

Abstract
Hemoprotein-catalyzed carbene and nitrene transformations have emerged as powerful tools for constructing complex molecules; they also nicely illustrate how new protein catalysts can emerge, evolve and diversify. These laboratory-invented enzymes exploit the ability of proteins to tame highly reactive carbene and nitrene species and direct their fates with high selectivity. New-to-nature carbene and nitrene transferases catalyze many useful reactions, including some that have no precedent using chemical methods. Here we cover recent advances in this field, including alkyne cyclopropenation, arene cyclopropanation, carbene C–H insertion, intramolecular nitrene C–H insertion, alkene aminohydroxylation, and primary amination. For such transformations, biocatalysts have exceeded the performance of reported small-molecule catalysts in terms of selectivity and catalyst turnovers. Finally, we offer our thoughts on using these new enzymatic reactions in chemical synthesis, integrating them into biological pathways and chemo-enzymatic cascades, and on their current limitations.
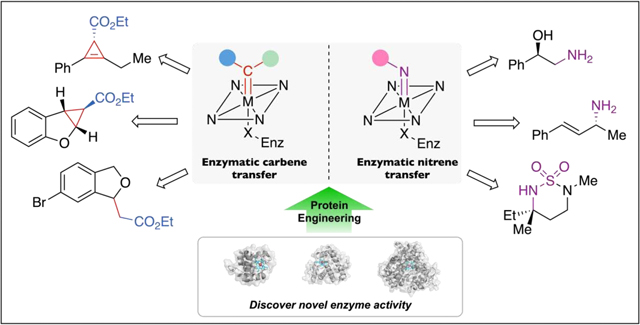
Graphical abstract
Introduction
This review covers recent contributions to development of two broad classes of new-to-nature enzymatic transformations of interest for chemical biology, biosynthesis, and biocatalysis. Expanding the repertoire of enzyme-catalyzed functions broadens the synthetic routes and products that can be made using biotechnology and opens new opportunities to exploit biocompatible and bioorthogonal enzymes in chemical biology and biotechnology. Some advances in new-to-nature biocatalysis have been covered in excellent reviews [1–3]. This review is limited to reactions involving transfer of carbene and nitrene intermediates that have been known and developed for decades using transition metal complexes and have been used industrially, for example, to prepare insecticides and antibiotics [4,5]; only recently have enzymes been engineered for these reactions. Almost all the newly-developed enzymatic carbene and nitrene transformations have no precedent in biology. Some have no precedent in chemistry and are possible only in a biocatalyst active site that can control and divert the reactive species along desired reaction pathways.
Heme-containing oxygenases are versatile biocatalysts for a broad range of transformations, such as hydroxylation, dealkylation, and C–C bond cleavage [6]. Most if not all of these processes rely on formation of a high-valent iron(IV)-oxo cation radical species known as Compound I [7] (Figure 1). The ability of these enzymes to tame highly reactive intermediates in active sites that force reactants to adopt desired conformations and orientations for controlled reactivities, while avoiding enzyme destruction, is extraordinary. This level of reactant and reaction manipulation far exceeds what small-molecule catalysts can achieve. The fact that nature can do this led researchers to ask whether other high energy intermediates such as metallocarbenes could be put to work for selective processes not previously discovered in natural systems [8].
Figure 1.

Metalloporphyrins are versatile scaffolds for oxene, carbene, and nitrene transfer reactions. The development of non-natural carbene and nitrene transferases was inspired by natural heme monooxygenases, with Compound I as the key catalytic intermediate [6,7]. These new activities were initially discovered by testing hemoproteins for the ability to mimic known activities of small-molecule metalloporphyrin catalysts [9,10].
Due to similarities in their electronic structures, carbenes and nitrenes can be viewed as “synthetic siblings” of oxene species (Figure 1) [9–11]. A variety of transformations involving carbene and nitrene intermediates, especially those that forge C–C and C–N bonds, are powerful tools for building complex molecular structures. Although highly enabling, these synthetic methods usually require noble metal catalysts, costly exogenous ligands, and harsh reaction conditions. Furthermore, low selectivity due to limited control of the highly reactive and undiscriminating carbene and nitrene species can be a problem. Biocatalysts performing these transformations could be a good alternative to existing synthetic methodologies, but no ‘carbene transferase’ has been discovered in nature, and only one enzyme has been proposed to catalyze a nitrene transfer reaction as its native function [12●]. It was not until 2013 that carbene and nitrene transferase activities were reported as promiscuous functions of hemoproteins that could be improved by engineering the protein [8,13,14].
The development of biocatalytic reactions is analogous to developing small-molecule-catalyzed transformations. Both start with identifying ‘hits’ from an existing library of catalysts. For biocatalysis, this library can be assembled by expressing gene sequences pulled from databases, from collections of expressed enzymes, or even from designed sequences. Once an enzyme is found that catalyzes the desired reaction at even a low level, its activity can often be improved by directed evolution.
Various technologies are available for facile generation of libraries of slightly modified genetically encoded protein catalysts, which can again be screened for improved activities in a directed evolution campaign [15,16]. In contrast, the throughput for screening small-molecule catalysts is low, because they often require lengthy syntheses. In addition to being easy to produce and modify, protein catalysts can be improved through iterative rounds of mutation and screening [17,18]. This reliable evolutionary optimization and enzymes’ inherent ‘tunability’ confer an advantage for highly selective catalysis. With a good optimization process, the bottleneck for developing an enzyme for a new transformation becomes finding a protein with some level of activity that can then be optimized. Unfortunately, screening collections of proteins for often-low levels of activity can be time-consuming and labor-intensive, and designing enzymes for new activities is still highly challenging [1].
The successful development of carbene and nitrene transferases relied on the inherent reaction and substrate promiscuity of the heme cofactor and the broad range of natural hemoproteins that can be tested for these novel activities. Since the initial reports, which recapitulated transformations already well-known for small-molecule catalysts, biocatalysis has pushed the boundaries of carbene and nitrene chemistry with increased efficiency and selectivity in known reactions and even several unprecedented chemical transformations [1,11]. We will describe the development trajectory of these new-to-nature enzymes, with emphasis on progress in the past two years. We will discuss how these biocatalytic systems might tackle synthetic challenges and what stands in the way of practical use.
New-to-nature carbene transfer reactions
Biocatalytic carbene transfer chemistry has experienced rapid growth and diversification in just a few years [1,11]. Discovery of this function [8] was inspired by the large repertoire of synthetic carbene chemistry: the first enzymes were found and engineered to catalyze reactions that mimicked synthetic methods [9]. The two main avenues for developing new carbene transferases have been (Figure 2a): 1) introducing new carbene precursors (carbene donors), such as fluorinated diazoalkanes and diazolactones [19–23], and 2) utilizing new carbene acceptors, such as alkynes, Si–H bonds, and C(sp3)–H bonds [24–29]. These reactions greatly expand nature’s repertoire of C–C and C–X bond-forming reactions. As more and more carbene transferases were created, interesting new promiscuous activities were discovered, which has led to novel transformations unfamiliar to the synthetic chemistry community, as discussed below.
Figure 2.

Hemoprotein-catalyzed carbene transfer reactions. (a) Selected carbene donors and acceptors in enzymatic carbene transfer reactions. All competent carbene donors are diazo compounds bearing an electron-withdrawing group adjacent to the diazo carbon [19–23]. Carbene acceptors range from σ-bonds [19,25,26●] to unsaturated π-systems [24●●,27●●,28]. Intramolecular carbene transfer reactions have also been developed to form ring structures [29●,35]. (b) X-ray crystal structure of carbene-bound Rma cytochrome c (1.29 Å, PDB ID: 6CUN). Hydrophobic amino acid residues interacting with the carbene species are highlighted in yellow [30●]. (c) Recent examples of enzymatic carbene transfer reactions that highlight the evolvability of hemoprotein scaffolds [24●●,27●●,28]. These include challenging transformations catalyzed by artificial metalloenzymes containing exogenously added Ir-porphyrin cofactors [38,39●].
Important mechanistic studies have also been performed for these engineered carbene transferases. An unprecedented carbine-bound Rhodothermus marinus (Rma) cytochrome c complex yielded an X-ray crystal structure of the iron- porphyrin carbene (IPC) intermediate (PDB ID: 6CUN) (Figure 2b) [30●]. This highly reactive carbene intermediate appears to be stabilized by five hydrophobic amino acid residues (T75, M76, P79, I83, M89) located 3.5–4.3 Å from the carbene in the enzyme active site. Further spectroscopic studies and computational modeling support the IPC intermediate having a singlet electronic state, which is influenced by the geometrical constraints and noncovalent interactions in the protein active site. These mechanistic studies are valuable for understanding how enzymes based on Rma cytochrome c tame unstable carbene species and control them for selective processes. In another study, Hilvert and coworkers obtained the crystal structure of an (Fe, N)-bridged IPC intermediate in an artificial hemoprotein, which contains a non-canonical N-methylhistidine as the axial ligand [31●]. It was suggested by quantum chemical calculation that the (Fe, N)-bridged carbene would equilibrate with the more reactive end-on form before carbene transfer.
Carbenes’ ability to react with unsaturated bonds has been utilized widely in synthetic chemistry to prepare strained ring systems [9]. Biocatalytic alkene cyclopropanation reactions have been developed by taking advantage of this reactivity [1,8,11]. Exploring other π-systems such as alkynes as carbene acceptors has allowed access to carbocycles with higher ring strain. Chen et al. engineered the first biocatalysts capable of forming cyclopropenes and bicyclobutanes from alkynes with excellent enantioselectivity and catalytic efficiency by directed evolution of a P411 enzyme (P450 engineered to have aserine axial ligand rather than cysteine) (Figure 2c) [24●●,28]. There are no reported small-molecule iron complexes that perform even racemic versions of these transformations: these new-to-nature activities are enabled by the protein scaffold. Notably, the enzymes’ lifetime of catalytic cyclopropenation, which is reflected in the catalyst total turnover number (TTN) and determines the maximum yield of products from a catalytic center, is much higher than that of reported transition-metal catalysts (usually less than one hundred TTN) [32]. The various P411-based carbene transferases only differ by a handful of mutations, demonstrating the ease of evolving for new functions.
Nucleophilic heterocycles were also shown to be competent carbene acceptors. Vargas et al. reported the cyclopropanation of benzofuran derivatives using engineered myoglobins (Figure 2c) [27●●]. The excellent stereoselectivity achieved (>99.9% de and >99.9% ee in all cases) surpasses that of previously investigated rhodium catalytic systems, although the substrates are not exactly the same [33]. Interestingly, the same myoglobin variant (Mb-H64G V68A) also catalyzes C3-alkylation of indoles. The different reaction outcomes from benzofuran and indole substrates were rationalized based on density functional theory (DFT) calculations: a zwitterionic intermediate appears to be favored for the indole substrate, while a concerted reaction mechanism is more likely for benzofuran. Computational studies performed on a model of histidine-ligated Fe-porphyrin with and without active site residues pointed to the protein’s role in differentiating these pathways. Another recent study showed that P411 enzymes can be also used for selective alkylation of indoles and pyrroles [34].
Metalloporphyrins containing metals such as iridium and ruthenium [35] are known to catalyze a broader range of transformations than their iron analogues. This encouraged researchers to investigate whether incorporating abiological metalloporphyrins into protein scaffolds that normally bind heme would enable the proteins to catalyze even more new transformations. In the past few years, significant progress has been made in the development of artificial metalloproteins (ArMs), especially those with Ir-porphyrin cofactors [35–38]. Integrating an artificial metalloporphyrin into a protein could combine the high selectivity imparted by a protein active site with the general activity of the transition-metal catalyst. In a recent study, Gu et al. achieved a biocatalytic carbene C(sp3)–H insertion reaction with regioselectivity dictated by a distal substituent on the molecule (Figure 2c) [39●]. The catalyst comprises an Ir-porphyrin cofactor (Ir(Me)-MPIX) and a P450 scaffold. Random mutagenesis and screening were used to generate several variants selective for producing either of the two regioisomers of the C–H alkylation product. The free Ir(Me)-MPIX cofactor catalyzes this reaction with lower activity and complete loss of regioselectivity, which indicates that the protein scaffold is the key to achieving selective C(sp3)–H insertion.
ArMs are attractive because they can impart selectivity, and in some instances perhaps greater activity, to a less-selective metal complex. However, the ArM systems have significant limitations. Using a noble metal such as Ir increases cost and complexity compared to fully genetically encoded proteins that use earth-abundant iron. Preparation of the artificial cofactors requires multiple synthetic steps, whereas iron-heme is made relatively cost free as part of cellular metabolism. Moreover, assembly of a protein containing an artificial cofactor is often problematic for whole-cell systems; this is usually done in vitro with partially purified apoprotein and cofactor. Directed evolution of ArMs is therefore still challenging.
Hartwig and coworkers established a biosynthetic pathway incorporating an Ir-containing cyclopropanating P450 enzyme to make a non-natural terpenoid. A specific heme transport system was co-expressed to enable transport of exogenous Ir-porphyrin into the cell where the ArM was assembled [40●].
Unconventional nitrene transfer reactions
Developing methods to construct C–N bonds is one of the major themes of modern synthetic chemistry [41]. Among the numerous methods to install nitrogen functional groups, nitrene transfer reactions are powerful thanks to the unique reactivity of nitrene intermediates, particularly their capacity to rapidly rearrange or insert into various bonds. However, achieving good selectivity for substrates with multiple reactive sites is difficult, especially when a highly reactive nitrene intermediate is involved [42]. Biocatalytic nitrene transformations have the potential to address these challenges [1,11]. Although enzymatic nitrene transfer was established only recently, impressive catalysts have been reported (Figure 3a).
Figure 3.

Hemoprotein-catalyzed nitrene transfer reactions. (a) Selected nitrene donors and acceptors in enzymatic nitrene transfer reactions. Azide-based nitrene precursors are widely used in abiological nitrene transformations. Nitrene sources that contain N–O bonds are also suitable reagents for enzymes [46●,47●]. The corresponding nitrene species can react with unsaturated π-systems, thioether-based nucleophiles, and C–H coupling partners [11,43–45●]. (b) Recent advances include development of novel synthetic methods [46●,47●] and applications in cell imaging [50●].
Direct insertion of nitrene species into C–H bonds is particularly attractive, because the selective functionalization of these ubiquitous bonds represents the most efficient way to build molecular complexity [42]. The first efforts to develop C–H aminases focused on using intramolecular processes to construct ring systems, such as benzosultams and oxazolidinones [13,14,43,44]. Although this nitrene C–H insertion strategy has been widely explored in organic synthesis to construct heterocycles, site-selectivity strongly favors activated C–H bonds. By evolving P411 variants for benzosultam formation, Hyster et al. identified a set of enzymes for benzylic and homobenzylic functionalization [43]. They selectively targeted amination to an inert homobenzylic C–H bond in the presence of much weaker benzylic C–H bonds (~13 kcal/mol difference in bond strength), something that has only been realized using a protein catalyst. A recent study by Yang et al. showed that P411 enzymes can direct nitrene species to insert into various types of C–H bonds with different bond strengths, including strong methyl C–H bonds [45●]. In this work, racemic substrates bearing tertiary C–H bonds were transformed to highly enantioenriched products. The stereoconvergent process suggested radical generation at the chiral center, which was further supported by DFT calculations.
Nitrene precursors for these enzymatic reactions typically contain an azido functional group, which can be explosive and dangerous on larger scale. Biocatalysts that use less hazardous precursors are thus attractive. To this end, nitrene sources bearing an N–O bond have been explored as alternatives to azides [46●,47●]. Efforts in this direction led to the successful incorporation of O-pivaloylhydroxylamine in hemoprotein-catalyzed amination reactions (Figure 3a). This hydroxylamine-based reagent is particularly interesting because it decomposes into an unprotected nitrene intermediate to add a free amino group to the substrate. Previously, this reagent had been utilized in alkene aminohydroxylation reactions catalyzed by iron phthalocyanine [48]. However, due to the high energy and small size of non-substituted nitrene species, asymmetric transformations involving this intermediate are extremely challenging and have not been achieved with small-molecule catalysts. Directed evolution of Rma cytochrome c variants generated enzymes that catalyze aminohydroxylation in an enantioselective fashion (Figure 3b) [46●]. This enzymatic transformation was performed at 2.0-mmol scale with 4-methoxystyrene in 61% isolated yield and 85% ee.
Chemical intuition can also guide discovery of biocatalytic reactions that go well beyond and are even unprecedented in the synthetic literature. Jia et al. recently reported P411-catalyzed enantioselective primary amination at benzylic and allylic positions using O-pivaloylhydroxyl- amine as the nitrene precursor (Figure 3b) [47●]. This biocatalytic amination reaction tolerates a broad scope of C–H coupling partners bearing primary, secondary, and tertiary C–H bonds. An asymmetric primary amination process is highly valuable considering the fundamental role of chiral amines in the biological world and in organic chemistry. There are no previous examples of free amination of C–H bonds involving unmasked nitrene species. Previous work on hemoprotein-catalyzed benzylic amination with tosyl azide [49] and aminohydroxylation involving the same nitrene intermediate [46●] paved the way for this remarkable enzymatic activity.
Biocatalytic and biocompatible nitrene transformations are already appearing in applications other than chemical synthesis. Azide reduction was a known side reaction in several enzymatic nitrene transfer processes [14,49]. O’Connor et al. designed an azide-based bioreductive dye that used this promiscuous activity of native cytochrome P450s for imaging hypoxia in a range of human cancer cell lines (Figure 3b) [50●]. At low oxygen concentrations, the enzymes effectively catalyze reduction of the aryl azide to an aniline derivative, which is a fluorogenic marker. This overlooked azide reduction activity of P450s could be valuable for other, future applications in chemical biology.
Conclusion and Outlook
In just a few years, explorations of new-to-nature carbene and nitrene transfer reactivities of hemoproteins have led to the discovery of a series of highly selective and efficient biocatalytic transformations. These processes have progressed from the initial demonstrations which mimicked known reactions of small-molecule catalysts to reactions that are wholly unprecedented in the literature. We anticipate that metalloproteins catalyzing many more new reactions will be discovered and developed in the next few years. Investigations into the mechanisms of carbene and nitrene transferases will improve our understanding of how metalloproteins can acquire these non-natural functions through controlling both the formation and fate of the reactive intermediates. Excitingly, non-heme iron proteins were recently demonstrated to function as promiscuous nitrene transferases [51,52]. With more coordination sites available at the iron center compared to hemoproteins, these enzymes are tunable through small-molecule ligands [51] and are potentially capable of abiological transformations that involve binding of more than one site of the metal.
Late-stage modification of drugs or their analogs is an attractive near-term application of nitrene and carbene transferases. Unlike many native enzymes, which have high specificity for only one or two substrates, hemoproteins, especially P450 enzymes, can accept a wide range of substrates, reflecting their roles in defense and detoxification of xenobiotic compounds [6]. We anticipate that some of these enzymes can be engineered to perform highly selective late-stage modifications with their new nitrene and carbene transferase capabilities.
The new-to-nature enzymes can be used together with small-molecule-catalysts or other enzymes in a cascade or stepwise fashion. A recent example incorporated a biocatalytic carbene C–H insertion reaction in the formal syntheses of the natural products (+)/(–)-cuspareine and (+)-lyngbic acid [26●]. Carbene and nitrene transferases could also be incorporated into novel metabolic pathways to produce natural product analogs [2]. For example, a non-natural terpenoid was prepared from glucose and diazoacetate through an artificial biosynthetic pathway containing an Ir-based ArM [40●] (vide supra).
Enzymatic nitrene and carbene transformations, however, are very new, and there is considerable room for improvement, especially if they are to be used at scale or incorporated into novel metabolic pathways. The use of unstable carbene and nitrene precursors, such as diazo and azido compounds, is appropriate for laboratory-scale research, but is not attractive for larger-scale syntheses due to cost and safety concerns. They are also toxic to microorganisms. To this end, introducing cheap and safe carbene and nitrene precursors, or making them in situ, will be necessary to realize the full potential of these biotransformations. Furthermore, side reactions with heme or protein side chains are a problem. Similar to the reactive oxygen species that eventually inactivate cytochrome P450s and other enzymes, the carbene and nitrene species are also effective inactivators. Studies revealed that nitrogen atoms on porphyrin and nucleophilic residues (H92, H100) of a P450 variant could react with iron-bound carbene intermediates [53]. Substitution to non-nucleophilic residues led to slightly improved performance. Similarly, nitrene transferases usually cannot tolerate high concentrations of the nitrene precursors used to date [47●]. It is possible that directed evolution can find sequences that effectively guard the protein from inactivation, just as natural evolution has done for enzymes that generate reactive oxygen species. Further evolution and innovative process design are required to achieve biocatalytic carbene and nitrene transfer processes with high working concentrations of substrates and satisfactory yields. Catalytic systems that can use less reactive carbene precursors are also desirable for large scale processes.
In conclusion, hemoprotein-catalyzed carbene and nitrene transformations have developed rapidly over the past several years. These enzymatic reactions can be used not only in synthesis, but also in other areas where the biocompatibility of the reactions and genetic encoding of the catalyst will enable applications in living organisms or in the presence of other enzymes. Furthermore, new carbene and nitrene transferases are proving to be good starting points for even more challenging transformations. Whole new families of enzymes discovered and engineered with inspiration from chemistry will soon marry synthetic chemists’ favorite transformations with the selectivity and efficiency of biological catalysis.
Highlights.
New-to-nature carbene and nitrene transfer enzymes inspired by chemical catalysis.
Engineered hemoproteins that catalyze these transformations with high selectivities tackle difficult synthetic challenges.
New classes of enzymatic transformations open opportunities for applications in chemical biology and biosynthesis.
Acknowledgements
This work was supported by the NSF Division of Molecular and Cellular Biosciences (2016137), the NIH National Institute of General Medical Sciences (R01GM138740), the U.S. Department of Energy (DE-SC0021141), the US Army Research Office Institute for Collaborative Biotechnologies (cooperative agreement W911NF-19–2-0026 and contract W911NF-19-D-0001), and the Jacobs Institute for Molecular Engineering for Medicine at Caltech. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding agencies. The authors thank Dr. Benjamin Levin, Dr. Sabine Brinkmann-Chen, Dr. Kai Chen, Dr. Yang Yang, Dr. David Miller, and Dr. Soumitra Athavale for helpful discussions and comments on the manuscript.
Footnotes
Conflict of interest statement
Nothing declared.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
● of special interest
●● of outstanding interest
References
- 1.Chen K, Arnold FH: Engineering new catalytic activities in enzymes. Nat Catal 2020, 3:203–213. [Google Scholar]
- 2.Erb TJ, Jones PR, Bar-Even A: Synthetic metabolism: metabolic engineering meets enzyme design. Curr Opin Chem Biol 2017, 37:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jeschek M, Panke S, Ward TR: Artificial metalloenzymes on the verge of new-to-nature metabolism. Trends Biotechnol 2018, 36:60–72. [DOI] [PubMed] [Google Scholar]
- 4.Noyori R: Asymmetric catalysis: science and opportunities (Nobel lecture). Angew Chem Int Ed 2002, 41:2008–2022. [PubMed] [Google Scholar]
- 5.Campos KR, Coleman PJ, Alvarez JC, Dreher SD, Garbaccio RM, Terrett NK, Tillyer RD, Truppo MD, Parmee ER: The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363:eaat0805. [DOI] [PubMed] [Google Scholar]
- 6.Schmitz LM, Rosenthal K, Lütz S: Recent advances in heme biocatalysis engineering. Biotechnol Bioeng 2019, 116:3469–3475.31483477 [Google Scholar]
- 7.Moody PCE, Raven EL: The nature and reactivity of ferryl heme in compounds I and II. Acc Chem Res 2018, 51:427–435. [DOI] [PubMed] [Google Scholar]
- 8.Coelho PS, Brustad EM, Kannan A, Arnold FH: Olefin cyclopropanation via carbene transfer catalyzed by engineered cytochrome P450 enzymes. Science 2013, 339:307–310. [DOI] [PubMed] [Google Scholar]
- 9.Doyle MP: Catalytic methods for metal carbene transformations. Chem Rev 1986, 86:919–939. [DOI] [PubMed] [Google Scholar]
- 10.Dequirez G, Pons V, Dauban P: Nitrene chemistry in organic synthesis: still in its infancy? Angew Chem Int Ed 2012, 51:7384–7395. [DOI] [PubMed] [Google Scholar]
- 11.Brandenberg OF, Fasan R, Arnold FH: Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr Opin Biotechnol 2017, 47:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●12.Tsutsumi H, Katsuyama Y, Izumikawa M, Takagi M, Fujie M, Satoh N, Shin-ya K, Ohnishi Y: Unprecedented cyclization catalyzed by a cytochrome P450 in benzastatin biosynthesis. J Am Chem Soc 2018, 140:6631–6639.Through analysis of the benzastatin biosynthetic gene cluster, the authors identified a cytochrome P450 enzyme which is proposed to catalyze nitrene transfer reactions to form aziridines. This work represents the first report of a natural nitrene transferase.
- 13.McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, Arnold FH: Enantioselective intramolecular C–H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew Chem Int Ed 2013, 52:9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh R, Bordeaux M, Fasan R: P450-catalyzed intramolecular sp3 C–H amination with arylsulfonyl azide substrates. ACS Catal 2014, 4:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Packer MS, Liu DR: Methods for the directed evolution of proteins. Nat Rev Genet 2015, 16:379–394. [DOI] [PubMed] [Google Scholar]
- 16.Qu G, Li A, Acevedo-Rocha CG, Sun Z, Reetz MT: The crucial role of methodology development in directed evolution of selective enzymes. Angew Chem Int Ed 2020, 59:13204–13231. [DOI] [PubMed] [Google Scholar]
- 17.Arnold FH, Volkov AA: Directed evolution of biocatalysts. Curr Opin Chem Biol 1999, 3:54–59. [DOI] [PubMed] [Google Scholar]
- 18.Reetz MT: Biocatalysis in organic chemistry and biotechnology: past, present, and future. J Am Chem Soc 2013, 135:12480–12496. [DOI] [PubMed] [Google Scholar]
- 19.Huang X, Garcia-Borràs M, Miao K, Kan SBJ, Zutshi A, Houk KN, Arnold FH: A biocatalytic platform for synthesis of chiral α- trifluoromethylated organoborons. ACS Cent Sci 2019, 5:270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen K, Zhang S-Q, Brandenberg OF, Hong X, Arnold FH: Alternate heme ligation steers activity and selectivity in engineered cytochrome P450-catalyzed carbene-transfer reactions. J Am Chem Soc 2018, 140:16402–16407. [DOI] [PubMed] [Google Scholar]
- 21.Zhang J, Huang X, Zhang RK, Arnold FH: Enantiodivergent α-amino C–H fluoroalkylation catalyzed by engineered cytochrome P450s. J Am Chem Soc 2019, 141:9798–9802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●22.Hock KJ, Knorrscheidt A, Hommelsheim R, Ho J, Weissenborn MJ, Koenigs RM: Tryptamine synthesis by iron porphyrin catalyzed C−H functionalization of indoles with diazoacetonitrile. Angew Chem Int Ed 2019, 58:3630–3634.An indole C3 alkylation reaction was accomplished with diazoacetonitrile. The final products can be further derivatized to make valuable tryptamine analogs. The authors showed that this reaction could be catalyzed by both an iron-complex (FeTPPCl) and a hemoprotein (YfeX).
- 23.Chandgude AL, Fasan R: Highly diastereo- and enantioselective synthesis of nitrile-substituted cyclopropanes by myoglobin-mediated carbene transfer catalysis. Angew Chem Int Ed 2018, 57:15852–15856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●●24.Chen K, Huang X, Kan SBJ, Zhang RK, Arnold FH: Enzymatic construction of highly strained carbocycles. Science 2018, 360:71–75.The authors reported the first biocatalytic reactions for cyclopropene and bicyclobutane formation from alkynes with excellent enantioselectivity. These strained carbocycles are challenging to synthesize via conventional methods.
- 25.Kan SBJ, Huang X, Gumulya Y, Chen K, Arnold FH: Genetically programmed chiral organoborane synthesis. Nature 2017, 552:132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●26.Zhang RK, Chen K, Huang X, Wohlschlager L, Renata H, Arnold FH: Enzymatic assembly of carbon–carbon bonds via iron-catalysed sp3 C–H functionalization. Nature 2019, 565:67–72.Using engineered P411 variants, the authors achieved intermolecular carbene C–H insertion with excellent chemo- and stereoselectivity. C–H bonds at benzylic, allylic, propargylic and nitrogen α-positions are all competent for this transformation. Formal syntheses of natural products utlizing this enzymatic method were also demonstrated.
- ●●27.Vargas DA, Khade RL, Zhang Y, Fasan R: Biocatalytic strategy for highly diastereo-and enantioselective synthesis of 2,3-dihydrobenzofuran-based tricyclic scaffolds. Angew Chem Int Ed 2019, 58:10148–10152.Myoglobins were reported to catalyze the cyclopropanation of benzofuran derivatives. Interestingly, the same myoglobin variant could also catalyze C3-alkylation of indoles. DFT calculations revealed that active site residues are important in favoring cyclopropanation over C3-alkylation for benzofuran substrates.
- 28.Chen K, Arnold FH: Engineering cytochrome P450s for enantioselective cyclopropenation of internal alkynes. J Am Chem Soc 2020, 142:6891–6895. [DOI] [PubMed] [Google Scholar]
- ●29.Chandgude AL, Ren X, Fasan R: Stereodivergent intramolecular cyclopropanation enabled by engineered carbene transferases. J Am Chem Soc 2019, 141:9145–9150.An intramolecular cyclopropanation reaction was developed with engineered myoglobins to access both enantiomers from the same substrates. Various cyclopropyl-γ-lactones can be prepared through this method with excellent yields and enantioselectivity.
- ●30.Lewis RD, Garcia-Borràs M, Chalkley MJ, Buller AR, Houk KN, Kan SBJ, Arnold FH: Catalytic iron-carbene intermediate revealed in a cytochrome c carbene transferase. Proc Natl Acad Sci USA 2018, 115:7308–7313.The X-ray crystal structure of carbene-bound Rma cytochrome c TDE variant was obtained. Spectroscopic studies combined with computation support the proposal that the iron porphyrin carbene (IPC) intermediate has a singlet ground electronic state.
- ●31.Hayashi T, Tinzl M, Mori T, Krengel U, Proppe J, Soetbeer J, Klose D, Jeschke G, Reiher M, Hilvert D: Capture and characterization of a reactive haem–carbenoid complex in an artificial metalloenzyme. Nat Catal 2018, 1:578–584.Myoglobin with N-methylhistidine as the axial ligand was found to be an efficient catalyst for alkene cyclopropanation. The authors also obtained the X-ray crystal structure of the IPC intermediate, which adopts a bridging Fe-C-N configuration.
- 32.Protopopova MN, Doyle MP, Mueller P, Ene D: High enantioselectivity for intermolecular cyclopropenation of alkynes by diazo esters catalyzed by chiral dirhodium(II) carboxamides. J Am Chem Soc 1992, 114:2755–2757. [Google Scholar]
- 33.Lehner V, Davies HML, Reiser O: Rh(II)-catalyzed cyclopropanation of furans and its application to the total synthesis of natural product derivatives. Org Lett 2017, 19:4722–4725. [DOI] [PubMed] [Google Scholar]
- 34.Brandenberg OF, Chen K, Arnold FH: Directed evolution of a cytochrome P450 carbene transferase for selective functionalization of cyclic compounds. J Am Chem Soc 2019, 141:8989–8995. [DOI] [PubMed] [Google Scholar]
- 35.Key HM, Dydio P, Clark DS, Hartwig JF: Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534:534–537. [DOI] [PubMed] [Google Scholar]
- 36.Davis HJ, Ward TR: Artificial metalloenzymes: challenges and opportunities. ACS Cent Sci 2019, 5:11120–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sreenilayam G, Moore EJ, Steck V, Fasan R: Metal substitution modulates the reactivity and extends the reaction scope of myoglobin carbene transfer catalysts. Adv Synth Catal 2017, 359:2076–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Natoli SN, Hartwig JF: Noble-metal substitution in hemoproteins: an emerging strategy for abiological catalysis. Acc Chem Res 2019, 52:326–335. [DOI] [PubMed] [Google Scholar]
- ●39.Gu Y, Natoli SN, Liu Z, Clark DS, Hartwig JF: Site-selective functionalization of (sp3)C−H bonds catalyzed by artificial metalloenzymes containing an iridium-porphyrin cofactor. Angew Chem Int Ed 2019, 58:13954–13960.Artificial metalloenzymes containing Ir-porphyrin cofactors were used to catalyze regioselective intermolecular C–H alkylation reactions. Random mutagenesis was used to identify enzyme pairs favoring each of the two regioisomers.
- ●40.Huang J, Liu Z, Bloomer BJ, Clark DS, Keasling JD, Hartwig JF: Artificial biosynthetic pathway for an unnatural terpenoid with an iridium-containing P450. ChemRxiv 2020, DOI: 10.26434/chemrxiv.11955174.v1.Using a heterologous heme transport system, the authors constructed an artificial biosynthetic pathway incorporating an Ir-porphyrin based metalloenzyme in Escherichia coli (E. coli).
- 41.Hili R, Yudin AK: Making carbon-nitrogen bonds in biological and chemical synthesis. Nat Chem Biol 2006, 2:284–287. [DOI] [PubMed] [Google Scholar]
- 42.Lescot C, Darses B, Collet F, Retailleau P, Dauban P: Intermolecular C–H amination of complex molecules: insights into the factors governing the selectivity. J Org Chem 2012, 77:7232–7240. [DOI] [PubMed] [Google Scholar]
- 43.Hyster TK, Farwell CC, Buller AR, McIntosh JA, Arnold FH: Enzyme-controlled nitrogen-atom transfer enables regiodivergent C–H amination. J Am Chem Soc 2014, 136:15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Singh R, Kolev JN, Sutera PA, Fasan R: Enzymatic C(sp3)–H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal 2015, 5:1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●45.Yang Y, Cho I, Qi X, Liu P, Arnold FH: An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3)–H bonds. Nat Chem 2019, 11:987–993.Nitrene intermediates were shown to insert intramolecularly into C–H bonds bearing different bond strengths, including strong methyl C–H bonds. This approach could be used to prepare a variety of synthetically useful structural motifs.
- ●46.Cho I, Prier CK, Jia Z, Zhang RK, Görbe T, Arnold FH: Enantioselective aminohydroxylation of styrenyl olefins catalyzed by an engineered hemoprotein. Angew Chem Int Ed 2019, 58:3138–3142.This cytochrome c-catalyzed olefin aminohydroxylation reaction marks the first use of an unsubstituted nitrene precursor in enzymatic reactions. An array of styrene derivates, including internal alkenes are suitable substrates.
- ●47.Jia Z-J, Gao S, Arnold FH: Enzymatic primary amination of benzylic and allylic C(sp3)–H bonds. J Am Chem Soc 2020, 142:10279–10283.The authors demonstrated biocatalytic primary amination with P411 variants, directly functionalizing benzylic and allylic C–H bonds. Highly valuable primary amines could be obtained in high synthetic yields and enantiopurity.
- 48.Legnani L, Morandi B: Direct catalytic synthesis of unprotected 2-amino-1-phenylethanols from alkenes by using iron(II) phthalocyanine. Angew Chem Int Ed 2016, 55:2248–2251. [DOI] [PubMed] [Google Scholar]
- 49.Prier CK, Zhang RK, Buller AR, Brinkmann-Chen S, Arnold FH: Enantioselective, intermolecular benzylic C–H amination catalysed by an engineered iron-haem enzyme. Nat Chem 2017, 9:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ●50.O’Connor LJ, Mistry IN, Collins SL, Folkes LK, Brown G, Conway SJ, Hammond EM: CYP450 enzymes effect oxygen-dependent reduction of azide-based fluorogenic dyes. ACS Cent Sci 2017, 3:20–30.An organic dye containing an azido group could be reduced to an aniline-based fluorogenic maker by P450 enzymes under low oxygen conditions. This reaction was used to image hypoxia in a range of human cancer cell lines.
- 51.Goldberg NW, Knight AM, Zhang RK, Arnold FH: Nitrene transfer catalyzed by a non-heme iron enzyme and enhanced by non-native small-molecule ligands. J Am Chem Soc 2019, 141:19585–19588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vila MA, Steck V, Giordano SR, Carrera I, Fasan R: C–H amination via nitrene transfer catalyzed by mononuclear non-heme iron-dependent enzymes. ChemBioChem 2020, 21:1981–1987. [DOI] [PubMed] [Google Scholar]
- 53.Renata H, Lewis RD, Sweredoski MJ, Moradian A, Hess S, Wang ZJ, Arnold FH: Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes. J Am Chem Soc 2016, 138:12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]