

SUMMARY
Dosage compensation in Drosophila melanogaster involves a 2-fold transcriptional upregulation of the male X chromosome, which relies on the X-chromosome-binding males-specific lethal (MSL) complex. However, how such 2-fold precision is accomplished remains unclear. Here, we show that a nuclear pore component, Mtor, is involved in setting the correct levels of transcription from the male X chromosome. Using larval tissues, we demonstrate that the depletion of Mtor results in selective upregulation at MSL targets of the male X, beyond the required 2-fold. Mtor and MSL components interact genetically, and depletion of Mtor can rescue the male lethality phenotype of MSL components. Using RNA fluorescence in situ hybridization (FISH) analysis and nascent transcript sequencing, we find that the effect of Mtor is not due to defects in mRNA export but occurs at the level of nascent transcription. These findings demonstrate a physiological role for Mtor in the process of dosage compensation, as a transcriptional attenuator of X chromosome gene expression.
Graphical Abstract
In brief
During dosage compensation, the expression from the single X chromosome of male flies is upregulated to match the two X’s of females. Aleman et al. show that a nuclear pore protein functions in achieving this transcriptional precision and restricts the upregulation of the male X to the required 2-fold.
INTRODUCTION
Dosage compensation (DC) is an essential process for equalizing gene expression between the sexes. Gene dosage in opposite sexes of many species differs due to the hemizygosity of the X chromosome in males as opposed to homozygosity of the X in females. Various species have acquired unique mechanisms for equalizing the expression of the X chromosomes, and a well-known paradigm of this process is the random inactivation of one of the female X chromosomes in mammals (Galupa and Heard, 2015). Unlike mammals, the single male X chromosome in Drosophila undergoes a 2-fold transcriptional upregulation to match gene expression levels produced from the 2 X chromosomes in the female (Ferrari et al., 2014). The transcriptional upregulation of the Drosophila male X depends on the binding and activity of the males-specific lethal (MSL) complex, which contains a histone acetyltransferase (HAT) males absent on the first (MOF) and 2 long non-coding RNAs (lncRNAs) roX1 and roX2 (Conrad and Akhtar, 2012; Gelbart and Kuroda, 2009). MOF is responsible for depositing high levels of the histone H4 lysine K16 acetylation (H4K16ac) mark along the compensated male X chromosome (Morales et al., 2004; Smith et al., 2000), although it is not entirely understood how the deposition of this mark supports the 2-fold transcriptional upregulation. Several recent studies aimed to elucidate the mechanism of the male X transcriptional upregulation by investigating which step of the transcriptional process is regulated by DC (Conrad et al., 2012; Ferrari et al., 2013; Larschan et al., 2011), and one model that emerges from these studies highlights transcription elongation as a key regulatory point (Ferrari et al., 2013). How this upregulation is restrained to 2-fold remains an open question, with far-reaching implications for our general understanding of chromosome-wide regulation and of transcriptional precision.
It has been hypothesized that additional biological machinery may regulate the 2-fold attenuation of gene expression in Drosophila DC (Mendjan and Akhtar, 2007). Biochemically, the components of other nuclear complexes have been identified as interacting partners of the MSL complex (Mendjan et al., 2006). One such factor is the component of the nuclear pore complex (NPC), nucleoporin (Nup) Megator (Mtor), which has been shown to co-immunoprecipitate with MOF (Mendjan et al., 2006) and to be preferentially enriched along the male X chromosome in S2 culture cells (Vaquerizas et al., 2010). However, the functional significance of these interactions has been debated—in particular, whether Mtor has any direct effects on dosage-compensated gene expression (Grimaud and Becker, 2009; Mendjan et al., 2006). Some of the conflicting results have been attributed to differences in the cell type and the experimental conditions used, and it presently remains unclear whether Mtor plays a functional role in DC.
Mtor is 1 of 30 Nups that make up the NPC, a massive protein complex that forms a nuclear membrane-embedded channel that mediates nuclear-cytoplasmic transport (Wente and Rout, 2010). The nuclear basket structure of the NPC comprises Mtor and contains extensive coiled-coil domains that can support the formation of long filaments (Qi et al., 2004; Snow and Paschal, 2014). In multiple systems, homologs of Mtor have been shown to have a substantial intranuclear presence, often as seemingly filamentous structures that can come in contact with chromatin (Arlucea et al., 1998; Fontoura et al., 2001; Kosova et al., 2000; Strambio-de-Castillia et al., 1999; Zimowska et al., 1997). The filamentous nature of Mtor is also consistent with the reported chromatin binding of Mtor in long continuous domains, termed nucleoporin-associated regions (NARs) in Drosophila cells (Vaquerizas et al., 2010). Functionally, yeast homologs of Mtor, Mlp1, and Mlp2 have been shown to regulate RNA biogenesis processes, such as mRNA quality control upon export (Fasken and Corbett, 2005; Galy et al., 2004), although the loss of Mlp1/2 does not appear to affect mRNA export itself (Kosova et al., 2000; Strambio-de-Castillia et al., 1999). Interestingly, Mlp1/2 have also been linked to the repression of transcription in response to aberrant mRNA export, implicating Mlp1/2 in the maintenance of proper gene expression levels (Vinciguerra et al., 2005).
Multiple NPC components have been shown to functionally contribute to gene regulation via physical interactions with chromatin, both at the nuclear periphery and in the nuclear interior (Ptak and Wozniak, 2016; Raices and D’Angelo, 2017; Sood and Brickner, 2014). For example, the loss of Drosophila Nups Nup98 and Sec13 can decrease the transcriptional output of developmental target genes (Capelson et al., 2010; Kalverda et al., 2010; Pascual-Garcia et al., 2014). A growing number of studies have also pointed to an alternative role of select NPC components in gene silencing (Jacinto et al., 2015; Labade et al., 2016; Van de Vosse et al., 2013). Mechanistically, Nups have been proposed to act as modulators of gene regulatory outputs, increasing the transcriptional response of inducible genes (Light et al., 2013; Pascual-Garcia et al., 2017; Raices et al., 2017; Taddei et al., 2006; Tan-Wong et al., 2009) or stabilizing silent states of heterochromatic regions (Gozalo et al., 2020; Iglesias et al., 2020). Although multiple Nups have been studied in this context, the precise roles of Mtor in gene expression have not been thoroughly explored.
Here, we set out to characterize the gene regulatory role of Mtor in the context of Drosophila DC. We performed many of our assays in the larval salivary gland, which is non-dividing, thus allowing functional separation from mitotic defects, previously associated with loss of Mtor (Lince-Faria et al., 2009; Qi et al., 2004). By specifically depleting Mtor in the salivary gland, we have uncovered a male-specific role for Mtor in the restriction of X chromosome-linked dosage-compensated gene expression to the required 2-fold level. This Mtor-driven modulation was found to occur specifically at the level of nascent transcription. In the larger sense, our findings identify a role for a structural nuclear basket component in controlling the correct level of chromosome-wide transcriptional output.
RESULTS
Loss of Mtor leads to overcompensation of X-linked genes in Drosophila males
To investigate the effects of Mtor on dosage-compensated gene expression in vivo, we generated Mtor RNAi mosaics in the salivary gland using the FLP-FRT and UAS-Gal4 systems (Figure 1A) (see STAR Methods). The use of mosaic tissue provided a powerful system since control cells could be compared directly to RNAi-depleted cells in the same tissue, bypassing sample and developmental variability. In this system, the presence of GFP marks nuclei with Mtor RNAi (Figures 1A and 1B), where we observed the robust depletion of Mtor, assessed by immunofluorescence (IF) with a previously characterized antibody to Mtor (Qi et al., 2004) (Figure 1A). Consistent with previous reports (Qi et al., 2004; Zimowska et al., 1997), we observed wild-type (WT) Mtor localized both at the nuclear periphery and intranuclearly (Figures 1A and S1A). To begin addressing the role of Mtor in DC and based on previous links of Mtor to RNA biogenesis (Galy et al., 2004; Vinciguerra et al., 2005), we performed single-molecule RNA fluorescence in situ hybridization (FISH) (smRNA FISH) for roX1, a lncRNA part of the MSL complex. Interestingly, although we did not observe drastic changes in roX1 targeting to the male X chromosome, we found that in addition to X chromosome localization, extranuclear soluble roX1 was present upon Mtor knockdown (KD) (Figure 1B). Consistently, the quantified X chromosome-associated roX1 fluorescence signal was not significantly changed, but the nuclear soluble roX1 signal was increased in Mtor-depleted nuclei (Figure 1C). To verify normal roX1 binding to the X chromosome, we performed smRNA FISH on polytene chromosome squashes and again found no difference in roX1 signal on the X chromosome. We did, however, observe an increase in roX1 signal associated with the autosomes, representative of the extra presence of roX1 in Mtor-depleted conditions (Figure S1C).
Figure 1. Loss of Mtor leads to overcompensation of X chromosome targets in Drosophila males.
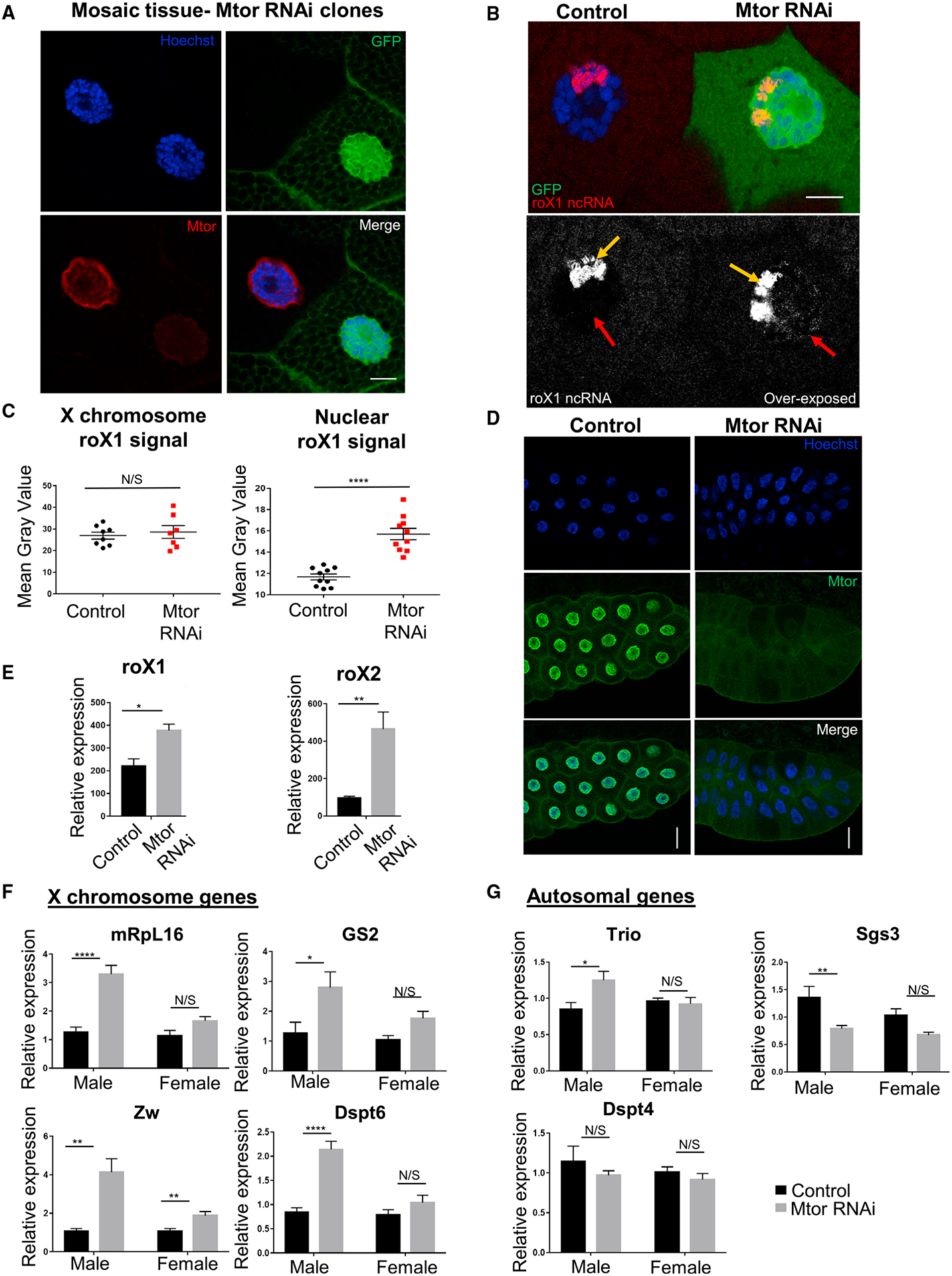
(A) IF staining of larval salivary gland nuclei with Mtor RNAi mosaics. Mtor-depleted nucleus is marked with GFP. Antibodies are as labeled. Details on genetics are outlined in text and STAR Methods. Scale bar, 12 μm.
(B) SmRNA FISH for roX1 lncRNA (red or white) in GFP-marked Mtor RNAi mosaic salivary glands. The bottom panel shows roX1 overexposed to reveal X chromosome-localized roX1 (yellow arrow) and nuclear soluble roX1 (red arrow). Scale bar, 12 μm.
(C) Image quantification of X chromosome-localized and nuclear soluble rox1. N = 7–10 images from 2 animals. Error bars represent SEMs here and in the rest of the figure; ****p < 0.0001, N/S p > 0.05, using a paired t test.
(D) Salivary gland depletion of Mtor, induced by Nub-Gal4. Control or WT is the Nub-Gal4 stock for all experiments unless otherwise specified. Antibodies are as labeled. Scale bar, 45 μm.
(E) Expression levels of roX1 and roX2 from male salivary glands via qRT-PCR, normalized to rp49 (here and for all qRT-PCR). N = 4 biological replicates per condition, 10 glands per replicate. *p < 0.05, **p < 0.01, using an unpaired t test.
(F) Expression levels of X chromosome genes via qRT-PCR from control or Mtor KD male and female larval salivary glands, normalized to female controls. N = 6 biological replicates per condition, 10 glands per replicate. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, N/S is p > 0.05, using 1-way ANOVA, followed by the Holm-Sidak test for multiple comparisons.
(G) Expression levels of autosomal genes via qRT-PCR from samples and with p values as described in (F).
To determine whether the observed extra presence of nuclear roX1 was due to its increased expression, we generated whole salivary gland KD of Mtor using the Nubbin-Gal4 (Nub-Gal4) driver, which resulted in a robust depletion of Mtor (Figures 1D and S1B). qRT-PCR experiments on the Mtor-depleted versus control salivary glands from male larvae for roX1 and roX2 revealed an increase in the relative expression of both (Figure 1E), providing an explanation for the increase in nuclear soluble roX1 seen by smRNA FISH (Figures 1B and 1C). We did not observe disruption of the general nuclear structure and integrity in Mtor-depleted salivary glands, as shown by normal protein import and chromatin targeting of the ecdysone receptor (EcR), which is imported into the nucleus in late 3rd instar larvae (Johnston et al., 2011), and of heterochromatin protein 1 (HP1), which serves as a marker of heterochromatin integrity (Powers and Eissenberg, 1993), upon Mtor KD (Figures S1D and S1E).
We next tested whether expression of the other X chromosome target genes was similarly affected and whether these differences in expression were sex specific. We performed qRT-PCR for X chromosome targets known to be dosage compensated (Chiang and Kurnit, 2003; Furuhashi et al., 2006) and for autosomal control genes in the salivary glands of both males and females in control and Mtor KD conditions. Upon Mtor KD, we observed a male-specific increase in X chromosome target gene expression in comparison to the more modest or not significant changes in the female (Figure 1F); tested autosomal genes did not follow the same male-specific trend (Figure 1G). Lastly, we tested for any male-specific phenotypes of Mtor at the organismal level, using adult Mtor RNAi mosaics (see Figures S1F and S1G for full genotypes). We induced Mtor KD by heat shock and performed fertility assays with the resulting F1 males and females compared to un-induced male controls. We observed a male-specific fertility defect in the induced Mtor RNAi mosaic flies, with 50% of such males being sterile (Figures S1F and S1G), supporting the male-specific role of Mtor in gene expression.
Depletion of Mtor results in selective upregulation of the male X chromosome
To determine the extent of male-specific overcompensation upon Mtor KD, we next performed RNA sequencing (RNA-seq) on male and female larval salivary glands in control and Nub-Gal4-driven Mtor KD conditions. When comparing differentially expressed genes (DEGs) in Mtor-depleted versus WT conditions in both sexes, we observed a widespread male-specific upregulation of gene expression on the X chromosome (Figure 2A). In contrast, female gene expression changes across the X chromosome were more equally divided between up- and downregulation (Figure 2A). Similarly, a genomic plot of all DEGs in the male revealed a prevailing pattern of upregulated genes across the X chromosome, which was unique in comparison to the autosomes (Figure 2B). The upregulation across the male X was robust and reproducible, as seen in the high degree of similarity between replicates (Figure 2C). In regions across the X where upregulation was occurring in the male, upregulation was also often observed in the female, but it was generally much less pronounced (Figure S2A), which is consistent with our qRT-PCR experiments (Figure 1F). Overall, the male X chromosome had the largest number of gene expression changes in the genome, and most of these were in the upregulated direction (Figure 2D), in comparison to autosomal changes and changes seen in the female. Significantly, a large number of autosomal genes remained unperturbed by Mtor RNAi in both sexes (examples in Figure S2B), arguing against a general disruption of nuclear processes. A comparison of DEGs in male versus female Mtor KD highlighted the unique upregulation of genes across the male X chromosome (since the majority of DEGs across the autosomes effectively canceled each other out) (Figures 2D and S2C), while the same comparison between male and female WT revealed very few differences (Figure S2D). These data reveal that one of the main effects of Mtor on gene expression is attenuation across a large portion of the male X chromosome, further linking Mtor to the process of DC.
Figure 2. Depletion of Mtor results in selective upregulation of the male X chromosome.
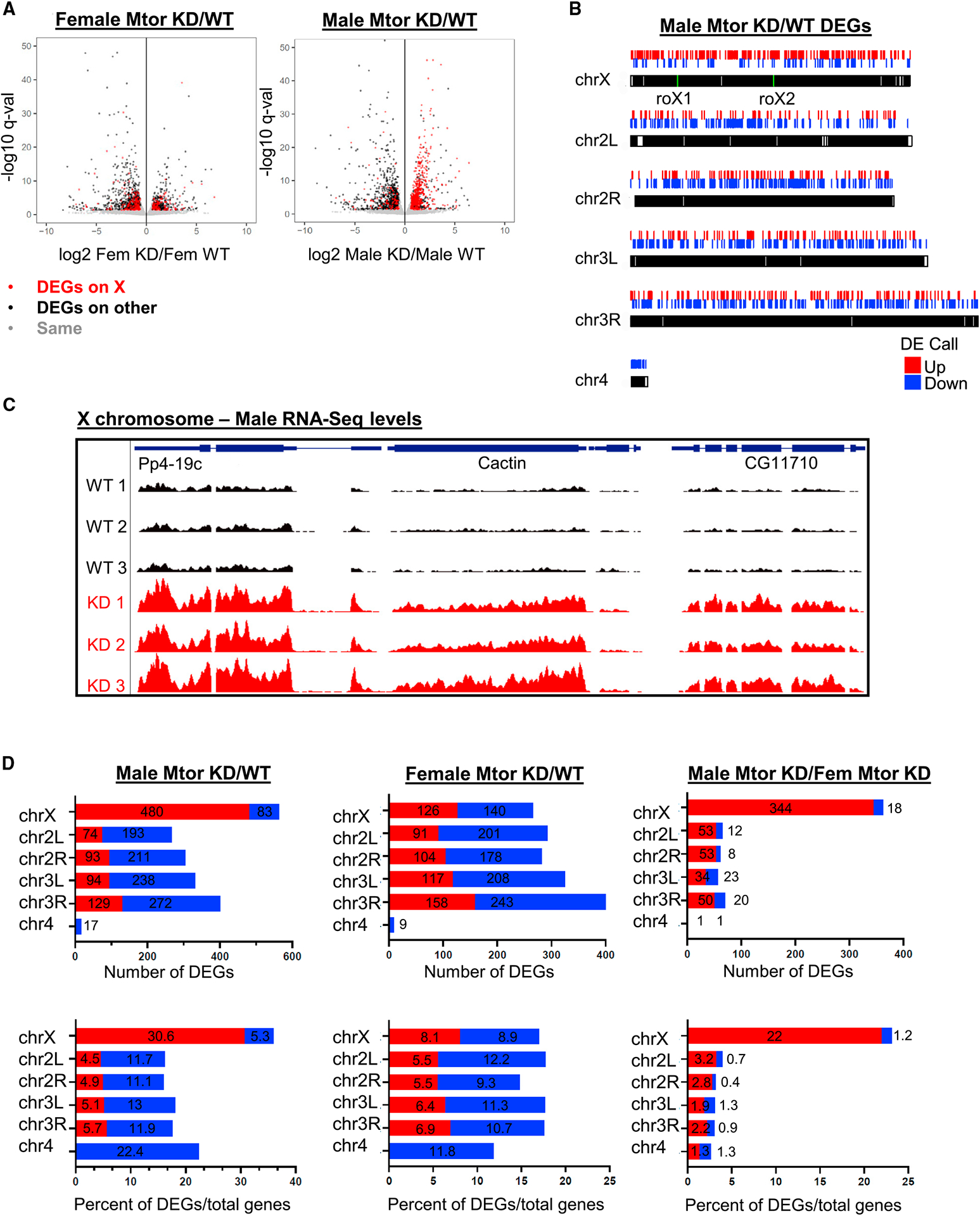
(A) Volcano plot showing differentially expressed genes (DEGs) in females and males in salivary glands with Nub-Gal4-driven Mtor RNAi (KD) relative to WT controls, assessed by RNA-seq. Differential gene analysis was performed using DESeq2. Genes with a false discovery rate (FDR) < 0.05 and fold change > 1.5 were defined as DEGs.
(B) Genomic plot showing DEGs, obtained from RNA-seq, along chromosomes in males. roX1 and roX2 locations are labeled in green.
(C) Genome browser (GB) snapshot of RNA-seq reads in control and Mtor KD male replicates at an X-linked region. Displayed region is 9.1 kb and scale is 0–50.
(D) Numbers of DEGs per chromosome (top row) and percentages of DEGs out of total number of genes per chromosome (bottom row) in Mtor KD versus control males and females, as well as Mtor KD males versus females, obtained from RNA-seq analysis.
Mtor-driven attenuation of the male X chromosome occurs in broad domains bound by MOF
As outlined above, we found a large fraction of genes on the male X chromosome to be upregulated in Mtor-depleted tissues, and we noticed that such genes tend to be upregulated in clustered groups, reminiscent of MSL binding. When we excluded singleton DEGs in our analysis and only plotted groups of ≥2 nearby DEGs, we still observed a widespread upregulation of gene expression across the male X (Figure 3A). When normalized to the total number of DEGs per chromosome, the male X still exhibited the highest fraction of upregulated grouped DEGs in Mtor KD (Figure S3A), suggesting that this pattern is not simply due to the number of genes on a chromosome. By genome-wide analysis, we found these regions of upregulation across the male X correlate with chromatin binding domains of MOF, the HAT of the MSL complex (Conrad et al., 2012) (Figures 3B and 3C). Consistently, we found that upregulated DEGs on the male X correlate with significantly higher MOF chromatin immunoprecipitation sequencing (ChIP-seq) signal than DEGs that are downregulated or unchanged (Figure S3B). Furthermore, X-linked genes that contained MOF binding were significantly more upregulated in Mtor-depleted males than genes that did not (Figures 3B, S3C, and S3D). We did not observe as much of a correlation between MOF binding and DEG upregulation when looking at the whole genome (Figure S3C), suggesting that the effect of Mtor on MOF/MSL targets is most pronounced on the X chromosome. We detected a similar correlation between X-linked DEGs and peaks of H4K16ac, the histone modification deposited by MOF (Figures S3E and S3F). In addition, we correlated the upregulated X-linked DEGs with high-affinity sites (HAS), defined by the Becker (Straub et al., 2008) and Kuroda (Alekseyenko et al., 2008) labs as sites that initially recruit the MSL complex. We found that ~60% of upregulated DEGs on the male X overlap with HAS as defined in these studies (Figure S3G), further supporting the notion that Mtor restricts the expression of chromatin domains bound by MSL.
Figure 3. Mtor-driven attenuation of the male X chromosome occurs in MOF-bound domains.
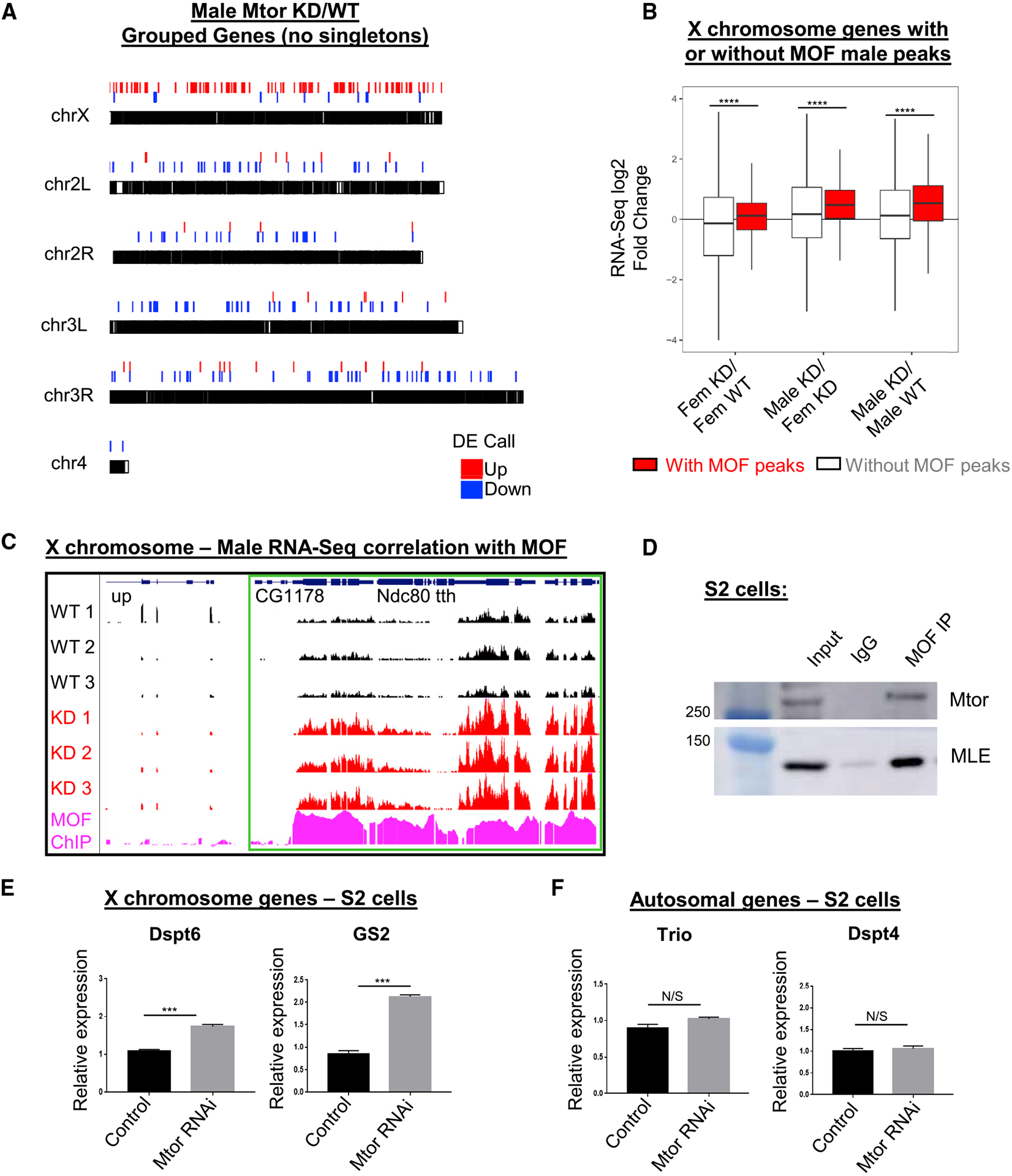
(A) Genomic plot of grouped DEGs (groups of ≥2, no singletons) in Mtor KD versus control males, from RNA-seq described in Figure 2.
(B) Boxplot showing correlation of MOF ChIP-seq (Conrad et al., 2012) with RNA-seq change for X-linked genes in male and female Mtor KD versus controls. Error bars represent SEMs; ****p < 0.0001, using 2-sample Wilcoxon tests.
(C) GB snapshot of RNA-seq tracks in control and Mtor KD male replicates, showing upregulated domain of 4 adjacent genes (outlined in green box), which corresponds to MOF ChIP-seq signal. The displayed region is 21.5 kb.
(D) IP with antibodies to MOF and control immunoglobulin Gs (IgGs) in S2 cells, western blotted as indicated.
(E) Expression levels of X chromosome targets via qRT-PCR from S2 cells in control and Mtor KD conditions. N = 3 biological replicates per condition. Error bars represent SEM; ***p < 0.001, N/S is p > 0.05, using an unpaired t test.
(F) Expression levels of autosomal targets via qRT-PCR from S2 cells described in (E).
It has been reported that MOF interacts with Mtor biochemically in both Drosophila and human cells (Mendjan et al., 2006), and we were able to confirm this interaction by co-immunoprecipitation (coIP) experiments in S2 cells, which have a male karyotype (Figure 3D). Importantly, qRT-PCR experiments also showed an upregulation of tested X-linked genes in S2 cells, treated with double-stranded RNA (dsRNA) against Mtor, as opposed to no effect on autosomal targets and relative to control cells, treated with dsRNA against gene White (dsWhite) (Figures 3E, 3F, and S3H). Although the observed increase in the expression of X targets was less pronounced than the increase seen in tissues, it was nonetheless reproducible and significant. These results support the notion that Mtor co-functions with MOF to restrict dosage-compensated expression across different cell types.
Mtor and MSL complex components cooperate in setting the levels of dosage-compensated expression
To determine whether Mtor cooperates with MSL components to regulate X gene expression, we performed genetic rescue experiments in the fly system. We combined the UAS-Mtor RNAi line with a UAS-driven MOF RNAi line or roX1/roX2 loss-of-function mutations (Apte et al., 2014) genetically, and tested whether combining these elements would rescue the Mtor phenotype of upregulation of X gene expression. The crossing schemes and their expected effects on male X gene expression are detailed in Figure 4A. To control for an additional UAS element, the Nub-Gal4 driver was also crossed to a UAS-Mtor RNAi;UAS-mCD8 RFP line, which exhibited similarly overcompensated X gene expression (Figures 4A and 4B). Mutations of MSL components are known to disrupt DC, leading to the under-compensation of X gene expression (Deng and Meller, 2006; Hamada et al., 2005; Straub et al., 2005). As expected, MOF RNAi or the roX1/roX2 mutations alone lowered X gene expression relative to control conditions, as assessed by qRT-PCR on the salivary glands of male larvae (Figures 4B, 4C, and S4A). Strikingly, combining Mtor RNAi with either MOF RNAi or roX1/roX2 resulted in a rescue of upregulated X gene expression to nearly WT levels (Figures 4B and 4C), demonstrating a genetic interaction between Mtor and MSL complex components.
Figure 4. Mtor and MSL components cooperate in vivo to set the levels of dosage-compensated expression.
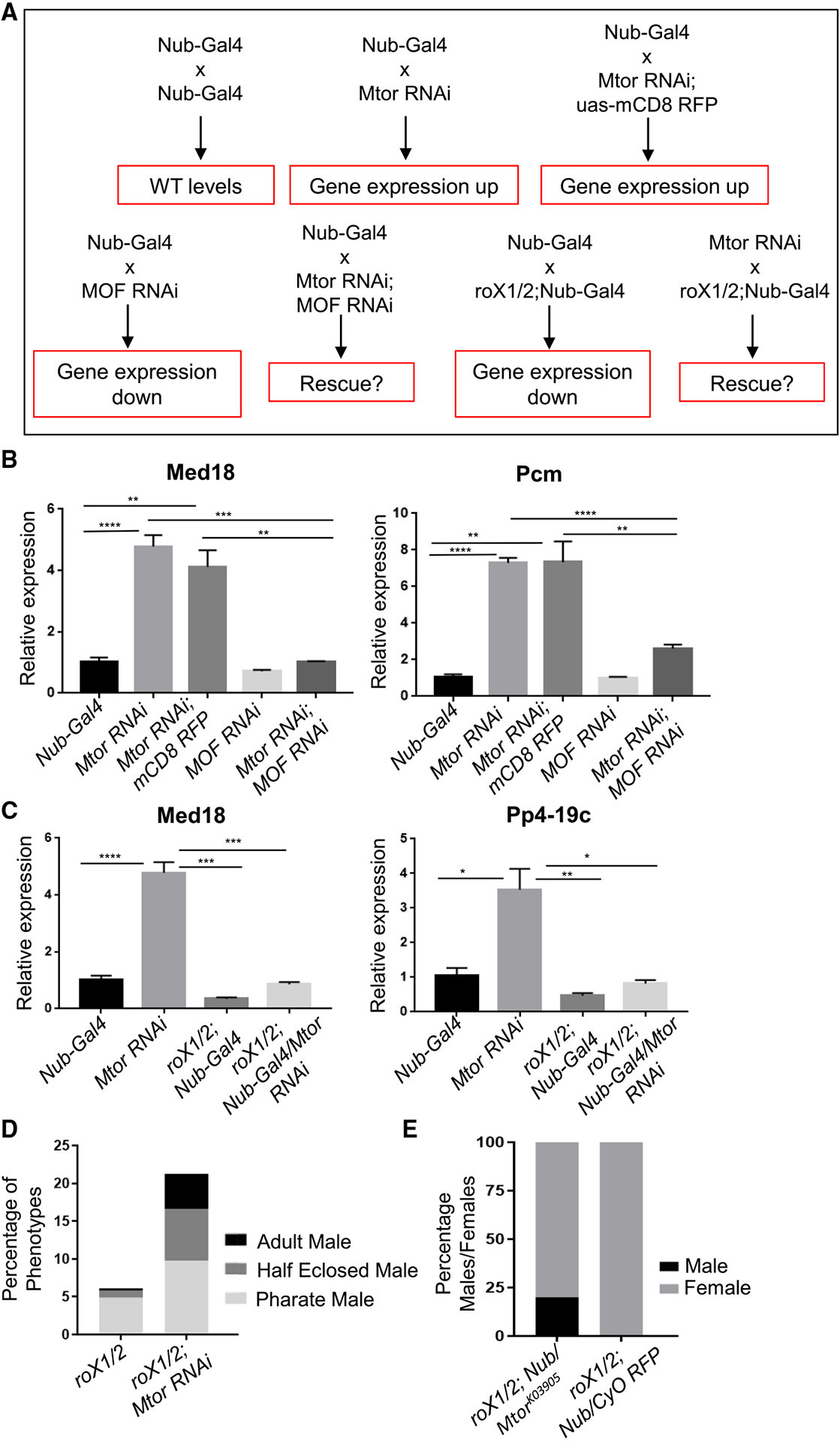
(A) Schematic of the genetic crosses used in the rescue experiments and the expected outcomes in male X chromosome expression (red boxes).
(B) Normalized expression levels of X chromosome targets via qRT-PCR on male salivary glands of indicated genotypes, for MOF/Mtor rescue experiments outlined in (A). N = 3–4 biological replicates, 10 salivary glands per replicate. Error bars represent SEMs. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001, using an unpaired t test.
(C) Normalized expression levels of X chromosome targets via qRT-PCR on male salivary glands of indicated genotypes, for rox1/2/Mtor rescue experiments outlined in (A). Biological replicates, SEMs, and p values as in (B).
(D) Adult viability outcomes of male F1 progeny from rescue experiments with Mtor RNAi and roX1/roX2 mutants, shown as percentage of total F1. See Figure S4C for F1 numbers and STAR Methods for details on scoring.
(E) Percentages of male and female F1 progeny from rescue experiments with MtorK03905 and roX1/roX2 mutants. See Figure S4D for F1 numbers and STAR Methods for details.
In these experiments, we also noticed that Mtor RNAi seemed to rescue the phenotype of roX1/roX2 mutants, bringing expression back up to WT levels (Figures 4C and S4B). We thus asked whether Mtor depletion can also rescue the known male lethality phenotype of roX1/roX2 mutants (Kuroda et al., 1991; Meller and Rattner, 2002). As previously reported (Apte et al., 2014; Meller and Rattner, 2002), we observed a pronounced male lethality in the roX1/roX2 mutant strain (see STAR Methods for details), such that only a very small proportion of males make it to the pupal-pharate or adult viable stages (Figures 4D and S4C). When the Nub-Gal4-driven Mtor RNAi was introduced into the roX1/roX2 background, we observed a dramatic increase in the survival of male progeny for both full viability and survival to pharate and pharate-to-adult transition (scored as “half-eclosed” males) stages (Figures 4D and S4C). In addition, we used a loss-of-function allele of Mtor (Qi et al., 2004), heterozygous over the CyO balancer marked with red fluorescent protein (RFP), and combined it with the same roX1/roX2 mutant line. In this genetic assay, the only males that we observed developing to the pharate stage were the roX1/roX2;Mtor/+ double mutants, which were RFP−, although similar numbers of RFP+ and RFP−females eclosed from these crosses (Figures 4E and S4D). These results demonstrate that Mtor depletion can partially rescue the male-specific lethality of classical MSL mutants. To our knowledge, there has only been one other report of a similar genetic rescue of MSL mutations by a non-MSL complex component, the overcompensating males (ocm) gene, whose function was similarly proposed to involve restricting the activity of MSL (Lim and Kelley, 2013).
Role of Mtor in dosage compensation is independent of H4K16 acetylation
Given that MOF deposits the H4K16ac mark, we next asked whether the mechanism by which Mtor affects gene expression was through modulating the levels of H4K16ac. To address this question, we stained the polytene chromosome for H4K16ac by IF in Mtor KD males and controls and found no obvious differences in the levels of H4K16ac on the X chromosome (Figure 5A). To confirm our assessment, we normalized H4K16ac fluorescence intensity, measured as mean gray value, on the X chromosome to autosomal signal on chromosome 3R (identified by EcR staining at 2 distinct puffs at the E74 and E75 loci; Capelson et al., 2010) (Figures 5A and 5B). This quantification approach showed the expected lack of enrichment on the X and an X:autosome intensity ratio of 1 in the female control, as opposed to the X:autosome intensity ratio of ~2 in both control and Mtor KD males (Figure 5B). Importantly, it confirmed no significant difference in the X:autosome H4K16ac ratio between control and Mtor KD males (Figure 5B). In support of this conclusion, we found no change in the levels of MOF itself on polytene chromosomes from Mtor-depleted males relative to controls (Figures 5C and 5D). To further confirm the lack of effect on H4K16ac, we assessed whether total H4K16ac levels were affected by Mtor depletion in male S2 cells. To this end, we extracted histones from S2 cells treated with dsWhite or dsMtor RNA and observed no significant difference in global levels of H4K16ac by western blotting, as normalized to the histone H3 antibody signal (Figures S5A–S5C). The lack of correlation between changes in fly X chromosome H4K16ac and X chromosome expression has been previously reported in other contexts (Sun and Birchler, 2009), which provides further validity to our results.
Figure 5. Role of Mtor in dosage compensation is independent of H4K16 acetylation.
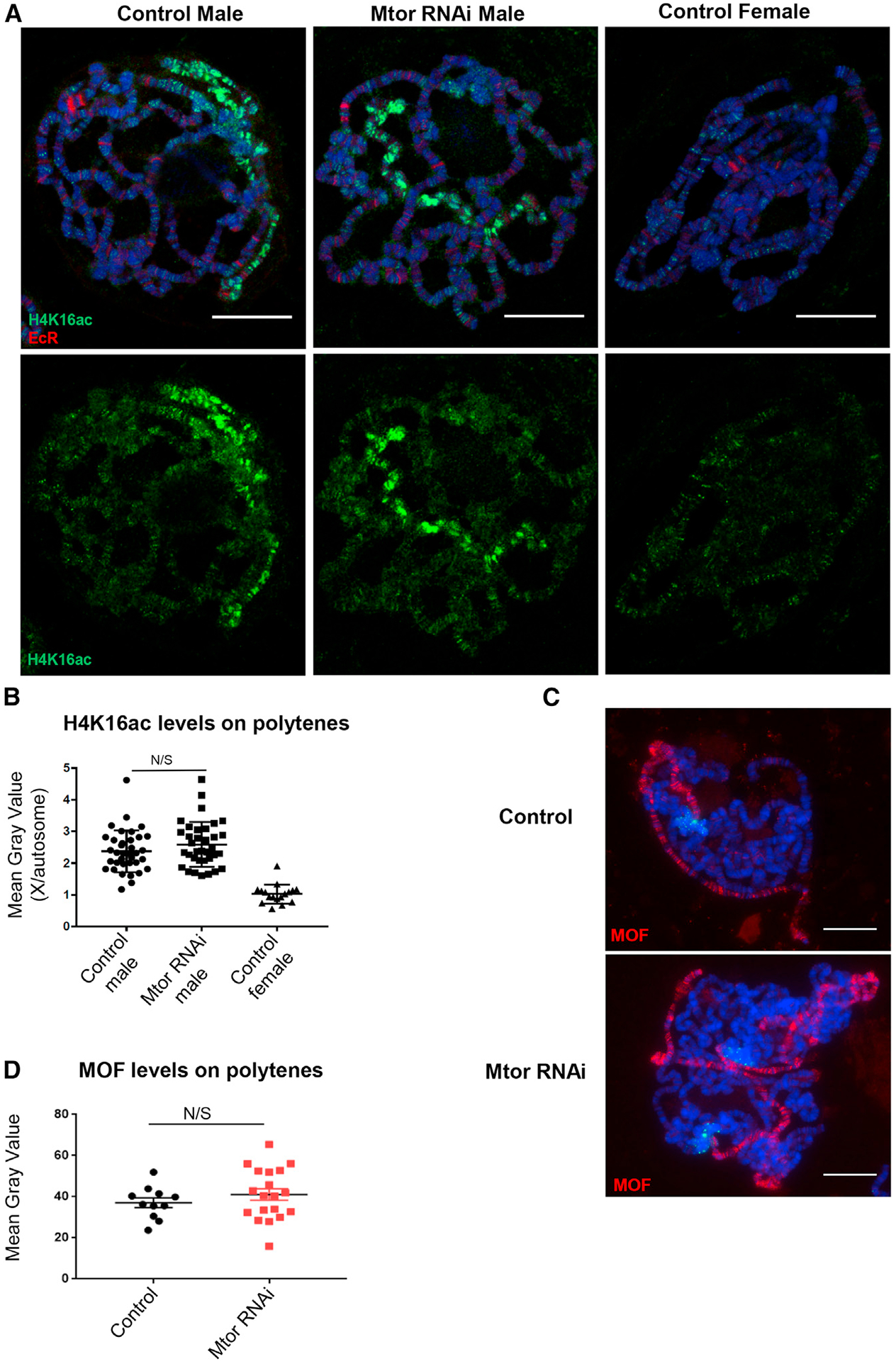
(A) Polytene chromosome from male and female control and male Nub-Gal4-driven Mtor RNAi larvae, stained with antibodies to H4K16ac (green) and EcR (red). Hoechst is in blue (here and the rest of the figure).
(B) Quantification of H4K16ac levels in polytene chromosomes, as a ratio of mean gray values for signal on X chromosome versus 3R autosome. N = 35–36 nuclei for males and 17 nuclei for females, from 4 biological replicates each. Error bars represent SEMs. N/S is p > 0.05, using an unpaired t test.
(C) Polytene chromosomes from control and Mtor RNAi male larvae, stained for MOF (red).
(D) Quantification of MOF levels on X chromosome as mean gray value of MOF signal. N = 11–19, from 3 biological replicates each. Error bars and p values as in (B).
Scale bars, 22 μm.
In addition, to test whether the uncovered function of Mtor may instead involve repressive histone marks (which would be expected to decrease in Mtor-depleted cells), we stained polytene chromosomes for H3K27me3 and H3K9me3 by IF in control and Mtor KD males (Figures S5D and S5E), quantified again as a ratio of mean gray values of X to autosomal signal (Figures S5F and S5G). No significant difference in the X:autosome ratio for H3K27me3 levels was observed between control and Mtor KD males (Figures S5D and S5F). Our quantification revealed a slight increase in the X:autosome ratio of H3K9me3 levels in Mtor-depleted males (Figure S5G), although such an increase was not discernable by eye. Interestingly, we also observed a similar increase in the X:autosome ratio of the H3K9me3 levels in females (Figure S5H). Since the H3K9me3 increase was found to occur in both sexes, we believe it is unlikely to explain the male-specific upregulation of the X chromosome observed upon Mtor loss. These results indicate that the role of Mtor in DC is likely independent of H4K16ac and of repressive marks H3K27me3 and H3K9me3.
Upregulation of the male X upon depletion of Mtor occurs at the level of transcription and not via changes in mRNA export
We next sought to determine whether the effect of Mtor on gene expression is exerted at the level of transcription or via post-transcriptional processes such as mRNA export. Homologs of Mtor in plants and human cells were found to affect mRNA export (Jacob et al., 2007; Skaggs et al., 2007), raising the possibility that the observed effects on X gene expression may be a consequence of misregulated mRNA transport and the resulting nuclear accumulation of mRNA. To distinguish between effects on transcription versus mRNA export, we used the smRNA FISH approach. Using exonic probes against 2 X chromosome target genes that showed upregulation in our RNA-seq datasets, Rpt6 and Pp4-19c (Figure 6A), we performed smRNA FISH in the Mtor RNAi mosaic salivary glands of males and observed a striking increase in Rpt6 and Pp4-19c mRNA in both the nucleus and cytoplasm in Mtor-depleted cells (Figure 6B). Consistent with previous observations, female Mtor RNAi mosaics showed a much more modest upregulation of these X chromosome targets (Figure S6A). We quantified mRNA density in both the nucleus and cytoplasm through the entire 3-dimensional (3D) space associated with each cell (see STAR Methods for further details). The calculated mRNA nuclear:cytoplasmic ratios were not found to be significantly different between control and Mtor-depleted conditions for Rpt6 and Pp4-19c (Figure 6C), demonstrating a lack of any obvious mRNA export defect.
Figure 6. Upregulation of the male X upon depletion of Mtor occurs at the level of transcription and not via changes in mRNA export.
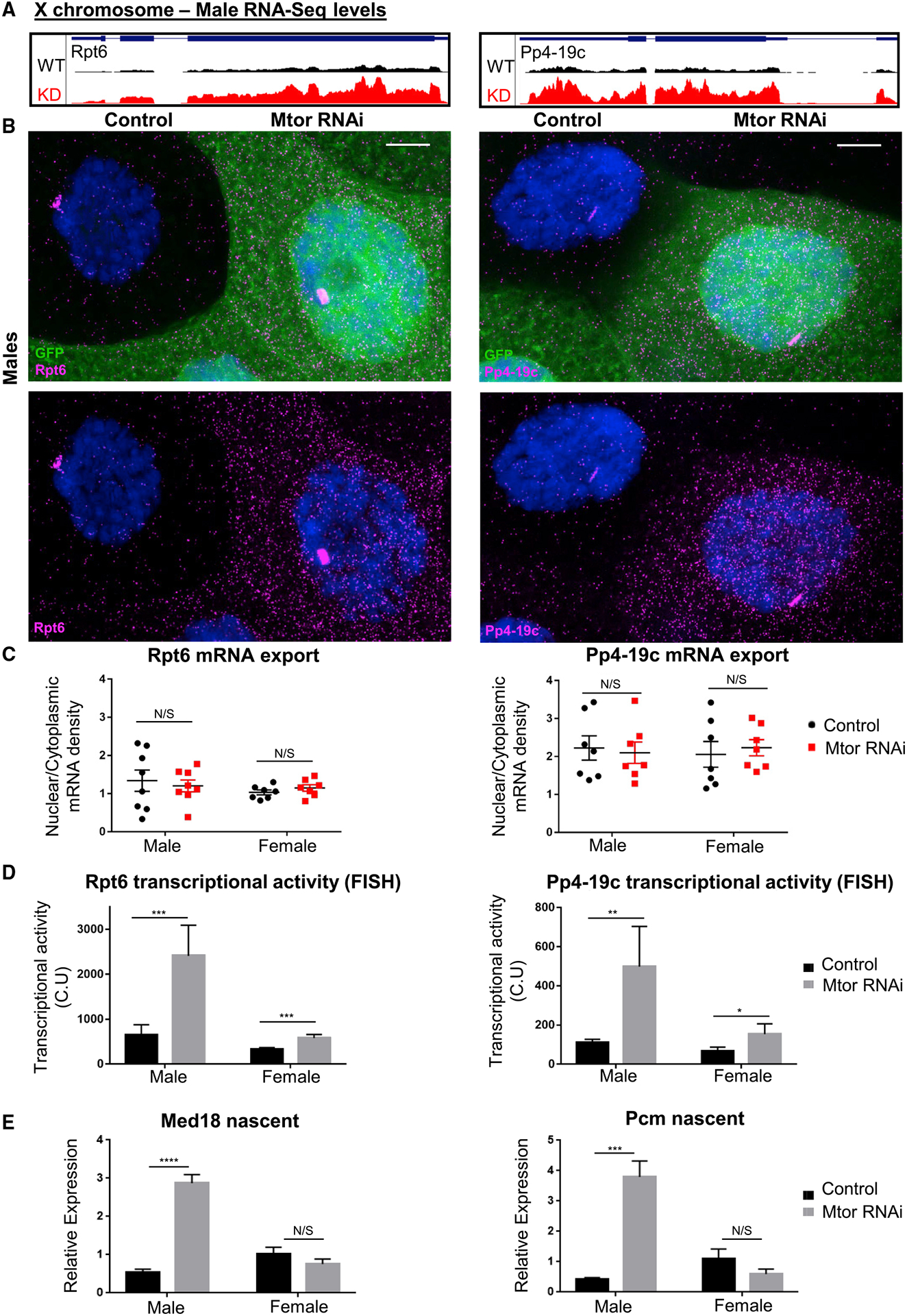
(A) GB snapshots of average RNA-seq levels at the X-linked genes selected for RNA FISH analysis, in control and Mtor KD males. The displayed region for Rpt6 is 1.6 kb and for Pp4-19c is 2.7 kb.
(B) SmRNA FISH for X chromosome targets Rpt6 and Pp4-19c in salivary gland nuclei from male larvae with induced Mtor RNAi mosaic system (GFP+ marks Mtor RNAi), as labeled and Hoechst in blue. Images are maximum projections spanning 5 μm. Scale bar, 12 μm.
(C) mRNA export quantification from smRNA FISH, as ratios of nuclear to cytoplasmic mRNA densities (see STAR Methods for details on image quantification). Each dot on each graph represents a single nucleus. N = 7–8, from 3 biological replicates each. Error bars represent SEMs. N/S is p > 0.05, using paired t tests.
(D) Quantification of nascent transcription site intensity in smRNA FISH. Transcriptional activity represented in CU (cytoplasmic units); see STAR Methods for details on quantification. N and error bars as in (C). *p < 0.05, **p < 0.01, and ***p < 0.001, using paired t tests.
(E) Nascent expression analysis via qRT-PCR of X chromosome targets in salivary glands of labeled genotypes (using exon-intron-spanning primers), normalized to female controls. N = 3 biological replicates, 10 salivary glands per sample. Error bars represent SEMs. ****p < 0.0001, N/S is p > 0.05, using a 1-way ANOVA, followed by the Holm-Sidak test for multiple comparisons.
We also observed an increase in the size and signal intensity of the nascent transcription sites for both genes upon Mtor KD, indicative of an increase in transcriptional activity (Figure 6B). The nascent site intensity was measured (see STAR Methods for further details) and found to be ~4 times higher in Mtor-depleted male nuclei relative to controls for both Rpt6 and Pp4-19c (Figures 6D and S6B). Interestingly, the transcriptional fold increase for both genes, as measured by smRNA FISH, was very similar to the fold increase for these genes in normalized RNA reads, detected by RNA-seq (Figure S6B), which validates these experimental approaches and supports our conclusion that Mtor exerts its effect at the level of transcription. Chromosome-wide, we found that the mean fold increase of X chromosome DEGs that were upregulated in the male upon Mtor depletion is 3.19 in males and only 1.87 in females (Figure S6C, as measured in reads per million, using the RNA-seq data).
To further validate these findings, we assessed the nascent transcription of additional X chromosome targets Med18 and Pcm via an alternative method of qRT-PCR using primers designed to span exon-intron junctions. Consistently, we observed a male-specific increase at the level of nascent transcription of both Med18 and Pcm, where Mtor KD males displayed a 3- to 4-fold transcriptional upregulation (Figure 6E). Female levels of nascent transcription were unchanged for these genes with Mtor KD. These data suggest that Mtor-dependent upregulation of X chromosome targets is a consequence of nascent transcription changes.
Mtor restrains dosage compensation at the level of nascent transcription
To further investigate nascent transcriptional changes upon Mtor depletion, we performed a genome-wide analysis in male S2 cells using transient transcriptome sequencing (TT-seq) that labels newly transcribed, nascent RNAs (Schwalb et al., 2016) (see Method details). TT-seq was performed in S2 cells, treated with dsWhite as the control, dsRNA against MOF (dsMOF), and dsMtor (Figures 7A–7C). To confirm that our TT-seq experiment successfully detected nascent transcripts, we assessed the fractions of intronic to exonic reads in each library and observed their ratios to be ~0.6 (Figure S7A), which is what has been previously reported for successful TT-seq experiments (in contrast to ratios of only 0.08 in standard RNA-seq) (Gregersen et al., 2020; Schwalb et al., 2016). Upon Mtor depletion, we observed an increase in TT-seq reads of select X chromosome target genes, which were downregulated upon MOF KD and which were similarly upregulated in the RNA-seq in Mtor-depleted salivary glands (Figures 7A, 7B, and S7B). On a genome-wide level, we detected an expected transcriptional downregulation of hundreds of genes across the X chromosome upon MOF KD (Figure 7B). In agreement with our previous findings, the most prevalent change we observed by TT-seq upon Mtor KD was an upregulation of a subset of X chromosome genes (Figure 7B). Furthermore, when we compared genes that were nascently upregulated upon Mtor depletion and downregulated upon MOF depletion with the MSL-2 ChIP signal (Straub et al., 2013), we found the MSL-2 signal to be significantly enriched at such overlapped X-linked DEGs, but not at genes downregulated upon Mtor depletion (Figure S7C). Consistently with previous qRT-PCR data from S2 cells (Figure 3E), the extent of upregulation upon Mtor depletion in S2 cells appears to be less dramatic than that observed in salivary glands, possibly due to differences between in vitro-propagated culture cells and the in vivo organism.
Figure 7. Mtor restrains dosage compensation at the level of nascent transcription.
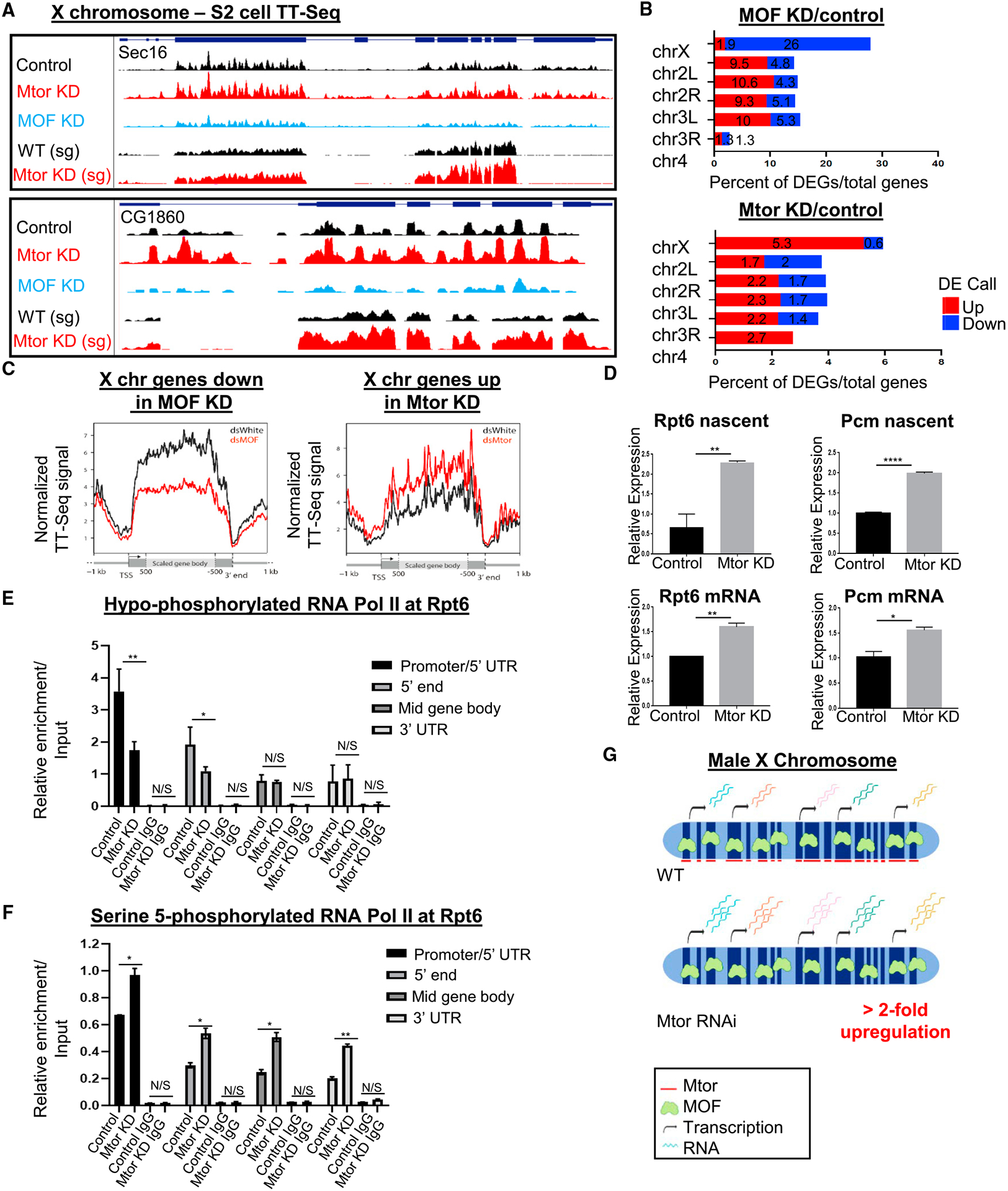
(A) GB snapshots of X chromosome genes Sec16 and CG1860, displaying average TT-seq reads from S2 cells in control and Mtor- or MOF-depleted conditions, and RNA-seq reads from WT and Mtor KD male salivary glands (bottom 2). For Sec16, the displayed region is 11.6 kb and for CG1860, it is 2.7 kb.
(B) Percentage of DEGs per chromosome upon MOF KD (top) and Mtor KD (bottom) relative to controls from TT-seq in S2 cells. DEG analysis was done as in Figure 2A.
(C) Metagene plots showing nascent transcript distribution along scaled X-linked genes that go down in MOF KD (left) or up in Mtor KD (right), obtained from TT-seq (see STAR Methods for analysis details).
(D) Expression analysis of X chromosome targets via qRT-PCR in control and Mtor KD S2 cells, for nascent transcript with exon-intron primers (top row) or mRNA with exon-exon primers (bottom row). N = 3 biological replicates. Error bars represent SEMs. *p < 0.05, **p < 0.01, and ****p < 0.0001, N/S is p > 0.05, using unpaired t tests.
(E) ChIP-qPCR for hypo-phosphorylated RNAPII (8WG16 antibody) at Rpt6 in control and Mtor KD S2 cells, with IgG controls. N = 4 biological replicates, error bars and p values as in (D).
(F) ChIP-qPCR for serine-5 phosphorylated RNAPII (CTD4H8 antibody) at Rpt6, conditions as in (E). N = 2 biological replicates, error bars and p values as in (D).
(G) Model of the role of Mtor in dosage-compensated gene expression. When Mtor is depleted, transcriptional upregulation of MOF/MSL X-linked targets exceeds the required 2-fold, pointing to the normal role of Mtor in restricting X chromosome DC. Image created with BioRender.com.
From the TT-seq results, we noticed that genes, which were upregulated upon Mtor depletion, exhibited a uniform degree of upregulation across the entire gene body, without any obvious enrichment at a particular gene end. To assess this distribution, we generated metagene profiles and observed a uniform increase in nascent RNA synthesis for X-linked genes upregulated in Mtor KD, starting at the transcription start site (TSS), and a similarly uniform decrease for X-linked genes downregulated in MOF KD (Figure 7C). To quantify these observations, we calculated the pausing index (PI) and elongation index (EdI) (as defined in Larschan et al., 2011) in both MOF- and Mtor-depleted conditions. Although TT-seq is not believed to be a suitable technique to detect RNA polymerase II (RNAPII) pausing (Schwalb et al., 2016), we aimed to use the PI and EdI as measurements of transcript enrichment at either the 5′ or 3′ end of the gene, and found that PI and EdI ratios of Mtor KD to control were not significantly different between the X chromosome and the autosomes (Figures S7D and S7E). These data suggest that nascent transcription is uniformly increased upon the loss of Mtor, and that this upregulation may occur at a step upstream of transcriptional pausing or elongation.
To gain more insight into which transcriptional step may be controlled by Mtor, we performed ChIP-qPCR experiments for 2 different forms of the C-terminal domain (CTD) of RNAPII, the hypo-phosphorylated form, associated with pre-initiation, and the serine-5 phosphorylated (Ser5P) form, associated with transcriptional pausing (Phatnani and Greenleaf, 2006). We confirmed upregulation in nascent transcription and mature mRNA expression for X chromosome genes Rpt6 and Pcm in S2 cells, using qRT-PCR with primers that spanned exon-intron boundaries or exon-exon junctions, respectively (Figure 7D). ChIP was performed at the Rpt6 and Pp4-19c genes at multiple positions in control and Mtor-depleted S2 cells (Figures 7E, 7F, S7F, and S7G). Interestingly, while we observed an expected increase in the Ser5P form of RNAPII across the Rpt6 gene upon Mtor KD (Figure 7F), we found a significant reduction in the hypo-phosphorylated form of RNAPII at the 5′ gene regions in Mtor-depleted conditions (Figure 7E). The same trends were observed at Pp4-19c (Figures S7F and S7G). These results suggest that normally Mtor may restrict gene expression by regulating the transition from hypo-phosphorylated to Ser5P RNAPII and thus the initial entry into transcription initiation. Without Mtor, the Ser5 phosphorylation of RNAPII appears to happen at aberrantly higher rates, pushing more RNAPII into the initiation state and leading to an overall increase in nascent transcript levels.
DISCUSSION
An outstanding question in the field of Drosophila DC is how the 2-fold transcriptional upregulation across the male X chromosome is achieved. Protein candidates, including the exosome machinery and Nups, have previously been hypothesized to be involved in regulation by the MSL complex (Mendjan and Akhtar, 2007; Mendjan et al., 2006), but the roles of these proteins had not been fully characterized within this context. Our presented results show that Nup Mtor is an attenuator of dosage-compensated gene expression levels, involved in setting the upregulation of the male X chromosome to 2-fold (Figure 7G). Previously, Mtor has been shown to be enriched on the male X chromosome via genome-wide ChIP studies (Vaquerizas et al., 2010). The MSL complex binds across the male X chromosome at specific dosage-compensated domains, where expression levels are increased but constrained to match, and not exceed gene expression levels coming from the 2 X chromosomes in the female. We propose that the MSL complex uses its interactions with Mtor to execute this constraint. Our model is supported by our finding that the effect of Mtor on X chromosome expression is dependent on MSL components and by the physical interaction of Mtor with MSL components that we and others have detected (Figure 3D; Mendjan et al., 2006). Significantly, we have shown that the effect of Mtor on gene expression occurs at the level of nascent transcription, such that the loss of Mtor leads to a uniform transcriptional upregulation across a subset of male X chromosome genes, which may be due to a misregulation of the RNAPII CTD Ser5 phosphorylation. These data define a role for Mtor as a negative modulator of transcriptional outputs, with this role being used in the process of DC.
Our finding of the role of Mtor as a transcriptional repressor is supported by previous findings in yeast that show that the loss of the homologs of Mtor can increase mRNA synthesis of select genes in response to export defects (Vinciguerra et al., 2005). We hypothesize that this role as a repressor of transcription may have evolved to be used by the MSL complex in Drosophila. Interestingly, while the loss of Mlp proteins does not lead to general mRNA export defects in yeast (Kosova et al., 2000; Strambio-de-Castillia et al., 1999), the loss of their plant homolog, the Arabidopsis thaliana tetratricopeptide repeat (AtTPR), does (Jacob et al., 2007). More recently, it has been demonstrated that auxin-induced depletion of the human homolog of Mtor, TPR, led to changes in both nascent transcription and mRNA export of a subset of genes (Aksenova et al., 2020). We did not identify any obvious mRNA export defects in Mtor-depleted conditions, at least for the X chromosome genes we tested (Figures 6B and 6C), suggesting that Drosophila Mtor is not essential for mRNA export and that its phenotype is not explained by an aberrant accumulation of nuclear mRNA. Presently, it cannot be ruled out that the effect of Mtor on gene expression does not also involve the quality control of mRNA, previously reported for Mlp1 (Galy et al., 2004), which is a possibility we plan to investigate in the future. In this manner, the reported connection of exosome components to the DC process (Mendjan et al., 2006) remains an intriguing possibility, and whether exosome-mediated degradation of extra mRNA produced as a result of DC is involved in Mtor-mediated attenuation is an interesting question for the future.
Although our genetic studies found that Mtor and the MSL complex work together to target the same process, we did not find the mechanism of action of Mtor to involve the H4K16ac modification. Loss of Mtor also did not lead to obvious defects in the targeting of MSL components to the X chromosome, which is in agreement with previously published findings (Grimaud and Becker, 2009). Although previous work reported conflicting results on the functional effect of Mtor on DC processes (Grimaud and Becker, 2009; Mendjan et al., 2006), we believe that the genome-wide and gene-specific analysis we report here, which was carried out both in the organism and in S2 culture cells, unambiguously identifies a functional role. One possibility for this discrepancy is that, as discussed above, the upregulation effect we identified was much more pronounced and widespread in tissues than in S2 cells, which were the primary model for functional studies in previous work (Mendjan et al., 2006).
How does Mtor regulate the transcriptional output of the X chromosome? The precise mechanism remains to be further explored, but our TT-seq analysis suggested that transcriptional steps such as escape from pausing and transcriptional elongation are affected equally by Mtor depletion. Significantly, our ChIP-qPCR experiments at X chromosome targets revealed not only higher levels of Ser5P RNAPII but also lower levels of the hypo-phosphorylated RNAPII. These results suggest that Mtor does not affect the initial recruitment of RNAPII but instead acts as a negative regulator of the first phosphorylation step of the CTD and of the transition into transcription initiation, such that a pre-initiation form of RNAPII is excessively pushed toward initiation upon the loss of Mtor. It is possible that Mtor exerts its regulation by preventing the recruitment of the transcription factor II human (TFIIH) complex, which contains the CDK7 kinase responsible for phosphorylating Ser5 (Nilson et al., 2015), or of other transcriptional machinery, and we plan to address these possibilities in the future. This regulatory mechanism will be particularly interesting to understand in light of the reported binding of Mtor along chromatin in continuous domains (Vaquerizas et al., 2010) and the reported long intranuclear filaments of Mtor homologs (Arlucea et al., 1998; Fontoura et al., 2001; Kosova et al., 2000; Strambio-de-Castillia et al., 1999; Zimowska et al., 1997). Such chromatin-associated filaments may help set a particular transcriptional output for an entire domain of expression. Elucidating these possibilities and the precise role of Mtor will undoubtedly shed light onto the mechanisms underlying DC and transcriptional modulation in general.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Requests for further information and resources should be directed to and will be fulfilled by the corresponding author, Dr. Maya Capelson (capelson@pennmedicine.upenn.edu or mcapelson@gmail.com).
Materials availability
All generated materials are available upon request and will be shared without restrictions.
Data and code availability
The GEO accession number for all sequencing data reported in this paper is GEO: GSE155323, containing the RNA-Seq datasets in Control and Mtor KD conditions from male and female larval salivary glands as well as TT-Seq datasets in Control, Mtor KD and MOF KD conditions in S2 cells. For the custom MATLAB pipeline used for quantification of RNA FISH data in Figure 6, please contact lead contact Dr. Maya Capelson (capelson@pennmedicine.upenn.edu or mcapelson@gmail.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Drosophila S2 cells were cultured at 25°C in Schneider’s medium (DGRC) supplemented with 10% heat inactivated fetal bovine serum (GIBCO) and antibiotics.
Fly lines
Drosophila were raised at 22°C on standard molasses fly food. Larvae were raised in undercrowded conditions and dissected at later wandering third instar stage, where larvae are minimally moving but anterior spiracles have not yet protruded. Fly lines in this study include the following Bloomington stocks: 5 (Oregon-R), (w118) 24265 (Mtor UAS-RNAi), 31401 (MOF UAS-RNAi), 86108 (Nub-Gal4), 27392 (UAS-mCD8 RFP), 10537 (MtorK03905 /CyO), 4411 (Act5c, FRT y+ FRT, Gal4, UAS-GFP). roX1SMC17A roX2Δw+ ; +/ CyO [w+roX1] (referred to as roX1/roX2 mutants in text) was a gift from Vicky Meller. y1, w*, hsFLP; Kr/CyO; MKRS/TM6B was a gift from Steve DiNardo.
METHOD DETAILS
Mtor RNAi mosaic generation and fertility scoring
Briefly, Mtor UAS-RNAi was combined with y1, w*, hsFLP; Kr/CyO; MKRS/TM6B to yield a balanced stock containing Mtor RNAi: y1,w*, hsFLP; Mtor RNAi/CyO; MKRS/+. Non-CyO, non-MKRS Mtor homozygotes from this line were crossed to the Act5c, FRT y+ FRT, Gal4, UAS-GFP line to obtain F1 for generation of Mtor RNAi mosaics. To induce mosaic/clone formation, F1 larvae were heat shocked at 37C for 30 mins on day 3 after initial mating day. Visual assessment of sufficient clone generation (GFP-marked Mtor RNAi nuclei) was verified in dissected salivary glands under a fluorescent compound microscope for subsequent antibody staining or RNA FISH experiments.
To assess fertility of the F1 Mtor clonal/mosaic adults, heat shocked (Mtor RNAi induced) or non-heat shocked (uninduced, control) individual adults were backcrossed to two or three Nub-Gal4 male or virgin female adults and fertility was assessed on day 7 by visual scoring of 3rd instar larva emergence. Percentage of flies that were sterile or fertile for each category was reported, in N = 3 independent rounds of clone generation and fertility assay matings.
Generation of roX1 and roX2/Mtor lines
Briefly, Nub-Gal4 was introduced into line roX1SMC17A roX2Δw+; +/ CyO [w+roX1] to yield the following stock: roX1SMC17A roX2Δw+ ; Nub-Gal4/ CyO[w+roX1]. Virgin females homozygous for Nub-Gal4, not carrying the rescuing roX1 transgene (roX1SMC17A roX2Δw ; Nub-Gal4) were crossed to Mtor RNAi males to achieve Mtor KD in the presence of roX1 and roX2 loss of function. Controls for these experiments used virgin females from roX1SMC17A roX2Δw+; Nub-Gal4 crossed to Nub-Gal4 males. Females from roX1SMC17A roX2Δw+; Nub-Gal4 were crossed with males from MtorK03905 / CyO RFP to yield the scored genotype categories of roX1SMC17A roX2Δw+ ; Nub-Gal4/ MtorK03905 or roX1SMC17A roX2Δw+ ; Nub-Gal4/CyO RFP.
Scoring of viability phenotypes – roX1/roX2/Mtor
Adult viability category descriptions and scoring for genetic rescue experiments with Mtor RNAi and roX1/roX2 null mutants were as follows: Pharate male denoted fully formed adults stuck in their pupal cases, identified as male via presence of sex combs; half-eclosed male denoted males that partially came out of their pupal case but died, also identified by their sex combs; adult male denoted males that fully eclosed from their pupal case, found alive or dead. Parents were removed from vials on day 7. Counting of F1’s began on day 10, consisting of fully eclosed flies (mostly females) and continued up until day 25. Development of pharate males was often delayed in comparison to females. See Figure S4B for table of numbers.
Adult viability outcomes of genetic rescue experiments with MtorK03905 loss of function mutation and roX1/roX2 null mutants were reported as percentage of males or females observed. Females scored were full adult females that eclosed. Males scored were pharate adults in both genotypes: roX1SMC17A roX2Δw+ ;Nub-Gal4/ MtorK03905 or roX1SMC17A roX2Δw+ ; Nub-Gal4/CyO RFP. Genotypes of pharate males were scored by presence or absence of RFP, which could be seen under a fluorescence dissecting scope. Parents were removed from vials on day 7. Counting of F1’s began on day 10, consisting of fully eclosed flies (mostly females) and continued up until day 25. See Figure S4C for table of numbers.
S2 cell RNA interference, lysate preparation and western blotting
dsRNA against Mtor, MOF and White genes were generated from PCR templates of fly genomic DNA using specific T7 primers, T7 flanking regions included in sequence: (Mtor-F 5′-ttaatacgactcactatagggagaTTGAAGCAGGATCTGCACAC-3′; Mtor-R 5′- ttaatacgactcactatagggagaTTTAATTTCGGAGATGCCCTG-3′; MOF-F 5′- ttaatacgactcactatagggagaCCAGCGACTTCTTTTCCTTG-3′; MO F-R 5′- ttaatacgactcactatagggagaTGCAGTTTGGCAACTACGAG-3′; White-F 5′-ttaatacgactcactatagggagaGATCCTGGCTGTCGGTGCTCA-3′; White-R 5′-ttaatacgactcactatagggagaGATCATCGGATAGGCAATCGC-3′). dsRNAs were synthesized using a Megascript T7 kit (Ambion) following the manufacturer’s instructions.
As seen in Figure S3H, S2 cell were subjected to 2 rounds of KD via RNAi treatment for a total of 6 days. 1.4 million cells were treated with dsRNA: 9ug of dsWhite or dsMOF and 12 ug of dsMtor at day 1, and then again after 48 hours. Cells were then collected approximately 72 hours later (a separate aliquot of these samples were also used to verify gene expression via qRT-PCR in Figures 3E, 3F, and 7D). Protein extracts were prepared by lysing cells in RIPA buffer (25 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% IGEPAL and 0.1% SDS) with 1mM of PMSF and Complete protease inhibitor cocktail (Roche) on ice for 30 min followed by brief centrifugation to remove debris. Protein concentration of the supernatant was quantified using BCA protein assay kit (ThermoFisher) and equal amounts of protein were loaded (20ug) and separated on 6% SDS-PAGE gels. Proteins were transferred to nitrocellulose membranes and incubated overnight at 4C using primary antibodies at the following dilutions: mouse anti-Mtor 1:200, rabbit anti-L7 Lamin DmO 1:1000. Membranes were then incubated with mouse HRP secondary antibody and Protein A secondary for 2 hr at room temperature and detected using ECL-Plus western blotting reagent (Amersham Biosciences).
Salivary gland lysate preparation and western blotting
Salivary glands from 50 animals per condition (control and Mtor KD) were collected on dry ice in Eppendorf tubes. 100 uL of PBS was added to each tube and glands were spun down gently at 300 g for 5 mins to get pellet of tissue to bottom of tube. PBS was carefully removed and RIPA buffer (150 mM NaCl, 1% IGEPAL, 0.5% NaDox, 0.1% SDS, 50 mM Tris pH 7.4, 0.5% Tween-20) with 1 mM PMSF and Complete protease inhibitor cocktail (Roche) was added to each tube. Tubes were incubated on ice for 5 mins. Glands were passed through an insulin needle until salivary gland particles were no longer seen (30–40 strokes). Tubes were incubated on ice for 10 mins then spun at 13,000 rpm for 10 mins at 4C. The supernatant was taken off and protein concentration was measured by Bradford assay. Equal amounts of protein were separated on a 6% SDS-PAGE gel, and western blotting conducted as described above. Primary antibodies were used at the following dilutions: mouse anti-Mtor 1:200, rabbit anti-MOF 1:2000.
Co-immunoprecipitation
Nuclei were purified from 1.4×108 cells by re-suspending in hypotonic buffer (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl and 0.5 mM EGTA) with 1 mM PMSF and Complete protease inhibitor cocktail (Roche), followed by 1 hr incubation on ice. Pelleted nuclei were incubated in lysis buffer (20 mM HEPES pH 7.9, 25% Glycerol, 0.42 M NaCl, 1.5 mM MgCl2 and 0.2 mM EGTA) with 1 mM PMSF and Complete protease inhibitor cocktail (Roche) for 30 min on ice and then diluted 1:3 with lysis buffer without NaCl. Protein extracts were pre-cleared and incubated overnight with the appropriate antibodies: 5.5 μL of rabbit anti-MOF antibody and 1 μL of the applicable IgG control. Finally, the antibody-protein complexes were incubated with 45 μL (as 50% slurry) of pre-blocked (PBS with 0.3% BSA) Protein A Dynabeads (Invitrogen) for 4 hr. The beads were washed 5 times with wash buffer (50 mM Tris-HCl pH 8, 150 mM NaCl, 0.2% Igepal and 1 mM EDTA) and boiled in SDS-loading buffer for western blot analysis. Proteins were then resolved on 6% SDS-PAGE gels, and western blotting conducted as described above. The primary antibodies used in western blotting were as follows: mouse anti-Mtor at 1:100, rabbit anti-MLE at 1:2000.
H4K16ac histone extraction and western blotting
11 million S2 cells per 10 cm dish were treated with 220 ug of the corresponding dsRNA (dsWhite or dsMtor) per plate for 72 hours. Samples were treated again with the same amounts of dsRNA for another 72 hours after the first round of KD (6 days total). Samples were collected and washed 2x in 1X PBS. Standard acid extraction of histones was then performed on this sample: 0.4 N H2SO4 was added for 3 hours, followed by TCA precipitation and resuspension in 100 uL H2O. Protein concentration was quantified using BCA protein assay kit (ThermoFisher) and equal amounts of protein were loaded (3 and 6 ug). and separated on 15% SDS-PAGE gels. Proteins were transferred to nitrocellulose membranes, probed with Ponceau then washed, and incubated overnight at 4C using primary antibodies at the following dilutions: rabbit anti-H4K16ac (sc-8662) 1:2000 and rabbit anti-Histone H3 (ab1791) 1:10,000 (which was probed for on same membrane, after membrane was stripped). Membranes were incubated with rabbit HRP secondary antibody at 1:5000 for 2 hr at room temperature and detected using ECL-Plus western blotting reagent (Amersham Biosciences).
Polytene chromosome preparation and immunofluorescence (IF) staining
Polytenes and semi-squashes were carried out as described in Kuhn et al. (2019). Briefly, salivary glands were dissected from wandering third instar D. melanogaster larvae in 0.1% PBS with Tween 20 (PBST), fixed in 2% PFA/45% acetic acid for 1 min at RT, squashed in a drop of 45% acetic acid between a Sigmacote (SL2; Sigma-Aldrich) coverslip and a poly-L-lysinated slide (Polysciences 22247) with a rubber hammer, and snap-frozen in liquid nitrogen; coverslips were flipped off, and slides were stored for 1 h in 0.1% PBST in a coplin jar before blocking in 3% BSA PBST for 30 min at RT and incubated overnight at 4°C in 30 μl in a blocking solution containing primary antibodies under a coverslip in a humid chamber. The following day they were washed three times for 10 min each in PBST, stained with secondary antibodies in blocking solution for 1 h at RT in the dark, and then washed three times for 10 min each time again before treatment with 10 μg/ml Hoechst stain in PBS for 2 min followed by a 5-min PBS wash before mounting in ProLong Gold Antifade (P36930; ThermoFisher), sealing with nail polish, and storage at 4°C. Slides were imaged within 1 wk of fixation. Widefield fluorescence imaging was conducted on a Leica DM6000 Microscope with PL APO 63 × /1.40–0.70 Oil objective using Type F Immersion Oil Leica 11513859, DFC365 FX Camera, and Leica LAS-X 3.3 Software. Confocal imaging was conducted at room temperature on a Leica TCS SP8 Confocal using PL APO 63 × /1.40 Oil objective, 2 × Zoom, Type F Immersion Oil Leica 11513859, and Leica Software LAS-X 3.3.
Semi-squashes used to better preserve nuclear shape to verify rim staining, as in Figure S1A, use an identical protocol as full squashes but with a 2-min fixation in 8% acetic acid/2% PFA (instead of 45% acetic acid/2%PFA above) and a 2% PFA droplet used on the coverslip, at which point the coverslip is not hammered but is gently moved ~1 mm in each direction two times before freezing. Antibodies and dilutions are listed in antibodies sub-section.
For H4K16ac, H3K27me3 and H3K9me3 stainings, polytene chromosome squashes required an alternative fixation protocol to prevent extraction of histones from chromatin: the protocol replaced standard fixation with a 30 s fixation in 2% PFA, followed by 2 min in 2% PFA/45% acetic acid, and a final placement into a drop of 45% acetic acid during squashing (similarly to as described in Kuhn et al., 2019). After flash freezing in liquid nitrogen, slides were kept at −20°C in 70% ethanol for ≥ 30 min before two quick rinses in PBST and the standard subsequent blocking and staining protocol.
Whole mount IF
Briefly, male larval heads (heads refers to anterior 1/3 of larvae) were dissected off in 1X PBS and inverted so that salivary glands (still attached) were exposed, and placed into an Eppendorf tube on ice containing 1X PBS. Tissue was fixed in 4% PFA in PEM buffer (100mM PIPES pH 7, 1mM EGTA pH 8, 2mM MgSO4) for 20 mins on a nutator at RT. After fixation, heads were washed 6x in 1mL BBT (1% (w/v) BSA, 0.1% (v/v) Triton X-100 in PBS). Heads were incubated overnight on a rotator in primary antibody diluted in BBT (Mtor, 1:30) at 4C. On the second day, heads were washed for 30 mins in BBT, changing buffer 6x. After washes, the appropriate secondary antibody, diluted in BBT was added and incubated overnight on a rotator at 4C. On day 3, heads were washed for 30 mins in 0.1% PBT, changing buffer 6x. DNA staining with Hoechst was carried out at 1:1000 in PBT on a covered nutator for 5 mins, then removed and washed with PBT. Samples were washed 3 more times with PBT. To mount samples, heads were transferred to a glass dissecting dish, salivary glands were separated off and placed onto a slide in a drop of vectashield, and a coverslip was placed on top. Slides were allowed to dry for ~30 mins before sealing with nail polish. Confocal imaging was conducted at room temperature on a Leica TCS SP8 Confocal using PL APO 20X, and Leica Software LAS-X 3.3.
Antibodies for IF
Primary antibodies and dilutions used were as follows: Rabbit anti-MOF (gift from Kuroda lab) at 1:100–1:200, Rabbit anti-H4K16ac (#8662 from Santa Cruz Biotechnology) at 1:100, Mouse anti-HP1 (C1A9 from the Developmental Studies Hybridoma Bank) at 1:200, Mouse anti-EcR (DDA2.7 from the Developmental Studies Hybridoma Bank) at 1:100, Mouse anti-Mtor (#12F10 from the Developmental Studies Hybridoma Bank) at 1:30, Chicken anti-GFP (#1020 from Aves Labs Inc.) at 1:500, Rabbit anti-H3K27me3 (#9733 from Cell Signaling) at 1:100, Rabbit anti-H3K9me3 (ab8898 from Abcam) at 1:100 and Hoechst DNA stain (H3570; ThermoFisher) at 1:1,000. Fluorescently conjugated secondary antibodies were as follows: ThermoFisher Alexa Fluor conjugates of goat anti-mouse, anti-rabbit, 488 and 568 at 1:300.
Whole mount single molecule RNA FISH: roX1
smRNA FISH probe for roX1 was generated and FISH protocol was carried out as detailed in Little and Gregor (2018). Briefly, male larval heads were dissected off in 1X PBS and inverted so that salivary glands (still attached) were exposed, and placed into an Eppendorf tube on ice containing 1X PBS. Tissue was fixed in 4% PFA in 1X PBS for 20 mins on a nutator at RT. Heads were washed 3x in 1X PBS. Heads were permeabilized overnight in methanol at −20C. To preserve GFP, heads were washed 1x in methanol for 5 mins and then incubated overnight in 100% ethanol. On day 2, heads were washed quickly 3x in 1XPBT and then in PBT for 20 mins on a nutator at RT. Heads were then washed quickly 2x in FISH wash buffer, and then in FISH wash buffer for 20 mins on a nutator at RT. During wash, diluted probe was preheated to 37C. roX1 probe was used at a 1:100 dilution in hybridization buffer. 100ul of diluted probe (probe + hybridization buffer) is needed to cover heads. Probe was then added after FISH wash buffer incubation. Tubes were incubated in a 37C rocking incubator that was dark, overnight. The next day, 1 mL of fresh, preheated FISH wash buffer was quickly added to sample after removing probe. A fresh aliquot of preheated FISH wash buffer was then added and incubated for 1 hour at 37C in the dark. After 1 hour, this wash buffer was removed and another wash was repeated for 1 more hour (2hr total wash time). 1 mL of PBT was then quickly added, followed by another quick wash in PBT. DNA staining with Hoechst was carried out at 1:1000 in PBT on a covered nutator for 5 mins, then removed and washed with PBT. Samples were washed 3 more times with PBT. To mount samples, a 1mL pipet tip was cut, rinsed several times in PBT, and heads were carefully transferred to a glass dissecting dish for salivary gland dissection with minimal light. Once glands were separated, they were placed onto a cleaned slide in a drop of vectashield and a coverslip was placed on top. Slides were allowed to dry for ~30 mins before sealing with nail polish. Confocal imaging was conducted at room temperature on a Leica TCS SP8 Confocal using PL APO 63 × /1.40 Oil objective, 1.6X Zoom, Type F Immersion Oil Leica 11513859, and Leica Software LAS-X 3.3.
Polytene smRNA FISH: roX1
smRNA FISH for roX1 in polytenes utilized the same probe used in whole mount RNA FISH. This protocol utilized the polytene chromosome squash protocol described above with the addition of the use of RNAsin Ribonuclease inhibitors (Promega) − 8 uL was added to 1mL of 45% Acetic Acid/2% PFA solution, and 10 uL was added to 5mL of 1X PBS used for dissecting. Dissecting dish was also cleaned with RNaseZAP spray (Sigma) between every animal, with fresh liquids being used between animals. Slides were permeabilized in methanol quickly and then stored in 100% ethanol overnight at −20C. The rest of the protocol was carried out as described in the whole mount RNA FISH for roX1 with the exception of these steps being performed on slides. To keep smaller amounts of liquid on slides, an Aqua-Hold Pap Pen was used (EMS), liquid was carefully aspirated from corners of coverslip boundaries, and probe was sealed onto slide with a coverslip using rubber cement. Slides were incubated overnight at 37C in a humid chamber. Widefield fluorescence imaging was conducted at room temperature on a Leica DM6000 Microscope with PL APO 63 × /1.40–0.70 Oil objective using Type F Immersion Oil Leica 11513859, DFC365 FX Camera, and Leica LAS-X 3.3 Software.
Whole mount smRNA FISH with Stellaris probes: Rpt6 and Pp4-19c
Custom Stellaris® FISH Probes were designed against Rpt6 and Pp4-19c by utilizing the Stellaris® FISH Probe Designer (Biosearch Technologies, Inc., Petaluma, CA) available online at http://www.biosearchtech.com/stellaris-designer. Larval heads were hybridized with the Rpt6 and Pp4-19c Stellaris FISH Probe sets, both labeled with Quasar® 570 Dye (Biosearch Technologies, Inc.), following the manufacturer’s instructions available online at http://www.biosearchtech.com/support/resources/stellaris-protocols, specifically, based on ‘Protocol for D. melanogaster Wing Imaginal Discs,” with some modifications for application in salivary glands. Briefly, as in the RNA FISH protocol used for roX1 staining, larval heads were dissected and fixed for 20 mins in 4% PFA in PBS, washed 2x with PBS, and permeabilized in 1 mL of 100% methanol at 4C overnight (needed to quench some GFP signal and reduce GFP background, GFP signal still remained for identification of KD nuclei). The next day, probe dilution was prepared by adding 1 uL of the probe stock solution to 100uL of Hybridization Buffer (Biosearch Technologies Cat# SMF-HB1-1), and vortexed and centrifuged. Diluted probe solution was preheated to 37C. 1mL of Wash Buffer A (Biosearch Technologies Cat# SMF-WA1-60) was added and incubated for 5 mins. 100 uL of diluted probes was then added to the sample and incubated in the dark at 37C overnight. The next day, probe was removed and 1 mL Wash Buffer A was added and incubated for 30 mins at 37C in the dark. This was then removed and Hoechst at (1:1000) in Wash Buffer A was added for 30 mins at 37C in the dark. 1 mL of Wash Buffer B (Biosearch Technologies Cat# SMF-WB1-20) was then added and incubated for 5 mins. Samples were then mounted as described in Method details for roX1 RNA FISH above. Confocal imaging was conducted at room temperature on a Leica TCS SP8 Confocal using PL APO 63 × /1.40 Oil objective, 1.6X Zoom, Type F Immersion Oil Leica 11513859, and Leica Software LAS-X 3.3.
RNA extraction and qRT-PCR expression analysis
Total RNA was isolated using TRIzol (Ambion) from salivary glands (10–12 animals per sample is sufficient) vortexed at 4°C for 2 h or from S2 cell pellets vortexed for 30 min, extracted with ethanol precipitation, and subsequently purified with PureLink RNA Kit columns (Invitrogen). 1 μg of the extracted RNA was used for first-strand cDNA synthesis using a one-step RT-PCR kit (QIAGEN). To measure mRNA levels, real-time qPCRs were performed on resulting cDNA using gene-specific primers, as listed in Table S1. Each RT-qPCR was repeated at least three times, the values were normalized to the Rp49 transcript. For X chromosome target genes where we compared both male and female expression, data were normalized to female controls. Error bars represented the standard error of the mean.
RNA isolation and library generation for RNA-seq
Ten salivary glands per sample were collected from wandering 3rd instar larvae of wild-type (Nub-Gal4 x w118) and Mtor KD flies in 1 mL of TRIzol (Ambion). Total RNA was purified as mention above and the quality of RNA was examined by running on agarose-formaldehyde gels. For library preparation, polyA+ RNA was isolated from 500 ng of total RNA using Oligo(dT)25 Dynabeads (Thermo Fisher) and constructed into strand-specific libraries using the dUTP method (Parkhomchuk et al., 2009). UTP-marked cDNA was end-repaired using end-repair mix (Enzymatics, MA), tailed with deoxyadenine using Klenow exo- (Enzymatics), and ligated to custom dualindexed adapters with T4 DNA ligase (Enzymatics). Libraries were size-selected with SPRIselect beads (Beckman Coulter, CA) and quantified by qPCR before and after amplification using NEB library quantification kit. Sequencing for this salivary gland RNA-Seq was performed on a NextSeq 500 (Illumina, CA).
TT-seq
TT-Seq in S2 cells was performed largely based on protocol described in Schwalb et al. (2016), with some adaptations for use in Drosophila cell culture. Briefly, 35 million cells were plated in 10 cm dishes and 700 ug of dsRNA was added per dish. 2–3 replicates per condition were run in parallel: dsWhite, dsMtor and dsMOF. KD proceeded for 72 hours and was then repeated with 700 ug dsRNA per condition. After 48 hours, cells were harvested as follows: 50 mM 4-thiouridine (4sU) (Sigma, T4509) stock solution in H2O was prepared ahead of time and stored at 4C protected from light. Dishes were treated and harvested in manageable batches of 3 by replicate to keep timing windows precise. For each plate, 500 uM of 4sU was added in dim light. Cells were incubated for 5 mins then collected. Cells were directly centrifuged after 20 mins at 1,500 RPM for 3 min at RT. Supernatant was removed and 1mL of Trizol was added per 20 million cells, samples were resuspended, rotated at 4C for 15 minutes in the dark for homogenization, flash frozen on dry ice and stored at −80C. Mammalian cells used for spike in (IMR90 cells here) were treated identically to S2 cells to obtain enough RNA for a 5% spike in. Total RNA was isolated using standard phenol/chloroform extraction, RNA was fragmented on a Covaris sonicator (300 ug S2 RNA plus 15 ug mammalian spike in per sample), using the following settings: 1 burst: 30 s ON / 30 s OFF at high settings (200 cycles/burst, Peak Incident Power 140 W, Duty Factor 5%). Fragmentation of RNA was assessed on formamide-agarose gels, revealing a smear roughly between 1.5 kb – 200bp, showing significant depletion of rRNA band(s). RNA was next denatured at 65C for 10 minutes. Samples were split in two into batches of 150ug RNA each and biotinylated with 200 ug of EZ-link HPDP Biotin (Thermo Fisher Scientific 21341), stock at 2mg/mL in DMF) in 1X Binding Buffer (100mM Tris pH7.5, 10mM EDTA pH 8.0) and 30% vol/vol DMF with a total reaction volume of 1mL, rotating in the dark at RT for 2 hr. Biotinylated RNA was then immediately precipitated with the addition of chloroform, followed by centrifugation to isolate the aqueous layer. 1/10 volume of 5 M NaCl and 1 volume of isopropanol were then added. This was spun at 15,000 rpm at 4C for 30 mins to pellet biotinylated RNA. Pellet was washed in cold 75% ethanol, spun again for 5 mins, supernatant removed without allowing pellet to dry, and RNA was resuspended in 100 uL H2O. To separate biotinylated RNA from unlabeled as follows: 100ul of Invitrogen Magnetic Streptavidin Beads (Invitrogen 65002) were washed 2x with 2 vol Wash Buffer (WB: 100mM Tris pH 7.5, 10mM EDTA pH 8, 1M NaCl, 0.1% Tween-20),and resuspended in 1 vol WB. 200uL biotinylated RNA was incubated at 65C for 10 minutes, placed on ice for 5 minutes, mixed with 100ul of prepared streptavidin beads, and incubated at 4C for 15min on rotor. Samples were placed on magnetic rack for 3 min, washed 3x with 900uL 65C WB, 2x with 900ul RT WB, re-suspended in 50ul 1X Turbo DNase Buffer, 1ul of DNase was added and samples were incubated at 37C for 30 minutes. Samples were washed 2x with 900uL RT WB, eluted twice in 100ul RT 100mM DTT, incubated 5 minutes, transferred to magnetic rack and pooled. RNA was then purified using SPRI select beads (in a non-size selecting manner using 50uL beads, 100uL sample, 130ul bead buffer (20% PEG-8000, 2.5 M NaCl, 10 mM Tris HCl pH7.5, 1 mM EDTA, 0.05% Tween-20) and 270ul isopropanol). Beads were washed 2x with 500ul RT 80% EtOH, eluted in 10ul TE pH7.4 and stored at −80C. Quality of labeled RNA was then assessed on a Bioanalyzer RNA Chip. Final concentration of labeled RNA purified was ~200 ng per sample. Lastly, samples were then processed with the same library protocol described for RNA-Seq in salivary glands, minus Poly A+ selection.
ChIP-qPCR
Knockdown conditions were as described for TT-Seq experiments. 10 cm dishes of S2 cells at 85%–95% confluency were cross-linked with 1% methanol-free formaldehyde and quenched with 0.125 M final concentration Glycine. Cells were then harvested and washed with PBS + 0.2mM PMSF. Cells were then treated with 1 mL of ChIP Buffer I (50 mM HEPES pH 7.6, 140 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 10% glycerol, 0.5% IGEPAL, 0.25% Triton X-100, and C0mplete protease inhibitors (11836170001), incubated on a rotator at 4 C for 10 min, and spun down at 400 rcf., 5 min 4°C. Pellets were resuspended in 1 mL of ChIP Buffer II (20 mM Tris pH 8.0, 200 mM NaCl, 1mM EDTA, pH 8.0, 0.5 mM EGTA, pH 8.0, and C0mplete protease inhibitors), incubated on a rotator at 4°C for 10 min, and spun down at 400 rcf., 5 min 4°C. Pellets were resuspended in 1 mL ChIP Buffer III (20 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 0.5% Sarkosyl, 0.1% Na-Deoxycholate (NaDOC) and C0mplete protease inhibitors) and sonicated in a S220 Covaris (15 min peak power 140, Duty Ratio 5, Cycles 200). Samples were transferred into 1.5 mL Lo-bind tubes, 1% Triton X-100 final volume was added and samples were spun down at max speed 10 min at 4°C. Supernatants were then quantified using a BCA assay. Beads were washed 2x with the PBS+BSA and then incubated at 4C on a rotator for 5h with 10 ul of appropriate RNAPII antibody. After 5 hours, 700 ug of protein was added. 10ul of RNAP II antibody was used in each IP (hypo-phosphorylated, 8WG16, sc-56767 or Serine-5 phosphorylated, CTDH8, sc-47701)/3ug of IgGs and Dilution Buffer (20 mM Tris, 100 mM NaCl, 1 mM EDTA, 0.5 mM EGTA) in a 1:2 ratio of lysate:dilution buffer. IPs were incubated on a rotator O/N at 4°C. 10% Input for samples were saved and stored at −80°C. 40ul of Dynabeads per IP were washed and then blocked in 0.3% BSA in PBS on a rotator O/N at 4°C. Beads were then washed twice in Dilution Buffer and added to the IPs and incubated on a rotator at 4°C for 3 hours. After incubation, beads were washed 4x in 1 mL of the following buffers: Low Salt Buffer (20 mM Tris-HCl pH 8, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100), High Salt Buffer (20 mM Tris-HCl, pH 8, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% TritonX- 100), 1X LiCl Buffer (10 mM Tris-HCl pH 8, 250 mM LiCl, 1% IGEPAL, 1% NaDOC, 1 mM EDTA), and TE50 (10 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA). The TE50 wash was repeated twice to ensure that all residual LiCl Buffer was removed. Beads were resuspended in 200ul of Elution Buffer (50mM Tris-HCl, pH 8.0, 10mM EDTA, 1% SDS) and eluted in a thermomixer at 65°C 600 rpm for 30 min. Samples (IPs and Inputs) were decrosslinked at 65°C. After de-crosslinking, equal volume TE (10 mM Tris-HCl pH 8.0, 1 mM EDTA) was added to samples and 0.2 mg/mL final concentration RNase A (Roche 10109169001) was added and incubated at 37°C followed by the addition of 0.2 mg/mL final concentration Proteinase K (Fisher Scientific BP1700-100) and incubation at 55°C. 1 X sample volume of Phenol/Chloroform/isoamyl alcohol was added, samples were incubated at RT and then spun down. 1 X sample volume of chloroform/isoamyl alcohol was added to the aqueous layer and samples were incubated at RT and then spun down at max speed. 0.1X sample volume of sodium acetate (pH 5.2, final concentration of 0.3 M), 1.5 μL glycogen (stock 20 mg/ ml, Roche) and 2.5 X sample volume of cold 100% ethanol was added to the aqueous layer and samples were mixed and incubated at −20°C. Samples were then spun down at max speed, 20 min 4°C, the DNA pellet was washed with 70% cold ethanol, and spun down at max speed, 10 min 4°C. Pellets were then quick-spun and residual ethanol was pipetted off. Pellets were then air-dried until all ethanol was removed. DNA pellets were then re-suspended in TE buffer and used for downstream qPCR analysis, using primers listed in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of ECR
All immunofluorescence quantification on polytene chromosomes described was carried out using FIJI. Background was subtracted and all chromosome arms were selected and included in the fluorescence intensity measurement, which was represented as integrated density to take into account the natural variation in the size of polytene chromosomes within each animal’s salivary gland. N = 7 nuclei per genotype from 3 biological replicates each.
Quantification of HP1
Background was subtracted and levels of HP1 in control and Mtor KD conditions were measured with an ROI encompassing all of the HP1 signal present at the chromocenter and represented as integrated density. Area for these same ROIs was also recorded and plotted. N = 11 nuclei per genotype from 3 biological replicates each.
Quantification of MOF
Background was subtracted and X chromosome associated mean gray value of MOF fluorescence intensity was measured by selecting the entire X chromosome. N = 11 nuclei for control males and 19 nuclei for Mtor RNAi males from 3 biological replicates each.
Quantification of H4K16ac, H3K27me3 and H3K9me3
Background was subtracted, and mean gray value was measured on the X chromosome as well as on chromosome 3R (which was identified via the two distinct bands of EcR at the ecdysone genes, E74 and E75). These mean gray values were then represented as a ratio of X/autosome signal intensity. For H4K16ac, N = 36 nuclei for control males, 35 nuclei for Mtor RNAi males, 17 nuclei for control females all from 4 biological replicates. For H3K27me3, N = 25 nuclei for control males and 30 nuclei for Mtor RNAi males from 4 biological replicates. For H3K9me3, N = 29 nuclei for control males and 32 nuclei for Mtor RNAi males from 6 biological replicates. N = 17 nuclei for control females and 22 for Mtor RNAi females from 3 biological replicates.
Whole mount smRNA FISH quantification: roX1
To quantify roX1 FISH signal, background was subtracted and mean gray value was measured for each nucleus in an average of 5 single z slices throughout entirety of Z stack (stacks on average are 30–40 slices). For X chromosome associated signal, an ROI was drawn around the visible X chromosome territory and mean gray value was measured within that ROI in different Z-slices. For nuclear soluble associated roX1 signal, an ROI was drawn to capture the entire nucleus minus the X chromosome territory. X chromosome plot has less measurements since roX1 X chromosome signal is limited to a smaller focal plane. n = 2 animals, both control and Mtor KD nucleus in same image were measured.
Polytene smRNA FISH quantification
Single molecule RNA FISH for roX1 in polytene squashes was carried out in Mtor RNAi mosaics. Background was subtracted, and mean gray value measurements were sampled using a set ROI at 15 non-overlapping positions along the X chromosome and at 15 different autosomal positions. N = 2 animals per condition, 4 total nuclei per condition quantified.
Single molecule RNA FISH analysis (Rpt6 and Pp4-19c)
Image segmentation
A custom MATLAB pipeline was used to analyze RNA FISH data for these two genes, as seen in Figures 6 and S6. Hoechst staining was used to segment 3D stacks into volumes representing the nucleoplasm of individual cells of the salivary gland using balanced histogram thresholding. 3D nuclear masks were morphologically closed to create contiguous objects comprised of pixels assigned to nucleoplasm. To ensure that only the nucleus interior was used for density calculations, 3D nuclear volumes were eroded by 3% of the mean nuclear diameter, which was determined as the maximum distance between edges of the bounding box describing the nucleus. Cytoplasmic volumes corresponding to each cell were determined by dynamic dilation of nuclear masks in three dimensions using increments of 3% of nuclear diameter until dilated objects overlapped. This was used to determine the distances between the edges of nuclei. One quarter of this distance was used to generate a 3D shell surrounding a modified nuclear mask. The modified nuclear mask was generated by dilating the eroded nuclear volumes by 6% of nuclear diameters. The newly dilated volume created a thin shell around the nuclear mask which functioned as a neutral volume of uncertainty with assigned to neither nucleoplasm nor cytoplasm. Masks were visually inspected to ensure nucleus and cytoplasmic assignments were made to the correct cells. All subsequent calculations were performed on the density of objects found in these volumes, i.e., counts of single mRNAs per cubic micron with the segmented volumes.
mRNA quantification
Detection of diffraction-limited puncta was performed as described (Little et al., 2013), producing a list of coordinates in three-dimensional space. A 2D circular Gaussian with the radius of the point spread function was fit to the mean image of all cytoplasmic objects to estimate the mean intensity of single mRNAs, as described (Little et al., 2015). Densities of punctae in cytoplasm or nucleoplasm was calculated as the count of objects within each 3D mask divided by the volume of the mask. mRNA export graphs represented nuclear and cytoplasmic mRNA densities as a ratio. To quantify the transcriptional output, two approaches were taken. First, for most cells the region of the nucleus containing the transcribing endoreplicated loci was readily apparent in the FISH image and could easily be segmented from the surrounding image using histogram thresholding. Second, in cases where the transcribing loci could not be identified, the brightest objects with fit intensities greater than four times the mean and present within 500 microns of each other were selected as actively transcribing loci. The selection of putative transcribing sites by both methods was visually inspected for accuracy. After selection, the summed intensity was determined and offset per pixel in the average surrounding pixels within a distance of 250 nm was estimated. After background subtraction, the total intensity was divided by the intensity of individual mRNAs, yielding the estimate of instantaneous transcriptional activity.
RNA-seq analysis
RNA-Seq data were aligned against reference genome (dm3) using STAR (v2.3.0e) with default parameters and maximum fragment size of 2kb (“–alignMatesGapMax 2000”). The resulting files were filtered for concordant, primary alignments using SAMtools (v1.1). The enrichment of different genomic regions were analyzed using RSeQC (v3.0.1). Reads were assigned to RefSeq genes using featureCounts (v1.6.2) and differential expression between groups were analyzed using DESeq2. Low count genes whose average read counts across all samples was less than 1 were removed. DEGs were defined by 5% FDR and 1.5 fold change. For visualization, alignment files were converted to bedgraph files using BEDtools (v2.27.1) and each library was normalized to 10 million reads. Replicate bedgraph files were averaged using UCSC toolkit’s bigWigMerge function.
TT-seq analysis
TT-Seq data were pre-processed in similar ways as RNA-Seq. Visualization tracks were also generated in the same way. When assigning reads to RefSeq genes, we included the whole gene body instead of only coding regions. Differential expression between groups were analyzed again using DESeq2. The enrichment of different genomic regions were analyzed using Deeptools (v2.5.7). Heatmaps and metaplots over selected sets of genes were also generated using Deeptools.
Pausing and elongation density index calculations
Pausing and elongation index calculations were based on the definitions given in Larschan et al. (2011). Briefly, Pausing Index (PI) was calculated by taking the ratio of read density within the first 500 bp downstream of the transcriptional start site to the read density within the next 25% of the gene body. The read density is calculated using Bwtool package (the “summary” function) which sums up signal in normalized bigwig files over each given region in a bed file.
To calculate Elongation density Index (EdI): The first 500 bp of the gene is excluded from this calculation to eliminate the effect of the large 5′ peak frequently associated with paused polymerase. The remainder of the gene is then split into two portions, the 5′ region and the 3′ region. EdI was defined as the ratio of the 5′ read density (first 25% of the gene after the first 500 bp) and the 3′ read density (the remaining 75%). The read density is calculated as it was done above.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Mtor | gift from Prof. K.M. Johansen (Iowa State University) and available at Developmental Studies Hybridoma Bank | Clone 12F10, RRID: AB_2721935 |
| Rabbit polyclonal anti-Lamin DmO | gift from Prof. P. Fisher (SUNY StonyBrook School of Medicine) | Clone L7 |
| Rabbit anti-MOF | Gift from Prof M.I. Kuroda (Harvard University) | N/A |
| Rabbit polyclonal anti-MLE | Gift from Prof M.I. Kuroda (Harvard University) | N/A, RRID: AB_2568421 |
| Rabbit polyclonal anti-H4K16ac | Santa Cruz Biotechnology, Inc. | Cat# sc-8662, RRID: AB_634804 |
| Rabbit polyclonal anti-Histone H3 | Abcam | Cat#ab1791, RRID:AB_302613 |
| Mouse monoclonal anti-HP1 | Developmental Studies Hybridoma Bank | Clone C1A9, RRID:AB_528276 |
| Mouse monoclonal anti-EcR | Developmental Studies Hybridoma Bank | Clone DDA2.7, RRID:AB_528209 |
| Chicken polyclonal anti-GFP | Aves Labs, Inc. | Cat#1020, RRID:AB_10000240 |
| Rabbit monoclonal anti-H3K27me3 | Cell Signaling Technology | Cat#9733, RRID:AB_2616029 |
| Rabbit polyclonal anti-H3K9me3 | Abcam | Cat#ab8898, RRID:AB_306848 |
| Goat anti-mouse IgG secondary Alexa Fluor 488 | Thermo Fisher Scientific | Cat # A32723, RRID:AB_2633275 |
| Goat anti-rabbit IgG secondary Alexa Fluor 488 | Thermo Fisher Scientific | Cat # A32731, RRID:AB_2633280 |
| Goat anti-mouse IgG secondary Alexa Fluor 568 | Thermo Fisher Scientific | Cat # A-11004, RRID:AB_2534072 |
| Goat anti-rabbit IgG secondary Alexa Fluor 568 | Thermo Fisher Scientific | Cat # A-11011, RRID:AB_143157 |
| Biological samples | ||
| Drosophila 3rd instar larval salivary glands | This study | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dynabeads Protein A | Thermo Fisher Scientific | Cat#10002D |
| Power SYBR Green PCR Master mix | Applied Biosystems | Cat#4367659 |
| Sigmacote | Sigma Aldrich | Cat#SL2 |
| Poly L-lysinate slides | Polysciences | Cat#22247 |
| ProLong Gold Antifade | Thermo Fisher Scientific | Cat# P36930 |
| VECTASHIELD® Antifade Mounting Medium | Vector Laboratories | Cat#H-1000-10 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#H3570 |
| Hybridization Buffer | Biosearch Technologies | Cat# SMF-HB1-1 |
| Wash Buffer A | Biosearch Technologies | Cat# SMF-WA1-60 |
| Wash Buffer B | Biosearch Technologies | Cat# SMF-WB1-20 |
| Dynabeads® Oligo (dT)25 | Thermo Fisher Scientific | Cat#61002 |
| SPRIselect beads | Beckman Coulter | Cat# B23317 |
| EZ-link HPDP Biotin | Thermo Fisher Scientific | Cat#21341 |
| 4-thiouridine (4sU) | Sigma-Aldrich | Cat# T4509 |
| Dynabeads MyOne Streptavidin C1 | Thermo Fisher Scientific | Cat#65002 |
| Critical commercial assays | ||
| NEBNext Library Quant Kit for Illumina | NEB | E7630L |
| OneStep RT-PCR kit | QIAGEN | Cat#210212 |
| MEGAscript T7 Transcription kit | Thermo Fisher Scientific | AM1334 |
| Pure Link RNA mini kit | Thermo Fisher Scientific | 12183018A |
| NextSeq 500/550 High Output v2 kit (75 cycles) | Illumina | FC-404-2005 |
| Bioanalyzer High Sensitivity RNA Analysis Kit | Agilent | Cat#5067-1513 |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat#23225 |
| Deposited data | ||
| RNA-Seq 3rd instar larval salivary glands | This study | GEO: GSE155323 |
| TT-Seq S2 cells | This study | GEO: GSE155323 |
| Experimental models: Cell lines | ||
| S2-DRSC | Drosophila Genomics Resource Center | Flybase: FBrf0024118 |
| Experimental models: Organisms/strains | ||
| Mtor RNAi line - w[1118]; Mi{GFP[E.3×P3] = ET1}side-VIII[MB03736] | Bloomington Drosophila Stock Center | #24265 |
| MOF RNAi line - y[1] v[1]; P{y[+t7.7] v [+t1.8] = TRiP.JF01707}attP2 | Bloomington Drosophila Stock Center | #31401 |
| Nubbin-Gal4 line - w[*]; P{w[nub.PK] = nub-GAL4.K}2 | Bloomington Drosophila Stock Center | #86108 |
| mCD8-RFP line - w[*]; P{w[+mC] = UAS-mCD8.ChRFP}3 | Bloomington Drosophila Stock Center | #27392 |
| Mtor allele - y[1] w[67c23]; P{w[+mC] = lacW}Mtor[k03905]/CyO | Bloomington Drosophila Stock Center | #10537 |
| Heatshock clone line (Act5c, FRT y+ FRT, Gal4, UAS-GFP) - y[1] w[*]; P{w[+mC] = AyGAL4}25 P{w[+mC] = UAS-GFP.S65T} Myo31DF[T2] | Bloomington Drosophila Stock Center | #4411 |
| Heatshock clone line - y1, w*, hsFLP; Kr/CyO; MKRS/TM6B | Gift from Prof. S. DiNardo (University of Pennsylvania) | N/A |
| roX1/2 allele - roX1SMC17A roX2Δw+; +/ CyO[w+roX1] | Gift from Prof. V. Meller (Wayne State University) | N/A |
| Oregon-R | Bloomington Drosophila Stock Center | #5 |
| w118 - w[118];Df(2R)H3D3/CyO | Bloomington Drosophila Stock Center | #31 |
| Oligonucleotides | ||
| Expression analysis primers | IDT | See Table S1 |
| dsRNA primers | IDT | See Table S1 |
| RNA FISH probes | Biosearch Technologies | See Table S1 |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://fiji.sc/ |
| Leica Application Suite X | Leica software | https://www.leica-microsystems.com/products/microscope-software/software-for-life-science-research/las-x-powerful-and-flexible/ |
| Prism | GraphPad software | https://www.graphpad.com/scientific-software/prism/ |
| Biorender | Biorender software | https://biorender.com/ |
| R | The R Project for Statistical Computing | https://www.r-project.org/ |
| STAR (v2.3.0e) | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| SAMtools (v1.1) | Li et al., 2009 | http://samtools.sourceforge.net/ |
| RSeQC (v3.0.1) | Wang et al., 2012 | http://rseqc.sourceforge.net/ |
| featureCounts (v1.6.2) | Liao et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| DESeq2 (v1.30.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| BEDtools (v2.27.1) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| Deeptools (v2.5.7) | Ramírez et al., 2016 | https://github.com/deeptools/deepTools |
| Python | Python Software Foundation | https://www.python.org |
| MATLAB | MATLAB software | https://www.mathworks.com/products/matlab.html |
Highlights.
In vivo loss of Nup Mtor leads to selective upregulation of the male X chromosome
Mtor-driven attenuation of the male X occurs in broad domains bound by MSL complex
Mtor interacts with MSL components genetically and rescues male-specific lethality
Mtor restrains dosage-compensated expression at the level of nascent transcription
ACKNOWLEDGMENTS
We are grateful to Mitzi Kuroda (Harvard University) for her gift of MOF and MLE antibodies. We thank Kristen Johansen (Iowa State University) and the Developmental Studies Hybridoma Bank for critical antibodies. We thank the Bloomington Drosophila Stock Center, Vicky Meller (Wayne State University), and Steve DiNardo (University of Pennsylvania) for key fly stocks, and the Raj Jain laboratory (University of Pennsylvania) and the University of Pennsylvania CDB Microscopy Core Facility for confocal microscope use. J.R.A. is supported by National Institutes of Health grant F31GM133161. M.C. is supported by National Institutes of Health grant R01GM124143.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109236.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY STATEMENT
We worked to ensure sex balance in the selection of non-human subjects. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. One or more of the authors of this paper received support from a program designed to increase minority representation in science.
REFERENCES
- Aksenova V, Smith A, Lee H, Bhat P, Esnault C, Chen S, Iben J, Kaufhold R, Yau KC, Echeverria C, et al. (2020). Nucleoporin TPR is an integral component of the TREX-2 mRNA export pathway. Nat. Commun 11, 4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee OK, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, and Kuroda MI (2008). A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134, 599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte MS, Moran VA, Menon DU, Rattner BP, Barry KH, Zunder RM, Kelley R, and Meller VH (2014). Generation of a useful roX1 allele by targeted gene conversion. G3 (Bethesda) 4, 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlucea J, Andrade R, Alonso R, and Aréchaga J (1998). The nuclear basket of the nuclear pore complex is part of a higher-order filamentous network that is related to chromatin. J. Struct. Biol 124, 51–58. [DOI] [PubMed] [Google Scholar]
- Capelson M, Liang Y, Schulte R, Mair W, Wagner U, and Hetzer MW (2010). Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell 140, 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang PW, and Kurnit DM (2003). Study of dosage compensation in Drosophila. Genetics 165, 1167–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, and Akhtar A (2012). Dosage compensation in Drosophila melanogaster: epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet 13, 123–134. [DOI] [PubMed] [Google Scholar]
- Conrad T, Cavalli FM, Vaquerizas JM, Luscombe NM, and Akhtar A (2012). Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science 337, 742–746. [DOI] [PubMed] [Google Scholar]
- Deng X, and Meller VH (2006). roX RNAs are required for increased expression of X-linked genes in Drosophila melanogaster males. Genetics 174, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasken MB, and Corbett AH (2005). Process or perish: quality control in mRNA biogenesis. Nat. Struct. Mol. Biol 12, 482–488. [DOI] [PubMed] [Google Scholar]
- Ferrari F, Plachetka A, Alekseyenko AA, Jung YL, Ozsolak F, Kharchenko PV, Park PJ, and Kuroda MI (2013). “Jump start and gain” model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Rep. 5, 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F, Alekseyenko AA, Park PJ, and Kuroda MI (2014). Transcriptional control of a whole chromosome: emerging models for dosage compensation. Nat. Struct. Mol. Biol 21, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontoura BM, Dales S, Blobel G, and Zhong H (2001). The nucleoporin Nup98 associates with the intranuclear filamentous protein network of TPR. Proc. Natl. Acad. Sci. USA 98, 3208–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuhashi H, Nakajima M, and Hirose S (2006). DNA supercoiling factor contributes to dosage compensation in Drosophila. Development 133, 4475–4483. [DOI] [PubMed] [Google Scholar]
- Galupa R, and Heard E (2015). X-chromosome inactivation: new insights into cis and trans regulation. Curr. Opin. Genet. Dev 31, 57–66. [DOI] [PubMed] [Google Scholar]
- Galy V, Gadal O, Fromont-Racine M, Romano A, Jacquier A, and Nehrbass U (2004). Nuclear retention of unspliced mRNAs in yeast is mediated by perinuclear Mlp1. Cell 116, 63–73. [DOI] [PubMed] [Google Scholar]
- Gelbart ME, and Kuroda MI (2009). Drosophila dosage compensation: a complex voyage to the X chromosome. Development 136, 1399–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozalo A, Duke A, Lan Y, Pascual-Garcia P, Talamas JA, Nguyen SC, Shah PP, Jain R, Joyce EF, and Capelson M (2020). Core Components of the Nuclear Pore Bind Distinct States of Chromatin and Contribute to Polycomb Repression. Mol. Cell 77, 67–81.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregersen LH, Mitter R, and Svejstrup JQ (2020). Using TTchem-seq for profiling nascent transcription and measuring transcript elongation. Nat. Protoc 15, 604–627. [DOI] [PubMed] [Google Scholar]
- Grimaud C, and Becker PB (2009). The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes Dev. 23, 2490–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada FN, Park PJ, Gordadze PR, and Kuroda MI (2005). Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev. 19, 2289–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias N, Paulo JA, Tatarakis A, Wang X, Edwards AL, Bhanu NV, Garcia BA, Haas W, Gygi SP, and Moazed D (2020). Native Chromatin Proteomics Reveals a Role for Specific Nucleoporins in Heterochromatin Organization and Maintenance. Mol. Cell 77, 51–66.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto FV, Benner C, and Hetzer MW (2015). The nucleoporin Nup153 regulates embryonic stem cell pluripotency through gene silencing. Genes Dev. 29, 1224–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob Y, Mongkolsiriwatana C, Veley KM, Kim SY, and Michaels SD (2007). The nuclear pore protein AtTPR is required for RNA homeostasis, flowering time, and auxin signaling. Plant Physiol. 144, 1383–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston DM, Sedkov Y, Petruk S, Riley KM, Fujioka M, Jaynes JB, and Mazo A (2011). Ecdysone- and NO-mediated gene regulation by competing EcR/Usp and E75A nuclear receptors during Drosophila development. Mol. Cell 44, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalverda B, Pickersgill H, Shloma VV, and Fornerod M (2010). Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell 140, 360–371. [DOI] [PubMed] [Google Scholar]
- Kosova B, Panté N, Rollenhagen C, Podtelejnikov A, Mann M, Aebi U, and Hurt E (2000). Mlp2p, a component of nuclear pore attached intranuclear filaments, associates with nic96p. J. Biol. Chem 275, 343–350. [DOI] [PubMed] [Google Scholar]
- Kuhn TM, Pascual-Garcia P, Gozalo A, Little SC, and Capelson M (2019). Chromatin targeting of nuclear pore proteins induces chromatin decondensation. J. Cell Biol 218, 2945–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda MI, Kernan MJ, Kreber R, Ganetzky B, and Baker BS (1991). The maleless protein associates with the X chromosome to regulate dosage compensation in Drosophila. Cell 66, 935–947. [DOI] [PubMed] [Google Scholar]
- Labade AS, Karmodiya K, and Sengupta K (2016). HOXA repression is mediated by nucleoporin Nup93 assisted by its interactors Nup188 and Nup205. Epigenetics Chromatin 9, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, and Kuroda MI (2011). X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Light WH, Freaney J, Sood V, Thompson A, D’Urso A, Horvath CM, and Brickner JH (2013). A conserved role for human Nup98 in altering chromatin structure and promoting epigenetic transcriptional memory. PLoS Biol. 11, e1001524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim CK, and Kelley RL (2013). The Drosophila over compensating males gene genetically inhibits dosage compensation in males. PLoS ONE 8, e60450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lince-Faria M, Maffini S, Orr B, Ding Y, Cláudia Florindo, Sunkel CE, Tavares A, Johansen J, Johansen KM, and Maiato H (2009). Spatiotemporal control of mitosis by the conserved spindle matrix protein Megator. J. Cell Biol 184, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SC, Tikhonov M, and Gregor T (2013). Precise developmental gene expression arises from globally stochastic transcriptional activity. Cell 154, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SC, and Gregor T (2018). Single mRNA molecule detection in Drosophila. Methods Mol. Biol 1649, 127–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little SC, Sinsimer KS, Lee JJ, Wieschaus EF, and Gavis ER (2015). Independent and coordinate trafficking of single Drosophila germ plasm mRNAs. Nat. Cell Biol 17, 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, and Rattner BP (2002). The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J. 21, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S, and Akhtar A (2007). The right dose for every sex. Chromosoma 116, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendjan S, Taipale M, Kind J, Holz H, Gebhardt P, Schelder M, Vermeulen M, Buscaino A, Duncan K, Mueller J, et al. (2006). Nuclear pore components are involved in the transcriptional regulation of dosage compensation in Drosophila. Mol. Cell 21, 811–823. [DOI] [PubMed] [Google Scholar]
- Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, and Becker PB (2004). Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J. 23, 2258–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilson KA, Guo J, Turek ME, Brogie JE, Delaney E, Luse DS, and Price DH (2015). THZ1 Reveals Roles for Cdk7 in Co-transcriptional Capping and Pausing. Mol. Cell 59, 576–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Garcia P, Jeong J, and Capelson M (2014). Nucleoporin Nup98 associates with Trx/MLL and NSL histone-modifying complexes and regulates Hox gene expression. Cell Rep. 9, 433–442. [DOI] [PubMed] [Google Scholar]
- Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, and Soldatov A (2009). Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Garcia P, Debo B, Aleman JR, Talamas JA, Lan Y, Nguyen NH, Won KJ, and Capelson M (2017). Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol. Cell 66, 63–76.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani HP, and Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936. [DOI] [PubMed] [Google Scholar]
- Powers JA, and Eissenberg JC (1993). Overlapping domains of the heterochromatin-associated protein HP1 mediate nuclear localization and heterochromatin binding. J. Cell Biol 120, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ptak C, and Wozniak RW (2016). Nucleoporins and chromatin metabolism. Curr. Opin. Cell Biol 40, 153–160. [DOI] [PubMed] [Google Scholar]
- Qi H, Rath U, Wang D, Xu YZ, Ding Y, Zhang W, Blacketer MJ, Paddy MR, Girton J, Johansen J, and Johansen KM (2004). Megator, an essential coiled-coil protein that localizes to the putative spindle matrix during mitosis in Drosophila. Mol. Biol. Cell 15, 4854–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M, and D’Angelo MA (2017). Nuclear pore complexes and regulation of gene expression. Curr. Opin. Cell Biol 46, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raices M, Bukata L, Sakuma S, Borlido J, Hernandez LS, Hart DO, and D’Angelo MA (2017). Nuclear Pores Regulate Muscle Development and Maintenance by Assembling a Localized Mef2C Complex. Dev. Cell 41, 540–554.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Skaggs HS, Xing H, Wilkerson DC, Murphy LA, Hong Y, Mayhew CN, and Sarge KD (2007). HSF1-TPR interaction facilitates export of stress-induced HSP70 mRNA. J. Biol. Chem 282, 33902–33907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, and Lucchesi JC (2000). The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol. Cell. Biol 20, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow CJ, and Paschal BM (2014). Roles of the nucleoporin Tpr in cancer and aging. Adv. Exp. Med. Biol 773, 309–322. [DOI] [PubMed] [Google Scholar]
- Sood V, and Brickner JH (2014). Nuclear pore interactions with the genome. Curr. Opin. Genet. Dev 25, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strambio-de-Castillia C, Blobel G, and Rout MP (1999). Proteins connecting the nuclear pore complex with the nuclear interior. J. Cell Biol 144, 839–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Neumann MF, Prestel M, Kremmer E, Kaether C, Haass C, and Becker PB (2005). Stable chromosomal association of MSL2 defines a dosage-compensated nuclear compartment. Chromosoma 114, 352–364. [DOI] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, and Becker PB (2008). The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet. 4, e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Zabel A, Gilfillan GD, Feller C, and Becker PB (2013). Different chromatin interfaces of the Drosophila dosage compensation complex revealed by high-shear ChIP-seq. Genome Res. 23, 473–485. 10.1101/gr.146407.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, and Birchler JA (2009). Interaction study of the male specific lethal (MSL) complex and trans-acting dosage effects in metafemales of Drosophila melanogaster. Cytogenet. Genome Res 124, 298–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A, Van Houwe G, Hediger F, Kalck V, Cubizolles F, Schober H, and Gasser SM (2006). Nuclear pore association confers optimal expression levels for an inducible yeast gene. Nature 441, 774–778. [DOI] [PubMed] [Google Scholar]
- Tan-Wong SM, Wijayatilake HD, and Proudfoot NJ (2009). Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 23, 2610–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Vosse DW, Wan Y, Lapetina DL, Chen WM, Chiang JH, Aitchison JD, and Wozniak RW (2013). A role for the nucleoporin Nup170p in chromatin structure and gene silencing. Cell 152, 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Suyama R, Kind J, Miura K, Luscombe NM, and Akhtar A (2010). Nuclear pore proteins nup153 and megator define transcriptionally active regions in the Drosophila genome. PLoS Genet. 6, e1000846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinciguerra P, Iglesias N, Camblong J, Zenklusen D, and Stutz F (2005). Perinuclear Mlp proteins downregulate gene expression in response to a defect in mRNA export. EMBO J. 24, 813–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: Quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wente SR, and Rout MP (2010). The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol 2, a000562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimowska G, Aris JP, and Paddy MR (1997). A Drosophila Tpr protein homolog is localized both in the extrachromosomal channel network and to nuclear pore complexes. J. Cell Sci 110, 927–944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number for all sequencing data reported in this paper is GEO: GSE155323, containing the RNA-Seq datasets in Control and Mtor KD conditions from male and female larval salivary glands as well as TT-Seq datasets in Control, Mtor KD and MOF KD conditions in S2 cells. For the custom MATLAB pipeline used for quantification of RNA FISH data in Figure 6, please contact lead contact Dr. Maya Capelson (capelson@pennmedicine.upenn.edu or mcapelson@gmail.com).