

Abstract
Nuclear and plastid (chloroplast) genomes experience different mutation rates, levels of selection, and transmission modes, yet key cellular functions depend on their coordinated interactions. Functionally related proteins often show correlated changes in rates of sequence evolution across a phylogeny [evolutionary rate covariation (ERC)], offering a means to detect previously unidentified suites of coevolving and cofunctional genes. We performed phylogenomic analyses across angiosperm diversity, scanning the nuclear genome for genes that exhibit ERC with plastid genes. As expected, the strongest hits were highly enriched for genes encoding plastid-targeted proteins, providing evidence that cytonuclear interactions affect rates of molecular evolution at genome-wide scales. Many identified nuclear genes functioned in post-transcriptional regulation and the maintenance of protein homeostasis (proteostasis), including protein translation (in both the plastid and cytosol), import, quality control, and turnover. We also identified nuclear genes that exhibit strong signatures of coevolution with the plastid genome, but their encoded proteins lack organellar-targeting annotations, making them candidates for having previously undescribed roles in plastids. In sum, our genome-wide analyses reveal that plastid–nuclear coevolution extends beyond the intimate molecular interactions within chloroplast enzyme complexes and may be driven by frequent rewiring of the machinery responsible for maintenance of plastid proteostasis in angiosperms.
Genome-wide evolutionary rate covariation analyses reveal potential novel plastid–nuclear interactions and suites of coevolving plastid proteostasis genes.

Introduction
Only a small fraction of the proteins that required plastid function are encoded by the plastid genome (plastome) itself (Timmis et al., 2004; van Wijk and Baginsky, 2011). The remaining plastid-localized proteins are encoded by the nuclear genome, translated in the cytosol, and imported into plastids [hereafter referred to as nucleus-encoded plastid-targeted (N-pt) proteins], where they often interact with the plastome and its gene products (Gould et al., 2008). These plastid–nuclear interactions are critical for overall fitness, as evidenced by the frequent role of plastid–nuclear incompatibilities in reproductive isolation (Schmitz-Linneweber et al., 2005; Greiner et al., 2011; Bogdanova et al., 2015; Barnard-Kubow et al., 2016; Zupoka et al., 2020).
One signature of proteins that are functionally related and/or coevolving is that they tend to exhibit correlated changes in their rates of sequence evolution across a phylogeny, which is known as evolutionary rate covariation (ERC) and can be quantified by comparing genetic distances or branch lengths of gene trees from two potentially interacting genes (Goh et al., 2000; Ramani and Marcotte, 2003; Sato et al., 2005; Clark and Aquadro, 2010; Clark et al., 2012; De Juan et al., 2013). The known physical interactions within “chimeric” plastid–nuclear complexes (i.e. those containing both plastome-encoded and N-pt proteins) have provided a valuable system to test and illustrate the principle that coevolution and functional interactions can result in ERC (Sloan et al., 2014a, 2014b; Zhang et al., 2015, 2016; Rockenbach et al., 2016; Weng et al., 2016; Williams et al., 2019).
In addition to probing known interactions, ERC has served as a powerful tool to scan entire genomes/proteomes to detect previously unrecognized functional relationships (Findlay et al., 2014; Raza et al., 2019), which do not always rely on direct physical interactions (Clark et al., 2012). For example, application of a genome-wide ERC scan in diverse insects with heterogeneous rates of mitochondrial genome evolution recovered novel mitonuclear interactions (Yan et al., 2019). However, despite strong evidence of correlated rates among known members of plastid–nuclear complexes, ERC analysis has not been applied on a genome-wide scale across diverse plant lineages, raising the intriguing possibility that we may have only scratched the surface with respect to the full breadth of plastid–nuclear interactions. A key barrier resides in the frequent occurrence of gene and whole-genome duplication in plants (Panchy et al., 2016; Wendel et al., 2018), which makes it inherently difficult to perform phylogenomic scans for ERC. Typical implementations of ERC analysis require one-to-one orthology in gene trees (Clark et al., 2012; Findlay et al., 2014; Wolfe and Clark, 2015; Yan et al., 2019), but gene duplication yields large gene families composed of sequences that share both orthology and paralogy (Bansal and Eulenstein, 2008; Stolzer et al., 2012). Outside of the context of ERC, numerous studies have overcome some of the challenges associated with phylogenomics in plants by carefully filtering gene families and/or extracting subtrees that represent mostly orthologs (Sanderson and McMahon, 2007; Duarte et al., 2010; De Smet et al., 2013; Sangiovanni et al., 2013; Forsythe et al., 2020). Nevertheless, these approaches cannot completely eliminate the pervasive effects of gene duplication and differential loss, so performing ERC analyses across diverse plant lineages requires a novel approach that can accommodate this recurring history.
ERC analyses have the potential to be especially powerful for probing plastid–nuclear interactions because the rate of plastome evolution can differ greatly across angiosperm species, with several lineages exhibiting extreme accelerations. Not surprisingly, angiosperms that lose photosynthetic function and transition to parasitic/heterotrophic lifestyles exhibit massive plastome decay and rapid protein sequence evolution (Wicke et al., 2016), in extreme cases, resulting in outright loss of the entire plastome (Molina et al., 2014). However, even among angiosperms that remain fully photosynthetic, there have been repeated accelerations in their rates of plastid gene evolution (Jansen et al., 2007; Guisinger et al., 2008; Knox, 2014; Sloan et al., 2014a, 2014b; Dugas et al., 2015; Nevill et al., 2019; Shrestha et al., 2019). These accelerations in angiosperms that retain a photosynthetic lifestyle can be highly gene-specific (Magee et al., 2010) and are often most pronounced in nonphotosynthetic genes, such as those that encode ribosomal proteins, RNA polymerase subunits, the plastid caseinolytic protease (Clp) subunit ClpP1, the acetyl-CoA carboxylase (ACCase) subunit AccD, and the essential chloroplast factors Ycf1 and Ycf2 (Guisinger et al., 2008; Sloan et al., 2014a, 2014b; Park et al., 2017; Shrestha et al., 2019). Accelerated protein sequence evolution has frequently been accompanied by other forms of plastome instability, including structural rearrangements and gene duplication (Guisinger et al., 2011; Knox, 2014; Sloan et al., 2014a, 2014b; Shrestha et al., 2019), as well as accelerated mitochondrial genome evolution in some cases (Cho et al., 2004; Parkinson et al., 2005; Jansen et al., 2007; Mower et al., 2007; Sloan et al., 2009; Park et al., 2017). Several explanations have been proposed for the cause of these cases of rapid plastome evolution, but they largely remain a mystery (Guisinger et al., 2008; Park et al., 2017; Williams et al., 2019). Discovering the full suite of nuclear genes that repeatedly co-accelerate with plastid genes may advance our understanding of this angiosperm evolutionary puzzle.
Here, we develop an approach to apply genome-wide ERC analyses across diverse angiosperms to identify hundreds of nuclear genes that exhibit signatures of ERC with the plastome. This set of genes is highly enriched for known N-pt genes with functions in several pathways that appear to be centered around maintenance of plastid protein homeostasis (proteostasis). We also observe strong signatures of plastid–nuclear ERC for more than 30 nucleus-encoded, nonplastid-targeted proteins, representing candidates for novel plastid–nuclear interactions. Together, our findings extend our understanding of the genome-wide landscape of plastid–nuclear interactions.
Results
Genome-wide ERC analyses detect correlated evolution between the plastome and N-pt genes
We sampled 20 angiosperm species to perform a genome-wide scan for plastid–nuclear ERC. Given that the signature of ERC relies on phylogenetic rate heterogeneity, we sampled species that are known to exhibit differences in evolutionary rate for at least some plastid genes, including seven representatives of accelerated lineages (Jansen et al., 2007; Guisinger et al., 2008; Knox, 2014; Sloan et al., 2014a, 2014b; Dugas et al., 2015; Nevill et al., 2019; Shrestha et al., 2019) and 13 species that exhibit the slow background rate of plastome evolution typical for most angiosperms (Figure 1; Supplemental Table S1). We did not include parasitic species with accelerated plastome evolution, as these represent special cases of plastid evolution associated with loss of photosynthetic function (Wicke et al., 2016). Because our ERC analysis employs a root-to-tip strategy for measuring branch lengths (described below), we avoided sampling pairs of species that are closely related to each other in order to minimize pseudoreplication caused by shared internal branches (Felsenstein, 1985; Yan et al., 2019). We included Amborella (Amborella trichopoda) and Chinese tulip tree (Liriodendron chinense) as outgroups. We chose to include two outgroups, so that gene families would contain an outgroup sequence even if gene loss occurred in one of the two species, allowing us to analyze a larger proportion of gene families. It should be noted that phylogenetic placement of magnoliids (including Liriodendron) with regard to the ingroup (eudicots and monocots) has been a topic of debate (Soltis et al., 1999; Zanis et al., 2002; Hilu et al., 2003; Qiu et al., 2005, 2006). However, large-scale analysis of the plastid genome resolved Liriodendron as an outgroup to a eudicot/monocot clade (Jansen et al., 2007). We partitioned the plastid-encoded proteins into seven functional categories: ClpP1 (Figure 1A), AccD (Figure 1B), Ycf1/Ycf2 (Figure 1C), Maturase K (MatK) (Figure 1D), photosynthesis (Figure 1E), ribosomal proteins (Figure 1F), and RNA polymerase (Figure 1G) (see also Supplemental Table S2).
Figure 1.

Trees based on plastome partitions. Branch length-optimized trees inferred from amino acid sequence alignments for plastid genes partitioned into seven functional categories (as described in Supplemental Table 2). Branch lengths are shown on the same scale for all trees to highlight differences in rates of amino acid evolution between partitions. Each plastome partition tree was used for ERC analysis against all nuclear gene trees.
We then applied a custom phylogenomic analysis pipeline to all nuclear genomes and transcriptomes (Figure 2). Our pipeline included steps designed to extract gene families sharing orthology in the presence of gene duplication and loss, yielding a filtered set of 7,929 gene trees with an average of 25.1 sequences per tree and 16.4 species per tree (Supplemental Figure S1). We executed our genome-wide scan for plastid–nuclear ERC by testing all possible 55,503 pairwise correlations between trees (7 plastome trees x 7,929 nuclear trees) based on normalized branch lengths to account for lineage-specific features that may affect rates across entire genomes (e.g. generation time) (Clark and Aquadro, 2010). To directly compare trees that can differ in topology, gene duplication, and species representation, we measured branch lengths for each species on each tree using a “root-to-tip” approach (Yan et al., 2019), in which we averaged the cumulative branch length of the path leading from the common ancestor of all monocots, and eudicots to each tip (gene copy) for each species (see Materials and methods section).
Figure 2.

Overview of the phylogenomic pipeline used to identify and analyze nuclear gene families. A, Flowchart depicting the steps leading up to ERC analyses. B, Steps of the extraction and quality-control procedure.
To illustrate the ERC principle, we highlighted the plastid Clp complex as a case study (Figure 3), which is composed of the plastid-encoded ClpP1 subunit and multiple N-pt subunits (Nishimura and van Wijk, 2015). This complex represents an effective positive control in the context of a genome-wide scan because it was previously shown to exhibit strong ERC signals among subunits (Rockenbach et al., 2016; Williams et al., 2019). The Clp complex core is composed of two heptameric rings: the R-ring and the P-ring. ClpP1 is part of the R-ring and interacts more closely with the other subunits in this ring (ClpR subunits) than with the subunits of the P-ring (ClpP subunits) (Nishimura and van Wijk, 2015). These core rings are also accompanied by a variety of accessory proteins (ClpC, ClpD, ClpF, ClpS, and ClpT subunits), allowing us to compare ERC results for N-pt genes with varying degrees of physical interaction. A mirrored tree diagram of ClpP1 and ClpR1 illustrated that branch lengths from corresponding species on the two trees exhibit strong ERC (R2 = 0.94; Figure 3, A and B). When extending this analysis to all nuclear genes, a genome-wide distribution of ERC results for ClpP1 revealed that 11 of the 13 known Clp proteins (or 85%) exhibit an uncorrected value of P <0.05. Further, all ClpR and ClpP subunits are present among the strongest ERC hits (top 2% of all genes analyzed), and all but one maintained genome-wide significance after correcting for multiple testing (Figure 3C). We also detected a general pattern of clustering of ERC values between ClpP1 and other Clp subunits that corresponds to the intimacy of their known interactions; ClpR subunits displayed the strongest ERC, followed by ClpP subunits, with the accessory Clp subunits showing the weakest signal.
Figure 3.

Case study of ERC between the plastid-encoded ClpP1 protease and nuclear gene trees. A, ClpP1 and ClpR1 gene trees, shown mirrored to highlight correlation of branch lengths. B, Linear regression analysis quantifying correlation of evolutionary rates between ClpP1 and ClpR1. Points represent normalized branch lengths estimated from ClpP1 (x-axis) and ClpR1 (y-axis) gene trees. Dotted line indicates best fit trend line. C, Results from ERC analyses of ClpP1 versus all nuclear genes. Each point represents P value and R2 values from a pairwise ERC analysis (Pearson correlation). ERC comparisons with negative slopes are not shown. Known Clp complex subunits encoded by nuclear genes are colored by their placement in the Clp structure (depicted in the legend), and includes ClpP1 to depict its position relative to other Clp subunits in the complex. Dashed lines indicate a raw P value of 0.05 (bottom) and a genome-wide significance at an FDR-corrected P value of 0.05 (top).
ClpP1 exhibited one of the most dramatic rate accelerations among plastome partitions (Figure 1A). Therefore, to assess how the magnitude of rate variation affected the statistical power of ERC, we also performed case studies (Supplemental Figure S2) for the plastid ribosome, which exhibited intermediate levels of acceleration (Figure 1F), and the photosynthesis partition, which showed less dramatic accelerations (Figure 1E). As observed in the Clp case study, these analyses detected significant ERC for much larger proportions of known interacting genes than would be expected by chance, but the degree of this enrichment for ERC signals was weaker and appeared to reflect the magnitude of rate variation in the corresponding plastome partition. For the plastid ribosome, 21 of the 34 nuclear genes (62%) had an uncorrected P <0.05 for ERC with the plastome ribosome partition, while 15 of 45 nuclear photosynthesis genes (33%) met this threshold for ERC with the plastome photosynthesis partition (Supplemental Figure S2). Overall, ERC appears to be sufficiently sensitive to detect functional plastid-nuclear interactions even with the background of a genome-wide scan.
We performed ERC analyses in parallel for each of the seven plastome partition trees shown in Figure 1 against normalized branch lengths from the nuclear trees (Supplemental Table S3). We determined that N-pt genes are highly significantly overrepresented in ERC hits for all plastome partitions, displaying roughly two-fold enrichment (Figure 4). We identified the subset of these genes that are known to directly physically interact with plastid-encoded proteins based on the CyMIRA classification (Forsythe et al., 2019) and observed an even higher degree of enrichment (approximately four- to eight-fold depending on the plastome partition). We also found correlations between plastome partitions and nuclear genes with mitochondrial function. Overall, nucleus-encoded, mitochondria-targeted (N-mt) proteins were significantly enriched among ERC hits for all plastome partitions with the exception of RNA polymerase and photosynthesis, although the effect size (approximately 1.5-fold) was smaller than for N-pt genes. N-mt proteins involved in direct physical interactions with mitochondrion-encoded proteins showed an increased degree of enrichment compared to all N-mt proteins (approximately two-fold), which was significant for all partitions. Proteins with dual localization to both plastids and mitochondria displayed a wider variance of enrichment with inconsistent significance, both of which may be related to the small sample size of this gene category. Finally, we found that genes annotated as localized to any parts of the cell other than the plastids or mitochondria are significantly depleted among ERC hits for all partitions (Figure 4). These results indicate that correlated plastid–nuclear evolution is pervasive across the nuclear genomes and that this signature is detectable by ERC.
Figure 4.
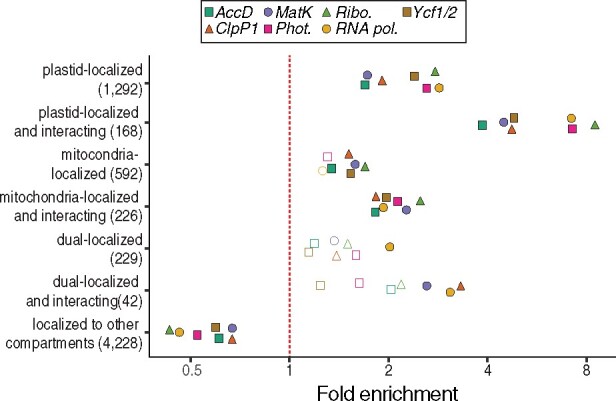
Subcellular localization and cytonuclear interactions of ERC hits. Proteins encoded by genes exhibiting signatures of coevolution with plastome partitions were analyzed for their localization and interactions as classified by the CyMIRA database (Forsythe et al., 2019). Categories indicating “interacting” refer to nucleus-encoded proteins predicted to directly physically interact with organelle-encoded proteins. The number of total genes in each category is indicated in parentheses. Statistical significance of enrichment/depletion (Fisher’s exact test) is indicated by filled points (P <0.05).
Functions associated with plastid proteostasis are highly enriched in ERC hits
Gene Ontology (GO) analyses of the ERC hits showed that several categories associated with plastid and mitochondrial functions were significantly enriched, while GO terms associated with other cellular compartments (e.g. “Nuclear” and “Endomembrane”) were significantly depleted (Figure 5). Combined with the targeting data presented above (Figure 4), these results reinforced the power of ERC in detecting cytonuclear interactions. Further, many of the enriched GO terms were more specifically connected to regulation of plastid proteostasis (Figure 5). For example, terms related to proteolytic activity (e.g. “protein quality control,” “chloroplastic Clp complex,” and “peptidase activity”) displayed some of the highest observed enrichments (more than eight-fold in some cases). This signature was further supported by detection of multiple subunits related to filamentation temperature-sensitive heterocomplex (FtsH) metalloproteases (Table 1). The translational machinery was also prominent: we detected enrichment for several related GO categories (e.g. “translation,” “ribosome biogenesis,” and “chloroplast rRNA processing”), and many individual genes that encode plastid ribosomal proteins or are involved in translation initiation/elongation (Table 1). The GO terms “protein transmembrane transport” and “protein localization to chloroplast” were also enriched, indicating genes involved in chloroplast protein import (Table 1). The above functions constitute key regulators of plastid proteostasis (Kim et al., 2013; Dogra et al., 2019), pointing to a possible driver of plastid–nuclear coevolution.
Figure 5.
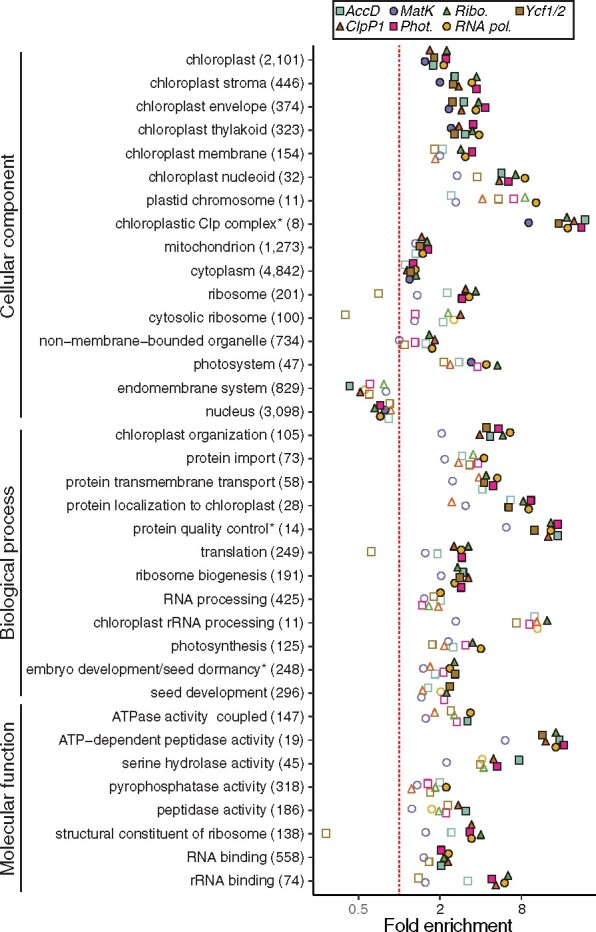
Functional enrichment of ERC hits. GO functional enrichment analyses were performed for ERC hits from each of the plastome partitions. Categories with significant enrichment/depletion in at least one partition are shown. Categories are grouped by type of GO annotation (cellular component, biological process, and molecular function). Some redundant or highly overlapping categories were removed (see Supplemental Data for full results). Asterisks indicate shortening of category name to fit figure dimensions. The number of total genes in each category is indicated in parentheses. Statistical significance of enrichment/depletion (Fisher’s exact test) is indicated by filled points (P <0.05). P values were corrected for multiple tests using FDR.
Table 1.
Organelle-localized strong ERC hits
| Plastome partition | Locus ID | Localization | Gene Symbol | TAIR Description | Slope | R 2 | Adj. p (Pearson) | Adj. p (Spearman) | Mult. reg. p | |
|---|---|---|---|---|---|---|---|---|---|---|
| Translation | Ribo., RNA pol., Phot., AccD, Ycf1/2, ClpP1 | At1g17220 | CP | FUG1 | Translation initiation factor 2, small GTP-binding protein | 1.44 | 0.95 | 9.27E–07 | 3.49E–13 | 1.21E–01 |
| Ribo., Phot. | At1g62750 | Dual | CPEF-G, SCO1 | Translation elongation factor EFG/EF2 protein | 2.61 | 0.78 | 2.35E–03 | 2.12E–01 | 5.03E–01 | |
| AccD | At5g67510 | CP | NA | Translation protein SH3-like family protein | 3.26 | 0.67 | 1.77E–02 | 7.08E–01 | 8.48E–02 | |
| Ribosomes | Ribo., RNA pol., Phot. | At4g34730 | CP | NA | ribosome-binding factor A family protein | 0.50 | 0.84 | 8.52E–04 | 8.30E–01 | 2.06E–02 |
| Ribo., ClpP1 | At2g33800 | CP | NA | Ribosomal protein S5 family protein | 0.41 | 0.76 | 5.31E–03 | 1.93E–01 | 5.04E–02 | |
| MatK | At5g02740 | MT | NA | Ribosomal protein S24e family protein | 0.33 | 0.81 | 7.67E–03 | 2.91E–13 | 1.33E–02 | |
| AccDa | At5g10360 | CP | EMB3010, RPS6B | Ribosomal protein S6e | 1.23 | 0.70 | 1.03E–02 | 6.91E–01 | 1.48E–02 | |
| Ribo., RNA pol. | At3g44890 | CP | RPL9 | ribosomal protein L9 | 0.75 | 0.71 | 1.15E–02 | 6.09E–01 | 9.37E–01 | |
| Ribo. | At5g40950 | CP | RPL27 | ribosomal protein large subunit 27 | 1.35 | 0.64 | 2.07E–02 | 7.26E–01 | 2.04E–01 | |
| ClpP1 | At1g64880 | MT | NA | Ribosomal protein S5 family protein | 3.73 | 0.60 | 4.56E–02 | 8.90E–01 | 1.67E–01 | |
| Clp | ClpP1,a AccD, Ribo., RNA pol., Phot.a | At1g49970 | CP | CLPR1, NCLPP5, SVR2 | CLP protease proteolytic subunit 1 | 1.46 | 0.94 | 4.70E–06 | 2.49E–13 | 8.26E–06 |
| ClpP1,a AccD,a Ribo., Phot. | At1g09130 | Dual | NA | ATP-dependent caseinolytic (Clp) protease/ crotonase family protein | 1.30 | 0.89 | 2.68E–05 | 2.49E–13 | 5.82E–05 | |
| ClpP1,a AccDa | At1g12410 | CP | CLPR2, NCLPP2, CLP2 | CLP protease proteolytic subunit 2 | 2.03 | 0.90 | 2.68E–05 | 5.44E–03 | 1.10E–03 | |
| AccD, Ribo., RNA pol., Phot., ClpP1,b Ycf1/2 | At5g45390 | CP | CLPP4, NCLPP4 | CLP protease P4 | 3.17 | 0.87 | 4.64E–04 | 5.93E–01 | 3.17E–03 | |
| ClpP1, AccD | At4g17040 | CP | CLPR4 | CLP protease R subunit 4 | 2.21 | 0.81 | 6.08E–04 | 2.49E–13 | 4.45E–03 | |
| AccD,b ClpP1 | At1g11750 | CP | CLPP6 | CLP protease proteolytic subunit 6 | 2.13 | 0.83 | 2.46E–03 | 6.00E–01 | 2.28E–03 | |
| ClpP1, Ycf1/2, Ribo. | At1g66670 | CP | CLPP3, NCLPP3 | CLP protease proteolytic subunit 3 | 2.62 | 0.76 | 4.52E–03 | 2.92E–01 | 6.46E–03 | |
| FtsH | AccD | At5g58870 | CP | FTSH9 | FTSH protease 9 | 9.22 | 0.75 | 7.52E–03 | 6.96E–01 | 1.37E–02 |
| Ribo., ClpP1,a AccD, RNA pol., Phot. | At5g53170 | Dual | FTSH11 | FTSH protease 11 | 1.47 | 0.76 | 3.27E–03 | 3.16E–01 | 1.05E–03 | |
| RNA-binding | RNA pol., Ribo.,b Phot. | At4g16390 | CP | SVR7 | pentatricopeptide (PPR) repeat-containing protein | 0.49 | 0.92 | 3.34E–05 | 4.50E–02 | 5.77E–04 |
| Ribo., Phot., RNA pol., AccD, Ycf1/2 | At5g66470 | CP | NA | RNA binding; GTP binding | 1.58 | 0.82 | 7.60E–04 | 2.47E–02 | 1.44E–01 | |
| AccD, Ycf1/2, Phot. | At4g31010 | MT | NA | RNA-binding CRS1 / YhbY (CRM) domain- containing protein | 8.39 | 0.87 | 9.07E–04 | 6.30E–01 | 9.42E–02 | |
| AccD, RNA pol. | At3g52150 | CP | NA | RNA-binding (RRM/RBD/RNP motifs) family protein | 2.33 | 0.87 | 1.65E–03 | 5.53E–01 | 4.25E–02 | |
| RNA pol., Ribo. | At3g23700 | CP | NA | Nucleic acid-binding proteins superfamily | 0.40 | 0.68 | 2.82E–02 | 4.16E–02 | 8.82E–03 | |
| RNA pol.b | At1g12800 | CP | NA | Nucleic acid-binding, OB-fold-like protein | 0.25 | 0.70 | 3.12E–02 | 4.50E–02 | 2.12E–02 | |
| RNA pol. | At2g20020 | CP | CAF1, | RNA-binding CRS1/YhbY (CRM) domain-containing protein | 0.72 | 0.61 | 3.40E–02 | 2.84E–01 | 4.68E–02 | |
| AccD | At5g14580 | MT | NA | Polyribonucleotide nucleotidyltransferase, putative | 9.88 | 0.62 | 6.86E–02 | 2.18E–13 | 9.07E–02 | |
| Import | AccD, ClpP1, RNA pol. | At1g06950 | CP | TIC110 | Translocon at the inner envelope membrane of chloroplasts 110 | 5.77 | 0.79 | 3.45E–03 | 5.53E–01 | 1.42E–02 |
| Ycf1/2, RNA pol.a | At5g22640 | CP | EMB1211 | Membrane Occupation and Recognition Nexus repeat-containing protein | 1.72 | 0.91 | 3.45E–04 | 1.12E–01 | 1.43E–03 | |
| AccDb | At5g03940 | CP | FFC, 54CP, CPSRP54, SRP54CP | Chloroplast signal recognition particle 54 kDa subunit | 15.57 | 0.82 | 1.65E–03 | 5.53E–01 | 3.84E–02 | |
| Ribo., Phot., RNA pol. | At4g26670 | Dual | NA | Mitochondrial import inner membrane translocase subunit Tim17/Tim22/Tim23 family protein | 0.45 | 0.72 | 5.14E–03 | 6.85E–01 | 7.42E–01 | |
| Ribo. | At3g23710 | CP | NA | Tic22-like family protein | 0.47 | 0.75 | 9.53E–03 | 2.91E–01 | 5.87E–01 | |
| Phot. | At3g04340 | CP | FTSHi5 | FTSH protease-like 5 | 0.09 | 0.71 | 4.89E–02 | 8.98E–01 | 1.37E–01 | |
| Ycf1/2b | At1g79560 | CP | EMB156, EMB36, EMB1047, FTSH12 | FTSH protease 12 | 3.31 | 0.87 | 7.86E–04 | 2.73E–01 | 2.09E–02 |
Arabidopsis Genome Initiative (AGI) locus identifiers are shown for nuclear genes showing significant ERC with plastome partition(s).
Significant ERC for the partition in multiple regression.
The partition was the only significant ERC under multiple regression. For genes that are hits in multiple plastome partitions, the slope, R2, and P-values for partition with the lowest Pearson P value are reported. Shown here is a subset of the complete set of 99 organelle-localized strong ERC hits. For full results see Supplemental Data. Phot, photosynthesis; Ribo, ribosomes; RNA pol, RNA polymerase; CP, chloroplast-localized protein; MT, mitochondrion-localized protein; NA, not available.
Interestingly, the only significantly enriched GO category that is not directly related to plastid or mitochondrion-localized function was “cytosolic ribosome,” which also has a clear role in translation. We found that each of the identified cytosolic ribosome gene families contained multiple Arabidopsis (Arabidopsis thaliana) paralogs, and we confirmed that these were bona fide cytosolic ribosomal subunits rather than misannotations of plastid ribosomal subunits in the GO classification scheme (Supplemental Figure S4; Bonen and Calixte, 2005; Creff et al., 2010; Tiller et al., 2012; Bieri et al., 2017; Boerema et al., 2018; Waltz et al., 2019). This result suggests that factors that affect the rate of evolution of plastid genes (and their N-pt interaction partners) may also act on cytosolic ribosomes, pointing to the potential regulation of plastid proteostasis via maintenance of cytonuclear stoichiometry (see Discussion section).
ERC analyses identify candidates for novel plastid functions
As previously mentioned, the individual hits with the strongest signatures of ERC were dominated by known N-pt or N-mt genes (76%; Table 1). These hits included 11 genes that have been annotated as organelle-localized but designated as “proteins of unknown function.” ERC for these genes provides evidence that may help resolve their roles in plastids. In addition, we observed 31 genes (24%) that are not annotated as plastid or mitochondrion-localized by CyMIRA (Forsythe et al., 2019) (Table 2). These are candidates for novel N-pt genes and may contribute to some of the functions described in the previous section. We discuss some of the most intriguing examples below, including potential novel plastid proteostasis regulators. In sum, our results highlight the specific pathways that exhibit plastid–nuclear ERC and reveal novel N-pt candidates, leading to new hypotheses to advance our understanding of the full scope of plastid–nuclear interactions and their effects on plant evolution.
Table 2.
Strong ERC hits lacking organelle-localized annotation
| Plastome Partition | Locus ID | Gene Symbol | TAIR Description | Slope | R 2 | Adj. P (Pearson) | Adj. P (Spearman) | Mult. reg. P |
|---|---|---|---|---|---|---|---|---|
| AccDa | At5g59860 | NA | RNA-binding (RRM/RBD/RNP motifs) family protein | 1.29 | 0.94 | 6.07E–04 | 6.51E–01 | 7.41E–06 |
| AccDa | At1g16750 | NA | Protein of unknown function, DUF547 | 2.18 | 0.94 | 1.65E–03 | 6.91E–01 | 3.32E–03 |
| Ycf1/2 | At1g04110 | SDD1 | Subtilase family protein | 0.91 | 0.94 | 1.71E–03 | 3.63E–01 | 1.66E–02 |
| Ycf1/2b | At5g22450 | NA | Unknown protein | 1.68 | 0.81 | 8.14E–03 | 1.92E–01 | 6.25E–02 |
| RNA pol.a | Os03g58204 | NA | NA | 0.39 | 0.75 | 8.50E–03 | 1.97E–01 | 6.91E–03 |
| Ribo. | At4g14100 | NA | Transferases, transferring glycosyl groups | 0.41 | 0.67 | 1.02E–02 | 7.52E–01 | 4.74E–01 |
| RNA pol. | At3g26618 | ERF1-3 | Eukaryotic release factor 1-3 | 0.54 | 0.68 | 1.08E-02 | 7.37E–01 | 6.40E–01 |
| ClpP1b | At1g09800 | NA | Pseudouridine synthase family protein | 5.28 | 0.74 | 1.23E–02 | 5.69E–01 | 1.53E–03 |
| Ycf1/2 | At4g25320 | NA | AT hook motif DNA-binding family protein | 0.75 | 0.75 | 2.22E–02 | 2.66E–01 | 2.61E–02 |
| Ycf1/2 | Os03g53360 | NA | NA | 1.59 | 0.88 | 2.22E–02 | 2.19E–01 | 2.14E–01 |
| AccD | At5g36000 | NA | BEST A.thaliana match: reduced male fertility | 0.18 | 0.80 | 2.65E–02 | 5.53E–01 | 8.69E–03 |
| Ycf1/2a | At1g55870 | PARN, AHG2 | Polynucleotidyl transferase, ribonuclease H-like superfamily protein | 2.61 | 0.73 | 2.85E–02 | 3.37E–01 | 4.26E–03 |
| ClpP1b | At2g16770 | bZIP23 | Basic-leucine zipper (bZIP) transcription factor family protein | 4.48 | 0.63 | 3.05E–02 | 7.41E–01 | 9.04E–04 |
| RNA pol., AccD, Ribo. | At4g19985 | NA | Acyl-CoA N-acyltransferases (NAT) superfamily protein | 0.42 | 0.66 | 3.20E–02 | 4.10E–01 | 7.14E–01 |
| RNA pol. | At1g69410 | ELF5A-3 | Eukaryotic elongation factor 5A-3 | 0.12 | 0.61 | 3.20E–02 | 8.76E–01 | 4.79E–01 |
| RNA pol. | At3g17880 | HIP, TDX, HIP2 | Tetratricopeptide domain-containing thioredoxin | 0.11 | 0.62 | 3.37E–02 | 7.47E–01 | 8.60E–02 |
| AccD | At4g19350 | EMB3006 | Embryo defective 3006 | 1.88 | 0.75 | 3.42E–02 | 7.16E–01 | 1.97E–02 |
| Ycf1/2b | Os03g26080 | NA | NA | 4.10 | 0.71 | 3.50E–02 | 2.20E–01 | 1.17E–01 |
| RNA pol. | At5g25840 | NA | Protein of unknown function (DUF1677) | 0.45 | 0.63 | 3.54E–02 | 2.60E–01 | 1.98E–01 |
| RNA pol. | At4g39920 | POR, TFCC | C-CAP/cofactor C-like domain-containing protein | 0.36 | 0.65 | 3.71E–02 | 5.08E–01 | 2.94E–01 |
| Ribo., RNA pol. | At1g71000 | NA | Chaperone DnaJ-domain superfamily protein | 0.62 | 0.62 | 3.93E–02 | 5.96E–01 | 5.68E–01 |
| AccD | At5g39420 | cdc2c | CDC2C | 4.59 | 0.74 | 3.95E–02 | 6.58E–01 | 4.11E–01 |
| RNA pol. | At1g03330 | NA | Small nuclear ribonucleoprotein family protein | 0.69 | 0.77 | 4.27E–02 | 2.18E–01 | 1.83E–01 |
| Ribo. | At2g03820 | NA | Nonsense-mediated mRNA decay NMD3 family protein | 0.19 | 0.59 | 4.34E–02 | 7.52E–01 | 1.06E–01 |
| RNA pol. | At5g26610 | NA | D111/G-patch domain-containing protein | 0.65 | 0.58 | 4.49E–02 | 5.07E–01 | 5.88E–01 |
| Phot., RNA pol. | At5g20040 | IPT9 | Isopentenyltransferase 9 | 0.35 | 0.63 | 4.74E–02 | 1.70E–01 | 1.61E–02 |
| AccD | At5g52860 | ABCG8 | ABC-2 type transporter family protein | 7.03 | 0.57 | 1.16E–01 | 2.18E–13 | 6.62E–02 |
| AccD | At2g28315 | NA | Nucleotide/sugar transporter family protein | 6.45 | 0.63 | 1.17E–01 | 2.18E–13 | 1.53E–01 |
| AccD | Os09g39370 | NA | NA | 1.68 | 0.59 | 2.70E–01 | 2.18E–13 | 1.51E–01 |
| MatK | At4g23330 | NA | BEST A. thaliana match: eukaryotic translation initiation factor 3A | 0.37 | 0.43 | 3.96E–01 | 2.91E–13 | 5.69E–01 |
AGI locus and Michigan State University (MSU) rice genome identifiers are shown for nuclear genes with significant ERC with plastome partition(s).
Significant ERC for the partition in multiple regression.
The partition was the only significant ERC under multiple regression. For genes that are hits in multiple plastome partitions, the slope, R2, and P values for partition with the lowest Pearson P value are reported. Rice IDs are shown for families in which Arabidopsis is not present. One ERC hit lacking an Arabidopsis and rice ID was omitted. For full results, see Supplemental Data. NA, not available.
Discussion
Genomic signatures of plastid–nuclear interactions can be detected with ERC in plants
ERC has revealed novel interactions in animals and fungi but, until now, has not been applied at broad phylogenetic scales in plants due to the prevalence of gene/genome duplication. We adapted existing techniques, initially developed with the stringent requirement of one-to-one orthology, to make them more tolerant of duplications, thus allowing us to analyze a substantial portion of plant nuclear genomes. Our pipeline (Figure 2) included several features tailored to the analysis of plant genomes. For example, our orthologous subtree extraction procedure identified subtrees with reduced paralogy compared to input trees, shifting the distribution of trees closer to one-to-one orthologous relationships without substantial loss of data (Supplemental Figure S1). In addition, our iterative gene tree/species tree (GT/ST) reconciliation approach resolved topological disagreements when they lacked phylogenetic support, thereby minimizing phylogenetic noise while retaining well-supported phylogenetic signatures. The typical implementations of ERC assume that every gene tree has the exact same sampling and topology (Clark and Aquadro, 2010; Clark et al., 2012; Findlay et al., 2014; Wolfe and Clark, 2015). However, this is rarely the case in datasets derived from plant genomes, as they are prone to topological variation introduced by internal duplications, incomplete lineage sorting, and differential gene loss (Degnan and Rosenberg 2009; Leebens-Mack et al., 2019), making it infeasible to compare individual branches in a one-to-one fashion between gene trees or to apply model-based evaluation of correlation from joint likelihoods (Clark and Aquadro, 2010). This challenge prompted us to apply a root-to-tip approach to calculating branch lengths. A drawback of this approach is that it introduces pseudoreplication by sampling shared internal branches multiple times (Felsenstein, 1985; Yan et al., 2019). We minimized this effect with our taxon-sampling by avoiding closely related species and, thus, approximating a “star-phylogeny” as closely as possible. Finally, when multiple paralogs were present in a gene tree, we averaged the branch lengths between all paralogs for a given species. This approach permitted us to accommodate localized duplication events within trees. Our results offer the proof-of-principle that ERC can be successfully extended to plant genomes at phylogenetic scales spanning angiosperm diversity, and likely further. While we focused on plastid–nuclear interactions, our results open the door to applying this method broadly to probe the entire plant interactome.
We used the plastid Clp, plastid ribosome, and photosynthetic enzyme complexes as case studies to assess the performance of ERC (Figure 3; Supplemental Figure S2). In all three cases, known interactors were enriched among the ERC hits, demonstrating the power of ERC to detect functional interactions. Each plastome partition also returned a number of ERC hits for genes that are not known interactors. Given that ERC has been demonstrated between nonphysically interacting but cofunctional genes (Clark et al., 2012), these genes may represent putative novel interactors. Indeed, the predominance of known N-pt proteins among these ERC hits indicates that ERC selectively returns genes with plastid functions (Figures 4 and 5), pointing to cofunctionality as a driver of ERC. However, it is also possible that a subset of the putative novel interactors is the result of noise rather than functional interaction. As such, there will be an obvious need for experimental validation of any newly identified interactions of interest.
Despite some uncertainty regarding interpretation of false positives, known interactions in our case studies do allow at least a rough assessment of the features that influence the power of ERC. The plastome partition trees used for each of these case studies exhibit a range of rate accelerations (Figure 1) that appear to roughly correlate with the predictive power of ERC, as ClpP1, ribosomes, and photosynthesis returned significant ERC hits for 85%, 61%, and 33% of known interactors, respectively. Further, unlike the Clp analysis, the strongest ERC hits for the plastid ribosome and photosynthetic enzymes were not known interactors. Therefore, the strength of signal may decline for plastome partitions that are more conserved in sequence and exhibit less rate variation across taxa.
Another factor that may limit the power of ERC is the extent to which functional rate covariation is concentrated on individual residues or individual proteins. This factor comes into play at two levels in our analysis. We inferred our nuclear gene trees from alignments of full protein sequences (trimmed to remove poorly aligned regions), meaning that branch length estimates are averaged across the entire length of proteins. If rate covariation was concentrated on a small number of residues (Madaoui and Guerois, 2008; Ovchinnikov et al., 2014), this averaging process would result in dilution of the true signal. Furthermore, our strategy of concatenating multiple plastid genes for some plastome partitions (Supplemental Table S2) holds similar risks of diluting or mixing signals. Conversely, an advantage of averaging across full-protein and concatenated alignments is that including more sequence data in an alignment may amplify signatures of functional covariation that are widespread but subtle. Further, combining individual sites into full-protein alignments and groups of known cofunctional plastid proteins into a concatenated alignment dramatically reduced the dimensionality of our pair-wise ERC comparisons, which is critical to scaling analyses of the whole genome. We reasoned that the advantages of using full-protein alignments and concatenating genes together outweigh the risks of signal dilution, especially given the evidence that ERC signature is often distributed along primary protein sequence, rather than being concentrated on individual residues (Clark et al., 2012). However, future analyses aimed at pinpointing the specific genes and residues that drive the broad signatures of ERC that we detect may provide further insight into the mechanisms of plastid–nuclear coevolution.
Taken together, our results illustrate the consequences of plastid–nuclear interactions on evolutionary rates at a genome-wide scale. However, it is important to consider the correlative nature of ERC and the fact that detected effects does not always imply direct functional interactions. For example, we observed significant enrichment of N-mt proteins among our ERC hits (albeit with a much weaker signal than for N-pt genes; Figure 4 and Table 1). Given that our ERC searches were seeded with plastome partitions, it is tempting to interpret these signals as evidence for cofunctionality or crosstalk between mitochondria and plastids. Although such factors may contribute to the observed N-mt signal, the rates of evolution of the plastid and mitochondrial genomes are known to be partially correlated with each other. Lineages such as Plantago, Silene, and Geraniaceae that exhibit rapid rates of plastome evolution in our sample (Figure 1) also have unusually rapidly evolving mitochondrial genomes (Cho et al., 2004; Parkinson et al., 2005; Jansen et al., 2007; Mower et al., 2007; Sloan et al., 2009; Seongjun Park et al., 2017). As such, we would expect overlap between ERC hits from the two genomes even in the absence of co-functionality between the mitochondria and plastids. Similarly, our plastome partitions do not evolve entirely independently of each other. Although the magnitudes of rate acceleration can vary greatly between genes (Figure 1; Guisinger et al., 2008; Sloan et al., 2014a, 2014b; Park et al., 2017; Shrestha et al., 2019), we observed significant ERC between all pairs of our plastome partition trees (Supplemental Table S4), thus limiting our ability to distinguish specific signatures of ERC for individual partitions. Consistent with this observation, we found overlap between the hits identified for each partition (Supplemental Figure 3, A and B). Multiple regression analyses provided some assistance in identifying the partitions making the strongest contributions to plastid–nuclear ERC (Supplemental Figure 3, C and D; Tables 1 and 2), but further investigation will be needed to tease apart the effects of correlated rates of evolution within and between organellar genomes in order to pinpoint the loci responsible for ERC with nuclear genes.
Networks of co-functional proteins are connected via their involvement in plastid proteostasis
ERC analyses point to plastid proteases, ribosomal proteins (subunits and binding/maturation factors), translation initiation/elongation factors, and proteins involved in protein import into plastids (Figure 4 and Table 1), all of which contribute to maintaining protein quality control, proteostasis, and the unfolded protein response (Kim et al., 2013; Dogra et al., 2019; Heinemann et al., 2020) (Supplemental Figure 5). Proteases exhibited some of the most striking signatures of ERC. In addition to Clp subunits, we observed strong ERC for FtsH7, FtsH9, and FtsH11. These proteins are thought to form two separate protease complexes, both of which localize to the plastid envelope (Ferro et al., 2003, 2010; Wagner et al., 2012). Interaction partners and substrates have been identified for FtsH11 (Adam et al., 2019), but very little is known about the function of the FtsH7/9 complex. These FtsH protease subunits do not appear to form a complex with any plastid-encoded protein, making them an example of correlated plastid–nuclear evolution in the absence of direct physical interaction. It is somewhat surprising that we did not observe significant ERC for other members of the gene family that comprise the thylakoid FtsH protease (FtsH1/2/5/8), considering that clp mutants are suppressors of the variegation phenotype seen in Arabidopsis variegated 2 (var2) mutant, which lacks a thylakoid-localized FtsH (Park and Rodermel, 2004; Yu et al., 2008). However, our results may be consistent with the prior observation that the expression of genes encoding thylakoid FtsH subunits is not affected by clp mutants, suggesting a lack of reciprocity in the interactions between Clp and the thylakoid FtsH protease (Kim et al., 2013). On the other hand, we did observe strong ERC for additional members of the FtsH family FtsH12 and FtsHi5, which form part of a complex that facilitates protein import across the inner membrane of the plastid, acting as an ATPase motor rather than a protease (Kikuchi et al., 2018). Plastid–nuclear ERC for this complex may result from the fact that it also contains plastid-encoded Ycf2 (another FtsH paralog) (Kikuchi et al., 2018). These and other genes involved in protein import (most notably, TRANSLOCON AT THE INNER ENVELOPE MEMBRANE OF CHLOROPLASTS 110 [Tic110]) (Table 1) point to the strong signature of plastid–nuclear evolution exhibited by import machinery, again highlighting the prominence of proteostasis pathways in our ERC hits.
We observed ERC for several plastid ribosomal subunits and other genes involved in plastid translation (Table 1). For example, SUPPRESSOR OF VARIEGATION 7 is a pentatricopeptide repeat protein that is involved in plastid rRNA processing, whose loss of function (like Clp subunits) suppresses the variegation phenotype of the var2 mutant (Liu et al., 2010), again pointing to functional connections between plastid translation and other proteostasis pathways. However, perhaps our most surprising piece of evidence for the role of translation in plastid–nuclear ERC is the association between ClpP1 and protein subunits of the cytosolic ribosome (Figure 4; Supplemental Figure 4). While ERC has been previously detected among cytonuclear subunits in plastid and mitochondrial ribosomes (Sloan et al., 2014a, 2014b; Weng et al., 2016), cytosolic ribosomes themselves have never been demonstrated to exhibit ERC with the mitochondrial or plastid genomes. Most of the plastid proteome is synthesized in the cytosol, meaning that the levels of N-pt and plastid-encoded proteins must be regulated to achieve stoichiometric balance for cytonuclear complexes (Colombo et al., 2016). In mitochondria, this balance is achieved through coordination of cytosolic and mitochondrial translation (Houtkooper et al., 2013; Couvillion et al., 2016). Recent evidence suggests that changes in cytosolic translation may have strong genetic interactions with the plastid proteostasis machinery. Specifically, mutation of a cytosolic ribosome subunit was shown to enhance the variegation phenotypes in var2 mutants (Wang et al., 2018). Given that disruption of plastid translation can suppress these same phenotypes (Yu et al., 2008; Liu et al., 2010; Zheng et al., 2016), it appears that ribosomes in both compartments play a key role in maintenance of plastid-nuclear stoichiometric balance. Additionally, we observed strong ERC for a putative tRNA pseudouridine synthase (At1g09800) that shows no evidence of either plastid or mitochondrial targeting (Table 2), suggesting that it likely modifies cytosolic tRNAs, again consistent with cytosolic translation being subject to plastid–nuclear selection. These results suggest that the effects of perturbation of plastid proteostasis may extend to cytosolic ribosomes, supporting a level of co-function-mediated ERC that spans cellular compartments.
Genes involved in various aspects of proteostasis appear to have been subject to accelerated protein evolution in independent angiosperm lineages. We propose that proteostasis systems have been perturbed in these lineages, causing shifts in selection that simultaneously affected numerous functionally related genes. Although the evolutionary events that may have led to these changes are unclear, one possible explanation may be related to the constant stoichiometric pressure plants experience in the face of nuclear gene/genome duplication (Birchler and Veitia, 2012; Sharbrough et al., 2017). Similarly, the susceptibility of plastomes to instability and rearrangements in certain angiosperm lineages (Jansen et al., 2007) may provide an initial trigger that elicits a series of coevolutionary responses. It has also been hypothesized that antagonistic interactions between the nucleus and selfish genetic elements in plastids may drive accelerated rates of evolution (Rockenbach et al., 2016; Sobanski et al., 2019). Finally, perturbations may be prompted by changes in abiotic or biotic stress, as many of the pathways that contribute to proteostasis are stress-responsive (e.g. the unfolded protein response to photooxidative stress) (Dogra et al., 2019; Heinemann et al., 2020). The cause of these perturbations may differ by lineage and disentangling them would reveal a critical driver of plant genome evolution. Regardless of the mechanisms, it is striking that the ripple effects are apparent across disparate pathways and cellular compartments and can be detected against the background of the entire genome in a large swath of plant diversity.
ERC points to novel plastid–nuclear interactions
Decades of proteomics research have led to the identification of over 2,400 plastid-localized proteins in Arabidopsis (http://ppdb.tc.cornell.edu; http://cymira.colostate.edu/). Yet, these proteins may only represent about 70% of the plastid proteome (Millar et al., 2006; van Wijk and Baginsky, 2011; Christian et al., 2020). Large-scale plastid proteome surveys are limited by ascertainment bias associated with protein accumulation level, tissue- and condition-specificity of accumulation/plastid-localization, and biochemical properties that influence mass spectrometry profiles (van Wijk and Baginsky, 2011). ERC offers an alternative line of evidence for plastid function/localization that is complementary to biochemical approaches and may not share the same biases. Our analyses returned several proteins that lack plastid-targeting annotations (Table 2) and represent candidates for novel N-pt proteins. For example, two of our strongest nonplastid-localized hits were annotated as RNA-binding (At5g59860) and Glycosylphosphatidylinositol (GPI)-anchored adhesin-like (At1g16750) proteins based on in silico domain predictions but are, otherwise, lacking in functional information. The signature of plastid–nuclear ERC that we observe for the genes in Table 2 suggested that they have experienced correlated changes in selection associated with accelerated plastome evolution. A natural hypothesis is that these are cryptic N-pt proteins that have evaded biochemical identification and curation in CyMIRA and its underlying databases (Forsythe et al., 2019). However, an alternative explanation is that they contribute to plastid function without localizing to plastids, similar to our hypothesis for cytosolic ribosomes and the pseudouridine synthase described above. A third possibility is that the proteins are plastid-localized in many plants but not in Arabidopsis, which is possible given the apparent lability of plastid targeting across plants (Christian et al., 2020; Costello et al., 2020). While each of these explanations comes with its own set of functional and evolutionary implications, future work to disentangle these alternative hypotheses will undoubtably advance our understanding of the full repertoire of plastid–nuclear interactions.
Materials and methods
Obtaining and processing sequence data
Our analysis was conducted on publicly available genomes and transcriptomes. We obtained the full set of 20 proteomes from several sources (Supplemental Table 1) and processed fasta files to add standardized sequence identifiers. For genome-based datasets that contained multiple splice variants per gene, we used only the first gene model (i.e. gene model ending in .1) and removed the others to avoid falsely defining splice variants as paralogs in gene family clustering.
Plastome gene datasets were extracted from GenBank files (see Supplemental Table 1) using a custom BioPerl script and manually curated to deal with missing annotations and inconsistent naming conventions. The corresponding protein sequences were either analyzed individually (ClpP1, AccD, and MatK) or concatenated from multiple plastid genes that are part of a common plastid complex and/or pathway (photosynthesis, ribosomes, RNA polymerase, and Ycf1/Ycf2) (Supplemental Table 2). The plastome sampling matched the nuclear proteome samples described above, except that no plastome sequence was available for New Guinea wattle (Acacia aulacocarpa), so we used the plastome of dune wattle (Acacia ligulata) in its place. The accD gene is missing from the plastome of rice (Oryza sativa) and great blue lobelia (Lobelia siphilitica), and ycf1 and ycf2 are missing from O. sativa and Madeira cranesbill (Geranium maderense). These species were omitted from the alignments and trees for AccD and Ycf1/Ycf2. Amino acid alignments based on plastome partitions were used to estimate branch lengths on a constraint tree with a topology based on Angiosperm Phylogeny Website (http://www.mobot.org/MOBOT/research/APweb) (Figure 1).
Gene family clustering, sequence alignment, and phylogenetic inference
We clustered homologous gene families using Orthofinder (v2.2.6) (Emms and Kelly, 2015) and performed multiple sequence alignment using the L-INS-i algorithm in MAFFT (v7.407) (Katoh and Standley, 2013). We used RAxML (v8.2.12) (Stamatakis, 2014) to infer maximum likelihood trees with 100 bootstrap replicates. Tree inference was performed using the command below for each gene. The -m argument indicates the model used (gamma distributed rate heterogeneity, empirical amino-acid frequencies, and the LG substitution model). The -p argument provides a seed for parsimony search. The -x argument provides a seed for rapid bootstrapping. The -# argument indicates the number of bootstrap replicates. The -f a argument implements rapid bootstrap analyses and best scoring tree search. The -T argument indicates the number of threads used for parallel computing.
raxmlHPC−PTHREADS−SSE3−s<input file name>−n<output file name>−m PROTGAMMALGF−p 12345−x 12345−# 100−f a−T 24.
For the step in which we optimized branch lengths on a constraint tree (see below), we used the following command, with -f e indicating parameter and branch-length optimization.
raxmlHPC−PTHREADS−SSE3−s<input file name>−n<output file name>−t name of constraint tree file>−m PROTGAMMALGF−p 12345−T 24−f e
Subtree extraction and quality control pipeline
ERC analyses are sensitive to false inferences of orthology. Particularly, treating cryptic out-paralogs as orthologs can alter branch length estimates (Smith and Hahn, 2020). While Orthofinder clusters sequences that share homology, these clusters do not always represent groups that share strict orthology. ERC analyses are also sensitive to poorly aligned sequences, which can result in long outlier branches on trees. To address these inherent challenges to genome-scale phylogenetic analyses, we built a pipeline to process nuclear gene trees and retain the portions of alignments and trees least likely to be affected by biasing factors. Our pipeline enlists several existing programs. In this section, we provide a summary of the steps in the pipeline and point the reader to subsequent sections for details on our application of individual components of the pipeline.
Step 1: Starting with the full gene trees, we performed GT/ST reconciliation in order to root the tree, rearrange poorly supported portions of the tree to conform with the species tree, and infer nodes in the tree that represent gene duplication rather than speciation.
Step 2: We used duplication information from Step 1 to extract subtrees representing orthology groups.
Step 3: We performed a second round of sequence alignment (using MAFTT as above) to generate alignments that contain only the sequences in subtrees.
Step 4: We trimmed these alignments to remove poorly aligned regions using GBLOCKS. We filtered out any alignments shorter than 50 amino acids in length, as well any alignments for which GBLOCKS trimming resulted in the removal of an entire sequence from the alignment.
Step 5: We inferred a new phylogeny for each subtree from the trimmed alignment using RAxML as above and again applied GT/ST reconciliation to the subtree trees to rearrange poorly supported nodes and root the tree.
Step 6: We used the reconciled versions of the gene trees (as constraint trees) and the trimmed version of the alignments to optimize final branch lengths for use in downstream ERC analyses.
Step 7: As a final means of quality control before performing ERC analyses, we assessed each tree to ask whether the ingroup formed a monophyletic clade in the branch-length-optimized tree. Those that were not monophyletic were pruned and rerooted in order to retain ingroup monophyly. We also filtered out trees with one very long outlier branch by removing any trees in which the longest branch was more than 10 times the length of the second longest branch.
GT/ST reconciliation
We used GT/ST reconciliation to reconstruct the history of gene duplication for each gene tree using Notung (v2.9) (Vernot et al., 2008; Stolzer et al., 2012). Briefly, Notung compares the topology of a gene tree inferred from an individual gene to the topology of a user-input species tree. We used the topology of the plastome trees described above as our species tree. Incongruencies between the gene tree and species tree were taken to be the result of historical gene duplication occurring at specific nodes of the tree. Notung uses a parsimony framework to reconcile these incongruences by inferring duplication and loss events along the gene tree to yield the most parsimonious series of duplication and loss events for each gene tree. Notung can also apply this logic to root unrooted gene trees by the most parsimonious root. Since topological incongruence is the signature by which Notung infers duplication events, inferences are sensitive to phylogenetic error, evidenced by branches with low bootstrap support. To avoid false inference of duplication from weakly supported branches, we made use of Notung’s option to only infer duplication supported by branches with bootstrap support of at least 80%.
We performed the rearranging step for each gene tree on the command line with the following command:
java -jar Notung-2.9.jar <path to gene tree file> -s <path to species tree file> –rearrange –threshold 80 –treeoutput nhx –nolosses –speciestag prefix –edgeweights name –outputdir <output directory>
We performed the rooting step for each gene tree with the following command:
java -jar Notung-2.9.jar <path to rearranged gene tree file> -s <path to species tree file> –root –treeoutput nhx –nolosses –speciestag prefix –edgeweights name –outputdir <output directory>
In both of the above commands, –treeoutput nhx indicates trees to be output in the newick extended format, which allows for the retention of duplication information. –nolosses indicates that loss information is omitted from the output file (but still included in the reconciliation process). –speciestag and –edgeweights instructs Notung where to find relevant information in the input file.
Orthologous subtree extraction
We used duplication information from Notung to extract portions of gene trees (i.e. subtrees) in which the taxa share orthology relationships to each other (as opposed to paralogy). We required that these subtrees contain at least one eudicot, one monocot, and one outgroup sequence (A. trichopoda or L. chinense). We required that at least ten species be represented in each subtree and the eudicot and monocot taxa in the subtree (i.e. the ingroup) form a monophyletic clade. To extract subtrees that fulfill these criteria, for each gene tree we started by iteratively splitting the tree at each node indicated as a duplication node by Notung and retaining the two daughter trees from the splits. Daughter trees were assessed independently and those that fulfilled the above criteria were retained, meaning that multiple subtrees were retained from an initial gene tree in some cases. The final subtrees retained after this process were non-overlapping subtrees containing at least ten taxa representing eudicots, monocots, and at least one outgroup with eudicots and monocots forming a monophyletic clade.
Multiple sequence alignment trimming with GBLOCKS
We used GBLOCKS (v0.91b) (Castresana, 2000) to trim poorly aligned regions of our alignments using the below command, with -b4 indicating the minimum length of the retained block, -b5 = h indicating that gaps are allowed in up to half of the total species, and -b2 indicating the minimum number of sequences for a flank position.
Gblocks<aln directory><aln file name>−b5=h−b4=5−b2= <half the total number of sequences>
Rerooting to retain ingroup monophyly following subtree phylogenetic inference
We realigned and inferred a new phylogeny for subtrees using the same methodology described above. In some cases, these new trees no longer placed eudicots and monocots (i.e. the ingroup) as a monophyletic group, which was a requirement of our downstream ERC analyses. This problem arose in trees in which there were multiple sequences from outgroup species and one or more of these taxa was nested within the ingroup causing the ingroup to be polyphyletic. For these trees, we identified the offending outgroup branches and pruned them from the tree. If A. trichopoda remained following pruning, we rooted on a branch leading to that species, choosing one at random if there were multiple A. trichopoda sequences. If no A. trichopoda branches remained, we rooted on L. chinense in a similar fashion.
ERC analysis
We obtained branch lengths for ERC analyses from rooted branch-length-optimized gene trees. The branch lengths for these trees were calculated with an LG substitution model, empirical amino acid frequencies, and gamma-distributed rate heterogeneity across sites (see RAxML command above). We used a root-to-tip method that measures the collective lengths of the path of branches from each ingroup tip to the node representing the most recent common ancestor of all ingroup tips, allowing for phylogeny-aware measurement of the amino acid substitutions in each lineage. We obtained these root-to-tip branch length measurements for all ingroup species for each gene tree using the dist.nodes() command from the Ape package (Paradis et al., 2004) in R. When multiple paralogs from a given species were present, the mean root-to-tip distance from all paralogs was used. When species were absent from trees, branch lengths were indicated as missing values for those species and excluded from ERC analysis for those genes. To account for lineage-specific differences in whole-genome rates of evolution, we normalized the branch length for each species by dividing the value of each tree by the average branch length for that species across all genes in our analysis. These normalized branch length values were used for pairwise ERC comparisons.
We compared each of the seven plastome partition trees against all nuclear trees. Each pairwise comparison comprised a correlation analysis of the branch lengths for each species in the plastid tree versus the branch lengths for the same species in the nuclear gene tree (see Figure 3 for visual depiction). For each pairwise comparison, we calculated Pearson and Spearman correlation coefficients. Because there is no clear biological expectation for significant inverse relationships in ERC, we only considered genes with positive correlations (slope >0) in downstream analyses. We adjusted P-values for multiple comparisons using the false discovery rate (FDR) method implemented with the p.adjust() function in R.
CyMIRA and GO functional enrichment analyses
In order to perform functional enrichment analyses, we needed a threshold to separate our “hits” from our background genes. We chose to make use of P-values from both Pearson and Spearman correlations as metrics because Pearson gains power from large branch lengths, potentially expected under true evolutionary co-acceleration, while Spearman is less sensitive to outlier branches. Any gene with a Pearson P ≤0.05 and a Spearman P ≤0.1 was considered as a “hit.” Our goal here was to identify the tail end of the distribution for the sake of functional enrichment analysis. A more stringent threshold was applied when assessing the significance of individual hits (Tables 1 and 2).
We used the Arabidopsis sequence identifiers present within gene families to probe functional enrichment of significant hits, based on localization/interaction annotations from CyMIRA and functional annotations from GO. We used the 7,929 genes in our filtered dataset as the background (rather than using the full Arabidopsis genome). For gene families that contained multiple Arabidopsis paralogs, we selected a single Arabidopsis paralog at random to represent the family. Families that did not contain any Arabidopsis sequences were omitted from this portion of the analysis. Fold enrichment was calculated as the number of observed hits in a category divided by the number of expected hits in a category, where expected is the proportion of the background in a category multiplied by the number of hits. The localization/interaction enrichment analyses were performed in R. GO enrichment analyses was performed using the PANTHER web-based tool (http://geneontology.org/) (database release from 10-08-2019). Significance of enrichment was assessed with Fisher’s Exact Test with an FDR correction for multiple comparisons.
Identification of genes displaying strong signatures of ERC
To identify individual genes displaying the strongest signatures of plastid–nuclear ERC, we applied more stringent criteria that considered Pearson and Spearman correlation P values in their raw and FDR-corrected forms. Our criterion for labeling a gene as a strong hit was that either the adjusted Pearson P value or the adjusted Spearman P value (or both) must be ≤0.05. Additionally, for the genes in which only one of the two adjusted P was ≤0.05, we also required that the raw Pearson and raw Spearman P value both be ≤0.05. This approach allowed us to incorporate information from both correlation coefficients and from FDR multiple test correction while still retaining power to detect the strongest hits. Genes passing these criteria are presented in Tables 1 and 2.
Multiple regression analyses
To investigate the relative contributions of each plastome partition to the evolutionary rates of each nuclear-encoded protein, we conducted a multiple regression analysis using branch lengths from our constructed trees. Due to the lack of accD in O. sativa and L. siphilitica and the lack of ycf1/ycf2 in O. sativa and G. maderense, we excluded branch lengths from those three species, which allowed us to include all seven plastome partitions. Each nuclear gene was analyzed separately, where the y values were the normalized branch lengths for each species for that particular gene and the x values were the normalized branch lengths for each plastome partition for each species. Any additional missing data led to removal of the involved species. Models were created using the lm() function in R with default parameters.
Data availability
Alignments and phylogenetic trees used in this analysis have been deposited at Dryad Digital Repository and can be accessed at: https://doi.org/10.5061/dryad.7h44j0zs3.
Code used to conduct this analysis is available at: https://github.com/EvanForsythe/Plastid_nuclear_ERC.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure S1. Taxon composition of trees and subtrees.
Supplemental Figure S2. ERC case studies for known plastid complexes.
Supplemental Figure S3. Plastome partition ERC result overlap and multiple regression analysis.
Supplemental Figure S4. Cytosolic ribosome subunits found to have significant ERC with ClpP1.
Supplemental Figure S5. Comparison of ERC hits to genes with altered expression in proteostasis mutants.
Supplemental Table S1. Proteome data sources.
Supplemental Table S2. Plastome partition multiple sequence alignments.
Supplemental Table S3. ERC hits identified for each plastome partition.
Supplemental Table S4. ERC comparisons among the seven plastome partitions.
Supplemental Data Set 1. List of hits.
Supplemental Data Set 2. GO enrichment analysis for AccD (biological processes).
Supplemental Data Set 3. GO enrichment analysis for AccD (molecular function).
Supplemental Data Set 4. GO enrichment analysis for AccD (cellular component).
Supplemental Data Set 5. GO enrichment analysis for ClpP1 (biological processes).
Supplemental Data Set 6. GO enrichment analysis for ClpP1 (molecular function).
Supplemental Data Set 7. GO enrichment analysis for ClpP1 (cellular component).
Supplemental Data Set 8. GO enrichment analysis for MatK (biological processes).
Supplemental Data Set 9. GO enrichment analysis for MatK (molecular function).
Supplemental Data Set 10. GO enrichment analysis for MatK (cellular component).
Supplemental Data Set 11. GO enrichment analysis for photosynthesis (biological processes).
Supplemental Data Set 12. GO enrichment analysis for photosynthesis (molecular function).
Supplemental Data Set 13. GO enrichment analysis for photosynthesis (cellular component).
Supplemental Data Set 14. GO enrichment analysis for ribosomes (biological processes).
Supplemental Data Set 15. GO enrichment analysis for ribosomes (molecular function).
Supplemental Data Set 16. GO enrichment analysis for ribosomes (cellular component).
Supplemental Data Set 17. GO enrichment analysis for RNA polymerase (biological processes).
Supplemental Data Set 18. GO enrichment analysis for RNA polymerase (molecular function).
Supplemental Data Set 19. GO enrichment analysis for RNA polymerase (cellular component).
Supplemental Data Set 20. GO enrichment analysis for Ycf1/Ycf2 (biological processes).
Supplemental Data Set 21. GO enrichment analysis for Ycf1/Ycf2 (molecular function).
Supplemental Data Set 22. GO enrichment analysis for Ycf1/Ycf2 (cellular component).
Supplementary Material
Acknowledgments
This work was supported by a National Science Foundation (NSF) grant (MCB-1733227) and graduate fellowships from NSF (DGE-1321845) and the National Institutes of Health (T32-GM132057). The authors thank M.P. Simmons and J.C. Havird for helpful discussions.
Conflict of interest statement. None declared.
Contributor Information
Evan S Forsythe, Department of Biology, Colorado State University, Fort Collins, Colorado 80523, USA.
Alissa M Williams, Department of Biology, Colorado State University, Fort Collins, Colorado 80523, USA.
Daniel B Sloan, Department of Biology, Colorado State University, Fort Collins, Colorado 80523, USA.
E.S.F., A.M.W., and D.B.S. conceived this work and performed analyses. E.S.F. drafted this manuscript with input from all authors.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: Evan S. Forsythe (esfors@rams.colostate.edu).
References
- Adam Z, Aviv-Sharon E, Keren-Paz A, Naveh L, Rozenberg M, Savidor A, Chen J (2019) The chloroplast envelope protease FTSH11––Interaction with CPN60 and identification of potential substrates. Front Plant Sci 10: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal MS, Eulenstein O (2008) The multiple gene duplication problem revisited. Bioinformatics 24: i132– i13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard-Kubow KB, So N, Galloway LF (2016) Cytonuclear incompatibility contributes to the early stages of speciation. Evolution 70: 2752–2766 [DOI] [PubMed] [Google Scholar]
- Bieri P, Leibundgut M, Saurer M, Boehringer D, Ban N (2017) The complete structure of the chloroplast 70S ribosome in complex with translation factor pY. EMBO J 36: 475–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Veitia RA (2012) Gene balance hypothesis: connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci USA 109: 14746–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boerema AP, Aibara S, Paul B, Tobiasson V, Kimanius D, Forsberg BO, Wallden K, Lindahl E, Amunts A (2018) Structure of the chloroplast ribosome with chl-RRF and hibernation-promoting factor. Nat Plants 4: 212–217 [DOI] [PubMed] [Google Scholar]
- Bogdanova VS, Zaytseva OO, Mglinets AV, Shatskaya NV, Kosterin OE, Vasiliev GV (2015) Nuclear-cytoplasmic conflict in pea (Pisum sativum L.) is associated with nuclear and plastidic candidate genes encoding acetyl-coA carboxylase subunits. PLoS One 10: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonen L, Calixte S (2005) Comparative analysis of bacterial-origin genes for plant mitochondrial ribosomal proteins. Mol Biol Evol 23: 701–712 [DOI] [PubMed] [Google Scholar]
- Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17: 540–552 [DOI] [PubMed] [Google Scholar]
- Cho Y, Mower JP, Qiu YL, Palmer JD (2004) Mitochondrial substitution rates are extraordinarily elevated and variable in a genus of flowering plants. Proc Natl Acad Sci USA 101: 17741–17746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian RW, Hewitt SL, Roalson EH, Dhingra A (2020) Genome-scale characterization of predicted plastid-targeted proteomes in higher plants. Sci Rep 10: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NL, Alani E, Aquadro CF (2012) Evolutionary rate covariation: a bioinformatic method that reveals co-functionality and co-expression of genes. Genome Res 22: 714–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark NL, Aquadro CF (2010) A novel method to detect proteins evolving at correlated rates: identifying new functional relationships between coevolving proteins. Mol Biol Evol 27: 1152–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Tadini L, Peracchio C, Ferrari R, Pesaresi P (2016) GUN1, a jack-of-all-trades in chloroplast protein homeostasis and signaling. Front Plant Sci 7: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello R, Emms DM, Kelly S (2020) Gene duplication accelerates the pace of protein gain and loss from plant organelles. Mol Biol Evol 37: 969–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvillion MT, Soto IC, Shipkovenska G, Churchman LS (2016) Synchronized mitochondrial and cytosolic translation programs. Nature 533: 499–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creff A, Sormani R, Desnos T (2010) The two Arabidopsis RPS6 genes, encoding for cytoplasmic ribosomal proteins S6, are functionally equivalent. Plant Mol Biol 73: 533–546 [DOI] [PubMed] [Google Scholar]
- De Juan D, Pazos F, Valencia A (2013) Emerging methods in protein co-evolution. Nat Rev Genet 14: 249–261 [DOI] [PubMed] [Google Scholar]
- De Smet R, Adams KL, Vandepoele K, Van Montagu MCE, Maere S, Van de Peer Y (2013) Convergent gene loss following gene and genome duplications creates single-copy families in flowering plants. Proc Natl Acad Sci USA 110: 2898–2903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan JH, Rosenberg NA (2009) Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol Evol 24: 332–340 [DOI] [PubMed] [Google Scholar]
- Dogra V, Duan J, Lee KP, Kim C (2019) Impaired PSII proteostasis triggers a UPR-like response in the var2 mutant of Arabidopsis. J Exper Botany 70: 3075–3088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte JM, Wall PK, Edger PP, Landherr LL, Ma H, Pires JC, Leebens-Mack J, dePamphilis CW (2010) Identification of shared single copy nuclear genes in Arabidopsis, Populus, Vitis and Oryza and their phylogenetic utility across various taxonomic levels. BMC Evol Biol 10: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas DV, Hernandez D, Koenen EJM, Schwarz E, Straub S, Hughes CE, Jansen RK, Nageswara-Rao M, Staats M, Trujillo JT, et al. (2015) Mimosoid legume plastome evolution: IR expansion, tandem repeat expansions, and accelerated rate of evolution in clpP. Sci Rep 5: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms DM, Kelly S (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J (1985) Phylogenies and the comparative method. Am Nat 125: 1–15 [Google Scholar]
- Ferro M, Brugière S, Salvi D, Seigneurin-Berny D, Court M, Moyet L, Ramus C, Miras S, Mellal M, Le Gall S, et al. (2010) AT-CHLORO, a comprehensive chloroplast proteome database with subplastidial localization and curated information on envelope proteins. Mol Cell Proteom 9: 1063–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferro M, Salvi D, Brugière S, Miras S, Kowalski S, Louwagie M, Garin J, Joyard J, Rolland N (2003) Proteomics of the chloroplast envelope membranes from Arabidopsis thaliana. Mol Cell Proteom 2: 325–345 [DOI] [PubMed] [Google Scholar]
- Findlay GD, Sitnik JL, Wang W, Aquadro CF, Clark NL, Wolfner MF (2014) Evolutionary rate covariation identifies new members of a protein network required for Drosophila melanogaster female post-mating responses. PLoS Genet 10: e1004108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ES, Nelson ADL, Beilstein MA (2020) Biased gene retention in the face of introgression obscures species relationships. Genome Biol Evol 12(9): 1646––1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ES, Sharbrough J, Havird JC, Warren JM, Sloan DB (2019) CyMIRA: the cytonuclear molecular interactions reference for Arabidopsis. Genome Biol Evol 11: 2194–2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh CS, Bogan AA, Joachimiak M, Walther D, Cohen FE (2000) Co-evolution of proteins with their interaction partners. J Mol Biol 299: 283–293 [DOI] [PubMed] [Google Scholar]
- Gould SB, Waller RF, McFadden GI (2008) Plastid evolution. Annu Rev Plant Biol 59: 491–517 [DOI] [PubMed] [Google Scholar]
- Greiner S, Rauwolf U, Meurer J, Herrmann RG (2011) The role of plastids in plant speciation. Mol Ecol 20: 671–691 [DOI] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2008) Genome-wide analyses of Geraniaceae plastid DNA reveal unprecedented patterns of increased nucleotide substitutions. Proc Natl Acad Sci USA 105: 18424–18429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisinger MM, Kuehl JV, Boore JL, Jansen RK (2011) Extreme reconfiguration of plastid genomes in the angiosperm family Geraniaceae: rearrangements, repeats, and codon usage. Mol Biol Evol 28: 583–600 [DOI] [PubMed] [Google Scholar]
- Heinemann B, Künzler P, Braun H.-P, Hildebrandt T (2020) Estimating the number of protein molecules in a plant cell: a quantitative perspective on proteostasis and amino acid homeostasis during progressive drought stress. Plant Physiol 10.1093/plphys/kiaa050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilu KW, Borsch T, Müller K, Soltis DE, Soltis PS, Savolainen V, Chase MW, Powell MP, Alice LA, Evans R, et al. (2003) Angiosperm phylogeny based on matK sequence information. Am J Botany 90: 1758–1776 [DOI] [PubMed] [Google Scholar]
- Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497: 451–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Müller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, et al. (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA 104: 19369–19374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi S, Asakura Y, Imai M, Nakahira Y, Kotani Y, Hashiguchi Y, Nakai Y, Takafuji K, Bédard J, Hirabayashi-Ishioka Y (2018) A Ycf2-FtsHi heteromeric AAA-ATPase complex is required for chloroplast protein import. Plant Cell 30: 2677–2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Olinares PD, Oh SH, Ghisaura S, Poliakov A, Ponnala L, van Wijk KJ (2013) Modified Clp protease complex in the ClpP3 null mutant and consequences for chloroplast development and function in Arabidopsis. Plant Physiol 162: 157–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox EB (2014) The dynamic history of plastid genomes in the Campanulaceae sensu lato is unique among angiosperms. Proc Natl Acad Sci USA 111: 11097–11102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leebens-Mack JH, Barker MS, Carpenter EJ, Deyholos MK, Gitzendanner MA, Graham SW, Grosse I, Li Z, Melkonian M, Mirarab S, et al. (2019) One thousand plant transcriptomes and the phylogenomics of green plants. Nature 16: 1–7 [Google Scholar]
- Liu X, Yu F, Rodermel S (2010) An Arabidopsis pentatricopeptide repeat protein, SUPPRESSOR OF VARIEGATION7, is required for FtsH-mediated chloroplast biogenesis. Plant Physiol 154: 1588–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaoui H, Guerois R (2008) Coevolution at protein complex interfaces can be detected by the complementarity trace with important impact for predictive docking. Proc Natl Acad Sci USA 105: 7708–7713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee AM,, Aspinall S, Rice DW, Cusack BP, Sémon M, Perry AS, Stefanović S, Milbourne D, Barth S, Palmer JD, et al. (2010) Localized hypermutation and associated gene losses in legume chloroplast genomes. Genome Res 20: 1700–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AH, Whelan J, Small I (2006) Recent surprises in protein targeting to mitochondria and plastids. Curr Opin Plant Biol 9: 610–615 [DOI] [PubMed] [Google Scholar]
- Molina J, Hazzouri KM, Nickrent D, Geisler M, Meyer RS, Pentony MM, Flowers JM, Pelser P, Barcelona J, Inovejas SA, et al. (2014) Possible loss of the chloroplast genome in the parasitic flowering plant Rafflesia lagascae (Rafflesiaceae). Mol Biol Evol 31: 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mower JP, Touzet P, Gummow JS, Delph LF, Palmer JD (2007) Extensive variation in synonymous substitution rates in mitochondrial genes of seed plants. BMC Evol Biol 7: 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevill PG, Howell KA, Cross AT, Williams AV, Zhong X, Tonti-Filippini J, Boykin LM, Dixon KW, Small I (2019) Plastome-wide rearrangements and gene losses in Carnivorous droseraceae. Genome Biol Evol 11: 472–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, van Wijk KJ (2015) Organization, function and substrates of the essential Clp protease system in plastids. BBA–Bioenergetics 1847: 915–930 [DOI] [PubMed] [Google Scholar]
- Ovchinnikov S, Kamisetty H, Baker D (2014) Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. ELife 2014: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchy N, Lehti-Shiu M, Shiu SH (2016) Evolution of gene duplication in plants. Plant Physiol 171: 2294–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K (2004) APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290 [DOI] [PubMed] [Google Scholar]
- Park S, Ruhlman TA, Weng ML, Hajrah NH, Sabir JSM, Jansen RK (2017) Contrasting patterns of nucleotide substitution rates provide insight into dynamic evolution of plastid and mitochondrial genomes of Geranium. Genome Biol Evol 9: 1766–1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Rodermel SR (2004) Mutations in ClpC2/Hsp100 suppress the requirement for FtsH in thylakoid membrane biogenesis. Proc Natl Acad Sci USA 101: 12765–12770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson CL, Mower JP, Qiu YL, Shirk AJ, Song K, Young ND, DePamphilis CW, Palmer JD (2005) Multiple major increases and decreases in mitochondrial substitution rates in the plant family Geraniaceae. BMC Evol Biol 5: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu YL, Dombrovska O, Lee J, Li L, Whitlock BA, Bernasconi-Quadroni F, Rest JS, Davis CC, Borsch T, Hilu KW, et al. (2005) Phylogenetic analyses of basal angiosperms based on nine plastid, mitochondrial, and nuclear genes. Int J Plant Sci 166: 815–842 [Google Scholar]
- Qiu YL, Li L, Hendry TA, Li R, Taylor DW, Issa MJ, Ronen AJ, Vekaria ML, White AM (2006) Reconstructing the basal angiosperm phylogeny: evaluating information content of mitochondrial genes. Taxon 55: 837–856 [Google Scholar]
- Ramani AK, Marcotte EM (2003) Exploiting the co-evolution of interacting proteins to discover interaction specificity. J Mol Biol 327: 273–284 [DOI] [PubMed] [Google Scholar]
- Raza Q, Choi JY, Li Y, O’Dowd RM, Watkins SC, Chikina M, Hong Y, Clark NL, Kwiatkowski AV (2019) Evolutionary rate covariation analysis of E-cadherin identifies Raskol as a regulator of cell adhesion and actin dynamics in Drosophila. PLoS Genet 15: 1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenbach K, Havird JC, Grey Monroe J, Triant DA, Taylor DR, Sloan DB (2016) Positive selection in rapidly evolving plastid-nuclear enzyme complexes. Genetics 204: 1507–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ, McMahon MM (2007) Inferring angiosperm phylogeny from EST data with widespread gene duplication. BMC Evol Biol 7: S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangiovanni M, Vigilante A, Chiusano M (2013) Exploiting a reference genome in terms of duplications: the network of paralogs and single copy genes in Arabidopsis thaliana. Biology 2: 1465–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Yamanishi Y, Kanehisa M, Toh H (2005) The inference of protein-protein interactions by co-evolutionary analysis is improved by excluding the information about the phylogenetic relationships. Bioinformatics 21: 3482–3489 [DOI] [PubMed] [Google Scholar]
- Schmitz-Linneweber C, Kushnir S, Babiychuk E, Poltnigg P, Herrmann RG, Maier RM (2005) Pigment deficiency in nightshade/tobacco cybrids is caused by the failure to edit the plastid ATPase α-subunit mRNA. Plant Cell 17: 1815–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharbrough J, Conover JL, Tate JA, Wendel JF, Sloan DB (2017) Cytonuclear responses to genome doubling. Am J Botany 104: 1–4 [DOI] [PubMed] [Google Scholar]
- Shrestha B, Weng ML, Theriot EC, Gilbert LE, Ruhlman TA, Krosnick SE, Jansen RK (2019) Highly accelerated rates of genomic rearrangements and nucleotide substitutions in plastid genomes of Passiflora subgenus Decaloba. Mol Phylogenet Evol 138: 53–64 [DOI] [PubMed] [Google Scholar]
- Sloan DB, Oxelman B, Rautenberg A, Taylor DR (2009) Phylogenetic analysis of mitochondrial substitution rate variation in the angiosperm tribe Sileneae. BMC Evol Biol 9: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan DB, Triant DA, Forrester NJ, Bergner LM, Wu M, Taylor DR (2014a) A recurring syndrome of accelerated plastid genome evolution in the angiosperm tribe Sileneae (Caryophyllaceae). Mol Phylogenet Evol 72: 82–89 [DOI] [PubMed] [Google Scholar]
- Sloan DB, Triant DA, Wu M, Taylor DR (2014b) Cytonuclear interactions and relaxed selection accelerate sequence evolution in organelle ribosomes. Mol Biol Evol 31: 673–682 [DOI] [PubMed] [Google Scholar]
- Smith ML, Hahn MW (2020) New approaches for inferring phylogenies in the presence of paralogs. BioRxiv 1: 6–8 [DOI] [PubMed] [Google Scholar]
- Sobanski J, Giavalisco P, Fischer A, Kreiner JM, Walther D, Aurel M, Pellizzer T, Golczyk H, Obata T, Bock R, et al. (2019) Chloroplast competition is controlled by lipid biosynthesis in evening primroses. 116: 5665–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltis PS, Soltis DE, Chase MW (1999) Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 402: 402–404 [DOI] [PubMed] [Google Scholar]
- Stamatakis A (2014) RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30: 1312–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzer M, Lai H, Xu M, Sathaye D, Vernot B, Durand D (2012) Inferring duplications, losses, transfers and incomplete lineage sorting with nonbinary species trees. Bioinformatics 28: i409–i415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller N, Weingartner M, Thiele W, Maximova E, Scho MA (2012) The plastid-specific ribosomal proteins of Arabidopsis thaliana can be divided into non-essential proteins and genuine ribosomal proteins. Plan J 69: 302–316 [DOI] [PubMed] [Google Scholar]
- Timmis JN, Ayliff MA, Huang CY, Martin W (2004) Endosymbiotic gene transfer: organelle genomes forge eukaryotic chromosomes. Nat Rev Genet 5: 123–135 [DOI] [PubMed] [Google Scholar]
- van Wijk KJ, Baginsky S (2011) Plastid proteomics in higher plants: current state and future goals. Plant Physiol 155: 1578–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernot B, Stolzer M, Goldman A, Durand D (2008) Reconciliation with non-binary species trees. J Comput Biol 15: 981–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R, Aigner H, Funk C (2012) FtsH proteases located in the plant chloroplast. Physiol Plantarum 145: 203–214 [DOI] [PubMed] [Google Scholar]
- Waltz F, Nguyen TT, Arrivé M, Bochler A, Chicher J, Hammann P, Kuhn L, Quadrado M, Mireau H, Yashem Y, et al. (2019) Small is big in Arabidopsis mitochondrial ribosome. Nat Plants 5: 106–117 [DOI] [PubMed] [Google Scholar]
- Wang R, Zhao J, Jia M, Xu N, Liang S, Shao J, Qi Y, Liu X, An L, Yu F (2018) Balance between cytosolic and chloroplast translation affects leaf variegation. Plant Physiol 176: 804–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel JF, Lisch D, Hu G, Mason AS (2018) The long and short of doubling down: polyploidy, epigenetics, and the temporal dynamics of genome fractionation. Curr Opin Genet Dev 49: 1–7 [DOI] [PubMed] [Google Scholar]
- Weng M, Ruhlman TA, Jansen RK (2016) Plastid––nuclear interaction and accelerated coevolution in plastid ribosomal genes in Geraniaceae. Genome Biol Evol 8: 1824–1838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicke S, Müller KF, DePamphilis CW, Quandt D, Bellot S, Schneeweiss GM (2016) Mechanistic model of evolutionary rate variation en route to a nonphotosynthetic lifestyle in plants. Proc Natl Acad Sci USA 113: 9045–9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams AM, Friso G, Wijk KJV, Sloan DB (2019) Extreme variation in rates of evolution in the plastid Clp protease complex. Plan J 98: 1–17 [DOI] [PubMed] [Google Scholar]
- Wolfe NW, Clark NL (2015) ERC analysis: web-based inference of gene function via evolutionary rate covariation. Bioinformatics 31: 3835–3837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Ye G, Werren JH (2019) Evolutionary rate correlation between mitochondrial-encoded and mitochondria-associated nuclear-encoded proteins in insects. Mol Biol Evol 36: 1022–1036 [DOI] [PubMed] [Google Scholar]
- Yu F, Liu X, Alsheikh M, Park S, Rodermel S (2008) Mutations in SUPPRESSOR OF VARIEGATION1, a factor required for normal chloroplast translation, suppress var2-mediated leaf variegation in Arabidopsis. Plant Cell 20: 1786–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanis MJ, Soltis DE, Soltis PS, Mathews S, Donoghue MJ (2002) The root of the angiosperms revisited. Proc Natl Acad Sci USA 99: 6848–6853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ruhlman TA, Sabir J, Blazier JC, Jansen RK (2015) Coordinated rates of evolution between interacting plastid and nuclear genes in Geraniaceae. Plant Cell 27: 563–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ruhlman TA, Sabir JSM, Blazier JC, Weng M, Park S, Jansen RK (2016) Coevolution between nuclear-encoded DNA replication. Recomb Repair Genes Plastid Genome Complex 8: 622–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng M, Liu X, Liang S, Fu S, Qi Y, Zhao J, Shao J, An L, Yu F (2016) Chloroplast translation initiation factors regulate leaf variegation and development. Plant Physiol 172: 1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupoka A, Kozula D, Schöttlera MA, Niehörstera J, Garbscha F, Liereb K, Malinovaa I, Bock R, Greiner S (2020) A photosynthesis operon in the chloroplast genome drives speciation in evening primroses. BioRvix doi.org/10.1101/2020.07.03.186627 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Alignments and phylogenetic trees used in this analysis have been deposited at Dryad Digital Repository and can be accessed at: https://doi.org/10.5061/dryad.7h44j0zs3.
Code used to conduct this analysis is available at: https://github.com/EvanForsythe/Plastid_nuclear_ERC.