

Abstract
Transcription factors (TFs) represent a major class of therapeutic targets for the treatment of human diseases including cancer. Although the biological functions and even crystal structures of many TFs have been clearly elucidated, there is still no viable approach to target the majority of TFs, thus rendering them undruggable for decades. PROTACs (proteolysis targeting chimeras) emerge as a powerful class of therapeutic modalities, which rely on induced protein—protein interactions between the proteins of interest (POIs) and E3 ubiquitin ligases to aid the degradation of POIs by the ubiquitin-proteasome system (UPS). Here, we report the development of a platform termed TF-PROTAC, which links an DNA oligonucleotide to an E3 ligase ligand via a click reaction, to selectively degrade the TF of interest. The selectivity of these TF-PROTACs depends on the DNA oligonucleotides utilized that can be specific to the TFs of interest. We have developed two series of VHL-based TF-PROTACs, NF-κB-PROTAC (dNF-κB) and E2F-PROTAC (dE2F), which effectively degrade endogenous p65 and E2F1 proteins in cells, respectively, and subsequently display superior antiproliferative effects in cells. Collectively, our results suggest that TF-PROTACs provide a generalizable platform to achieve selective degradation of TFs and a universal strategy for targeting most “undruggable” TFs.
Graphical Abstract

INTRODUCTION
Genetic regulation relies largely on the sequence-specific transcription factors (TFs) that recognize short DNA segments, also known as TF binding motifs, which typically locate in the enhancer and promotor regions of respective genes.1,2 These TFs promote or repress genetic transcription and play pivotal roles in the development of various human diseases, including cancer.3,4 The cancer dependency map project (DepMap) uncovered that TFs represent a major class of essential genes that maintain the proliferation and tumorigenesis of cancer cells, indicating that many TFs are potential therapeutic targets for cancer.5 To date, major efforts have been devoted to targeting TFs using small molecule inhibitors (SMIs),6 including NF-κB,7,8 STAT3/5,9–12 and MYC13,14, as well as the well-characterized nuclear receptors AR15–17 and ER.18 However, the majority of TFs cannot be effectively targeted by SMIs for potential therapeutic intervention.
TFs, in general, lack active sites or allosteric regulatory pockets that normally exist in kinases or other enzymes, thereby making it difficult for them to be targeted by SMIs.2 Given the key role of TFs in binding with specific DNA sequence to regulate genetic transcription, it is rational to expect that the TF DNA binding motif could, in theory, define the biological and biochemical specificity for different TFs.19 Indeed, a large number of studies have theoretically and/or experimentally defined the unique DNA binding motif for most of TFs.20 In the human genome, there are approximately 1600 putative TFs, which could be classified into tens of families largely based on their distinct DNA binding domains, such as C2H2, bZIP, bHLH, and Homeodomain.21 Several low-throughput and high-throughput methods including protein binding microarray (PBM), SELEX, DAP-seq, HiTS-FLIP, EMSA, and ChIP have been used to define the specific DNA sequence or motif that binds each individual TF.21–24 To date, the DNA binding motifs have been successfully defined for more than 600 putative human TFs, by experimental approaches,21–24 and more than 1300, by theoretical methods,20 presenting an invaluable treasure of information that may aid the development of TFs-targeting therapies.
By the hijacking of an endogenous E3 ubiquitin ligase and the ubiquitin-proteasome system (UPS), the proteolysis targeting chimera (PROTAC) technology is suitable for targeting any intracellular protein of interest (POI) for proteasomal degradation, including TFs.25,26 Recently, RNA-PROTACs have also been developed for the targeted degradation of the RNA-binding proteins (RBPs),27 but this approach has several shortcomings such as the instability of RNA oligomer and the requirement of RNA secondary structure for its proper interaction with RBPs. Compared with RNA, the DNA oligomer is relatively more stable and more importantly, the DNA binding specificity of TFs is much better-defined than the RNA binding specificity of RBPs. Moreover, a DNA-based PROTAC, TRAFTAC, has been recently developed by using a CRISPR-guided method, which unfortunately relies on the ectopic expression of the Cas-9 protein in cells, limiting its utilities in general.28
Herein, we report the development of a generalizable platform termed TF-PROTAC to selectively degrade the TFs of interest. We conjugate commercially available azide-modified DNA oligomers (N3-ODN) to the bicyclooctyne (BCN)-modified VHL ligands with various linkers (VHLL-X-BCN) via a copper-free strain-promoted azide—alkyne cycloaddition (SPAAC) reaction (Figures 1 and S1). After a simple purification process to remove the excess ligand VHLL-X-BCN, the TF-PROTACs are ready for transfection into cells and can potentially induce the targeted degradation of specific TFs of interest, based on the selective recognition/binding of the DNA oligomers by the TFs (Figure 1).
Figure 1.
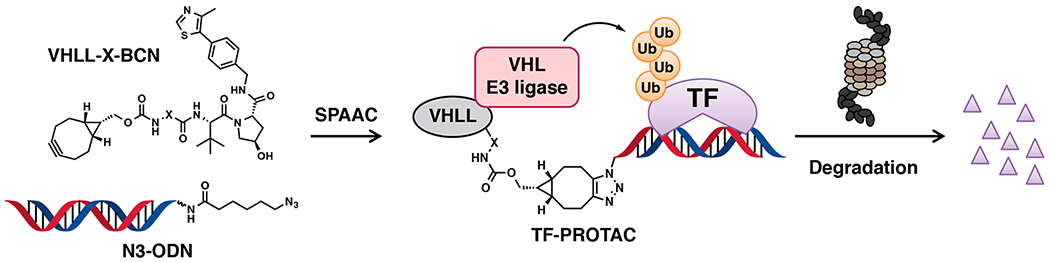
A schematic diagram of the TF-PROTAC strategy. A BCN-modified VHL ligand (VHLL-X-BCN) is incorporated onto an azide-modified DNA oligomer (N3-ODN) via a copper-free strain-promoted azide–alkyne cycloaddition (SPAAC) reaction, forming a TF-PROTAC to recruit the VHL E3 ubiquitin ligase to ubiquitinate the transcription factor (TF) of interest, which is subsequently degradated by the 26S proteasome.
RESULTS AND DISCUSSION
Design and Synthesis of dNF-κB.
To experimentally evaluate this unique PROTAC platform, we first chose the canonical NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) as a target since the DNA sequence (or binding motif) recognized by NF-κB is well-defined and has been extensively validated in vitro and in vivo.29,30 The sequence (or motif) binding to NF-κB, GGGRNNYYCC (R is purine, Y is pyrimidine, N is any base) is shared by many of its transcriptional targets, such as TNFα and HIV-1 (Figure S2A).29,30 Thus, we generated a single strand DNA oligonucleotide, 5′-TGGGGACTTTCCAGTTTCTGGAAAGTCCCCA-3′ (NF-κB decoy, hereafter termed as NF-κB-ODN),31 which forms a double-strand hairpin structure via intradimerization and therefore confers resistance to exonucleases in cells32 (Figure S2B,C). For the purpose of conjugation of an E3 ligase ligand on the end of NF-κB-ODN, an azide-modified version of NF-κB-ODN (hereafter termed as N3-NF-κB-ODN) was designed by incorporating an azide group onto the 5′ end of NF-κB-ODN through the 5′ amino modifier C6 (Figures 2A, S2A,C). As expected, the N3-NF-κB-ODN forms homodimer and oligomer via interdimerization, while only the annealing step ensures the entire intradimerization process (Figure S2D). To determine whether NF-κB-ODN binds with NF-κB properly, we also synthesized a biotin-modified version of NF-κB-ODN (hereafter termed as Biotin-NF-κB-ODN, Figure S2A–C). As expected, Biotin-NF-κB-ODN was capable of binding with RelA/p65, the DNA binding subunit of NF-κB heterodimer29,30 (Figure S2E,F), demonstrated by a streptavidin pulldown assay. Moreover, excess NF-κB-ODN or N3-NF-κB-ODN competed with Biotin-NF-κB-ODN for the binding with p65 (Figures 2B and S2E), indicating that the azide modification will be unlikely to affect the binding ability of NF-κB-ODN with NF-κB in this experimental setting.
Figure 2.
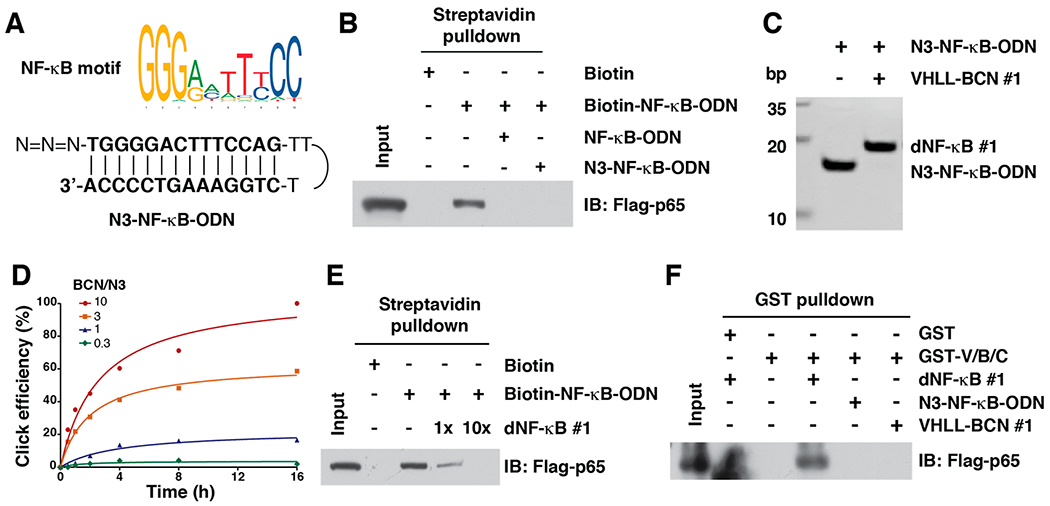
In vitro click reaction that leads to the generation of TF-PROTACs to target NF-κB. (A) A schematic diagram for the NF-κB motif and N3-NF-κB-ODN. (B) N3-NF-κB was capable of binding with NF-κB subunit p65. (C) Incorporation of VHLL-BCN #1 onto the N3-NF-κB-ODN led to an increase of molecular weight of 664 Da, which can be clearly separated by 20% native PAGE. (D) The click efficiency between VHLL-BCN #1 and N3-NF-κB-ODN with different ratios. A mixture of VHLL-BCN #1 and N3-NF-κB-ODN (50 μM) was incubated in PBS at 37 °C for indicated time points, followed by separation via 20% native PAGE. (E) dNF-κB #1 competed with Biotin-NF-κB for binding to RelA/p65. (F) dNF-κB #1 induced the VHL-TF-PROTAC-p65 ternary complex formation, which was determined by a GST pulldown assay.
In order to synthesize the TF-PROTACs bearing a DNA oligomer binding motif, we employed a robust biorthogonal reaction, copper-free strain-promoted azide—alkyne cycloaddition (SPAAC) reaction. First, we conjugated a BCN group with a VHL ligand (VH-032) via different linkers (hereafter termed as VHLL-X-BCN, Figure 1 and Scheme S1). A SPAAC reaction between the BCN-modified VHL ligands and N3-TF-ODN under physiological conditions afforded TF-PROTACs (Figure S3A). To assess the efficiency of the SPAAC reaction for N3-ODN, we monitored the amount of the product generated by the click reaction between VHLL-BCN #1 (X is CH2) and N3-NF-κB-ODN under physiological conditions (PBS, 37 °C). Notably, after the incorporation of VHLL-BCN #1 to N3-NF-κB-ODN, the molecule weight will increase 664 Da, which can be clearly separated by 20% native polyacrylamide gel electrophoresis (PAGE, Figures 2C, S3B,C). As expected, increasing the amount of VHLL-BCN #1 and the reaction time improved the efficiency of the SPAAC reaction. Furthermore, as showed in Figure 2D, the click efficiency could be obtained up to more than 60% with 10-fold excess of VHLL-BCN #1 after a 4 h reaction (Figures 2D and S3C). Moreover, the click reaction product VHLL-NF-κB-ODN (hereafter termed as dNF-κB) can compete with Biotin-NF-κB for the binding to p65 (Figure 2E), indicating that dNF-κB #1 retains the ability to bind with NF-κB. Furthermore, we determined whether dNF-κB #1 is capable of recruiting the VHL E3 ubiquitin ligase to the POI, NF-κB. To this end, recombinant GST-VHL protein was purified from E. coli on GST-agarose beads, which was further incubated with cell lysis derived from HEK293T cells that ectopically expressed p65 protein in the presence of dNF-κB #1 (Figure S4A–C). As expected, dNF-κB #1 mediated the interaction between p65 and VHL E3 ubiquitin ligase (Figure 2F), suggesting that TF-PROTAC might be used for degradation of p65. However, we found that dNF-κB #1 could not efficiently promote the degradation of p65 in cells, which prompted us to further optimize the length and type of the linker between the VHL ligand and DNA oligomer for achieving effective destruction of NF-κB in cells.
dNF-κB Degrades p65 in Cells.
Thus, we further designed and synthesized another 17 BCN-modified VHL ligands, with linkers that are different in length and/or polarity (Figure 3A). Similar to VHLL-BCN #1, these 17 modified VHL ligands could be chemically clicked onto azide-modified DNA oligomers under physiological conditions (Figure S5A). Notably, the click efficiency of VHLL-BCN #2 - #5 and #11 - #18 was as high as VHLL-BCN #1 (Figures 3B, S5B,C). However, we noticed that the click efficiency gradually reduced when the length of the alkyl chain increased (Figures 3B, S5B,C). Importantly, when transfected into HeLa cells with liposome, five of the 18 dNF-κB led to a reduction of endogenous p65 protein abundance to less than 50% in comparison to control cells (Figure 3C), with dNF-κB #15 and #16 being the most effective. Moreover, in vivo pulldown assay and ubiquitination assay indicated that, consistent with their NF-κB degradation effect, these five dNF-κBs induced the interaction between VHL E3 ubiquitin ligase and p65 (Figure S5D) and efficient ubiquitination of p65 in cells (Figure 3D). Furthermore, in an unbiased global proteomic study using quantitative mass spectrometry, we found that dNF-κB #16 led to significant degradation of p65 (Figure 3E). Moreover, the majority of the 31 proteins with a reduced expression level in dNF-κB-treated samples, out of more than 4100 detected proteins, had a physical or a functional relationship with NF-κB/p65 (Figures 3E and S6), including the p65-binding partners IκBβ33 and CDK6,34 suggesting that this TF-PROTAC is relatively selective for p65.
Figure 3.
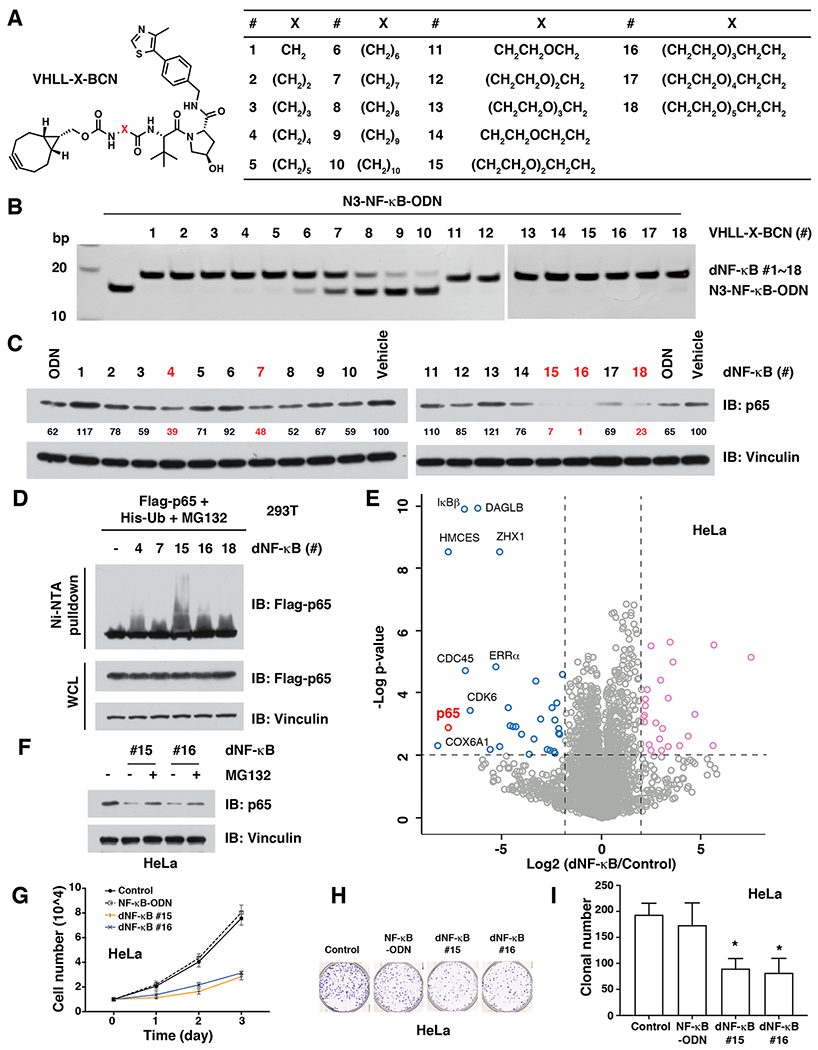
dNF-κB promotes the targeted degradation of the NF-κB transcription factor in cells. (A) Design of a series of 18 BCN-modified VHL ligands with various linkers between BCN and the VHL ligand. (B) In vitro click of VHLL-X-BCN (#1 - #18, 150 μM) with N3-NF-κB-ODN (50 μM), which can be separated by native PAGE. (C) Western blots for p65 in HeLa cells after treated with 10 μg/mL of dNF-κB (#1 - #18) for 12 h. (D) dNF-κB (#4, #7, #15, #16, and #18) led to the ubiquitination of p65 in cells. HEK293T cells were transfected with Flag-p65 for 24 h and then transfected with 10 μg/mL of indicated dNF-κB for another 12 h, followed by Western blotting for Flag-p65 in Ni-NTA pulldown and whole cell lysis (WCL). (E) Comparison of proteomic changes after treatment with dNF-κB #16 or control in HeLa cells. Dotted lines indicate either 50% loss or 2-fold increase of the protein level (x axis) and p = 0.01 (y axis). (F) The proteasome inhibitor MG132 blocked the degradation of endogenous p65 by dNF-κB (#15 and #16) in HeLa cells. HeLa cells were transfected with 10 μg/mL of indicated dNF-κB and then treated with 10 μM MG132, followed by Western blotting for p65. (G) dNF-κB (#15 and #16, 10 μ/mL) repressed the proliferation of HeLa cells. (H—I) dNF-κB (#15 and #16, 10 μg/mL) reduced the tumorigenesis of HeLa cells in a colony formation assay. *: p < 0.05.
To determine whether the degradation of p65 is due to an on-target effect of the TF-PROTAC rather than a nonspecific effect from the VHLL-BCN, we treated HeLa cells with several VHLL-BCN compounds alone and found that these VHLL-BCN compounds were incapable of reducing p65 (Figure S7A). Furthermore, the degradation of endogenous p65 by NF-κB degraders could be blocked by cotreatment with either the proteasome inhibitor MG132 (Figure 3F) or the VHL ligand VH-032 (Figure S7B). Consistent with these results, we also found that dNF-κB could not degrade p65 in HeLa cells after depletion of the endogenous VHL E3 ubiquitin ligase (Figure S7C). In addition, because the hydroxyl group in the VHL ligand moiety of dNF-κB is critical for binding with the VHL E3 ubiquitin ligase, we synthesized a negative control VHLL-BCN #16-NC by inversion of the stereochemistry of the hydroxyl group from R to S (Figure S7D) and found that the negative control dNF-κB #16-NC, resulting from the SPAAC reaction of VHLL-BCN #16-NC and N3-NF-κB-ODN, could not degrade p65 (Figure S7E). These results together indicate that these TF-PROTACs degrade p65 in a VHL- and proteasome-dependent manner. Lastly, treatment with dNF-κB #15 and #16, but not NF-κB-ODN, inhibited the proliferation (Figure 3G) and tumorigenesis ability (Figure 3H–I), presumably via promoting the degradation of the oncogenic TF NF-κB.
Design and Synthesis of dE2F.
To determine whether the TF-PROTAC platform could be applied to other transcription factors besides NF-κB, we further designed a double strand DNA 15-mers containing an E2F binding motif (TTTG/CG/CCGC) from MYC promoter,35 in which the sense chain is 5′-CTAGATTTCCCGCG-3′ and the antisense chain is 5′-CTAGCGCGGAAAT-3′,36,37 hereafter named as E2F-ODN (Figure S8A). Similarly, we also designed an azide-modified E2F-ODN (hereafter termed as N3-E2F-ODN) and biotin-modified E2F-ODN (hereafter termed as Biotin-E2F-ODN; Figures 4A and S8A,B). After annealing in vitro, the sense and antisense oligomers formed a double stand heterodimer, which could be quality controlled by separation in native PAGE (Figure 4B). Using a competitive binding assay, we found that N3-E2F-ODN was bound with the E2F1 transcription factor as efficiently as E2F-ODN in this experimental setting (Figures 4C and S8C,D).
Figure 4.
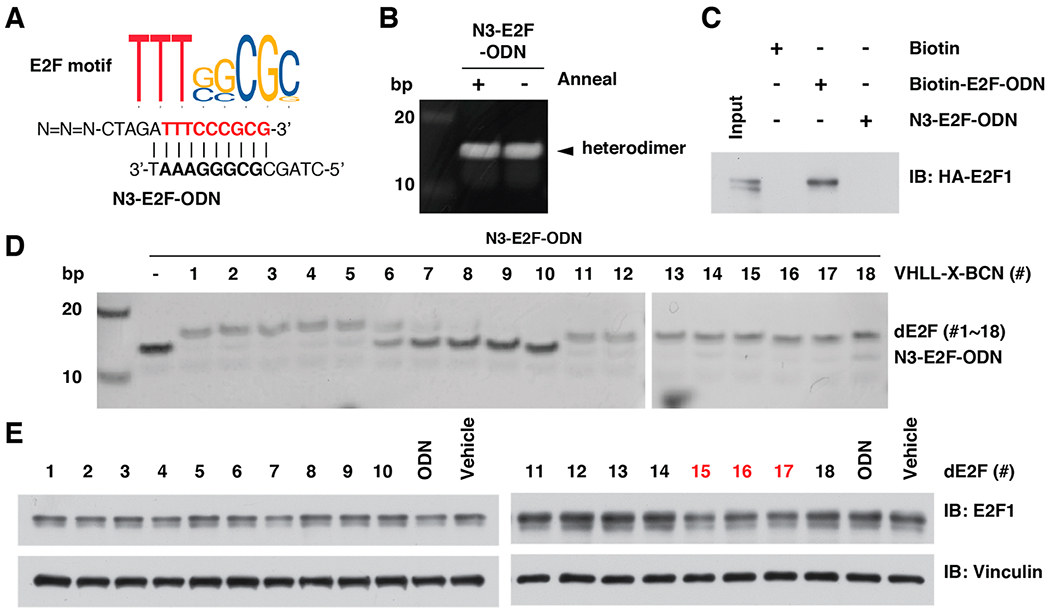
Design and in vitro click of dE2F. (A) A schematic diagram for the E2F motif and the sequence of N3-E2F-ODN. (B) Annealing of double strain N3-E2F-ODN. (C) N3-E2F-ODN was capable of binding with E2F1 transcription factor. (D) In vitro click of VHLL-BCN (#1 - #18, 150 μM) with N3-E2F-ODN (50 μM), which can be separated by 20% native PAGE. (E) Western blotting for E2F1 in HeLa cells after being treated with 10 μg/mL of dE2F (#1 - #18) for 12 h.
dE2F Degrades E2F1 in Cells.
Next, we measured the click efficiency between the 18 BCN-modified VHL ligands (VHLL-BCN #1 - #18) and N3-E2F-ODN (Figures 4D and S9A). Notably, VHLL-BCN #1 - #5 and #11 - #18 were efficiently clicked with N3-E2F-ODN, while VHLL-BCN #6 - #10 did not (Figure 4D). After transfection of these dE2F into HeLa cells with liposome, the in-cell degradation results indicated that two of the 18 dE2F1 (#16 and #17) led to a noticeable reduction of endogenous E2F1 protein abundance (Figures 4E and 5A), while the VHLL-BCN compounds (#15 – #18) could not (Figure S9B). Consistent with these results, dE2F1 (#16 and #17) induced the interaction between the VHL E3 ubiquitin ligase and E2F1 (Figure S9C), which led to the ubiquitination of E2F1 in cells (Figure 5B). Moreover, the degradation of E2F1 by dE2F #16 and #17 could be blocked by the proteasome inhibitor MG132 (Figure 5C), the VHL ligand VH-032 (Figure S9D), or depletion of the endogenous VHL E3 ligase (Figure S9E), indicating that these E2F1 TF-PROTACs function in a VHL- and proteasome-dependent manner. Consistent with these results, treatment with dE2F #16 or #17 inhibited the proliferation (Figure 5D) and tumorigenesis ability of HeLa cells (Figure 5E,F).
Figure 5.

dE2F promotes degradation of the E2F1 transcription factor in HeLa cells. (A) dE2F #16 and #17 degraded E2F1 in HeLa cells in a concentration-dependent manner. HeLa cells were transfected with 10 or 25 μg/mL of dE2F #16 or #17 for 12 h, followed by Western blotting for E2F1. (B) dE2F led to ubiquitination of E2F1 in cells. HeLa cells were transfected with HA-E2F1 for 24 h, then treated with 25 μg/mL of dE2F #16 or #17 for 12 h, followed by Western blotting for HA-E2F1 in Ni-NTA pulldown and WCL. (C) The proteasome inhibitor MG132 blocked the degradation of endogenous E2F1 by dE2F #16 and #17 in cells. HeLa cells were transfected with 25 μg/mL indicated dE2F1 and then treated with 30 μM MG132, followed by Western blotting for E2F1. (D) dE2F1 (#16 and #17, 25 μg/mL) repressed the proliferation of HeLa cells. (E, F) dE2F (#16 and #17, 25 μg/mL) reduced the tumorigenesis of HeLa cells in a colony formation assay. **, ***: p < 0.01, p < 0.001.
CONCLUSION
In summary, we designed, synthesized, characterized, and validated proof-of-concept TF-PROTACs targeting NF-κB and E2F. Our lead dNF-κB and dE2F induced TF-degrader-E3 ligase ternary complex formation and effectively degraded p65 and E2F1, respectively, in a VHL E3 ligase- and proteasome-dependent manner in cells. Moreover, these TF-PROTACs effectively inhibited the proliferation and tumorigenesis in cells, while the corresponding DNA oligomers did not. Collectively, our results strongly suggest that this TF-PROTAC platform can be a universal strategy for targeting “undruggable” TFs with a known motif, thereby significantly increasing the druggable target spectrum with enormous potential therapeutic benefits for patients with various diseases including cancer.
EXPERIMENTAL METHODS
General Chemistry Methods.
Common materials or reagents were purchased from commercial sources and used without further purification. High-performance liquid chromatography (HPLC) spectra were acquired using an Agilent 1200 Series system with DAD detector for all the intermediates and final products below. Chromatography was performed on a 2.1 × 150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High-resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Ultraperformance liquid chromatography (UPLC) spectra for compounds were acquired using a Waters Acquity I-Class UPLC system with a PDA detector. Chromatography was performed on a 2.1 Å~ 30 mm ACQUITY UPLC BEH C18 1.7 μm column with water containing 3% acetonitrile, 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.8 mL/min. The gradient program was as follows: 1–99% B (1–1.5 min), and 99–1% B (1.5–2.5 min). Nuclear Magnetic Resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) and 151 MHz for carbon (13C NMR); chemical shifts are reported in (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 220 or 254 nm. Samples were injected onto a Phenomenex Luna 250 × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 40 mL/min. A linear gradient was used with 10% of acetonitrile in H2O (with 0.08% NH4HCO3) (B) to 100% of acetonitrile (A). HPLC was used to test the purity of target compounds. All of the final compounds had >96% purity using the HPLC methods described above. The 18 VHL ligand liners, VHLL 1–18, were synthesized according to the published procedures.38 VH-032 was synthesized according to the published procedures.39
Oligomer Synthesis.
The single strand oligonucleotides containing the NF-κB binding motif, namely, NF-κB-ODN, was synthesized based on the HIV κB site.29,30 The sequence of 31-mer NF-κB-ODN is 5′-TGGGGACTTTCCAGTTTCTGGAAAGTCCCCA-3′ in which the κB site is underlined. Moreover, the azide modification version of NF-κB-ODN, namely, N3-NF-κB-ODN was further synthesized by incorporating an azide group on the 5′ end of NF-κB-ODN through the 5′ amino modifier C6. Similarly, the Biotin-NF-κB-ODN was synthesized by the addition of a biotin to the 5′-end of NF-κB-ODN using a spacer of NH2-(CH2)2-O-(CH2)2-OH. The double-stained 15-mers oligonucleotide, namely, E2F-ODN, contains the E2F binding motif derived from MYC promoter.35–37 The sense chain is 5′-CTAGATTTCCCGCG-3′ and the antisense chain is 5′-CTAGCGCGGAAAT-3′, in which the E2F binding site is underlined. Furthermore, N3-E2F-ODN and Biotin-E2F-ODN were synthesized by the addition of an azide group or biotin on the 5′-end of the sense stain oligomer.
All oligomers were synthesized by Integrated DNA Technologies, Inc. The unmodified oligomers were purified by standard desalting, while both azide- and biotin-modified oligomers were purified by HPLC. The single NF-κB-ODN (unmodified and modified) were annealed by heating the solution to 95 °C for 5 min, and cooling it to room temperature at 5 °C per min. Similarly, the sense and antisense E2F oligomers were mixed at a 1:1 ratio and the mixture was annealed before use.
In Vitro Copper-Free Strain-Promoted Azide—Alkyne Cycloaddition (SPAAC) Reaction.
For incorporation of the VHL ligand onto the oligomers, BCN-modified VHL ligands (VHLL-X-BCN) were incubated with either N3-NF-κB-ODN (50 μM) or the azide-modified sense stain of E2F oligomer (50 μM) in phosphate buffered saline (PBS) buffer at 37 °C for indicated time periods. The reaction mixtures were further purified by the Nucleotide Removal Kit (QIAGEN), followed by annealing as described above.
Native DNA Polyacrylamide Gel Electrophoresis (PAGE).
Oligomers (0.5-1 μg) were separated by 20% native polyacrylamide gel electrophoresis (PAGE) at 100 V for 1 h, followed by incubation in 0.2% EtBr solution in 1 X Tris-boric acid-EDTA (TBE, pH 8.3) buffer that consisted of 89 mM Tris, 89 mM boric acid, 2 mM EDTA. The native gels contained 20% acrylamide and 2.5% glycerol, 0.075 ammonium persulfate (APS), and 0.05% tetramethylethylenediamine (TEMED) in 0.5 X Tris-boric acid-EDTA (TBE) buffer. Finally, the gels were imaged with UV illumination with the ChemiDoc Touch Imaging System (Bio-Rad).
Cell Culture and Treatment.
Human embryonic kidney 293T (HEK293T) and HeLa cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (FBS), 100 units/mL of penicillin and 100 μg/mL streptomycin. For the ectopic expression of transcription factors, Flag-p65 and HA-E2F1 were transfected into HEK293T cells and harvested for lysis 48 h after transfection. For TF-PROTACs treatment, HeLa cells in a 12-well plate were transfected with individual TF-PROTACs for 12 h, followed by harvest for Western blot analysis. For proteasome or VHL ligand inhibition assay, cells were treated with respective TF-PROTACs, together with either 30 μM of MG132 (BML-Pl102, ENZO Life Sciences) or 10 μM of VHL ligand (VH-032) for 12 h. For depletion of endogenous VHL E3 ligase, two sgRNA for VHL E3 ligase were synthesized and inserted into lenti-CRISPR-V2 construct.40 The sgVHL lentivirus was generated in the HEK293T cells as previously described41,42 for infection of HeLa cells overnight, followed by selection with puromycin for 72 h.
Streptavidin–Biotin Pulldown Assay.
HEK293T cells that ectopically expressed either Flag-p65 or HA-E2F1 were lysed by EBC buffer (50 mM Tris pH 7.5, 120 mM NaCl, 0.5% NP-40) that was supplemented with protease inhibitors (Pierce) and phosphatase inhibitors (phosphatase inhibitor cocktail set I and II, Calbiochem). The protein concentrations of the lysates were measured using the Bio-Rad protein assay reagent on a Beckman Coulter DU-800 spectrophotometer. The cell lysates (1 mg) were further incubated with biotin-ODN (10 μg) at 4 °C for 3 h, followed by the addition of 10 μL of Streptavidin agarose beads (Thermo Fisher) for another 1 h. The beads were washed 4 times with NETN buffer (100 mM NaCl, 20 mM Tris-Cl, pH 8.0, 0.5 mM EDTA, 0.5% NP-40), boiled in SDS loading buffer, further separated by 10% SDS-PAGE, and blotted with individual antibody.
In Vivo Ubiquitination Assay.
Denatured in vivo ubiquitination assays were performed as previously described.41 Briefly, HEK293T cells were first transfected with Flag-p65 or HA-E2F1 for 24 h, followed by additional transfection of individual TF-PROTAC for another 12 h. Before been harvested, HEK293T cells were treated with 30 μM of MG132 for 4 h. After being harvested in denatured buffer A (6 M guanidine-HCl, pH 8.0, 0.1 M Na2HPO4/NaH2PO4, and 10 mM imidazole) and sonication for 10 s, cell lysis was incubated with Ni-nitrilotriacetic acid (NTA) matrices under rotation for 3 h at room temperature. The pulldown products were washed sequentially twice in buffer A, twice in buffer A/TI mixture (buffer A, buffer TI = 1:3, v/v), and once in buffer TI (25 mM tris-HCl, pH 6.8, and 20 mM imidazole). The ubiquitinated proteins were finally separated by 7% SDS-PAGE and blotted with individual antibody.
Western Blot Assay.
Cells were lysed in EBC buffer supplemented with protease inhibitors cocktail and phosphatase inhibitors, and the protein concentrations were measured as described above. The lysates (40 μg protein) were then resolved by 10% SDS-PAGE at 130 V for 80 min, immunoblotted with indicated antibodies at 4 °C overnight, washed 4 times with Tris-buffered saline with 0.1% Tween-20 (TBST), incubated with secondary antibody for 1 h at room temperature, and then washed 4 times with TBST buffer. Anti-p65 (#8742, 1:1000) and E2F1 (#3742, 1:500) antibodies were purchased from Cell Signaling Technologies. Anti-HA antibody (#901513, 1:1000) was purchased from Biolegend. Anti-Flag (F3165, 1: 5000), antivinculin antibody (V-4505, 1: 50000), peroxidase-conjugated antimouse secondary antibody (A-4416, 1:3000), and peroxidase-conjugated antirabbit secondary antibody (A-4914, 1:3000) were purchased from Sigma. All primary antibodies were diluted in 5% bovine serum albumin (BSA) in TBST buffer, and secondary antibodies were diluted in 5% nonfat milk in TBST buffer.
Purification of GST-VHL/Elongin B/C Complex.
pGEX-GST-VHL/ElonginB construct was kindly gifted by Dr. William Kaelin, Jr. at the Dana-Farber Cancer Institute, Harvard Medical School. pGEX-GST-Elongin C plasmid was generated by the subcloning of Elongin C CDS into the BamH1 and XhoI sites of pGEX-4T1-GST vector. The GST-VHL/ElonginB/C complex was purified as described.43 Briefly, BL21(DE3) E. coli was cotransfected with pGEX-GST-VHL/ElonginB and pGEX-GST-ElonginC plasmid. Protein expression was induced by 100 μM isopropyl beta-D-thiogalactopyranoside (IPTG, Fisher Scientific) at 18 °C overnight. Then, the bacterial culture was centrifuged and sonicated in PBS buffer with a protease inhibitor cocktail. Protein lysates were further purified with glutathione-Sepharose beads (GE17–0756–05) and washed with PBS for 5 times.
In Vitro GST Pulldown Assay.
Glutathione-Sepharose beads loaded with GST-VHL E3 ligase complex were incubated at 4 °C with a cell lysate (1 mg total protein) derived from HEK293T cells that were ectopically transfected with an individual transfection factor. After incubation for 3 h, TF-PROTACs (10 μg) were added into the mixture for another 2 h, followed by washing with an NETN buffer thrice. The pulldown products were further separated by 10% SDS-PAGE and blotted for indicated proteins.
Proteomics Sample Preparation.
HeLa cells were treated with either vehicle or dNF-κB #16 for 12 h, washed twice with cold PBS, and then harvested in PBS to pellet the cells. Cell pellets were resuspended in lysis buffer (8 M Urea, 50 mM Tris-HCl, pH 8.0), reduced with dithiothreitol (5 mM final) for 30 min at room temperature, and alkylated with iodoacetamide (15 mM final) for 45 min in the dark at room temperature. Samples were diluted 4-fold with digestion buffer (25 mM Tris-HCl, pH 8.0, 1 mM CaCl2) and digested with trypsin at 1:100 ratio (trypsin/protein, w/w) overnight at room temperature. Peptides were desalted on homemade C18 stagetips. There were two biological samples at each condition.
Mass Spectrometry Analysis.
Dried peptides were dissolved in 0.1% formic acid and 2% acetonitrile. Peptide concentration was measured with a Pierce Quantitative Colorimetric Peptide Assay (Thermofisher). A 0.5 μg portion of peptides was analyzed on a Q-Exactive HF-X coupled with an Easy nanoLC 1200 (Thermo Fisher Scientific, San Jose, CA). Peptides were loaded on to a nanoEase MZ HSS T3 Column (100 Å, 1.8 μm, 75 μm × 150 mm, Waters). Analytical separation of all peptides was achieved with a 130 min gradient. A linear gradient of 5 to 30% buffer B over 100 min and 30% to 45% buffer B over 20 min was executed at a 250 nL/min flow rate followed by a ramp to 100% B in 1 min and a 9 min wash with 100% B, where buffer A was aqueous 0.1% formic acid, and buffer B was 80% acetonitrile and 0.1% formic acid. LC-MS experiments were also carried out in a data-dependent mode with full MS (externally calibrated to a resolution of 60000 at m/z 200) followed by high energy collision-activated dissociation-MS/MS of the top 15 most intense ions with a resolution of 15000 at m/z 200. High energy collision-activated dissociation-MS/MS was used to dissociate peptides at a normalized collision energy of 27 eV in the presence of nitrogen bath gas atoms. Dynamic exclusion was 30 s. Each sample was subjected to two replicate technical LC-MS analyses.
Raw Proteomics Data Processing and Analysis.
Mass spectra were processed, and peptide identification was performed using the MaxQuant software version 1.6.10.43 (Max Planck Institute, Germany). Protein database searches were performed against the UniProt human protein sequence database (UP000005640). A false discovery rate (FDR) for both peptide-spectrum match (PSM) and protein assignment was set at 1%. Search parameters included up to two missed cleavages at Lys/Arg on the sequence, oxidation of methionine, and protein N-terminal acetylation as a dynamic modification. Carbamidomethylation of cysteine residues was considered as a static modification. Peptide identifications were reported by filtration of reverse and contaminant entries and assignment to their leading razor protein. Data processing and statistical analysis were performed on Perseus (Version 1.6.10.50). Quantitation using iBAQ intensity (Intensity-Based Absolute Quantification) was performed with a p-value of 0.01 to report statistically significant abundance fold-changes.
Cell Proliferation and Clonal Formation Assay.
For the cell growth curve assay, HeLa cells (10000/well) in a 12-well plate were transfected with individual TF-PROTAC; then the cells were trypsined and the cell numbers were counted at the indicated time. For the clonal formation assay, HeLa cells were transfected with individual TF-PROTAC for 12 h, followed by being further plated into a 6-well plate (1000/well). Three weeks later, the cells were fixed in fixation buffer (acetic acid/methanol = 1:7) and stained with 0.4% crystal violet in 20% ethanol. Then, the clonal numbers were quantified by ImageJ software.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the NIH grants R35CA253027 (W.W.), R01CA218600 (J.J.), R01CA230854 (J.J.), R01GM122749 (J.J.), R01CA260666 (J.J.), and P30CA196521 (J.J.). This work utilized the AVANCE NEO 600 MHz NMR Spectrometer System that was upgraded with funding from a National Institutes of Health SIG grant 1S10OD025132-01A1. We thank Wei lab members for critical reading of the manuscript, and members of the Wei and Jin laboratories for helpful discussions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c03852.
Compound synthesis and experimental details (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.1c03852
The authors declare the following competing financial interest(s): W.W. is a co-founder and stockholder of the Rekindle Thera-peutics. J.J. is a co-founder, equity shareholder and consultant of Cullgen, Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, and Cullgen, Inc. All other authors declare no competing interests.
Contributor Information
Jing Liu, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
He Chen, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
H. Ümit Kaniskan, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Ling Xie, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Xian Chen, Department of Biochemistry and Biophysics, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences and Oncological Sciences, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York 10029, United States.
Wenyi Wei, Department of Pathology, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts 02215, United States.
REFERENCES
- (1).Mitchell PJ; Tjian R, Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 1989, 245 (4916), 371–378. [DOI] [PubMed] [Google Scholar]
- (2).Lambert SA; Jolma A; Campitelli LF; Das PK; Yin Y; Albu M; Chen X; Taipale J; Hughes TR; Weirauch MT, The human transcription factors. Cell 2018, 172 (4), 650–665. [DOI] [PubMed] [Google Scholar]
- (3).Darnell JE Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2 (10), 740–749. [DOI] [PubMed] [Google Scholar]
- (4).Bushweller JH, Targeting transcription factors in cancer - from undruggable to reality. Nat. Rev. Cancer 2019, 19 (11), 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tsherniak A; Vazquez F; Montgomery PG; Weir BA; Kryukov G; Cowley GS; Gill S; Harrington WF; Pantel S; Krill-Burger JM; Meyers RM; Ali L; Goodale A; Lee Y; Jiang G; Hsiao J; Gerath WFJ; Howell S; Merkel E; Ghandi M; Garraway LA; Root DE; Golub TR; Boehm JS; Hahn WC, Defining a cancer dependency map. Cell 2017, 170 (3), 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Chen A; Koehler AN, Transcription factor inhibition: lessons learned and emerging targets. Trends Mol. Med 2020, 26 (5), 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gilmore TD; Herscovitch M, Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25 (51), 6887–6899. [DOI] [PubMed] [Google Scholar]
- (8).Gupta SC; Sundaram C; Reuter S; Aggarwal BB, Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim. Biophys. Acta, Gene Regul Mech 2010, 1799 (10–12), 775–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Furqan M; Akinleye A; Mukhi N; Mittal V; Chen Y; Liu D, STAT inhibitors for cancer therapy. J. Hematol Oncol 2013, 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Schust J; Sperl B; Hollis A; Mayer TU; Berg T, Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol 2006, 13 (11), 1235–1242. [DOI] [PubMed] [Google Scholar]
- (11).Song H; Wang R; Wang S; Lin J, A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (13), 4700–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wei N; Li J; Fang C; Chang J; Xirou V; Syrigos NK; Marks BJ; Chu E; Schmitz JC, Targeting colon cancer with the novel STAT3 inhibitor bruceantinol. Oncogene 2019, 38 (10), 1676–1687. [DOI] [PubMed] [Google Scholar]
- (13).Yin X; Giap C; Lazo JS; Prochownik EV, Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 2003, 22 (40), 6151–6159. [DOI] [PubMed] [Google Scholar]
- (14).Han H; Jain AD; Truica MI; Izquierdo-Ferrer J; Anker JF; Lysy B; Sagar V; Luan Y; Chalmers ZR; Unno K; Mok H; Vatapalli R; Yoo YA; Rodriguez Y; Kandela I; Parker JB; Chakravarti D; Mishra RK; Schiltz GE; Abdulkadir SA, Small-molecule MYC inhibitors suppress tumor growth and enhance immunotherapy. Cancer Cell 2019, 36 (5), 483–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Watson PA; Arora VK; Sawyers CL, Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15 (12), 701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wong YN; Ferraldeschi R; Attard G; de Bono J, Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat. Rev. Clin. Oncol 2014, 11 (6), 365–376. [DOI] [PubMed] [Google Scholar]
- (17).Kono M; Fujii T; Lim B; Karuturi MS; Tripathy D; Ueno NT, Androgen receptor function and androgen receptor-targeted therapies in breast cancer: a review. JAMA Oncol. 2017, 3 (9), 1266–1273. [DOI] [PubMed] [Google Scholar]
- (18).Riggs BL; Hartmann LC, Selective estrogen-receptor modulators — mechanisms of action and application to clinical practice. N. Engl. J. Med 2003, 348 (7), 618–629. [DOI] [PubMed] [Google Scholar]
- (19).Fornes O; Castro-Mondragon JA; Khan A; Van der Lee R; Zhang X; Richmond PA; Modi BP; Correard S; Gheorghe M; Baranaţć D, JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2019, 48 (1), 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Pujato M; Kieken F; Skiles AA; Tapinos N; Fiser A, Prediction of DNA binding motifs from 3D models of transcription factors; identifying TLX3 regulated genes. Nucleic Acids Res. 2014, 42 (22), 13500–13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gerstein MB; Kundaje A; Hariharan M; Landt SG; Yan KK; Cheng C; Mu XJ; Khurana E; Rozowsky J; Alexander R; Min R; Alves P; Abyzov A; Addleman N; Bhardwaj N; Boyle AP; Cayting P; Charos A; Chen DZ; Cheng Y; Clarke D; Eastman C; Euskirchen G; Frietze S; Fu Y; Gertz J; Grubert F; Harmanci A; Jain P; Kasowski M; Lacroute P; Leng JJ; Lian J; Monahan H; O’Geen H; Ouyang Z; Partridge EC; Patacsil D; Pauli F; Raha D; Ramirez L; Reddy TE; Reed B; Shi M; Slifer T; Wang J; Wu L; Yang X; Yip KY; Zilberman-Schapira G; Batzoglou S; Sidow A; Farnham PJ; Myers RM; Weissman SM; Snyder M, Architecture of the human regulatory network derived from ENCODE data. Nature 2012, 489 (7414), 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Berger MF; Philippakis AA; Qureshi AM; He FS; Estep PW 3rd; Bulyk ML, Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol 2006, 24 (11), 1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Weirauch MT; Yang A; Albu M; Cote AG; Montenegro-Montero A; Drewe P; Najafabadi HS; Lambert SA; Mann I; Cook K; Zheng H; Goity A; van Bakel H; Lozano JC; Galli M; Lewsey MG; Huang E; Mukherjee T; Chen X; Reece-Hoyes JS; Govindarajan S; Shaulsky G; Walhout AJM; Bouget FY; Ratsch G; Larrondo LF; Ecker JR; Hughes TR, Determination and inference of eukaryotic transcription factor sequence specificity. Cell 2014, 158 (6), 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jolma A; Yan J; Whitington T; Toivonen J; Nitta KR; Rastas P; Morgunova E; Enge M; Taipale M; Wei G; Palin K; Vaquerizas JM; Vincentelli R; Luscombe NM; Hughes TR; Lemaire P; Ukkonen E; Kivioja T; Taipale J, DNA-binding specificities of human transcription factors. Cell 2013, 152 (1–2), 327–339. [DOI] [PubMed] [Google Scholar]
- (25).Dale B; Cheng M; Park K-S; Kaniskan HÜ; Xiong Y; Jin J, Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer, 2021, in press. DOI: 10.1038/s41568-021-00365-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu J; Ma J; Liu Y; Xia J; Li Y; Wang ZP; Wei W, PROTACs: a novel strategy for cancer therapy. Semin. Cancer Biol 2020, 67 (2), 171–179. [DOI] [PubMed] [Google Scholar]
- (27).Ghidini A; Clery A; Halloy F; Allain FHT; Hall J, RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem., Int. Ed 2021, 60 (6), 3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Samarasinghe KTG; Jaime-Figueroa S; Burgess M; Nalawansha DA; Dai K; Hu Z; Bebenek A; Holley SA; Crews CM, Targeted Degradation of Transcription Factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol 2021, 28 (5), 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nabel G; Baltimore D, An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature 1987, 326 (6114), 711–713. [DOI] [PubMed] [Google Scholar]
- (30).Bielinska A; Shivdasani RA; Zhang LQ; Nabel GJ, Regulation of gene expression with double-stranded phosphorothioate oligonucleotides. Science 1990, 250 (4983), 997–1000. [DOI] [PubMed] [Google Scholar]
- (31).Govan JM; Lively MO; Deiters A, Photochemical control of DNA decoy function enables precise regulation of nuclear factor kappaB activity. J. Am. Chem. Soc 2011, 133 (33), 13176–13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Maksimenko AV; Gottikh MB; Helin V; Shabarova ZA; Malvy C, Physico-chemical and biological properties of antisense phosphodiester oligonucleotides with various secondary structures. Nucleosides Nucleotides 1999, 18 (9), 2071–2091. [DOI] [PubMed] [Google Scholar]
- (33).Malek S; Huang DB; Huxford T; Ghosh S; Ghosh G, X-ray crystal structure of an IκBβ•NF-κB p65 homodimer complex. J. Biol. Chem 2003, 278 (25), 23094–23100. [DOI] [PubMed] [Google Scholar]
- (34).Handschick K; Beuerlein K; Jurida L; Bartkuhn M; Muller H; Soelch J; Weber A; Dittrich-Breiholz O; Schneider H; Scharfe M; Jarek M; Stellzig J; Schmitz ML; Kracht M, Cyclin-dependent kinase 6 is a chromatin-bound cofactor for NF-κB-dependent gene expression. Mol. Cell 2014, 53 (2), 193–208. [DOI] [PubMed] [Google Scholar]
- (35).Thalmeier K; Synovzik H; Mertz R; Winnacker EL; Lipp M, Nuclear factor E2F mediates basic transcription and transactivation by E1a of the human MYC promoter. Genes Dev. 1989, 3 (4), 527–536. [DOI] [PubMed] [Google Scholar]
- (36).Morishita R; Gibbons GH; Horiuchi M; Ellison KE; Nakama M; Zhang L; Kaneda Y; Ogihara T; Dzau VJ, A gene therapy strategy using a transcription factor decoy of the E2F binding site inhibits smooth muscle proliferation in vivo. Proc. Natl. Acad. Sci. U. S. A 1995, 92 (13), 5855–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ehsan A; Mann MJ; Dell’Acqua G; Tamura K; Braun-Dullaeus R; Dzau VJ, Endothelial healing in vein grafts: proliferative burst unimpaired by genetic therapy of neointimal disease. Circulation 2002, 105 (14), 1686–1692. [DOI] [PubMed] [Google Scholar]
- (38).Liu J; Plewe MB; Wang J; Han X; Chen L, Preparation of pyrazolopyridines and related heterocycles as CBP and p300 degradation bivalent compds for treatment of diseases. PCT, WO 2020173440 A1, 2020.
- (39).Galdeano C; Gadd MS; Soares P; Scaffidi S; Van Molle I; Birced I; Hewitt S; Dias DM; Ciulli A, Structure-guided design and optimization of small molecules targeting the protein-protein interaction between the von Hippel-Lindau (VHL) E3 ubiquitin ligase and the hypoxia inducible factor (HIF) alpha subunit with in vitro nanomolar affinities. J. Med. Chem 2014, 57 (20), 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sanjana NE; Shalem O; Zhang F, Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 2014, 11 (8), 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Liu J; Chen H; Ma L; He Z; Wang D; Liu Y; Lin Q; Zhang T; Gray N; Kaniskan HU; Jin J; Wei W, Light-induced control of protein destruction by opto-PROTAC. Sci. Adv 2020, 6 (8), eaay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Liu J; Chen H; Liu Y; Shen Y; Meng F; Kaniskan HU; Jin J; Wei W, Cancer selective target degradation by folate-caged PROTACs. J. Am. Chem. Soc 2021, 143 (19), 7380–7387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Miller F; Kentsis A; Osman R; Pan ZQ, Inactivation of VHL by tumorigenic mutations that disrupt dynamic coupling of the pVHL•hypoxia-inducible transcription factor-1α complex. J. Biol. Chem 2005, 280 (9), 7985–7996. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.