

Abstract
Microglia are the resident immune cells of the central nervous system (CNS) and are increasingly recognized as critical players in development, brain homeostasis, and disease pathogenesis. The lifespan, maintenance, proliferation, and turnover of microglia are important factors that regulate microglial behavior and affect their roles in the CNS. However, emerging evidence suggests that microglia are morphologically and phenotypically distinct in different brain areas, at different ages, and during disease. Ongoing research focuses on understanding how microglia acquire specific phenotypes in response to extrinsic cues in the environment and how phenotypes are specified by intrinsic properties of different populations of microglia. With the development of pharmacological and genetic tools that allow the investigation of microglia in vivo, there have been considerable advances in understanding molecular signatures of both homeostatic microglia and those reacting to injury and disease. Here, we review the master gene regulators that define microglia as well as discuss the evidence that microglia are heterogeneous and fall into distinct clusters that display specific intrinsic properties and perform unique tasks in different settings. Taken together, the information presented supports the idea that microglia morphology and transcriptional heterogeneity should be considered when studying the complex nature of microglia and their roles in brain health and disease.
Keywords: microglia, heterogeneity, development, microbiota, depletion, repopulation
Graphical Abstract
Overview of Microglia History
In 1919, Pio Del Rio Hortega was the first to distinguish between astrocytes, microglia, and oligodendrocytes (Del Rio-Hortega, 1932). Hortega took advantage of the tools present at that time to capture the physical landscape of the brain. He illustrated microglia in drawings and described them as “cells with short prolongations and enlarged cell bodies (Del Rio-Hortega, 1932).” Hortega’s description of microglia holds to this day, almost a century later. It took many decades before neuroscience research switched gears to focus on the role of glia in the healthy and diseased brain. Initially, microglia were regarded as docile spectators of the brain parenchyma (Lawson et al., 1992). They were thought to be derived from the neuroectoderm, but this view was later revised due to evidence that showed a monocytic blood origin through fate-mapping experiments (Alliot et al., 1991; Alliot et al., 1999; Ginhoux et al., 2010; Kierdorf et al., 2013; Butovsky et al., 2014). Over the last three decades, microglia’s role in health and disease has started to be unraveled. During development, microglia function in synapse tagging and elimination and regulate the number of neurons (Stevens et al., 2007; Schafer & Stevens, 2010; Paolicelli et al., 2011; Schafer et al., 2012; Lehrman et al., 2018). Not only do microglia phagocytose apoptotic cells, but they also control the fate of neurons and their progenitors (Paolicelli et al., 2011). Throughout life, microglia continuously monitor the brain parenchyma and respond rapidly to injuries (Davalos et al., 2005; Nimmerjahn et al., 2005). The role of microglia as active surveillants and their role as first responders to injury is their most characterized features. In disease, microglia take on either a damaging or a restorative role in the CNS (De Lucia et al., 2016; Krasemann et al., 2017). New research is beginning to characterize other aspects of microglial behavior, such as their role in synaptic plasticity (Tremblay et al., 2010; Miyamoto et al., 2016; Sipe et al., 2016). Finally, as new research is focused on the physiological roles of microglia, this is an especially interesting time to examine what is known about their potential to self-renew and maintain homeostasis in the adult brain (Zhan et al., 2019; Mendes et al., 2020; Zhan et al., 2020).
Origin of Microglia, Brain Colonization, and Development
Microglia enter the brain during early development around embryonic day 8 in the mouse (Alliot et al., 1991; Kurz & Christ, 1998; Alliot et al., 1999; Ginhoux et al., 2010; Katsumoto et al., 2014; Buttgereit et al., 2016; Matcovitch-Natan et al., 2016). Shortly after this time, the blood-brain barrier (BBB) is formed, and peripheral macrophages are largely excluded from the brain (Ben-Zvi et al., 2014; Sohet et al., 2015). Fate mapping experiments reveal that microglia emerge from early erythromyeloid precursors via a PU.1 (Ginhoux et al., 2010; Kierdorf et al., 2013) pathway and are regulated by colony- stimulating factor receptor 1 (CSF1R) in the embryonic yolk sac (YS) (Ginhoux et al., 2010; Kierdorf et al., 2013) (Fig. 1). In mice, RUNX1 (runt-related transcription factor 1) is a marker for myeloid precursors in the extraembryonic YS that appear before embryonic day 8 (Ginhoux et al., 2010; Kierdorf et al., 2013; Kierdorf & Prinz, 2013; Crotti & Ransohoff, 2016; Matcovitch-Natan et al., 2016) (Fig. 1). This progenitor is believed to generate the entire microglial adult population independent of bone marrow-derived cells (Ginhoux et al., 2010; Kierdorf et al., 2013). These c-KIT+ stem cells depend on the transcription factor PU.1, interferon regulator factor 8 (IRF8), and colony- stimulating factor 1 receptor (CSF1R) to develop into mature CD45-/C-kit-/CX3CR1+ cells that then acquire their identity as microglial cells (Ginhoux et al., 2010). The early colonization of the developing brain by microglia precedes neurogenesis and other cellular processes, suggesting that microglia may be important in mediating early homeostatic events (Alliot et al., 1991; Ginhoux et al., 2010; Squarzoni et al., 2014; Matcovitch-Natan et al., 2016) (Fig. 1). As progenitors in the CNS undergo massive proliferative expansion to produce the essential number of neurons and microglia, microglia enhance proliferation and stimulate differentiation of different CNS populations (Buchsbaum & Cappello, 2019; Bae et al., 2020). Microglia can also promote and inhibit neurogenesis via receptors such as C1q (Paolicelli et al., 2011; Bialas & Stevens, 2013) or neurotrophic factors (Paolicelli et al., 2011; Schafer et al., 2012; Parkhurst et al., 2013; Hong et al., 2016b; Miyamoto et al., 2016) (Fig. 1). Once nascent neural networks form, excessive synaptic connections are subsequently removed by microglia during a period of activity-dependent refinement (Stevens et al., 2007; Bialas & Stevens, 2013). Microglia also interact with other glia and stimulate oligodendrocyte development, (Domingues et al., 2016), and astrocyte differentiation (Schafer & Stevens, 2013; Sierra et al., 2014; Liddelow et al., 2017).
Figure 1: Microglia development follows a stepwise program.
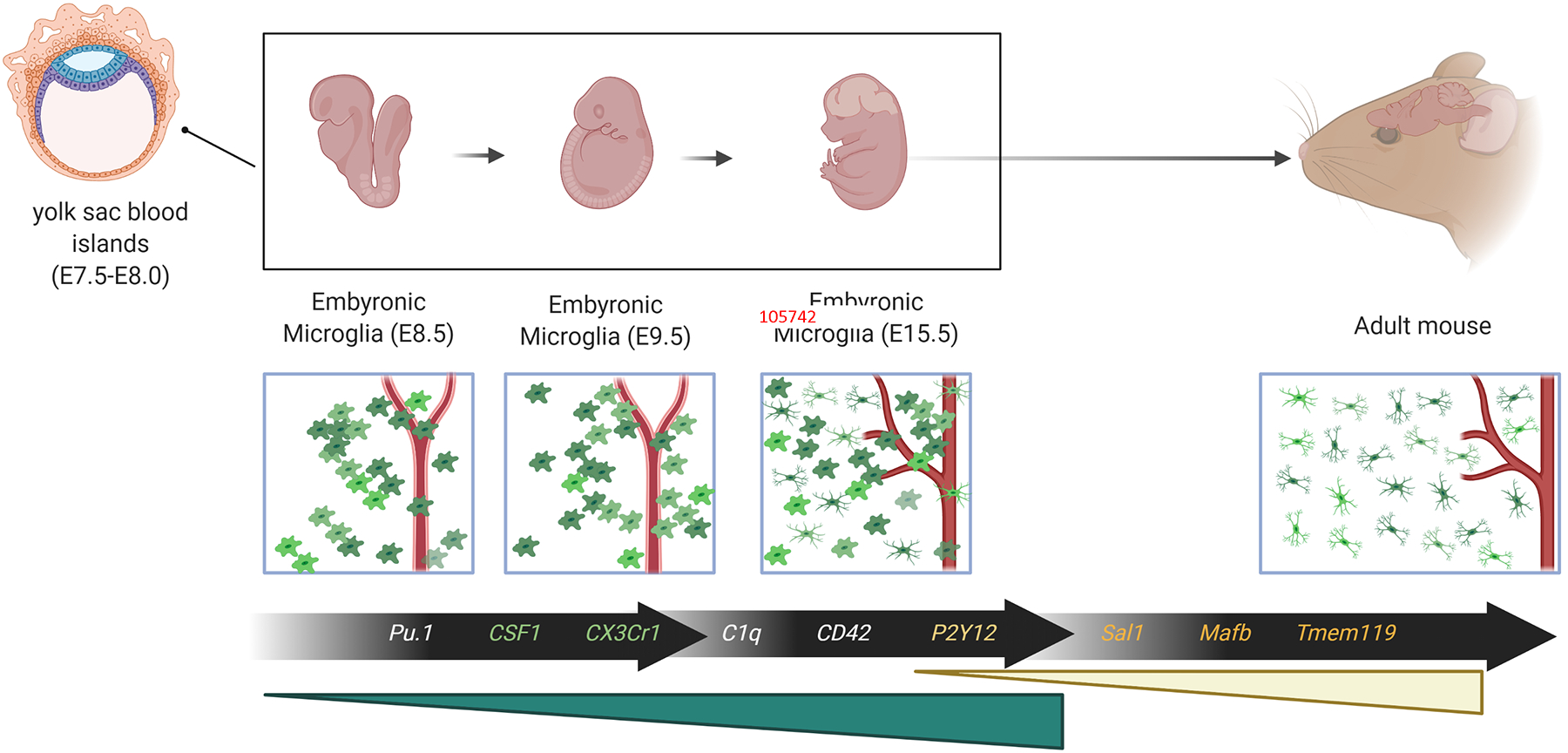
Primitive macrophages exit the yolk sac blood islands at the onset of blood circulation and colonize the brain at E9.5 in the mouse. These cells go on to give rise to microglia. The blood barrier is formed at around E13.5, and this isolates the developing brain. During this time, embryonic microglia expand and colonize the CNS until they acquire their mature characteristics in adulthood. Several genes regulate the development of microglia at different stages. During the early stages, CSF1 and PU.1 are responsible for the development and survival of microglia. During the later stages of microglia development and maturation, genes such as Sal1, TGFβ, and Mafb contribute to microglia homeostasis.
Microglial ontogeny and maturation in the adult brain
Baseline microglia maintenance in the adult brain
After their initial colonization of the brain, microglia appear to self-maintain without the contribution of peripheral monocytes (Ajami et al., 2007; Hashimoto et al., 2013). Maintenance of the microglial population may be accomplished by the slow turnover of long-lived cells in the mouse and the human (Askew et al., 2017; Reu et al., 2017); however, there is still controversy surrounding the self-renewal rate in the brain (Fuger et al., 2017; Reu et al., 2017). Recent reports estimate that a complete renewal of the population occurs once every 96 days in mice and that 2% of the population of microglia turnover in the human brain at any one time (Askew et al., 2017; Tay et al., 2017). Additionally, in vivo tracking of microglia reveals a stable cell density throughout the lifetime, with stochastic proliferation and apoptosis to maintain the microglial niche (Tay et al., 2017). This complex maintenance of the microglia throughout life under basal conditions is sustained through a constant but small spatial translocation of microglia, which is mediated by P2Y12 (Eyo et al., 2018). More recently, pharmacological and genetic methods of depleting microglia and forcing repopulation (Parkhurst et al., 2013; Elmore et al., 2014; Bruttger et al., 2015) have been used to understand the kinetics of microglia self-renewal in the adult brain.
Microglia Depletion
Although embryonic microglia origin and birth (Ginhoux et al., 2013; Kierdorf et al., 2013) have widely been studied using fate-mapping experiments, microglial ontogeny and maturation in the adult brain and retina are only now started to be elucidated (Parkhurst et al., 2013; Elmore et al., 2014; Askew et al., 2017; Fuger et al., 2017; Huang et al., 2018a; Huang et al., 2018b). Models that deplete the microglial niche provide an opportunity to study adult microglial turnover and maintenance (Zhang et al., 2018; Zhan et al., 2019; Mendes et al., 2020; Zhan et al., 2020). The depletion of microglia has been demonstrated in both the embryo and in the adult brain (Varvel et al., 2012; Parkhurst et al., 2013; Elmore et al., 2014; Bruttger et al., 2015; Elmore et al., 2015). Repopulation follows very rapidly after depletion to reinstate the niche (Elmore et al., 2015), testifying as to the robust nature of microglia and their effort to restore homeostasis. There are three main mechanisms which have been considered by which microglial density and spatial orientation is rapidly restored following depletion: first, peripheral bone marrow-derived cells could infiltrate the parenchyma and then differentiate into microglia-like cells; second, a special microglia-like progenitor remaining after depletion could be responsible for generating new microglia; and third, microglia that remain during depletion could proliferate rapidly to fill the empty niche (Waisman et al., 2015). For this review, we will briefly discuss how several different depletion paradigms have been used to address the mechanisms behind microglial repopulation.
Genetic and pharmacological methods of microglia depletion
Several genetic methods have been developed that make microglia vulnerable to certain toxins, leading to depletion of this cell population. The most popular of these is the CX3Cr1 creERT: iDTR transgenic mouse, which specifically expresses the diphtheria toxin receptor, which is usually absent in mice, in microglia (but not in macrophages if toxin injection is appropriately timed). This allows for specific ablation of microglia following tamoxifen and diphtheria toxin injections (Parkhurst et al., 2013; Bruttger et al., 2015). Using this approach, a 95% reduction in the number of microglial cells was reported 3 days after diphtheria toxin injection, with cell numbers returning to baseline 14 days after injection (Bruttger et al., 2015). However, this method reported a robust neuroinflammatory response due to genetic ablation. This cytokine storm was not observed in Parkhurst et al.(Parkhurst et al., 2013), which used a similar mouse model. This suggests that there is some variability in this mouse model of depletion that should be taken into consideration for future experiments.
An alternate pharmacological microglial depletion method has used inhibition of CSF1R to deplete microglia and macrophages in the CNS (Elmore et al., 2015). CSF-1 is expressed exclusively by microglia in the CNS under physiological conditions (Ginhoux et al., 2010; Elmore et al., 2015), and signaling through its receptor regulates microglia proliferation, survival, and development (Elmore et al., 2014; Elmore et al., 2015). Therefore, the administration of a CSF1R inhibitor reduces the number of microglia and macrophages without affecting surrounding cells (Elmore et al., 2014; Dagher et al., 2015; Elmore et al., 2015; Spangenberg et al., 2016). PLX3397 and PLX5622 are small CSF1R inhibitors that can cross the BBB and deplete microglia in the brain and retina in a matter of days (Elmore et al., 2014; Dagher et al., 2015; Huang et al., 2018a). These inhibitors have been widely used to investigate microglia origin and maturation in the healthy adult brain and various disease models.
Interestingly, CSF1R has two ligands - CSF1 and IL-34, which have a similar structure but different binding sites, and both regulate the migration and the colonization of microglial precursors. Global deletion of CSF1R leads to brain abnormalities, postnatal growth retardation, osteoporosis and is usually fatal (Dai et al., 2002). In mice, pharmacological depletion of CSF1 and the use of a CSF1 antibody in the brain show that this factor is also required for prenatal microglia development and colonization in the brain (Wu et al., 2018). The Il34-csf1ra cytokine signaling pathway regulates microglia colonization in the forebrain and retina in zebrafish (Dai et al., 2002; Wang et al., 2012; De et al., 2014; Wu et al., 2018) and may be required to maintain adult microglia (Greter et al., 2012). Finally, Csf1R ligands IL-34 and CSF1 are differentially required for microglia maintenance in the brain (Easley-Neal et al., 2019). While genetic and pharmacologic methods of depletion eliminate microglia throughout the brain, blockade of CSF1 signaling depletes microglia in white matter, while blockade of IL-34 depletes gray matter microglia. Also, conditional depletion of CSF1 from the brain results in a disruption in cerebellar microglia homeostasis and morphology while the microglia in the cortex remain intact (Easley-Neal et al., 2019; Kana et al., 2019). This provides for the exciting idea that identified microglial populations could be targeted explicitly for depletion from specific brain regions or subareas to dissect the roles of heterogeneous microglial subsets in different brain functions.
Rapid repopulation
Microglia repopulation of the brain is remarkably rapid, reaching control numbers in less than a week even when depletion is over 90% efficient (Parkhurst et al., 2013; Elmore et al., 2014; Bruttger et al., 2015; Mendes et al., 2020). In the CX3Cr1 creERT: iDTR system, this repopulation was shown to be driven exclusively by brain resident microglia, rather than an influx of peripheral monocytes if the BBB remained intact, despite the neuroinflammatory environment generated by depletion (Bruttger et al., 2015). An influx of peripheral cells has also not been observed following pharmacological depletion, which does not cause an overt neuroinflammatory response (Dagher et al., 2015; Elmore et al., 2015). In another genetic model using the CX3Cr1CreER/+R26DTA/+ mice, however, the microglia niche was repopulated by a combination of the local proliferation of microglia and infiltration of macrophages even in the absence of BBB breakdown (Lund et al., 2018). Interestingly, while new microglia generated in the brain adopt expression patterns of endogenous microglia, engrafting macrophages that enter the brain in the presence or absence of BBB breakdown adopt a microglia-like phenotype but remain distinct in terms of their gene expression (Bruttger et al., 2015; Lund et al., 2018). Thus, microglia generally repopulate rapidly without the contribution of peripheral cells. However, in certain circumstances, such as radiation exposure, engraftment from circulating cells is possible, but in such cases, the engrafting cells never fully adopt microglial phenotypes (Bruttger et al., 2015).
Initial studies that examined the proliferation of microglia within the brain following depletion suggested that microglial repopulation was driven by nestin-expressing microglial progenitor cells that proliferated and migrated to fill the niche (Bruttger et al., 2015; Elmore et al., 2015). This suggested that a special progenitor cell that was resistant to depletion may be responsible for microglial generation throughout life. However, fate mapping and in vivo experiments showed that microglial repopulation was solely due to residual microglial cells that remain after depletion (Tay et al., 2017; Zhan et al., 2019). These residual microglia, along with the newly-born microglia they generate, transiently express nestin, and this expression decreases once the number of microglia returns to baseline (Huang et al., 2018b). In fact, the microglia remaining after depletion have a remarkable capacity to proliferate, acquiring baseline numbers in as little as 3 days (Dagher et al., 2015). Microglial proliferation at baseline and in response to depletion has been imaged in vivo, showing that microglia acquire an elongated, double-soma, or “doublet” morphology before separating into two cells (Fuger et al., 2017; Mendes et al., 2020). Overall, this evidence supports the idea that microglia have a remarkable capacity to self-renew.
The mechanisms that govern microglial quiescence and proliferation are still not well understood. Bruttger et al. showed that IL-1 signaling actively regulates microglia repopulation following genetic depletion (Bruttger et al., 2015). Another study suggested that NF-KB signaling contributes to the rapid repopulation of microglia (Zhan et al., 2019), although it is difficult to pinpoint how NF-KB signaling modulates microglia repopulation because it has roles in so many other processes in the brain (Leonardo et al., 2015). More work needs to be done to identify specific downstream regulators in this signaling pathway that directly affect adult microglia ontogeny. Most recently, Mac2+ progenitor-like-cells were described as a key population that made up the remaining cells after depletion and contributed to the generation of newly-born microglia in the adult brain (Zhan et al., 2020). This is an interesting possibility because Galactin-3 or (Mac-2) controls microglia phenotype and is required for microglial activation (Lalancette-Hebert et al., 2012). This suggests that a specific subpopulation of microglia that has been described in early development and disease states (Lalancette-Hebert et al., 2012) (see below) may have unique roles in the adult brain in homeostatic conditions. Finally, our recent work shows that microglial proliferation after depletion in the visual cortex proceeds normally in the absence of P2Y12. Overall, it appears that P2Y12 is critical for microglia translocation and landscape organization under basal conditions (Eyo et al., 2018) but plays a minor role in adult microglia ontogeny and maturation in vivo (Eyo et al., 2018; Mendes et al., 2020). It is plausible that an interplay of several intrinsic factors, including the ones highlighted above, is working together to repopulate the brain. These could synergize with extrinsic factors provided by other cell types that also respond to decreases in microglial numbers. Whether different subtypes of microglia or microglia in different brain areas use different molecular cues and intrinsic machinery to effect rapid division is currently unknown.
Fast maturation of Microglia
While newly-born/repopulated microglia rapidly (within days of repopulation) take on mature characteristics, including a ramified morphology, efficient surveillance of the parenchyma, and sensing and chemotaxis to purines (Elmore et al., 2015; Mendes et al., 2020), there is also evidence that these newly-born microglia take on an immature molecular signature similar to that of early postnatal microglia (Matcovitch-Natan et al., 2016; Zhan et al., 2019). Single-cell RNA sequencing of microglia at different stages of repopulation revealed a stepwise maturation of newly-born microglia, with the early-stage newly born/repopulated microglia (around 4 days of repopulation) closely resembling the early postnatal microglia cluster (postnatal day 4 (P4)). Newly-born microglia at 4 days of repopulation in the adult brain share 24% of genes with postnatal day 4 early postnatal microglia observed in development. Newly born microglia at this stage of repopulation downregulate genes known to be important for microglial maturation and maintenance of mature phenotypes such as TGF- β, Mafb, P2Y12, and Tmem119 (Butovsky et al., 2014; Bennett et al., 2016; Matcovitch-Natan et al., 2016; Zhan et al., 2019). Also, triggering receptor expressed on myeloid cells 2 (Trem2), a disease-associated microglial gene, was differentially expressed at this stage (Hammond et al., 2019). This suggests that newly-born/repopulated microglia in the adult brain express genes associated with a transcriptomic signature that overlaps with early postnatal microglia and differs from non-depleted adult microglia (Matcovitch-Natan et al., 2016; Zhan et al., 2019). This finding is surprising since the newly-born microglia at day 4 have proliferated to control densities, resemble control microglia morphologically, and achieve equal cell-to-cell spacing (Mendes et al., 2020). Several other genes are also upregulated at this stage to promote repopulation. For instance, cycle-related genes such as CdC20 and Cdc25b were highly expressed in newly-born microglia but returned to homeostatic levels very quickly (Hammond et al., 2019; Zhan et al., 2019). This suggests that newly-born microglia re-enter the cell cycle early in the microglia repopulation stage (Zhan et al., 2019). Overall, this indicates a dissociation between molecular and morphological phenotypes of microglia during repopulation. Fast maturation of expression follows a stepwise program like what we see in the embryonic development of microglia, however, this does not affect microglial morphology or surveillance of the brain, which recovers rapidly and does not follow developmental phenotypes. This may have implications for understanding microglial phenotypes in the mature brain – whereby similar morphologies and dynamics may not necessarily indicate similarities in expression patterns and functions.
The fast acquisition of mature microglial dynamics could be regulated in part by epigenetic mechanisms. Cells can acquire behaviors based on signals in their local microenvironment that persist and alter their roles and orientation in the brain. Microglia are exquisitely responsive to their environment and can adapt depending on their history of inflammatory exposure, analogous to “innate immune memory” in the peripheral immune system. It is likely that this ‘microglia memory’ plays a part in repopulation in the adult brain (Boraschi & Italiani, 2018). Epigenetic modifications are possibly stored in individual newly-born/repopulated microglia cells that acquire these changes from their parent microglia. In development, deletion of the class 1 histone deacetylases (Hdac1 and Hdac2) led to deficits in microglia development. Pro-apoptotic and cell cycle genes were hyperacetylated in the absence of HDACs, which led to an increase in microglial death (Datta et al., 2018). Therefore, as observed in development, epigenetic modifications may regulate the balance between death and division, thus dictating how fast microglia repopulate and may regulate gene expression in a way that affects the acquisition of mature characteristics. If passed to daughter cells, such epigenetic signatures could either preserve heterogeneity by maintaining phenotypes acquired in different microglial populations or decreased heterogeneity if cells with different epigenetic signatures are more or less susceptible to depletion and/or death in the normal lifecycle of microglia.
Sustained depletion and its effects on repopulation
While microglia have a remarkable capacity to rapidly self-renew, there may be limits to this process. Continuous and repeated depletion cycles of microglia in the adult brain lead to deficits in repopulation (Najafi et al., 2018). Whereas microglia repopulate the brain fully after one cycle of depletion, additional cycles result in only partial repopulation, despite the continued survival of a subset of microglia during depletion (Najafi et al., 2018). Surprisingly, a number of genes known to be highly expressed in microglia, such as P2ry12, Tmem110, and Tgfbr1, were not expressed after a few cycles of microglia depletion due to the deficit in the repopulation of microglial cells (Najafi et al., 2018). This suggests that microglia may have a limited number of divisions they can undergo in the adult brain. Alternatively, subsequent depletions rounds may affect the survival of different microglia subtypes, possibly depleting the population responsible for rapid repopulation. Interesting, several oligodendrocyte precursor cells (OPC)-expressed genes were also downregulated with microglia elimination, supporting the idea that adult microglia directly affect OPC population and maintenance (Hagemeyer et al., 2017; Miron, 2017; Najafi et al., 2018). The lack of microglial repopulation after several cycles of depletion is reminiscent of OPC regeneration (Miron, 2017). This may be another piece of the puzzle, suggesting that interactions with other cell types may contribute to the microglial potential for proliferation.
Roles of Microglia at the Synapse
Some of the most complex aspects of nervous system function are the formation and refinement of neural circuits. Microglia sculpt developing axons and their postsynaptic targets and promote organization and reorganization of neural networks (Schafer et al., 2012; Hughes & Appel, 2020). Through the lifespan, communication between microglia and neurons is controlled by a complex signaling network (Sun & Barres, 2016; Singhvi & Shaham, 2019). Several factors that modulate this interaction are critical to adaptive and innate immunity and are located on the synapse, allowing for active regulation of circuit development and plasticity by microglia (Eyo & Wu, 2013; Sheridan & Murphy, 2013; Sipe et al., 2016; Stowell et al., 2019). Under basal conditions, microglia react rapidly to neuronal activity by modulating physical contact with synaptic structures (Wake et al., 2009; Tremblay et al., 2010; Sipe et al., 2016). These physical contacts influence synaptic remodeling and turnover of dendritic spines. Microglia can impact both synaptic formation and outgrowth and eliminate excess synapses from the CNS through phagocytosis (Stevens et al., 2007; Schafer et al., 2012; Bialas & Stevens, 2013; Sierra et al., 2014).
Several lines of evidence suggest that microglial recruitment to synapses is dependent on neuronal activity. Early work using electron microscopy and in vivo two-photon imaging showed that physical interactions between neurons and microglia in the visual cortex were altered by inhibition or retinal activity (Wake et al., 2009) and dark adaptation and subsequent light exposure (Tremblay et al., 2010). These manipulations that alter neuronal activity in the visual cortex also changed the number of phagocytic inclusions (Tremblay et al., 2010). Manipulating the activity of retinogeniculate projections also alters microglia-mediated synaptic elimination in the visual thalamus, as microglia preferentially phagocytose less active presynaptic inputs (Schafer et al., 2012). In the zebrafish, microglia are attracted to neurons with enhanced activity, and interestingly, removal of microglia results in enhanced spontaneous activity suggesting an activity-mediated loop between microglia and neurons (Li et al., 2012). While the mechanisms that may mediate this activity-dependence are not entirely clear, it has been shown that activation of dendritic N-methyl-D-aspartate receptors (NMDA) on neurons by exogenous ATP application or seizure induction in vivo could trigger microglial recruitment to neurons (Dissing-Olesen et al., 2014; Eyo et al., 2014). To further understand the diversity in microglial pathways that modulate physical interactions between microglia and neurons, we review some of the main candidates that have been described in different forms of plasticity across the lifespan.
Fractalkine
Many lines of evidence suggest that synaptic activity can modulate the phagocytic activity of microglia and that microglial synaptic phagocytosis may be an important part of synaptic pruning. Phagocytic removal of synapses in the hippocampus is dependent on fractalkine signaling, and mice lacking the only known fractalkine receptor, CX3CR1, have an increased density and delayed maturation of dendritic spines in CA1 pyramidal neurons compared to controls (Paolicelli et al., 2011) (Fig. 2). This impacts the development of interconnectivity between brain areas leading to deficits in behavior (Gunner et al., 2019). While some of these deficits may be indirectly affected by microglial colonization of the brain, which is thought to be driven by fractalkine signaling (Paolicelli et al., 2011; Hoshiko et al., 2012), synapse elimination in the barrel cortex is dependent on direct CX3CR1 signaling. In the developing barrel cortex, thalamocortical synapses are also immature in CX3CR1KO mice (Hoshiko et al., 2012). CX3CR1, CX3CL1, and ADAM10 (a metalloprotease known to cleave CX3CL1) mediate the engulfment of thalamocortical (TC) synapses (Gunner et al., 2019). This suggests an activity-dependent molecular mechanism by which neurons, which release fractalkine (CX3CL1) in an activity-dependent fashion, communicate with microglia (which express CX3CR1) to refine synapses during development (Fig. 2). It is important to note that synaptic plasticity does not always require fractalkine signaling. For instance, ocular dominance plasticity, which occurs in the visual cortex in early adolescence in mice and requires intact microglia, does not require CX3CR1 (Lowery et al., 2017; Schecter et al., 2017). Whether specific microglial subtypes require fractalkine for their roles at synapses or whether fractalkine is required at specific developmental stages in certain brain areas or neuronal networks in which plasticity occurs is currently unknown. High expression of CX3CR1 is a common feature of all microglia, suggesting that the production, processing, and release of fractalkine by neurons and astrocytes may regulate this pathway’s function in plasticity.
Figure 2: Microglial roles at the Synapse.
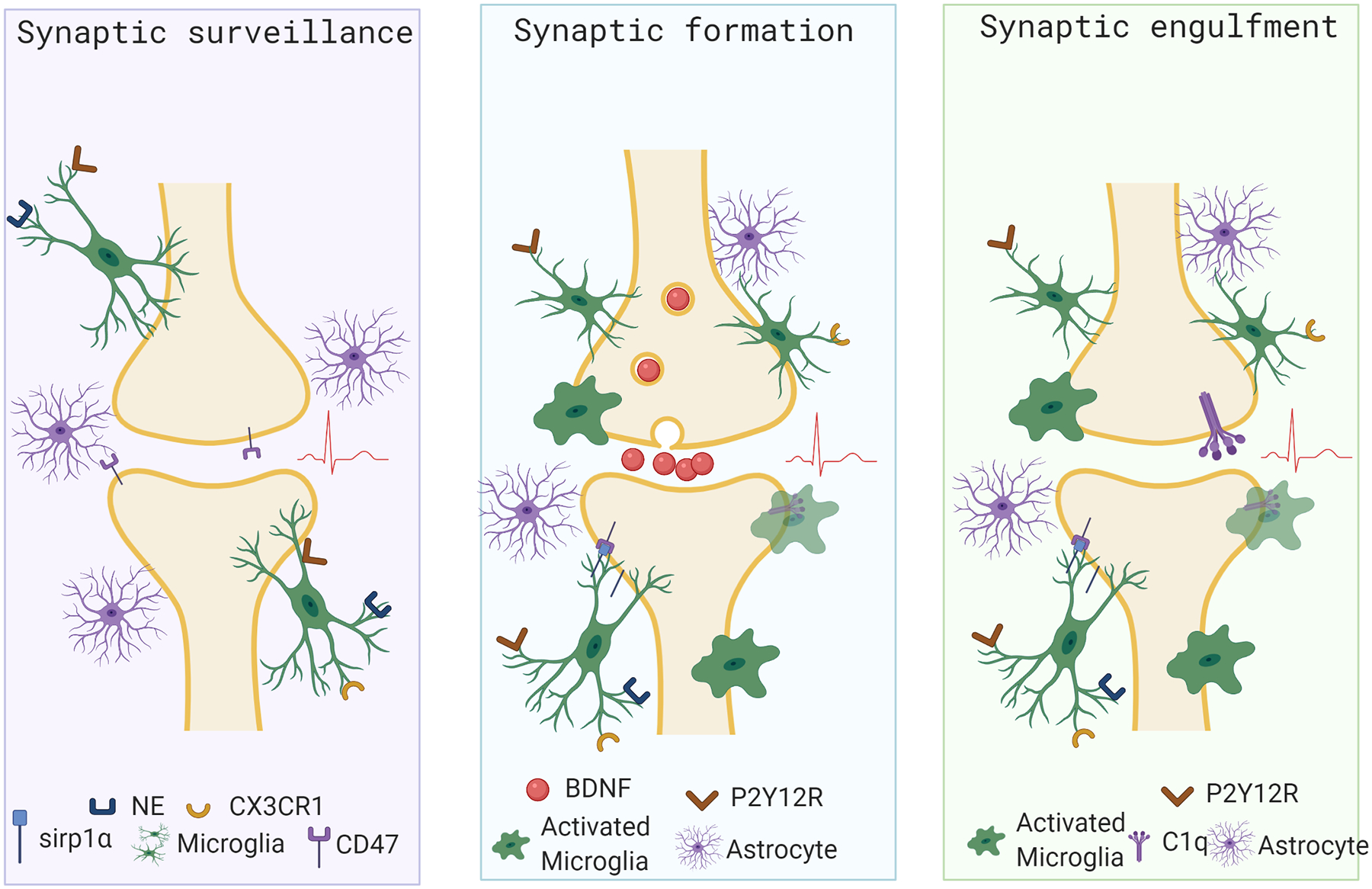
Recent studies have highlighted three functions of microglia at the synapse. Microglia actively survey the synapse through the extension and retraction of their processes. Microglia contribute to the formation of new synapses in the brain. Finally, microglia engage in synaptic engulfment and pruning during early development and throughout life.
Purines
Purinergic signaling is a form of extracellular signaling mediated by purine nucleotides such as ATP and ADP. The activation of purinergic receptors regulates the functions of many different cell types. ATP can be released in an activity-dependent manner from synapses and glia (Sipe et al., 2016) and activate purinergic receptors on microglia that modulate intracellular Ca++ levels (Haynes et al., 2006; Swiatkowski et al., 2016; Mildner et al., 2017). Interactions between purinergic receptors, cytokine signaling, and growth factors regulate synaptic development (Fields & Burnstock, 2006). Signaling through different purinergic receptors and its effects on development is an area currently being explored. The field’s focus has shifted to P2Y receptors and their function in the direct regulation of synaptic transmission. Recently, P2Y12 has been demonstrated to facilitate microglial roles in synaptic remodeling through ATP-mediated intercellular communication (Eyo & Wu, 2013; Eyo et al., 2014; Sipe et al., 2016).
The purinergic receptor P2Y12 is primarily expressed by microglia in the CNS and is one of the signature genes that define the microglial sensome (Haynes et al., 2006; Butovsky et al., 2014; Mildner et al., 2017; Eyo et al., 2018) (Fig. 2). P2Y12 signaling is necessary for ocular dominance plasticity (ODP) during the critical period of primary visual cortex (V1) development, a synaptic remodeling that requires an imbalance of activity in the neural pathways that serve the two eyes (Sipe et al., 2016). In vitro and in vivo studies have demonstrated that P2Y12 is an important mediator of microglial dynamics and is highly sensitive to ADP (Davalos et al., 2005). This may be a direct example of how immune signaling is reused in the context of microglial roles at synapses, as ATP-sensing through P2Y12 mediates the early injury response in microglia (Haynes et al., 2006).
In control animals, an ocular dominance (OD) shift in mice during the critical period consists of two stages: a loss of responsiveness to the deprived eye during the first three days of monocular deprivation (MD) and a subsequent gain of responsiveness to the open eye occurring over the next few days (Frenkel & Bear, 2004). Emerging evidence suggests that microglia can eliminate synapses through phagocytosis, raising the question of whether microglia eliminate synapses following MD and thereby contribute to ODP. While pharmacological and genetic disruption of P2Y12 did not affect the development of the normal strong bias towards the contralateral eye that is observed in naïve animals, ODP was disrupted after the deprivation of the contralateral eye (Sipe et al., 2016), implicating P2Y12 signaling as an integral and necessary mediator of ocular dominance plasticity in V1. Deprivation-induced microglial recruitment to synapses and increased phagocytic activity were also disrupted by the loss of P2Y12. There are still many unanswered questions regarding how purinergic signaling modulates ODP in V1. Specifically, mechanisms for the release of ATP are not yet clearly defined, and the specific role of purinergic signaling in plasticity has not been explored. Given the link between NMDA activation and P2Y12-mediated chemotaxis (Dissing-Olesen et al., 2014; Eyo et al., 2014), P2Y12 activation may recruit microglia to dendrites to facilitate synaptic removal.
Norepinephrine
Locus coeruleus (LC) noradrenergic neurons send projections throughout the brain. LC neurons play a major role in stress, attention, arousal, and learning in many different brain areas (Heneka et al., 2010; O’Donnell et al., 2012; Gyoneva & Traynelis, 2013). Therefore, it is not a surprise that these neurons have a widespread impact on microglial function (O’Donnell et al., 2012). NE is known to be anti-inflammatory and increases microglial phagocytosis in the context of Alzheimer’s disease (AD) (Heneka et al., 2010). However, NE’s role extends to modulating microglia in developing and adult animals in the context of plasticity, as has recently been described (Liu et al., 2019; Stowell et al., 2019) (Fig. 2). Cortical microglia primarily express β2 adrenergic receptors at rest and α2A receptors are expressed in the activated state (Tanaka et al., 2002; Gyoneva & Traynelis, 2013; Liu et al., 2019). These adrenergic receptors modulate microglial surveillance, affecting the interactions between microglia and other cells such as neurons (Eyo et al., 2014; Miyamoto et al., 2016; Stowell et al., 2019). Increases in NE release, or direct stimulation of β2 adrenergic receptors, causes microglial process retraction, a decrease in surveillance, and fewer interactions between microglia and dendritic spines (Liu et al., 2019; Stowell et al., 2019). Prolonged β2 adrenergic stimulation also impairs experience-dependent plasticity in the visual cortex, but only if microglial β2 adrenergic receptors are intact (Stowell et al., 2019). Because of NE’s roles in mediating wakefulness, microglia interactions with synapses and thus their roles in synaptic plasticity are likely altered depending on the vigilance state of the animal (Liu et al., 2019; Stowell et al., 2019).
Additionally, given that NE had similar effects on microglia in different cortical regions at different developmental ages, β2 adrenergic signaling may be a common, ubiquitous mechanism regulating microglial function throughout the brain. It is also likely that receptors such as P2Y12 and β2 adrenergic receptors work in unison to regulate microglia roles in neurotransmission. Both these receptors are G-protein coupled, acting through Gi and Gs subunits, respectively, and thus engaging antagonistic downstream pathways to regulate the dynamic shifts in microglia dynamics and surveillance. While the push-pull relationship between P2Y12 and β2 adrenergic receptors has been explored (Gyoneva & Traynelis, 2013), it is likely that other G-protein coupled receptors interact in this context to aid microglia in navigating their complex and ever-changing environment.
Complement
The complement cascade is a complex signaling system composed of an array of proteins activated through partial cleavage (Carroll, 2004). The expression of the complement proteins was, until recently, thought to be restricted largely to cells of the immune system. It is now clear that complement ligands and receptors are widespread throughout many tissue and cell types (Stephan et al., 2012). Additionally, brain cells such as astrocytes, microglia, and surprisingly even neurons express complement proteins. It appears that the role of the complement cascade in clearing debris and protecting against autoimmunity is conserved in the periphery and centrally.
Complement proteins are widely expressed throughout the developing CNS, and the classical complement cascade plays a large role in microglial-mediated engulfment of synapses (Schafer et al., 2012; Wu et al., 2015). Immunohistochemistry experiments have identified C1q and downstream complement protein C3 on subsets of dendrites and axons throughout the postnatal brain (Stevens et al., 2007) (Fig. 2). The complement cascade appears to be particularly important to the active remodeling of synaptic circuits by microglia and astrocytes in the early postnatal period in mice (Stevens et al., 2007; Tremblay et al., 2010; Paolicelli et al., 2011). During early development, retinal ganglion cells (RGCs) make transient connections with relay neurons in the dorsal lateral geniculate nucleus (dLGN) of the thalamus, and complement regulates the remodeling and elimination of weak synapses between these two partners (Schafer et al., 2012). Neuroanatomical tracings of retinogeniculate projections and electrophysiological recordings in dLGN neurons showed that, in the absence of key complement proteins such as C1q and C3, mice had deficits in synaptic refinement and elimination (Stevens et al., 2007). Microglia-mediated engulfment of RGCs is highly regulated, and signaling between CR3, and its ligand, the complement protein C3, underlie synaptic pruning. Microglia continue to express the initiating molecule of the complement cascade, C1q, in the mature brain and are the only resident brain cells to express CR3 (Ransohoff & Perry, 2009). Early changes in complement signaling result in sustained defects in synaptic connectivity, as evidenced by increased numbers of synapses in the adult visual cortex of mice deficient in C3 or CR3 (Schafer et al., 2012). Whether complement has direct roles in physiological synaptic refinement after the early developmental period is currently unknown.
Phosphatidylserine
When cells undergo apoptosis, they externalize phosphatidylserine as a signal to phagocytes to remove the dying cells. Surprisingly, microglia phagocytosis and synaptic refinement in the non-injured developing brain also depend on phosphatidylserine (Scott-Hewitt et al., 2020). Microglia specific deletion of Gpr56 (a splicing isoform) results in a deficit in microglia synapses pruning (Li et al., 2020; Scott-Hewitt et al., 2020), whereby excess synapses are not removed through microglial phagocytosis. This raises the interesting idea that microglial phagocytosis of synapses is driven by a complex set of activity-driven signals that must be tightly controlled and present at the exact specific time and location to elicit the appropriate development of neural circuits. Understanding the temporal aspect of this interaction may give insight into microglia-mediated mechanisms of circuit maintenance.
CD47
Complement and phosphatidylserine signaling serves to tag weak synapses for microglial engulfment in the visual cortex and hippocampus, suggesting that these mechanisms may be common “eat me” signals in all regions of the CNS (Mallat et al., 2005; Tremblay et al., 2010; Paolicelli et al., 2011; Schafer et al., 2012; Li et al., 2020; Scott-Hewitt et al., 2020). However, it remains unclear what preferentially determines the elimination of a synapse. Because synapses are eliminated based on their activity, there is likely an activity-dependent process that either drives or interacts with complement-based tagging (Lehrman et al., 2018). Most recently, the CD47-SIRPα signaling pathway was identified as a “don’t eat me” signal that protects synapses from elimination during development (Lehrman et al., 2018) (Fig. 2). The “don’t eat me” signal is found on more active synapses, and its loss results in increased phagocytic activity of microglia and increased synaptic pruning. In the absence of CD47-SIRPα the activity-dependence of synaptic pruning is disrupted.
BDNF
Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family and is a key regulator in the neuroimmune axis (Bessis et al., 2007) and synaptogenesis and synaptic plasticity (Coull et al., 2005; Ethell & Ethell, 2007). Although neurons are the major source of BDNF in the adult brain (Altar, 1999), BDNF is also expressed in oligodendrocytes, astrocytes, and microglia (Parkhurst et al., 2013). Microglial contribution to synaptic plasticity and function was demonstrated through an inducible conditional knockout mouse line, where BDNF was depleted from microglia (Parkhurst et al., 2013). Mice lacking microglial BDNF displayed a decrease in synaptic proteins, deficits in spine formation, and a deficit in a motor learning task, suggesting an important role for microglial derived BDNF in modulating synaptic plasticity (Parkhurst et al., 2013). Given the widespread expression of BDNF, this suggests that the local release of BDNF and possibly other growth factors and cytokines from microglia may impact the gain, loss, and remodeling of individual synapses. The mechanisms microglia use to influence their roles at the synapse in different contexts will be the focus of many studies going forward.
Distinct subtypes of microglia may have specific roles in different brain areas and developmental time periods
Traditionally, microglia were considered a homogeneous population, which was supported by the fact that they originated from a uniform progenitor pool that invaded the brain early in development and populated all brain regions. Although differences in morphology between white and gray matter microglia were appreciated in early studies (Del Rio-Hortega, 1932; Lawson et al., 1990; Easley-Neal et al., 2019), only recently has there been a renewed interest in understanding microglial heterogeneity. It is becoming clear that microglial distribution differs by brain region and that microglial morphology is remarkably fluid, changing both during physiological conditions and injury. Microglia exhibit regional differences in self-renewal and turnover rates under normal physiological conditions and with external stimuli (Askew et al., 2017; Ayata et al., 2018; Huang et al., 2018a; Zhang et al., 2018; Mendes et al., 2020). Specifically, self-renewal rates and morphology of newly-born microglia differ in the cerebellum, cortex, and retina. Recently there has been a lot of evidence suggesting that microglia are also heterogeneous in their molecular signatures.
One of the most distinct microglial phenotypes can be found in the cerebellum, an evolutionarily old brain area. Cerebellar microglia are physiologically distinct from cortical microglia populations both in terms of their expression patterns and their morphology and dynamic behavior (Ashwell, 1990; Lawson et al., 1990; Grabert et al., 2016; Ayata et al., 2018; Stowell et al., 2018). RNAseq experiments showed that cortical and striatal microglia transcriptomes were closely related, while a more distinct transcriptome was found for hippocampal and cerebellar microglia (Grabert et al., 2016; Ayata et al., 2018; Li et al., 2019). Several lines of evidence suggest that cerebellar microglia exist in a more immmunovigilent state than microglia in other brain areas. The cerebellar microglia transcriptome is characterized by higher expression of genes related to immune function compared to cortical or even hippocampal microglia (Li et al., 2019), and is subject to epigenetic regulation, which increases phagocytic activity (Ayata et al., 2018). This is further reflected in the dynamics of cerebellar microglia, which are much less complex morphologically and are diffusely distributed. Cerebellar microglia also surveil the healthy cerebellum, in part, by translocating their cell body (Stowell et al., 2018), a phenomenon more common in macrophages and largely absent in microglia in other parts of the brain (Eyo et al., 2018).
Interestingly this transcriptional and morphological phenotype is distinct from an activated state, suggesting that microglia in the cerebellum are a separate functional cell type rather than existing in a chronic inflammatory state. However, it is important to note that in the cortex, a distinct subset of microglia is characterized by the expression of immunoregulatory molecules such as CD47 and CD300a, regulators that are known to regulate myeloid cell response. This subset is much less prominent in the cerebellum, suggesting that immune processes are likely regulated differently in the two regions (Grabert et al., 2016; Li et al., 2019).
The differences between microglial phenotypes in the cerebellum, hippocampus, and cortex are surprising, given that all these microglial originate from the same precursor cells. Microglial heterogeneity may arise from cues in the brain environment, especially given the immense sensitivity of microglia to environmental signals. For instance, cerebellar microglia may become immunologically primed by the environment in which they develop, which is characterized by enhanced neuronal apoptosis compared to cortical regions (Ayata et al., 2018). In the basal ganglia, region-specific microglial phenotypes emerge during the second postnatal week once microglia have colonized and matured in their new environment (De Biase et al., 2017). Despite the proximity of basal ganglia nuclei, microglia are characterized by different densities, morphologies, lysosomal content, electrophysiological properties, and expression profiles (especially those related to metabolism and phagocytosis). Interestingly, microglial densities are co-regulated with the density of astrocytes, again suggesting extrinsic cues and interactions between different cell types in guiding microglial identities (De Biase & Bonci, 2019). These cues may also originate from outside of the brain. There is increasing evidence to suggest that microbiota in the gut are vital regulators of the CNS innate immune system. Microglia in mice housed in a germ-free environment, a manipulation that substantially changes the gut microbiome, show deficits in numbers and display an immature phenotype that leads to impaired immune responses (Erny et al., 2015). Bacterial-derived short-chain fatty acids (SCFA) are key to microglia homeostasis, and deficiency in SCFA receptor FFAR2 elicits similar defects in microglia maturation and function (Erny et al., 2015). The richness and diversity of the gut microbiota may play a role in conferring heterogeneity to microglia during development, providing a critical way for environmental factors to impact brain development.
Single-cell transcriptomic analyses of microglia reveal different microglial subsets
With the advent of single-cell RNA sequencing (scRNAseq), the traditional classification of brain cell types and subtypes is changing. This technique has delineated distinct clusters of microglia that likely have different behaviors and functions in the brain. In the mouse brain, 7–9 microglial clusters have been identified (Hammond et al., 2019; Li et al., 2019; Masuda et al., 2019; Sankowski et al., 2019). While distinct, many of these clusters show significant overlap, underscoring the malleable nature of microglia that exist in a continuum of functional states (Hammond et al., 2019). Many of these clusters represent microglia across different developmental timelines and brain areas, but there is evidence to suggest that heterogeneity may also exist within a single brain region (Hammond et al., 2019; Li et al., 2019). However, the potential significance of these clusters for normal CNS function is not yet clear, partly because functional studies need to be performed to characterize these clusters and because of the current focus of studies on differential gene expression between control and disease tissues rather than within the healthy brain. ScRNAseq results have confirmed other transcriptomic studies that suggest that there are a number of microglial “master gene regulators” that define all microglial populations. These master genes are expressed during the early stages of life through aging. Their expression contributes to the proper development and function of both the microglial population and the brain as a whole (Hammond et al., 2019; Li et al., 2019). Canonical microglial genes include Fcrls, P2ry12, Cx3cr1, Trem2, C1qa and Slc2a5 (Hammond et al., 2019; Li et al., 2019). These proteins are primarily associated with maintaining homeostasis and performing phagocytic roles in the brain, defining the common function of all microglial populations.
Microglial diversity during development.
ScRNAseq studies suggest that microglia are more diverse during early development and became less heterogeneous with time. During murine mid-gestation and early postnatal life (between embryonic day 14.5 and postnatal day 5), a subset of distinct genes peak, including Arg1, Rrm2, Ebec2, Cenpa, Fabp5, Spp1, Hmox1, Ms4a7 (Hammond et al., 2019; Li et al., 2019). Most of these genes allow microglia to sense changes in their environment, respond to injury, and transition from a surveillant state to a more activated state (Hammond et al., 2019). This may be the time for microglia to interact with their environment and accrue brain-region specific phenotypes. Although a number of distinct microglia subpopulations were identified during early development, we will focus our attention on two clusters: Cluster 3 and 4 defined by Hammond et al. (Hammond et al., 2019). Microglia in cluster 3 are almost exclusively found in the embryonic and early postnatal brain and are characterized by unique expression of Fabp5 (Fatty acid Binding Protein 5), Mif (migration inhibitor factor), lactate dehydrogenase A (Ldha), and triosephosphate isomerase (Tp1) (Hammond et al., 2019). Mif and Fabp5 are both linked to cell growth, motility, inflammation, and immunomodulation, which are all functions critical to the proper development of microglia in the brain. In fact, several genes, such as thymosin Beta 4 X-Linked (Tmsb4x), Profilin 1 (Pfn1), and Cofilin 1 (Cfl1) involved in actin cytoskeleton dynamics were differentially upregulated. Other enriched genes were associated with glycolysis, suggesting a unique metabolic profile of developing microglia. Fth1, Ftl1, and Met1 genes also peak during this period and are critical for homeostasis (Hammond et al., 2019). Finally, ribosomal component genes such as Rps14, Rps18, Rps35, and Rps29 were upregulated (Hammond et al., 2019; Li et al., 2019). Another subset of microglia that is found in early developmental periods has been termed proliferative-region-associated microglia (PAM) (Li et al., 2019). These early postnatal microglia, which are phagocytic and prevalent in the olfactory bulbs and white matter track, shared similar gene signatures with degenerative disease-associated microglia (DAM), which have been identified in disease models (Li et al., 2019). This is also true of the early postnatal microglia described above (cluster 3 from (Hammond et al., 2019)), which express DAM genes such as Spp1, Gpnmb, Igf1, Clec7a, Lpl, Cd9, Cd63, Lgal3, Fabp5, Itgax, Apoe and Tyrobp (Hammond et al., 2019; Li et al., 2019). These genes may dictate the ameboid microglia phenotype of early development when microglia work to clear dying cells.
Microglial gene expression in adulthood and aging
As microglia mature, heterogeneity decreases, and the core microglial genes are expressed universally (Hammond et al., 2019; Li et al., 2019). These include P2ry12 (Mildner et al., 2017), Mafb (Matcovitch-Natan et al., 2016), Sal1 (Buttgereit et al., 2016), and Tmem119 (Bennett et al., 2016), distinguishing microglia from macrophages. These transcripts are expressed at much lower levels or not at all in the developing brain, suggesting that microglia acquire a universal phenotype that impacts their function as they mature. In addition, almost all microglia express immune-regulatory signature genes such as Slc2a5, Fcrls, Trem2, and C1qa (Hammond et al., 2019; Li et al., 2019), reflecting their immune roles in the adult brain. Unlike the clusters and genes identified at the younger ages (embryonic to P30), adult microglia (P100; clusters 7a-c from (Hammond et al., 2019)) were not defined by specific genes but rather expressed the core microglial genes as described above.
As microglia age (post-natal day 540 (P540) in mice), a unique subset of genes arises. In mice, Ccl4, a chemokine also known as a macrophage inflammatory protein, was enriched in aged microglia (Hammond et al., 2019). This chemokine regulates trafficking and communication with other immune cells, possibly contributing to inflammation in the aged brain. PAM microglia, characterized by expression of Spp1 and Gpnm, that are prominent in development, are also present in the aged brain (Li et al., 2019). At the morphological level, PAM microglia are ameboid with thicker primary branches and larger cell bodies. They express genes associated with a phagocytic phenotype, but unlike DAM, they do not depend on TREM2 or Apoe (Keren-Shaul et al., 2017) to phagocytose (Li et al., 2019). This means that during aging and disease, microglia take on an inflammatory phenotype and are generally primed (Spittau, 2017).
Cerebellar microglia also appear to be more sensitive to aging and disease processes than their cortical counterparts. Aging differentially regulates a specific cluster of genes exclusively in the cerebellum (Grabert et al., 2016; Li et al., 2019). Immune-related antigen processing and clearance genes such as Celf1, Cd2ap, and Epha1 show a significantly higher expression in the cerebellum during late-onset Alzheimer’s disease (AD) compared to other cortical regions, even though the cerebellum is largely spared of pathology in this disease. In addition, homeostatic microglia genes such a Clu, Mef2c, and Bin1 were significantly lower in the cerebellum (Chappell et al., 2018). This suggests that many immunoregulatory pathways are differentially regulated by brain region during aging and require a specific subset of genes to function. This highlights the cerebellum as an area that contains a distinct subset of microglia developmentally and throughout the lifespan. Cerebellar heterogeneity in gene expression is potentially relevant to the pathogenies of AD and could be an area of interest for exploring neuroprotective pathways or biomarkers of the disease.
Microglia heterogeneity and Alzheimer’s Disease
Microglia are observed to play an important role in neurological diseases such as Alzheimer’s disease (AD), Multiple Sclerosis (MS), Frontotemporal dementia, and Parkinson’s disease (PD) (Bolmont et al., 2008; Ransohoff & El Khoury, 2015; Spittau, 2017). Microglia are described to be beneficial and detrimental at varying stages of pathology in these different diseases. To avoid trying to integrate a very large and complex literature, this review focuses on the role of microglia in AD to illustrate the possible roles of heterogeneous populations of microglia in the diseased brain (Fig. 3). It is still unclear whether or how microglia tailor their response to disease and whether distinct populations of microglia exist in pathological tissues. Pathways expressed in development are often re-expressed in disease, as detailed above. Not only do diseased microglia have morphological similarities with microglia in development, but they also re-express developmental markers such as Apoe, Lpl, and Spp1. Their transcription states do not overlap, however, showing that they are not reverting to a developmental phenotype (Hammond et al., 2019). The diversity of microglial phenotypes in the context of disease is an important area of study. A deep understanding of this field will allow us to define the very complex roles that may be carried out by different populations of microglia to facilitate or protect against disease progression.
Figure 3: Microglia in Alzheimer’s Disease.
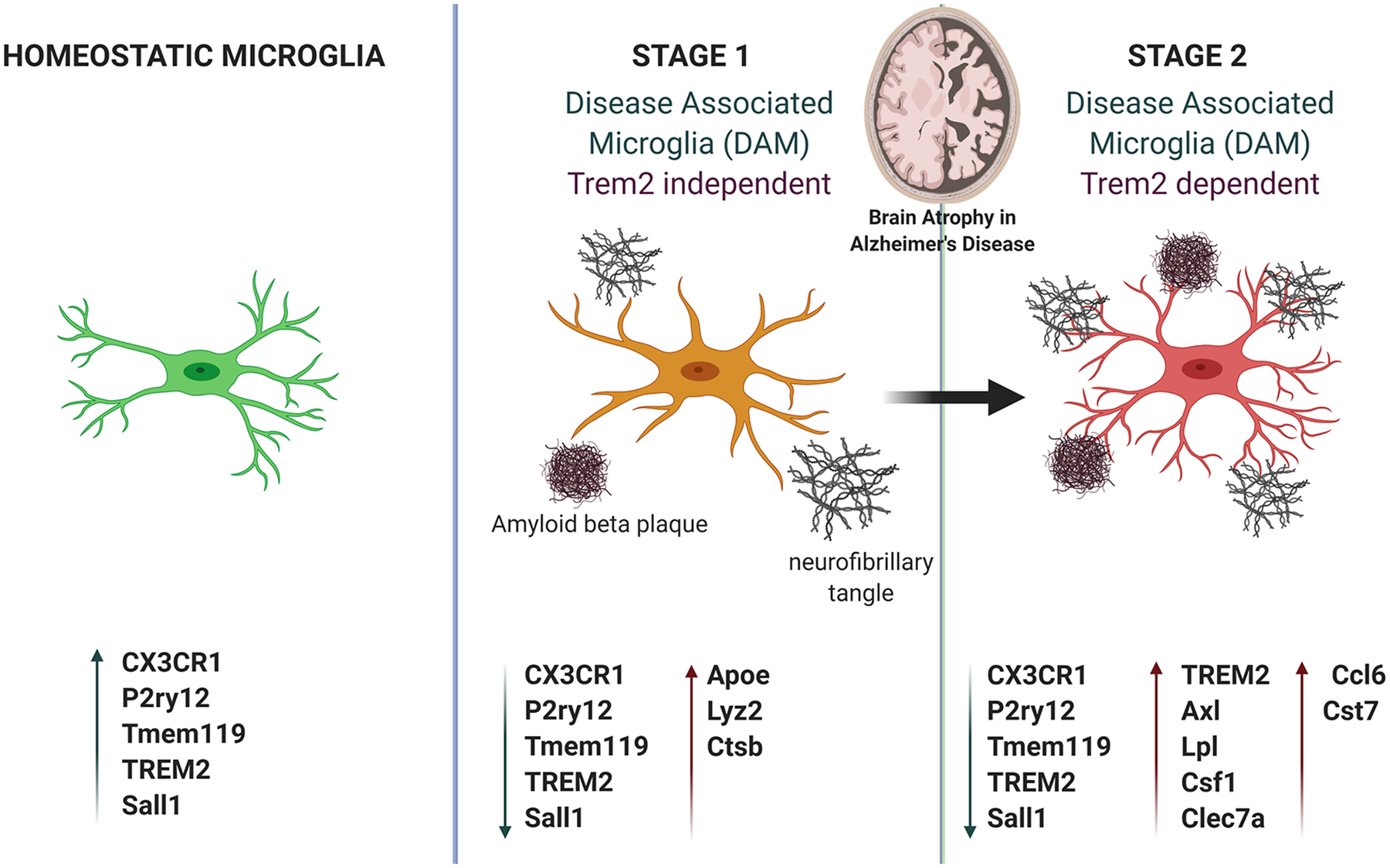
A schematic describing the changes that occur with the onset of Alzheimer’s disease. In the healthy brain, tau protein stabilizes the healthy microtubule. Homeostatic microglia express canonical genes such as CX3CR1 and P2ry12. However, with age and in the Alzheimer’s brain, tau proteins form tangles, and the microtubules disintegrate. At this stage (stage 1), microglia transition to an activated state, surround diffuse amyloid-beta plaques and interact with neurofibrillary tangles. Disease associated microglia (DAM) are upregulated expressing genes such as Apoe Lyz2 and Ctsb. In stage 2, more DAM defining genes such as Trem2, Axl, and Lpl is upregulated. In both stages, canonical microglial gene expression is downregulated.
AD is a neurodegenerative disease characterized by progressive loss of memory and other cognitive functions (McManus & Heneka, 2017). Pathological characteristics of the AD brain are amyloid-β (Aβ) accumulation, neurofibrillary tangles (NFT), synaptic loss, neurodegeneration (Hardy & Selkoe, 2002; Crotti & Ransohoff, 2016; Spangenberg et al., 2016), and neuroinflammation (Streit et al., 2004; Cherry et al., 2014a; b; Rivera-Escalera et al., 2014; Heneka et al., 2015; Calsolaro & Edison, 2016; Domingues et al., 2017; Dionisio-Santos et al., 2019) (Fig. 3). Microglia typically accumulate around senile plaques and can modulate Aβ processing (Heneka et al., 2015; Keren-Shaul et al., 2017). Microglia secrete proteolytic enzymes that degrade Aβ and express receptors that promote the clearance and phagocytosis of Aβ (Wang et al., 2015). It is becoming clear that microglia have a large effect on AD progression. Recent transcriptome-wide studies of bulk ex vivo human microglia suggest that microglia change with age and have profiles enriched for disease genes (Sankowski et al., 2019). Additionally, SNPs that confer AD risk disproportionally affect microglial function (Nott et al., 2019).
While microglia play a large role in AD progression, it is not clear if microglial responses are beneficial or detrimental in the context of AD. In fact, many of the studies provide contradictory information. Some studies report that complement inhibition results in an increase in amyloid pathology and that C3 and CR3 contribute to Aβ phagocytosis, while others show that elimination of CR3 or inhibition of C3 in microglia decrease Aβ and/or plaques in the brain (Webster et al., 2000; Stephan et al., 2012; Shi et al., 2015; Hong et al., 2016a; Shi et al., 2017). In addition to complement, TREM2, a cell surface receptor expressed in myeloid cells, including microglia, was shown to modulate inflammation and phagocytosis in AD (Wang et al., 2016). TREM2 expression is increased in plaque-associated microglia and is required for complete activation of DAM (Jay et al., 2015). Despite evidence to suggest an important role for TREM2 in AD, we still do not know explicitly whether it has a beneficial or detrimental effect. TREM2-deficient APP mice, a common model of human AD, show a decrease in amyloid burden (Kang et al., 2018b). However, TREM2 deficiency can also increase hippocampal Aβ peptide (Jay et al., 2015). Variants of APOE (Apolipoprotein E), specifically APOEε4, is a major genetic risk factor for AD (Kang et al., 2018a). Individuals with a single copy of APOEε4 have a 3-fold increase risk of AD, and two copies are equivalent to a 12-fold increase (Calsolaro & Edison, 2016). TREM2 is described to bind directly to APOE and modulate AD pathology (Krasemann et al., 2017). Kraesmann et al. describe a TREM2-ApoE connection, where TREM2 interactions with APOE led to a switch from a homeostatic to a microglial neurodegenerative phenotype (MGnD) (Krasemann et al., 2017). MGnD microglia have decreased expression of homeostatic genes and increased expression of inflammatory genes and have largely been thought to be pathology-driving (Krasemann et al., 2017). Overall, these studies show a complex role for microglial mechanisms in Aβ pathology in AD. It is likely that some of these contradictory findings result from the heterogeneity in microglial phenotypes whereby subsets of microglia differentially impact disease progression. It is also likely that the impact of microglial activity depends on the stage of the pathogenesis, and this appears to be true for the role of TREM2 (Wang et al., 2016; Krasemann et al., 2017; Zhao et al., 2018).
While various subtypes of microglia associated with the disease have been identified, it is also still an open question whether the phenotypes adopted by microglia in AD brains, such as DAM and MGnD, are adaptive or maladaptive. The DAM profile was enriched in microglia that were in contact with amyloid plaques (Keren-Shaul et al., 2017), and loss of function mutations in the genes that define this profile promote AD (Keren-Shaul et al., 2017; Li et al., 2019). DAM microglia contain Aβ aggregates suggesting that the DAM profile is protective by increasing microglial phagocytosis of Aβ. However, an alternate possibility is that DAM are deleterious and propagate disease phenotypes, and in fact, MGnD are similar in transcriptional makeup and phenotype to DAM microglia. The MGnD microglia associate with neuritic plaques in the cortex during the early stages of the disease and are thought to contribute to the progression of AD (Krasemann et al., 2017). Whether MGnD and DAM microglia represent two different subpopulations of microglia that respond differently to disease, pathology will need to be established. To get to the root of this question, future experiments will need to target and delete individual DAM and MGnD associated genes and determine the effects of this deletion on disease progression. If methods can be established to deplete these two populations separately, this could also address their roles in AD, although the large overlap between expression profiles would make this approach difficult to implement.
Treatment with a CSF1R inhibitor (PLX) to deplete all microglia, irrespective of phenotype, protects against synapse loss in AD mouse models (Spangenberg et al., 2016; Rice et al., 2017), suggesting that at least one prominent AD-related microglial population contributes to synapse elimination. However, the impact of microglia depletion on behavior is inconclusive. Some studies see marked improvements in cognition (Dagher et al., 2015), while others see no change with depletion of microglia (Rice et al., 2015; Spangenberg et al., 2016). The differences may be due to methodological variation, the stage of pathology, and the mouse models used (Dagher et al., 2015; Spangenberg et al., 2016). Despite the many studies describing microglial roles in amyloid processing, most studies have reported that the formation and maintenance of Aβ plaques are not affected by microglia depletion (Grathwohl et al., 2009; Rice et al., 2015; De Lucia et al., 2016; Spangenberg et al., 2016). However, longer periods of depletion may be necessary to observe an effect on Aβ deposition in the brain. A recent study that used time-lapse imaging to track individual plaques reported that plaque size increased ~13% over 1 week in microglia-depleted brains (Zhao et al., 2017). Finally, partial microglia depletion prevented microglia plaque interaction and improved cognition in the 3xTG-AD mouse model (Dagher et al., 2015). The study suggests that Aβ levels and plaque loads were not altered by partial microglia depletion, but that behavior may have been improved by selectively depleting those microglia that associate with plaques (Dagher et al., 2015). Overall, the depletion studies imply that synapse loss around plaques is dynamically regulated by microglia, and depletion of microglia may rescue the synapse loss observed in the pathogenesis of AD. The use of new tools developed to selectively deplete different microglial subtypes will allow future dissection of how different microglial phenotypes contribute to disease.
Additionally, the roles of microglia in AD are likely modulated by environmental factors. For instance, it will be important to determine the role of the microbiota in shaping microglial profiles. Microglial development and immune capabilities are shaped by the microbiome, and these effects may not be restricted to development, as recent experiments have identified key changes in the gut of AD patients and animal models, which could have a profound effect on microglia. For example, the expression of the NLRP3 inflammasome was increased in the intestines and blood of AD mice, and this change in peripheral inflammation could mediate changes in gut microbiota (Hu et al., 2016; Pistollato et al., 2016; Jiang et al., 2017). In fact, in another study, rRNA gene sequencing was used to isolate DNA from fecal samples to compare the composition of the gut microbiome in participants with and without a diagnosis of AD (Vogt et al., 2017). Overall, the gut microbiome of AD participants had decreased microbial richness and diversity and a distinct composition compared to controls. This suggests that early changes in the gut microbiota may play a role in disease progression through immune activation and systemic inflammation (Hu et al., 2016; Jiang et al., 2017). These microbiome changes could have a profound role in altering the function of microglia in AD, although this idea requires more investigation.
Concluding remarks
Microglia have critical roles in brain homeostasis and disease. Here, we summarize evidence showing that the number of microglia, morphology, and molecular signatures are heterogeneous in different regions of the brain. This points to diversity in the function of microglia in these regions. Recent advances in RNA sequencing, in combination with in vivo imaging, provide compelling evidence for the rapid and heterogeneous dynamics of microglia. Discrete subclusters of microglia have been identified throughout the brain. These clusters may develop, self-renew, and respond to inflammation in a time- and-context-dependent manner independent of other subtypes in the adjacent milieu. However, the functional aspects of the microglial subtypes are not yet defined. More needs to be done to identify what the clusters mean throughout the brain. It is plausible that targeting microglial subtypes in specific regions of the brain may be the answer to treating neurodegenerative disease. As mentioned above in the review, there are still aspects of microglial heterogeneity that need further investigation. For example, it is still unclear how the microbiota may be contributing to the heterogeneous clusters we see in development and during disease. Another outstanding question is how microglia progenitors may be contributing the regional heterogeneity. Advances in sequencing techniques and MerFISH technology will allow us to dig deeper into the remaining questions regarding microglia heterogeneity.
Acknowledgments:
This work is supported by grants from the National Institute of Health (NIH): F99 NS108486-02 (to M.S.M), RO1s NS114480, AA027111 and R21 NS099973 (A.K.M), NSF 1557971 (A.K.M.), AARG-NTF-19-619116 and grants from the Schmitt and Mangurian foundations and the Rochester Center for Alzheimer’s Disease Research.
Abbreviations
- AD
Alzheimer’s Disease
- Apoe
Apolipoprotein E
- Arg1
Arginase 1
- ATP
adenosine triphosphate
- BDNF
Brain-derived neurophic factor
- Bin1
Myc box-dependent-interacting protein 1
- C1qa
Complement C1q A Chain
- C3
Complement component 3
- Ccl4
Macrophage inflammatory protein- 1β
- Cd2ap
CD2 Associated Protein
- CD300a
Cluster of Differentiation 300A
- CD47
thrombospondin-1
- Cd63
tetraspanin 63
- Cd9
tetraspanin 9
- Celf1
CUGBP Elav-like family member 1
- Cenpa
centrome protein A
- Cfl1
Cofilin 1
- Clec7a
C-type lectin/C-type lectin-like domain (CTL/CTLD)
- Clu
Clusterin precursor
- Cr3
Macrophage-1 antigen
- CSF1R
Colony stimulating factor 1 receptor
- Cx3Cr1
fractalkine
- DAM
Disease associated Microlgia
- dLGN
dorsal lateral geniculate nucleus
- Ebec2
- Epha1
EPH Receptor A1
- Fabp5
Fatty Acid Binding Protein 5
- Fcrls
Fc receptor-like molecule
- Fth1
ferritin heavy chain
- Ftl1
Ferritin light chain 1
- Gpnmb
Transmembrane glycoprotein NMB
- Hdac1
Histone Deacetylase 1
- Hmox1
Heme Oxygenase 1
- Igf1
Insulin like growth factor 1
- IL-34
Interleukin 34
- Itgax
Integrin alpha X
- LC
Locus coeruleus
- Ldha
Lactate Dehydrogenase A
- Lgal3
Galectin-3
- Lpl
Lipoprotein Lipase
- Mafb
MAF BZIP Transcription Factor B
- MD
Monocular deprivation
- Mef2c
Myocyte-specific enhancer factor 2C
- MerFISH
Multiplexed error-robust fluorescence in situ hybridization
- Met1
MET1 methyltransferase 1
- MGnD
microglial neurodegenerative phenotype
- Mif
Macrophage migration inhibitory factor
- Ms4a7
Membrane Spanning 4-Domains A7
- NE
norepinephrine
- NF-KB
Nuclear Factor Kappa-light-chain-enhancer of activated B cells
- NMDA
N-methyl-D-aspartate receptor
- OD
Ocular dominance
- ODP
ocular dominance plasticity
- OPC
oligodendrocyte progenitor cells
- P2Y12
purinergic receptors
- P30
Post natal day 30
- Pfn1
Profilin-1
- RGC
Retinal Ganglion cell
- RNAseq
RNA sequencing
- Rps
Ribosomal proteins (RPs)
- Rrm2
Ribonucleotide reductase subunit M2
- Slc2a5
Solute carrier family 2 member 5
- Spp1
Secreted Phosphoprotein 1
- TGF- β
Transforming growth factor beta
- Tmem110
transmembrane protein 110
- Tmsb4x
Thymosin beta 4
- Tp1
Topoisomerase 1
- TREM2
Triggering receptor expressed on myeloid cells 2
- Tyrobp
Transmembrane Immune signaling adaptor
- V1
visual cortex
Biographies


Footnotes
Conflict of Interest
The authors declare no conflicts of interest.
Data Accessibility
No primary data is presented in this review article.
References
- Ajami B, Bennett JL, Krieger C, Tetzlaff W & Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci, 10, 1538–1543. [DOI] [PubMed] [Google Scholar]
- Alliot F, Godin I & Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res, 117, 145–152. [DOI] [PubMed] [Google Scholar]
- Alliot F, Lecain E, Grima B & Pessac B (1991) Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci U S A, 88, 1541–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altar CA (1999) Neurotrophins and depression. Trends Pharmacol Sci, 20, 59–61. [DOI] [PubMed] [Google Scholar]
- Ashwell K (1990) Microglia and cell death in the developing mouse cerebellum. Brain Res Dev Brain Res, 55, 219–230. [DOI] [PubMed] [Google Scholar]
- Askew K, Li K, Olmos-Alonso A, Garcia-Moreno F, Liang Y, Richardson P, Tipton T, Chapman MA, Riecken K, Beccari S, Sierra A, Molnar Z, Cragg MS, Garaschuk O, Perry VH & Gomez-Nicola D (2017) Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Rep, 18, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayata P, Badimon A, Strasburger HJ, Duff MK, Montgomery SE, Loh YE, Ebert A, Pimenova AA, Ramirez BR, Chan AT, Sullivan JM, Purushothaman I, Scarpa JR, Goate AM, Busslinger M, Shen L, Losic B & Schaefer A (2018) Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci, 21, 1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae SH, Yoo MR, Kim YY, Hong IK, Kim MH, Lee SH & Kim DY (2020) Brain-derived neurotrophic factor mediates macrophage migration inhibitory factor to protect neurons against oxygen-glucose deprivation. Neural Regen Res, 15, 1483–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H & Gu C (2014) Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature, 509, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG & Barres BA (2016) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A, 113, E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bessis A, Bechade C, Bernard D & Roumier A (2007) Microglial control of neuronal death and synaptic properties. Glia, 55, 233–238. [DOI] [PubMed] [Google Scholar]
- Bialas AR & Stevens B (2013) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci, 16, 1773–1782. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M & Calhoun ME (2008) Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci, 28, 4283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boraschi D & Italiani P (2018) Innate Immune Memory: Time for Adopting a Correct Terminology. Front Immunol, 9, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Woertge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, Mack M, Pinteaux E, Mueller W, Zipp F, Binder H, Bopp T, Prinz M, Jung S & Waisman A (2015) Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity, 43, 92–106. [DOI] [PubMed] [Google Scholar]
- Buchsbaum IY & Cappello S (2019) Neuronal migration in the CNS during development and disease: insights from in vivo and in vitro models. Development, 146. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP & Weiner HL (2014) Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci, 17, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B & Greter M (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol, 17, 1397–1406. [DOI] [PubMed] [Google Scholar]
- Calsolaro V & Edison P (2016) Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement, 12, 719–732. [DOI] [PubMed] [Google Scholar]
- Carroll MC (2004) The complement system in regulation of adaptive immunity. Nat Immunol, 5, 981–986. [DOI] [PubMed] [Google Scholar]
- Chappell S, Patel T, Guetta-Baranes T, Sang F, Francis PT, Morgan K & Brookes KJ (2018) Observations of extensive gene expression differences in the cerebellum and potential relevance to Alzheimer’s disease. BMC Res Notes, 11, 646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA & O’Banion MK (2014a) Are “resting” microglia more “m2”? Front Immunol, 5, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry JD, Olschowka JA & O’Banion MK (2014b) Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. Journal of neuroinflammation, 11, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K, Gravel C, Salter MW & De Koninck Y (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature, 438, 1017–1021. [DOI] [PubMed] [Google Scholar]
- Crotti A & Ransohoff RM (2016) Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity, 44, 505–515. [DOI] [PubMed] [Google Scholar]
- Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, West BL & Green KN (2015) Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. Journal of neuroinflammation, 12, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XM, Ryan GR, Hapel AJ, Dominguez MG, Russell RG, Kapp S, Sylvestre V & Stanley ER (2002) Targeted disruption of the mouse colony-stimulating factor 1 receptor gene results in osteopetrosis, mononuclear phagocyte deficiency, increased primitive progenitor cell frequencies, and reproductive defects. Blood, 99, 111–120. [DOI] [PubMed] [Google Scholar]
- Datta M, Staszewski O, Raschi E, Frosch M, Hagemeyer N, Tay TL, Blank T, Kreutzfeldt M, Merkler D, Ziegler-Waldkirch S, Matthias P, Meyer-Luehmann M & Prinz M (2018) Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity, 48, 514–529.e516. [DOI] [PubMed] [Google Scholar]
- Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML & Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci, 8, 752–758. [DOI] [PubMed] [Google Scholar]
- De Biase LM & Bonci A (2019) Region-Specific Phenotypes of Microglia: The Role of Local Regulatory Cues. Neuroscientist, 25, 314–333. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Schuebel KE, Fusfeld ZH, Jair K, Hawes IA, Cimbro R, Zhang HY, Liu QR, Shen H, Xi ZX, Goldman D & Bonci A (2017) Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron, 95, 341–356 e346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De I, Nikodemova M, Steffen MD, Sokn E, Maklakova VI, Watters JJ & Collier LS (2014) CSF1 overexpression has pleiotropic effects on microglia in vivo. Glia, 62, 1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lucia C, Rinchon A, Olmos-Alonso A, Riecken K, Fehse B, Boche D, Perry VH & Gomez-Nicola D (2016) Microglia regulate hippocampal neurogenesis during chronic neurodegeneration. Brain Behav Immun, 55, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Rio-Hortega P (1932) Microglia. In Cytology and cellular pathology of the nervous system (ed. Penfield W), Hoeber, New York., pp. pp. 483–534. [Google Scholar]
- Dionisio-Santos DA, Olschowka JA & O’Banion MK (2019) Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation, 16, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dissing-Olesen L, LeDue JM, Rungta RL, Hefendehl JK, Choi HB & MacVicar BA (2014) Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J Neurosci, 34, 10511–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues C, da Cruz ESOAB & Henriques AG (2017) Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr Alzheimer Res, 14, 870–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingues HS, Portugal CC, Socodato R & Relvas JB (2016) Oligodendrocyte, Astrocyte, and Microglia Crosstalk in Myelin Development, Damage, and Repair. Front Cell Dev Biol, 4, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easley-Neal C, Foreman O, Sharma N, Zarrin AA & Weimer RM (2019) CSF1R Ligands IL-34 and CSF1 Are Differentially Required for Microglia Development and Maintenance in White and Gray Matter Brain Regions. Front Immunol, 10, 2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Lee RJ, West BL & Green KN (2015) Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation. PLoS One, 10, e0122912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, Kitazawa M, Matusow B, Nguyen H, West BL & Green KN (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82, 380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erny D, Hrabe de Angelis AL, Jaitin D, Wieghofer P, Staszewski O, David E, Keren-Shaul H, Mahlakoiv T, Jakobshagen K, Buch T, Schwierzeck V, Utermohlen O, Chun E, Garrett WS, McCoy KD, Diefenbach A, Staeheli P, Stecher B, Amit I & Prinz M (2015) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci, 18, 965–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethell IM & Ethell DW (2007) Matrix metalloproteinases in brain development and remodeling: synaptic functions and targets. J Neurosci Res, 85, 2813–2823. [DOI] [PubMed] [Google Scholar]
- Eyo UB, Mo M, Yi MH, Murugan M, Liu J, Yarlagadda R, Margolis DJ, Xu P & Wu LJ (2018) P2Y12R-Dependent Translocation Mechanisms Gate the Changing Microglial Landscape. Cell Rep, 23, 959–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB, Peng JY, Swiatkowski P, Mukherjee A, Bispo A & Wu LJ (2014) Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. Journal of Neuroscience, 34, 10528–10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyo UB & Wu LJ (2013) Bidirectional microglia-neuron communication in the healthy brain. Neural Plast, 2013, 456857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD & Burnstock G (2006) Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci, 7, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel MY & Bear MF (2004) How monocular deprivation shifts ocular dominance in visual cortex of young mice. Neuron, 44, 917–923. [DOI] [PubMed] [Google Scholar]
- Fuger P, Hefendehl JK, Veeraraghavalu K, Wendeln AC, Schlosser C, Obermuller U, Wegenast-Braun BM, Neher JJ, Martus P, Kohsaka S, Thunemann M, Feil R, Sisodia SS, Skodras A & Jucker M (2017) Microglia turnover with aging and in an Alzheimer’s model via long-term in vivo single-cell imaging. Nat Neurosci, 20, 1371–1376. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM & Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F, Lim S, Hoeffel G, Low D & Huber T (2013) Origin and differentiation of microglia. Front Cell Neurosci, 7, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabert K, Michoel T, Karavolos MH, Clohisey S, Baillie JK, Stevens MP, Freeman TC, Summers KM & McColl BW (2016) Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat Neurosci, 19, 504–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grathwohl SA, Kalin RE, Bolmont T, Prokop S, Winkelmann G, Kaeser SA, Odenthal J, Radde R, Eldh T, Gandy S, Aguzzi A, Staufenbiel M, Mathews PM, Wolburg H, Heppner FL & Jucker M (2009) Formation and maintenance of Alzheimer’s disease beta-amyloid plaques in the absence of microglia. Nat Neurosci, 12, 1361–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Lelios I, Pelczar P, Hoeffel G, Price J, Leboeuf M, Kundig TM, Frei K, Ginhoux F, Merad M & Becher B (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity, 37, 1050–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner G, Cheadle L, Johnson KM, Ayata P, Badimon A, Mondo E, Nagy MA, Liu L, Bemiller SM, Kim KW, Lira SA, Lamb BT, Tapper AR, Ransohoff RM, Greenberg ME, Schaefer A & Schafer DP (2019) Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat Neurosci, 22, 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S & Traynelis SF (2013) Norepinephrine modulates the motility of resting and activated microglia via different adrenergic receptors. J Biol Chem, 288, 15291–15302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeyer N, Hanft KM, Akriditou MA, Unger N, Park ES, Stanley ER, Staszewski O, Dimou L & Prinz M (2017) Microglia contribute to normal myelinogenesis and to oligodendrocyte progenitor maintenance during adulthood. Acta Neuropathol, 134, 441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond TR, Dufort C, Dissing-Olesen L, Giera S, Young A, Wysoker A, Walker AJ, Gergits F, Segel M, Nemesh J, Marsh SE, Saunders A, Macosko E, Ginhoux F, Chen J, Franklin RJM, Piao X, McCarroll SA & Stevens B (2019) Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity, 50, 253–271.e256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J & Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS & Merad M (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity, 38, 792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB & Julius D (2006) The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat Neurosci, 9, 1512–1519. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT & Kummer MP (2015) Neuroinflammation in Alzheimer’s disease. Lancet Neurol, 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Nadrigny F, Regen T, Martinez-Hernandez A, Dumitrescu-Ozimek L, Terwel D, Jardanhazi-Kurutz D, Walter J, Kirchhoff F, Hanisch UK & Kummer MP (2010) Locus ceruleus controls Alzheimer’s disease pathology by modulating microglial functions through norepinephrine. Proc Natl Acad Sci U S A, 107, 6058–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ & Stevens B (2016a) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science, 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Dissing-Olesen L & Stevens B (2016b) New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol, 36, 128–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshiko M, Arnoux I, Avignone E, Yamamoto N & Audinat E (2012) Deficiency of the microglial receptor CX3CR1 impairs postnatal functional development of thalamocortical synapses in the barrel cortex. J Neurosci, 32, 15106–15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang T & Jin F (2016) Alzheimer’s disease and gut microbiota. Sci China Life Sci, 59, 1006–1023. [DOI] [PubMed] [Google Scholar]
- Huang Y, Xu Z, Xiong S, Qin G, Sun F, Yang J, Yuan TF, Zhao L, Wang K, Liang YX, Fu L, Wu T, So KF, Rao Y & Peng B (2018a) Dual extra-retinal origins of microglia in the model of retinal microglia repopulation. Cell Discov, 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Xu Z, Xiong S, Sun F, Qin G, Hu G, Wang J, Zhao L, Liang YX, Wu T, Lu Z, Humayun MS, So KF, Pan Y, Li N, Yuan TF, Rao Y & Peng B (2018b) Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci, 21, 530–540. [DOI] [PubMed] [Google Scholar]
- Hughes AN & Appel B (2020) Microglia phagocytose myelin sheaths to modify developmental myelination. Nat Neurosci, 23, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL, Cotleur AC, Butovsky O, Bekris L, Staugaitis SM, Leverenz JB, Pimplikar SW, Landreth GE, Howell GR, Ransohoff RM & Lamb BT (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med, 212, 287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Li G, Huang P, Liu Z & Zhao B (2017) The Gut Microbiota and Alzheimer’s Disease. J Alzheimers Dis, 58, 1–15. [DOI] [PubMed] [Google Scholar]
- Kana V, Desland FA, Casanova-Acebes M, Ayata P, Badimon A, Nabel E, Yamamuro K, Sneeboer M, Tan IL, Flanigan ME, Rose SA, Chang C, Leader A, Le Bourhis H, Sweet ES, Tung N, Wroblewska A, Lavin Y, See P, Baccarini A, Ginhoux F, Chitu V, Stanley ER, Russo SJ, Yue Z, Brown BD, Joyner AL, De Witte LD, Morishita H, Schaefer A & Merad M (2019) CSF-1 controls cerebellar microglia and is required for motor function and social interaction. J Exp Med, 216, 2265–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Ebbert MTW, Baker KE, Cook C, Wang X, Sens JP, Kocher JP, Petrucelli L & Fryer JD (2018a) Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J Exp Med, 215, 2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SS, Kurti A, Baker KE, Liu CC, Colonna M, Ulrich JD, Holtzman DM, Bu G & Fryer JD (2018b) Behavioral and transcriptomic analysis of Trem2-null mice: not all knockout mice are created equal. Hum Mol Genet, 27, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsumoto A, Lu H, Miranda AS & Ransohoff RM (2014) Ontogeny and functions of central nervous system macrophages. J Immunol, 193, 2615–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, Itzkovitz S, Colonna M, Schwartz M & Amit I (2017) A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell, 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Kierdorf K, Erny D, Goldmann T, Sander V, Schulz C, Perdiguero EG, Wieghofer P, Heinrich A, Riemke P, Holscher C, Muller DN, Luckow B, Brocker T, Debowski K, Fritz G, Opdenakker G, Diefenbach A, Biber K, Heikenwalder M, Geissmann F, Rosenbauer F & Prinz M (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat Neurosci, 16, 273–280. [DOI] [PubMed] [Google Scholar]
- Kierdorf K & Prinz M (2013) Factors regulating microglia activation. Front Cell Neurosci, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O’Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C & Butovsky O (2017) The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity, 47, 566–581.e569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz H & Christ B (1998) Embryonic CNS macrophages and microglia do not stem from circulating, but from extravascular precursors. Glia, 22, 98–102. [PubMed] [Google Scholar]
- Lalancette-Hebert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, Sato S & Kriz J (2012) Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci, 32, 10383–10395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH, Dri P & Gordon S (1990) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience, 39, 151–170. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, Perry VH & Gordon S (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience, 48, 405–415. [DOI] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C & Stevens B (2018) CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron, 100, 120–134.e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardo CC, Mendes M, Ahmad AS & Dore S (2015) Efficacy of prophylactic flavan-3-ol in permanent focal ischemia in 12-mo-old mice. Am J Physiol Heart Circ Physiol, 308, H583–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, Gulati G, Bennett ML, Sun LO, Clarke LE, Marschallinger J, Yu G, Quake SR, Wyss-Coray T & Barres BA (2019) Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron, 101, 207–223.e210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Chiou B, Gilman CK, Luo R, Koshi T, Yu D, Oak HC, Giera S, Johnson-Venkatesh E, Muthukumar AK, Stevens B, Umemori H & Piao X (2020) A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. Embo j, 39, e104136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Du XF, Liu CS, Wen ZL & Du JL (2012) Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental cell, 23, 1189–1202. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B & Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YU, Ying Y, Li Y, Eyo UB, Chen T, Zheng J, Umpierre AD, Zhu J, Bosco DB, Dong H & Wu LJ (2019) Neuronal network activity controls microglial process surveillance in awake mice via norepinephrine signaling. Nat Neurosci, 22, 1771–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery RL, Tremblay ME, Hopkins BE & Majewska AK (2017) The microglial fractalkine receptor is not required for activity- dependent plasticity in the mouse visual system. Glia, 65, 1744–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund H, Pieber M, Parsa R, Han J, Grommisch D, Ewing E, Kular L, Needhamsen M, Espinosa A, Nilsson E, Overby AK, Butovsky O, Jagodic M, Zhang XM & Harris RA (2018) Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat Commun, 9, 4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat M, Marin-Teva JL & Cheret C (2005) Phagocytosis in the developing CNS: more than clearing the corpses. Curr Opin Neurobiol, 15, 101–107. [DOI] [PubMed] [Google Scholar]
- Masuda T, Sankowski R, Staszewski O, Bottcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, Reinacher PC, Michel A, Sure U, Gold R, Grun D, Priller J, Stadelmann C & Prinz M (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature, 566, 388–392. [DOI] [PubMed] [Google Scholar]
- Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, Ben-Yehuda H, David E, Zelada Gonzalez F, Perrin P, Keren-Shaul H, Gury M, Lara-Astaiso D, Thaiss CA, Cohen M, Bahar Halpern K, Baruch K, Deczkowska A, Lorenzo-Vivas E, Itzkovitz S, Elinav E, Sieweke MH, Schwartz M & Amit I (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science, 353, aad8670. [DOI] [PubMed] [Google Scholar]
- McManus RM & Heneka MT (2017) Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther, 9, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes MS, Atlas J, Brehm Z, Ladron de Guevara Ruiz A, McCall M & Majewska AK (2020) In vivo imaging of the kinetics of microglial self-renewal and maturation in the adult visual cortex. BioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mildner A, Huang H, Radke J, Stenzel W & Priller J (2017) P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia, 65, 375–387. [DOI] [PubMed] [Google Scholar]
- Miron VE (2017) Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. Journal of leukocyte biology, 101, 1103–1108. [DOI] [PubMed] [Google Scholar]
- Miyamoto A, Wake H, Ishikawa AW, Eto K, Shibata K, Murakoshi H, Koizumi S, Moorhouse AJ, Yoshimura Y & Nabekura J (2016) Microglia contact induces synapse formation in developing somatosensory cortex. Nat Commun, 7, 12540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najafi AR, Crapser J, Jiang S, Ng W, Mortazavi A, West BL & Green KN (2018) A limited capacity for microglial repopulation in the adult brain. Glia, 66, 2385–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F & Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, Keulen Z, Pasillas MP, O’Connor C, Nickl CK, Schafer ST, Shen Z, Rissman RA, Brewer JB, Gosselin D, Gonda DD, Levy ML, Rosenfeld MG, McVicker G, Gage FH, Ren B & Glass CK (2019) Brain cell type-specific enhancer-promoter interactome maps and disease- risk association. Science, 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell J, Zeppenfeld D, McConnell E, Pena S & Nedergaard M (2012) Norepinephrine: a neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem Res, 37, 2496–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D & Gross CT (2011) Synaptic pruning by microglia is necessary for normal brain development. Science, 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR & Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell, 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistollato F, Sumalla Cano S, Elio I, Masias Vergara M, Giampieri F & Battino M (2016) Role of gut microbiota and nutrients in amyloid formation and pathogenesis of Alzheimer disease. Nutr Rev, 74, 624–634. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM & El Khoury J (2015) Microglia in Health and Disease. Cold Spring Harb Perspect Biol, 8, a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM & Perry VH (2009) Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol, 27, 119–145. [DOI] [PubMed] [Google Scholar]
- Reu P, Khosravi A, Bernard S, Mold JE, Salehpour M, Alkass K, Perl S, Tisdale J, Possnert G, Druid H & Frisen J (2017) The Lifespan and Turnover of Microglia in the Human Brain. Cell Rep, 20, 779–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RA, Pham J, Lee RJ, Najafi AR, West BL & Green KN (2017) Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia, 65, 931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice RA, Spangenberg EE, Yamate-Morgan H, Lee RJ, Arora RP, Hernandez MX, Tenner AJ, West BL & Green KN (2015) Elimination of Microglia Improves Functional Outcomes Following Extensive Neuronal Loss in the Hippocampus. J Neurosci, 35, 9977–9989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Escalera F, Matousek SB, Ghosh S, Olschowka JA & O’Banion MK (2014) Interleukin-1beta mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer’s disease. Neurobiol Dis, 69, 124–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankowski R, Bottcher C, Masuda T, Geirsdottir L, Sagar, Sindram E, Seredenina T, Muhs A, Scheiwe C, Shah MJ, Heiland DH, Schnell O, Grun D, Priller J & Prinz M (2019) Mapping microglia states in the human brain through the integration of high-dimensional techniques. Nat Neurosci, 22, 2098–2110. [DOI] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA & Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron, 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP & Stevens B (2010) Synapse elimination during development and disease: immune molecules take centre stage. Biochem Soc Trans, 38, 476–481. [DOI] [PubMed] [Google Scholar]
- Schafer DP & Stevens B (2013) Phagocytic glial cells: sculpting synaptic circuits in the developing nervous system. Curr Opin Neurobiol, 23, 1034–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schecter RW, Maher EE, Welsh CA, Stevens B, Erisir A & Bear MF (2017) Experience-Dependent Synaptic Plasticity in V1 Occurs without Microglial CX3CR1. J Neurosci, 37, 10541–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott-Hewitt N, Perrucci F, Morini R, Erreni M, Mahoney M, Witkowska A, Carey A, Faggiani E, Schuetz LT, Mason S, Tamborini M, Bizzotto M, Passoni L, Filipello F, Jahn R, Stevens B & Matteoli M (2020) Local externalization of phosphatidylserine mediates developmental synaptic pruning by microglia. Embo j, 39, e105380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan GK & Murphy KJ (2013) Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol, 3, 130181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B & Lemere CA (2017) Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC, Caldarone BJ, Stevens B & Lemere CA (2015) Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci, 35, 13029–13042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Beccari S, Diaz-Aparicio I, Encinas JM, Comeau S & Tremblay ME (2014) Surveillance, phagocytosis, and inflammation: how never-resting microglia influence adult hippocampal neurogenesis. Neural Plast, 2014, 610343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhvi A & Shaham S (2019) Glia-Neuron Interactions in Caenorhabditis elegans. Annu Rev Neurosci, 42, 149–168. [DOI] [PubMed] [Google Scholar]
- Sipe GO, Lowery RL, Tremblay ME, Kelly EA, Lamantia CE & Majewska AK (2016) Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nature communications, 7, 10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohet F, Lin C, Munji RN, Lee SY, Ruderisch N, Soung A, Arnold TD, Derugin N, Vexler ZS, Yen FT & Daneman R (2015) LSR/angulin-1 is a tricellular tight junction protein involved in blood- brain barrier formation. J Cell Biol, 208, 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL & Green KN (2016) Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain, 139, 1265–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spittau B (2017) Aging Microglia-Phenotypes, Functions and Implications for Age-Related Neurodegenerative Diseases. Front Aging Neurosci, 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F & Garel S (2014) Microglia modulate wiring of the embryonic forebrain. Cell Rep, 8, 1271–1279. [DOI] [PubMed] [Google Scholar]
- Stephan AH, Barres BA & Stevens B (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci, 35, 369–389. [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW & Barres BA (2007) The classical complement cascade mediates CNS synapse elimination. Cell, 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Stowell RD, Sipe GO, Dawes RP, Batchelor HN, Lordy KA, Whitelaw BS, Stoessel MB, Bidlack JM, Brown E, Sur M & Majewska AK (2019) Noradrenergic signaling in the wakeful state inhibits microglial surveillance and synaptic plasticity in the mouse visual cortex. Nat Neurosci, 22, 1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowell RD, Wong EL, Batchelor HN, Mendes MS, Lamantia CE, Whitelaw BS & Majewska AK (2018) Cerebellar microglia are dynamically unique and survey Purkinje neurons in vivo. Dev Neurobiol, 78, 627–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ, Mrak RE & Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation, 1, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LO & Barres BA (2016) Glia Get Neurons in Shape. Cell, 165, 775–776. [DOI] [PubMed] [Google Scholar]
- Swiatkowski P, Murugan M, Eyo UB, Wang Y, Rangaraju S, Oh SB & Wu LJ (2016) Activation of microglial P2Y12 receptor is required for outward potassium currents in response to neuronal injury. Neuroscience, 318, 22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka KF, Kashima H, Suzuki H, Ono K & Sawada M (2002) Existence of functional beta1- and beta2-adrenergic receptors on microglia. J Neurosci Res, 70, 232–237. [DOI] [PubMed] [Google Scholar]
- Tay TL, Mai D, Dautzenberg J, Fernandez-Klett F, Lin G, Sagar, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, Margineanu A, Sporbert A, Steinmetz LM, Pospisilik JA, Jung S, Priller J, Grun D, Ronneberger O & Prinz M (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nat Neurosci, 20, 793–803. [DOI] [PubMed] [Google Scholar]
- Tremblay ME, Lowery RL & Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol, 8, e1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvel NH, Grathwohl SA, Baumann F, Liebig C, Bosch A, Brawek B, Thal DR, Charo IF, Heppner FL, Aguzzi A, Garaschuk O, Ransohoff RM & Jucker M (2012) Microglial repopulation model reveals a robust homeostatic process for replacing CNS myeloid cells. Proc Natl Acad Sci U S A, 109, 18150–18155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, Bendlin BB & Rey FE (2017) Gut microbiome alterations in Alzheimer’s disease. Sci Rep, 7, 13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waisman A, Ginhoux F, Greter M & Bruttger J (2015) Homeostasis of Microglia in the Adult Brain: Review of Novel Microglia Depletion Systems. Trends Immunol, 36, 625–636. [DOI] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S & Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci, 29, 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Tan MS, Yu JT & Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med, 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, Barrow AD, Diamond MS & Colonna M (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol, 13, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, Cella M, Grutzendler J, DeMattos RB, Cirrito JR, Holtzman DM & Colonna M (2016) TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med, 213, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG & Tenner AJ (2000) Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol, 161, 127–138. [DOI] [PubMed] [Google Scholar]
- Wu S, Xue R, Hassan S, Nguyen TML, Wang T, Pan H, Xu J, Liu Q, Zhang W & Wen Z (2018) Il34-Csf1r Pathway Regulates the Migration and Colonization of Microglial Precursors. Dev Cell, 46, 552–563.e554. [DOI] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA & Stevens B (2015) Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol, 36, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Fan L, Kodama L, Sohn PD, Wong MY, Mousa GA, Zhou Y, Li Y & Gan L (2020) A MAC2-positive progenitor-like microglial population is resistant to CSF1R inhibition in adult mouse brain. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan L, Krabbe G, Du F, Jones I, Reichert MC, Telpoukhovskaia M, Kodama L, Wang C, Cho SH, Sayed F, Li Y, Le D, Zhou Y, Shen Y, West B & Gan L (2019) Proximal recolonization by self-renewing microglia re-establishes microglial homeostasis in the adult mouse brain. PLoS Biol, 17, e3000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Zhao L, Wang X, Ma W, Lazere A, Qian HH, Zhang J, Abu-Asab M, Fariss RN, Roger JE & Wong WT (2018) Repopulating retinal microglia restore endogenous organization and function under CX3CL1-CX3CR1 regulation. Sci Adv, 4, eaap8492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Hu W, Tsai J, Li W & Gan WB (2017) Microglia limit the expansion of beta-amyloid plaques in a mouse model of Alzheimer’s disease. Mol Neurodegener, 12, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Pina-Crespo JC, Zhang M, Zhang N, Chen X, Bu G, An Z, Huang TY & Xu H (2018) TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron, 97, 1023–1031 e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]