents used were dried with a PureSolv solvent purification system prior to use. All chemical reagents were purchased from commercial sources and used without further purification. Thin layer chromatography (TLC) was performed on aluminum-backed silica gel, 200 μm, F254. Column chromatography was performed on flash grade silica gel, 40–63 μm, with a Teledyne ISCO Combiflash Rf purification system.
NMR spectra were recorded using an Agilent 400-MR 400 MHz, a Varian Inova 400 MHz or Bruker Avance II 500. 1H NMR chemical shifts are reported in ppm with the solvent resonance as an internal standard (CD3OD: 4.87 ppm; (CD3)2CO: 2.05 ppm; CH3CN: 1.94 ppm). 13C NMR, chemical shifts are reported in ppm with the solvent resonance as the internal standard (CD3OD: 49.00 ppm; (CD3)2CO: 206.26 ppm; CH3CN: 118.26 ppm). Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t= triplet, q = quartet, br = broad, m= multiplet), coupling constants (Hz), and integration. Rotamers and tautomers are denoted by an asterisk (*). Note about NMR characterizations: These compounds form a mixture of tautomers and rotamers in solution that cause significant line broadening in both 1H and 13C NMR. In some cases, peak integration is complex, and some peaks were too broad to detect. Analytical characterizations were performed via HPLC and high-resolution mass spectroscopy (HRMS) on thermo Electron TSQ triple quadrupole mass spectrometer equipped with an ESI source. All compounds tested in biological assays are >95% pure.
Synthesis of 5,6-Dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (7).
In a 500 mL round-bottom flask equipped with a condenser, a mixture of 1,2,5-oxadiazole-3,4-diamine (6, 50.0 g, 500 mmol) and oxalic acid (49.6 g, 551 mmol) in aq. HCl (250 mL, 10% v/v) was heated in a sand bath to reflux for 4 h. The resulting mixture was allowed to cool in an ice bath and the precipitate was filtered, rinsed with water (20 mL) and then ether (2 × 150 mL), and collected to yield [1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diol (55.2 g, 72%) as a colorless solid: 1H NMR ((CD3)2CO, 400 MHz) δ 11.65 (br s, 2 H); 13C NMR ((CD3)2CO, 100 MHz) δ 153.9, 144.8.71
In a 500 mL two-neck round-bottom flask equipped with a glass stopper and a condenser connected to an aq. Na2CO3 trap, a mixture of the diol (37.0 g, 240 mmol) and PCl5 (120 g, 576 mmol) in POCl3 (45 mL) was heated to 95 °C for 2 h. The mixture cooled to rt and then to 5–10 °C in an ice bath. The reaction mixture was slowly poured onto ice cold water (3 × 250 mL beakers with 100 mL H2O each), not allowing the temperature of the water to rise above 25 °C (monitored by thermometer in the water bath). The colorless precipitate was filtered, rinsed with water, and dissolved in acetone (ca. 200 mL). Water (3 × volume of acetone) was added to the organic solution to promote precipitation and the colorless solid was filtered, collected, and dried under vacuum with P2O5 as a desiccant to yield 7 (35.1 g, 77%) as a colorless solid: 13C NMR ((CD3)2CO, 100 MHz) δ 155.9, 151.9. Note: DMSO-d6 should not be used as it will hydrolyze the product back to the diol. Analytical data matches with the literature.72–73
General Procedure A:
Synthesis of Symmetrical Derivatives 8
5,6-Dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (7, 1 equiv.) was dissolved in THF (0.6 M solution). The desired substituted arylamine (3 equiv.) was then added and the reaction mixture was heated to reflux overnight. After which, the resulting mixture was concentrated under reduced pressure, and the residue was purified by column chromatography on silica gel to yield the title compound.
General Procedure B:
Synthesis of unsymmetrical derivatives 10 via one-pot reaction.
5,6-Dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (7, 1 equiv.) was dissolved in THF (0.52 M solution) and cooled to 0 °C. Then, 2-fluoroaniline (0.9 equiv.) was added dropwise and allowed to stir for 10 min. Triethylamine (1 equiv.) was then added dropwise and allowed to stir at 0 °C for an additional 1 h. The second desired substituted arylamine was then added dropwise and allowed to stir for 10 min. Triethylamine (1 equiv.) was then added dropwise, and the reaction was then allowed to warm to room temperature and allowed to stir overnight. After which, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography to yield the title compound.
Synthesis of Unsymmetrical Derivatives 10 via isolation and purification of intermediate 9.
5,6-Dichloro-[1,2,5]oxadiazolo[3,4-b]pyrazine (7, 1 equiv.) was dissolved in THF (0.6 M solution). 2-Fluoroaniline (2 equiv.) was then added and the reaction mixture was heated to reflux overnight. After which, the solvent was removed under reduced pressure, and the residue was purified by silica gel column chromatography using a dichloromethane/methanol solvent system to obtain 9. The intermediate was in turn dissolved in THF (0.6 M). The desired alkylamine was added dropwise at room temperature and allowed to stir for 1 h. The resulting reaction mixture was concentrated under reduced pressure, and the residue was purified via silica gel chromatography using a dichloromethane/methanol solvent system to obtain the unsymmetrical arylamine/alkylamine compounds 10.
N5,N6-di-o-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8a).
Synthesized by general procedure A. 35%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.36 (s, 2H), 7.58–7.52 (m, 2H), 7.37 – 7.26 (m, 6H), 2.34 (s, 6H); 13C NMR (101 MHz, (CD3)2CO) δ 152.7*, 152.3*, 151.1*, 150.9*, 136.3, 135.3, 131.6, 128.4, 127.5, 127.3, 18.1; HRMS (ESI+): Calcd for C18H17N6O+ [M+H]+: 333.1464, found 333.1470.
N5,N6-di-m-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8b).
Synthesized by general procedure A. 60%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.64 (brs, 2H), 7.74 – 7.38 (brm, 4H), 7.32 (t, J = 7.7 Hz, 2H), 7.03 (d, J = 7.5 Hz, 2H), 2.37 (s, 6H); HRMS (ESI+): Calcd for C18H17N6O+ [M+H]+: 333.1464, found 333.1464.
N5,N6-di-p-tolyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8c).
Synthesized by general procedure A. 69%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.56 (brs, 2H), 7.75 – 7.47 (brm, 4H), 7.25 (d, J = 8.1 Hz, 4H), 2.34 (s, 6H); HRMS (ESI+): Calcd for C18H17N6O+ [M+H]+: 333.1464, found 333.1441.
N5,N6-bis(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8d).
Synthesized by general procedure A. 16%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.03 – 8.64 (m, 2H), 7.28 – 6.97 (m, 8H), 3.92 (s, 6H); HRMS (ESI+): Calcd for C18H17N6O3+ [M+H]+: 365.1362, found 365.1345.
N5,N6-bis(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8e).
Synthesized by general procedure A. 32%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.64 (brs, 2H), 7.80–7.24 (brm, 4H), 7.34 (t, J = 8.1 Hz, 2H), 6.80 – 6.75 (m, 2H), 3.82 (s, 6H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 161.3, 130.7, 114.5, 111.1, 108.1, 55.6; HRMS (ESI+): Calcd for C18H17N6O3+ [M+H]+: 365.1362, found 365.1370.
N5,N6-bis(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8f).
Synthesized by general procedure A. 47%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.35 (s, 2H), 7.91 – 7.43 (brm, 4H), 7.03 – 6.98 (m, 4H), 3.83 (s, 6H); HRMS (ESI+): Calcd for C18H17N6O3+ [M+H]+: 365.1362, found 365.1367.
N5,N6-bis(2-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8g).
Synthesized by general procedure A. 54%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.61 (brs, 2H), 7.95 – 7.06 (brm, 4H), 7.32 (t, J = 8.1 Hz, 2H), 6.80 – 6.71 (m, 2H) 4.08 (q, J = 7.0 Hz, 2H), 1.38 (t, J = 7.0 Hz, 6H); HRMS (ESI+): Calcd for C20H21N6O3+ [M+H]+: 393.1675, found 393.1674.
N5,N6-bis(3-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8h).
Synthesized by general procedure A. 27%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.67 (brs, 2H), 7.85 – 7.00 (brm, 4H), 7.32 (t, J = 8.1 Hz, 2H), 6.78 – 6.74 (brm, 2H), 4.08 (q, J = 7.0 Hz, 4H), 1.38 (t, J = 7.0 Hz, 6H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 160.6, 130.7, 114.4, 111.7, 108.7, 64.1, 15.1; HRMS (ESI+): Calcd for C20H21N6O3+ [M+H]+: 393.1675, found 393.1656.
N5,N6-bis(4-ethoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8i).
Synthesized by general procedure A. 73%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.15 (brs, 2H), 8.01 – 7.50 (brm, 4H), 7.04 – 6.94 (m, 4H), 4.07 (q, J = 7.0 Hz, 4H), 1.38 (t, J = 6.9 Hz, 6H); 13C NMR (101 MHz, (CD3)2CO) δ 157.3, 151.0, 148.3, 131.7, 124.8 (br), 115.4, 64.2, 15.1; HRMS (ESI+): Calcd for C20H21N6O3+ [M+H]+: 393.1675, found 393.1673.
3,3’-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))diphenol (8j).
Synthesized by general procedure A. 43%, light yellow solid. 1H NMR (500 MHz, (CD3)2CO) δ 8.71 (brs, 2H), 7.89 – 7.35 (brm, 1H), 7.35 – 6.95 (brm, 2H), 7.23 (t, J = 7.9 Hz, 2H), 6.70 – 6.66 (m, 2H); 13C NMR (126 MHz, (CD3)2CO) δ 158.9, 150.9 (br), 150.6 (br), 140.4 (br), 130.7, 113.5, 112.8, 109.4; HRMS (ESI+): Calcd for C16H13N6O3+ [M+H]+: 337.1049, found 337.1044.
N,N’-(([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))bis(4,1-phenylene))bis(2,2,2-trifluoroacetamide) (8k).
Synthesized by general procedure A. 22%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.38 (s, 2H), 9.61 (s, 2H), 8.00 – 7.94 (m, 4H), 7.86 – 7.81 (m, 4H); 13C NMR (101 MHz, (CD3)2CO) δ 155.6 (q, JCF = 37.3 Hz), 152.3*, 152.3*, 150.8, 149.9*, 149.8*, 135.5*, 135.4*, 135.1*, 135.0*, 124.4*, 124.3*, 122.0*, 121.9*, 116.9 (q, JCF = 288 Hz); 19F NMR (376 MHz, (CD3)2CO) δ −76.58 (s, 6F); HRMS (ESI+): Calcd for C20H13F6N8O3+ [M+H]+: 527.1015, found 527.1009.
N5,N6-bis(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8l).
Synthesized by general procedure A. 20%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.23 (brs, 2H), 8.12 (brs, 2H), 7.54–7.46 (m, 4H), 7.38–7.29 (m, 2H); 19F NMR (376 MHz, (CD3)2CO) δ −58.69 (s, 6F); HRMS (ESI−): Calcd for C18H9F6N6O3− [M-H]: 471.0646, found 471.0657.
N5,N6-bis(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8m).
Synthesized by general procedure A. 69%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.00 (brs, 2H), 8.01–7.64 (brm, 4H), 7.58 (t, J = 8.1 Hz, 2H), 7.21–7.13 (m, 2H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 150.4, 131.6, 121.5 (q, JCF = 256 Hz), 121.1, 117.7, 114.9; 19F NMR (376 MHz, (CD3)2CO) δ −58.47 (s, 6F); HRMS (ESI+): Calcd for C18H11F6N6O3+ [M+H]+: 473.0797, found 473.0773.
N5,N6-bis(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8n).
Synthesized by general procedure A. 40%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.09 (brs, 2H), 8.03 – 7.55 (brm, 4H), 7.49 (d, J = 8.5 Hz, 4H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 149.9 (br), 146.4, 138.7 (br), 124.0, 122.8, 121.5 (q, JCF = 255 Hz); 19F NMR (376 MHz, (CD3)2CO) δ −58.78 (s, 6F); HRMS (ESI+): Calcd for C18H11F6N6O3+ [M+H]+: 473.0797, found 473.0792.
N5,N6-bis(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8o).
Synthesized by general procedure A. 52%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.40 (s, 2H), 8.03 – 7.76 (m, 6H), 7.64 (t, J = 7.7 Hz, 2H); 19F NMR (376 MHz, (CD3)2CO) δ −61.18 (s, 6F); HRMS (ESI+): Calcd for C18H11F6N6O+ [M+H]+: 441.0899, found 441.0892.
N5,N6-bis(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8p).
Synthesized by general procedure A. 50%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.11 (brs, 2H), 8.28 – 7.84 (brm, 4H), 7.70 (t, J = 7.9 Hz, 2H), 7.55 (d, J = 7.8 Hz, 2H); 19F NMR (376 MHz, (CD3)2CO) δ −63.20 (s, 6F); HRMS (ESI+): Calcd for C18H11F6N6O + [M+H]+: 441.0899, found 441.0887.
N5,N6-bis(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8q).
Synthesized by general procedure A. 63%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.17 (brs, 2H), 8.05 – 7.84 (brm, 4H), 7.80 (d, J = 8.3 Hz, 4H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 148.8 (br), 145.2 (br), 127.3 (q, JCF = 4.0 Hz), 126.9 (q, JCF = 5.6 Hz), 125.4 (q, JCF = 244 Hz), 122.6; 19F NMR (376 MHz, (CD3)2CO) δ −62.53 (s, 6F); HRMS (ESI+): Calcd for C18H11F6N6O + [M+H]+: 441.0899, found 441.0875.
4,4’-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))dibenzonitrile (8r).
Synthesized by general procedure A. 19%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.78 (s, 2H), 8.25 – 8.17 (m, 4H), 7.92 – 7.85 (m, 4H); 13C NMR (101 MHz, (CD3)2CO) δ 152.3*, 152.1*, 150.9*, 150.0*, 142.4, 134.1*, 134.0*, 123.9*, 123.8*, 119.3, 109.6; HRMS (ESI+): Calcd for C18H11N8O+ [M+H]+: 355.1050, found 355.1040.
3,3’-([1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diylbis(azanediyl))dibenzonitrile (8s).
Synthesized following general procedure 1. Yellow solid (34.4 mg, 14%); 1H NMR (400 MHz, (CD3)2CO) δ 10.18 (s, 2H), 8.33 – 7.83 (m, 4H), 7.68 (td, J = 7.7, 0.6 Hz, 2H), 7.61 (dt, J = 7.7, 1.1 Hz, 2H); HRMS (ESI−) m/z calcd for C18H11N8O [M-H]−: 355.1050, found 355.1046.
N5,N6-bis(4-ethylphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8t).
Synthesized following general procedure A. 3%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 9.61 (s, 2H), 7.65 (s, 4H), 7.31 – 7.26 (m, 4H), 2.66 (q, J = 7.6 Hz, 4H), 1.24 (t, J = 7.6 Hz, 6H); HRMS (ESI+) m/z calcd for C20H21N6O [M+H]+: 361.1771, found 361.1768.
N5,N6-bis(3,4-dimethylphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8u).
Synthesized following general procedure A. 4%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 9.55 (s, 2H), 7.47 (s, 4H), 7.18 (d, J = 8.0 Hz, 2H), 2.26 (d, J = 7.3 Hz, 12H); HRMS (ESI+) m/z calcd for C20H21N6O [M+H]+: 361.1771, found 361.1769.
N5,N6-bis(3-bromo-4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8v).
Synthesized following general procedure A. 34%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 10.20 (s, 2H), 8.15 (s, 2H), 7.80 (s, 2H), 7.58 (dd, J = 8.8, 1.4 Hz, 2H); HRMS (ESI+) m/z calcd for C18H9Br2F6N6O3 [M+H]+: 628.9002, found 628.8975.
N5,N6-bis(2-fluoro-4-(pent-1-yn-1-yl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8w).
Synthesized following general procedure A. 1%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 9.85 (s, 2H), 8.10 (s, 2H), 7.37 – 7.25 (m, 4H), 2.42 (t, J = 7.0 Hz, 4H), 1.62 (h, 4H), 1.05 (t, J = 7.4 Hz, 6H); HRMS (ESI+) m/z calcd for C26H23F2N6O [M+H]+: 473.1895, found 473.1887.
N5,N6-bis(4-bromo-2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8x).
Synthesized following general procedure A. 10%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 9.95 (s, 2H), 7.90 (s, 2H), 7.55 (dd, J = 10.2, 2.2 Hz, 2H), 7.51 – 7.48 (m, 2H); HRMS (ESI+) m/z calcd for C16H9Br2F2N6O [M+H]+: 496.9167, found 498.9165.
N5,N6-bis(2-fluoro-4-iodophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8y).
Synthesized following general procedure A. 8%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 10.21 (s, 2H), 7.92 – 7.52 (m, 6H); HRMS (ESI+) m/z calcd for C16H9F2I2N6O [M+H]+: 592.8889, found 592.8866.
N5,N6-bis(2-fluoro-4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8z).
Synthesized following general procedure A. Yellow solid (10.2 mg, 3%); 1H NMR (400 MHz, (CD3)2CO) δ 10.94 (s, 1H), 9.88 (s, 1H), 8.05 (s, 2H), 7.39 (dd, J = 10.8, 2.6 Hz, 2H), 7.33 (d, 2H); HRMS (ESI+) m/z calcd for C18H9F8N6O3 [M+H]+: 509.0603, found 509.0592.
N5,N6-bis(2,4,6-trifluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8aa).
Synthesized following general procedure A. Yellow solid (63.1 mg, 23% yield); 1H NMR (400 MHz, (CD3)2CO) δ 11.16 (s, 1H), 9.49 (s, 1H), 7.22 – 7.06 (m, 4H); HRMS (ESI+) m/z calcd for C16H7F6N6O [M+H]+: 413.0580, found 413.0598.
N5,N6-bis(3,5-difluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ab).
Synthesized following general procedure A. 14%, yellow solid: 1H NMR ((CD3)2CO, 400 MHz) δ 10.43 – 9.63 (brm, 2H), 7.82 – 7.03 (brm, 4H), 6.89 – 6.81 (m, 2H). HRMS (ESI+) m/z calcd for C16H9F4N6O [M+H]+: 377.0768, found 377.0764.
N5,N6-bis(4-iodo-2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ac).
Synthesized following general procedure A. Yellow solid (55.1 mg, 12%); 1H NMR (400 MHz, (CD3)2CO) δ 11.32 (s, 2H), 9.95 (s, 2H), 7.88 (dd, J = 8.4, 1.9 Hz, 2H), 7.84 (s, 2H); HRMS (ESI+) m/z calcd for C18H9F6I2N6O3 [M+H]+: 724.8724, found 724.8715.
N5,N6-bis(2-fluoro-4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ad).
Synthesized following general procedure A. 3%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 11.26 (s, 1H), 9.97 (s, 1H), 8.21 (s, 2H), 7.70 (s, 2H), 7.67 (s, 2H). HRMS (ESI+) m/z calcd for C18H9F8N6O [M+H]+: 477.0705, found 477.0701
N5,N6-bis(3-fluoro-4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ae).
Synthesized following general procedure A. 1H NMR ((CD3)2CO, 400 MHz) δ 10.67 – 10.04 (brm, 2H), 7.92 – 7.74 (brm, 2H), 7.81 (t, 2 H, J = 8.4 Hz), 7.70 – 7.57 (brm, 2H); 19F ((CD3)2CO, 376 MHz) δ −61.22 (d, 6 F), −114.5 – −114.73 (m, 2 F). HRMS (ESI+) m/z calcd for C18H9F8N6O [M+H]+: 477.0705, found 477.0689.
N5,N6-bis(2-fluoro-5-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8af).
Synthesized using General Procedure A. 1H NMR ((CD3)2CO, 400 MHz, asterisk denotes tautomer peaks) δ 11.57 – 10.94* (brs, 1H), 10.22 – 9.68* (brs, 1H), 8.86 – 7.82 (brm, 2H), 7.68 – 7.62 (m, 2H), 7.55 (t, 2H, J = 9.8 Hz). HRMS (ESI+) m/z calcd for C18H9F8N6O [M+H]+: 477.0705 found 477.0711.
N5,N6-bis(3,5-bis(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine. (8ag).
Synthesized using Generic Procedure A. 1H NMR ((CD3)2CO, 400 MHz, asterisk denotes tautomer peaks) δ 11.45 – 10.84* (brs, 1H), 10.59 – 10.10 (brs, 1H), 8.80 – 7.98 (brm, 4H), 7.87 (s, 2H). HRMS (ESI−) m/z calcd for C20H7F12N6O [M-H]−: 575.0495, found 575.0509.
N5,N6-di-tert-butyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ah).
Synthesized by general procedure A. 79%, white solid. 1H NMR (400 MHz, (CD3)2CO) δ 6.78 (s, 2H), 1.50 (s, 18H); 13C NMR (101 MHz, (CD3)2CO) δ 150.3, 149.1, 54.6, 28.4; HRMS (ESI+): Calcd for C12H21N6O+ [M+H]+: 265.1777, found 265.1776.
N5,N6-dicyclopropyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ai).
Synthesized by general procedure A. 33%, white solid. 1H NMR (400 MHz, (CD3)2CO) δ 7.32 (s, 2H), 3.01 (tt, J = 7.3, 3.9 Hz, 2H), 0.89 – 0.82 (m, 4H), 0.64 – 0.57 (m, 4H); 13C NMR (101 MHz, (CD3)2CO) δ 151.6, 150.9, 25.5, 6.9; HRMS (ESI+): Calcd for C10H13N6O+ [M+H]+: 233.1151, found 233.1137.
N5,N6-dicyclopentyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8aj).
Synthesized by general procedure A. 40%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 7.16 (brs, 2H), 4.54 – 4.44 (m, 2H), 2.16 – 2.05 (m, 4H), 1.74 – 1.49 (m, 12H); 13C NMR (101 MHz, (CD3)2CO) δ 151.4, 149.3, 54.5, 32.8, 24.6; HRMS (ESI+): Calcd for C14H21N6O+ [M+H]+: 289.1777, found 289.1774.
N5,N6-dicyclohexyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8ak).
Synthesized by general procedure A. 72%, clear solid. 1H NMR (400 MHz, (CD3)2CO) δ 7.10 – 7.01 (m, 2H), 4.18 – 4.07 (m, 2H), 2.13 – 2.06 (m, 4H), 1.82 – 1.73 (m, 4H), 1.71 – 1.63 (m, 2H), 1.47 – 1.36 (m, 4H), 1.31 – 1.13 (m, 6H); 13C NMR (101 MHz, (CD3)2CO) δ 151.3, 148.7, 51.8, 32.8, 26.3, 25.8; HRMS (ESI+): Calcd for C16H25N6O+ [M+H]+: 317.2090, found 317.2089.
5,6-dimorpholino-[1,2,5]oxadiazolo[3,4-b]pyrazine (8al).
Synthesized by general procedure A. 95%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 3.81 (brs, 16H); 13C NMR (101 MHz, (CD3)2CO) δ 153.9, 151.6, 66.6, 49.3; HRMS (ESI+): Calcd for C12H17N6O3 [M+H]+: 293.1362, found 293.1361.
N5,N6-bis(2-fluorophenyl)-N5,N6-dimethyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (8am).
Synthesized following general procedure A. 34%, yellow solid: 1H NMR (400 MHz, (CD3)2CO) δ 7.53 – 7.45 (m, 4H), 7.42 – 7.35 (m, 2H), 7.32 – 7.26 (m, 2H), 3.57 (d, J = 0.5 Hz, 6H); 19F NMR (376 MHz, (CD3)2CO) δ −123.40 (ddd, J = 10.5, 7.9, 4.9 Hz); 13C NMR (126 MHz, (CD3)2CO) δ 158.24 (d, JCF = 248.4 Hz), 154.54, 152.28 (d, JCF = 17.4 Hz), 151.49, 134.19 (d, JCF = 11.8 Hz), 131.06 (d, JCF = 8.1 Hz), 129.04, 126.51 (d, JCF = 4.1 Hz), 117.95 (d, JCF = 19.9 Hz), 43.02 (d, JCF = 2.7 Hz); HRMS (ESI+): Calcd for C18H15F2N6O+ [M+H+]+: 369.1275, found 369.1275.
N5-(2-fluorophenyl)- N6-phenyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10a).
Synthesized by general procedure B. 44%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.44 – 9.05 (brm, 2H), 7.76 (brs, 3H), 7.45 (t, J = 7.8 Hz, 2H), 7.35 – 7.14 (m, 4H); 19F NMR (376 MHz, CH3CN) δ −122.05 – −130.05 (m, 1F); HRMS (ESI+): Calcd for C16H12FN6O + [M+H]+: 323.1057, found 323.1051.
N5-(2-fluorophenyl)- N6-(3-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10b).
Synthesized by general procedure B. 42%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.09 (brs, 2H), 7.95 – 7.65 (m, 2H), 7.63 – 7.51 (m, 1H), 7.47 (q, J = 7.7 Hz, 1H), 7.31 – 7.23 (m, 3H), 7.00 – 6.95 (m, 1H); 13C NMR (126 MHz, (CD3)2CO) δ 163.8 (d, JCF = 243 Hz), 155.1 (d, JCF = 245 Hz), 149.0 (br), 142.7 (br), 131.5 (d, JCF = 9.5 Hz), 127.2 (d, JCF = 7.3 Hz), 127.1 (d, JCF = 6.80 Hz), 125.8 (d, JCF = 3.7 Hz), 125.0, 124.6, 118.0 (d, JCF = 2.8 Hz), 116.8 (d, JCF = 19.8 Hz), 112.1 (d, JCF = 21.3 Hz), 109.2 (d, JCF = 25.9 Hz); 19F NMR (376 MHz, (CD3)2CO) δ −113.18 – −113.24 (m, 1F), −125.3 (brm, 1F); HRMS (ESI+): Calcd for C16H11F2N6O + [M+H]+: 341.0962, found 341.0956.
N5-(2-fluorophenyl)- N6-(4-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10c).
Synthesized by general procedure B. 28%, off-white solid. 1H NMR (500 MHz, (CD3)2CO) δ 10.82 (s, 1H), 9.72 (s, 1H), 8.28 – 7.56 (brm, 4H), 7.36 – 7.17 (m, 4H); 19F NMR (376 MHz, (CD3)2CO) δ −118.35 – −120.14 (m, 1F), −129.66 – −129.73 (m, 1F); HRMS (ESI+): Calcd for C16H11F2N6O + [M+H]+: 341.0962, found 341.0967.
N5-(2,3-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10d).
Synthesized by general procedure B. 32%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.91 (s, 1H), 9.75 (s, 1H), 8.64 – 8.03 (m, 1H), 7.86 – 7.42 (m, 1H), 7.35 – 7.22 (m, 4H), 7.22 – 7.13 (m, 1H); 19F NMR (376 MHz, (CD3)2CO) δ −139.18 – −139.86 (m, 1F), −148.96 – −154.99 (m, 2F); HRMS (ESI+): Calcd for C16H10F3N6O + [M+H]+: 359.0868, found 359.0858.
N5-(2,4-difluorophenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10e).
Synthesized by general procedure B. 16%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.90 (s, 1H), 9.76 (s, 1H), 8.43 – 7.45 (brm, 2H), 7.34 – 7.25 (m, 2H), 7.24 – 7.16 (m, 2H), 7.17 – 7.08 (m, 2H); 19F NMR (376 MHz, (CD3)2CO) δ −114.32 – −116.24 (m, 2F), −119.15 – −121.61 (m, 1F); HRMS (ESI+): Calcd for C16H10F3N6O + [M+H]+: 359.0868, found 359.0862.
N5-(2-fluorophenyl)- N6-(o-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10f).
Synthesized by general procedure B. 17%, off-white solid. 1H NMR (500 MHz, (CD3)2CO) δ 9.67 – 9.54 (brm, 1H), 8.63 (brs, 1H), 7.47 – 7.41 (m, 1H), 7.40 – 7.36 (m, 2H), 7.35 – 7.25 (m, 5H), 2.28 (s, 3H); 19F NMR (376 MHz, CH3CN) δ −122.81 – −123.46 (m, 1F); HRMS (ESI+): Calcd for C17H14FN6O+ [M+H]+: 337.1213, found 337.1200.
N5-(2-fluorophenyl)-N6-(m-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10g).
Synthesized by general procedure B. 43%, yellow solid. 1H NMR (400 MHz, CH3CN) δ 9.10 (brs, 2H), 7.70 – 7.54 (brs, 1H), 7.52 – 7.39 (m, 2H), 7.34 (t, J = 8.1 Hz, 1H), 7.31 – 7.24 (m, 3H), 7.07 (d, J = 7.5 Hz, 1H), 2.39 (s, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −118.84 – −127.22 (m, 1F); HRMS (ESI+): Calcd for C17H14FN6O + [M+H]+: 337.1213, found 337.1203.
N5-(2-fluorophenyl)-N6-(p-tolyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10h).
Synthesized by general procedure B. 40%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.91 (brs, 1H), 9.77 (brs, 1H), 8.09 – 7.55 (brm, 2H), 7.49 – 7.18 (m, 6H), 2.35 (s, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −120.34 – −128.87 (m, 1F); HRMS (ESI+): Calcd for C17H14FN6O + [M+H]+: 337.1213, found 337.1201.
N5-(2-fluorophenyl)-N6-(2-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10i).
Synthesized by general procedure B. 55%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.05 (s, 1H), 8.78 (s, 1H), 7.33 – 7.22 (m, 5H), 7.22 – 7.12 (m, 2H), 7.08 (app. t, J = 7.5 Hz, 1H), 3.97 (s, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −124.05 – −125.57 (m, 1F); HRMS (ESI−): Calcd for C17H12FN6O2− [M-H]−: 351.1011, found 351.1015.
N5-(2-fluorophenyl)-N6-(3-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10j).
Synthesized by general procedure B. 50%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.80 (s, 1H), 9.71 (s, 1H), 7.86 – 7.40 (brm, 2H), 7.35 (t, J = 8.1 Hz, 1H), 7.31 – 7.22 (m, 4H), 3.83 (s, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −124.34 – −126.26 (brm, 1F); HRMS (ESI−): Calcd for C17H12FN6O2− [M-H]−: 351.1011, found 351.1003.
N5-(2-fluorophenyl)- N6-(4-methoxyphenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10k).
Synthesized by general procedure B. 38%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.65 (brs, 1H), 9.65 (s, 1H), 8.20 – 7.46 (brm, 3H), 7.31 – 7.22 (m, 3H), 7.16 – 6.90 (m, 2H), 3.83 (s, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −124.80 (s, 1F); HRMS (ESI+): Calcd for C17H14FN6O2+ [M+H]+: 353.1162, found 353.1177.
N5-(2-ethoxyphenyl)- N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10l).
Synthesized by general procedure B. 39%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.81 (s, 1H), 10.20 (s, 1H), 8.76 (s, 1H), 7.36 (brs, 1H), 7.31 – 7.20 (m, 3H), 7.20 – 7.04 (m, 3H), 4.22 (q, J = 7.0 Hz, 2H), 1.42 (t, J = 6.9 Hz, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −123.40 – −126.20 (brm, 1F); HRMS (ESI+): Calcd for C18H16FN6O2+ [M+H]+: 367.1319, found 367.1322.
N5-(3-ethoxyphenyl)- N6 -(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10m).
Synthesized by general procedure B. 22%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.76 (brs, 1H), 9.77 (brs, 1H), 7.95 – 7.41 (brm, 2H), 7.33 (t, J = 8.0 Hz, 1H), 7.31 – 7.23 (m, 4H), 6.77 (app d, J = 6.9 Hz, 1H), 4.09 (q, J = 7.0 Hz, 2H), 1.39 (t, J = 7.0 Hz, 3H); 19F NMR (376 MHz, CH3CN) δ −122.42 – −130.56 (brm, 1F); HRMS (ESI+): Calcd for C18H16FN6O2+ [M+H]+: 367.1319, found 367.1324.
N5-(4-ethoxyphenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10n).
Synthesized by general procedure B. 55%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.64 (brs, 2H), 7.80 (brs, 3H), 7.32 – 7.22 (m, 3H), 7.03 – 6.96 (m, 2H), 4.08 (q, J = 7.0 Hz, 2H), 1.38 (t, J = 7.0 Hz, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −124.93 – −127.24 (brm, 1F); HRMS (ESI+): Calcd for C18H16FN6O2+ [M+H]+: 367.1319, found 367.1313.
3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol (10o).
Synthesized by general procedure B. 33%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 9.42 (s, 1H), 8.27 – 8.17 (m, 1H), 7.42 – 7.27 (m, 3H), 7.19 (t, J = 8.1 Hz, 1H), 6.73 (t, J = 2.2 Hz, 1H), 6.71 – 6.66 (m, 1H), 6.65 – 6.60 (m, 1H), 5.00 (s, 2H); 13C NMR (101 MHz, (CD3)2CO) δ 156.4 (d, JCF = 247 Hz), 156.0, 153.6, 152.1, 151.3, 150.7, 148.6, 130.9, 128.3 (d, JCF = 7.8 Hz), 126.7, 126.1 (d, JCF = 11.3 Hz), 125.5 (d, JCF = 3.8 Hz), 116.6 (d, JCF = 19.5 Hz), 113.6, 110.1, 108.0; 19F NMR (376 MHz, (CD3)2CO) δ −124.79 – −124.95 (m, 1F); HRMS (ESI+): Calcd for C16H12FN6O2 + [M+H]+: 339.1006, found 339.1004.
2,2,2-Trifluoro-N-(4-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenyl)acetamide (10p).
Synthesized by general procedure B. 18%, yellow solid. 1H NMR (400 MHz, CH3CN) δ 9.23 (brs, 2H), 7.87 – 7.46 (brm, 6H), 7.36 – 7.21 (m, 3H); 19F NMR (376 MHz, CH3CN) δ −76.46 – −76.75 (m, 3F), −125.89 (s, 1F); HRMS (ESI+): Calcd for C18H12F4N7O2 + [M+H]+: 434.0989, found 434.0988.
N5-(2-fluorophenyl)- N6-(2-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10q).
Synthesized by general procedure B. 14%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 11.10 (brs, 1H), 9.88 (brs, 1H), 7.56 – 7.40 (m, 2H), 7.39 – 7.23 (m, 6H); 19F NMR (376 MHz, (CD3)2CO) δ −58.48 (s, 3F), −123.69 – −131.84 (brd, 1F); HRMS (ESI+): Calcd for C17H11F4N6O2+ [M+H]+: 407.0880, found 407.0897.
N5-(2-fluorophenyl)-N6-(3-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10r).
Synthesized by general procedure B. 59%, yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.02 (brs, 2H), 8.08 – 7.65 (brm, 3H), 7.58 (t, J = 8.2 Hz, 1H), 7.33 – 7.22 (m, 3H), 7.20 – 7.15 (m, 1H); 13C NMR (101 MHz, (CD3)2CO) δ some carbons are not visible 155.12 (d, JCF = 245 Hz), 150.3, 131.5, 127.2 (br), 125.8, 125.1 (br), 121.5 (q, JCF = 256 Hz), 121.0, 117.7, 116.8 (q, JCF = 19.3 Hz), 114.9; 19F NMR (376 MHz, (CD3)2CO) δ −58.45 (s, 3F), −123.31 – −127.07 (m, 1F); HRMS (ESI+): Calcd for C17H11F4N6O2+ [M+H]+: 407.0880, found 407.0887.
N5-(2-fluorophenyl)-N6-(4-(trifluoromethoxy)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10s).
Synthesized by general procedure B. 28%, light yellow solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.96 (brs, 1H), 9.80 (brs, 1H), 8.36 – 7.53 (brm, 3H), 7.44 – 7.41 (m, 2H), 7.32 – 7.23 (m, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −59.45 (s, 3F), −122.88 – −126.12 (m, 1F); HRMS (ESI+): Calcd for C17H11F4N6O2+ [M+H]+: 407.0880, found 407.0872.
N5-(2-fluorophenyl)- N6-(2-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10t).
Synthesized by general procedure B. 27%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 11.13 (s, 1H), 9.82 (s, 1H), 8.67 (s, 1H), 7.79 (d, J = 7.9 Hz, 1H), 7.71 (t, J = 7.7 Hz, 1H), 7.51 – 7.19 (m, 5H); 19F NMR (376 MHz, (CD3)2CO) δ −62.71 (s, 3F), −131.01 – −131.82 (m, 1F); HRMS (ESI+): Calcd for C17H11F4N6O+ [M+H]+: 391.0930, found 391.0932.
N5-(2-fluorophenyl)- N6-(3-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10u).
Synthesized by general procedure B. 49%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.82 (brs, 1H), 9.90 (brs, 1H), 8.49 – 7.92 (brm, 3H), 7.70 (t, J = 8.0 Hz, 1H), 7.55 (d, J = 7.8 Hz, 1H), 7.36 – 7.19 (m, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −63.03 – −63.34 (m, 3F), −125.05 – −125.39 (m, 1F); HRMS (ESI+): Calcd for C17H11F4N6O + [M+H]+: 391.0930, found 391.0917.
N5-(2-fluorophenyl)- N6-(4-(trifluoromethyl)phenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10v).
Synthesized by general procedure B. 44%, off-white solid. 1H NMR (400 MHz, (CD3)2CO) δ 10.97 (brs, 1H), 9.87 (brs, 1H), 8.60 – 7.85 (brm, 3H), 7.80 (d, J = 8.4 Hz, 2H), 7.35 – 7.22 (m, 3H); 19F NMR (376 MHz, (CD3)2CO) δ −62.52 (s, 3F), −125.19 (s, 1F); HRMS (ESI+): Calcd for C17H10F4N6NaO [M+Na]+: 413.0750, found 413.0740.
N5-(3,5-bis(trifluoromethyl)phenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10w).
Synthesized using General Procedure B. 1H NMR ((CD3)2CO, 400 MHz, asterisk denotes tautomer peaks) δ 11.29 – 10.62* (brs, 1H), 10.32 – 9.54 (brs, 1H), 8.91 – 8.04 (brm, 3H), 7.85 (s, 1H), 7.34 – 7.25 (m, 3H). HRMS (ESI+) m/z calcd for C18H10F7N6O [M+H]+: 459.0799, found 459.0796.
N5-(2-fluoro-3-(trifluoromethyl)phenyl)-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10x).
Synthesized using General Procedure B. 1H NMR ((CD3)2CO, 400 MHz, asterisk denotes tautomer peaks) δ 11.32 – 10.84* (brs, 1H), 9.95 – 9.56 (brs, 1H), 8.57 – 7.67 (brm, 2H), 7.59 (t, 1H, J = 7.7 Hz), 7.50 (t, 1H, J = 7.7 Hz), 7.35 – 7.24 (m, 3H). HRMS (ESI+) m/z calcd for C17H10F5N6O [M+H]+: 409.0831, found 409.0833.
4-Fluoro-3-((6-((2-fluorophenyl)amino)-[1,2,5]oxadiazolo[3,4-b]pyrazin-5-yl)amino)phenol (10y).
Synthesized by general procedure B. 22%, off-white solid. 1H NMR (400 MHz, CH3CN) δ 8.79 (brs, 1H), 8.05 (td, J = 8.1, 2.4 Hz, 1H), 7.41 – 7.24 (m, 3H), 7.12 (dd, J = 11.0, 8.8 Hz, 1H), 6.79 (dd, J = 7.6, 2.9 Hz, 1H), 6.62 – 6.56 (m, 1H), 4.49 (brs, 2H); 19F NMR (376 MHz, CH3CN) δ −124.06 – −124.63 (m, 1F), −138.91 – −139.40 (m, 1F); HRMS (ESI+): Calcd for C16H11F2N6O2+ [M+H]+: 357.0912, found 357.0878.
N5-(2-fluorophenyl)- N6-(pyridin-2-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10z).
Synthesized by general procedure B. 12%, off-white solid. 1H NMR (400 MHz, CH3CN) δ 10.16 (s, 1H), 8.65 (t, J = 8.0 Hz, 1H), 8.18 (d, J = 5.7 Hz, 1H), 8.05 – 7.99 (m, 1H), 7.44 (d, J = 8.7 Hz, 1H), 7.37 – 7.11 (m, 5H); 19F NMR (376 MHz, CH3CN) δ −129.95 – −130.15 (m, 1F); HRMS (ESI+): Calcd for C15H11FN7O + [M+H]+: 324.1009, found 324.0997.
N5-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10aa).
At 0 °C, 9 (1 equiv.) was dissolved in acetonitrile (0.125 M). While stirring, 4 mL of 14.5 M ammonium hydroxide were slowly added. The mixture was stirred for 16 h. Afterwards, the resulting mixture was concentrated under vacuum to obtain the desired product quantitatively. 1H NMR (CD3OD, 400 MHz) δ 7.76 – 7.65 (brm, 1 H), 7.35 – 7.21 (m, 3 H); 19F (CD3OD, 376 MHz) δ −123.91 (s, 1 F). HRMS (ESI+) m/z calcd for C10H8N6OF [M+H]+: 247.0738 found 247.0738.
N5-ethyl-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10ab)
Synthesized using General Procedure B. 92%, yellow solid: 1H NMR (CDCl3, 400 MHz) δ 8.57 (bs, 1H), 7.45 – 7.35 (m, 1H), 7.23 – 7.14 (m, 2H), 7.05 – 6.96 (m, 1H), 3.65 (q, 2H, J = 6.7 Hz), 1.69 (brs, 1H), 1.35 (t, 3H, J = 7.3 Hz); 13C NMR (CDCl3, 100 MHz) δ 153.1, 152.1, 149.9, 142.3, 136.8, 132.2, 126.8, 125.5, 123.3, 117.2, 36.6, 13.9. HRMS (ESI+) m/z calcd for C12H12FN6O [M+H]+: 275.1051, found 275.1051.
N5-(2-fluorophenyl)-N6-propyl-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10ac).
Synthesized using General Procedure B. 77%, yellow solid: 1H NMR (CDCl3, 400 MHz) δ 8.35 (bs, 1H), 7.49 – 7.39 (m, 1H), 7.25 – 7.14 (m, 3H), 7.06 – 6.98 (m, 1H), 3.59 (q, 2H, J = 7.0 Hz), 1.76 (h, 2H, J = 7.4 Hz), 1.04 (t, 3H, J = 7.4 Hz)19F NMR ((CD3)2CO, 376 MHz) δ −123.18 - −123.28 (m, 1F). HRMS (ESI+) m/z calcd for C13H14FN6O [M+H]+: 289.1208, found 289.1200.
N5-(2-fluorophenyl)-N6-(3,3,3-trifluoropropyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10ad).
Synthesized using General Procedure B. 96%, yellow solid: 1H NMR ((CD3)2CO, 400 MHz) δ 8.91 (bs, 1H), 8.40 – 8.01 (m, 1H), 7.93 – 7.65 (m, 1H), 7.44 – 6.96 (m, 3H), 3.98 – 3.89 (m, 2H), 2.86 – 2.71 (m, 2H) HRMS (ESI+) m/z calcd for C13H11F4N6O [M+H]+: 343.0925, found 343.0936.
N5-cyclohexyl-N6-(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10ae).
Synthesized using General Procedure B. 91%, yellow solid: 1H NMR ((CD3)2CO, 400 MHz, δ 7.74 – 7.34 (brm, 2H), 7.32 – 7.19 (m, 2H), 4.25 – 4.11 (brm, 1H), 2.20 – 2.09 (m, 2H), 1.87 – 1.75 (m, 2H), 1.74 – 1.65 (m, 1H), 1.54 – 1.38 (m, 4H), 1.32 – 1.22 (m, 1H); 19F NMR ((CD3)2CO, 3.76 MHz) δ −123.13 (s, 1 F). HRMS (ESI+) m/z calcd for C16H18FN6O [M+H]+: 329.1521, found 329.1526.
N5-(2-fluorophenyl)-N6-(1-methylpiperidin-4-yl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (10af).
Synthesized using General Procedure B. 85%, yellow solid: 1H NMR ((CD3)2CO, 400 MHz) δ 7.74 (brs, 1H), 7.51 (m, 1H), 7.31 – 7.17 (m, 2H), 4.21 – 4.11 (m, 1H), 2.94 (d, 2H, J = 11.7 Hz), 2.31 (s, 3H), 2.26 (t, 2H, J = 12.3 Hz), 2.17 – 2.08 (m, 2H), 1.84 – 1.71 (m, 2H); 19F NMR ((CD3)2CO, 376 MHz) δ −123.27 (s, 1 F). HRMS (ESI+) m/z calcd for C16H19FN7O [M+H]+: 344.1630, found 344.1642.
Oxygen Consumption Rate Seahorse Assay.
Oxygen consumption rate (OCR) was measured using an Agilent Seahorse XF24 or XFe96 Analyzer (Agilent Technologies, Santa Clara, CA). L6 myoblasts were seeded in a Seahorse 24 or 96-well tissue culture plate at a density of 3.5×104 cells/well. The cells were then allowed to adhere overnight. Prior to the assay, the media was changed to unbuffered DMEM containing pyruvate and glutamine (Gibco #12800–017, pH = 7.4 at 37°C) and the cells were equilibrated for 1hr at 37°C without CO2. Compounds were injected during the assay and OCR was measured using 2 min measurement periods. Cells were treated with a single drug concentration per well and measured over a 90 min period. 2 wells were used per condition and, where applicable, results from multiple plates were averaged together. The first three measurements after injection for each concentration were averaged to produce a dose curve. EC50 values were calculated using GraphPad Prism’s non-linear regression built-in equation, Y=Bottom + (X^Hillslope)*(Top-Bottom)/(X^HillSlope + EC50^HillSlope), with the Bottom constrained to the 100% baseline. Area under curve (AUC) values were also calculated using the same software.
STAM Mouse Study.
The STAM mouse model was performed by SMC Laboratories, Inc (Tokyo, Japan). Compound 10b was administered in diet by premixing high-fat diet (HFD, 57 kcal% fat, Cat# HFD32, CLEA Japan Inc., Japan) at SMC Laboratories. Telmisartan (Micardis®) was purchased from Boehringer Ingelheim GmbH (Germany) and dissolved in pure water. NASH was induced in 24 male mice by a single subcutaneous injection of 200 μg streptozotocin (Sigma-Aldrich, USA) solution 2 days after birth and feeding with HFD after 4 weeks of age. From 6 weeks of age, 10b was administered orally by feeding with HFD to achieve a dose of 25 mg/kg/d, and Telmisartan was administered orally in a volume of 10 mL/kg to achieve a dose of 10 mg/kg/d. The viability, clinical signs and behaviour were monitored daily. Body weight was recorded before the treatment. Food intake was measured for each cage of 4 mice every 3–4 days. Mice were observed for significant clinical signs of toxicity, moribundity and mortality approximately 60 minutes after each administration. The animals were sacrificed at 9 weeks of age (Day 21) by exsanguination through direct cardiac puncture under isoflurane anaesthesia.
Non-fasting blood glucose was measured in whole blood using Stat Strip glucose meter (NIPRO CORPORATION, Japan). For plasma biochemistry, non-fasting blood was collected in polypropylene tubes with anticoagulant (Novo-Heparin, Mochida Pharmaceutical Co. Ltd., Japan) and centrifuged at 1,000 xg for 15 minutes at 4°C. The supernatant was collected and stored at −80°C until use. Plasma ALT level was measured by FUJI DRI-CHEM 7000 (Fujifilm, Japan).
For liver samples, the left lateral lobe was collected and cut into 6 pieces. Two pieces of left lateral lobe, left and right medial lobes, and caudate lobe were snap frozen in liquid nitrogen and stored at −80°C. The other 2 pieces of left lateral lobe were fixed in Bouin’s solution and then embedded in paraffin. Samples were stored at room temperature for histology. The remaining pieces of left lateral lobe were embedded in O.C.T. compound and quick frozen in liquid nitrogen. Samples were stored at −80°C. The right lobe was snap frozen in liquid nitrogen and stored at −80°C for liver biochemistry.
Liver total lipid-extracts were obtained by Folch’s method. Liver samples were homogenized in chloroform-methanol (2:1, v/v) and incubated overnight at room temperature. After washing with chloroform-methanol-water (8:4:3, v/v/v), the extracts were evaporated to dryness, and dissolved in isopropanol. Liver triglyceride content was measured by Triglyceride E-test (Wako Pure Chemical Industries, Ltd., Japan).
For histopathology staining, sections were cut from paraffin blocks of liver tissue prefixed in Bouin’s solution and stained with Lillie-Mayer’s Hematoxylin (Muto Pure Chemicals Co., Ltd., Japan) and eosin solution (Wako Pure Chemical Industries). NAFLD Activity score (NAS) was calculated according to the criteria of Kleiner.79 To visualize collagen deposition, Bouin’s fixed liver sections were stained using picro-Sirius red solution (Waldeck, Germany). For quantitative analysis of fibrosis areas, bright field images of Sirius red-stained sections were captured around the central vein using a digital camera (DFC295; Leica, Germany) at 200-fold magnification, and the positive areas in 5 fields/section were measured using ImageJ software (National Institute of Health, USA).
The animals were maintained in a SPF facility under controlled conditions of temperature (23 ± 2°C), humidity (45 ± 10%), lighting (12-hour artificial light and dark cycles; light from 8:00 to 20:00) and air exchange. A high pressure was maintained in the experimental room to prevent contamination of the facility. All animals used in the study were housed and cared for in accordance with the Japanese Pharmacological Society Guidelines for Animal Use.
Plasma assays for STAM model.
Plasma triglyceride was measured by a colorimetric assay through reaction with GPO reagent (Pointe Scientific T7532) according to the manufacturer’s protocol. A standard curve was constructed using serial dilutions of glycerol standard (Sigma G7793). In brief, samples were incubated with GPO at 37°C for 5–20 min until absorbance values at 500 nm had stabilized. Sample plasma triglyceride concentrations were determined through interpolation from the standard curve.
Plasma cholesterol was measured by a colorimetric assay using Infinity Cholesterol Liquid Stable Reagent (ThermoFisher TR13421) according to the manufacturer’s protocol. A standard curve was constructed using serial dilutions of cholesterol standard (Pointe Scientific C7509). In brief, samples were incubated with Infinity Cholesterol Reagent at 37°C for 5–20 min until absorbance values at 500 nm had stabilized. Absorbance at 660 nm was subtracted from absorbance at 500 nm to correct for background. Sample plasma cholesterol concentrations were determined through interpolation from the standard curve.
Plasma AST was measured using Infinity AST (GOT) Liquid Stable Reagent (ThermoFisher TR70121) according to the manufacturer’s protocol. In brief, samples were incubated with the Infinity AST Reagent at 37°C and absorbance was measured at 340 nm every minute for 20 minutes. Activity in U/L was calculated by multiplying the change in absorbance per minute by a correction factor, which was defined as total volume of reaction divided by the product of the molar absorption coefficient of NADH, the sample volume added and the pathlength of absorption.
Western blots for STAM livers.
End point frozen liver tissue was homogenized in RIPA buffer (150 mM sodium chloride, 1.0% NP-40 (Sigma I8896), 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 50 mM Tris, pH 8.0) powdered in liquid nitrogen using a tissue pulverizer (Cellcrusher, USA), then homogenized with a motorized pellet pestle homogenizer in PBS containing protease inhibitors (Sigma, S8830-2TAB, Australia), and phosphatase inhibitors (Sigma, P5726–1ML, Australia) using a mortar and pestle cooled in dry ice. Homogenate was probe sonicated for 120 seconds and then centrifuged (20,000 xg, 10 minutes). Supernatant was collected and protein concentration was quantified using the Pierce Bicinchoninic Acid (BCA) Protein Assay Kit according to the manufacturer’s instructions (ThermoScientific, 23225, Australia).
Protein lysates were mixed with 5 × Laemmli buffer (250 mM Tris (pH 6.8), 25% RO water 10 % (w/v) SDS, 25 % (v/v) glycerol, 0.2 % w/v bromophenol blue, and 5% (v/v) β-mercaptoethanol) and were heated for 5 minutes at 95°C. Protein lysates (15 μg) were resolved on Any kD™ Mini-Protean TGX Precast gels (Bio-Rad) and electro-transferred to nitrocellulose membrane. As a positive control, protein lysate from NMuLi cells treated with 5 μM FCCP for 1 hour to induce AMPK activation was run concurrently. Protein transfer was confirmed using Ponceau S staining. Membranes were blocked with 10% (w/v) skim milk in TBST (Tris-buffered saline with 0.1% (v/v) Tween 20) for 1.5 hours.
Proteins were detected with rabbit antibodies; phospho-AMPKα (Thr172) (Cell Signaling Technology, 2535, USA), Acetyl-coA Carboxylase (ACC) (Cell Signaling Technology, 3676, USA), phospho-ACC (Ser79) (Cell Signaling Technology, 3661, USA), and mouse antibodies AMPK (Santa Crus, sc-74461, USA), 14-3-3 (Santa, Cruz, sc-1657, USA), overnight at 4°C. Primary antibodies were detected with donkey anti-mouse IgG (AlexaFluor790) (Abcam ab186699) or anti-rabbit IgG (AlexaFluor680) (Abcam ab186692) and membranes were scanned on the LI-COR ODYSSEY CLx System (LI-COR, Lincoln, NE, USA). Densitometry was used to calculate the ratio of phosphorylated protein to total protein. Where phosphorylated and total proteins were run on the different gels (ACC), bands were first normalized to density of loading control 14-3-3.
Tissue distribution.
Acute dose studies of 10b were performed at UNSW. All mouse experiments were approved by the UNSW Animal Care and Ethics Committee (project approval 17–66B). Mice were purchased from Australian BioResources (Moss Vale, NSW, Australia). Mice were housed at 22°C in a light/dark cycle of 12 hours. Unless otherwise stated, mice were provided with ad libitum access to water and standard chow diet (Gordons Specialty Feeds, NSW, Australia).
Male C57BL/6 mice at 12 weeks of age were given a bolus of 10b by oral gavage at a dose of 50 mg/kg body weight. Compound 10b was delivered in a mixture containing 90% (v/v) methylcellulose (Sigma, M0512), 2% (v/v) Tween80 (Sigma, P6474) and 8% (v/v) DMSO (Sigma, D5879). Mice were euthanized by cervical dislocation after 1 h, and tissues were dissected, rinsed in PBS and snap-frozen in liquid nitrogen. To process the tissues for liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis, frozen tissues were powdered in liquid nitrogen using a tissue pulverizer (Cellcrusher, USA), then homogenized with a motorized pellet pestle homogenizer. Homogenate was centrifuged (1,000 xg for 10 minutes) and supernatant collected, and added to a solution of 90% (v/v) acetonitrile and 10% (v/v) methanol to extract 10b. The solution was briefly vortexed then centrifuged (18,000 xg for 10 minutes) and supernatant was collected in auto-sampler vials for analysis. Standards were prepared alongside samples by spiking known concentrations (0.1, 1, 10 and 100 ng) of 10b into vehicle treated tissue or serum samples prior to extraction.
LC-MS/MS was performed on a Shimadzu Prominence LCMS-8030 (Shimazdu, Japan). Chromatographic separation was achieved using an ACUITY UPLC BEH, C18 column (Waters, WT186002350, USA). Mobile phase A consisted of 0.1% v/v formic acid in HLPC grade water. Mobile phase B consisted of 0.1% v/v formic acid in acetonitrile. The analyte was eluted with a gradient of 5–80% mobile phase B at a flow rate of 0.4 mL/minute with 10 μL injection volume electrosprayed into the mass spectrometer. ESI was performed in positive mode. Transition of m/z 341 > 162, and 6-minute retention time was used to identify 10b. Quantification was determined by measuring peak areas using LabSolutions Software on the instrument. Concentration of test samples was interpolated from a standard curve derived from the integrated intensity values of standards. Quantified values were normalized to tissue weight.
Body temperature assessment.
Male C57BL/6 mice at 11 weeks of age were given a bolus of 10b by oral gavage at a dose of up to 100 mg/kg body weight, or vehicle. Compound 10b was delivered in a mixture containing 90% (v/v) methylcellulose (Sigma, M0512), 2% (v/v) Tween80 (Sigma, P6474) and 8% (v/v) DMSO (Sigma, D5879). Core body temperature was measured with a rectal probe thermometer (Braintree, TW2-107) at the time points shown.
Gene expression.
Total RNA was extracted from liver tissue using TRI Reagent® according to the manufacturer’s protocol (Sigma-Aldrich, St. Louis, MO, USA). RNA was eluted in 20–50 μL of RNase-free water. RNA quantity and quality were assessed spectrophotometrically using a Nanodrop-1000 (Thermo Fisher Scientific, Wilmington, DE, USA). Analysis of the expression profiles of 7 genes related to fibrosis and 3 housekeeping genes was performed using a Custom NanoString nCounter® panel (NanoString Technologies). The fibrosis genes probed in this custom panel include matrix metallopeptidase 2 (Mmp2, NM_008610.3), tissue inhibitor matrix metalloproteinase 1 (Timp1, NM_001044384.1), collagen 1A1 (Col1a1, NM_007742.4); transforming growth factor β1 (Tgfb1, NM_011577.2), alpha-smooth muscle actin (a-SMA or Acta2, NM_007392.3), cluster of differentiation 146 (Cd146, NM_023061.2), and glial fibrillary acidic protein (Gfap, NM_001131020.1). The housekeeping genes include Ribosomal protein, large, P0 (Rplp0, NM_007475.5), Ribosomal protein L19 (Rpl19, NM_009078.2), and TATA box binding protein (Tbp, NM_013684.3).
In detail, 300 ng of RNA from each sample was hybridised with the nCounter® Custom CodeSet. All procedures related to the NanoString assay, including sample hybridization, detection and scanning were performed by the Ramaciotti Centre for Genomics following the manufacturer’s instructions. Normalization and analysis of NanoString data was carried out using nSolver (V4.0). Raw NanoString counts for each mRNA were subjected to technical normalization using the geometric mean counts obtained from the three in-built positive-control probe sets with the highest counts. Following this, counts for each mRNA were normalised using the geometric mean counts obtained from the three housekeeping genes included in the CodeSet. Inter-PlexSet variability was accounted for by calibration with a reference sample that was run across all eight PlexSets. The statistical significance of differences in the number of normalised counts of mRNA measured in treatment groups compared to the vehicle was determined using one-way ANOVA with Dunnett’s post-hoc test in GraphPad Prism (V8.1.2).
Statistical analysis.
All data are presented as the mean ± standard error of the mean (SEM). Statistical testing was carried out using Prism (v.8.1.2; GraphPad Software), where the threshold for significance was designated as p<0.05, compared to vehicle. For normally distributed data, differences between groups were examined using a One-Way Analysis of Variance (ANOVA) with Dunnett’s post-hoc test for multiple comparisons. For non-parametric data, the Kruskal-Wallis test was conducted with Dunn’s post-hoc test for multiple comparisons. For normally distributed data measured over time with missing values, differences between groups were examined using by Two-Way Repeated Measures ANOVA with Dunnett’s post-hoc test for multiple comparisons.
Supplementary Material
ACKNOWLEDGMENTS
We thank Continuum Biosciences, Inc., Commonwealth of Virginia’s Alzheimer’s and Related Diseases Research Award Fund, and VBHRC Virginia Catalyst for financial support. JSR and CCG are supported by a training grant from NIH (R25GM072767).
ABBREVIATIONS
- ATP
adenosine triphosphate
- pmf
proton motive force
- UCP
uncoupling protein
- T2D
type 2 diabetes
- NASH
nonalcoholic steatohepatitis
- FDA
Food and Drug Administration
- DNP
2,4-Dinitrophenol
- NAFLD
nonalcoholic fatty liver disease
- FCCP
carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone
- SAR
structure–activity relationship
- STZ
streptozotocin, STAM, Stelic animal model
Footnotes
Supporting Information
Additional figures illustrating oxygen consumption rate of 10b in rat primary hepatocytes, body temperature data, plasma lipids and AST from the STAM mouse model, and protein expression results from STAM mouse liver tissues.
The authors declare the following competing financial interest(s): W.L.S. and K.L.H. are inventors of small molecule mitochondrial uncouplers that are licensed to Continuum Biosciences, Inc.
References
- 1.Mitchell P, Chemiosmotic Coupling in Oxidative and Photosynthetic Phosphorylation. BBA-Bioenergetics 2011, 1807, 1507–1538. [DOI] [PubMed] [Google Scholar]
- 2.Khakh BS; Burnstock G, The Double Life of Atp. Sci. Am 2009, 301, 84–90, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loomis WF; Lipmann F, Reversible Inhibition of the Coupling between Phosphorylation and Oxidation. J. Biol. Chem 1948, 173, 807–808. [PubMed] [Google Scholar]
- 4.Weinbach EC; Garbus J, Mechanism of Action of Reagents That Uncouple Oxidative Phosphorylation. Nature 1969, 221, 1016–1018. [DOI] [PubMed] [Google Scholar]
- 5.Rolfe DF; Brand MD, Contribution of Mitochondrial Proton Leak to Skeletal Muscle Respiration and to Standard Metabolic Rate. Am. J. Physiol.-Cell Ph 1996, 271, C1380–C1389. [DOI] [PubMed] [Google Scholar]
- 6.Busiello RA; Savarese S; Lombardi A, Mitochondrial Uncoupling Proteins and Energy Metabolism. Front. Physiol 2015, 6, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erlanson-Albertsson C, The Role of Uncoupling Proteins in the Regulation of Metabolism. Acta Physiol. Scand 2003, 178, 405–412. [DOI] [PubMed] [Google Scholar]
- 8.Ricquier D; Bouillaud F, Mitochondrial Uncoupling Proteins: From Mitochondria to the Regulation of Energy Balance. J. Physiol 2000, 529, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rousset S; Alves-Guerra M-C; Mozo J; Miroux B; Cassard-Doulcier A-M; Bouillaud F; Ricquier D, The Biology of Mitochondrial Uncoupling Proteins. Diabetes 2004, 53, S130. [DOI] [PubMed] [Google Scholar]
- 10.Krauss S; Zhang C-Y; Lowell BB, The Mitochondrial Uncoupling-Protein Homologues. Nat. Rev. Mol. Cell Biol 2005, 6, 248–261. [DOI] [PubMed] [Google Scholar]
- 11.Gimeno RE; Dembski M; Weng X; Deng N; Shyjan AW; Gimeno CJ; Iris F; Ellis SJ; Woolf EA; Tartaglia LA, Cloning and Characterization of an Uncoupling Protein Homolog: A Potential Molecular Mediator of Human Thermogenesis. Diabetes 1997, 46, 900. [DOI] [PubMed] [Google Scholar]
- 12.Nedergaard J; Golozoubova V; Matthias A; Asadi A; Jacobsson A; Cannon B, Ucp1: The Only Protein Able to Mediate Adaptive Non-Shivering Thermogenesis and Metabolic Inefficiency. BBA-Bioenergetics 2001, 1504, 82–106. [DOI] [PubMed] [Google Scholar]
- 13.Echtay KS; Roussel D; St-Pierre J; Jekabsons MB; Cadenas S; Stuart JA; Harper JA; Roebuck SJ; Morrison A; Pickering S; Clapham JC; Brand MD, Superoxide Activates Mitochondrial Uncoupling Proteins. Nature 2002, 415, 96–99. [DOI] [PubMed] [Google Scholar]
- 14.Divakaruni AS; Brand MD, The Regulation and Physiology of Mitochondrial Proton Leak. Physiology 2011, 26, 192–205. [DOI] [PubMed] [Google Scholar]
- 15.Peter K; Robert SP; Ratnasamy S; Nandita S; Peter JOB, Mechanism of Mitochondrial Uncouplers, Inhibitors, and Toxins: Focus on Electron Transfer, Free Radicals, and Structure -Activity Relationships. Curr. Med. Chem 2005, 12, 2601–2623. [DOI] [PubMed] [Google Scholar]
- 16.Terada H, Uncouplers of Oxidative Phosphorylation. Environ. Health Persp 1990, 87, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Childress ES; Alexopoulos SJ; Hoehn KL; Santos WL, Small Molecule Mitochondrial Uncouplers and Their Therapeutic Potential. J. Med. Chem 2018, 61, 4641–4655. [DOI] [PubMed] [Google Scholar]
- 18.Harper JA; Dickinson K; Brand MD, Mitochondrial Uncoupling as a Target for Drug Development for the Treatment of Obesity. Obes. Rev 2001, 2, 255–265. [DOI] [PubMed] [Google Scholar]
- 19.Kalinovich AV; Shabalina IG, Novel Mitochondrial Cationic Uncoupler C4R1 Is an Effective Treatment for Combating Obesity in Mice. Biochemistry-Moscow 2015, 80, 620–628. [DOI] [PubMed] [Google Scholar]
- 20.Suzuki T; Kikuchi H; Ogura M; Homma MK; Oshima Y; Homma Y, Weight Loss by PPC-1, a Novel Small Molecule Mitochondrial Uncoupler Derived from Slime Mold. PLoS ONE 2015, 10, e0117088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tseng Y-H; Cypess AM; Kahn CR, Cellular Bioenergetics as a Target for Obesity Therapy. Nat. Rev. Drug Discov 2010, 9, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perry Rachel J.; Kim T; Zhang X-M; Lee H-Y; Pesta D; Popov Violeta B.; Zhang D; Rahimi Y; Jurczak Michael J.; Cline Gary W.; Spiegel David A.; Shulman Gerald I., Reversal of Hypertriglyceridemia, Fatty Liver Disease, and Insulin Resistance by a Liver-Targeted Mitochondrial Uncoupler. Cell Metab. 2013, 18, 740–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao H; Zhang Y; Zeng X; Shulman GI; Jin S, Niclosamide Ethanolamine–Induced Mild Mitochondrial Uncoupling Improves Diabetic Symptoms in Mice. Nat. Med 2014, 20, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perry RJ; Zhang D; Zhang X-M; Boyer JL; Shulman GI, Controlled-Release Mitochondrial Protonophore Reverses Diabetes and Steatohepatitis in Rats. Science 2015, 347, 1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanemoto N; Okamoto T; Tanabe K; Shimada T; Minoshima H; Hidoh Y; Aoyama M; Ban T; Kobayashi Y; Ando H; Inoue Y; Itotani M; Sato S, Antidiabetic and Cardiovascular Beneficial Effects of a Liver-Localized Mitochondrial Uncoupler. Nat. Commun 2019, 10, 2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Figarola JL; Singhal P; Rahbar S; Gugiu BG; Awasthi S; Singhal SS, Coh-Sr4 Reduces Body Weight, Improves Glycemic Control and Prevents Hepatic Steatosis in High Fat Diet-Induced Obese Mice. PLoS ONE 2013, 8, e83801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fu YY; Zhang M; Turner N; Zhang LN; Dong TC; Gu M; Leslie SJ; Li JY; Nan FJ; Li J, A Novel Chemical Uncoupler Ameliorates Obesity and Related Phenotypes in Mice with Diet-Induced Obesity by Modulating Energy Expenditure and Food Intake. Diabetologia 2013, 56, 2297–2307. [DOI] [PubMed] [Google Scholar]
- 28.Kalinovich AV; Mattsson CL; Youssef MR; Petrovic N; Ost M; Skulachev VP; Shabalina IG, Mitochondria-Targeted Dodecyltriphenylphosphonium (C12TPP) Combats High-Fat-Diet-Induced Obesity in Mice. Int. J. Obesity 2016, 40, 1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuel VT; Liu Z-X; Qu X; Elder BD; Bilz S; Befroy D; Romanelli AJ; Shulman GI, Mechanism of Hepatic Insulin Resistance in Non-Alcoholic Fatty Liver Disease. J. Biol. Chem 2004, 279, 32345–32353. [DOI] [PubMed] [Google Scholar]
- 30.Berry BJ; Trewin AJ; Amitrano AM; Kim M; Wojtovich AP, Use the Proton Motive Force: Mitochondrial Uncoupling and Reactive Oxygen Species. J. Mol. Biol 2018, 430, 3873–3891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demine S; Renard P; Arnould T, Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cadenas S, Mitochondrial Uncoupling, ROS Generation and Cardioprotection. BBA-Bioenergetics 2018, 1859, 940–950. [DOI] [PubMed] [Google Scholar]
- 33.Baffy G, Mitochondrial Uncoupling in Cancer Cells: Liabilities and Opportunities. BBA-Bioenergetics 2017, 1858, 655–664. [DOI] [PubMed] [Google Scholar]
- 34.Urra FA; Muñoz F; Córdova-Delgado M; Ramírez MP; Peña-Ahumada B; Rios M; Cruz P; Ahumada-Castro U; Bustos G; Silva-Pavez E; Pulgar R; Morales D; Varela D; Millas-Vargas JP; Retamal E; Ramírez-Rodríguez O; Pessoa-Mahana H; Pavani M; Ferreira J; Cárdenas C; Araya-Maturana R, Fr58P1A; a New Uncoupler of OXPHOS That Inhibits Migration in Triple-Negative Breast Cancer Cells Via Sirt1/AMPK/B1-Integrin Pathway. Sci. Rep 2018, 8, 13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar R; Coronel L; Somalanka B; Raju A; Aning OA; An O; Ho YS; Chen S; Mak SY; Hor PY; Yang H; Lakshmanan M; Itoh H; Tan SY; Lim YK; Wong APC; Chew SH; Huynh TH; Goh BC; Lim CY; Tergaonkar V; Cheok CF, Mitochondrial Uncoupling Reveals a Novel Therapeutic Opportunity for P53-Defective Cancers. Nat. Commun 2018, 9, 3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang Y; Nasiri AR; Damsky WE; Perry CJ; Zhang X-M; Rabin-Court A; Pollak MN; Shulman GI; Perry RJ, Uncoupling Hepatic Oxidative Phosphorylation Reduces Tumor Growth in Two Murine Models of Colon Cancer. Cell Rep. 2018, 24, 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harper ME; Bevilacqua L; Hagopian K; Weindruch R; Ramsey JJ, Ageing, Oxidative Stress, and Mitochondrial Uncoupling. Acta Physiol. Scand 2004, 182, 321–331. [DOI] [PubMed] [Google Scholar]
- 38.Caldeira da Silva CC; Cerqueira FM; Barbosa LF; Medeiros MHG; Kowaltowski AJ, Mild Mitochondrial Uncoupling in Mice Affects Energy Metabolism, Redox Balance and Longevity. Aging Cell 2008, 7, 552–560. [DOI] [PubMed] [Google Scholar]
- 39.Padalko VI, Uncoupler of Oxidative Phosphorylation Prolongs the Lifespan of Drosophila. Biochemistry-Moscow 2005, 70, 986–989. [DOI] [PubMed] [Google Scholar]
- 40.Antonenko YN; Denisov SS; Khailova LS; Nazarov PA; Rokitskaya T; Tashlitsky VN; Firsov AM; Korshunova GA; Kotova EA, Alkyl-Substituted Phenylamino Derivatives of 7-Nitrobenz-2-Oxa-1,3-Diazole as Uncouplers of Oxidative Phosphorylation and Antibacterial Agents: Involvement of Membrane Proteins in the Uncoupling Action. BBA-Biomembranes 2017, 1859, 377–387. [DOI] [PubMed] [Google Scholar]
- 41.Enoch SJ; Schultz TW; Popova IG; Vasilev KG; Mekenyan OG, Development of a Decision Tree for Mitochondrial Dysfunction: Uncoupling of Oxidative Phosphorylation. Chem. Res. Toxicol 2018, 31, 814–820. [DOI] [PubMed] [Google Scholar]
- 42.Parascandola J, Dinitrophenol and Bioenergetics: An Historical Perspective. Mol. Cell. Biochem 1974, 5, 69–77. [DOI] [PubMed] [Google Scholar]
- 43.Tainter ML; Stockton AB; Cutting WC, Use of Dinitrophenol in Obesity and Related Conditions: A Progress Report. JAMA 1933, 101, 1472–1475. [Google Scholar]
- 44.Grundlingh J; Dargan PI; El-Zanfaly M; Wood DM, 2,4-Dinitrophenol (DNP): A Weight Loss Agent with Significant Acute Toxicity and Risk of Death. J. Med. Toxicol 2011, 7, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geisler GJ, 2,4 Dinitrophenol as Medicine. Cells 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bianchetti A; Pugliatti C; Jori A, On the Hyperthermia Induced by 2,4-Dinitrophenol. Pharmacology 1967, 17, 401–408. [DOI] [PubMed] [Google Scholar]
- 47.McFee RBC, T. R.; McGuigan MA; Reynolds SA; Bellanger P, Dying to Be Thin: A Dinitrophenol Related Fatality. Vet. Hum. Toxicol 2004, 46, 251–254. [PubMed] [Google Scholar]
- 48.Poole FE; Haining RB, Sudden Death from Dinitrophenol Poisoning: Report of a Case with Autopsy. JAMA 1934, 102, 1141–1147. [Google Scholar]
- 49.Bettman JW, Experimental Dinitrophenol Cataract. Am. J. Ophthalmol 1946, 29, 1388–1395. [DOI] [PubMed] [Google Scholar]
- 50.Horner WD, Cataract Following Di-Nitrophenol Treatment for Obesity. JAMA Ophthalmol. 1936, 16, 447–461. [Google Scholar]
- 51.Horner WD, A Study of Dinitrophenol and Its Relation to Cataract Formation. Trans Am. Ophthalmol. Soc 1941, 39, 405–437. [PMC free article] [PubMed] [Google Scholar]
- 52.Rodin FH, Cataracts Following the Use of Dinitrophenol: A Summary of Thirty-Two Cases. Cal West Med 1936, 44, 276–279. [PMC free article] [PubMed] [Google Scholar]
- 53.Patricia CJ; St. John, J. B., Effects of 2,4-Dinitrophenol on Membrane Lipids of Roots. Plant Physiol. 1982, 70, 858–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brismar T; Collins VP, Effect of External Cation Concentration and Metabolic Inhibitors on Membrane Potential of Human Glial Cells. J. Physiol 1993, 460, 365–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buckler KJ; Vaughan-Jones RD, Effects of Mitochondrial Uncouplers on Intracellular Calcium, Ph and Membrane Potential in Rat Carotid Body Type I Cells. J. Physiol 1998, 513, 819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juthberg SKA; Brismar T, Effect of Metabolic Inhibitors on Membrane Potential and Ion Conductance of Rat Astrocytes. Cell. Mol. Neurobiol 1997, 17, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Draber W; Büchel KH; Schäfer G, Quantitative Structure-Activity Studies of Hydrazones, Uncouplers of Oxidative Phosphorylation. In Z. Naturforsch. B, 1972; Vol. 27, p 159. [DOI] [PubMed] [Google Scholar]
- 58.Heytler PG; Prichard WW, A New Class of Uncoupling Agents — Carbonyl Cyanide Phenylhydrazones. Biochem. Biophys. Res. Commun 1962, 7, 272–275. [DOI] [PubMed] [Google Scholar]
- 59.Antalík M; Šturdík E; Sulo P; Propperová A; Mihalovová E; Podhradský D; Dzurila M, Uncoupling Effect of Protonophoric and Nonprotonophoric Analogs of Carbonyl Cyanide Phenylhydrazone on Mitochondrial Oxidative Phosphorylation. Gen. Physiol. Biophys 1988, 7, 517–528. [PubMed] [Google Scholar]
- 60.Park K-S; Jo I; Pak Y; Bae S-W; Rhim H; Suh S-H; Park S; Zhu M; So I; Kim K, Fccp Depolarizes Plasma Membrane Potential by Activating Proton and Na+ Currents in Bovine Aortic Endothelial Cells. Pflügers Archiv 2002, 443, 344–352. [DOI] [PubMed] [Google Scholar]
- 61.To M-S; Aromataris EC; Castro J; Roberts ML; Barritt GJ; Rychkov GY, Mitochondrial Uncoupler Fccp Activates Proton Conductance but Does Not Block Store-Operated Ca2+ Current in Liver Cells. Arch. Biochem. Biophys 2010, 495, 152–158. [DOI] [PubMed] [Google Scholar]
- 62.Chen W; Mook RA; Premont RT; Wang J, Niclosamide: Beyond an Antihelminthic Drug. Cell. Signal 2018, 41, 89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li Y; Li P-K; Roberts MJ; Arend RC; Samant RS; Buchsbaum DJ, Multi-Targeted Therapy of Cancer by Niclosamide: A New Application for an Old Drug. Cancer Lett. 2014, 349, 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Amireddy N; Puttapaka SN; Vinnakota RL; Ravuri HG; Thonda S; Kalivendi SV, The Unintended Mitochondrial Uncoupling Effects of the Fda-Approved Anti-Helminth Drug Nitazoxanide Mitigates Experimental Parkinsonism in Mice. J. Biol. Chem 2017, 292, 15731–15743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Younossi Z; Tacke F; Arrese M; Chander Sharma B; Mostafa I; Bugianesi E; Wai-Sun Wong V; Yilmaz Y; George J; Fan J; Vos MB, Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- 66.Chalasani N; Younossi Z; Lavine JE; Charlton M; Cusi K; Rinella M; Harrison SA; Brunt EM; Sanyal AJ, The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [DOI] [PubMed] [Google Scholar]
- 67.Romero FA; Jones C; Xu Y; Fenaux M; Halcomb RL, The Race to Bash Nash: Emerging Targets and Drug Development in a Complex Liver Disease. J. Med. Chem 2020. [DOI] [PubMed] [Google Scholar]
- 68.Kenwood BM; Weaver JL; Bajwa A; Poon IK; Byrne FL; Murrow BA; Calderone JA; Huang L; Divakaruni AS; Tomsig JL; Okabe K; Lo RH; Coleman GC; Columbus L; Yan Z; Saucerman JJ; Smith JS; Holmes JW; Lynch KR; Ravichandran KS; Uchiyama S; Santos WL; Rogers GW; Okusa MD; Bayliss DA; Hoehn KL, Identification of a Novel Mitochondrial Uncoupler That Does Not Depolarize the Plasma Membrane. Mol. Metab 2014, 3, 114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kenwood BM; Calderone JA; Taddeo EP; Hoehn KL; Santos WL, Structure–Activity Relationships of Furazano[3,4-b]Pyrazines as Mitochondrial Uncouplers. Bioorg. Med. Chem. Lett 2015, 25, 4858–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Konstandaras N; Matto L; Bhadbhade M; Hunter L; Harper JB, Synthesis and Determination of the Pka Values of a Series of Bis(Anilino)Furazano[3, 4-b]Pyrazines. ChemistrySelect 2017, 2, 7018–7023. [Google Scholar]
- 71.Gasco A; Ruà G; Menziani E; Nano GM; Tappi G, Studies in the Chemistry of 1,2,5-Oxadiazole. I. Synthesis of Some Furazanopyrazines from 3,4-Diamino-1,2,5-Oxadiazole. J. Heterocycl. Chem 1969, 6, 769–770. [Google Scholar]
- 72.Starchenkov IB; Andrianov VG, Chemistry of Furazano[3,4-b]Pyrazines. Chem. Heterocycl. Compd 1997, 33, 1219–1233. [Google Scholar]
- 73.Thottempudi V; Yin P; Zhang J; Parrish DA; Shreeve J. n. M., 1,2,3-Triazolo[4,5,-E]Furazano[3,4,-B]Pyrazine 6-Oxide—a Fused Heterocycle with a Roving Hydrogen Forms a New Class of Insensitive Energetic Materials. Chem.–Eur. J 2014, 20, 542–548. [DOI] [PubMed] [Google Scholar]
- 74.Divakaruni AS; Paradyse A; Ferrick DA; Murphy AN; Jastroch M, Chapter Sixteen - Analysis and Interpretation of Microplate-Based Oxygen Consumption and Ph Data. In Methods in Enzymology, Murphy AN; Chan DC, Eds. Academic Press: 2014; Vol. 547, pp 309–354. [DOI] [PubMed] [Google Scholar]
- 75.Alam S; Kabir J; Mustafa G; Gupta U; Hasan S; Alam A, Effect of Telmisartan on Histological Activity and Fibrosis of Non-Alcoholic Steatohepatitis: A 1-Year Randomized Control Trial. Saudi J. Gastroenterol 2016, 22, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ejima C; Kuroda H; Ishizaki S, A Novel Diet-Induced Murine Model of Steatohepatitis with Fibrosis for Screening and Evaluation of Drug Candidates for Nonalcoholic Steatohepatitis. Physiol. Rep 2016, 4, e13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hirata T; Tomita K; Kawai T; Yokoyama H; Shimada A; Kikuchi M; Hirose H; Ebinuma H; Irie J; Ojiro K; Oikawa Y; Saito H; Itoh H; Hibi T, Effect of Telmisartan or Losartan for Treatment of Nonalcoholic Fatty Liver Disease: Fatty Liver Protection Trial by Telmisartan or Losartan Study (FANTASY). Int. J. Endocrinol 2013, 2013, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Abulizi A; Perry RJ; Camporez JPG; Jurczak MJ; Petersen KF; Aspichueta P; Shulman GI, A Controlled-Release Mitochondrial Protonophore Reverses Hypertriglyceridemia, Nonalcoholic Steatohepatitis, and Diabetes in Lipodystrophic Mice. FASEB J. 2017, 31, 2916–2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kleiner DE; Brunt EM; Van Natta M; Behling C; Contos MJ; Cummings OW; Ferrell LD; Liu Y-C; Torbenson MS; Unalp-Arida A; Yeh M; McCullough AJ; Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research, N., Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.