

Abstract
As non-“self” macromolecules, biotherapeutics can trigger an immune response that can reduce drug efficacy, require patients to be taken off therapy, or even cause life-threatening reactions. To enable flexible and facile design of protein biotherapeutics while reducing the prevalence of T cell epitopes that drive immune recognition, we have integrated into the Rosetta protein design suite a new scoring term that allows design protocols to account for predicted or experimentally-identified epitopes in the optimized objective function. This flexible scoring term can be used in any Rosetta design trajectory, can be targeted to specific regions of a protein, and can be readily extended to work with a variety of epitope predictors. By performing extensive design runs with varied design parameter choices for three case study proteins as well as a larger diverse benchmark, we show that the incorporation of this scoring term enables effective exploration of an alternative, deimmunized sequence space to discover diverse proteins that are potentially highly deimmunized while retaining physical and chemical qualities similar to those yielded by equivalent non-deimmunizing sequence design protocols.
INTRODUCTION
In recent years, there has been a large growth in the use of therapeutic proteins to treat a wide range of diseases,1,2 including cancer,3–6 autoimmune disorders and rheumatoid arthritis,7–11 hematological disorders,12,13 and others. While this growth has demonstrated the utility and versatility of biotherapeutics when compared to conventional small molecule therapeutics, it has also highlighted a major drawback in using macromolecules as pharmaceutical agents: the recognition of macromolecules by the immune system can frequently lead to an anti-drug response that can neutralize the therapeutic molecule or even lead to life-threatening allergic reactions.14 In order to overcome this limitation, a critical area of research has been the development of methods to deimmunize potential therapeutics early in the drug development process by identifying and eliminating the immunogenic epitopes that drive detrimental immune responses.15–25 In addition to the development of animal models of the human immune system17,23,26 and sensitive ex vivo immunoassays to assess immunogenicity across a diverse panel of human donors,16,18,23,27 computational methods that can both identify15,28,29 and remove15,18,30,31 immunogenic epitopes have become important tools in the therapeutic design process.
The immune response against an exogenous protein such as a biotherapeutic begins when it is taken up by professional antigen presenting cells (APCs), which express class II major histocompatibility complex (MHC-II) proteins. In the APCs, the exogenous protein is proteolyzed into short peptides of about 13–25 residues.32,33 Those peptides that are bound by MHC-II, with a 9-residue “core” peptide engaging MHC-II’s peptide binding cleft, are transported to the cell’s surface for inspection by T cells. If a cognate T cell receptor recognizes the presented peptide-MHC complex, the T cell can be activated against the non-“self” protein and can give help to B cells that are also primed against the protein, thereby enabling the maturation of high-affinity class-switched anti-biotherapeutic antibodies.34 A rational approach to protein deimmunization is thus to identify peptides in the therapeutic that have a high propensity for binding MHC-II and to introduce mutations therein that reduce this binding. MHC-II is genetically encoded, and the peptide-binding propensities manifested across human allelic diversity are highly degenerate, with a relatively small number of alleles sufficient to represent recognition patterns across most of the human population.35–37 This highly predictable upstream bottleneck thus provides a readily engineerable target by which to achieve deimmunization.
To realize this rational deimmunization approach, it is necessary to predict which peptides are likely to be recognized by MHC-II and which mutations may reduce this binding while maintaining protein stability and therapeutic activity. The last twenty years have seen improvements in the number and sophistication of epitope prediction tools,15,28,29 including those leveraging immunopeptidomics data,38,39 as well as the curation of an increasingly large number of epitope data supporting better training of these tools.40 At the same time, general protein design strategies have matured, and synthetic biology approaches have greatly decreased in cost and accessibility, making rational protein design campaigns a reality.41–43 These advances have reinforced the need to seamlessly integrate computationally-driven deimmunization in the design, development, and engineering of all manner of protein therapeutics. Computational protein design methods that selectively explore the non-immunogenic sequence space compatible with a desired therapeutic function could preemptively mitigate the future risk of adverse immune responses, while side-stepping the need for time-consuming and labor-intensive experimentally-driven efforts. Computational protein design methodologies have been previously developed30,31 and successfully applied18,23 to a number of therapeutics; however, these platforms are focused specifically on mutagenic deimmunization, while a fully featured computational design platform would support a highly diverse set of design tasks in which accounting for immunogenicity is one of many goals to be achieved.
Here, we present a state-of-the-art method for computationally-driven deimmunization implemented by the integration of epitope prediction methods into the premiere protein design suite, Rosetta.41 Rosetta performs sequence design and side-chain rotamer optimization using a Monte Carlo-based module called the “packer” to find a set of amino acid types and side-chain conformations minimizing an objective function (typically the Rosetta energy function). Because the Rosetta energy function is a weighted sum of scoring terms that can be calculated independently, this function can be augmented with additional non-energetic scoring terms to guide design towards some additional objective (multi-objective optimization). We have integrated deimmunization as an additional objective during design by implementing a new scoring term, mhc_epitope, within Rosetta. While epitope prediction methods have previously been used in Rosetta,15,44 these methods either were never integrated into the publicly available Rosetta suite44, or they relied on specialized protocols preventing multi-objective sequence optimization and limiting their general applicability.15 Since the inclusion of mhc_epitope allows deimmunization to be performed using the packer, it can be easily incorporated into any existing protein design pipeline. In addition, the modular organization of our code allows mhc_epitope to work with multiple epitope predictors and promises easy integration of newly developed predictors in the future. At the time of writing, mhc_epitope enables straightforward deimmunization using the ProPred predictor,28 and also provides a means to integrate with state-of-the-art command-line and web based predictors, such as NetMHCII,29 using pre-computed databases of immunogenic peptide predictions. Experimental data from the Immune Epitope Database40 can likewise be incorporated to guide designs away from experimentally validated epitopes. Finally, Rosetta’s existing SVM-based predictor,15 “nmer,” has been integrated into mhc_epitope, allowing it to also be used with the Rosetta packer in any design context.
The mhc_epitope scoring term will greatly simplify computational deimmunization campaigns by combining Rosetta’s ability to design stable, functional protein variants with a variety of epitope predictors in a single step. We have benchmarked mhc_epitope in order to identify some recommended settings for typical protein deimmunization projects, highlighting the goal of achieving deimmunized, native-like designs with similar quality metrics to equivalent Rosetta designs without deimmunization. While we have focused on MHC-II for the common context of extracellular biotherapeutics, the framework can be readily adapted to support MHC-I for intracellular proteins (e.g., in gene therapy).45,46 We also expect that de novo protein design projects undertaken using Rosetta will be able to use mhc_epitope to avoid immunogenic designs in the first place.
METHODS
Implementation of MHCEpitopeEnergy.
Physics-based and nonphysical guidance scoring terms within Rosetta are computed internally by modules called “energy methods.” We have developed a new energy method in Rosetta called “MHCEpitopeEnergy” that allows a user to incorporate epitope prediction in any Rosetta design strategy through a scoring term called “mhc_epitope.” As illustrated in Figure 1A, the process considers all constituent peptides within a protein, evaluating each separately for its propensity to bind and/or be displayed by MHC. Since it remains difficult to predict how peptides will be proteolytically processed before loading onto MHC,39 we follow the approach of most epitope predictors by considering every fixed-length peptide fragment, typically either 9 residues (the core unit binding MHC) or 15 residues (allowing for N- and C-terminal “overhangs”) in length using a “sliding window” approach (Figure 1A). Each peptide is passed to one or more epitope predictors (subclasses of class MHCEpitopePredictor), which score the epitope’s propensity to bind and/or be presented. The sum of scores for all predictors over all peptides then represents the penalty added to the Rosetta energy for a particular protein sequence.
Figure 1.
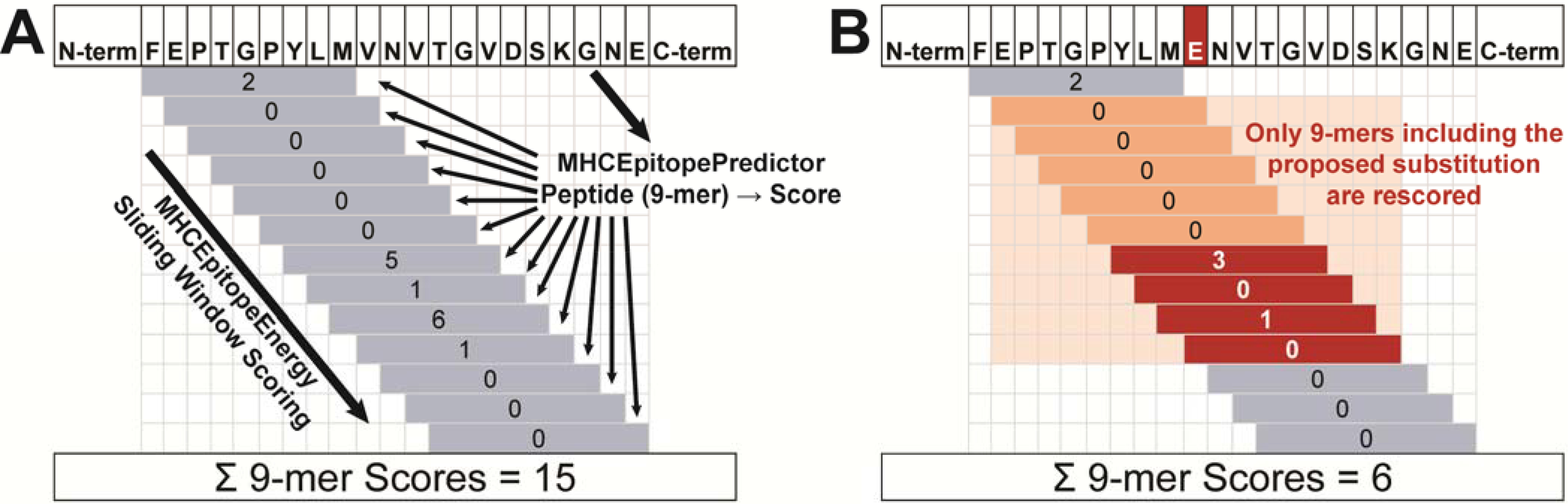
MHCEpitopeEnergy scoring overview. (A) In initial scoring, a sliding window approach enumerates peptide sequences of a specified length and passes them to an MHCEpitopePredictor, which scores each peptide. The scores are cached and summed to get the total mhc_epitope score. (B) During packing, when an amino acid substitution is considered (red “E” in sequence), the peptides that include that residue (orange shading) are re-scored. Scores that change (red) are updated in the cache, as is the total mhc_epitope score.
Because this evaluation is not pairwise-decomposable, we take advantage of the new “design-centric guidance scoring term” framework47–49 in Rosetta to evaluate the immunogenicity penalty as a function of the entire protein sequence during sequence design using the packer. When the mhc_epitope scoring term is set to a non-zero weight, this activates the MHCEpitopeEnergy energy method, which in turn evaluates a scoring penalty based on the MHCEpitopePredictor class selected by the user.
During a Rosetta design simulation (i.e., during a packing trajectory), when the packer considers an amino acid substitution at a particular position, only those peptides whose sequences change as a result of the substitution are considered for evaluating the change in the immunogenicity score. Note that this approach depends only on the amino acid sequence, and thus changes in rotamer packing that preserve amino acid identity do not affect the score. To implement efficient re-scoring, a cache of scores is maintained, one for each peptide for each predictor. Peptides affected by the proposed substitution are re-scored and the cache is updated (Figure 1B). The total mhc_epitope score is updated by the difference between the former and current scores for the affected peptides, allowing scoring to proceed with only a modest slowdown in computation time compared to a normal Rosetta rotamer packing run (roughly 10–20% with mid-sized proteins and the ProPred Predictor). As mhc_epitope scores can only change with a sequence change, the energy method is disabled during gradient-based Rosetta energy minimization, which affects conformation only.
Since MHCEpitopeEnergy is evaluated using a packer-compatible scoring term, it can in principle be integrated into any design calculation available in the Rosetta design suite by using a custom scoring function. The scoring term is compatible with symmetric structures that are used to model homo-oligomeric proteins and with structures that contain non-protein residues, such as DNA, RNA, and small molecule ligands. Proteins containing non-canonical amino acids are also supported, but since epitope predictors typically are not designed to score peptides containing these amino acids, they do not to contribute the mhc_epitope score.
The different methods for identifying epitope sequences are implemented as separate subclasses of the MHCEpitopePredictor class to allow straightforward extension of this method. Any algorithm that takes a fixed length peptide sequence and returns a score can be readily implemented as an MHCEpitopePredictor, with the only other requirement being to create a user-facing configuration interface in the MHCEpitopeEnergySetup class.
We have currently implemented three main Predictors. Here, we present results using the ProPred28 Predictor and using NetMHCII29 by way of the database-based External/PreLoaded Predictors. The former is an older, but very rapid, matrix-based means of evaluating peptide immunogenicity. The latter uses a pre-computed database of individual peptide scores, which can be calculated using advanced epitope prediction methods, including NetMHCII29 as presented here. Predictors like NetMHCII are too slow to be used to evaluate peptide scores during a packing trajectory, thus necessitating pre-computed databases. Likewise, any command-line or web-based predictor could be used to generate a database of scores to use during design. Additionally, External/PreLoaded Predictors allow the scoring and thereby deletion of experimentally-validated epitopes from the Immune Epitope Database (IEDB, https://www.iedb.org/),40 and we provide tools to manage the importing of such data. In addition to these Predictors, we have also implemented a support vector machine-based Predictor that utilizes the “nmer” scoring term; this epitope prediction method was already implemented in Rosetta,15 but was not packer-compatible and thus could not be used during a standard design calculation prior to its integration with MHCEpitopeEnergy.
Usage and Configuration.
We have provided detailed documentation for the use and configuration of mhc_epitope on the Rosetta Commons documentation site (https://www.rosettacommons.org/docs/latest/rosetta_basics/scoring/MHCEpitopeEnergy). In brief, there are two requirements to enable deimmunization in a design trajectory: (1) the mhc_epitope scoring term must be set to a non-zero weight in the Rosetta scoring function used during design, and (2) a “.mhc” configuration file must be provided. This file is used to indicate which Predictor should be used, and to set various options for the Predictor. We have ensured that the mhc_epitope scoring term is fully configurable in both the RosettaScripts50 XML and the PyRosetta51 Python user interfaces for Rosetta. In RosettaScripts,50 the weight and configuration file can be set directly in the <SCOREFXNS> block. In PyRosetta,51 the settings can either be set by passing the configuration command line flags to the init() function, or by using the EnergyMethodOptions.set_mhc_epitope_setup_files() function. For other Rosetta applications, the specific program’s documentation should be consulted for scoring function customization.
In order to accommodate cases in which a user only wants to deimmunize part of a structure (e.g. a binding protein being designed in complex with its target, where deimmunization of only the binding protein is desired), or in which a user would like to deimmunize with multiple Predictors/configurations simultaneously, we have implemented a Rosetta “mover” (a type of Rosetta module that modifies a structure) that adds MHCEpitopeEnergy constraints to a structure (AddMHCEpitopeConstraintMover, documented at https://www.rosettacommons.org/docs/latest/scripting_documentation/RosettaScripts/Movers/movers_pages/AddMHCEpitopeConstraintMover). The additional Predictors/configurations added by this mover work alongside the “global” configuration provided to the scoring function, if any. If the constraint is added using a residue selector, only the selected portion of the structure will be deimmunized with that configuration. Note that a scoring function with a non-zero mhc_epitope weight must be used during design for the constraint to have an effect on a design trajectory. The scoring term’s weight is combined multiplicatively with a weight that is set in the constraint mover.
We created a base deimmunization protocol, implemented as a Rosetta Scripts50 XML script, which can be can be found at rosetta_scripts_scripts/scripts/public/protein_design/deimmunization/mhc_epitope_design.xml in the RosettaCommons rosetta_scripts_scripts GitHub repository (initial commit: 931b264). The repository can be found in public releases of Rosetta at Rosetta/main/rosetta_scripts_scripts. This protocol performs three cycles of all-residue fixed-backbone design followed by minimization, and can be varied by modifying the scoring function and its settings, as well as by applying a “resfile” (a packer configuration file) to alter design space sampled by Rosetta. All design trajectories apply a “FavorNative” penalty to avoid spurious mutations: each residue that is mutated from its native identity will result in a penalty of 1.5 Rosetta energy units. At the end of the protocol, a number of metrics are evaluated to be used as indicators of design quality.
Applications and Benchmarking.
We applied our protocol to three sets of targets: (1) three “case study” proteins that we use to establish reasonable parameters for protein deimmunization, (2) a benchmark set of 54 diverse, soluble proteins that was previously used in a similar study,44 and (3) a set of three targets that have been previously deimmunized and experimentally validated. Prior to deimmunizing the target proteins, solvent molecules were removed from the structure which was then idealized in Rosetta using the FastRelax protocol, the REF2015 scoring function,52 and backbone coordinate constraints. We then applied our base deimmunization protocol, as described above. The various settings that we evaluated throughout the study are summarized in Table S1. In addition to running the protocol as described, we also ran the equivalent protocol with the mhc_epitope weight set to 0 in order to allow for a direct comparison of design quality. For each of the protocol settings that were tested, 1000, 240, and 500 decoys were generated for the case study, benchmark, and experimentally validated protein targets, respectively.
Following deimmunization, all designs were re-scored using ProPred28 (Southwood allele set35) and NetMHCII29 (Paul allele set37) using the mhc_score.py companion script available in the Rosetta tools repository (located in Rosetta/main/tools by default) in the mhc_energy_tools subdirectory.
In order to restrict the design space sampled by Rosetta based on evolutionary constraints, we generated a position-specific scoring matrix (PSSM) for the sequences of all target proteins using three iterations of PSI-BLAST.53 From these PSSMs, we generated resfiles that restricted design space to permit only the native residue plus any residue that has a base 2 log-odds scores in the PSSM above a threshold value; we tested thresholds of 1, 2, or 3. The resfiles were generated using the mhc_gen_db.py companion script available in the Rosetta tools repository under mhc_energy_tools.
In order to evaluate the performance of MHCEpitopeEnergy using NetMHCII as a Predictor, we enumerated all 15mer peptides allowable for our three case study proteins, when restricting allowed amino acids to those with base-2 log-odds scores of 3 or greater in the PSSM. We generated a pre-computed database by scoring these peptides with NetMHCII29 (Paul37 allele set).
RESULTS & DISCUSSION
Having implemented a scoring term enabling prediction and deletion of epitopes as part of Rosetta-based protein design, we wanted to investigate the impact of deimmunization parameters on the resulting designs and ultimately establish a “best practices” protocol for protein deimmunization using MHCEpitopeEnergy. In particular, we sought to establish design parameters in which the predicted immunogenicity of the target is reduced while minimally perturbing other metrics of design quality, as compared to equivalent design trajectories performed without deimmunization.
In the following results, we first thoroughly assess the effects of design parameters on deimmunization of three case study proteins, evaluating if and how the settings affect resulting design quality. We then apply inferred “best practices” settings to deimmunize a larger benchmark set of 54 targets, investigating the general trends of deimmunization and design quality over these diverse proteins. We next assess the impact of the epitope predictor choice, in particular evaluating the extent to which deimmunizing for one predictor achieves deimmunization for another. Finally, in order to explore the diversity of “deimmunized sequence space” around a target, we compare deimmunized designs emerging from this protocol with some deimmunized variants that have previously been experimentally evaluated.
MHCEpitopeEnergy Case Studies.
Mirroring the approach used in a previous study,44 we investigated various strategies for deimmunizing proteins with three representative proteins: human erythropoietin (PDB ID 1EER, 166 amino acids),54 staphylokinase (PDB ID 2SAK, 121 amino acids),55 and a designed hemagglutinin binding protein (PDB ID 3R2X, 93 amino acids).56 To evaluate design quality, in addition to Rosetta’s REF2015 score and packing quality (“PackStat”)57 score, we also assess changes in the number of buried unsatisfied hydrogen bonds and in the number of charged residues (since a preference for negatively charged residues over hydrophobic residues has frequently been observed with various epitope predictors).15,29,44 We also monitor sequence recovery, since deimmunizing a protein with a minimal number of mutations should result in more functional and stable designs.
Since the Rosetta energy function is a sum of independent terms combined linearly, each weighted by a constant coefficient, we first explored the effect of varying the weighting coefficient for the mhc_epitope scoring term. Since introducing charged residues is an effective means to reduce MHC-II binding, we then evaluated our ability to avoid over-charging the protein by separately restricting the number of positively- and negatively-charged residues. Finally, to utilize more conservative mutations in deimmunization design (which can indirectly constrain charged residues), we investigated reducing design space with an evolutionary-derived position-specific scoring matrix (PSSM).
mhc_epitope weight.
First, we considered an ideal weight for the mhc_epitope scoring term itself (Table S1, protocol 1) and monitored its effect on the key metrics described above (Figure 2, Figure S1, and Figure S2). As expected for a multi-objective optimization problem, higher mhc_epitope weights sacrificed some predicted thermodynamic stability, as measured by the REF2015 score (A panels), in order to achieve decreased epitope scores (B panels), reflecting that the designs were deimmunized according to the ProPred predictor. Almost all designs (with or without a nonzero mhc_epitope weight) still yielded better REF2015 scores than relaxation of the wild-type (with no design), and the differences among the designs’ scores were generally relatively modest compared to the differences from the wild-type score. We noted that using an mhc_epitope weight of less than 0.5 was largely ineffective at reducing the unweighted ProPred score. Conversely, weights greater than 1.5 yielded diminishing returns with respect to the ProPred score while negatively impacting the REF2015 score. While the PackStat score (C panels), the number of buried unsatisfied hydrogen bonds (D panels), and the number of positive charges (E panels) remained largely unchanged, we did note a considerable increase in the number of negative charges (F panels) as the mhc_epitope weight increased. We concluded that for most applications using the ProPred predictor, an mhc_weight of between 0.5–1.5 would be appropriate and decided to use a weight of 1.0 for subsequent analyses. Deimmunization problems that require more aggressive deimmunization at the expense of stability may warrant a higher weight, whereas problems in which minimally impacting the protein’s stability is critical may consider a smaller weight. It should also be noted that the raw mhc_epitope score can scale with the number of alleles, so a Predictor that uses many alleles may require a smaller mhc_epitope weight, and vice versa.
Figure 2.
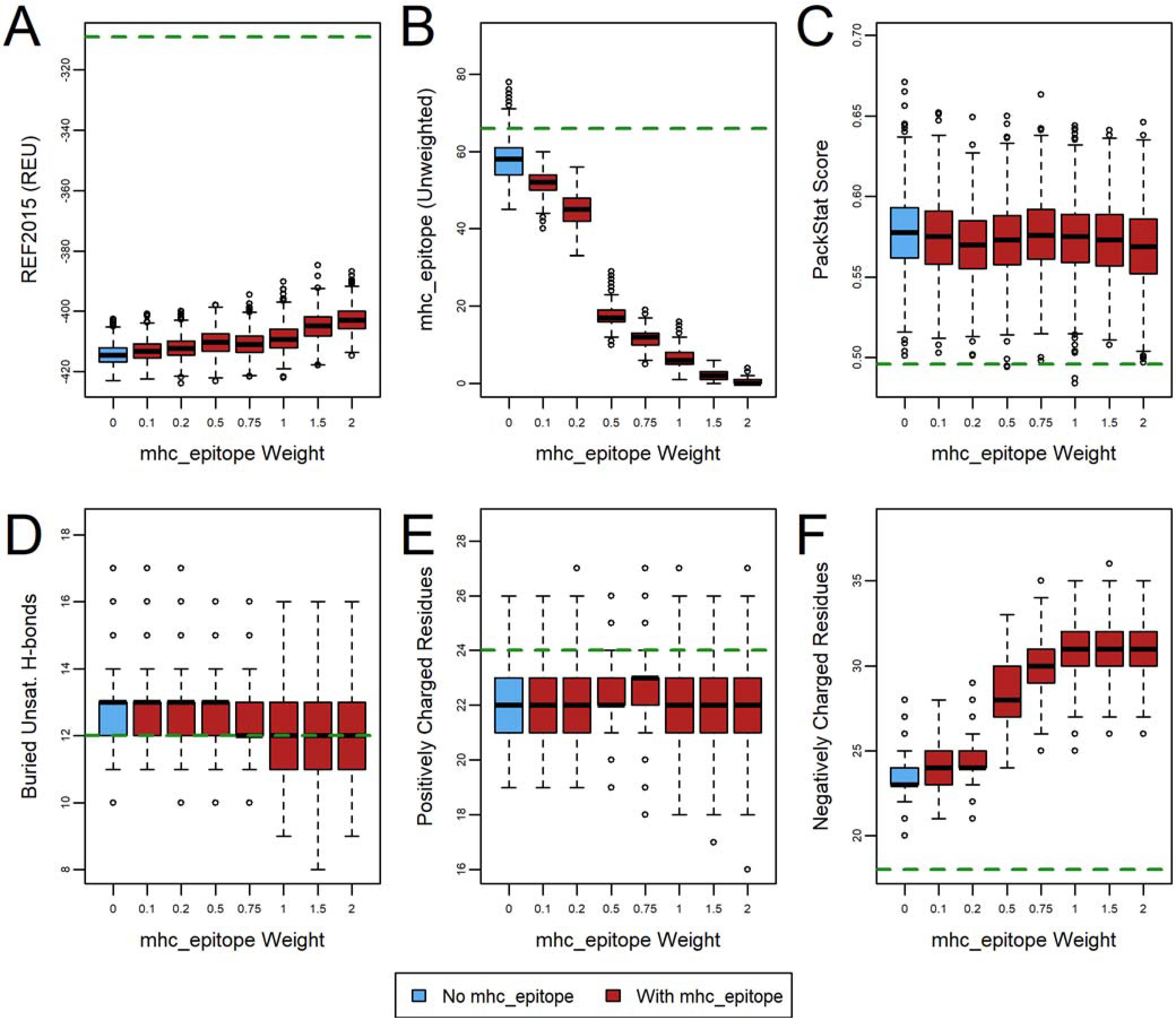
The effect of mhc_epitope weight on deimmunization of human erythropoietin (PDB ID 1EER). Designs (n=1000) were generated with fixed backbone Rosetta design with varying mhc_epitope weights, and the effect on six metrics evaluated: (A) REF2015 score, (B) unweighted mhc_epitope score, (C) PackStat score, and the number of (D) buried unsatisfied hydrogen bonds, (E) positively charged residues, and (F) negatively charged residues. Weights of 3.0, 5.0, and 10.0 were also tested, but were similar to a weight of 2.0 and are thus omitted for clarity. The green dashed line indicates the wild-type value (relaxed, but with no design). See also Figure S1 and Figure S2 for corresponding plots for other protein targets.
Restricting Changes in Protein Charge.
For several common MHC alleles, bound peptides tend to have few negatively charged residues, and thus many epitope predictors, including ProPred, tend to predict peptides that include negatively charged residues to be non-immunogenic. Consequently, unrestricted deimmunization has previously been seen to introduce many negatively charged residues and increase the number of charged residues over non-polar residues,15,29,44 an effect we observed here as well (Figure 2, Figure S1, and Figure S2, F panels). Rosetta has another design guidance scoring term, called aa_composition,47 that allows the amino acid composition to be restricted, so we investigated the impact of using this term alongside mhc_epitope on the number of charges.
To avoid simply introducing balanced charge-changing mutations, we chose to restrain the total number of positively and negatively charged residues individually, rather than the net charge (Table S1, protocol 2). We configured the aa_composition guidance scoring term so that, if the number of charged residues of a particular type increased or decreased by three residues or fewer, the unweighted penalty was equal to the change in the number of charges (i.e., an increase of two negatively charged residues would result in a penalty of 2). If the change was four or more, the penalty increased quadratically (Figure 3, Figure S3, and Figure S4). In this case, there was a slight deterioration in the REF2015 and ProPred scores, though these were largely within the spread of scores seen in the absence of any composition constraints (i.e., weight 0). As expected, with weight 0.5 or 1.0, the number of charges of each type were maintained at levels much closer to wild-type, while PackStat and the number of buried unsatisfied hydrogen bonds were largely unchanged. These results suggest that protein sequence space is large enough to accommodate epitope deletion while maintaining the overall charge composition of the native protein.
Figure 3.
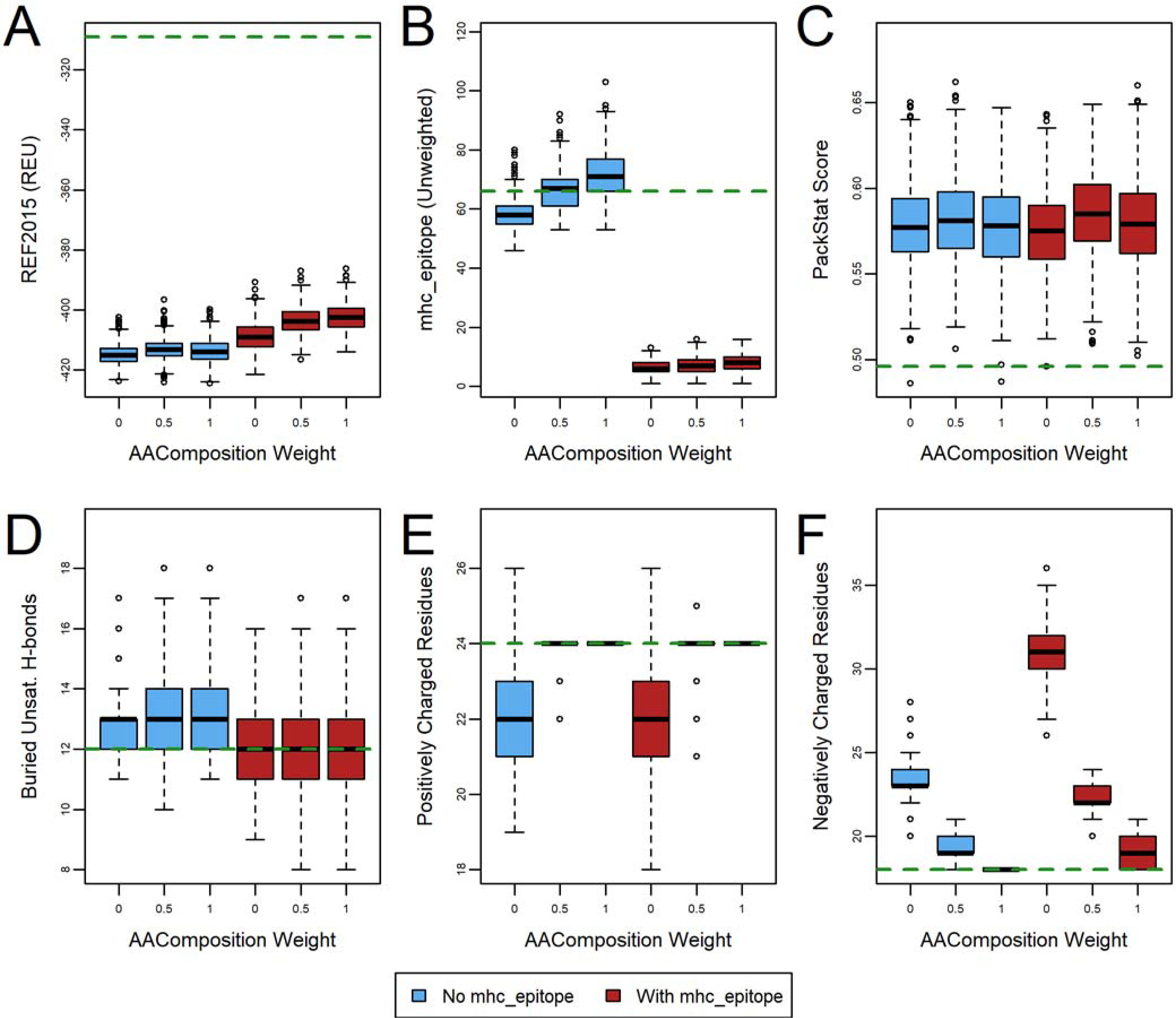
The effect of charged-based amino acid composition weight on deimmunization of human erythropoietin (PDB ID 1EER). Designs (n=1000) were generated with (red) and without (blue) an mhc_epitope weight of 1.0 turned on. The green dashed line indicates the wild-type value (relaxed, but with no design). The effect on six metrics is evaluated: (A) REF2015 score, (B) unweighted mhc_epitope score, (C) PackStat score, and the number of (D) buried unsatisfied hydrogen bonds, (E) positively charged residues, and (F) negatively charged residues. See also Figure S3 and Figure S4 for corresponding plots for other protein targets.
Evolutionary Conservation Constraint using a PSSM.
One strategy to minimally disrupt a protein’s function and stability during design is to restrict design space by allowing only substitutions that are observed in the sequence record.58 This strategy fortuitously also greatly reduces computation time. We restricted design space to include only the native residue plus any residue that has a base 2 log-odds scores in the PSSM above a threshold, and we explored thresholds of 1, 2, or 3 (Figure 4, Figure S5, and Figure S6, Table S1, protocol 3). Note that residue types that are not included in the allowed design space are not sampled by Rosetta at all, as opposed to being given a penalty during scoring. As expected, restricting design space using the PSSM leads to a smaller improvement in the REF2015 score than unrestricted design. In the case of benchmark protein 2SAK, the most stringent PSSM settings (thresholds of 2 or 3) combined with turning on ProPred deimmunization resulted in a moderate deterioration in REF2015 compared to wild-type, though a more moderate threshold of 1 resulted in scores that were comparable to the native score and similar to the scores obtained without deimmunization. Likewise, the mhc_epitope scores were slightly worse when PSSM-based design restrictions were used, though in all cases substantially better than the wild-type. The other metrics (PackStat score, buried unsatisfied hydrogen bonds, and number of charge residues) appeared to show only slight variation when applying PSSM restrictions that were not consistent from protein to protein. These also did not generally show large differences when comparing designs with and without deimmunization turned on. These results suggest that while the improvement in REF2015 over wild-type scores is somewhat diminished as compared to design without PSSM restrictions, orthogonal metrics that we are not specifically optimizing behave comparably. Since using evolutionary information to guide design is generally seen to improve the likelihood of obtaining stable, functional designs,59 the slightly worse REF2015 and mhc_epitope scores are likely an acceptable trade-off for many applications.
Figure 4.
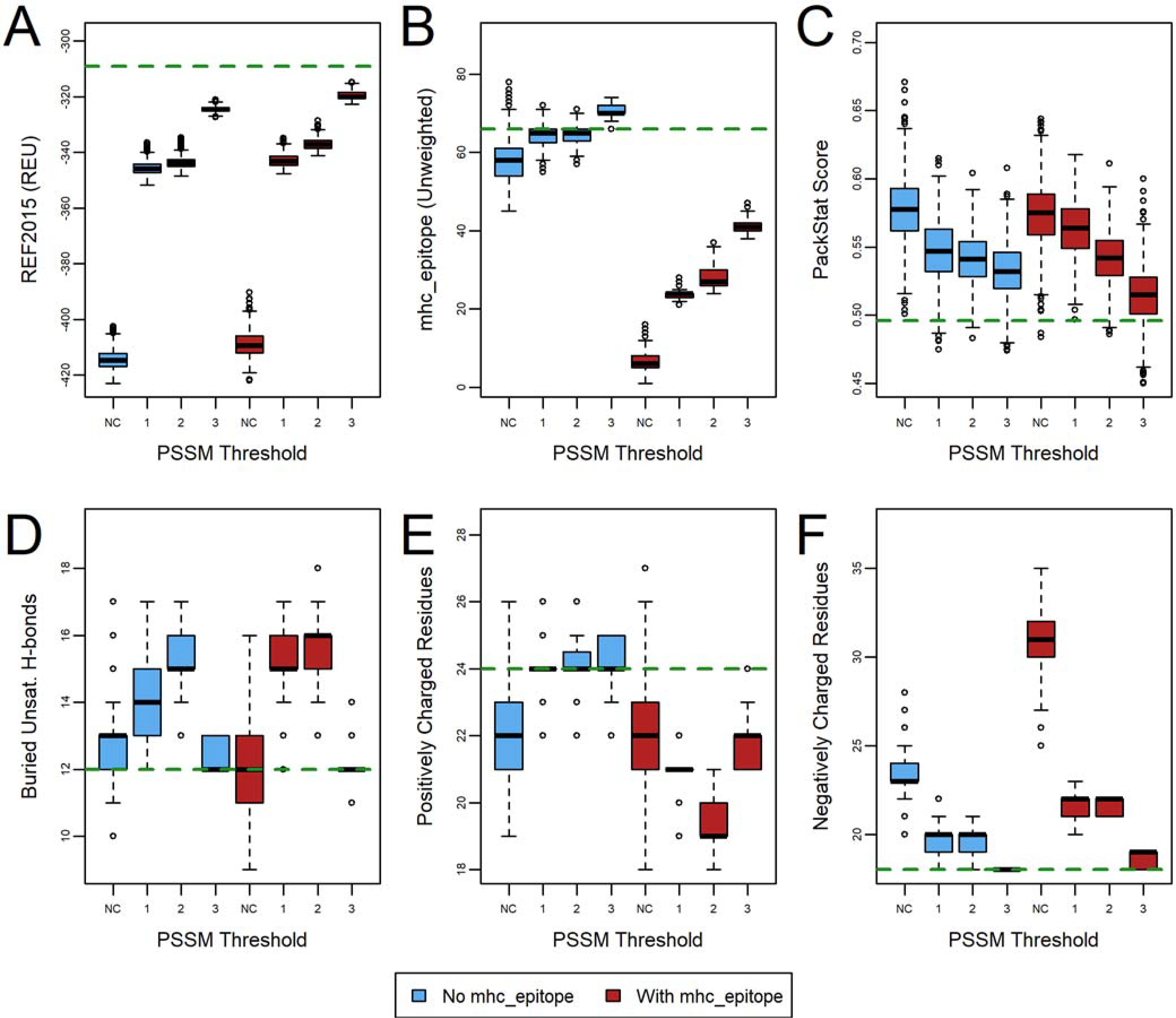
The effect of PSSM-based design restrictions on deimmunization of human erythropoietin (PDB ID 1EER). Designs (n=1000) were generated with (red) and without (blue) an mhc_epitope weight of 1.0 turned on after restricting designable residue identities to those with a base 2 log-odds score of 1, 2, or 3, or with no constraints (NC). The green dashed line indicates the wild-type value (relaxed, but with no design). The effect on six metrics is evaluated: (A) REF2015 score, (B) unweighted mhc_epitope score, (C) PackStat score, and the number of (D) buried unsatisfied hydrogen bonds, (E) positively charged residues, and (F) negatively charged residues. See also Figure S5 and Figure S6 for corresponding plots for other protein targets.
Combining charge constraints and PSSM-based design restrictions.
To further investigate the effects of the PSSM-based design restrictions, we combined the charge constraints with the PSSM restrictions (Figure 5, Figure S7, and Figure S8, Table S1, protocol 4). Interestingly, the only metric that showed substantial difference was the smaller variation in the number of positive and negative charges (panels E and F). The energy, deimmunization, packing quality, and number of buried unsatisfied hydrogen bonds (panels A-D) tended to show very similar trends regardless of the use of the charge constraints. It is also worth noting that the effect of the charge constraints was itself more muted than in the absence of PSSM-based restrictions, suggesting that a degree of charge constraint is embedded in the PSSM restrictions. As evolution is likely to show a tendency to maintain charged positions in a protein, it is somewhat unsurprising that the PSSM restrictions and charge constraints have similar effects on charge composition.
Figure 5.
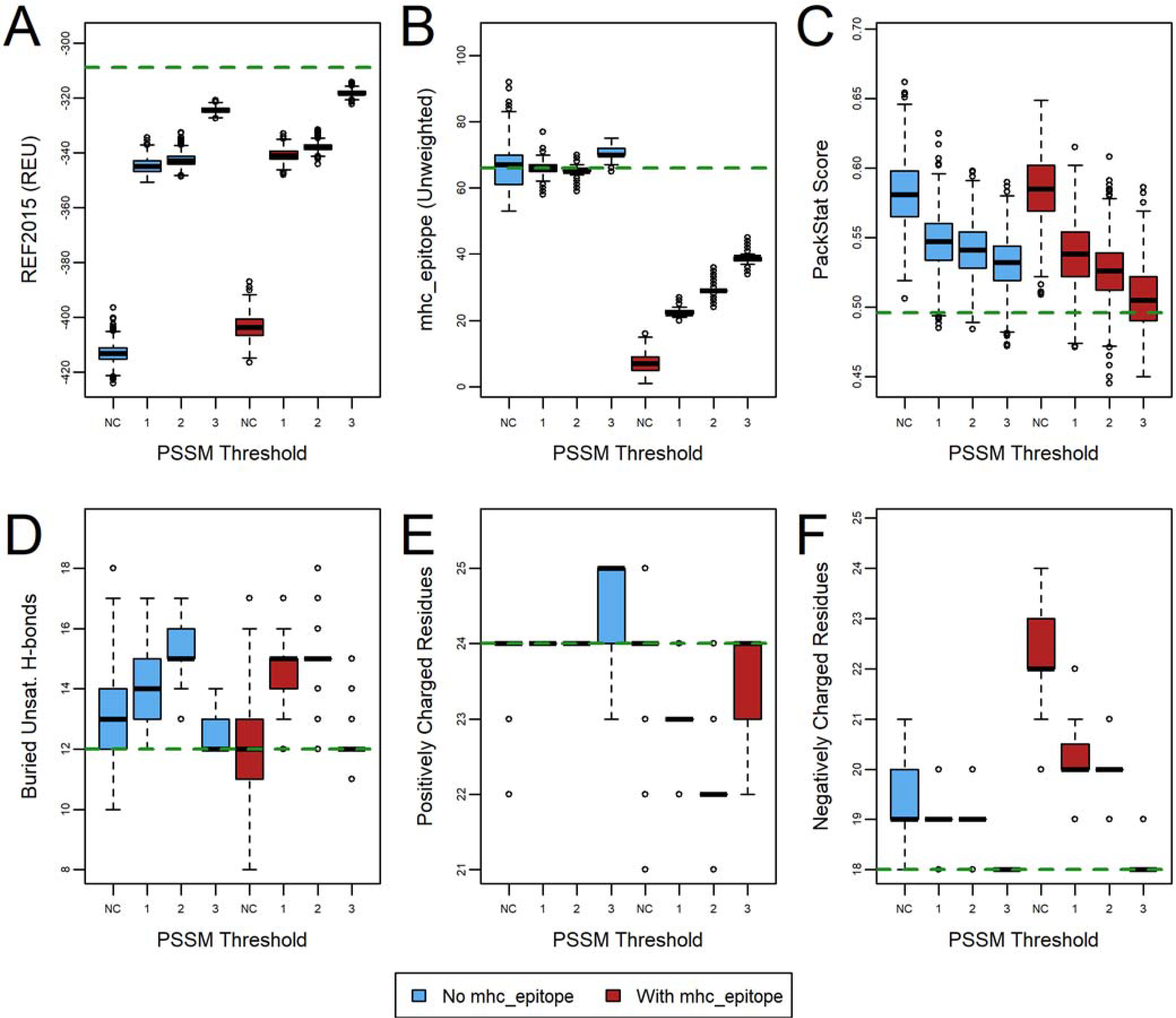
The effect of combined charged-based amino acid constraints and PSSM restrictions on deimmunization of human erythropoietin (PDB ID 1EER). Designs (n=1000) were generated with an mhc_epitope weight of 1.0 (red) and 0.0 (blue), and with an amino acid composition constraint weight of 0.5, after restricting designable residue identities to those with a base 2 log-odds score of 1, 2, or 3, or with no constraints (NC). The green dashed line indicates the wild-type value (relaxed, but with no design). The effect on six metrics is evaluated: (A) REF2015 score, (B) unweighted mhc_epitope score, (C) PackStat score, and the number of (D) buried unsatisfied hydrogen bonds, (E) positively charged residues, and (F) negatively charged residues. See also Figure S7 and Figure S8 for corresponding plots for other protein targets.
When examining the sequence recovery for these designs, we noted that the use of the mhc_epitope scoring term resulted in only a small decrease in sequence recovery as compared to the use of identical design parameters except mhc_epitope (Figure S9). It should also be noted that these sequence recovery values were quite high. We can thus conclude that while deimmunization using ProPred does increase the number of mutations, the difference is small when compared to the equivalent, non-deimmunizing design trajectories.
MHCEpitopeEnergy Benchmark and Scientific Test.
It is clear that the specific configuration to be used for protein deimmunization needs to be tailored to the specific goals of the design problem. Having said that, the case studies suggest settings that are an appropriate starting point for a “naïve” deimmunization problem: a moderate mhc_epitope weight of 1.0, restraining the number of positive and negative charges to near wild-type values, and the use of moderate, evolutionary-based sequence constraints. To evaluate the general utility of these settings, we used this protocol (Table S1, protocol 6) to deimmunize a set of 54 diverse, soluble proteins of a range of sizes, in which the crystal structures were solved at less than 2 Å resolution and did not contain ligands, non-canonical amino acids, or discontinuous chains. This benchmark set was previously used in a similar deimmmunizaton study.44 The results were analyzed to determine the extent to which the metrics we monitored in our three case study proteins behaved similarly in the diverse benchmark set.
Examination of the Pareto front of REF2015 vs. mhc_epitope scores for this benchmark set, along with the case study proteins from the previous section, demonstrated the expected trade-off between these two metrics (Figure 6). When examining the six metrics over a range of protein sizes, we saw no clear trends (Figure S10), suggesting that mhc_epitope deimmunization produces similar results on a range of proteins, irrespective of protein size.
Figure 6.
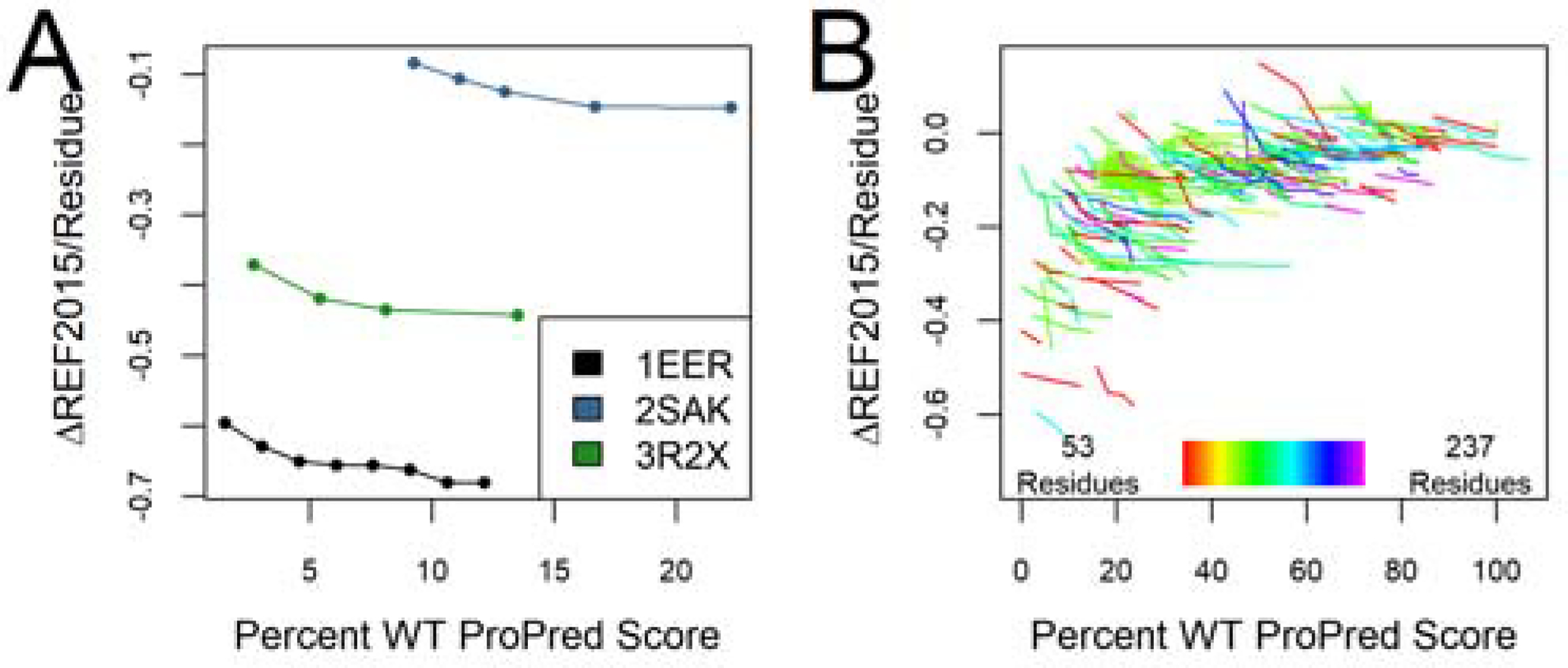
Pareto optimal set of deimmunized designs show the tradeoff between Rosetta score and ProPred score. (A) The Pareto optimal set of the three case study proteins is shown (n=1000/target). (B) The Pareto optimal set of all benchmark proteins is shown, with each protein indicated as a different line (n=240/target). Lines are color-coded according to the protein size.
In order to maintain an up-to-date record of MHCEpitopeEnergy’s performance as Rosetta continues to be developed, we have implemented a reduced form of this benchmark as a Rosetta “scientific test”60 called scientific.mhc_epitope_energy. This test runs regularly on the Rosetta Benchmark server (https://benchmark.graylab.jhu.edu/), testing the most recent version of Rosetta. The scripts and data used to run the benchmark are bundled with Rosetta and can be found in Rosetta/main/tests/scientific/tests/mhc_epitope_energy/.
Comparison of ProPred- and NetMHCII-based Deimmunization.
While NetMHCII is considered to be one of the leading epitope predictors currently available, it is considerably slower than ProPred, precluding its direct use as a packer-compatible predictor that can be evaluated at each step in a design trajectory. Our implementation of a pre-computed database Predictor as a means of using NetMHCII during design overcomes this problem, but due to the combinatorial explosion in the number of possible peptides that must be pre-scored according to the number of mutations considered, requires either a highly targeted approach or a very restrictive PSSM. We thus sought to evaluate whether NetMHCII-based deimmunization provided a significant improvement over the less restricted ProPred-based deimmunization evaluated prior to this point. First, to evaluate the relative effectiveness of the ProPred and NetMHCII Predictors at reducing each other’s scores, we deimmunized our case study proteins with either ProPred or a pre-computed NetMHCII database, with amino acid constraints and PSSM-based restrictions applied, and then scored designs using the Rosetta REF2015 energy function, ProPred, and NetMHCII (Figure S11–Figure S13, Table S1, protocol 5). As one would expect, deimmunization using ProPred decreases the final ProPred score more than the final NetMHCII score, and deimmunization using NetMHCII decreases the final NetMHCII score more than the final ProPred score. Nevertheless, both scores are reduced to some degree from their wild-type values during deimmunization, regardless of which Predictor is used during the design trajectory.
Since post hoc scoring with NetMHCII is computationally feasible, but NetMHCII-based deimmunization design trajectories require precomputed databases with limited sequence coverage for computational tractability, we further explored the extent to which ProPred-based deimmunization design could be used to produce designs with reduced NetMHCII scores. Figure 7 shows the correlation between ProPred and NetMHCII scores of the ProPred-deimmunized designs, with scores reported as a fraction of the scores of the native protein.
Figure 7.
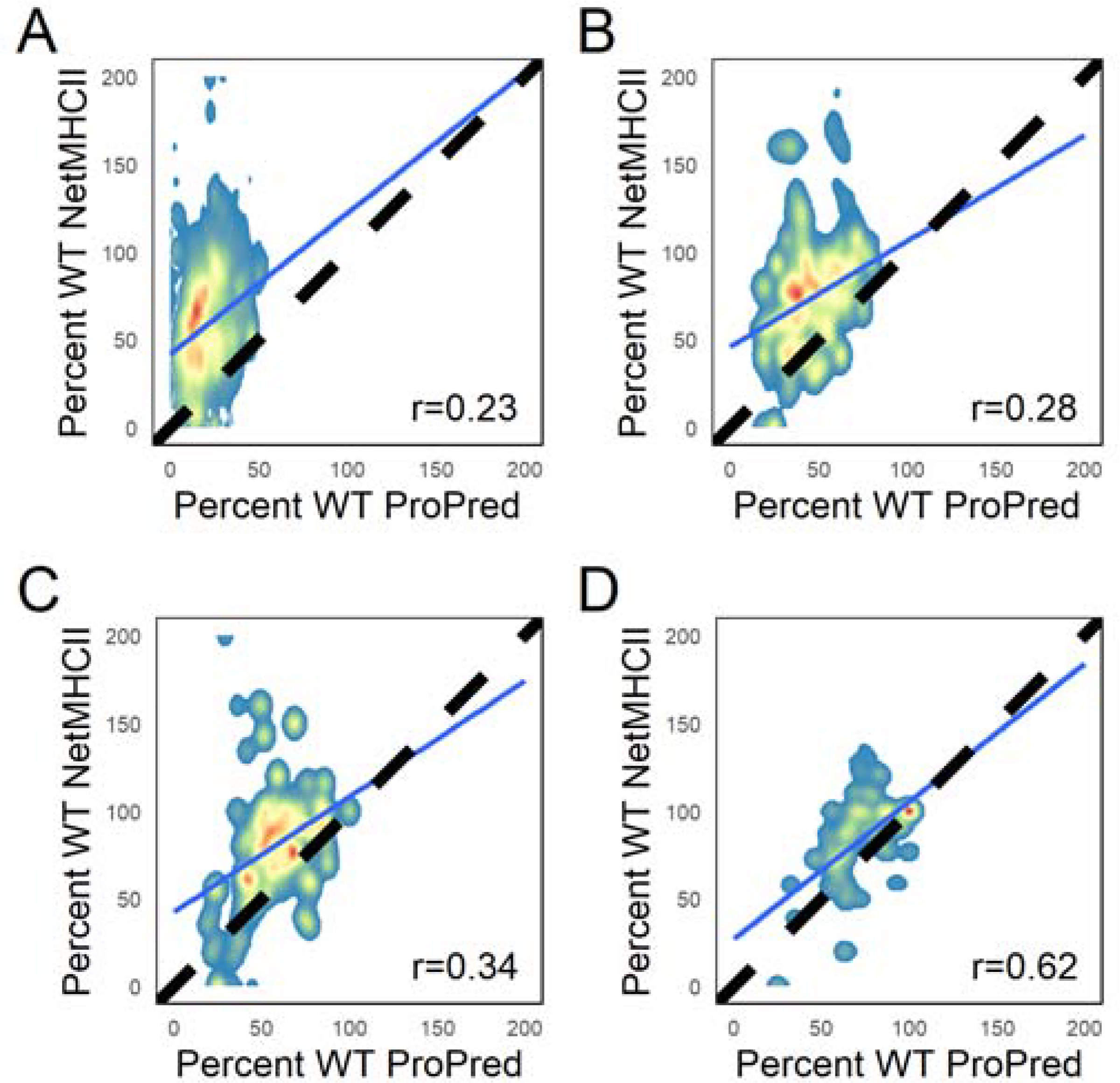
Correlation between ProPred scores (Southwood35 allele set) and NetMHCII scores (Paul37 allele set) in ProPred-deimmunized designs with varying PSSM restriction thresholds (n=240/target). In (A), no PSSM restrictions were applied. In the remaining plots, a base 2 log odds threshold of (B) 1, (C) 2, and (D) 3 was applied to limit allowed amino acid types at each position during design. The dashed line indicates designs with identical percent wild-type ProPred and NetMHCII scores. The Pearson correlation between the two scores for each threshold is given in the bottom right, and the line of best fit is shown in blue.
When used with more stringent PSSM thresholds, we see some correlation between the two scores (Pearson correlation of 0.23 without PSSM restrictions, and correlations of 0.28, 0.34, and 0.62 for PSSM thresholds of 1, 2, and 3, respectively). To more directly answer the question of whether the drop in ProPred score can be used to predict a low NetMHCII score, we defined designs with NetMHCII scores below a specified threshold as “sufficiently deimmunized.” The analysis was repeated twice, with that threshold set to either 33% or 50% of wild-type values. Using ROC and precision-recall analyses calculated using the pROC61 and PRROC62 R packages, respectively, we then examined whether the drop in ProPred score could be used to discriminate between “sufficiently” and “insufficiently” deimmunized designs (Figure 8, Figure S14–Figure S17). The precision-recall analysis demonstrates that the best ProPred designs are typically also the best NetMHCII designs, though it should be noted that particularly when using high PSSM thresholds, the “sufficiently deimmunized” designs are a very small fraction of total designs. The ROC analysis shows that using a lower NetMHCII threshold to define a “hit” or using a more stringent PSSM threshold results in the ProPred score being a better predictor of the NetMHCII scores. In this case, the analysis may be skewed by the very large imbalance between the small number of NetMHCII “hits” and the large number of “misses.”
Figure 8.
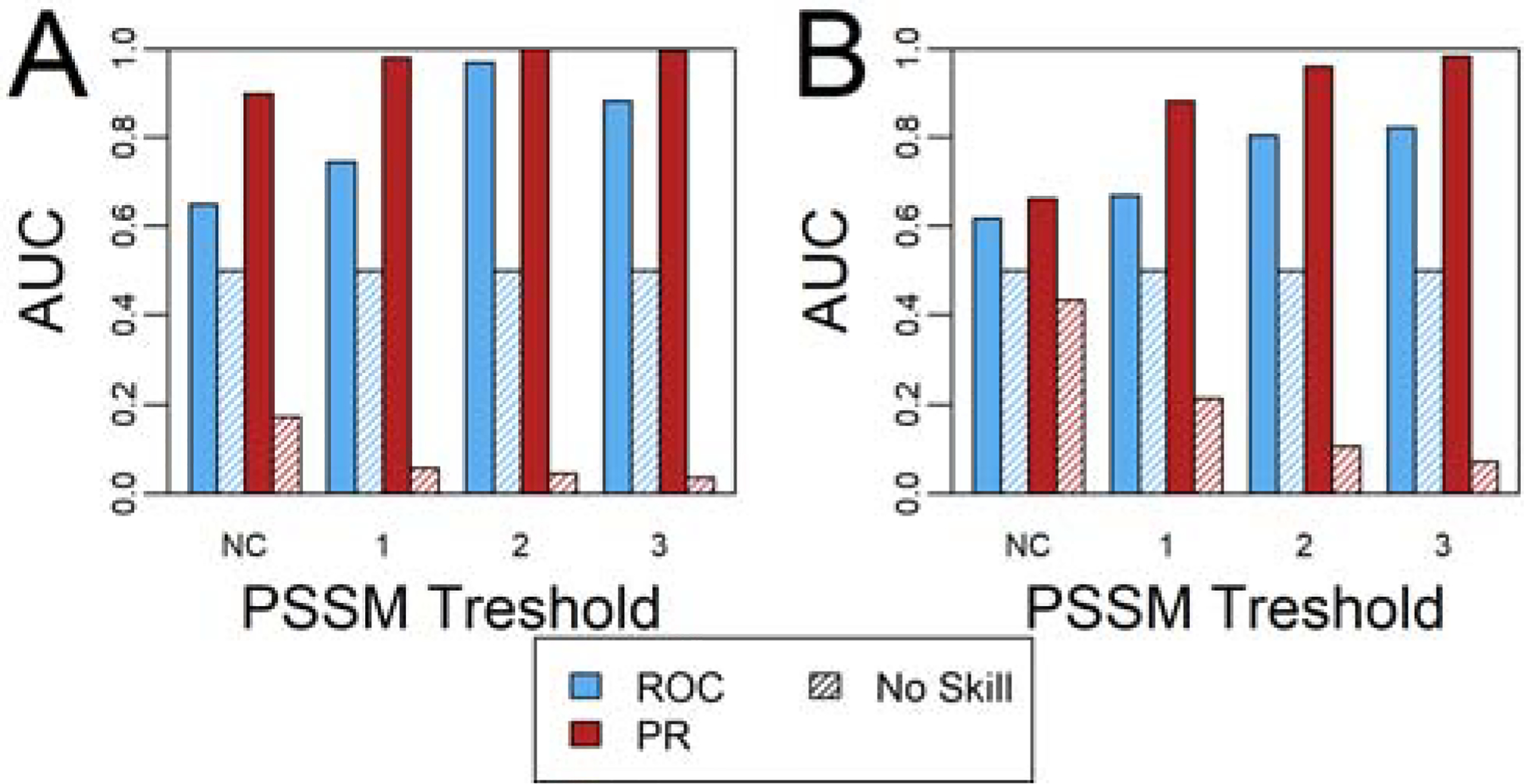
ROC and PR analysis of the ProPred score’s utility to design variants with favorable NetMHCII scores. A NetMHCII score that is (A) 33% and (B) 50% of the wild-type score were considered to be “favorable” scores in this analysis. Designs with no PSSM restrictions and base 2 log odds constraints of 1, 2, and 3 were considered separately. See also Figure S14–Figure S17.
From a user’s perspective, these results show that deimmunizing using MHCEpitopeEnergy with ProPred as the Predictor is a reasonable strategy to obtain designs that score well in terms of NetMHCII, particularly if designs are to be subsequently downselected upon re-scoring with NetMHCII. In cases where a very stringent deimmunization strategy is needed, doing an initial round of design using ProPred followed by design using NetMHCII that targets the remaining hotspots would be an efficient strategy to reduce NetMHCII scores, depending on the specifics of the design target. When considering the best ProPred-deimmunized designs for each benchmark target, as assessed based on the REF2015 and NetMHCII scores, the number of hotspot regions decreased in 29 of 55 targets in the benchmark set, frequently down to 0 hotspots (Figure S18A). Figure S18B shows a comparison of wild-type and variant NetMHCII scores after ProPred-based deimmunization of P99 β-lactamase (PDB ID 1XX2), highlighting that ProPred-based design eliminated some NetMHCII hotspots while leaving others unchanged. These results demonstrate the utility of following ProPred-based deimmunization with targeted NetMHCII deimmunization for certain design problems.
Recapitulation of Experimentally-Validated Deimmunized Proteins.
Finally, we compared deimmunized designs emerging from this approach with those obtained from previous deimmunization efforts. It is unrealistic to expect that our protocol would capture the same mutations as performed in the original experiments, since design space is very vast, and there is almost inevitably a number of deimmunized, stable, and functional sequences within that space. We would, however, expect that changes should be made in the same epitopes that have previously been found to reduce immunogenicity. Furthermore, the Rosetta trajectories can provide further insights into the diversity in the deimmunized sequence space available “near” wild-type. To that end, we used our “standard” protocol (Table S1, protocol 7) on three notable, experimentally deimmunized proteins: Pseudomonas exotoxin A,63,64 P99 β-lactamase from Enterobacter cloacae,18 and superfolder green fluorescent protein (sfGFP).65 We also ran identical design trajectories with deimmunization turned off to differentiate mutations introduced solely to optimize the REF2015 energy (present in both design trajectories) from those introduced to decrease immunogenicity (different variants in the deimmunization and non-deimmunization trajectories).
In the absence of evolutionary PSSM-based restrictions and with mhc_epitope turned on, Rosetta preferentially introduced mutations in all of the peptides previously targeted by experimentally validated mutations as compared to standard fixed backbone design (Figure 9, Figure S19–Figure S21). Imposing the evolutionary-based restrictions did cause Rosetta to “miss” some of the epitopes, though this occurred in a minority of cases except when the most stringent restrictions were applied. These results demonstrate that our methodology is able to target epitopes that were specifically found in experiments to yield less immunogenic variants. These results also suggest that running simulations with and without deimmunization and at various PSSM thresholds may be effective at prioritizing variants for experimental validation, both for evaluating individual variants or for limiting combinatorial expansion during library-based screening.
Figure 9.
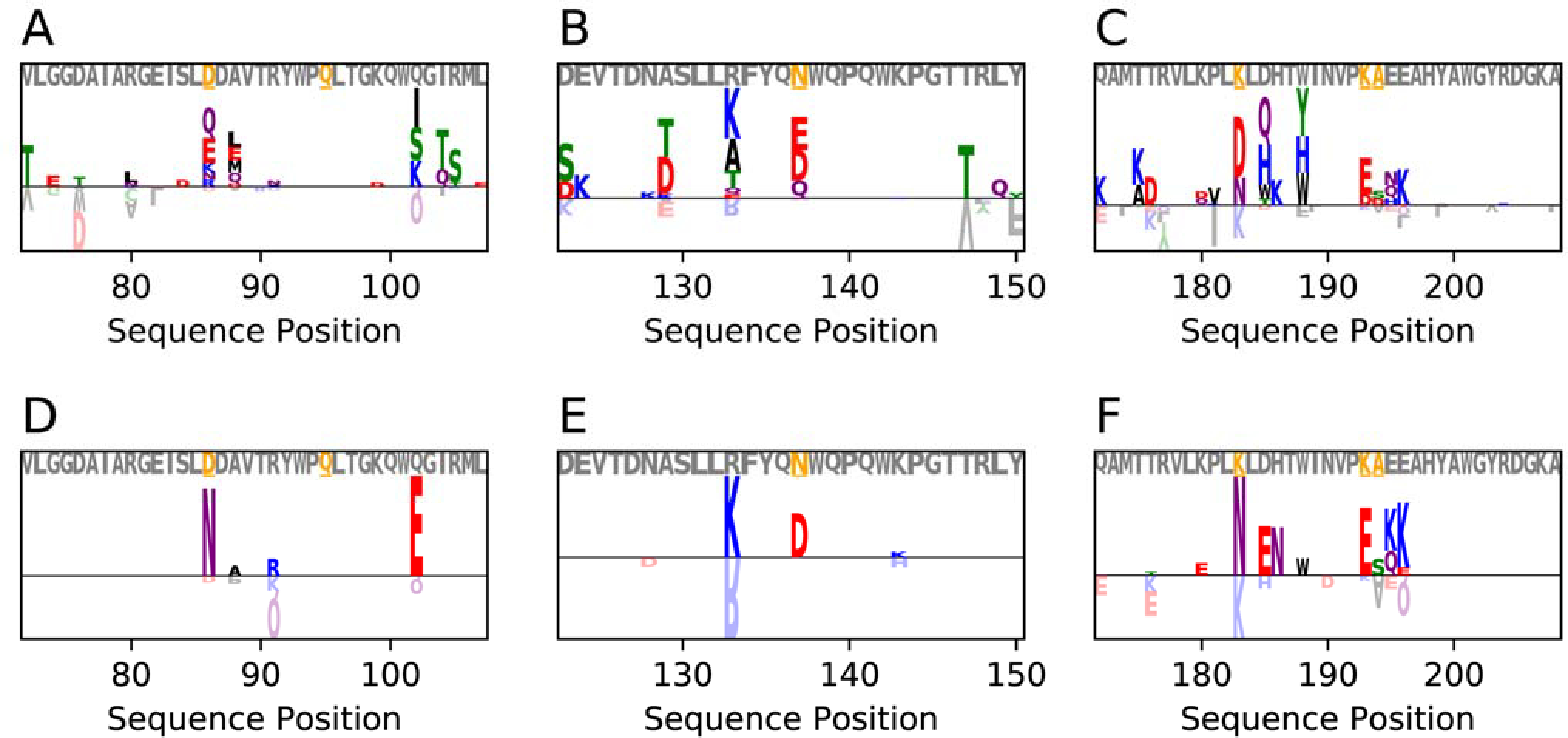
Logoplots showing the relative frequency of mutations for experimentally validated immunogenic peptides in deimmunized and non-deimmunized designs of P99 β-lactamase (PDB ID 1XX2). Letters above the horizontal line are seen more frequently in deimmunized designs (n=500), whereas letters below the horizontal line are seen more frequently in the standard Rosetta designs (n=500). Grey lettering above the logoplot indicates the native sequence, with the orange, underlined residues indicating positions that have been mutated in experimentally validated deimmunization campaigns.18 Designs with no PSSM-based restrictions are shown in panels A-C, and designs with base 2 log-odds PSSM-based restrictions of 1 for the same epitopes are shown in panels D-F. See also Figure S19–Figure S21 for corresponding plots for other protein targets.
More generally, examination of mutation patterns observed in experimentally-identified epitope regions demonstrate that performing deimmunization using our mhc_epitope approach leads to an increase in the number and diversity of variants, as compared to the same design strategy without mhc_epitope. This suggests that mhc_epitope is likely overcoming the memory of native residue identities imposed by the “FavorNative” constraint, as well as evolutionary and charge constraints, in highly immunogenic regions, with only minor effects on other metrics as compared to equivalent non-deimmunizing designs (Figure S22–Figure S24).
MHCEpitopeEnergy Is Readily Customizable.
Our approach in this study has been largely focused on establishing a “basic” protocol from which users can initiate deimmunized protein design campaigns. Our integration with the RosettaScripts framework and modular implementation of MHCEpitopePredictors, however, allows for straightforward customization towards more sophisticated design approaches. Of note, the MHCEpitopeConstraint framework allows the use of multiple Predictors simultaneously (e.g. ProPred28 deimmunization alongside deletion or avoidance of immunogenic peptides from the IEDB database40). Residue selectors allow selective application of these constraints to specific residues. For example, known or predicted hotspot epitopes could be targeted for deimmunization, or the propensity of MHC-II ligands to be more accessible for proteolysis in processing66–68 could be utilized to selectively target the highest risk regions. Alternatively, the “xform” option allows the mhc_epitope score to be linked to the wild-type sequence or a fixed, “acceptable” baseline per-residue score, thus avoiding spurious mutations to sequences with low, but non-zero, immunogenicity scores.
New sequence-based epitope prediction methods can also be readily added to the MHCEpitopeEnergy framework as they are developed. In cases where epitope prediction can be easily stored in a peptide sequence/score database, this can be implemented as an additional External/Preloaded Predictor database. Alternatively, prediction algorithms can easily be incorporated by implementing a new MHCEpitopePredictor class in Rosetta’s C++ codebase. For example, a prototype Tensorflow69 based Predictor has already been developed.
CONCLUSION
Protein deimmunization is becoming increasingly important as the biologics sector grows and seeks to utilize the myriad of functions that proteins can perform in therapeutic contexts. The challenges in developing stable, functional, and deimmunized proteins are considerable, especially when considering the high cost of screening proteins for immunogenicity. A number of specialized computational tools have been developed to address this need; however, a flexible package within a premiere protein design suite remained an unmet need.
MHCEpitopeEnergy attempts to fill this role by integrating leading epitope predictors with one of the most successful protein design packages, Rosetta. Unlike previously described deimmunization tools in Rosetta, MHCEpitopeEnergy can be integrated into any protocol that uses Rosetta’s standard Monte Carlo-based rotamer packing and sequence design algorithm. In addition, integration of new epitope predictors, as they are developed, is straightforward owing to its modular class organization.
We have also developed a benchmark protocol which acts as a starting point for users wishing to employ this tool. These results highlight that we can obtain similar quality designs with and without deimmunization based on sequence and structural metrics, and that these trends hold across a larger benchmark set. In addition, epitopes that have been validated experimentally in protein deimmunization campaigns are efficiently targeted using these protocols. Owing to the simple integration of epitope predictors with the core Rosetta design machinery, we expect that MHCEpitopeEnergy will greatly simplify workflows by eliminating the need for iterative design/prediction cycles, and by allowing existing design protocols that have been employed for a design objective to be adapted for simultaneous deimmunization. In this way, we expect that this will ease the design of functional, stable, and deimmunized variants of important protein therapeutics.
Supplementary Material
ACKNOWLEDGMENT
We would like to thank the Khare and Bailey-Kellogg labs for helpful discussions. We would also like to thank Indigo King for insight into the “nmer” SVM predictor. This work was funded in part by grants from the National Institutes of Health (National Institute of General Medical Sciences) 2R01GM098977 to CBK and 1R01GM132565 to SDK. VKM was supported by the Simons Foundation. BJY was awarded fellowships from the Canadian Institutes of Health Research and the Mistletoe Research Fellowship. The authors acknowledge the Office of Advanced Research Computing (OARC) at Rutgers, The State University of New Jersey for providing access to the Amarel cluster and associated computing resources that have contributed to the results reported here. Development of the design-centric guidance scoring term computational framework was carried out in part using an award of computer time provided to VKM by the Innovative and Novel Computational Impact on Theory and Experiment (INCITE) program. This research used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357.
Footnotes
In addition to his faculty position at Dartmouth, Chris Bailey-Kellogg is a co-founder and manager of Stealth Biologics, LLC, a Delaware biotechnology company. His potential conflicts of interest are managed by Dartmouth, and he affirms that this work is free of bias. Vikram Khipple Mulligan is a co-founder and shareholder of Menten AI, a Delaware biotechnology company. He too affirms that this work is free of bias.
The remaining authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. The Supporting Information is available free of charge at https://pubs.acs.org:
Supporting Table and Figures providing additional details about the deimmunization protocol and additional analyses of deimmunized protein data (PDF).
DATA AND SOFTWARE AVAILABILITY
MHCEpitopeEnergy and all related tools are available as part of the Rosetta Software Suite. Rosetta is available to all non-commercial users for free at www.rosettacommons.org, and is licensed for commercial use through a fee paid to UW CoMotion (https://els2.comotion.uw.edu/product/rosetta).
REFERENCES
- (1).Leader B; Baca QJ; Golan DE Protein Therapeutics: A Summary and Pharmacological Classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- (2).Sauna ZE; Lagassé HAD; Alexaki A; Simhadri VL; Katagiri NH; Jankowski W; Kimchi-Sarfaty C Recent Advances in (Therapeutic Protein) Drug Development. F1000Research. Faculty of 1000 Ltd February 7, 2017, p 113. 10.12688/f1000research.9970.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Coiffier B; Lepage E; Brière J; Herbrecht R; Tilly H; Bouabdallah R; Morel P; Van Den Neste E; Salles G; Gaulard P; Reyes F; Lederlin P; Gisselbrecht C CHOP Chemotherapy plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med 2002, 346, 235–242. 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- (4).Hurwitz H; Fehrenbacher L; Novotny W; Cartwright T; Hainsworth J; Heim W; Berlin J; Baron A; Griffing S; Holmgren E; Ferrara N; Fyfe G; Rogers B; Ross R; Kabbinavar F Bevacizumab plus Irinotecan, Fluorouracil, and Leucovorin for Metastatic Colorectal Cancer. N. Engl. J. Med 2004, 350, 2335–2342. 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- (5).Clavell LA; Gelber RD; Cohen HJ; Hitchcock-Bryan S; Cassady JR; Tarbell NJ; Blattner SR; Tantravahi R; Leavitt P; Sallan SE Four-Agent Induction and Intensive Asparaginase Therapy for Treatment of Childhood Acute Lymphoblastic Leukemia. N. Engl. J. Med 1986, 315, 657–663. 10.1056/NEJM198609113151101. [DOI] [PubMed] [Google Scholar]
- (6).Goldman SC; Holcenberg JS; Finklestein JZ; Hutchinson R; Kreissman S; Leonard Johnson F; Tou C; Harvey E; Morris E; Cairo MS A Randomized Comparison between Rasburicase and Allopurinol in Children with Lymphoma or Leukemia at High Risk for Tumor Lysis. Blood 2001, 97, 2998–3003. 10.1182/blood.V97.10.2998. [DOI] [PubMed] [Google Scholar]
- (7).Genovese MC; Becker J-C; Schiff M; Luggen M; Sherrer Y; Kremer J; Birbara C; Box J; Natarajan K; Nuamah I; Li T; Aranda R; Hagerty DT; Dougados M Abatacept for Rheumatoid Arthritis Refractory to Tumor Necrosis Factor α Inhibition. N. Engl. J. Med 2005, 353, 1114–1123. 10.1056/NEJMoa050524. [DOI] [PubMed] [Google Scholar]
- (8).Olsen NJ; Stein CM New Drugs for Rheumatoid Arthritis. New England Journal of Medicine. Massachusetts Medical Society May 20, 2004, pp 2167–2179+2226. 10.1056/NEJMra032906. [DOI] [PubMed] [Google Scholar]
- (9).Weinblatt ME; Keystone EC; Furst DE; Moreland LW; Weisman MH; Birbara CA; Teoh LA; Fischkoff SA; Chartash EK Adalimumab, a Fully Human Anti-Tumor Necrosis Factor α Monoclonal Antibody, for the Treatment of Rheumatoid Arthritis in Patients Taking Concomitant Methotrexate: The ARMADA Trial. Arthritis Rheum. 2003, 48, 35–45. 10.1002/art.10697. [DOI] [PubMed] [Google Scholar]
- (10).Maini R; St Clair EW; Breedveld F; Furst D; Kalden J; Weisman M; Smolen J; Emery P; Harriman G; Feldmann M; Lipsky P Infliximab (Chimeric Anti-Tumour Necrosis Factor α Monoclonal Antibody) versus Placebo in Rheumatoid Arthritis Patients Receiving Concomitant Methotrexate: A Randomised Phase III Trial. Lancet 1999, 354, 1932–1939. 10.1016/S0140-6736(99)05246-0. [DOI] [PubMed] [Google Scholar]
- (11).Present DH; Rutgeerts P; Targan S; Hanauer SB; Mayer L; van Hogezand RA; Podolsky DK; Sands BE; Braakman T; DeWoody KL; Schaible TF; van Deventer SJH Infliximab for the Treatment of Fistulas in Patients with Crohn’s Disease. N. Engl. J. Med 1999, 340, 1398–1405. 10.1056/NEJM199905063401804. [DOI] [PubMed] [Google Scholar]
- (12).Lincoff AM; Bittl JA; Harrington RA; Feit F; Kleiman NS; Jackman JD; Sarembock IJ; Cohen DJ; Spriggs D; Ebrahimi R; Keren G; Carr J; Cohen EA; Betriu A; Desmet W; Kereiakes DJ; Rutsch W; Wilcox RG; De Feyter PJ; Vahanian A; Topol EJ Bivalirudin and Provisional Glycoprotein IIb/IIIa Blockade Compared with Heparin and Planned Glycoprotein IIb/IIIa Blockade during Percutaneous Coronary Intervention: REPLACE-2 Randomized Trial. J. Am. Med. Assoc 2003, 289, 853–863. 10.1001/jama.289.7.853. [DOI] [PubMed] [Google Scholar]
- (13).The GUSTO Investigators. An International Randomized Trial Comparing Four Thrombolytic Strategies for Acute Myocardial Infarction. N. Engl. J. Med 1993, 329, 673–682. 10.1056/nejm199309023291001. [DOI] [PubMed] [Google Scholar]
- (14).Sauna ZE; Lagassé D; Pedras-Vasconcelos J; Golding B; Rosenberg AS Evaluating and Mitigating the Immunogenicity of Therapeutic Proteins. Trends Biotechnol. 2018, 36, 1068–1084. 10.1016/j.tibtech.2018.05.008. [DOI] [PubMed] [Google Scholar]
- (15).King C; Garza EN; Mazor R; Linehan JL; Pastan I; Pepper M; Baker D Removing T-Cell Epitopes with Computational Protein Design. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 8577–8582. 10.1073/pnas.1321126111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Mazor R; Vassall AN; Eberle JA; Beers R; Weldon JE; Venzon DJ; Tsang KY; Benhar I; Pastan I Identification and Elimination of an Immunodominant T-Cell Epitope in Recombinant Immunotoxins Based on Pseudomonas Exotoxin A. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, E3597–603. 10.1073/pnas.1218138109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cantor JR; Panayiotou V; Agnello G; Georgiou G; Stone EM Engineering Reduced-Immunogenicity Enzymes for Amino Acid Depletion Therapy in Cancer. In Methods in Enzymology; Academic Press Inc., 2012; Vol. 502, pp 291–319. 10.1016/B978-0-12-416039-2.00015-X. [DOI] [PubMed] [Google Scholar]
- (18).Salvat RS; Verma D; Parker AS; Kirsch JR; Brooks SA; Bailey-Kellogg C; Griswold KE Computationally Optimized Deimmunization Libraries Yield Highly Mutated Enzymes with Low Immunogenicity and Enhanced Activity. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E5085–E5093. 10.1073/pnas.1621233114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Griswold KE; Bailey-Kellogg C Design and Engineering of Deimmunized Biotherapeutics. Curr. Opin. Struct. Biol 2016, 39, 79–88. 10.1016/J.SBI.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Jankowski W; McGill J; Lagassé HAD; Surov S; Bembridge G; Bunce C; Cloake E; Fogg MH; Jankowska KI; Khan A; Marcotrigiano J; Ovanesov MV; Sauna ZE Mitigation of T-Cell Dependent Immunogenicity by Reengineering Factor VIIa Analogue. Blood Adv. 2019, 3, 2668–2678. 10.1182/bloodadvances.2019000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lamberth K; Reedtz-Runge SL; Simon J; Klementyeva K; Pandey GS; Padkjær SB; Pascal V; León IR; Gudme CN; Buus S; Sauna ZE Post Hoc Assessment of the Immunogenicity of Bioengineered Factor VIIa Demonstrates the Use of Preclinical Tools. Sci. Transl. Med 2017, 9. 10.1126/scitranslmed.aag1286. [DOI] [PubMed] [Google Scholar]
- (22).Dillon RL; Chooniedass S; Premsukh A; Adams GP; Entwistle J; MacDonald GC; Cizeau J Trastuzumab-DeBouganin Conjugate Overcomes Multiple Mechanisms of T-DM1 Drug Resistance. J. Immunother 2016, 39, 117–126. 10.1097/CJI.0000000000000115. [DOI] [PubMed] [Google Scholar]
- (23).Zhao H; Brooks SA; Eszterhas S; Heim S; Li L; Xiong YQ; Fang Y; Kirsch JR; Verma D; Bailey-Kellogg C; Griswold KE Globally Deimmunized Lysostaphin Evades Human Immune Surveillance and Enables Highly Efficacious Repeat Dosing. Sci. Adv 2020, 6, eabb9011. 10.1126/sciadv.abb9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Tangri S; Mothé BR; Eisenbraun J; Sidney J; Southwood S; Briggs K; Zinckgraf J; Bilsel P; Newman M; Chesnut R; LiCalsi C; Sette A Rationally Engineered Therapeutic Proteins with Reduced Immunogenicity. J. Immunol 2005, 174, 3187–3196. 10.4049/jimmunol.174.6.3187. [DOI] [PubMed] [Google Scholar]
- (25).Warmerdam PAM; Plaisance S; Vanderlick K; Vandervoort P; Brepoels K; Collen D; De Maeyer M Elimination of a Human T-Cell Region in Staphylokinase by T-Cell Screening and Computer Modeling. Thromb. Haemost 2002, 87, 666–673. 10.1055/s-0037-1613064. [DOI] [PubMed] [Google Scholar]
- (26).Allen TM; Brehm MA; Bridges S; Ferguson S; Kumar P; Mirochnitchenko O; Palucka K; Pelanda R; Sanders-Beer B; Shultz LD; Su L; PrabhuDas M Humanized Immune System Mouse Models: Progress, Challenges and Opportunities. Nat. Immunol 2019, 20, 770–774. 10.1038/s41590-019-0416-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Oseroff C; Sidney J; Kotturi MF; Kolla R; Alam R; Broide DH; Wasserman SI; Weiskopf D; McKinney DM; Chung JL; Petersen A; Grey H; Peters B; Sette A Molecular Determinants of T Cell Epitope Recognition to the Common Timothy Grass Allergen. J. Immunol 2010, 185, 943–955. 10.4049/jimmunol.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Singh H; Raghava GPS ProPred: Prediction of HLA-DR Binding Sites. Bioinformatics 2001, 17, 1236–1237. 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- (29).Jensen KK; Andreatta M; Marcatili P; Buus S; Greenbaum JA; Yan Z; Sette A; Peters B; Nielsen M Improved Methods for Predicting Peptide Binding Affinity to MHC Class II Molecules. Immunology 2018, 154, 394–406. 10.1111/imm.12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Choi Y; Verma D; Griswold KE; Bailey-Kellogg C EpiSweep: Computationally Driven Reengineering of Therapeutic Proteins to Reduce Immunogenicity While Maintaining Function. In Methods in Molecular Biology; Samish I, Ed.; 2017; pp 375–398. 10.1007/978-1-4939-6637-0_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Parker AS; Choi Y; Griswold KE; Bailey-Kellogg C Structure-Guided Deimmunization of Therapeutic Proteins. J. Comput. Biol 2013, 20, 152–165. 10.1089/cmb.2012.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Rudensky AY; Preston-Hurlburt P; Hong SC; Barlow A; Janeway CA Sequence Analysis of Peptides Bound to MHC Class II Molecules. Nature 1991, 353, 622–627. 10.1038/353622a0. [DOI] [PubMed] [Google Scholar]
- (33).Chicz RM; Urban RG; Lane WS; Gorga JC; Stern LJ; Vignali DAA; Strominger JL Predominant Naturally Processed Peptides Bound to HLA-DR1 Are Derived from MHC-Related Molecules and Are Heterogeneous in Size. Nature 1992, 358, 764–768. 10.1038/358764a0. [DOI] [PubMed] [Google Scholar]
- (34).Sethu S; Govindappa K; Alhaidari M; Pirmohamed M; Park K; Sathish J Immunogenicity to Biologics: Mechanisms, Prediction and Reduction. Archivum Immunologiae et Therapiae Experimentalis. Springer October 29, 2012, pp 331–344. 10.1007/s00005-012-0189-7. [DOI] [PubMed] [Google Scholar]
- (35).Southwood S; Sidney J; Kondo A; del Guercio M-F; Appella E; Hoffman S; Kubo RT; Chesnut RW; Grey HM; Sette A Several Common HLA-DR Types Share Largely Overlapping Peptide Binding Repertoires. J. Immunol. 1998, 160, 3363–3373. [PubMed] [Google Scholar]
- (36).Greenbaum J; Sidney J; Chung J; Brander C; Peters B; Sette A Functional Classification of Class II Human Leukocyte Antigen (HLA) Molecules Reveals Seven Different Supertypes and a Surprising Degree of Repertoire Sharing across Supertypes. Immunogenetics 2011, 63, 325–335. 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Paul S; Lindestam Arlehamn CS; Scriba TJ; Dillon MBC; Oseroff C; Hinz D; McKinney DM; Carrasco Pro S; Sidney J; Peters B; Sette A Development and Validation of a Broad Scheme for Prediction of HLA Class II Restricted T Cell Epitopes. J. Immunol. Methods 2015, 422, 28–34. 10.1016/j.jim.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Chen B; Khodadoust MS; Olsson N; Wagar LE; Fast E; Liu CL; Muftuoglu Y; Sworder BJ; Diehn M; Levy R; Davis MM; Elias JE; Altman RB; Alizadeh AA Predicting HLA Class II Antigen Presentation through Integrated Deep Learning. Nat. Biotechnol. 2019, 37, 1332–1343. 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Racle J; Michaux J; Rockinger GA; Arnaud M; Bobisse S; Chong C; Guillaume P; Coukos G; Harari A; Jandus C; Bassani-Sternberg M; Gfeller D Robust Prediction of HLA Class II Epitopes by Deep Motif Deconvolution of Immunopeptidomes. Nat. Biotechnol 2019, 37, 1283–1286. 10.1038/s41587-019-0289-6. [DOI] [PubMed] [Google Scholar]
- (40).Vita R; Mahajan S; Overton JA; Dhanda SK; Martini S; Cantrell JR; Wheeler DK; Sette A; Peters B The Immune Epitope Database (IEDB): 2018 Update. Nucleic Acids Res. 2019, 47, D339–D343. 10.1093/nar/gky1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Leman JK; Weitzner BD; Lewis SM; Adolf-Bryfogle J; Alam N; Alford RF; Aprahamian M; Baker D; Barlow KA; Barth P; Basanta B; Bender BJ; Blacklock K; Bonet J; Boyken SE; Bradley P; Bystroff C; Conway P; Cooper S; Correia BE; Coventry B; Das R; De Jong RM; DiMaio F; Dsilva L; Dunbrack R; Ford AS; Frenz B; Fu DY; Geniesse C; Goldschmidt L; Gowthaman R; Gray JJ; Gront D; Guffy S; Horowitz S; Huang P-S; Huber T; Jacobs TM; Jeliazkov JR; Johnson DK; Kappel K; Karanicolas J; Khakzad H; Khar KR; Khare SD; Khatib F; Khramushin A; King IC; Kleffner R; Koepnick B; Kortemme T; Kuenze G; Kuhlman B; Kuroda D; Labonte JW; Lai JK; Lapidoth G; Leaver-Fay A; Lindert S; Linsky T; London N; Lubin JH; Lyskov S; Maguire J; Malmström L; Marcos E; Marcu O; Marze NA; Meiler J; Moretti R; Mulligan VK; Nerli S; Norn C; Ó’Conchúir S; Ollikainen N; Ovchinnikov S; Pacella MS; Pan X; Park H; Pavlovicz RE; Pethe M; Pierce BG; Pilla KB; Raveh B; Renfrew PD; Burman SSR; Rubenstein A; Sauer MF; Scheck A; Schief W; Schueler-Furman O; Sedan Y; Sevy AM; Sgourakis NG; Shi L; Siegel JB; Silva D-A; Smith S; Song Y; Stein A; Szegedy M; Teets FD; Thyme SB; Wang RY-R; Watkins A; Zimmerman L; Bonneau R Macromolecular Modeling and Design in Rosetta: Recent Methods and Frameworks. Nat. Methods 2020, 17, 665–680. 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Weng L; Spoonamore JE Droplet Microfluidics-Enabled High-Throughput Screening for Protein Engineering. Micromachines MDPI AG November 1, 2019, p 734. 10.3390/mi10110734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hughes RA; Ellington AD Synthetic DNA Synthesis and Assembly: Putting the Synthetic in Synthetic Biology. Cold Spring Harb. Perspect. Biol. 2017, 9, a023812. 10.1101/cshperspect.a023812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Choi Y; Griswold KE; Bailey-Kellogg C Structure-Based Redesign of Proteins for Minimal T-Cell Epitope Content. J. Comput. Chem. 2013, 34, 879–891. 10.1002/jcc.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Crudele JM; Chamberlain JS Cas9 Immunity Creates Challenges for CRISPR Gene Editing Therapies. Nat. Commun 2018, 9, 9–11. 10.1038/s41467-018-05843-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Pien GC; Basner-Tschakarjan E; Hui DJ; Mentlik AN; Finn JD; Hasbrouck NC; Zhou S; Murphy SL; Maus MV; Mingozzi F; Orange JS; High KA Capsid Antigen Presentation Flags Human Hepatocytes for Destruction after Transduction by Adeno-Associated Viral Vectors. J. Clin. Invest 2009, 119, 1688–1695. 10.1172/JCI36891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hosseinzadeh P; Bhardwaj G; Mulligan VK; Shortridge MD; Craven TW; Pardo-Avila F; Rettie SA; Kim DE; Silva DA; Ibrahim YM; Webb IK; Cort JR; Adkins JN; Varani G; Baker D Comprehensive Computational Design of Ordered Peptide Macrocycles. Science 2017, 358, 1461–1466. 10.1126/science.aap7577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rocklin GJ; Chidyausiku TM; Goreshnik I; Ford A; Houliston S; Lemak A; Carter L; Ravichandran R; Mulligan VK; Chevalier A; Arrowsmith CH; Baker D Global Analysis of Protein Folding Using Massively Parallel Design, Synthesis, and Testing. Science 2017, 357, 168–175. 10.1126/science.aan0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mulligan VK; Workman S; Sun T; Rettie S; Li X; Worrall LJ; Craven TW; King DT; Hosseinzadeh P; Watkins AM; Renfrew PD; Guffy S; Labonte JW; Moretti R; Bonneau R; Strynadka NCJ; Baker D Computationally Designed Peptide Macrocycle Inhibitors of New Delhi Metallo-β-Lactamase 1. Proc. Natl. Acad. Sci 2021, 118, e2012800118. 10.1073/pnas.2012800118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fleishman SJ; Leaver-Fay A; Corn JE; Strauch E-M; Khare SD; Koga N; Ashworth J; Murphy P; Richter F; Lemmon G; Meiler J; Baker D RosettaScripts: A Scripting Language Interface to the Rosetta Macromolecular Modeling Suite. PLoS One 2011, 6, e20161. 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chaudhury S; Lyskov S; Gray JJ PyRosetta: A Script-Based Interface for Implementing Molecular Modeling Algorithms Using Rosetta. Bioinformatics. Oxford Academic January 7, 2010, pp 689–691. 10.1093/bioinformatics/btq007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; Labonte JW; Pacella MS; Bonneau R; Bradley P; Dunbrack RL; Das R; Baker D; Kuhlman B; Kortemme T; Gray JJ The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Schäffer AA; Aravind L; Madden TL; Shavirin S; Spouge JL; Wolf YI; Koonin EV; Altschul SF Improving the Accuracy of PSI-BLAST Protein Database Searches with Composition-Based Statistics and Other Refinements. Nucleic Acids Res. 2001, 29, 2994–3005. 10.1093/nar/29.14.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Syed RS; Reid SW; Li C; Cheetham JC; Aoki KH; Liu B; Zhan H; Osslund TD; Chirino AJ; Zhang J; Finer-Moore J; Elliott S; Sitney K; Katz BA; Matthews DJ; Wendoloski JJ; Egrie J; Stroud RM Efficiency of Signalling through Cytokine Receptors Depends Critically on Receptor Orientation. Nature 1998, 395, 511–516. 10.1038/26773. [DOI] [PubMed] [Google Scholar]
- (55).Rabijns A; De Bondt HL; De Ranter C Three-Dimensional Structure of Staphylokinase, a Plasminogen Activator with Therapeutic Potential. Nat. Struct. Biol 1997, 4, 357–360. 10.1038/nsb0597-357. [DOI] [PubMed] [Google Scholar]
- (56).Fleishman SJ; Whitehead TA; Ekiert DC; Dreyfus C; Corn JE; Strauch E-M; Wilson IA; Baker D Computational Design of Proteins Targeting the Conserved Stem Region of Influenza Hemagglutinin. Science 2011, 332, 816–821. 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Sheffler W; Baker D RosettaHoles: Rapid Assessment of Protein Core Packing for Structure Prediction, Refinement, Design and Validation. Protein Sci. 2008, 18, 229–239. 10.1002/pro.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Swint-Kruse L Using Evolution to Guide Protein Engineering: The Devil IS in the Details. Biophysical Journal. Biophysical Society July 12, 2016, pp 10–18. 10.1016/j.bpj.2016.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Goldenzweig A; Goldsmith M; Hill SE; Gertman O; Laurino P; Ashani Y; Dym O; Unger T; Albeck S; Prilusky J; Lieberman RL; Aharoni A; Silman I; Sussman JL; Tawfik DS; Fleishman SJ Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 2016, 63. 10.1016/j.molcel.2016.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Koehler Leman J; Lyskov S; Lewis S; Adolf-Bryfogle J; Alford RF; Barlow K; Ben-Aharon Z; Farrell D; Fell J; Hansen WA; Harmalkar A; Jeliazkov J; Kuenze G; Krys JD; Ljubetic A; Loshbaugh AL; Maguire J; Moretti R; Mulligan VK; Nguyen PT; O’Connor S; Roy Burman SS; Smith ST; Teets F; Tiemann JK; Watkins A; Woods H; Yachnin BJ; Bahl CD; Bailey-Kellogg C; Baker D; Das R; DiMaio F; Khare SD; Kortemme T; Labonte JW; Lindorff-Larsen K; Meiler J; Schief W; Schueler-Furman O; Siegel J; Stein A; Yarov-Yarovoy V; Kuhlman B; Leaver-Fay A; Gront D; Gray JJ; Bonneau R Ensuring Scientific Reproducibility in Biomacromolecular Modeling via Extensive, Automated Benchmarks. Nat. Commun 2021, Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Robin X; Turck N; Hainard A; Tiberti N; Lisacek F; Sanchez J-C; Müller M PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinformatics 2011, 12, 77. 10.1186/1471-2105-12-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Grau J; Grosse I; Keilwagen J PRROC: Computing and Visualizing Precision-Recall and Receiver Operating Characteristic Curves in R. Bioinformatics 2015, 31, 2595–2597. 10.1093/bioinformatics/btv153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Jørgensen R; Merrill AR; Yates SP; Marquez VE; Schwan AL; Boesen T; Andersen GR Exotoxin A-EEF2 Complex Structure Indicates ADP Ribosylation by Ribosome Mimicry. Nature 2005, 436, 979–984. 10.1038/nature03871. [DOI] [PubMed] [Google Scholar]
- (64).Yates SP; Taylor PL; Jørgensen R; Ferraris D; Zhang J; Andersen GR; Merrill AR Structure-Function Analysis of Water-Soluble Inhibitors of the Catalytic Domain of Exotoxin A from Pseudomonas Aeruginosa. Biochem. J 2005, 385, 667–675. 10.1042/BJ20041480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Pédelacq J-D; Cabantous S; Tran T; Terwilliger TC; Waldo GS Engineering and Characterization of a Superfolder Green Fluorescent Protein. Nat. Biotechnol 2006, 24, 79–88. 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- (66).Jørgensen KW; Buus S; Nielsen M Structural Properties of MHC Class II Ligands, Implications for the Prediction of MHC Class II Epitopes. PLoS One 2010, 5, e15877. 10.1371/journal.pone.0015877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Mettu RR; Charles T; Landry SJ CD4+ T-Cell Epitope Prediction Using Antigen Processing Constraints. J. Immunol. Methods 2016, 432, 72–81. 10.1016/j.jim.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Moss DL; Park HW; Mettu RR; Landry SJ Deimmunizing Substitutions in Pseudomonas Exotoxin Domain III Perturb Antigen Processing without Eliminating T-Cell Epitopes. J. Biol. Chem 2019, 294, 4667–4681. 10.1074/jbc.RA118.006704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Abadi M; Agarwal A; Barham P; Brevdo E; Chen Z; Citro C; Corrado GS; Davis A; Dean J; Devin M; Ghemawat S; Goodfellow I; Harp A; Irving G; Isard M; Jia Y; Jozefowicz R; Kaiser L; Kudlur M; Levenberg J; Mané D; Monga R; Moore S; Murray D; Olah C; Schuster M; Shlens J; Steiner B; Sutskever I; Talwar K; Tucker P; Vanhoucke V; Vasudevan V; Viégas F; Vinyals O; Warden P; Wattenberg M; Wicke M; Yu Y; Zheng X TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems; Software available from tensorflow.org., 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MHCEpitopeEnergy and all related tools are available as part of the Rosetta Software Suite. Rosetta is available to all non-commercial users for free at www.rosettacommons.org, and is licensed for commercial use through a fee paid to UW CoMotion (https://els2.comotion.uw.edu/product/rosetta).