

Abstract
Rationale:
Cleavage of the extra-cellular domain of the (pro)renin receptor (PRR) yields a soluble fragment (sPRR). Although changes in plasma sPRR levels have been reported in hypertension, the causal role of sPRR in blood pressure (BP) regulation is unknown.
Objective:
Determine the role of sPRR in BP regulation at baseline and following Ang-II induced hypertension.
Methods and Results:
CRISPR-Cas9 was used to mutate the cleavage site of the PRR such that sPRR is not generated. Because the gene encoding PRR is on the X-chromosome and male mutant sPRR mice are infertile, only male mice were studied. Mutant sPRR mice had virtually undetectable plasma sPRR levels compared to littermate controls. Mutant sPRR mice had normal survival and development and no apparent histological abnormalities in the kidney, heart or aorta despite lower body weight. During normal Na+ intake, no differences in food or water intake, urinary water or Na+ excretion, or acid-base status were observed between control and mutant sPRR mice. Compared to controls, mutant sPRR mice had lower BP at baseline and an attenuated hypertensive response to 2 weeks of Ang-II infusion (400 ng/kg/min) which was partially reversed by infusion of mouse recombinant sPRR. Mutant sPRR mice also had lower albuminuria, renal tubular injury and oxidative stress relative to control mice post Ang-II infusion. Further, mesenteric arteries from mutant sPRR mice displayed reduced Ang-II-induced vasocontraction and greater acetylcholine, but not sodium nitroprusside, evoked vasorelaxation under baseline conditions.
Conclusions:
Loss of sPRR reduces BP at baseline and decreases Ang-II induced hypertension and renal injury. These effects of sPRR loss are associated with greater endothelium-dependent but not independent vasorelaxation of resistance-sized arteries.
Subject Terms: Basic Science Research, High Blood Pressure, Hypertension, Nephrology and Kidney, Vascular Biology
Keywords: (pro)renin receptor, PRR, soluble PRR, CRISPR-Cas9, blood pressure, renin-angiotensin system, hypertension, kidney, hypertension
Graphical Abstract
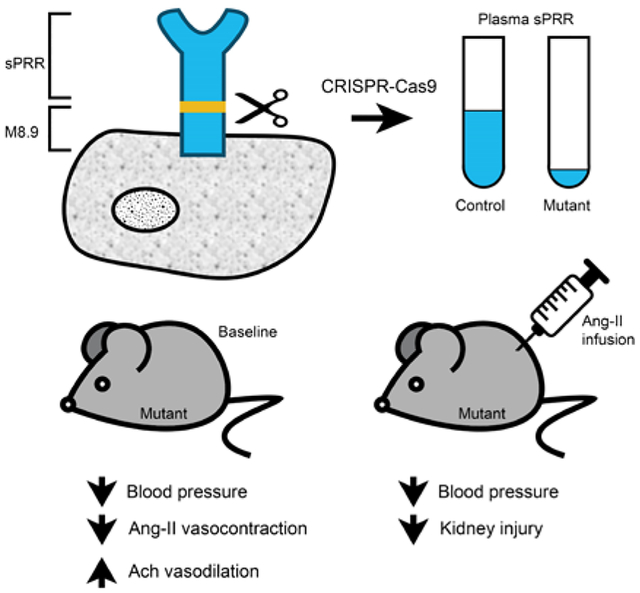
Elevated plasma sPRR levels have been reported in hypertension and kidney disease. However, the causal role of sPRR in BP regulation is unknown. We developed a novel mouse model with mutation in the cleavage site of the PRR such that sPRR is not generated. Mice with loss of sPRR have lower BP at baseline and following Ang-II infusion and attenuated kidney injury. sPRR effects on BP are mediated via renin angiotensin aldosterone system (RAAS) independent signaling through vascular endothelium dependent factors. Further, infusion of recombinant sPRR partially reverses the hypotension suggesting that sPRR likely plays a direct role in modulating BP.
INTRODUCTION
The renin angiotensin aldosterone system (RAAS) plays an essential role in maintaining blood pressure (BP) and Na+ homeostasis. The (pro)renin receptor (PRR) is a recently discovered component of the RAAS1 and is involved in the pathogenesis of hypertension2, 3. The PRR can exist as the full-length form, bound to the cell membrane, or be cleaved to generate a soluble PRR (sPRR) and M8.9 fragment2, 3.
PRR regulation of cell function depends on whether it is intact or cleaved into its constituent forms. Binding of prorenin to the PRR induces non-proteolytic activation of prorenin to mediate angiotensinogen cleavage while renin bound to the PRR has 4-fold higher catalytic efficiency compared to unbound renin, ultimately leading to increased angiotensin-II (Ang-II) synthesis1. Independent of Ang-II generation, prorenin/renin binding to the PRR activates intracellular signaling pathways such as mitogen activated kinase (p38 MAPK) and extracellular signal-regulated kinase (ERK1/2)1, 4, 5. The PRR is also involved in the Wnt/β catenin signaling cascade6, a function not contingent on prorenin/renin binding. Further, the M8.9 fragment is thought to serve as an accessory subunit of the vacuolar ATPase and is involved in lysosomal acidification7, 8. The sPRR, on the other hand, retains the ability to bind prorenin/renin and can potentially activate both Ang-II dependent and/or independent cell signaling pathways. Despite considerable advances in uncovering PRR function, the regulation and functional importance of the sPRR remains largely unknown.
Recent evidence suggests that site-1 protease and furin cleave the PRR sequentially at the Golgi apparatus and trans-Golgi network to generate the sPRR which is then secreted extracellularly9–11. Elevated plasma sPRR levels have been described in pre-eclampsia, heart failure and kidney disease12–17. Similarly, mice with elevated plasma sPRR levels are hypertensive18–20 and show activation of RAAS18 and autonomic nervous system pathways18–20. Additionally, rats with Ang-II infused hypertension have increased urinary sPRR levels21.
Based on these above findings, we hypothesized that the sPRR regulates BP via regulation of renal and cardiovascular function. To investigate this, we developed mice with absence of sPRR via site directed mutagenesis of the PRR cleavage site using Clustered Regularly Interspaced Short Palindromic Repeats – CRISPR associated genes 9 (CRISPR-Cas9). Herein, we show that mice lacking sPRR have lower BP at baseline and in Ang-II induced hypertension with attenuated kidney injury. We also observed attenuated Ang-II induced vasocontraction together with greater endothelium-dependent but not independent vasorelaxation in resistance arteries from mutant sPRR mice as compared to control mice. Collectively, these findings suggest that sPRR likely mediates BP via RAAS independent signaling.
METHODS
Data Availability.
The data supporting findings of this study are available from the corresponding author upon reasonable request.
Generation of mutant sPRR mice.
Generation of the sPRR occurs by sequential processing of the PRR by furin and site-1 protease9–11. Therefore, we used CRISPR-Cas9 (Cyagen Inc, Santa Clara, CA) to target both cleavage sites of the PRR so that sPRR generation is completely blocked. Briefly, candidate guide RNAs were designed to target and introduce two point mutations (R276A and R279A) into exon 8 of the ATP6AP2 gene. The resultant homology-directed repair (AGG>GCG) alters the furin and site-1 protease sites of the PRR and prevents cleavage of sPRR. Cas9 mRNA and gRNAs with targeting vector and donor oligo were co-injected into mouse embryos on a C57BL/6J background. The pups were genotyped by PCR and sequencing performed to identify founder mice with the mutated cleavage sites. Off-target analyses identified five potential sites, none of which were found to be altered in the founder mice.
Plasma sPRR levels were used to confirm absence of sPRR and measured using enzyme immunoassay (IBL America, Minneapolis, MN). Littermates without the mutation were used as controls. Details on genotyping are provided in the data supplement.
Histology and Immunofluorescence.
Mice were euthanized at 2 months of age for histological studies. Immunofluorescence for LAMP-2 (1:50; catalog no. ABL-93, DSHB, Iowa City, IA) was performed on deparaffinized kidney, heart and aorta sections. To assess cardiac myocyte area, deparaffinized heart sections were stained with wheat germ agglutinin (WGA, Alexa Fluor 488, catalog no: W11621, Thermo Fisher) and cardiomyocyte cross sectional area (μm2) was quantified using CellSens Dimension software. Further details are provided in the data supplement.
Body composition and indirect colorimetry analysis.
Body composition and indirect colorimetry analysis was performed at the Metabolic Phenotyping Core at the University of Utah in conscious control and mutant sPRR mice between 2–3 months of age.
Metabolic balance studies.
Control and mutant sPRR mice (n=10/group) were given 9 ml of a gelled normal Na+ diet (Test Diet no. 7551, St. Louis, MO) for 3 days with free access to water. Mice were placed in metabolic cages for measurement of food and water intake, body weight, and 24-h urine collection. At the end of the metabolic balance studies, mice were sacrificed and plasma and tissue harvested for further analyses.
Blood and urine assays.
Blood pH, electrolytes, blood urea nitrogen and hemoglobin were analyzed using the iSTAT analyzer (Abbott Laboratories, Abbott Park, IL). Urinary Na+ and K+ were determined using an EasyVet Analyzer (Medica, Bedford, MA). Urine osmolality were measured using Osmett II (Precision System, Natick, MA). Urine AVP was assayed using an enzyme immunoassay (EIA) (Enzo Life Sciences, Farmingdale, NY). Plasma angiotensinogen was measured using EIA (IBL America, Minneapolis, MN). Plasma renin concentration was measured as the amount of Ang-I generated after incubation with excess porcine angiotensinogen using the Ang-I EIA kit (Peninsula Laboratories, San Carlos, CA). Plasma and urine aldosterone were measured using EIA (Enzo Life Sciences, Farmingdale, NY).
Angiotensin measurements.
In a separate group of mice (n=10/group), angiotensin peptides were analyzed in equilibrated heparin plasma samples and whole kidneys using LC-MS/MS (Attoquant Diagnostics, Vienna, Austria) as previously described18. Quantification of angiotensin peptides was performed for Ang-I, Ang-II, Ang-III (2–8), Ang-IV (3–8), Ang 1–7, and Ang 1–5.
Blood pressure monitoring.
BP was recorded via telemetry (TA11-PAC10; Data Sciences International, St. Paul, MN) as described in the Data supplement.
Angiotensin II infusion.
After 3 days of continuous BP monitoring, mini-osmotic pumps (Alzet model 1002, Durect, Cupertino, CA) were placed subcutaneously in between the scapulae under isoflurane anesthesia. Ang-II was infused for 14 days at 400 ng·kg−1·min−1 and BP was recorded continuously for 14 days on a normal-Na+ diet. At the end of the BP studies, mice were allowed to recover for 6 weeks during which BP returned to basal levels. A second mini-osmotic pump containing Ang-II was implanted under isoflurane anesthesia, taking care to avoid the previous pump. Mice were allowed to recover for 18 hours and then placed in metabolic cages for 8 days for urine collection. Metabolic balance studies were limited to the first 8 days of Ang-II infusion so that any differences in electrolyte excretion are apparent before the mice reach steady state. Cumulative Na+ and K+ excretion was calculated as continuous summation by each consecutive day. Urine albumin was measured using an EIA kit (Exocell, Philadelphia, PA) while plasma blood urea nitrogen was determined using a quantitative colorimetric method (BioAssay Systems, Hayward, CA). Specific EIA kits were used to measure urine kidney injury molecule-1 (KIM-1, Abcam) and thiobarbituric acid reactive substances (TBARS, Cayman Chemical, Ann Arbor, MI).
Echocardiography.
Transthoracic echocardiography was performed on control and mutant sPRR mice at baseline and on days 12–13 during the second Ang-II infusion by the Small Animal Ultrasound Core at the University of Utah.
Recombinant sPRR infusion.
In a separate group of mice, after recovery following placement of telemetry devices, mouse recombinant sPRR (30 μg·kg−1·min−1 (residues 18–276, Genscript)18 was infused via mini-osmotic pumps. Mice were randomized to receive either vehicle or recombinant sPRR. BP was monitored for 4 days and a second osmotic mini-pump placed for Ang-II infusion at 400 ng·kg−1·min−1. BP was recorded continuously for 12 days on a normal-Na+ diet.
Mesenteric artery vasoreactivity.
Detailed experimental protocol for mesenteric artery vasoreactivity is described in the Data supplement. Vasocontractile responses to potassium chloride (KCl, 20–100 mM), phenylephrine (PE, 10−8-10−5 M) and Ang II (10−9-10−5 M) and vasorelaxation to acetylcholine (10−8-10−5 M) and sodium nitroprusside (10−9-10−4 M) was examined. Values from two mesenteric artery segments per mouse were averaged23.
Since we observed blunted Ang-II induced vasoconstriction in mesenteric arteries from mutant sPRR mice, additional experiments were performed to identify potential mechanisms. In a separate group of control and mutant sPRR mice, mesenteric arteries were isolated and prepared as described. After determining vasocontractile responses to KCl (30 minutes later), mesenteric arteries from both groups were incubated i) NG- Methyl-L-Arginine acetate (L-NMMA, 1 mmol/L; to inhibit nitric oxide synthase); (ii) indomethacin (10 μmol/L; to inhibit products of cyclooxygenase metabolism); (iii) apamin (1 μmol/L; to inhibit small conductance Ca2+ -activated K+ channels) + charybdotoxin (100 nmol/L; to inhibit intermediate and large conductance Ca2+ -activated K+ channels); or (iv) or tempol (100 μmol/L; a superoxide dismutase mimetic). After treatment with each compound for 30-min, Ang II-induced vasocontraction was evaluated.
Quantitation of Ang-II receptor mRNA.
RNA was isolated from mesenteric arteries of control and mutant sPRR mice and relative expression of Ang-II receptor Type IA, Type IB and Type II using TaqMan gene expression assay (Probe cat. Mm00616371-m1, Mm02620758-s1, Mm00431727-g1 respectively).
Statistical analysis.
No mice were excluded from the analyses. GraphPad Prism 9 was used to perform all statistical analysis. All results are expressed as means ± SE. For BP, mixed effects model with Geisser-Greenhouse correction was used to compare differences over time. Student’s unpaired t-test (2 groups) and one-way ANOVA with Tukey post-hoc test (>2 groups) was used to examine differences in BP at pre-determined time points (baseline, days 7 and 14 of infusion). Two-way ANOVA with Bonferroni correction was used to examine differences in vascular reactivity and post-Ang-II infusion parameters between control and mutant sPRR mice. For all other parameters, all groups were assessed for normal distribution with Shapiro-Wilk Test (p<0.05). Upon confirmation of normal distribution, the unpaired 2-tailed Student’s t test was used to compare differences between control and mutant sPRR mice. When normal distribution was not confirmed or when n<6/group, Mann-Whitney nonparametric tests were used to compare differences between groups. Repeated measures ANOVA with Bonferroni correction was used to examine differences in cumulative urinary Na+ and K+ excretion between control and mutant sPRR mice. The criterion for significance was p ≤ 0.05.
RESULTS
Generation of mutant sPRR mice.
Using CRISPR-Cas9, point mutations were introduced in Exon 8 of the ATP6AP2 gene such that both furin and site-1 protease cleavage sites were altered (Figure 1A). Since the ATP6AP2 gene is located on the X-chromosome, male mice are hemizygous while female mice are heterozygous for the cleavage site mutation. Female mutant sPRR mice were bred with wild-type male mice on C57BL/6J background as male mutant mice were found to be infertile.
Figure 1.
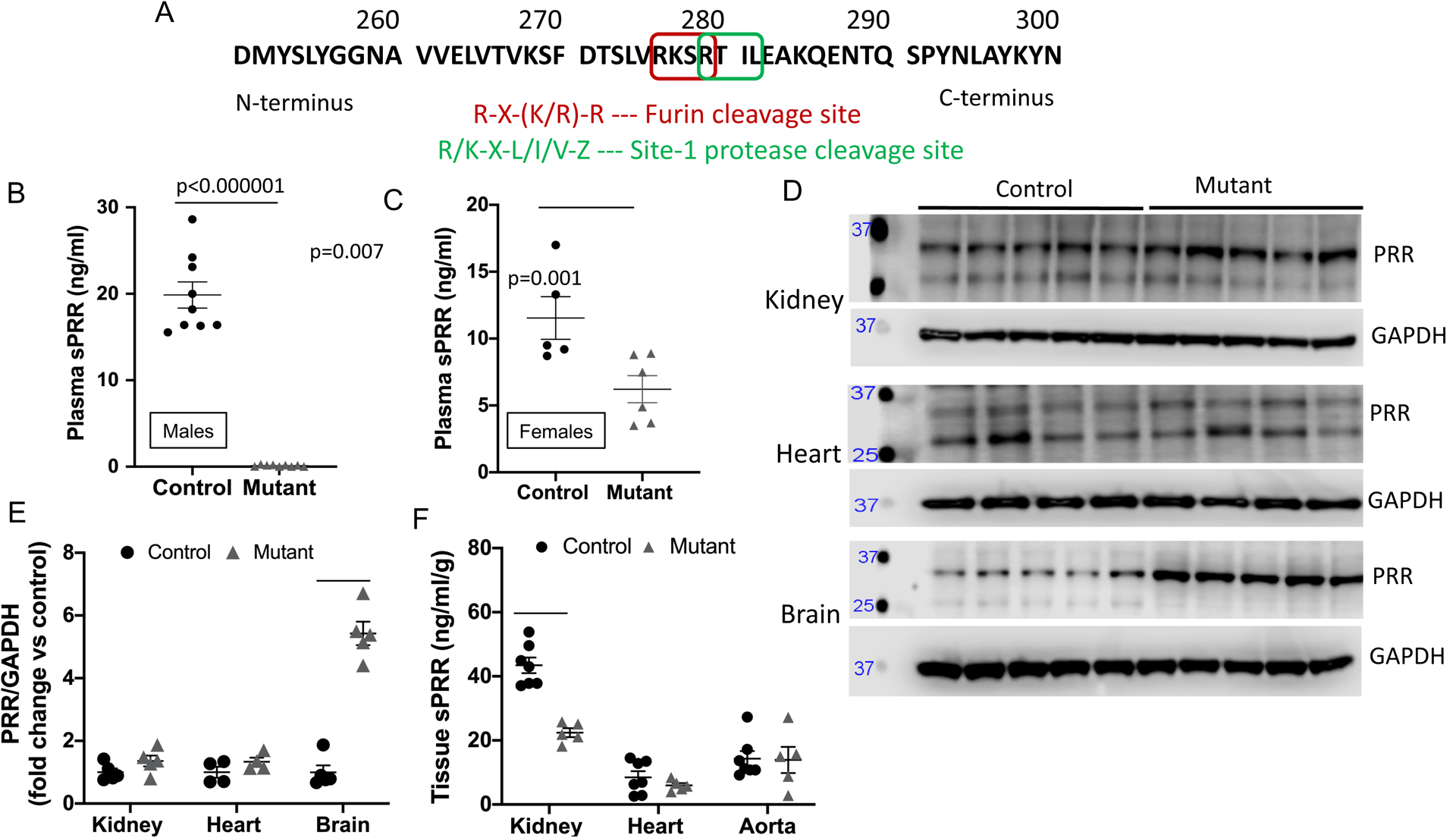
A: Protein sequence of the (pro)renin receptor with cleavage sites and sequence motifs. R – arginine, X – any amino acid, K – lysine, L – Leucine, I – Isoleucine, V- Valine, Z – Leucine or Threonine; B: plasma sPRR levels by EIA in male control (n=9) and mutant sPRR mice (n=10); C: plasma sPRR levels by EIA in female control and mutant sPRR mice (n=5/group; D: western blot of full length PRR in whole kidney (n=5/group), heart (n=4/group), brain (n=5/group); E: densitometry of full length PRR in whole kidney, heart and brain; F: tissue sPRR in whole kidney, heart and aorta by EIA. P value calculated using Student’s unpaired t-test for Figure 1B and Mann-Whitney non-parametric test for all other comparisons.
Mutant sPRR mice were born at the expected frequency and survived up to 24 months of age although body weight was lower in mutant sPRR mice. Compared to littermate controls, plasma sPRR was essentially undetectable in male mutant sPRR mice (Figure 1B) while female mutant sPRR mice had a 40% reduction in plasma sPRR (Figure 1C) levels. Hence, all further studies were performed using male mice to allow clear interpretation of the role of sPRR in BP regulation.
Mutant sPRR mice had lower body weight compared to controls, while basal plasma electrolytes and blood gas values were similar between the two groups (Online Table I). Since previous studies with constitutive deletion of the PRR resulted in abnormal lysosomal acidification24–27 and the M8.9 fragment is associated with the vacuolar ATPase, autophagy was examined in control and mutant sPRR mice for lysosomal associated membrane protein-2 (LAMP2) and p62. Immunofluorescence for LAMP-2 was similar between control and mutant sPRR mice in the kidney, heart and aorta (Online Figure IA). Similarly, abundance of p62 were comparable between control and mutant sPRR mice in whole kidney lysates and heart (Online Figure IC–D). Abundance of full length intact PRR was not different between control and mutant sPRR mice in the kidney or heart (Figure 1D–E); however, mutant sPRR mice had significantly increased abundance of PRR in the brain compared to controls (Figure 1D–E). Tissue sPRR by EIA was reduced by 50% in whole kidney but not heart or aorta (Figure 1F). It is important to note that sPRR measurements by EIA does not differentiate between full length intact PRR and sPRR at a tissue level; hence, the 50% reduction in kidney sPRR levels are presumably due to measurement of intact PRR levels at the tissue level (p values in Figure 1E–F are not corrected for multiple testing). No gross anatomical abnormalities were observed in the heart, kidney or aorta between control and mutant sPRR mice (Online Figure IB).
Compared to controls, mutant sPRR mice had higher lean body mass without significant differences in fat or fluid mass by NMR analyses (Online Figure IIA). Indirect colorimetry using CLAMS metabolic chambers demonstrated comparable food and water intake (Online Figure IIB–C) and energy expenditure (Online Figure IID–G) in control and mutant sPRR mice during active and inactive periods. Tissue weights for kidney, liver and epididymal fat were lower in mutant mice compared to controls (Online Table II).
Effects of sPRR deficiency on Na+ and water balance and RAAS activity.
On a normal Na+ diet, food intake was slightly lower in mutant sPRR mice compared to control mice (2.7 ± 0.1 vs 3.1 ± 0.2 g/day). However, as noted above, no differences in food intake were observed during indirect colorimetry. No apparent differences were noted in water intake, urine volume or urinary excretion of Na+ or K+ (Figure 2). Likewise, urinary osmolality and AVP excretion were not statistically different between the two groups (Figure 2).
Figure 2.
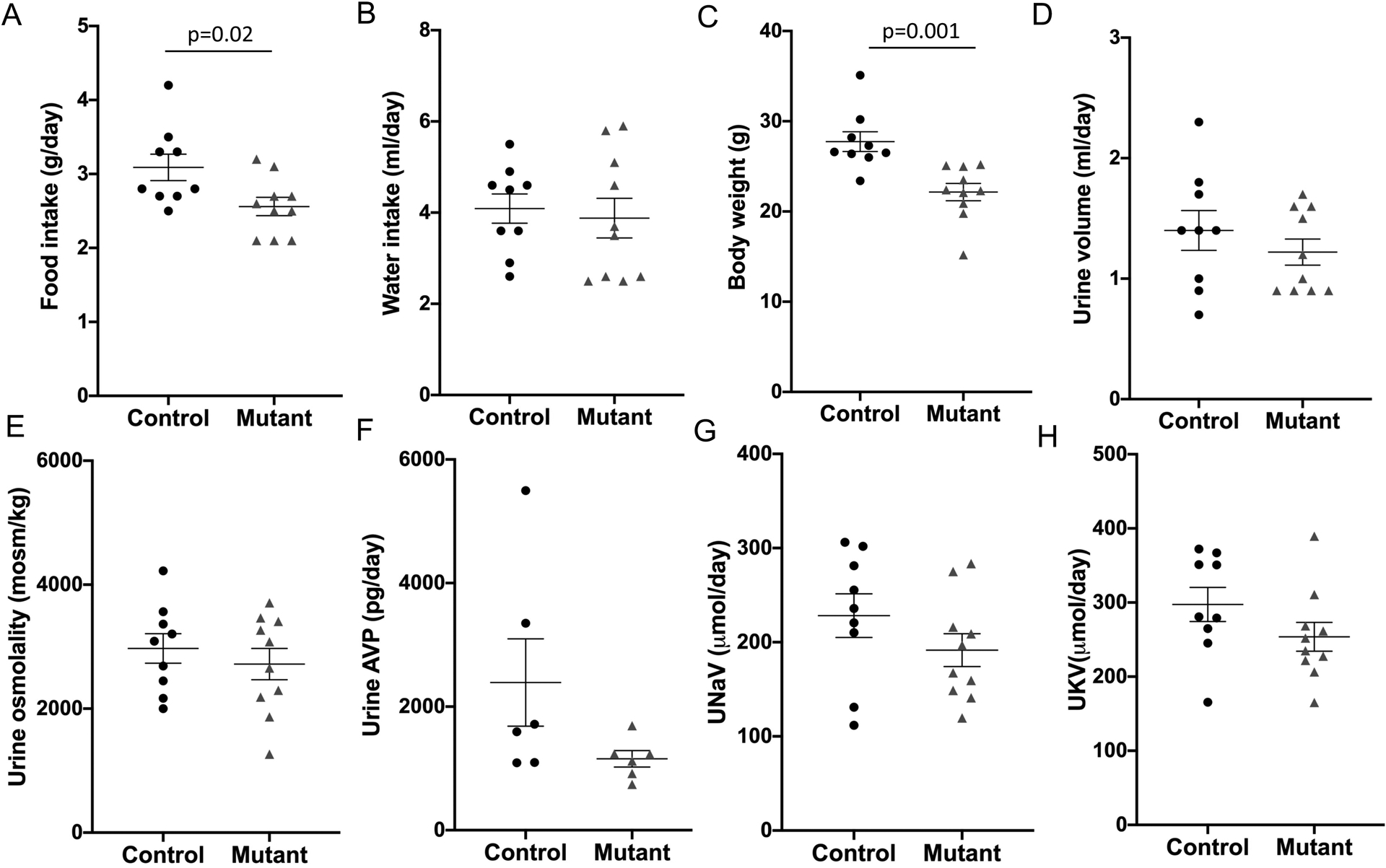
Baseline parameters in control (n=9) and mutant sPRR mice (n=10). A: food intake, B: water intake, C: body weight, D: urine volume, E: urine osmolality, F: urinary vasopressin excretion, G: 24-hour urinary Na+ excretion and H: 24-hour urinary K+ excretion; P value calculated using Student’s unpaired t-test for all comparisons except Figure 2D where Mann-Whitney non-parametric test was used.
Compared to control mice, plasma (Figure 3A) and kidney Ang peptides (Figure 3B) were not statistically different between control and mutant sPRR mice. Plasma angiotensinogen levels were lower in the mutant sPRR mice while plasma renin concentration and plasma aldosterone levels were comparable between the two groups (Figure 3 C–E). Similarly, urinary aldosterone excretion was not different between control and mutant sPRR mice (Control: 21.1 ± 8.6 vs mutant sPRR mice: 21.7 ± 5.1 ng/day).
Figure 3.
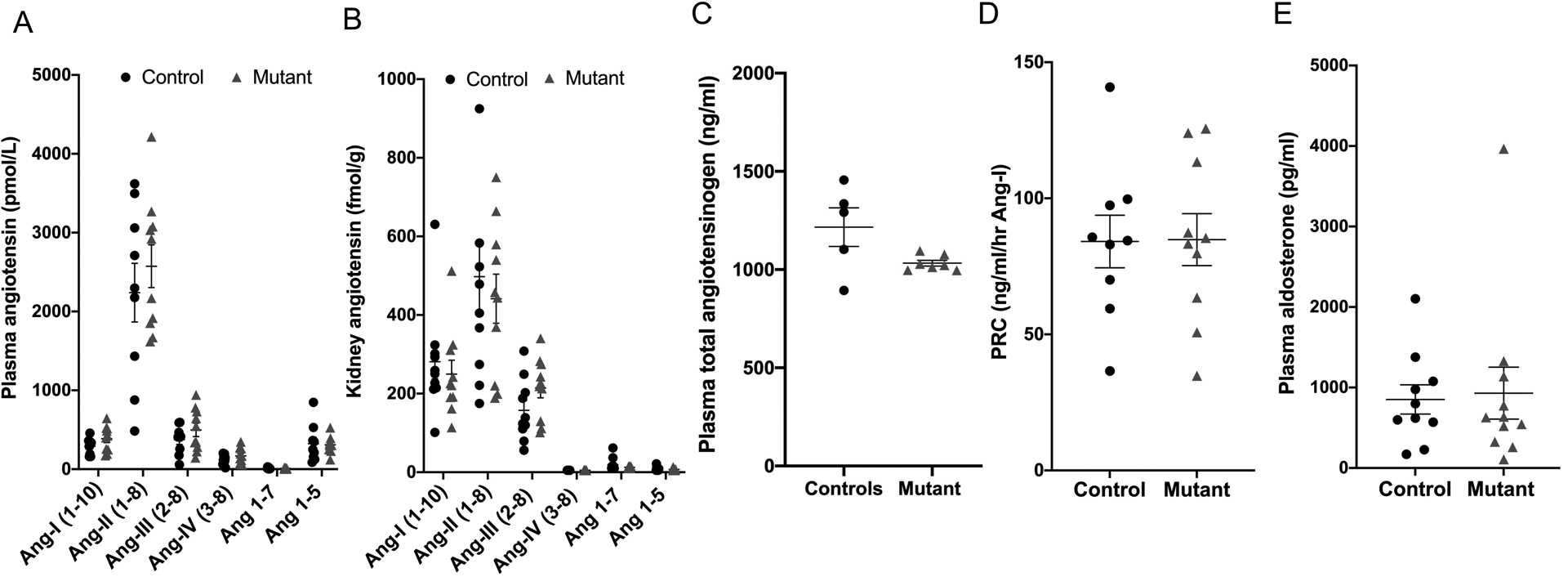
RAAS components in control (n=9) and mutant sPRR mice (n=10) at baseline conditions. A: plasma angiotensin peptide levels; B: kidney angiotensin peptide levels by LC-MS; C: plasma angiotensinogen; D: plasma renin concentration; E: plasma aldosterone levels. Comparisons between control and mutant sPRR mice performed using Student’s unpaired t-test except Figure 3E where Mann-Whitney non-parametric test was used.
Effects of sPRR deficiency on BP and cardiac function.
At baseline, mutant sPRR mice had lower systolic BP compared to control mice (Figure 4A). To determine if differences in baseline BP are associated with altered cardiac function, echocardiography was performed in anesthetized control and mutant sPRR mice. After accounting for differences in body weight, cardiac functional parameters including cardiac output, ejection fraction, fractional shortening, stroke volume, end systolic and end diastolic left ventricular volume and E/A were not different between the two groups (Table 1).
Figure 4.
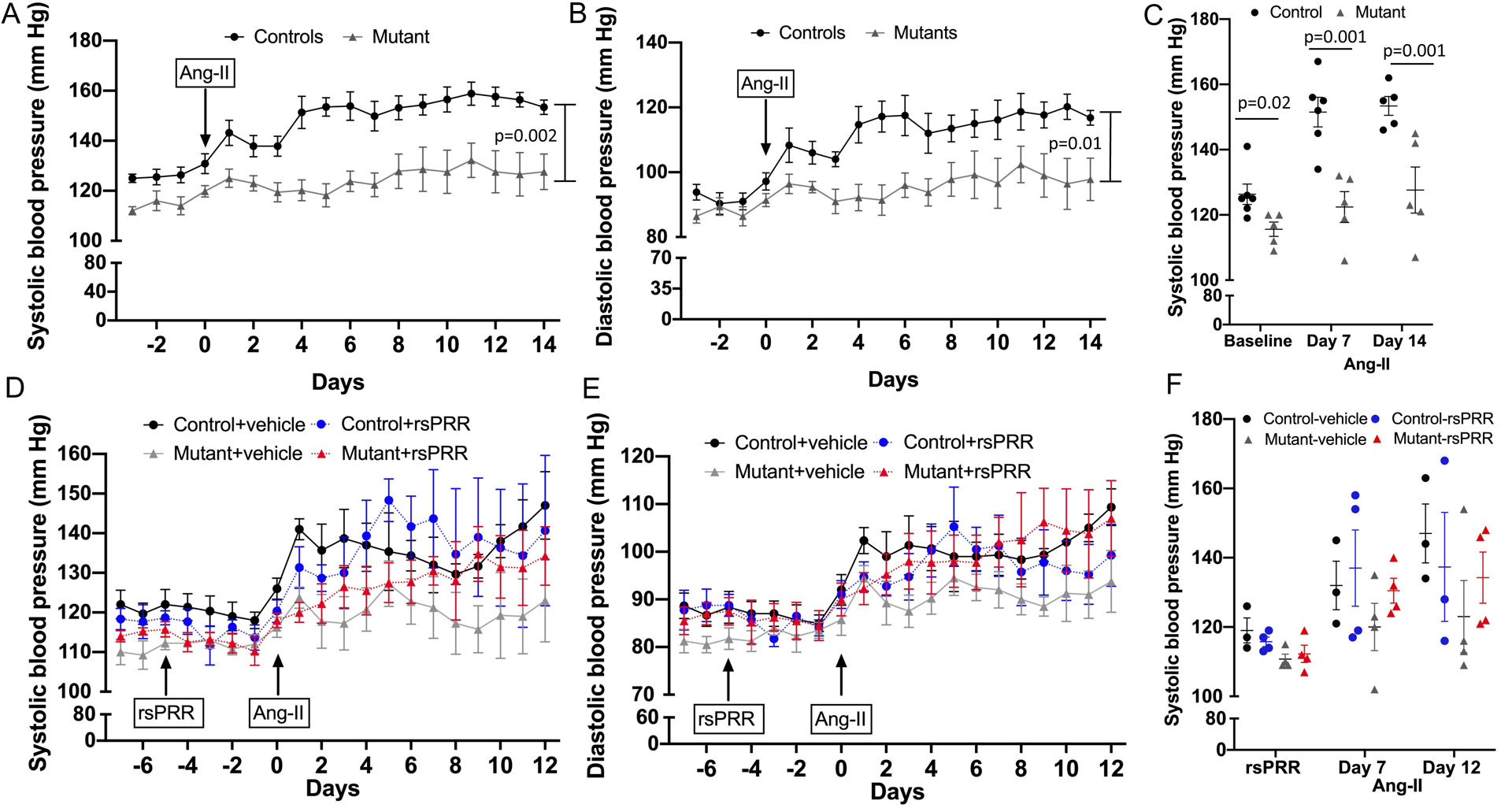
Blood pressure by telemetry in control (n=6) and mutant sPRR mice (n=5). A-C: at baseline and following Ang-II infusion at 400 ng/kg/min for 14 days. A: 24 hour systolic blood pressure; B: 24 hour diastolic blood pressure, *P <0.001 by mixed effects model; C: systolic blood pressure at baseline, days 7 and 14 following Ang-II infusion, P value calculated using Student’s unpaired t-test at each time point; D-F: following mouse recombinant sPRR infusion (rsPRR, 30μg/kg/day) and concurrent Ang-II infusion at 400 ng/kg/min (control n=3, all other groups n=4/group). D: 24 hour systolic blood pressure; E: 24 hour diastolic blood pressure using mixed effects model; F: systolic blood pressure following recombinant sPRR prior to and after Ang-II infusion using one-way ANOVA with Tukey post-hoc test at each time point.
Table 1.
Echocardiography in control & mutant sPRR mice at baseline and following 12-days of Ang-II infusion.
| Control (N=8) | Mutant (N=9) | Control (N=6) | Mutant (N=8) | |
|---|---|---|---|---|
| Heart rate (bpm) | 365 ± 9 | 339 ± 13 | 361 ± 18 | 359 ± 12 |
| Cardiac output (ml/min/g) | 0.4 ± 0.02 | 0.4 ± 0.02 | 0.3 ± 0.05 | 0.4 ± 0.03 |
| Ejection fraction (%) | 47.1 ± 2.6 | 45.6 ± 1.9 | 44.9 ± 5.9 | 48.6 ± 2.8 |
| Fractional shortening (%) | 23.8 ± 3.5 | 22.3 ± 1.0 | 22.4 ± 3.5 | 24.2 ± 1.6 |
| Left ventricular mass (mg/g) | 2.9 ± 0.1 | 3.3 ± 0.2 * | 3.0 ± 0.1 | 3.3 ± 0.1 |
| Stroke volume (μl/g) | 1.0 ± 0.04 | 1.0 ± 0.07 | 0.9 ± 0.09 | 1.1 ± 0.06 * |
| Volume (end-diastolic, μl/g) | 2.2 ± 0.2 | 2.3 ± 0.2 | 2.0 ± 0.09 | 2.3 ± 0.09 * |
| Volume (end-systolic, μl/g) | 1.2 ± 0.2 | 1.2 ± 0.1 | 1.1 ± 0.2 | 1.2 ± 0.1 |
| E/A | 1.8 ± 0.1 | 1.5 ± 0.1 | 2.0 ± 0.3 | 1.7 ± 0.1 |
P <0.05 vs control mice by Student’s unpaired t-test.
Interestingly, LV mass was significantly higher in mutant sPRR mice compared to controls (Table 1). Histological examination of cardiac tissue and aorta did not demonstrate any changes between control and mutant sPRR mice (Online Figure IB). Further, cardiomyocyte size appeared to be similar between control and mutant sPRR (Online Figure IE–F). No differences in activation of ERK1/2 pathway were noted between the two groups in kidney, heart or mesenteric arteries (Online Figure III).
Effects of sPRR deficiency in Ang-II infused hypertension.
To further characterize the role of sPRR in BP regulation, control and mutant sPRR mice were infused with Ang-II at 400 ng/kg/min for 14 days, a dose shown to produce consistent and modest elevation in BP without end-organ damage in our studies and others28, 29. Systolic BP increased in control mice from baseline of 125 ± 3 mm Hg to 138 ± 4 mm Hg on day 2 of Ang-II infusion and remained elevated throughout the first Ang-II infusion period (Day 7: 150 ± 6; Day 14: 158 ± 4 mm Hg). In contrast, mutant sPRR mice had an attenuated hypertensive response (SBP: baseline 116 ± 4; Day 2: 123 ± 3; Day 7: 122 ± 5; Day 14: 128 ± 7 mm Hg; Figure 4A, 4C). Similar trends were observed in diastolic BP (Figure 4B). No differences in heart rate were observed between control and mutant sPRR mice.
In a separate group of control and mutant sPRR mice, systolic and diastolic BP remained unchanged with infusion of mouse recombinant sPRR (30 μg·kg−1·min−1) compared to vehicle infusion (Figure 4D–F). Concurrent Ang-II infusion increased SBP in control mice (Day 7: vehicle 132 ± 5 vs recombinant sPRR 144 ± 10 mm Hg) and mutant sPRR mice with recombinant sPRR infusion (Day 7: 131 ± 4 mm Hg) but not mutant sPRR mice with vehicle infusion (Day 7: 121 ± 8 mm Hg) (Figure 4D, 4F). Infusion of recombinant sPRR did not increase BP more than Ang-II alone in control mice (Day 12: sPRR infusion 141 ± 18 vs vehicle 147 ± 8) while mutant sPRR mice with recombinant sPRR infusion tended to have higher BP than mutant sPRR mice with vehicle infusion (Day 12: sPRR infusion 134 ± 7 vs vehicle 123 ± 9 mm Hg, Figure 4F).
After a recovery period of 6 weeks during which BP returned to pre-Ang-II levels, the first group of control and mutant sPRR mice (without recombinant sPRR infusion) were infused with a second dose of Ang-II at 400 ng/kg/min for 14 days. This protocol allows accurate, independent evaluation of BP and metabolic changes in response to Ang-II infusion within the same mouse (i.e., one cannot perform metabolic cages while simultaneously measuring BP due to the stressful effects of metabolic cages). BP was not measured during the second phase of Ang-II infusion. Metabolic balance studies were performed for the first 8 days of Ang-II infusion; results from days 2 and 7 are presented to allow examination of immediate and post-equilibrium changes in Na+ and water excretion. Compared to control mice, mutant sPRR mice had no differences in food and water intake following Ang-II infusion (Figure 5A and B) with significantly lower body weight (Figure 5C). Notably, urine volume was significantly lower in mutant sPRR mice on days 2 and 7 following Ang-II infusion (Figure 5D) with no differences in urine osmolality (Figure 5E) as compared to control mice. No differences in urine AVP excretion (Figure 5F) was observed on days 2 or 7 of Ang-II infusion. Further, cumulative urinary Na+ and K+ excretion, after adjusting for body weight, were identical between control and mutant sPRR mice (Figure 5G and H).
Figure 5.
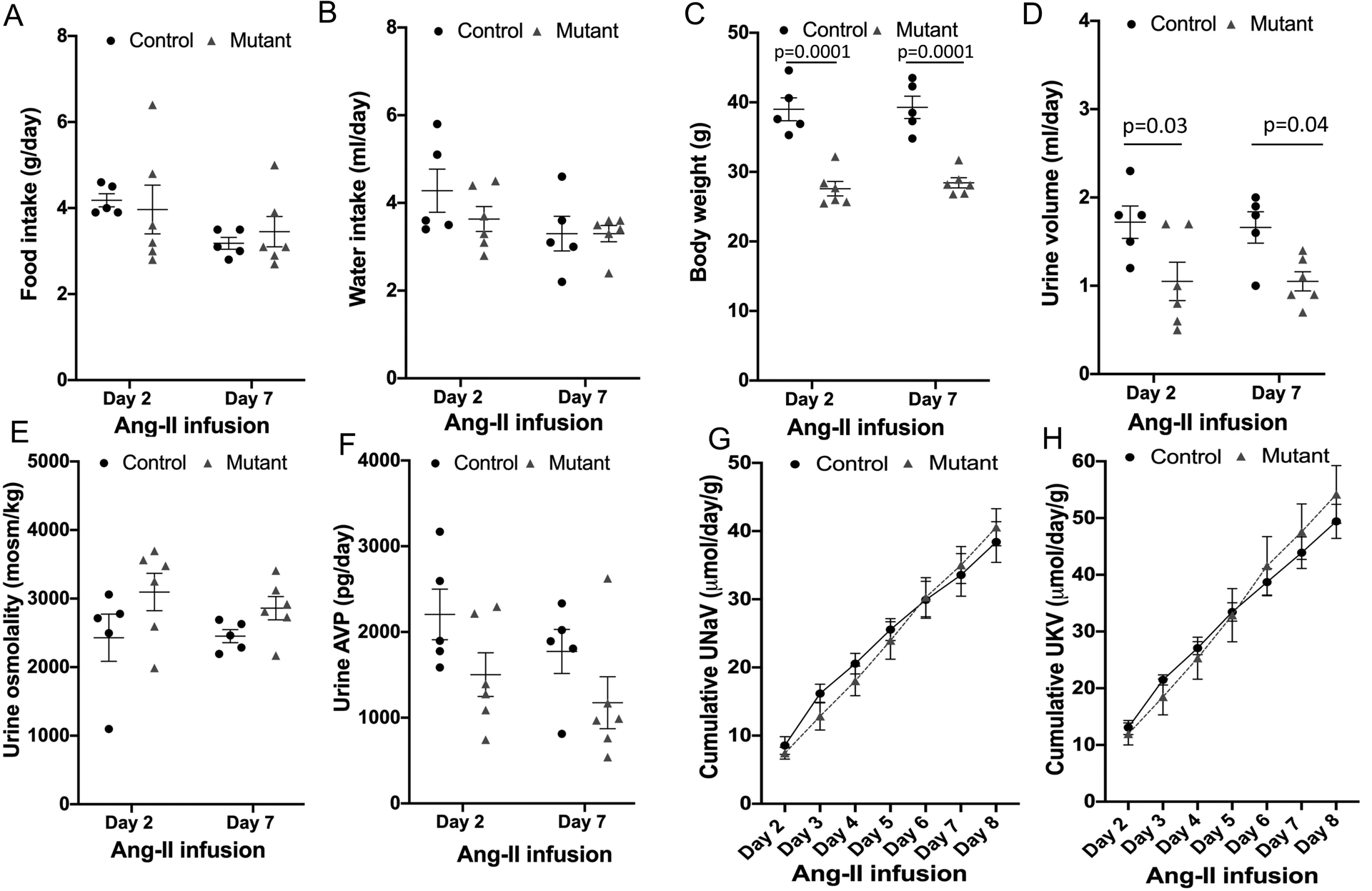
Post Ang-II infusion parameters in control (n=6) and mutant sPRR mice (n=5). A: food intake, B: water intake, C: body weight, D: urine volume, E: urine osmolality, F: urinary vasopressin excretion, G: cumulative urinary Na+ excretion adjusted for body weight and H: cumulative urinary K+ excretion adjusted for body weight; P value calculated using two-way ANOVA with Bonferroni correction for Figures 5A-F and repeated measures ANOVA with Bonferroni correction for Figures 5G-H.
Although GFR was comparable between the two groups post Ang-II infusion (Figure 6A), renal histology demonstrated tubular vacuolation in the cortex suggestive of renal injury in control mice (Figure 6B). No such changes were observed in mutant sPRR mice. Urinary albumin excretion was also significantly reduced in mutant mice compared to control mice (Figure 6 C). Urinary KIM-1 levels were not statistically different between control and mutant sPRR mice (Figure 6D). In contrast, urinary TBARS, a marker of oxidative stress, was significantly lower in mutant sPRR mice compared to controls post Ang-II infusion (Figure 6E). Blood urea nitrogen following Ang-II infusion was comparable between control and mutant sPRR mice (Figure 6F). Echocardiography following Ang-II infusion demonstrated that cardiac output, ejection fraction, fractional shortening, left ventricular mass and E/A were not different between control and mutant sPRR mice (Table 1); however, stroke volume and left ventricular end diastolic volume were higher in the mutant sPRR mice, compared to control mice (Table 1). No apparent differences were observed in kidney-to-body weight ratio (controls: 6.0% ± 0.3 vs mutant sPRR: 6.1% ± 0.4) or heart-to-body weight ratio (controls: 5.2 ± 0.2% vs mutant sPRR: 5.6 ± 0.3%) between the two groups.
Figure 6.
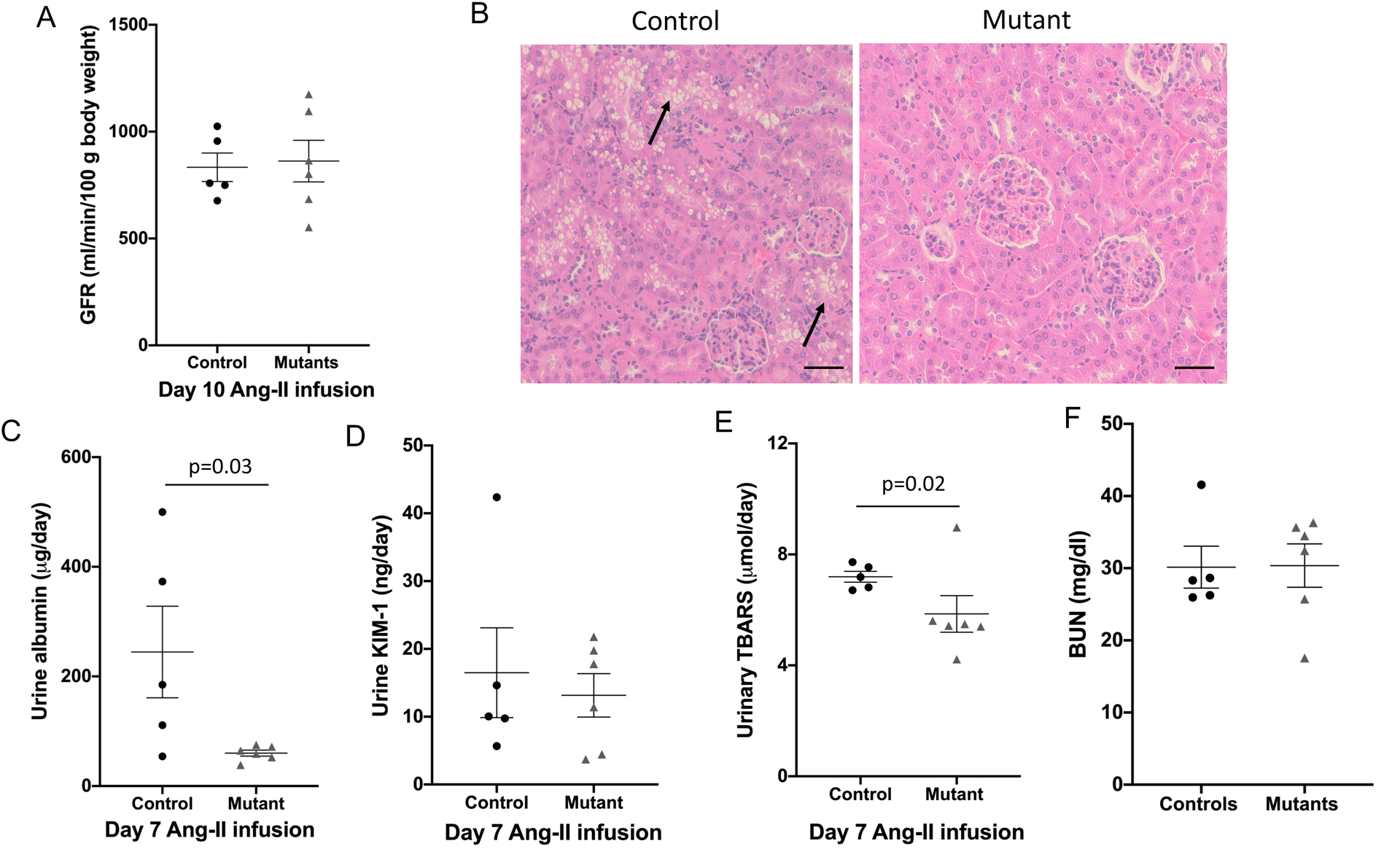
Kidney functional parameters in control (n=6) and mutant sPRR mice (n=5) following Ang-II infusion at 400 ng/kg/min. A: glomerular filtration rate determined by FITC- sinistrin at day 10 of Ang-II infusion; B: hematoxylin/eosin staining of kidney sections show extensive tubular vacuolization in control mice (arrows) but not mutant mice, images representative of 5 mice/group, scale bar = 50 μm; C: 24-hour urine albumin excretion; D: 24-hour urine kidney injury molecule (KIM-1) excretion; E: 24 hour urine thiobarbituric acid reactive substances (TBARS) excretion on day 7 of Ang-II infusion; F: blood urea nitrogen at sacrifice; Comparison between control and mutant sPRR mice was performed using Mann-Whitney non-parametric test.
Effects of sPRR deficiency on vascular function.
In order to determine if the differences in baseline BP were associated with changes in vascular function, vascular reactivity was examined in mesenteric arteries under baseline conditions using isometric procedures. No differences were detected between control and mutant sPRR mice with regard to resting internal diameter, vessel length, maximal KCl-evoked tension development during the determination of Lmax, and resting tension at Lmax (Online Table III).
No differences in vasocontraction were observed between control and mutant sPRR mice in response to KCl (Figure 7A), which depolarizes the vascular smooth muscle cell membrane potential to open voltage-gated Ca2+ channels. Similarly, vasocontractile responses to phenylephrine, which activates α1-adrenergic receptors, was comparable between arteries from the two groups (Figure 7B). In contrast, relative to control mice, mutant sPRR mice had a reduced vasoconstrictor response to Ang-II at low doses (10−8 and 10−7 M) but not at higher doses (Figure 7C).
Figure 7.
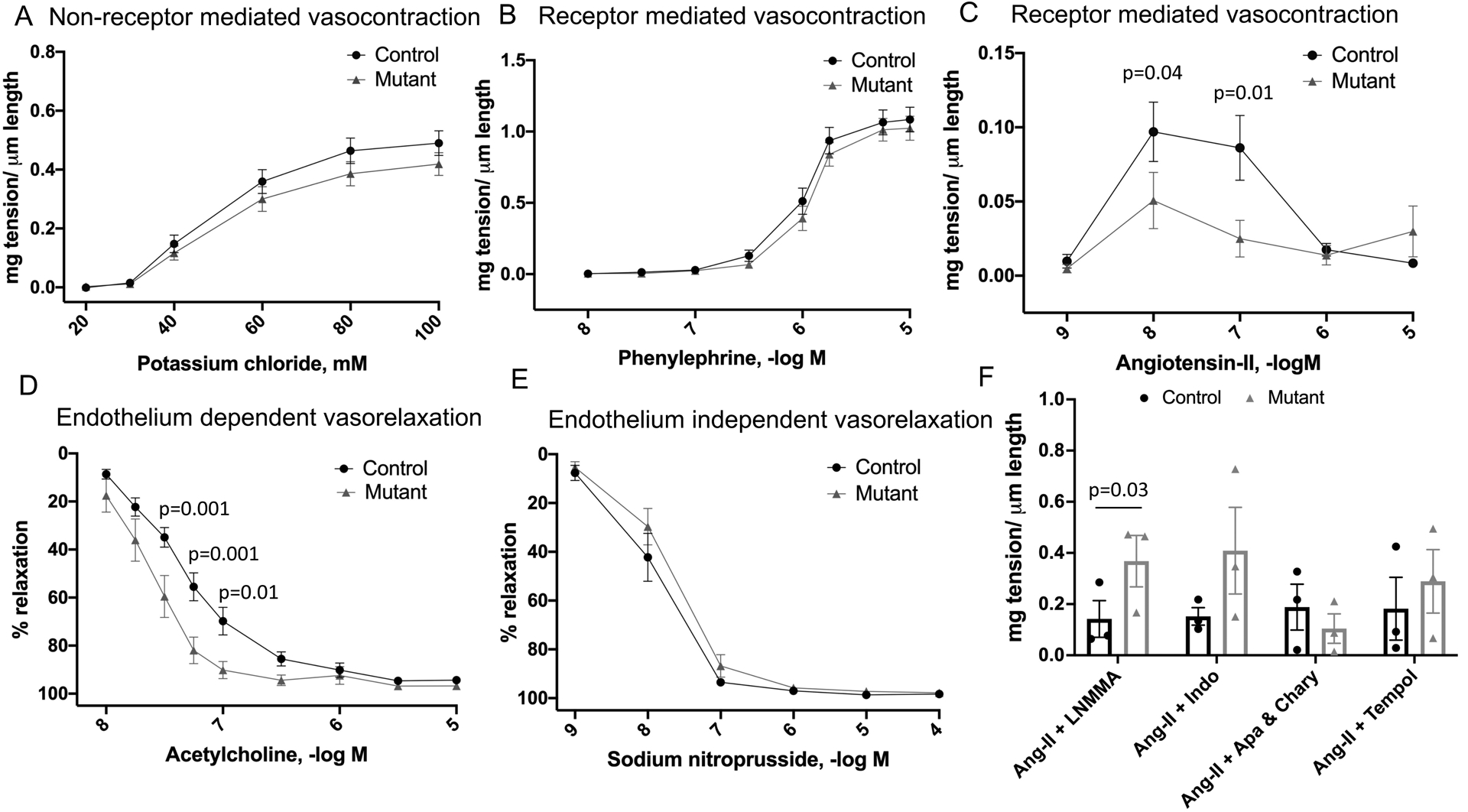
A-E: Vascular reactivity in mesenteric arteries by wire myography in control and mutant sPRR mice at basal conditions (n=5 mice/group); Comparison between control and mutant sPRR mice using two-way ANOVA with Bonferroni correction; F: Vascular reactivity to Ang-II at 10−7 M in mesenteric arteries in control and mutant sPRR mice (n=3 mice/group). Mesenteric arteries were pre-treated with NG- Methyl-L-Arginine acetate (L-NMMA, NOS inhibitor), indomethacin (COX inhibitor), apamin + charybdotoxin (EDHF blocker) or tempol (SOD mimetic). Results displayed are average of two mesenteric artery segments obtained from each mouse. Comparison between control and mutant sPRR mice was performed using Mann-Whitney non-parametric test.
Acetylcholine binds to M3 muscarinic receptors on the vascular endothelium to increase intracellular Ca2+ which in turn activates constitutive type III nitric oxide (NO) synthase (NOS). After stable precontraction with phenylephrine that was not different between the two groups, acetylcholine evoked vasorelaxation was significantly greater in arteries from mutant sPRR compared to control mice (Figure 7D). Sodium nitroprusside activates guanylyl cyclase to relax vascular smooth muscle in a manner that is not dependent on substances released from the endothelium. After precontraction using phenylephrine, sodium nitroprusside evoked vasodilation was similar in mesenteric arteries from control and mutant sPRR mice (Figure 7E).
We then examined if nitric oxide, products of cyclooxygenase metabolism, endothelium-derived hyperpolarizing factors or superoxide might be contributing to the blunted Ang-II vasocontractile response in mutant sPRR mice. Relative to controls, pre-treatment with L-NMMA caused greater vasocontraction in mutant sPRR mice at an Ang-II dose of 10−7 M (Figure 7F). Pre-treatment with indomethacin, apamin and charybdotoxin or tempol caused similar degrees of vasocontraction in control and mutant sPRR mice (Figure 7F). No differences were observed in Ang-II receptor expression in mesenteric arteries isolated from control and mutant sPRR mice (Online Figure IV).
DISCUSSION
Using a novel mouse model of sPRR deficiency, the current study reports the following key findings: 1) sPRR production depends entirely on the furin/site-1 protease cleavage sites in the PRR; 2) sPRR does not contribute to circulating Ang-II synthesis; 3) loss of sPRR decreases BP at baseline; 4) absence of sPRR attenuates Ang-II induced hypertension and renal injury; 5) sPRR deficiency reduces the vasocontractile response to Ang-II and enhances the vasodilatory response to acetylcholine; and 6) mutation of the PRR furin/site-1 protease cleavage site does not lead to lysosomal dysfunction and associated cell toxicity.
At baseline, compared to controls, mutant sPRR mice had no differences in Ang peptides, plasma renin concentration or plasma/urine aldosterone levels. This would indicate that plasma sPRR has little role in plasma Ang-I generation and has minimal effects on other RAAS components under physiological circumstances. A key point is that mutant sPRR mice are relatively hypotensive despite no changes in plasma and kidney levels of Ang peptides. While Ang peptide levels were not assessed in all organs, these findings raise the possibility that sPRR deficiency lowers BP through mechanisms independent of alterations in Ang peptide levels. In support of this notion, male mice with systemic infusion of sPRR are hypertensive and have enhanced vascular sympathetic tone without changes in the systemic RAAS19. In addition, treatment of cultured collecting duct cells with recombinant sPRR increased trans-epithelial current within 2 minutes (reflective of epithelial Na+ channel [ENaC] activity) via Nox4 activation, while chronic (24 hours) treatment increased ENaC abundance and activity via the Wnt/β catenin signaling30; all of this is clearly independent of the RAAS since no RAAS components were present in the culture system. Taken together, these studies suggest that, while no cognate receptor for sPRR has been identified, sPRR may directly act on cells, independent of modifying Ang peptide levels, to elicit specific biological responses.
The relative hypotension in mutant sPRR mice under baseline conditions was not associated with impaired cardiac function or lower circulating RAAS components (plasma renin concentration, Ang-II or aldosterone). However, reduced Ang-II mediated vasocontraction was observed in mesenteric arteries from mutant sPRR mice compared to controls. This response was specific to Ang-II receptor mediated activation because no differences were observed to a non-receptor mediated (KCl) or alternative receptor-mediated (phenylephrine) vasocontractile agonist. Further, because responses to acetylcholine were greater in arteries from mutant sPRR mice relative to control mice, but vasorelaxation to sodium nitroprusside was similar, we hypothesized that substances such as nitric oxide, cyclooxygenase products, or endothelium-derived hyperpolarizing factors might contribute to the altered vascular reactivity. Our results demonstrate that mutant sPRR may have greater endothelium derived nitric oxide generation and/or cyclooxygenase products leading to a blunted Ang-II mediated vasocontractile response. Whether this is the case and how this occurs will be the focus of future studies. In particular, it will be important to determine if sPRR, independent of RAAS components, directly modifies endothelial cell function.
The reduced hypertensive response to Ang-II in mutant sPRR mice was similar to that previously reported in mice with nephron-wide or collecting duct-specific deletion of the intact PRR31–33. Rats with overexpression of the human intact PRR in vascular smooth muscle are hypertensive34, however mice with vascular smooth muscle-specific intact PRR deletion did not have altered BP35. In addition, mice with global or cardiac-specific intact PRR overexpression did not have altered BP or cardiac function36, 37. It is important, however, to be careful in comparing overexpression/deletion studies of intact PRR vs sPRR. Loss of intact PRR affects cell surface PRR expression as well as sPRR levels. Further, intact PRR deletion clearly impairs lysosomal function24–27 thereby complicating interpretation. Importantly, infusion of recombinant sPRR in mutant sPRR mice with Ang-II infusion tended to increase BP to levels similar to control mice with Ang-II infusion indicating that sPRR likely plays a direct role in modulating BP.
Urinary Na+ excretion was not different between mutant sPRR and control mice under baseline conditions although this is not surprising given that the mice should have been in steady-state Na+ balance. Of greater significance is that no differences in urinary Na+ excretion between mutant sPRR and control mice were observed during the first week of Ang-II administration, a time period when clear differences in BP occurred between the two groups. Thus, while small differences in urinary Na+ excretion may have occurred, no evidence was found that kidney Na+ excretion was of primary importance in mediating the reduced Ang-II hypertensive response in the mutant sPRR mice. Relevant to this, only one study to date has examined the role of sPRR in Na+ transport30; as discussed earlier, recombinant sPRR increased ENaC activity in cultured collecting duct cells30. In addition, nephron-specific deletion of the intact PRR enhanced urinary Na+ excretion associated with reduced ENaC abundance and activity33, however the specificity of this response is uncertain given possible effects on lysosomal function.
In the current study, lack of sPRR did not affect water metabolism under normal physiological conditions as evidenced by no apparent effect on AVP levels, urine volume and urine osmolality during ad lib water intake. Our findings are consistent with Gatineau et al, wherein infusion of mouse recombinant sPRR in obese male mice did not alter AVP levels, urine volume or urine osmolality19. In contrast, under the same dietary conditions, mice with nephron-specific intact PRR deletion manifested marked polyuria and hyposthenuria8, 38. Further, systemic infusion of recombinant sPRR into mice with renal tubular or collecting duct specific PRR knockout attenuated the urinary concentration defect39. Similarly, mice with renal tubule specific deletion of site-1 protease (protease partly responsible for generating the sPRR) recapitulate the urinary concentration defect which is restored by infusion of recombinant sPRR39. Finally, as alluded to earlier, Lu et al found that recombinant sPRR stimulates Wnt/βcatenin signaling in collecting duct cells leading to enhanced aquaporin gene transcription40. The reasons for these discordant results are uncertain, however may relate to differences between the models (sPRR vs. intact PRR deletion, administration of potentially supra-physiologic amounts of sPRR as well as use of recombinant sPRR generated from rat39 versus mouse18). Of note, mutant sPRR mice had enhanced antidiuresis during Ang-II administration; this result was surprising given the lower Ang-II elicited BP elevation in mutant sPRR mice as well as the previous studies reporting that sPRR per se is antidiuretic. Clearly, further examination of sPRR and water metabolism is necessary.
Mutant sPRR mice had lower albuminuria, oxidative stress and reduced renal tubular injury in response to Ang-II. This may simply reflect lower BP although differences in intrinsic renal responses to Ang-II cannot be excluded. Notably, plasma sPRR levels have been reported to be elevated in conditions with impaired renal function (heart failure and chronic kidney disease12–15, 17), although this relationship is associative and not yet been proven to be causal.
There are potential limitations to interpretation of the current study. First, since mutant sPRR males are infertile and the ATP6AP2 gene is on the X chromosome, it is not possible to obtain hemizygous mutant sPRR females. While studying heterozygous mutant sPRR females will ultimately be of interest, the lack of hemizygous mutant sPRR female mice makes analysis of sex differences in sPRR biology challenging. Second, male mutant sPRR mice manifest a complex phenotype, including lower body weights (potentially indicating metabolic effects), infertility and possibly other features. Thus, it is possible that loss of sPRR could be exerting effects on the cardiovascular and renal systems through as yet undetected means. In this regard, the PRR is expressed in multiple organs including the kidney, heart, vascular smooth muscle, adipose tissue, brain, liver and placenta1. Hence, mutation in the cleavage site of the PRR might affect sPRR generation in multiple tissues (although which tissues generate sPRR, or which tissues contribute to circulating sPRR, is incompletely understood). Third, loss of sPRR and the attendant 8.9 kD fragment might impair lysosomal acidification as occurs with the absence of intact PRR8, 24–27, thereby leading to generalized cellular toxicity. However, mutant sPRR mice demonstrated no evidence of lysosomal dysfunction or altered systemic acid-base status. This result not only helps validate the current mouse model, but also suggests that the intact PRR per se may be able to function as an accessory protein to the vacuolar H+-ATPase; the current study was not designed to explore this issue in detail but provides impetus for further investigations in this area.
In summary, loss of sPRR lowers baseline BP, attenuates Ang-II induced hypertension and renal injury, and promotes pro-hypotensive vascular responsiveness. These effects appear to be largely independent of changes in plasma and renal Ang peptides and occur in the absence of apparent lysosomal injury. Key remaining questions relate to the mechanisms by which sPRR acts upon cells (particularly in terms of RAAS-independent actions and cellular targets), what other forms of hypertension sPRR may be involved in (e.g., L-NAME or DOCA-salt hypertension), and whether sPRR represents a viable target for drug intervention in hypertension and kidney disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The PRR ((pro)renin receptor) is a multifunctional protein that is involved in the pathogenesis of hypertension.
The extracellular domain of the PRR is cleaved to form a soluble fragment, termed soluble PRR (sPRR); elevated plasma sPRR levels have been described in hypertension, heart failure and kidney disease.
sPRR can potentially activate both angiotensin-II (Ang-II) dependent and/or independent cell signaling pathways.
What New Information Does This Article Contribute?
Loss of sPRR in mice decreases blood pressure (BP) at baseline and attenuates Ang-II induced hypertension and renal injury.
sPRR does not contribute to circulating Ang-II synthesis; sPRR deficiency reduces the vasocontractile response to Ang-II and enhances the vasodilatory response to acetylcholine.
sPRR production depends entirely on the furin/site-1 protease cleavage sites in the PRR and mutation in this site does not lead to lysosomal dysfunction.
ACKNOWLEDGEMENTS
We acknowledge the University of Utah HSC Cores, in particular, the DNA sequencing core (directed by Mr. Derek Warner), Small animal ultrasound core (directed by Dr. Kevin Whitehead) and Metabolic phenotyping core (directed by Dr. Anil Laxman) for their assistance with this project.
SOURCES OF FUNDING
This research was supported in part by funding from the American Diabetes Association (1-19-JDF-035) and Pittsburgh Center for Kidney Research (P30 DK079307) to NR, National Heart, Lung, and Blood Institute grant P01 HL136267 to DEK, University of Utah Graduate Research Fellowship and American Heart Association Predoctoral Fellowship 20PRE35110066 to JMC, American Heart Association Grant in Aid 16GRNT31050004, National Institute of Aging grant RO3AGO52848 and National Heart, Lung, and Blood Institute grant RO1HL141540 to JDS.
Nonstandard Abbreviations and Acronyms:
- RAAS
Renin angiotensin aldosterone system
- PRR
Prorenin receptor
- sPRR
Soluble prorenin receptor
- Ang-II
Angiotensin-II
- ERK1/2
extracellular signal-regulated kinase
- LAMP-2
lysosomal associated membrane protein-2
- AVP
Arginine vasopressin
- KIM-1
Kidney injury molecule
- TBARS
thiobarbituric acid reactive substances
- NMR
Nuclear magnetic resonance
Footnotes
Publisher's Disclaimer: This article is published in its accepted form. It has not been copyedited and has not appeared in an issue of the journal. Preparation for inclusion in an issue of Circulation Research involves copyediting, typesetting, proofreading, and author review, which may lead to differences between this accepted version of the manuscript and the final, published version.
DISCLOSURES
None
SUPPLEMENTAL MATERIALS
REFERENCES
- 1.Nguyen G, Delarue F, Burckle C, Bouzhir L, Giller T and Sraer JD. Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. J Clin Invest. 2002;109:1417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ichihara A and Yatabe MS. The (pro)renin receptor in health and disease. Nat Rev Nephrol. 2019;15:693–712. [DOI] [PubMed] [Google Scholar]
- 3.Ramkumar N and Kohan DE. The (pro)renin receptor: an emerging player in hypertension and metabolic syndrome. Kidney Int. 2019;95:1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feldt S, Batenburg WW, Mazak I, et al. Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension. 2008;51:682–8. [DOI] [PubMed] [Google Scholar]
- 5.Saris JJ, t Hoen PA, Garrelds IM, Dekkers DH, den Dunnen JT, Lamers JM and Jan Danser AH. Prorenin induces intracellular signaling in cardiomyocytes independently of angiotensin II. Hypertension. 2006;48:564–71. [DOI] [PubMed] [Google Scholar]
- 6.Cruciat CM, Ohkawara B, Acebron SP, Karaulanov E, Reinhard C, Ingelfinger D, Boutros M and Niehrs C. Requirement of prorenin receptor and vacuolar H+-ATPase-mediated acidification for Wnt signaling. Science. 2010;327:459–63. [DOI] [PubMed] [Google Scholar]
- 7.Ludwig J, Kerscher S, Brandt U, Pfeiffer K, Getlawi F, Apps DK and Schagger H. Identification and characterization of a novel 9.2-kDa membrane sector-associated protein of vacuolar proton-ATPase from chromaffin granules. J Biol Chem. 1998;273:10939–47. [DOI] [PubMed] [Google Scholar]
- 8.Trepiccione F, Gerber SD, Grahammer F, et al. Renal Atp6ap2/(Pro)renin Receptor Is Required for Normal Vacuolar H+-ATPase Function but Not for the Renin-Angiotensin System. J Am Soc Nephrol. 2016;27:3320–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cousin C, Bracquart D, Contrepas A, Corvol P, Muller L and Nguyen G. Soluble form of the (pro)renin receptor generated by intracellular cleavage by furin is secreted in plasma. Hypertension. 2009;53:1077–82. [DOI] [PubMed] [Google Scholar]
- 10.Fang H, Xu C, Lu A, Zou CJ, Xie S, Chen Y, Zhou L, Liu M, Wang L, Wang W and Yang T. (Pro)renin receptor mediates albumin-induced cellular responses: role of site-1 protease-derived soluble (pro)renin receptor in renal epithelial cells. Am J Physiol Cell Physiol. 2017;313:C632–C643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakagawa T, Suzuki-Nakagawa C, Watanabe A, Asami E, Matsumoto M, Nakano M, Ebihara A, Uddin MN and Suzuki F. Site-1 protease is required for the generation of soluble (pro)renin receptor. J Biochem. 2017;161:369–379. [DOI] [PubMed] [Google Scholar]
- 12.Amari Y, Morimoto S, Nakajima F, Ando T and Ichihara A. Serum Soluble (Pro)Renin Receptor Levels in Maintenance Hemodialysis Patients. PloS one. 2016;11:e0158068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gong L, Zhang S, Li L, Gao X, Wang D, Wu D, Wang K and Liu Y. Elevated plasma soluble (pro)renin receptor levels are associated with left ventricular remodeling and renal function in chronic heart failure patients with reduced ejection fraction. Peptides. 2019;111:152–157. [DOI] [PubMed] [Google Scholar]
- 14.Hamada K, Taniguchi Y, Shimamura Y, et al. Serum level of soluble (pro)renin receptor is modulated in chronic kidney disease. Clin Exp Nephrol. 2013;17:848–56. [DOI] [PubMed] [Google Scholar]
- 15.Morimoto S, Ando T, Niiyama M, et al. Serum soluble (pro)renin receptor levels in patients with essential hypertension. Hypertens Res 2014;37:642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Narita T, Ichihara A, Matsuoka K, Takai Y, Bokuda K, Morimoto S, Itoh H and Seki H. Placental (pro)renin receptor expression and plasma soluble (pro)renin receptor levels in preeclampsia. Placenta. 2016;37:72–8. [DOI] [PubMed] [Google Scholar]
- 17.Fukushima A, Kinugawa S, Homma T, Masaki Y, Furihata T, Abe T, Suga T, Takada S, Kadoguchi T, Okita K, Matsushima S and Tsutsui H. Increased plasma soluble (pro)renin receptor levels are correlated with renal dysfunction in patients with heart failure. Int J Cardiol. 2013;168:4313–4. [DOI] [PubMed] [Google Scholar]
- 18.Gatineau E, Cohn DM, Poglitsch M, Loria AS, Gong M and Yiannikouris F. Losartan prevents the elevation of blood pressure in adipose-PRR deficient female mice while elevated circulating sPRR activates the renin-angiotensin system. Am J Physiol Heart Circ Physiol. 2019;316:H506–H515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatineau E, Gong MC and Yiannikouris F. Soluble Prorenin Receptor Increases Blood Pressure in High Fat-Fed Male Mice. Hypertension. 2019;74:1014–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu CH, Mohammadmoradi S, Thompson J, Su W, Gong M, Nguyen G and Yiannikouris F. Adipocyte (Pro)Renin-Receptor Deficiency Induces Lipodystrophy, Liver Steatosis and Increases Blood Pressure in Male Mice. Hypertension. 2016;68:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonzalez AA, Lara LS, Luffman C, Seth DM and Prieto MC. Soluble form of the (pro)renin receptor is augmented in the collecting duct and urine of chronic angiotensin II-dependent hypertensive rats. Hypertension. 2011;57:859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schock-Kusch D, Geraci S, Ermeling E, et al. Reliability of transcutaneous measurement of renal function in various strains of conscious mice. PLoS One. 2013;8:e71519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petersen C, Bharat D, Cutler BR, Gholami S, Denetso C, Mueller JE, Cho JM, Kim JS, Symons JD and Anandh Babu PV. Circulating metabolites of strawberry mediate reductions in vascular inflammation and endothelial dysfunction in db/db mice. Int J Cardiol. 2018;263:111–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kinouchi K, Ichihara A, Sano M, et al. The (pro)renin receptor/ATP6AP2 is essential for vacuolar H+-ATPase assembly in murine cardiomyocytes. Circ Res. 2010;107:30–4. [DOI] [PubMed] [Google Scholar]
- 25.Oshima Y, Kinouchi K, Ichihara A, et al. Prorenin receptor is essential for normal podocyte structure and function. J Am Soc Nephrol. 2011;22:2203–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Riediger F, Quack I, Qadri F, et al. Prorenin receptor is essential for podocyte autophagy and survival. J Am Soc Nephrol. 2011;22:2193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendling O, Champy MF, Jaubert S, et al. Atp6ap2 ablation in adult mice impairs viability through multiple organ deficiencies. Sci Rep. 2017;7:9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonzalez-Villalobos RA, Satou R, Ohashi N, et al. Intrarenal mouse renin-angiotensin system during ANG II-induced hypertension and ACE inhibition. Am J Physiol Renal Physiol. 2010;298:F150–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramkumar N, Stuart D, Rees S, Hoek AV, Sigmund CD and Kohan DE. Collecting duct-specific knockout of renin attenuates angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2014;307:F931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang F, Luo R, Peng K, Liu X, Xu C, Lu X, Soodvilai S and Yang T. Soluble (pro)renin receptor regulation of ENaC involved in aldosterone signaling in cultured collecting duct cells. Am J Physiol Renal Physiol. 2020;318:F817–F825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng K, Lu X, Wang F, Nau A, Chen R, Zhou SF and Yang T. Collecting duct (pro)renin receptor targets ENaC to mediate angiotensin II-induced hypertension. Am J Physiol Renal Physiol. 2017;312:F245–F253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prieto MC, Reverte V, Mamenko M, et al. Collecting duct prorenin receptor knockout reduces renal function, increases sodium excretion, and mitigates renal responses in ANG II-induced hypertensive mice. Am J Physiol Renal Physiol. 2017;313:F1243–F1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramkumar N, Stuart D, Mironova E, Bugay V, Wang S, Abraham N, Ichihara A, Stockand JD and Kohan DE. Renal tubular epithelial cell prorenin receptor regulates blood pressure and sodium transport. Am J Physiol Renal Physiol. 2016;311:F186–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burckle CA, Jan Danser AH, Muller DN, Garrelds IM, Gasc JM, Popova E, Plehm R, Peters J, Bader M and Nguyen G. Elevated blood pressure and heart rate in human renin receptor transgenic rats. Hypertension. 2006;47:552–6. [DOI] [PubMed] [Google Scholar]
- 35.Kurauchi-Mito A, Ichihara A, Bokuda K, Sakoda M, Kinouchi K, Yaguchi T, Yamada T, Sun-Wada GH, Wada Y and Itoh H. Significant roles of the (pro)renin receptor in integrity of vascular smooth muscle cells. Hypertens Res. 2014;37:830–5. [DOI] [PubMed] [Google Scholar]
- 36.Mahmud H, Candido WM, van Genne L, et al. Cardiac function and architecture are maintained in a model of cardiorestricted overexpression of the prorenin-renin receptor. PloS one. 2014;9:e89929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosendahl A, Niemann G, Lange S, et al. Increased expression of (pro)renin receptor does not cause hypertension or cardiac and renal fibrosis in mice. Lab Invest. 2014;94:863–72. [DOI] [PubMed] [Google Scholar]
- 38.Ramkumar N, Stuart D, Calquin M, Quadri S, Wang S, Van Hoek AN, Siragy HM, Ichihara A and Kohan DE. Nephron-specific deletion of the prorenin receptor causes a urine concentration defect. Am J Physiol Renal Physiol. 2015;309:F48–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang F, Xu C, Luo R, Peng K, Ramkumar N, Xie S, Lu X, Zhao L, Zuo CJ, Kohan DE and Yang T. Site-1 protease-derived soluble (pro)renin receptor targets vasopressin receptor 2 to enhance urine concentrating capability. JCI Insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu X, Wang F, Xu C, Soodvilai S, Peng K, Su J, Zhao L, Yang KT, Feng Y, Zhou SF, Gustafsson JA and Yang T. Soluble (pro)renin receptor via beta-catenin enhances urine concentration capability as a target of liver X receptor. Proc Natl Acad Sci U S A. 2016;113:E1898–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting findings of this study are available from the corresponding author upon reasonable request.