

Abstract
Photosystem II (PSII) uses solar energy to oxidize water and delivers electrons for life on Earth. The photochemical reaction center of PSII is known to possess two stationary states. In the open state (PSIIO), the absorption of a single photon triggers electron-transfer steps, which convert PSII into the charge-separated closed state (PSIIC). Here, by using steady-state and time-resolved spectroscopic techniques on Spinacia oleracea and Thermosynechococcus vulcanus preparations, we show that additional illumination gradually transforms PSIIC into a light-adapted charge-separated state (PSIIL). The PSIIC-to-PSIIL transition, observed at all temperatures between 80 and 308 K, is responsible for a large part of the variable chlorophyll-a fluorescence (Fv) and is associated with subtle, dark-reversible reorganizations in the core complexes, protein conformational changes at noncryogenic temperatures, and marked variations in the rates of photochemical and photophysical reactions. The build-up of PSIIL requires a series of light-induced events generating rapidly recombining primary radical pairs, spaced by sufficient waiting times between these events—pointing to the roles of local electric-field transients and dielectric relaxation processes. We show that the maximum fluorescence level, Fm, is associated with PSIIL rather than with PSIIC, and thus the Fv/Fm parameter cannot be equated with the quantum efficiency of PSII photochemistry. Our findings resolve the controversies and explain the peculiar features of chlorophyll-a fluorescence kinetics, a tool to monitor the functional activity and the structural-functional plasticity of PSII in different wild-types and mutant organisms and under stress conditions.
The closed-state of photosystem II possesses a hitherto unrecognized structural and functional plasticity and upon illumination assumes a light-adapted charge-separated state.

Introduction
Photosystem II (PSII) is a multisubunit enzyme that catalyzes the oxidation of water and the reduction of plastoquinone (Nelson and Yocum, 2006). In the open-state PSII (PSIIO), electron transfer, upon the absorption of one photon, starts with the formation of the primary radical pair P680+Pheo−, which is stabilized via the re-oxidation of Pheo− by the first, stable quinone acceptor molecule, QA, leading to a charge-separated state P680+. (Here, P680 and Pheo, following the conventional notations, refer to the primary electron donor from which charge separation starts, irrespective of its molecular identity, and pheophytin-aD1, respectively.) In consecutive steps, P680+ is re-reduced by the redox-active tyrosine (YZ), which then oxidizes the Mn4CaO5 cluster, producing the S2 state of the oxygen-evolving complex (OEC). This generates a closed-state of PSII (PSIIC) with QA reduced. In this state, the light energy absorbed can produce only a rapidly recombining species, the P680+Pheo− radical pair (Sipka et�al., 2019). PSIIC persists for several hundred microseconds, until the electron is transferred from to QB, the secondary quinone acceptor (Shlyk-Kerner et�al., 2006). When this step is blocked by PSII inhibitor molecules, such as 3-(3′,4′ dichlorophenyl)-1,1′dimethylurea (DCMU), its lifetime becomes considerably longer, and PSII may assume a stationary state since the charge recombination between and is a thermally assisted de-trapping reaction (Tyystjarvi and Vass, 2004).
The activity of PSII is routinely tested using chlorophyll-a (Chl-a) fluorescence induction kinetics (Papageorgiou and Govindjee (2004) and references therein). To characterize the dark-to-light transition of PSII, either the yield or the intensity of the fluorescence emission is monitored. In both cases, the fluorescence levels rise from the minimum to the maximum, from Fo to Fm, or from O to P, respectively. The fast fluorescence transients usually contain intermediary states, J and I; these are absent when the electron transfer from to QB is inhibited, and then the O-to-P rise assumes a sigmoidal shape. According to the mainstream model, to reach Fm (or P) it is necessary, and sufficient, to have QA completely reduced in all the active PSII centers (Duysens and Sweers, 1963; Stirbet and Govindjee, 2012). Fluorescence lifetime measurements confirmed that the transition from Fo to Fm induced by the reduction of QA, upon continuous illumination, effectively corresponds to an increase in the lifetime (Holzwarth et�al., 1985; Hodges and Moya, 1986; Roelofs et�al., 1992) and, therefore, in the fluorescence yield resulting from center closure. In this framework, the Fv/Fm parameter represents an estimation of the maximal quantum efficiency of PSII (Fv = Fm − Fo; Butler, 1978; Duysens, 1978; Genty et�al., 1989). The multiphasic (O–J–I–P) rise is explained by whole-chain electron-transfer reactions affecting the reduction state of QA (Strasser et�al., 2004; Stirbet and Govindjee, 2012), and the sigmoidal rise is ascribed to an energetic coupling (connectivity) between PSII units (Joliot and Joliot, 1964; Lavergne and Trissl, 1995; Stirbet, 2013). Despite the success of this widely used “QA model”, it is not free of controversy (Joliot and Joliot, 1979; Vredenberg, 2011; Schansker et�al., 2014; Magyar et�al., 2018; Laisk and Oja, 2020). Also, in some cyanobacterial mutants (Vavilin et�al., 1999) and wild-type green algal cells (Treves et�al., 2016) no or very poor correlation is observed between the measured Fv/Fm parameter and the oxygen evolution activity of PSII.
The main problem with the QA model, i.e. that the redox state of QA accounts in full for the changes in fluorescence yield upon center closure, is that Fm cannot be reached upon reducing QA with a single-turnover saturating flash (STSF), and several additional flashes are required to complement Fv (Joliot and Joliot, 1979; Magyar et�al., 2018). Also, to generate the O-to-P rise, multiple turnover saturating flashes (MTSFs) are required (Delosme, 1967; Schansker et�al., 2011; Magyar et�al., 2018). Recent investigations revealed the occurrence of conformational changes associated with Fv and uncovered the requirement of relatively long waiting times between the STSFs, hence the existence of rate-limiting steps that cannot be accounted for either by rapid photochemical reactions or by heterogeneity of PSII (Lavergne and Trissl, 1995) as discussed by Magyar et�al. (2018). Evidently, after closing PSII, all STSFs (or surplus excitations) act on PSIIC. Hence, the gradual fluorescence rise, observed upon exposing PSIIC to a train of STSFs, strongly suggests the stepwise formation of a previously unidentified state of PSII—the light-adapted charge-separated state (PSIIL), which is characterized here at physiological and cryogenic temperatures. The PSIIC-to-PSIIL transition appears to be driven by transient, light-induced, electric fields but with rate-limiting processes, waiting times between effective excitations involved, which might be linked to dielectric relaxation. We show that PSIIL can be generated both in the monomeric and dimeric forms of PSII, demonstrating the occurrence of the sigmoidal rise of fast Chl-a fluorescence in the absence of connectivity between PSII units. Further, we provide irrevocable experimental evidence showing that the Fv/Fm parameter cannot be used to determine the quantum efficiency of PSII photochemistry. Our findings resolve the controversies regarding the origin of Fv and lay the foundations for a deeper understanding of the structural and functional changes during the dark-to-light transition of PSII in different organisms and mutants, as well as upon exposing plants to different stress conditions.
Results
Stepwise light-induced generation of PSIIL from PSIIC: Chl-a fluorescence transients
To investigate the possibility that in the dimeric PSII core complex (PSII CC) the charge-separated states of the two monomers influence each other, we compared the Chl-a fluorescence induction of DCMU-treated dimeric and monomeric PSII CC. The kinetics of the fast-fluorescence intensity transients of monomeric and dimeric complexes were virtually indistinguishable (Figure�1). Moreover, monomers, similarly to dimers, displayed a gradual rise of the fluorescence elicited by a train of STSFs (Supplemental Figure 1). These results indicate the absence of significant cooperativity between two monomers in the PSII CC of Thermosynechococcus (T.) vulcanus. Since the physiological state of PSII is a dimer (Shen, 1998), all further experiments using PSII CC were performed on its dimeric form.
Figure 1.
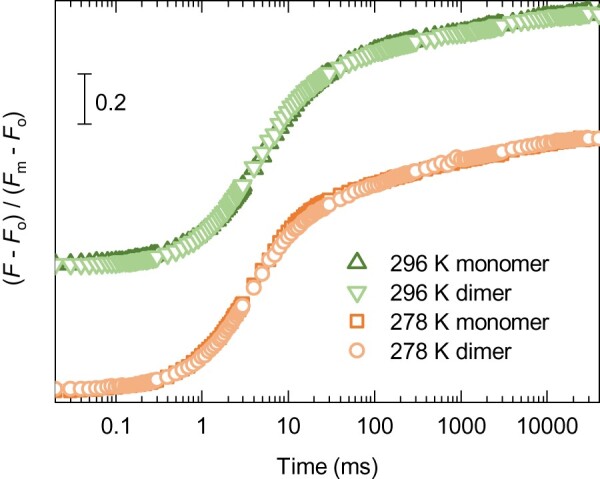
Fast Chl-a fluorescence rise of DCMU-treated solubilized monomeric and dimeric PSII core complexes of T. vulcanus at 278 and 296 K. The photon flux density of the excitation was 3500 �mol photons m−2 s−1. (Note: curves at 296 K are upshifted for simpler comparison).
To verify earlier conclusions that it is possible to increase the Chl-a fluorescence yield in the presence of reduced QA (PSIIC) and without inducing additional stable charge separation (Joliot and Joliot, 1979; Magyar et�al., 2018; Sipka et�al., 2019), we performed experiments on PSII CCs in which QA was pre-reduced with 2-mM dithionite. This treatment also prevented the formation of the S2 state of the OEC, as evidenced by the absence of thermoluminescence (data not shown). (In the presence of DCMU alone, T. vulcanus PSII CC gives rise to the so-called Q-band due to recombination; Shen and Inoue, 1993.) Also, because of the prereduction of QA, the so-called C550 absorption band shift (Butler and Okayama, 1971) was not observed (Supplemental Figure 2). C550 is diagnostic of the presence of , via detecting the effect of local electric field on Pheo, an electrochromic band shift of the Qx transition of PheoD1 (�rsk�ld et�al., 2003). As shown in Figure�2A, despite the pre-reduced QA, the train of STSFs was still capable of inducing fluorescence increments, gradually reaching Fm (PSIIL).
Figure 2.

Effects of dithionite and FeCy on the variations of the STSF-induced Chl-a fluorescence yield of DCMU-treated PSII CC of T. vulcanus at 278 K. A, Kinetic traces, normalized to Fm−Fo, in the absence and presence of 2-mM dithionite, recorded with 1.6-kHz sampling rate; STSFs were applied 500 ms apart; at the end, three 200-ms long blue laser flashes were fired to ensure the saturation. B, Variations of the fluorescence yield parameters relative to the Fm level of the control (DCMU-treated PSII CC) in the absence and presence of 2-mM dithionite or 2-mM FeCy. F1, F2, and F3 denote the levels after the first, second, and third STSF, respectively. C, Dependence of the F1/Fm and Fv/Fm ratios on the concentration of dithionite.
To avoid the possible involvement of alternative reductive electron-transfer pathways, we also recorded STSF-induced Chl-a fluorescence transients of DCMU-treated PSII CC in the presence of ferricyanide (FeCy; Figure�2B). Secondary electron transfer from cytochrome b559 (Cyt b559) to P680+ (Tracewell and Brudvig, 2008) and to tyrosine-D or the S2 state of the OEC (Feyziyev et�al., 2013) have earlier been shown to occur at cryogenic temperatures. Absorption transient measurements were carried out between 410 and 460 nm and 525 and 565 nm to estimate the significance of these reactions at room temperature. Our data show that electron donation from Cyt b559 occurs in <10% of the RCs, probably due to partial impairment of the donor side in some PSII CCs (Supplemental Figure 3). At the same time, FeCy exerted little effect on Fv. Similar to the DCMU-treated control (Magyar et�al., 2018), the first STSF, which closed PSII, induced an Fo–F1 fluorescence rise (F1 < Fm) but additional flashes were required to increase the fluorescence in a stepwise manner (producing F1–F2, F2–F3, etc. increments) before eventually, reaching Fm (Figure�2B).
Thus, it can be concluded that, albeit the overall fluorescence yields were affected by these treatments, the fluorescence increments were present in all cases. Very similar data were obtained at cryogenic temperatures and on isolated thylakoid membranes (Supplemental Figure 4—showing the STSF-induced Chl-a fluorescence transients of DCMU-treated thylakoid membranes at 213 K in the absence and presence of dithionite or FeCy). Hence, neither the prereduction of QA nor the oxidation of potentially active reducing components of PSII CC and the thylakoid membranes prevent the fluorescence increments. By using C550 and ΔA515 measurements on PSII CC of T. vulcanus and Spinacia oleracea thylakoid membranes, respectively, we confirmed the conclusion of (Joliot and Joliot, 1979) that the second STSF produced no additional stable charge separation (Supplemental Table 1), despite the fact that sufficiently long waiting times were allowed to induce the F1–F2 increments.
The STSF-induced fluorescence increments could only be halted in the presence of high concentrations of dithionite (Figure�2C), which are known to pre-reduce Pheo (Klimov et�al., 1977; Barber and Melis, 1990), and thus prevent the formation of the P680+Pheo− radical pair. Hence, these data show that the flash-induced generation of the radical pair is a necessary condition to generate the fluorescence increments in PSIIC and thus to allow the light-induced formation of PSIIL. It is interesting to note that dithionite, while reducing (2 mM) or essentially eliminating (20 mM) Fv, also modulated the overall Chl-a fluorescence yield of PSII CC (Supplemental Table 2); at the same time, in spinach thylakoid membranes 2-mM dithionite exerted very little effect on Fm, <10% increase compared to the control (Supplemental Figure 4C). (This difference between PSII CC and thylakoids might be due to different penetration ability of dithionite in the absence and presence of the bilayer lipid membrane.) These data strongly suggest that, similar to Chl-a (Connolly et�al., 1982), the fluorescence yield depends on the physico-chemical environment of the emitter molecules.
Conformational changes and charge stabilization associated with the PSIIC–PSIIL transition: rapid-scan FTIR
Our earlier experiments have indicated the involvement of conformational changes associated with the fluorescence increments following the closure of PSII (Schansker et�al., 2011; Magyar et�al., 2018). To obtain information on the nature of these changes, rapid-scan Fourier transform infrared (FTIR) difference spectroscopy (Mezzetti and Leibl, 2017) experiments were performed on DCMU-treated PSII CC. Light-induced FTIR difference spectroscopy is a widely used technique to study the mechanism of photo-induced reactions in proteins, as it makes it possible to visualize changes not only in cofactors but also in the protein. Time-resolved FTIR difference spectra have the additional advantage of monitoring the kinetic evolution of these changes. Flash-induced time-resolved FTIR difference spectra were recorded after exposing the sample to either 1 or 20 STSFs (Figure�3, A–C) and prominent changes in the 1,800–1,200 cm−1 region were observed. In this region, several marker bands have been identified in the past, such as the positive band at 1,478 cm−1 (semiquinone ; Berthomieu et�al., 1990) or the negative band at 1,401 cm−1 (marker for the S1 → S2 transition in the Mn cluster; Noguchi, 2007). Furthermore, in FTIR, difference spectra changes in the so-called amide I region (1690–1610 cm−1) are indicative of conformational changes in the protein.
Figure 3.

Time-resolved rapid-scan FTIR difference spectra and relaxation kinetics following 1 or 20 STSFs (saturating intensity 20 mJ, 532 nm, 7-ns laser flash) excitation of DCMU-treated PSII core particles at 303 K. Spectra recorded at different times, as indicated after one (A) or 20 (B) flash(es). C, Spectra of transients induced by 1 or 20 STSFs averaged as indicated by the label in the figure; and normalized to the band pair at 1,364/1,401 cm−1, characteristic of the OEC (upper trace); and the PSIIL–PSIIC double-difference spectra (lower trace). D, Relaxation kinetics of the FTIR signal at 1,401 cm−1, characteristic of the Mn cluster, and E, at 1,478 cm−1, characteristic of the , after 1 and 20 flashes. The repetition rate of the train of laser flashes was 10 s−1.
The double-difference spectra of the two transients revealed evident changes in the amide I region (Figure�3C). These data provide direct experimental evidence for the involvement of conformational changes associated with the F1–Fm fluorescence increment in the presence of DCMU. Earlier studies using time-resolved femtosecond serial X-ray crystallography have uncovered light-induced conformational changes at the QB–nonheme-iron region and the OEC (Kern et�al., 2013; Kupitz et�al., 2014; Suga et�al., 2017). These reactions are blocked in the presence of DCMU, and thus our data show that the structural dynamics of PSII is not confined to the OEC and the QB pocket.
Our measurements also revealed that at physiological temperature, 303 K, the relaxation of the FTIR signal after 20 STSFs was considerably slower than after one flash (cf. Figure�3, A and B), suggesting that the charge-separated state in PSIIL is more stable than in PSIIC. Indeed, the decay of both of the 1,401 cm−1 signal originating from the S2 state of the OEC (Onoda et�al., 2000), and the positive band 1,478 cm−1, characteristic of were slowed down by about a factor of three in PSIIL compared to PSIIC (cf. Figure�3D). This conclusion is very similar to that obtained under comparable conditions on the stability of charge-separated state in purple bacterial reaction centers (bRCs; Malferrari et�al., 2013). It should be emphasized that the fact that the OEC signal at 1,401 cm−1 decays with the same kinetics of the signal at 1,478 cm−1 shows that in both cases (one flash and 20 STSFs) the predominant relaxation process is the S2 → S1QA charge recombination.
Distribution of the excitation energy in PSIIO, PSIIC, and PSIIL: 80 K fluorescence emission spectroscopy
Low-temperature (80 K) fluorescence emission spectroscopy experiments were performed to characterize the distribution of excitation energy in PSII CC in the open, closed, and light-adapted charge-separated states. At and near liquid N2 temperatures PSII CC has been shown to exhibit two main emission bands, at around 685 and 695 nm, originating from red-shifted Chl-a molecules located in the inner antenna complexes CP43 and CP47, respectively (Andrizhiyevskaya et�al., 2005). The spectral shape is determined by a set of rate constants and reaction routes, exciton relaxations and exciton transfers between the reaction center (RC) and the two antenna complexes as well as the processes between domains of the protein complexes (Shibata et�al., 2013).
Figure�4A shows the spectral variations during the Chl-a fluorescence emission in PSII CC during the induction caused by a series of sub-saturating light pulses. As shown in Figure�4B, the emission spectra of PSIIO and PSIIL—corresponding to Fo (approximately) and Fm—display considerably different shapes, wherein the ratio F685/F695 significantly increased. Accordingly, the Fv/Fm spectrum exhibited a peak around 685 nm (Figure�4C). These changes cannot be assigned solely to the reduction of QA. The Fo–F1 transition hardly affected the spectral distribution (Figure�4D) and the fluorescence yield, which increased by only about 10% compared to Fm. As shown in Supplemental Figure 5A, STSFs at 80 K were capable of generating sizeable fluorescence increase but the increments were much smaller than at higher temperatures, and Fm was not reached even after 500 flashes. This can be explained by the increased rigidity of the protein matrix of PSII CC at low temperature. This notion is supported by the data in Supplemental Figure 5B, showing that while at 233 K 20 STSFs nearly saturated Fv, at 173 K the same train of flashes generated a fluorescence yield which was only about 60% of Fm.
Figure 4.

Spectral variations associated with Fv of DCMU-treated PSII CC T. vulcanus at 80 K—comparison of Fo, F1 and Fm. A, Fluorescence emission spectra measured in response to a train of 450 nm sub-saturating flashes from a pulsed LED recorded during Chl-a fluorescence induction at 80 K; the first spectrum (orange) represents the Fo and the last one represents the Fm condition. B, Area-normalized fluorescence spectra—corresponding to Fo and Fm states. C, The spectral distribution of the calculated Fv/Fm ratio. D, Area-normalized fluorescence emission spectra before and after pre-illumination with a STSF, corresponding to Fo and F1, respectively. Shaded areas represent the SD of the measurements.
The above data clearly indicate that the most significant changes in the excitation energy distribution (irrespectively of the underlying processes which determine the exact spectral profile of the fluorescence emission at 80 K) occur during the PSIIC–PSIIL transition, rather than upon the PSIIO–PSIIC step. Remarkably, PSIIL relaxes even at 80 K (Supplemental Figure 6), in accordance with our earlier observation on an intact leaf at 77 K (Magyar et�al., 2018). The results are fully consistent with our earlier findings that, at cryogenic temperatures, only a small fraction of Fv arises from the closure of PSII (see also Fv with pre-reduced QA). It is also important to point out that the spectral variations reflect reorganizations, which affect the rates of energy or electron transfer (Shibata et�al., 2013) in the PSIIL state. At cryogenic temperatures, equilibration with the low-energy Chls at CP47, responsible for the emission at 695 nm is incomplete. Under these partial equilibration conditions, the RC can trap excitations from CP43 more effectively than from CP47. Consequently, if excitation trapping by the RC is slower from CP47, the F685/F695 nm ratio is expected to increase. It is an interesting observation, in line with the involvement of conformational changes, that PSII CC appears to possess “memory” regarding its pre-illumination history at higher, but still cryogenic temperatures, where no relaxation of occurs. As shown in Supplemental Figure 7, the F685/F695 ratio of the Fm spectra at 80 K can be enhanced by exposing the sample to a pre-illumination period at 233 K. This effect is reminiscent of the so-called Kleinfeld effect in bRC (Kleinfeld et�al., 1984). It is equally interesting that the spectral distributions of the Fm states at 80 and 90 K resemble those in the Fo states at 90 and 100 K, respectively (Supplemental Figure 8), suggesting a complex energetic landscape linked to energy migration barriers that are very sensitive to the bath temperature, pointing also toward a possible role for local heat effects due to thermal dissipation (Cseh et�al., 2000).
Changes in excitation kinetics upon PSIIC–PSIIL transition: time-resolved fluorescence spectroscopy
As shown above, the transition from PSIIC to PSIIL causes larger changes in the Chl-a fluorescence yield and emission spectrum than QA reduction does. To further examine the possible origin of the fluorescence changes, we measured the picosecond fluorescence decay kinetics of PSII CC in dark-adapted open state (Fo), dark-adapted closed state (F1), and light-adapted state (Fm). At room temperature, we obtained essentially identical kinetics in Fo and Fm conditions as previously published by different groups (Szczepaniak et�al., 2009; Caffarri et�al., 2011; van der Weij-de Wit et�al., 2011; Supplemental Figure 9). In Fo condition, the decay could be described by two main lifetimes of ∼40 and 200 ps and an average fluorescence lifetime of 110 ps, whereas in Fm conditions decay lifetimes from 100 ps to 4 ns were resolved and the average lifetime increased to ∼0.9 ns. To record the decay kinetics of PSIIC (F1), STSFs were applied shortly before the excitation pulses eliciting the fluorescence emission—in practice the pre-illumination was done just outside the sample chamber ∼4 s before the circulated sample solution reaches the measuring beam. To minimize the reoxidation of QA, the experiments were performed at 278 K in the presence of DCMU. Control Fo and Fm measurements were done under the same conditions but without the STSFs (Fo) or with additional preillumination (Fm). Despite the presence of a fraction of reopened RCs (resulting in a shorter average lifetime at Fm), the results showed that the sample was close to the ‘real’ Fo and Fm states (Figure�5) with an Fv/Fm ratio of 0.75 calculated from the average fluorescence lifetimes. While the fluorescence decay was much slower in Fm, the F1 kinetics appeared to be closer to Fo than Fm (Figure�5A), confirming that the F1–Fm fluorescence increment is associated with changes in the excited-state lifetime of Chl. The kinetics at Fo and F1 could be described with a similar set of exponential decay lifetimes but the relative contribution of slower decay components (600 ps and 1.7 ns) increased in F1, so that the average lifetime increased from 110–160 to 220–360 ps, depending on the exact excitation conditions. In the Fm (PSIIL) state, displaying an average lifetime of 570 ps, all decay lifetimes were longer than in F1 and the relative amplitudes of long-lived components grew further (Figure�5B).
Figure 5.

Fluorescence decay kinetics of DCMU-treated PSII CC of T. vulcanus under Fo, F1, and Fm conditions. A, Fluorescence decays recorded at 278 K at 685 nm, excited at 632 nm. The slower decay under Fm conditions corresponds to the higher fluorescence yield. When the RCs are closed by a STSF, the decay (F1) is only slightly slower compared to Fo. B, Distribution of fluorescence lifetime components by their relative amplitudes (the lifetimes, in ps, are indicated above the bars). Inset: average fluorescence lifetimes. C, Gibbs free energy differences (ΔG) between different compartments of kinetic models—with three radical pairs—found for PSII core complex in both charge-separated (F1, left) and light-adapted charge-separated (Fm, right) state (all rates are given in ns−1).
The data confirm that the changes in the fluorescence yield upon converting PSIIC to PSIIL are caused by altered Chl-a excitation decay kinetics and indicate the presence of an efficient de-excitation channel in PSIIC other than the reduction of QA. The most likely mechanism of de-excitation in F1 is nonradiative recombination of transiently generated radical pairs.
Currently there are several models for the excitation migration and trapping dynamics in PSII that fit the experimental data (van Amerongen and Croce, 2013). Irrespective of the specifics of the kinetic model, if one considers dynamic equilibration between antenna exciton states and charge-separated states, then the overall excitation decay, and hence the fluorescence yield, depends on the electron-transfer reactions in the RC—the conventional explanation for the variable fluorescence. To gain further insight into the process, we modeled the F1 and Fm fluorescence decays with a kinetic scheme comprising a sequence of four reversible steps (Figure�5). Global analysis of the spectrally resolved fluorescence kinetics showed that all lifetime components have virtually the same spectral shapes, indicating that the emission is from the same equilibrated pool of antenna pigments (Supplemental Figure 9). We further assume that the resolved decay lifetimes (50 ps and longer) are due to RC dynamics. Hence, only the first state (AntRC*), representing excited states, is emissive, whereas the following steps represent non-fluorescent charge-separated states (radical pairs RP1–3). In such case, all rate constants (forward and backward) can be independently determined by fitting the calculated time-dependent population of AntRC* to the fluorescence decay kinetics. The models fit the experimental data (Supplemental Figure 10) with residuals and χ2 statistics equal to a free four-exponential fit (which has the same number of free parameters). The errors of the rate constants were estimated from the diagonal elements of the covariance matrix of the fit parameters as and found to be within 3%–9%.
Antenna fluctuations could contribute to the kinetics on shorter timescales (Chmeliov et�al., 2014) and, as pointed out above, the conditions to some extent deviate from the “true” F1 and Fm; therefore, we emphasize on the trend rather than the exact parameter values. Further, there can be different interpretations regarding the nature of the radical pair states, including different oxidation-reduction states of the RC cofactors as well as protein structural dynamics on a picosecond–nanosecond timescale (Szczepaniak et�al., 2009; Chmeliov et�al., 2014). Regardless of the assignment, we observe that the forward reactions are slowed down in Fm compared to F1, shifting the equilibrium to the antenna exciton states and increasing the fluorescence lifetime and yield (Figure�5C). Thus, the time-resolved fluorescence data are in full agreement with the change in the steady-state fluorescence emission spectra upon F1–Fm transition. Correspondingly, the free energy gaps between all reaction intermediates are smaller in Fm than in F1. Since QA is reduced in both states, we must assume that the differences in the kinetics are caused by the local reconfiguration of the protein environment. For example, displacement and reorientation of polar residues can shift the midpoint potentials of the RC redox factors, altering the Gibbs free energy of forward electron transfer as well as charge recombination reactions. Such rearrangements will also affect exciton states with charge-transfer character or mixed exciton–charge-transfer states, key players in the primary photochemistry of PSII (Romero et�al., 2017), whose energy is strongly polarity-sensitive.
Discussion
Main attributes of the light-adapted charge- separated state of PSII
Our results point to the existence of a newly identified state, PSIIL, distinct from the dark-adapted closed-state PSIIC. The PSIIC–PSIIL transition is characterized by two quite peculiar features. First, in contrast to the PSIIO–PSIIC transition, which is generated by a single-photon absorption, PSIIL can be attained only in a stepwise manner, via multiple excitations of the sample following the closure of PSII (Figures�1, 2, and 4, and Supplemental Figures 4 and 5; see also Joliot and Joliot, 1979; Magyar et�al., 2018). It is important to stress that during PSIIC–PSIIL transition, no additional stable charge separation can occur and the transition is driven by a series of rapidly recombining P680+Pheo− radical pairs (Magyar et�al., 2018; Sipka et�al., 2019). The other unique feature of this transition is that a radical pair, when generated “too early”, remains ineffectual. In other terms, the generation of the P680+Pheo− radical pair is only a necessary but not sufficient condition for the F1–Fm (PSIIC–PSIIL) transition. The excitations promoting the transition must arrive with sufficiently long waiting times (Δτ) with respect to each other. This rate limitation has been shown to depend strongly on the temperature, with Δτ values four to six orders of magnitude larger than the lifetime of the primary radical pair (Magyar et�al., 2018; Sipka et�al., 2019).
Compared to PSIIC, PSIIL possesses several distinctive attributes. Beside the higher fluorescence of PSIIL (Fm) compared to F1 in PSIIC (Figures�1–2), which is paralleled by an increase in the fluorescence decay lifetimes (Figure�5), and therefore an increase in fluorescence yield, PSIIL and PSIIC display distinct 80 K emission spectra (Figure�4). The latter is an indication of a change in the energy landscape of trapping/detrapping of excitations in the core antenna. The imposed energetic bottlenecks are very sensitive to temperature, as shown by the changes in emission band-shape upon cooling at cryogenic temperature even in a narrow temperature range (e.g. 80–100 K). These observations point to reorganizations in the protein matrix in the vicinity of the RC. Further, as revealed by our rapid-scan FTIR spectroscopy data, at noncryogenic temperatures the protein conformational states are also different in the two charge-separated states (Figure�3). Although PSIIL can be induced at cryogenic temperatures, similar to PSIIC, only PSIIL retains its capability of relaxing—albeit very slowly at low temperatures (Figure�4). In general, PSIIC and PSIIL exhibit different temperature dependences (Magyar et�al., 2018).
Physical mechanism
With regard to the underlying physical mechanism, in accordance with our earlier work (Magyar et�al., 2018), we propose that dielectric relaxation processes play key roles in the PSIIC–PSIIL transitions. Dielectric relaxations in the protein matrix of PSII CC evidently occur in response to the stationary and transient electric fields around and the P680+Pheo− radical pair, respectively (Supplemental Figure 11). This mechanism, akin to solvent polarization, is very similar to that offered to explain the light-induced conformational changes in bRCs. Conformational changes in bRC, affecting the electron transfer kinetics, have been reported by different authors (Kleinfeld et�al., 1984; Nabedryk et�al., 1990; K�lm�n and Mar�ti, 1997; Abgaryan et�al., 1998; Nagy et�al., 2008). In the bRC, the electric field generated by light-induced charge separation has been proposed to perturb the molecular order, via dielectric relaxation (Kleinfeld et�al., 1984; Nabedryk et�al., 1990). Our rapid-scan FTIR data strongly suggest that the structural dynamics of PSII closely resembles that of the bRC (Mezzetti et�al., 2002; Mezzetti and Leibl, 2017), the ancestor of PSII (Cardona et�al., 2012). The structural dynamics can also be related to the photoinduced volume contraction via electrostriction, observed both in the bRC and PSII CC (Hou et�al., 2001).
Dielectric relaxation processes with a broad range of lifetimes, and different dominance at different temperature intervals have been shown to occur in hydrated proteins (Nakanishi and Sokolov, 2015). In the temperature range allowing motions in the protein moiety, the relaxation processes are manifested in changes that can be detected by time resolved FTIR measurements (Figure�3). At lower temperatures, other, slower relaxation processes may explain our observations. In general, electric fields have been shown to affect the functioning of enzymes (Fried and Boxer, 2017), and that of PSII in particular (Dau and Sauer, 1992; Vredenberg, 2011; Laisk and Oja, 2020), also at cryogenic temperatures (Knox et�al., 1984). It is to be noted here that the stationary and transient local electric fields in the PSII RC are at least an order of magnitude stronger than the uniform transmembrane fields in thylakoid membranes (Zim�nyi and Garab, 1989)—generated during the operation of the vectorial electron transport (Laisk and Oja, 2020) or exposed by ion gradients (Witt, 1979; Dau and Sauer, 1992). A nonconflicting hypothesis is that local-heat effects (Cseh et�al., 2000), due to dissipation accompanying the recombination of charges of the primary radical pair, facilitate the conformational transitions. Nevertheless, the explanation based on the physical mechanism of dielectric relaxation, with or without the local heat, must be complemented in order to satisfy the special conditions for the generation of the PSIIL state. In particular, there are rate limitations in Chl-a fluorescence, i.e. P680+Pheo− induces increments only if it is generated by a sufficiently long waiting time after the reduction of or after the consecutive steps. The occurrence of waiting times suggests that slow relaxation processes in the protein matrix play an important role. Because of the rigidity of the protein matrix, the relaxation appears to be incomplete (leading only to the Fo–F1 rise), and further excitations are required to gradually induce Fm. Such excitation can be achieved by generating the P680+Pheo− radical pairs. The sequence of putative events following the reduction of QA is schematically illustrated in Figure�6.
Figure 6.

Schematic illustration of the behavior of the dielectric matrix in stationary and transient electric fields—leading to the formation of the PSIIL state. A, Charge-separated closed state of dark-adapted PSII, generated by a STSF; B, perturbation of the stationary field by a transient charge separation; C, fully relaxed charge-separated state (PSIIL) formed after several transient charge separation events. The circles represent the primary redox cofactors in the RC and the schematic dipoles illustrate the readjustment of the dielectric matrix to the local electric field, represented by the field vectors (arrowed lines). For the corresponding potential profiles, see Supplemental Figure 11.
The ns transient electric field of the primary radical pair strongly perturbs the local electric field around : on the QA side of the radical pair, the negative potential will be transiently increased by the presence of Pheo−; also, on the donor side, the weak positive potential, arising from , will be transiently “replaced” by a very strong positive potential due to P680+ (for a schematic illustration, see Supplemental Figure 9). It is reasonable to assume that this nonequilibrium configuration of the dielectric matrix relaxes orders of magnitude slower than the transient field itself. One can also argue that this perturbation facilitates to complete the dielectric relaxation around . Elucidation of the molecular and physical mechanisms is beyond the scope of the present study. Nevertheless, in broad terms, the observed stabilization of the charge-separated state by multiple STSFs and the strong temperature dependence of the STSF-induced Chl-a fluorescence increments are in harmony with the mechanism outlined above.
Physiological role of the formation of PSIIL: Chl-a fluorescence transients
Photosystem II is the engine of life: by oxidizing water, it provides photosynthesis with an unlimited supply of reducing equivalents to reduce carbon dioxide to carbohydrates, which is the energetic basis of virtually all life on Earth (Barber, 2004). For these reasons, monitoring PSII activity is important in most plant physiology studies. Such investigations are routinely performed by measuring the Chl-a fluorescence induction, a noninvasive measuring technique that can be applied on leaves, algal, or cyanobacterial cells under a variety of physiologically important experimental and environmental conditions, as well as on isolated thylakoids and PSII particles. Chl-a fluorescence is one of the most widely employed techniques in plant biology, and the Fv/Fm parameter is certainly the most used Chl fluorescence parameter in the world. The main purpose of measuring this parameter is to determine the maximum quantum efficiency of PSII photochemistry, which is typically attained in dark-adapted samples when the plastoquinone pool is fully oxidized.
The key assumptions behind equating the Fv/Fm ratio with the maximum quantum efficiency of PSII photochemistry are that at Fo PSII centers are maximally quenched by photochemistry (open centers) and at Fm the photochemical quenching is absent (closed centers; Stirbet and Govindjee, 2012). The latter condition is often equaled to the QA being reduced. However, now irrevocable experimental evidence shows that the reduction of QA alone is not a sufficient requisite for attaining Fm. Clearly, Fm cannot be reached by a STSF, despite closing all PSII (Figure�2, see also Joliot and Joliot 1979; Magyar et�al., 2018; Sipka et�al., 2019; Laisk and Oja, 2020). Also, the corresponding Fv/Fm values of PSIIC, i.e. measured after the first STSF inducing stable charge separation in all active centers under physiologically relevant conditions, are typically not higher than 0.5 (Figure�2), and can be much lower at cryogenic temperatures (Figure�4). In contrast, the quantum efficiency of the stable charge separation in PSII is thought to be close to unity at all temperatures (Romero et�al., 2017). Further, closing the RC by chemically pre-reducing QA does not prevent the light-induced Chl-a fluorescence transitions (Figure�2). For these reasons, the physical mechanism of Fv must be laid on new grounds, which beside the reduction of QA consider the reconfiguration of PSII to a functional state in which stable photochemistry is actually prevented. Thus, our data strongly suggest that the Fv/Fm parameter is determined, in large part, by the structural dynamics of PSII. The strong correlation between the photochemical efficiency of PSII and the magnitude of the Fv/Fm parameter observed using MTSFs and continuous wave irradiation (Genty et�al., 1989; Papageorgiou and Govindjee, 2004) indicates that the structural dynamics are part of PSII functionality under physiological conditions. At the same time, estimation of both the maximal photochemical efficiency of PSII and, particularly, of the QA (or plastoquinone) redox level by STSFs would be biased. However, even this correlation cannot be taken for granted, as shown by reported cases in which efficient functioning of PSII is not accompanied by sizeable Fv (Vavilin et�al., 1999; Treves et�al., 2016). It remains to be investigated if the deficiency of Fv in these cases is due to the lack of structural dynamics upon the PSIIC–PSIIL transition, or, what seems more likely, due to a different organization of the pigment molecules or of the protein matrix. In general, testing the functional activity of PSII by using the Fv/Fm parameter requires a cautious approach and in special cases, e.g. under stress conditions or in mutants, independent investigations should be carried out to reveal the underlying physical mechanisms and the physiological status of PSII.
Another physiologically important attribute of oxygenic photosynthetic organisms is their capability of quenching the excess excitation energy, i.e. the fraction of absorbed light which cannot be utilized for photosynthesis, by a process called nonphotochemical quenching (NPQ) of the first singlet-excited state of Chl-a, aka NPQ of Chl-a fluorescence (Demmig-Adams et�al., 2014; Ruban, 2016). To determine the magnitude and kinetics of NPQ, the most commonly used Chl-a fluorescence measuring and imaging techniques are based on generating the maximum fluorescence levels—using MTSFs—before, during, and after the actinic illumination. This is performed with the aim to close PSII and to account for the contribution of the photochemical quenching due to stable charge separation. However, since both MTSFs and the actinic illumination evidently lead to the formation of PSIIL, rather than merely generating and retaining PSIIC, contributions from factors other than processes in the antenna cannot be a priori ruled out. In fact, it has recently been shown that the magnitude of NPQ depends strongly on the state (Fo versus Fm) of PSII (Farooq et�al., 2018).
Another parameter of physiological importance, which is frequently derived from the sigmoidal rise of fluorescence in the presence of DCMU, is the connectivity of PSII units (Joliot and Joliot, 1964; Stirbet, 2013). It is clear, however, that sigmoidal rises, observed on isolated monomeric and dimeric PSII CCs (Figure�1), originate from several successive photoreactions spaced by waiting times between consecutive excitations, as already pointed out by Magyar et�al. (2018 ; see also Schansker et�al., 2011; Vredenberg, 2011).
Last but not least, we would like to stress that Chl-a fluorescence transitions cannot be understood without taking into account the rate-limiting steps in the fluorescence induction, i.e. the waiting times (Δτ) between excitations that are required for the effectiveness of excitations to induce fluorescence increments after closing PSII (Magyar et�al., 2018). This is a somewhat odd but crucial novel experimental observation by Magyar et�al. (2018), which is nevertheless fully consistent with earlier observations (Joliot and Joliot, 1979; France et�al., 1992). It must also be emphasized here that Δτ does not mean gating of the primary charge separation events, i.e. the generation of the P680+Pheo− radical pair (Sipka et�al., 2019). In fact, the fluorescence increments during the PSIIC–PSIIL transition appear to be driven by the rapidly recombining local electric-field transients due to these radical pairs—but only if they are generated with a sufficiently long waiting time. Qualitatively, the Δτ-dependent fluorescence increments offer explanation on the peculiarity of the fast (O–J–I–P) Chl-a fluorescence induction kinetics of PSII in the absence of PSII inhibitor and under physiologically relevant conditions. The conundrum concerning these transitions had been that only the initial (O–J) step of these transitions, the so-called photochemical phase, could be ascribed to photochemistry (Delosme, 1967), and the nature of the remaining (J–I–P) phases, originally termed thermal phase, had remained elusive. Some data suggested an origin in conformational changes (cf. Schansker et�al. (2014) and references therein), while others linked it to the operation of the linear electron transport chain, and the reduction of the entire intersystem electron transport system (Stirbet and Govindjee, 2012). As already briefly outlined by Magyar et�al. (2018), by using the condition of waiting time, we can at least qualitatively explain why the so-called photochemical, O–J phase in the O–J–I–P transient does not reach the P level even in extremely high light (Schansker et�al., 2011). In this case, the rate-limited rise of the fluorescence emission competes with the re-oxidation of by QB (Shlyk-Kerner et�al., 2006), which reopens PSII, and thus leads to a dip (J–I phase). The rise is then resumed in the I–P phase, upon the re-reduction of QA, and the reduction of the entire electron transport chain—in good accordance with the explanation by Stirbet and Govindjee (2012) as well as the finding that the I–P phase depends on the activity of photosystem I (Schansker et�al., 2005; Ceppi et�al., 2012).
In conclusion, here we have shown that in addition to the two well-known stationary states of functionally active PSII, the open and closed states, PSIIO and PSIIC, respectively, this photosystem can also assume light-adapted charge-separated state, PSIIL. Formation of this latter state is shown to facilitate stabilization of the charge-separated state. The PSIIC–PSIIL transition is associated with a large part of Fv, the variable Chl-a fluorescence, and appears to reflect the structural dynamics of PSII. These data cannot be reconciled with the most widely accepted model, the QA model, according to which Fv originates solely form the PSIIO-to-PSIIC transition. Accordingly, the Fv/Fm parameter should not be equated with the quantum efficiency of PSII photochemistry while this parameter might still be used to monitor the functioning of PSII. Estimation of QA and the PQ redox pool by fluorescence-based methods might suffer from even larger interpretation biases and should therefore be adopted with caution to compare the activity of PSII on the same organism and under comparable conditions. Our data also show that the sigmoidal rise of the Chl-a fluorescence induction, in the presence of PSII inhibitor, should not be used to measure the connectivity of PSII units. By using the rate-limiting step in Fv identified earlier, and taking into account the “competing” event, the reopening of PSII upon the reoxidation of QA by QB, we offer a qualitative explanation of the fast (O–J–I–P) Chl-a fluorescence transient which, with its I–P transition in agreement with the generally accepted view within the frameworks of the QA model, depends on the functioning of the entire linear electron transport system. Regarding the physical mechanisms, our data strongly suggest that intense stationary and transient local electric fields and dielectric relaxation processes play key roles. Elucidation of the nature of the evidently subtle reorganizations and of the underlying physical mechanisms—especially during the waiting times—will most certainly provide further crucial information on the structural dynamics of PSII.
Materials and methods
Source material
Thermosynechococcus vulcanus, a thermophilic cyanobacterial strain isolated from a hot spring in Yunomine, Japan (Koike and Inoue, 1983), was grown as a batch culture. Spinach (S. oleracea) was purchased from a local growers’ market.
Growth conditions
Thermosynechococcus vulcanus cells were grown in BG11 medium (pH 7) at 323 K under continuous illumination with a white fluorescent lamp at intensity of 50–100 μmol photons m−2 s−1 photon flux density (Shen et�al., 2011). Cultures were aerated on a gyratory shaker operating at 120–150 r.p.m. to exponential growth phase.
Sample preparation
Thylakoid membranes were isolated from fresh spinach (S. oleracea) leaves essentially as described earlier (Chylla et�al., 1987), with minor modifications. Briefly, deveined leaves were homogenized in a resuspension medium containing 50-mM tricine (pH 7.5), 400-mM sorbitol, 5-mM KCl, 2-mM MgCl2, filtered through a nylon mesh and the supernatant was centrifuged for 7 min at 6,000g. The pellet was resuspended in a hypotonic medium containing 50-mM Tricine (pH 7.5), 5-mM KCl, and 5-mM MgCl2, followed by the immediate addition of the resuspension medium supplemented with 800 mM Sorbitol before centrifugation for 7 min at 6,000g. The pellet was finally resuspended in the resuspension medium and stored in liquid nitrogen at a concentration of 2–3 mg mL−1 Chl. Identical results were obtained with fresh preparations.
PSII core complexes of T. vulcanus were isolated as described earlier (Shen and Inoue, 1993; Shen and Kamiya, 2000; Kawakami and Shen, 2018). For all the experiments, the isolated PSII CCs were diluted in a reaction buffer medium containing 5% glycerol, 20-mM MES (pH 6.0), 20-mM NaCl, 3-mM CaCl2.
Fluorescence yield measurements
Relative fluorescence yields were measured using a PAM (Pulse Amplitude Modulation) 101 fluorometer (Walz, Effeltrich, Germany). The frequency of the modulated measuring light (low intensity, nonactinic) was 1.6 kHz. Variable fluorescence was induced by STSFs (Xe flashes, Excelitas LS-1130-3 Flashpac with FX-1163 Flashtube with reflector) of 1.5-μs duration at half-peak intensity. The sample was placed at the sample holder of a thermoluminescence apparatus in order to control the temperature. The timing of the flashes was controlled by using a home-designed programmable digital pulse generator. The decays of each measurement were recorded by using a National Instrument data acquisition device (DAQ 6001) via custom-designed LabVIEW software. Least-squares optimization was used to estimate the decay parameters. The optimization algorithms were implemented in Matlab (The MathWorks, Natick, MA, USA). For Chl-a fluorescence transient measurements the Chl concentration of the thylakoid membranes were diluted to ∼100 μg mL−1 in resuspension medium; the PSII CC to ∼25–50 μg mL−1 in reaction buffer. DCMU was dissolved in dimethyl sulfoxide (DMSO) and added to all samples immediately before the fluorescence measurements at a final concentration of 40 μM (the final concentration of DMSO did not exceed 1%). Before the measurements, the samples were dark adapted for 5 min at room temperature. In the case of dithionite, after 2 min of dark adaptation with the dithionite, DCMU was added, and the sample was dark adapted for 5 min before the measurement. In the case of FeCy, first DCMU was added to avoid the Ikegami-Katoh effect (Ikegami and Katoh, 1973), and after 5-min dark adaptation 2-mM FeCy was also added to the mixture.
FTIR measurements
For FTIR measurements, PSII CCs (corresponding to Chl concentrations between 2.5 and 4 mg mL−1, depending on the batch used) were prepared as thin paste squeezed between two CaF2 windows to yield an absorbance in the amide I region of the spectrum of 0.7–0.9 OD. The sample cell was sealed with silicone grease. To block the QA−to−QB electron transfer, DCMU was added to the PSII CC suspension. The sample was kept in a nitrogen cryostat (Oxford Instruments) to keep the temperature at 303 K stable during the measurements. The sample was stabilized in the dark for more than 2 h before the spectra were recorded.
Time-resolved rapid-scan FTIR difference spectra were recorded using a Bruker IFS88 FTIR spectrometer equipped with a photoconductive MCT-A (Mercury Cadmium Telluride - narrow band) detector at 4 cm−1 resolution and OPUS software, following the approach of Mezzetti and co-workers (Mezzetti et�al., 2002). Spectra were recorded in time windows of different durations (increasing with the time distance from the flash or from the 20 flashes sequence); this allowed recording the decay of the signal with a better signal-to-noise ratio (S/N). The photoreaction was triggered by one (or 20, fired at 10 Hz) saturating flash from a frequency-doubled Nd:YAG laser (Quantel) delivering 20-mJ pulses of 7-ns width. Synchronization between laser flashes and the recording of interferograms was checked on a digital oscilloscope. The temperature was set using a nitrogen flux Oxford Instrument cryostat.
The results from several cycles (obtained on at least three different samples) were averaged to improve the S/N ratio. At 303 K, an appropriate delay time between cycles (10 min and 20 min after 1 and 20 flashes, respectively) allowed a complete recovery of the RC neutral state after the light-induced charge separation.
Steady-state absorption spectroscopy
Absorption spectra, in the range of 350–750 nm, were recorded at room temperature with a Thermo Evolution 500 spectrophotometer. PSII CC samples in solution were diluted in reaction buffer to an absorbance of 1.0 at the red maximum. Measurements were performed in a semi-micro quartz cell of 1-cm optical path length.
Flash-induced absorption change measurements
The kinetic traces of absorption changes were detected by a home-constructed single-beam kinetic spectrophotometer according to (B�chel and Garab, 1995; Sipka et�al., 2018). The kinetics of absorption changes were induced by STSFs (Xe flashes of Excelitas LS-1130-3 Flashpac with FX-1163 Flashtube with reflector) of 1.5-μs duration at half-peak intensity. The actinic flashes were passed through a Schott RG630 filter. For the measuring light, a Schott KL 2500 LED lamp was used. A monochromator (Bausch & Lomb with a concave holographic grating) was used to disperse the measuring light. The monochromatic, transmitted measuring light was detected by a photomultiplier (EMI 9558 B, protected by a Corning 4-96 filter) which was connected to a differential amplifier and to a digital oscilloscope (Analog Discovery 2 100-MSPS USB Oscilloscope). For the synchronization of the flash lamps during the measurements, the shutter and the oscilloscope were controlled by a programmable digital pulse generator (BNC 577, Berkeley Nucleonics Corp) via custom-designed LabVIEW software. To increase the S/N ratio, 8–128 kinetic traces were averaged depending on the required S/N. Samples were placed in a semi-micro quartz cuvette (of 10-mm optical pathlength and 2-mm width). The STSF-induced energization of the thylakoid membranes was monitored by the electrochromic band shift of the carotenoids at 515 nm. The electrochromic signal at a given wavelength was calculated from the kinetic traces at 2 ms after the flash. On PSII CC, flash-induced absorption changes were monitored between 410 and 460 nm and 515 and 565 nm. All measurements were carried out at room temperature. Thylakoid membranes were suspended in the resuspension medium; PSII CCs were suspended in the reaction buffer; in all cases, the samples were anaerobically dark adapted for 5 min prior to measurement.
Low-temperature fluorescence spectroscopy
Steady-state fluorescence emission spectra were recorded in the 80–300 K range on a high-sensitivity fluorescence set-up, taking into account the spectral/temporal variation of fluorescence yield due to photochemical trapping and other processes on nonphotochemical origin of isolated PSII CC. The laboratory-assembled fluorimeter was equipped with a shutter-protected liquid nitrogen cooled charge-coupled device (CCD) camera (Princeton Applied Research, LN/CCD-ST138), coupled to a spectrometer (SpectraPro-300i, Princeton; Nematov et�al., 2017). An OG570 (Schott) bandpass filter was placed before the spectrograph to reduce scattered and stray excitation light. The fluorescence excitation source was a LED (LumiLeds LXML-PR02-1050), controlled by a laboratory-assembled driver/pulser, which allowed synchronization with the detection camera electronic control unit (Princeton Applied Research, ST138) that has its built-in triggering capability, as well as the synchronization of an additional pulsed light source (Xe-Flashlamp) for single-turnover flash (STF) excitation. The LED pulse-length, repetition rate, and intensity were also controlled by the laboratory-assembled pulser/driver. The LED pulse intensity was further attenuated by neutral density filters to approx. 0.14-�mol photons s−1 m−2. Spectra were acquiring by setting the LED flash within the opening time of CCD-protecting shutter (software controlled, Roper Instrument, WinSpec32), whereas STFs were fired during the shutter dark time, and proved by successive LED pulses, Spectra collected close to Fo condition (Rizzo et�al., 2014; Remelli and Santabarbara, 2018) was tested either with 1-, 2-, 5-, and 10-ms LED pulse duration; there was no difference in the spectra, but in the S/N. To obtain F1 or Fo (approx.), the spectra were measured after the first LED pulse excitation with or without pre-STSF illumination, respectively; for STSF excitations an Excelitas LS-1130-3 Flashpac with FX-1163 Flashtube with reflector of 1.5-μs duration at half-peak intensity was used. For F1 and Fo, only the first spectra of each measurement were averaged (n = 8). Fm was reached with multiple Xe-flash illuminations and/or continuous (∼100 s) LED excitation; on the plateau, about hundred spectra were averaged to obtain the Fm spectra. Spectra were acquired at a resolution of 0.25 nm per pixel and corrected for the wavelength sensitivity of the detector. Samples were diluted to a Chl-a concentration of 5 μg mL−1; DCMU was added at 40 μM final concentration. For low temperature measurements the samples, held in 1-cm pathlength plastic cuvette, were suspended, immediately before cooling, in a buffer containing 60% (w/v) glycerol as a cryoprotectant to obtain transparent matrixes. The plastic cuvette was placed in a flow cryostat (Oxford mod. Optistat CF) equipped with temperature control unit (ICT-503, Oxford Instruments).
Time-resolved fluorescence
Fluorescence decays were measured at 278 and 293 K by time-correlated single-photon counting using an instrument described earlier (Akhtar et�al., 2020). Excitation pulses centered at 632 nm at 20 MHz repetition rate and ∼0.1-pJ energy, were obtained from a Fianium WhiteLase Micro (NKT Photonics, UK) supercontinuum laser. The PSII CC suspension was diluted to an absorbance of 0.03 at the excitation wavelength in reaction buffer supplemented with 20-μM DCMU in a 3-mm pathlength flow cell. The sample was continuously circulated during the measurement to avoid repeated excitation of the same sample volume. Fluorescence decays were recorded at 685 nm and binned in 4 ps time channels. The total instrument response function (IRF) width was ∼50 ps measured using 1% Ludox (colloidal silica) as scattering medium. The fluorescence lifetimes were determined by multiexponential fitting of the fluorescence decay kinetics combined with iterative re-convolution with the IRF using MATLAB routines created in-house.
To keep the RC open in the presence of DCMU, an open flow system was used, wherein the sample passed only once through the excitation beam at a flow rate of 200 μL s−1. Under these conditions, approximate Fo level was achieved. Baseline measurements were performed without DCMU and adding 25-�M dichlophenolindophenol and 0.5-mM FeCy to make sure that RCs are fully open. For the approximate F1 level, STSFs were applied (Excelitas LS-1130-3 Flashpac with FX-1163 Flashtube with reflector) at 2-Hz repetition rate corresponding to a single flash per sample passage. For measurements of light-adapted closed-state PSII (Fm level), the sample was pre-illuminated with a background light from a Schott KL 2500 LED lamp.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure 1. Kinetic traces (A) and parameters (B) of STSF-induced Chl-a fluorescence yield transients of DCMU-treated dimeric and monomeric PSII CCs of T. vulcanus at 278 K.
Supplemental Figure 2. Amplitudes of the light-induced C550 absorbance transients of DCMU-treated PSII CC of T. vulcanus in the absence and presence of 2-mM dithionite.
Supplemental Figure 3. STSF-induced transient absorption spectra of DCMU-treated PSII CC of T. vulcanus in the presence and absence of 2-mM ferricyanide (FeCy) 2 ms after the excitation.
Supplemental Figure 4. Effects of dithionite and ferricyanide on the STSF-induced Chl-a fluorescence induction of DCMU-treated isolated spinach thylakoid membranes at 213 K.
Supplemental Figure 5. Chl-a fluorescence induction kinetics of DCMU-treated PSII CCs of T. vulcanus at 80 K (A) and at 173 and 233 K (B).
Supplemental Figure 6. Relaxation of Fm of F695 and F685 of DCMU-treated PSII CC of T. vulcanus at 80 K.
Supplemental Figure 7. Variations of the 80 K Fm fluorescence emission spectra of DCMU-treated PSII CC of T. vulcanus exposed to different preillumination protocols at 233 K.
Supplemental Figure 8. Comparison of Fo and Fm fluorescence emission spectra, normalized at 695 nm, of DCMU-treated PSII CC of T. vulcanus at different temperatures, as indicated.
Supplemental Figure 9. Decay-associated spectra of PSII CC of T. vulcanus obtained from four-exponential global analysis of fluorescence decays measured by TCSPC at 293 K.
Supplemental Figure 10. Kinetic model fitting of the fluorescence decays of PSII CC recorded by TCSPC at 278 K and emission wavelength 680 nm in F1 and Fm conditions.
Supplemental Figure 11. Overall structure of PSII core complex from T. vulcanus (PDB: 5GTH) and schematic representation of the stationary (A) and transient (B) electric field, and the superposition (C) of the two fields due to the presence of and the P680+Pheo− radical pair, respectively—characterized by equipotential profiles.
Supplemental Table 1. STSF-induced absorption changes of PSII CC of T. vulcanus and spinach thylakoid membranes in the presence of 40-μM DCMU and 2-mM FeCy with and without waiting times (Δτ) between flashes.
Supplemental Table 2. Effects of dithionite on the variations of the STSF-induced Chl-a fluorescence yields of DCMU-treated PSII CC of T. vulcanus at 278 K.
Supplementary Material
Acknowledgments
The authors are indebted to Profs. Govindjee, P. Joliot, A.W. Rutherford, T. Noguchi, and A.R. Holzwarth for helpful discussions. The authors also benefited from numerous stimulating discussions with Profs. A. D�r and L. Zim�nyi. We thank to Dr. W. Leibl for his help in configuring the FTIR experiments and for critical reading of the manuscript and to Dr. L. Kov�cs for recording the thermoluminescence glow curves on PSII CC.
Funding
The authors acknowledge the support from the Hungarian Ministry of Innovation and Technology, National Research, Development and Innovation Fund (OTKA grants KH-124985 and K-128679 to G.G.; PD-121225 to M.M.; and NN-124904 to P.H.L., who also used support from the grant 2018-1.2.1-NKP-2018-00009.) G.G. also acknowledges the support from T�T 2018-2.1.14-T�T-CN-2018-00004 from the Hungarian Ministry of Innovation and Technology, the Czech Science Foundation (GA ČR 19-13637S), and the E�tv�s Lor�nd Research Network (ELKH K�-37/2020). M.M. and G.S. acknowledge the support from COST Actions CM1306 STSM Grant (ref. No.: 40047) and CA15126 STSM Grant (ref. No.: 41468), respectively. S.S. obtained support from the Grant Fondazione Cariplo (CYAO Project) Grant Number 2016–0667. J.-R.S., G.H., Q.Z., and Y.X. acknowledge the support from a National Key R&D Program of China (2017YFA0503700), a Strategic Priority Research Program of the Chinese Academy of Sciences (XDB17000000) and a National Natural Science Foundation of China (31470339). P.A. used funds from a grant to ELI-ALPS project (GINOP-2.3.6-15-2015-00001), which is supported by the European Union and co-financed by the European Regional Development Fund. G.G. would like to dedicate this paper to the memory of his friend and colleague, Jacques Breton, whose hospitality in the lab and in his home are remembered with sentiments.
Conflict of interest statement. None declared.
G.G. conceived the study together with G.S., M.M, J.-R.S., and P.H.L. The Chl-a fluorescence and the flash-induced absorption kinetic devices were modified/constructed by G.S., and the measurements were performed and analyzed by G.S. and M.M. The low-temperature fluorescence emission spectroscopy measurements were carried out and analyzed by G.S. and S.S. The time-resolved fluorescence spectroscopy experiments were configured by P.H.L., G.S., and P.A. who also carried out the measurements and performed the model calculations; the electric field calculations were carried out by G.S. The rapid-scan FTIR spectroscopy measurements were performed and analyzed by M.M. and A.M. Whole cells and isolated PSII CC of T. vulcanus were provided by J.-R.S., Q.Z., Y.X., and G.H. The paper was written by G.G., G.S., M.M., P.H.L., P.A., S.S., A.M., and J.-R.S.
The authors responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) are: Győző Garab (garab.gyozo@brc.hu) and Petar H. Lambrev (lambrev.petar@brc.hu).
References
- Abgaryan GA, Christophorov LN, Goushcha AO, Holzwarth AR, Kharkyanen VN, Knox PP, Lukashev EA (1998) Effects of mutual influence of photoinduced electron transitions and slow structural rearrangements in bacterial photosynthetic reaction centers. J Biol Phys 24: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar P, Nowakowski PJ, Wang W, Do TN, Zhao S, Siligardi G, Garab G, Shen JR, Tan HS, Lambrev PH (2020) Spectral tuning of light-harvesting complex II in the siphonous alga Bryopsis corticulans and its effect on energy transfer dynamics. Biochim Biophys Acta Bioenerg 1861: 148191. [DOI] [PubMed] [Google Scholar]
- Andrizhiyevskaya EG, Chojnicka A, Bautista JA, Diner BA, van Grondelle R, Dekker JP (2005) Origin of the F685 and F695 fluorescence in photosystem II. Photosynth Res 84: 173–180 [DOI] [PubMed] [Google Scholar]
- �rsk�ld SP, Masters VM, Prince BJ, Smith PJ, Pace RJ, Krausz E (2003) Optical spectra of synechocystis and spinach photosystem II preparations at 1.7 K: identification of the D1-pheophytin energies and stark shifts. J Am Chem Soc 125: 13063–13074 [DOI] [PubMed] [Google Scholar]
- Barber J (2004) Engine of life and big bang of evolution: a personal perspective. Photosynth Res 80: 137. [DOI] [PubMed] [Google Scholar]
- Barber J, Melis A (1990) Quantum efficiency for the photoaccumulation of reduced pheophytin in Photosystem II. Biochim Biophys Acta 1020: 285–289 [Google Scholar]
- Berthomieu C, Nabedryk E, Mantele W, Breton J (1990) Characterization by FTIR spectroscopy of the photoreduction of the primary quinone acceptor QA in photosystem II. FEBS Lett 269: 363–367 [DOI] [PubMed] [Google Scholar]
- Butler WL (1978) Energy distribution in the photochemical apparatus of photosynthesis. Annu Rev Plant Physiol 29: 345–378 [Google Scholar]
- Butler WL, Okayama S (1971) The photoreduction of C550 in chloroplasts and its inhibition by lipase. Biochim Biophys Acta 245: 237–239 [DOI] [PubMed] [Google Scholar]
- B�chel C, Garab G (1995) Electrochromic absorbency changes in the chlorophyll-c-containing alga Pleurochloris-Meiringensis (Xanthophyceae). Photosynth Res 43: 49–56 [DOI] [PubMed] [Google Scholar]
- Caffarri S, Broess K, Croce R, van Amerongen H (2011) Excitation energy transfer and trapping in higher plant Photosystem II complexes with different antenna sizes. Biophys J 100: 2094–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardona T, Sedoud A, Cox N, Rutherford AW (2012) Charge separation in photosystem II: a comparative and evolutionary overview. Biochim Biophys Acta Bioenerg 1817: 26–43 [DOI] [PubMed] [Google Scholar]
- Ceppi MG, Oukarroum A, Cicek N, Strasser RJ, Schansker G (2012) The IP amplitude of the fluorescence rise OJIP is sensitive to changes in the photosystem I content of leaves: a study on plants exposed to magnesium and sulfate deficiencies, drought stress and salt stress. Physiol Plant 144: 277–288 [DOI] [PubMed] [Google Scholar]
- Chmeliov J, Trinkunas G, van Amerongen H, Valkunas L (2014) Light harvesting in a fluctuating antenna. J Am Chem Soc 136: 8963–8972 [DOI] [PubMed] [Google Scholar]
- Chylla RA, Garab G, Whitmarsh J (1987) Evidence for slow turnover in a fraction of photosystem II complexes in thylakoid membranes. Biochim Biophys Acta 894: 562–571 [Google Scholar]
- Connolly JS, Samuel EB, Janzen AF (1982) Effects of solvent on the fluorescence properties of bacteriochlorophyll a. Photochem Photobiol 36: 565–574 [Google Scholar]
- Cseh Z, Rajagopal S, Tsonev T, Busheva M, Papp E, Garab G (2000) Thermooptic effect in chloroplast thylakoid membranes. Thermal and light stability of pigment arrays with different levels of structural complexity. Biochemistry 39: 15250–15257 [DOI] [PubMed] [Google Scholar]
- Dau H, Sauer K (1992) Electric-field effect on the picosecond fluorescence of Photosystem-II and its relation to the energetics and kinetics of primary charge separation. Biochim Biophys Acta 1102: 91–106 [Google Scholar]
- Delosme R (1967) Study of the induction of fluorescence in green algae and chloroplasts at the onset of an intense illumination. Biochim Biophys Acta 143: 108–128 [DOI] [PubMed] [Google Scholar]
- Demmig-Adams B, Stewart JJ, Burch TA, Adams WW 3rd (2014) Insights from placing photosynthetic light harvesting into context. J Phys Chem Lett 5: 2880–2889 [DOI] [PubMed] [Google Scholar]
- Duysens LMN, Sweers HE (1963) Studies on Microalgae and Photosynthetic Bacteria. Japanese Society of Plant Physiologists, University of Tokyo Press, Tokyo [Google Scholar]
- Duysens LN (1978) Transfer and trapping of excitation energy in photosystem II. Ciba Found Symp 61: 323–340 [DOI] [PubMed] [Google Scholar]
- Farooq S, Chmeliov J, Wientjes E, Koehorst R, Bader A, Valkunas L, Trinkunas G, van Amerongen H (2018) Dynamic feedback of the photosystem II reaction centre on photoprotection in plants. Nat Plants 4: 225–231 [DOI] [PubMed] [Google Scholar]
- Feyziyev Y, De�k Z, Styring S, Bern�t G (2013) Electron transfer from Cyt b559 and tyrosine-D to the S2 and S3 states of the water oxidizing complex in photosystem II at cryogenic temperatures. J Bioenerg Biomembr 45: 111–120 [DOI] [PubMed] [Google Scholar]
- France LL, Geacintov NE, Breton J, Valkunas L (1992) The dependence of the degrees of sigmoidicities of fluorescence induction curves in spinach-chloroplasts on the duration of actinic pulses in pump-probe experiments. Biochim Biophys Acta 1101: 105–119 [Google Scholar]
- Fried SD, Boxer SG (2017) Electric fields and enzyme catalysis. Annu Rev Biochem 86: 387–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genty B, Briantais JM, Baker NR (1989) The relationship between the quantum yield of photosynthetic electron-transport and quenching of chlorophyll fluorescence. Biochim Biophys Acta 990: 87–92 [Google Scholar]
- Hodges M, Moya I (1986) Time-resolved chlorophyll fluorescence studies of photosynthetic membranes - resolution and characterization of 4 kinetic components. Biochim Biophys Acta 849: 193–202 [Google Scholar]
- Holzwarth AR, Wendler J, Haehnel W (1985) Time-resolved picosecond fluorescence-spectra of the antenna chlorophylls in chlorella-vulgaris - resolution of Photosystem-I fluorescence. Biochim Biophys Acta 807: 155–167 [Google Scholar]
- Hou JM, Boichenko VA, Diner BA, Mauzerall D (2001) Thermodynamics of electron transfer in oxygenic photosynthetic reaction centers: volume change, enthalpy, and entropy of electron-transfer reactions in manganese-depleted photosystem II core complexes. Biochemistry 40: 7117–7125 [DOI] [PubMed] [Google Scholar]
- Ikegami I, Katoh S (1973) Studies on chlorophyll fluorescence in chloroplasts II. Effect of ferricyanide on the induction of fluorescence in the presence of 3-(3,4-dichlorophenyl)-1,1-dimethylure. Plant Cell Physiol 14: 829–836 [Google Scholar]
- Joliot A, Joliot P (1964) �tude cin�tique de la r�action photochimique lib�rant l’oxyg�ne au cours de la photosynth�se. CR Acad Sci Paris 258: 4622–4625 [PubMed] [Google Scholar]
- Joliot P, Joliot A (1979) Comparative study of the fluorescence yield and of the C550 absorption change at room temperature. Biochim Biophys Acta 546: 93–105 [DOI] [PubMed] [Google Scholar]
- K�lm�n L, Mar�ti P (1997) Conformation-activated protonation in reaction centers of the photosynthetic bacterium Rhodobacter sphaeroides. Biochemistry 36: 15269–15276 [DOI] [PubMed] [Google Scholar]
- Kawakami K, Shen JR (2018) Purification of fully active and crystallizable photosystem II from thermophilic cyanobacteria. Methods Enzymol 613: 1–16 [DOI] [PubMed] [Google Scholar]
- Kern J, Alonso-Mori R, Tran R, Hattne J, Gildea RJ, Echols N, Glockner C, Hellmich J, Laksmono H, Sierra RG, et al. (2013) Simultaneous femtosecond X-ray spectroscopy and diffraction of Photosystem II at room temperature. Science 340: 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinfeld D, Okamura MY, Feher G (1984) Electron-transfer kinetics in photosynthetic reaction centers cooled to cryogenic temperatures in the charge-separated state: evidence for light-induced structural changes. Biochemistry 23: 5780–5786 [DOI] [PubMed] [Google Scholar]
- Klimov VV, Klevanik AV, Shuvalov VA, Kransnovsky AA (1977) Reduction of pheophytin in the primary light reaction of photosystem II. FEBS Lett 82: 183–186 [DOI] [PubMed] [Google Scholar]
- Knox PP, Venediktov PS, Kononenko AA, Garab GI, Faludidaniel A (1984) Role of electric polarization in the thermo-luminescence of chloroplasts. Photochem Photobiol 40: 119–125 [Google Scholar]
- Koike H, Inoue Y (1983) Preparation of oxygen-evolving photosystem II particles from a thermophilic blue-green alga. InInoue Y, Crofts AR, Govindjee, Murata N, Renger G, Satoh K, eds, The Oxygen Evolving System of Photosynthesis. San Diego, CA: Academic Press, pp 257–263 [Google Scholar]
- Kupitz C, Basu S, Grotjohann I, Fromme R, Zatsepin NA, Rendek KN, Hunter MS, Shoeman RL, White TA, Wang D, et al. (2014) Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 513: 261–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laisk A, Oja V (2020) Variable fluorescence of closed photochemical reaction centers. Photosynth Res 143: 335–346 [DOI] [PubMed] [Google Scholar]
- Lavergne J, Trissl HW (1995) Theory of fluorescence induction in Photosystem-II - derivation of analytical expressions in a model including exciton-radical-pair equilibrium and restricted energy-transfer between photosynthetic units. Biophys J 68: 2474–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magyar M, Sipka G, Kov�cs L, Ughy B, Zhu Q, Han G, Špunda V, Lambrev PH, Shen JR, Garab G (2018) Rate-limiting steps in the dark-to-light transition of Photosystem II - revealed by chlorophyll-a fluorescence induction. Sci Rep 8: 2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malferrari M, Mezzetti A, Francia F, Venturoli G (2013) Effects of dehydration on light-induced conformational changes in bacterial photosynthetic reaction centers probed by optical and differential FTIR spectroscopy. Biochim Biophys Acta Bioenerg 1827: 328–339 [DOI] [PubMed] [Google Scholar]
- Mezzetti A, Leibl W (2017) Time-resolved infrared spectroscopy in the study of photosynthetic systems. Photosynth Res 131: 121–144 [DOI] [PubMed] [Google Scholar]
- Mezzetti A, Nabedryk E, Breton J, Okamura MY, Paddock ML, Giacometti G, Leibl W (2002) Rapid-scan Fourier transform infrared spectroscopy shows coupling of GLu-L212 protonation and electron transfer to QB in Rhodobacter sphaeroides reaction centers. Biochim Biophys Acta 1553: 320–330 [DOI] [PubMed] [Google Scholar]
- Nabedryk E, Bagley KA, Thibodeau DL, Bauscher M, M�ntele W, Breton J (1990) A protein conformational change associated with the photoreduction of the primary and secondary quinones in the bacterial reaction center. FEBS Lett 266: 59–62 [DOI] [PubMed] [Google Scholar]
- Nagy L, Mar�ti P, Terazima M (2008) Spectrally silent light induced conformation change in photosynthetic reaction centers. FEBS Lett 582: 3657–3662 [DOI] [PubMed] [Google Scholar]
- Nakanishi M, Sokolov AP (2015) Dielectric spectroscopy of hydrated biomacromolecules. In V Raicu, Y Feldman, eds, Dielectric Relaxation in Biological Systems. Oxford University Press, Oxford [Google Scholar]
- Nelson N, Yocum CF (2006) Structure and function of photosystems I and II. Annu Rev Plant Biol 57: 521–565 [DOI] [PubMed] [Google Scholar]
- Nematov S, Casazza AP, Remelli W, Khuvondikov V, Santabarbara S (2017) Spectral dependence of irreversible light-induced fluorescence quenching: chlorophyll forms with maximal emission at 700-702 and 705-710nm as spectroscopic markers of conformational changes in the core complex. Biochim Biophys Acta Bioenerg 1858: 529–543 [DOI] [PubMed] [Google Scholar]
- Noguchi T (2007) Light-induced FTIR difference spectroscopy as a powerful tool toward understanding the molecular mechanism of photosynthetic oxygen evolution. Photosynth Res 91: 59–69 [DOI] [PubMed] [Google Scholar]
- Onoda K, Mino H, Inoue Y, Noguchi T (2000) An FTIR study on the structure of the oxygen-evolving Mn-cluster of Photosystem II in different spin forms of the S2 state. Photosynth Res 63: 47–57 [DOI] [PubMed] [Google Scholar]
- Papageorgiou GC, Govindjee (2004) Chlorophyll a Fluorescence: A Signature of Photosynthesis. Springer, Dordrecht, The Netherlands [Google Scholar]
- Remelli W, Santabarbara S (2018) Excitation and emission wavelength dependence of fluorescence spectra in whole cells of the cyanobacterium Synechocystis sp. PPC6803: influence on the estimation of Photosystem II maximal quantum efficiency. Biochim Biophys Acta Bioenerg 1859: 1207–1222 [DOI] [PubMed] [Google Scholar]
- Rizzo F, Zucchelli G, Jennings R, Santabarbara S (2014) Wavelength dependence of the fluorescence emission under conditions of open and closed Photosystem II reaction centres in the green alga Chlorella sorokiniana. Biochim Biophys Acta Bioenerg 1837: 726–733 [DOI] [PubMed] [Google Scholar]
- Roelofs TA, Lee CH, Holzwarth AR (1992) Global target analysis of picosecond chlorophyll fluorescence kinetics from pea-chloroplasts - a new approach to the characterization of the primary processes in Photosystem-II alpha-units and beta-units. Biophys J 61: 1147–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero E, Novoderezhkin VI, van Grondelle R (2017) Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543: 355–365 [DOI] [PubMed] [Google Scholar]
- Ruban AV (2016) Nonphotochemical chlorophyll fluorescence quenching: mechanism and effectiveness in protecting plants from photodamage. Plant Physiol 170: 1903–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schansker G, Toth SZ, Strasser RJ (2005) Methylviologen and dibromothymoquinone treatments of pea leaves reveal the role of photosystem I in the Chl a fluorescence rise OJIP. Biochim Biophys Acta 1706: 250–261 [DOI] [PubMed] [Google Scholar]
- Schansker G, T�th SZ, Holzwarth AR, Garab G (2014) Chlorophyll a fluorescence: beyond the limits of the QA model. Photosynth Res 120: 43–58 [DOI] [PubMed] [Google Scholar]
- Schansker G, T�th SZ, Kov�cs L, Holzwarth AR, Garab G (2011) Evidence for a fluorescence yield change driven by a light-induced conformational change within photosystem II during the fast chlorophyll a fluorescence rise. Biochim Biophys Acta Bioenerg 1807: 1032–1043 [DOI] [PubMed] [Google Scholar]
- Shen J-R (1998) Possible functional differences between dimer and monomer of Photosystem II complex. InGarab G, ed, Photosynthesis: Mechanisms and Effects: Volume I–V: Proceedings of the XIth International Congress on Photosynthesis, Budapest, Hungary, 17–22 August 1998, pp. 941–944 [Google Scholar]
- Shen JR, Inoue Y (1993) Binding and functional-properties of two new extrinsic components, cytochrome c-550 and a 12-kDa Protein, in cyanobacterial photosystem II. Biochemistry 32: 1825–1832 [DOI] [PubMed] [Google Scholar]
- Shen JR, Kamiya N (2000) Crystallization and the crystal properties of the oxygen-evolving photosystem II from Synechococcus vulcanus. Biochemistry 39: 14739–14744 [DOI] [PubMed] [Google Scholar]
- Shen JR, Kawakami K, Koike H (2011) Purification and crystallization of oxygen-evolving photosystem II core complex from thermophilic cyanobacteria. Methods Mol Biol 684: 41–51 [DOI] [PubMed] [Google Scholar]
- Shibata Y, Nishi S, Kawakami K, Shen JR, Renger T (2013) Photosystem II does not possess a simple excitation energy funnel: time-resolved fluorescence spectroscopy meets theory. J Am Chem Soc 135: 6903–6914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlyk-Kerner O, Samish I, Kaftan D, Holland N, Sai PSM, Kless H, Scherz A (2006) Protein flexibility acclimatizes photosynthetic energy conversion to the ambient temperature. Nature 442: 827–830 [DOI] [PubMed] [Google Scholar]
- Sipka G, Kis M, Mar�ti P (2018) Characterization of mercury(II)-induced inhibition of photochemistry in the reaction center of photosynthetic bacteria. Photosynth Res 136: 379–392 [DOI] [PubMed] [Google Scholar]
- Sipka G, M�ller P, Brettel K, Magyar M, Kov�cs L, Zhu QJ, Xiao YA, Han GY, Lambrev PH, Shen JR, et al. (2019) Redox transients of P680 associated with the incremental chlorophyll-a fluorescence yield rises elicited by a series of saturating flashes in diuron-treated photosystem II core complex of Thermosynechococcus vulcanus. Physiol Plant 166: 22–32 [DOI] [PubMed] [Google Scholar]
- Stirbet A (2013) Excitonic connectivity between photosystem II units: what is it, and how to measure it? Photosynth Res 116: 189–214 [DOI] [PubMed] [Google Scholar]
- Stirbet A, Govindjee (2012) Chlorophyll a fluorescence induction: a personal perspective of the thermal phase, the J-I-P rise. Photosynth Res 113: 15–61 [DOI] [PubMed] [Google Scholar]
- Strasser RJ, Tsimilli-Michael M, Srivastava A (2004) Analysis of the chlorophyll a fluorescence transient. InPapageorgiou GC, Govindjee, eds, Chlorophyll a Fluorescence: A Signature of Photosynthesis. Springer, Dordrecht, The Netherlands, pp 463–495 [Google Scholar]
- Suga M, Akita F, Sugahara M, Kubo M, Nakajima Y, Nakane T, Yamashita K, Umena Y, Nakabayashi M, Yamane T, et al. (2017) Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 543: 131–135 [DOI] [PubMed] [Google Scholar]
- Szczepaniak M, Sander J, Nowaczyk M, M�ller MG, R�gner M, Holzwarth AR (2009) Charge separation, stabilization, and protein relaxation in photosystem II core particles with closed reaction center. Biophys J 96: 621–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracewell CA, Brudvig GW (2008) Multiple redox-active chlorophylls in the secondary electron-transfer pathways of oxygen-evolving Photosystem II. Biochemistry 47: 11559–11572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treves H, Raanan H, Kedem I, Murik O, Keren N, Zer H, Berkowicz SM, Giordano M, Norici A, Shotland Y, et al. (2016) The mechanisms whereby the green alga Chlorella ohadii, isolated from desert soil crust, exhibits unparalleled photodamage resistance. New Phytol 210: 1229–1243 [DOI] [PubMed] [Google Scholar]
- Tyystjarvi E, Vass I (2004) Light emission as a probe of charge separation and recombination in the photosynthetic apparatus: relation of prompt fluorescence to delayed light emission and thermoluminescence. InPapageorgiou GC, Govindjee, eds, Chlorophyll a Fluorescence. Advances in Photosynthesis and Respiration. Springer, Dordrecht, The Netherlands, pp 363–388 [Google Scholar]
- van Amerongen H, Croce R (2013) Light harvesting in Photosystem II. Photosynth Res 116: 251–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Weij-de Wit CD, Dekker JP, van Grondelle R, van Stokkum IHM (2011) Charge separation is virtually irreversible in Photosystem II core complexes with oxidized primary quinone acceptor. J Phys Chem A 115: 3947–3956 [DOI] [PubMed] [Google Scholar]
- Vavilin DV, Ermakova-Gerdes SY, Keilty AT, Vermaas WF (1999) Tryptophan at position 181 of the D2 protein of photosystem II confers quenching of variable fluorescence of chlorophyll: implications for the mechanism of energy-dependent quenching. Biochemistry 38: 14690–14696 [DOI] [PubMed] [Google Scholar]
- Vredenberg W (2011) Kinetic analyses and mathematical modeling of primary photochemical and photoelectrochemical processes in plant photosystems. Biosystems 103: 138–151 [DOI] [PubMed] [Google Scholar]
- Witt HT (1979) Energy-conversion in the functional membrane of photosynthesis - analysis by light-pulse and electric pulse methods - central role of the electric-field. Biochim Biophys Acta 505: 355–427 [DOI] [PubMed] [Google Scholar]
- Zim�nyi L, Garab G (1989) Configuration of the electric-field and distribution of ions in energy transducing biological membranes: model calculations in a vesicle containing discrete charges. J Theor Biol 138: 59–76 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.