

Abstract
The phenotypic consequences of the addition or subtraction of part of a chromosome is more severe than changing the dosage of the whole genome. By crossing diploid trisomies to a haploid inducer, we identified 17 distal segmental haploid disomies that cover ∼80% of the maize genome. Disomic haploids provide a level of genomic imbalance that is not ordinarily achievable in multicellular eukaryotes, allowing the impact to be stronger and more easily studied. Transcriptome size estimates revealed that a few disomies inversely modulate most of the transcriptome. Based on RNA sequencing, the expression levels of genes located on the varied chromosome arms (cis) in disomies ranged from being proportional to chromosomal dosage (dosage effect) to showing dosage compensation with no expression change with dosage. For genes not located on the varied chromosome arm (trans), an obvious trans-acting effect can be observed, with the majority showing a decreased modulation (inverse effect). The extent of dosage compensation of varied cis genes correlates with the extent of trans inverse effects across the 17 genomic regions studied. The results also have implications for the role of stoichiometry in gene expression, the control of quantitative traits, and the evolution of dosage-sensitive genes.
The addition of various chromosome arms to haploid maize predominantly causes inverse modulations of gene expression across the genome.
Introduction
Genome imbalance is the phenomenon that changing individual chromosomes (aneuploidy) has a more severe effect on the phenotype than changing a whole set of chromosomes (ploidy). Such phenotypes were first reported a century ago in Jimson weed (Datura stramonium) where trisomies showed different growth compared to normal diploid plants (Blakeslee et al., 1920; Blakeslee, 1921, 1934). Reports from other organisms suggest the commonality of this phenomenon (e.g. Bridges, 1925; Schinzel, 2001). Investigations into the mechanism of genome imbalance revealed that the phenotypic effect may result from dosage-sensitive genes from the varied chromosome or chromosomal segment (Birchler and Newton, 1981; Birchler et al., 2001; Bastiannse et al., 2019).
Monitoring the expression of genes at the RNA or protein levels in aneuploidy compared with euploidy in Drosophila as well as plants has shown that genes on the varied arm showed a dosage effect in which the amount of gene output is proportional to the altered dosage (Grell, 1962; Carlson, 1972; O’Brien and Gethmann, 1973). However, the expression of some genes on a varied arm showed no change, a phenomenon known as dosage compensation (Birchler, 1979, 1981; Birchler and Newton, 1981; Birchler et al., 1990; Guo and Birchler, 1994; Sun et al., 2013a, 2013b). Whether a dosage effect or compensation occurred depended on the specific region examined and its length. Modulations of gene expression on the unvaried chromosomes showed that an inverse effect (a decrease in gene expression with increased dosage) was more prevalent than a direct effect (an increase in gene expression with increased dosage; Birchler, 1979; Birchler and Newton, 1981; Rabinow et al., 1991; Guo and Birchler, 1994; Sun et al., 2013a, 2013b).
The inverse and direct effects of aneuploids were revealed to have a single gene basis by systematically dissecting the modifiers that regulate the white eye color reporter gene in Drosophila (Rabinow et al., 1991; Sabl and Birchler, 1993; Birchler et al., 2001). There are 47 known dosage-dependent modifiers recovered over a long period of screening (Birchler et al., 2001), with the majority showing a negative effect on white, while others have a positive effect. The identification of the molecular function of these regulators revealed that most of the genes were transcription factors (TFs), signal transduction components, or chromatin modifiers (Birchler et al., 2001). A dosage-regulatory model was applied to explain how there can be such a large number of modifiers of a single reporter gene. In other words, a regulatory system operates in a hierarchy with one regulator controlling downstream regulators, which, in turn, control other effectors ultimately reaching the reporter white eye color gene (Birchler et al., 2001). These aforementioned studies showed that the inverse effect was observed on the protein, RNA, and phenotypic levels and that it can be caused by single genes encoding TFs or signal transduction components.
Based on the analogous effects observed in aneuploids from various organisms, the Gene Balance Hypothesis (GBH) was formulated to explain the mechanism of the genome imbalance (Birchler et al., 2005; Birchler and Veitia, 2007, 2010, 2012). The GBH posits that altering the stoichiometry of members of macromolecular complexes or multicomponent interactions will affect the function of the whole as a result of the kinetics and mode of assembly. It is noteworthy that TFs, signal transduction components, and chromatin modifiers are enriched for subunits of macromolecular complexes (Birchler et al., 2001; Boell et al., 2013; Kondrashov and Koonin, 2004; Seidman and Seidman, 2002; Stranger et al., 2007). Specific examples illustrate the role of stoichiometry (Clark et al., 2020; Stahl et al., 2013). The GBH is also supported by evolutionary genomics studies that found that genes for members of macromolecular structures and interactions were selectively retained during genome fractionation following whole-genome duplication (WGD; Wolfe and Shields, 1997; Simillion et al., 2002; Papp et al., 2003; Blanc and Wolfe, 2004; Maere, et al., 2005; Aury et al., 2006; Blomme et al., 2006; Freeling and Thomas, 2006; Thomas et al., 2006; McGrath et al., 2014; Gout and Lynch, 2015; Defoort et al., 2019; Shi et al., 2020), suggesting that the genes involved in the multicomponent interactions are dosage-sensitive and there would be negative fitness consequences if one member of a balanced duplicated pair is deleted. In contrast to WGD, segmental duplications have an underrepresentation of these classes of genes (Freeling et al., 2008; Tasdighian et al., 2017), which is consistent with a role of genomic balance in their fate.
Genome-wide transcriptome analysis of aneuploids from Arabidopsis, Drosophila, and maize revealed the dosage effect or compensation in cis and global gene modulation in trans (Sun et al., 2013a, 2013b; Hou et al., 2018; Johnson et al., 2020). Re-analysis of expression data from yeast disomies (Torres et al., 2007) and mouse trisomies (Williams et al., 2008), which previously normalized the expression of the varied chromosome to the remainder of the genome, revealed prevalent inverse modulations in trans (Hou et al., 2018) that were obscured by the initial data treatment. Indeed, one hundred years ago, Blakeslee (1921) mapped a gene for purple pigment production to a chromosome in Datura and found that trisomies for every other chromosome would modulate the pigment levels in trans. The inverse effect on a subset of autosomal genes in humans has been found in a dosage series of the sex chromosomes (Raznahan et al., 2018; Zhang et al., 2020).
In maize, the genome can be broken into smaller segments, allowing the global modulation from smaller varied regions to be examined. Also, the ability to produce disomic haploids at a reasonable frequency by crossing diploid aneuploids by haploid inducers allowed us to determine how the greater dosage imbalance modulates gene expression at the genome-wide scale. Classical examples of haploid disomy in Datura illustrate the strong phenotypic effect (Satina et al., 1937). Here, we identified 17 segmental haploid disomies that covered ∼80% of the maize genome. The identification of disomies revealed that the higher dose change (2X) of even smaller chromosomal segments has a strong detrimental phenotypic impact. The RNA-seq analysis revealed that dosage compensation is a common phenomenon among all disomies in addition to the dosage effect for genes on the varied segment. Intriguingly, trans-acting effects are prevalent in the unvaried genomic regions, which, for most of the disomies, show an inverse effect as the major response to imbalance. The results have implications for the mechanisms of gene expression, the genetic control of quantitative traits, and the evolution of dosage-sensitive duplicate genes.
Results
Haploid induction and disomy identification
The genetic system we used to generate the dosage series in maize involved translocations between the supernumerary B chromosome and the normal A chromosomes, which have dominant anthocyanin pigment genes or their transposable element markers (Beckett, 1991; Alfenito and Birchler, 1993). The property of nondisjunction of the B chromosome at the second pollen mitosis, which gives rise to the two sperm in a pollen grain, allow the B-A translocation lines to generate zero, one, or two copies of the B-A chromosome in different sperm. After fertilizing female gametes from corresponding testers, which have a recessive anthocyanin marker, there will be one dose (hypoploid; monosomy; A, A-B), two doses (euploid heterozygote; diploid; A, A-B, B-A), and three doses (hyperploid heterozygote; trisomy; A, A-B, B-A, B-A) of the respective chromosome arm in the progeny. Female hyperploid heterozygotes were further chosen and crossed with male haploid inducer lines (∼10% induction rate) to generate haploids among which about half have haploid disomic embryos.
A collection of B-A translocations covers 16 maize chromosomal regions (Birchler and Alfenito, 1993). These B-A translocation lines were crossed with tester lines to produce segmental trisomies. Female hyperploid heterozygotes, which show purple embryo and yellow endosperm, were chosen from the progeny and crossed with one of two male haploid inducer lines, RWSK; R1-navajo (R1-nj); c1-I or RWS; green fluorescent protein (GFP); R1-nj (Deimling et al., 1997; Yu and Birchler, 2016). Haploid kernels either showed purple embryo and yellow endosperm when using RWSK; R1-nj; c1-I (Figure 1) or absence of GFP signal in roots when the RWS; GFP; R1-nj was used (Supplemental Figure 1). Because female hyperploid heterozygotes produce balanced (A-B, B-A) and unbalanced (A, B-A) gametes, the haploid kernels would be either euploid or disomic, respectively. The disomic kernels usually germinated later than the euploid and some disomic seedlings died within one week after germination but most survived. To verify that the plants were indeed disomy for a particular chromosomal arm, each individual was karyotyped via root tip metaphase chromosome spreads using specific probes that can distinguish each maize A chromosome and the B chromosome (Supplemental Figure 2). All disomies from each translocation had the chromosome constitution of A and B-A except TB-6Lc. TB-6Lc disomy had 1 copy of 6Lc-B (A-B) and an iso-chromosome for 6L that contains the B centromere. Previous results revealed that parts of the B centromere were lost in this isochromosome, suggesting that the chromosome was derived from mis-division of B-6Lc (B-A) in the B centromeric region and the isochromosome had 2 copies of 6L (Supplemental Figure 2). During the identification of TB-2Sa, in addition to recovering the disomy TB-2Sa, we also found TB-2Sa_deletion disomies for which the short arm tip of chromosome 2 was deleted (Supplemental Figure 2).
Figure 1.

Haploid induction. Haploid induction occurs by crossing a male haploid inducer (RWSK C1-I; R-nj) to a female hyperploid heterozygote, for example, TB-1La (1, 1La-B, B-1La, B-1La). The number or letter before the hyphen denotes the arm has a centromere. The number “1” refers to chromosome 1. The uppercase letter “L” denotes the long arm and the lowercase “a” represents the first B-A translocation produced for that chromosome (chromosome 1 in this case; The “b” designation represents the second B-A translocation recovered for that chromosome, e.g. TB-1Sb). A dominant anthocyanin marker gene, Bz2, is on the 1L arm (long arm of chromosome 1) that allows purple anthocyanin production. RWSK contains C1-I that can inhibit the expression of the normal C1 allele and thus can silence anthocyanin production. Following meiosis, the TB-1La heterozygote produces two major kinds of female gametes, balanced egg (1La-B, B-1La) and imbalanced egg (1, B-1La; producing disomy after fertilization). After double fertilization, diploid kernels will have yellow embryos and yellow endosperms because the expression of C1 is inhibited by the C1-I from RWSK, which blocks the anthocyanin pathway. If the chromosomes from RWSK are eliminated in the embryo, via haploid induction, the haploid kernels have purple embryos and yellow endosperms, which are distinguishable from the diploid kernels.
Detrimental effect of genome imbalance on the phenotype
The imbalance of the genome has a detrimental effect on developmental processes (Lee et al., 1996a, 1996b). Our study showed that abnormal phenotypes, such as decreased plant stature, were found in the disomic seedlings as early as 2 weeks after germination (Figure 2; Supplemental Figure 3A). In addition, some of the disomic plants also showed curly leaves (TB-4Sa) and yellowish-green leaves (e.g. TB-3La and TB-5Lb) at 45 days after seed germination (Figure 2; Supplemental Figure 3B). To determine the size of the varied chromosome arm in each translocation, we downloaded the genomic DNA-sequencing data of disomy from NCBI (NCBI-SRA, BioProject PRJNA633287) and performed copy number variation analysis (Supplemental Table 1). There were two ways to determine the copy number variation: (1) count the mapped reads both from genic and intergenic regions (bin size of 1 Mb); (2) count reads only from genic regions (per gene basis). The first method was used to confirm that only the portion of the A chromosome arm associated with the B-A translocation was varied. To this end, reads from disomy were compared to the control and the ratio plotted along each of the maize chromosomes. The corresponding chromosome arm in each translocation, for example, chromosome 4S in TB-4Sa, showed the copy number change but the ratios also fluctuate in some regions on other chromosomes, such as chromosome 1 (Supplemental Figure 4A). This result is probably due to linkage disequilibrium of the sections from nonrecurrent parents still present in our materials even though they have been backcrossed to W22 for seven generations. Next, we calculated the ratios on a per gene basis. As expected, for example, TB-4Sa disomy only showed a changed copy number on chromosome 4 compared to other chromosomes (Supplemental Figure 4B). The latter method was further applied to other disomies (Figure 3 and Table 1). The number of the cis genes is generally positively correlated with the detrimental effect of the gene dosage effect on the phenotype. For example, TB-3La disomy plants have a severely affected phenotype while the phenotype of TB-3Sb was less severely affected (Supplemental Figure 3; Table 1).
Figure 2.

Example disomic phenotypes. Disomic plants of TB-4Sa (A) and TB-5Lb (B) 45 days after germination are shown on the right with the W22 haploid plants, which contain a balanced chromosome set on the left. Both TB-4Sa and TB-5Lb disomies exhibit decreased plant stature. In addition to decreased stature, other adverse effects of genome imbalance also occur, such as curly leaf in TB-4Sa disomy and yellowish green leaf in TB-5Lb disomy. The ruler denotes 1 m. The full set is shown in Supplemental Figure 3.
Figure 3.

Gene copy number analysis. All disomies or monosomy and haploid controls were subjected to copy number analysis using the read counts from gene regions. The read counts were normalized by RPKM before calculating the ratio. For each chromosome, disomies contain one copy of the A chromosome and the B-A chromosome. TB-6Lc has the chromosome constitution of one copy of 6Lc-B and chromosome 6 as a monosomy for 6L to determine the breakpoint. Only the chromosome with a copy number change was displayed in each disomy based on the copy number variation results. Each data point represents one gene by the order of their positions on each chromosome as plotted on the x-axis. The y-axis denotes log ratio of normalized DNA counts with base 2.
Table 1.
The number of cis genes and the size of the cis region in each disomic line
| B-A translocation | Cis region size | Cis gene number |
Cis gene names |
|
|---|---|---|---|---|
| First cis gene | Last cis gene | |||
| TB-1Sb | 130.5 Mb | 2,462 | Zm00004b000001 | Zm00004b002462 |
| TB-1La | 145.7 Mb | 3,164 | Zm00004b002705 | Zm00004b005868 |
| TB-2Saa | 107.5 Mb | 2,176 | Zm00004b005869 (first region) | Zm00004b007874 (first region) |
| Zm00004b007977 (second region) | Zm00004b008146 (second region) | |||
| TB-2Sa_deletiona | 61.9 Mb | 740 | Zm00004b007305 (first region) | Zm00004b007874 (first region) |
| Zm00004b007977 (second region) | Zm00004b008146 (second region) | |||
| TB-3Sb | 37.6 Mb | 917 | Zm00004b015566 | Zm00004b016482 |
| TB-3La | 152.5 Mb | 3,109 | Zm00004b016969 | Zm00004b020077 |
| TB-4Sa | 103.3 Mb | 1,622 | Zm00004b020078 | Zm00004b021699 |
| TB-4Lb | 84.4 Mb | 2,055 | Zm00004b022488 | Zm00004b024542 |
| TB-5Sc | 83.8 Mb | 2,084 | Zm00004b010828 | Zm00004b012911 |
| TB-5Lb | 70 Mb | 1,965 | Zm00004b013601 | Zm00004b015565 |
| TB-6Lc | 112.1 Mb | 2,499 | Zm00004b028951 | Zm00004b031449 |
| TB-7Sc | 56 Mb | 876 | Zm00004b034624 | Zm00004b035499 |
| TB-7Lb | 95.1 Mb | 2,061 | Zm00004b035669 | Zm00004b037729 |
| TB-8Lc | 92 Mb | 2,354 | Zm00004b025927 | Zm00004b028280 |
| TB-9Sd | 57 Mb | 1,035 | Zm00004b031450 | Zm00004b032484 |
| TB-9Lc | 83.5 Mb | 1,951 | Zm00004b032673 | Zm00004b034623 |
| TB-10L19 | 98 Mb | 1,832 | Zm00004b038475 | Zm00004b040648 |
Both TB-2Sa and TB-2Sa_deletion have two noncontinuous regions. The TB-2Sa_deletion has a deleted part in the first region in TB-2Sa.
Transcriptome size in disomies
To investigate the molecular mechanism of gene dosage effects, RNA of leaf tissue (45 days after germination) from disomies and haploid controls was extracted and subjected to RNA sequencing (RNA-seq). Principal Component Analysis (PCA) of the biological replicates in each category showed that all 17 disomy groups were separated from the respective haploid control, which revealed that RNA expression showed a distinctive expression pattern in each group comparison (Supplemental Figure 5 and Supplemental Data Set 1).
We further verified the gene expression results from RNA-seq data using a droplet digital PCR (ddPCR) assay to estimate the total transcriptome size by determining selected gene expression relative to the respective DNA amounts in total nucleic acid (TNA; RNA plus gDNA) isolations. These determinations were then assessed relative to the respective RNA-seq data to estimate transcriptome size (Coate and Doyle, 2010). Aneuploidy presents challenges for these estimates and such determinations have not previously been made. These estimates might reveal substantial global effects, which we explored further by calculating the relative expression per cell for eight randomly selected genes (Supplemental Figure 6). To calculate this, for each gene, the Absolute Quantification (ABS) value of using cDNA-specific primers from the ddPCR assay was compared to the ABS value of using gDNA-specific primers to obtain the expression of a gene per genome. The relative expression per genome was obtained by comparing the disomy to haploid. Because the cis genes have two doses of gDNA, while the trans genes only have one, the relative expression per cell for cis genes is computed by multiplying the relative expression per genome by two. In contrast, for the trans genes, the relative expression per genome is equal to the relative expression per cell. Normalization methods such as reads/fragments per kilobase per million (RPKM or FPKM) are widely used in RNA-seq data analysis (Marioni et al., 2008; Mortazavi et al., 2008); apparent gene expression differences between two samples are actually differences in expression per unit of RNA or “per transcriptome.” The comparison between our ddPCR assay (relative expression per cell) and RNA-seq data (relative expression per transcriptome) showed most of the disomies do not exhibit strong differences between the two methods (Supplemental Figure 6). However, in TB-1La, TB3La, TB4Lb, and TB6Lc, the expression of the eight genes measured by ddPCR approximated half of the expression from the RNA-seq data. This result suggests there is a substantial transcriptome size change in these disomies. Coate and Doyle (2010) calculated the relative transcriptome size by comparing the relative expression per cell to the relative expression per transcriptome (RNA-seq data) for each gene.
Before we tested this possibility, we first validated whether the ddPCR method can be used to calculate the transcriptome size. A companion paper (Shi et al., 2021) reports measurements of the relative transcriptome size using ploidy series data including haploid, diploid, triploid, and tetraploid. The same eight genes were used to perform the ddPCR assay and compared to the relative expression of the corresponding genes from RNA-seq (the relative gene expression per transcriptome) in the haploid/diploid, triploid/diploid, and tetraploid/diploid data. The results showed that there is an approximately linear correlation of the transcriptome size and ploidy change in haploid/diploid (down to 0.5) and triploid/diploid (up to 1.5), and a higher change in tetraploid/diploid (up to 3.8), which suggests the ddPCR method can be used for measuring transcriptome size.
We applied this method in our study and found that most disomies have only a slight increase (TB-1Sb, TB-2Sa, TB-2Sa deletion, TB-4Sa, TB9-Sd, and TB-10L19, range from 1.08 to 1.32) or decrease (TB-3Sb, TB-5Lb, TB-7Sc, TB-7Lb, TB-8Lc, and TB-9Lc, range from 0.72 to 0.94) of transcriptome size estimates (Figure 4). However, four regions, TB-1La (0.62), TB-3La (0.56), TB-4Lb (0.45), and TB-6Lc (0.31), showed a more dramatic effect suggesting an overall reduction in transcriptome size based on this assay.
Figure 4.
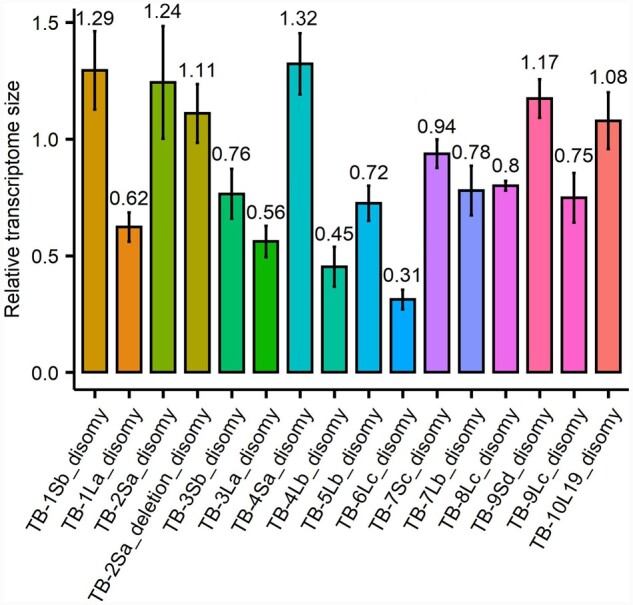
Transcriptome size measurement. There are eight independent estimates (eight genes) of transcriptome size in each disomy by comparing the relative expression per cell (disomy/haploid) from ddPCR to the relative expression per transcriptome (disomy/haploid) from RNA-seq data. The x axis is the average value of eight estimates and the bar is standard deviation across the eight estimates.
Cis genes display a spectrum from dosage effect to dosage compensation and trans genes exhibit inverse effects in most disomies
To further examine how the genome imbalance affects the global expression trend, ratio distributions were performed to determine both cis and trans effects of disomies on gene expression. To this end, read counts were averaged across biological replicates. Lowly expressed genes were filtered and ratios between the compared genotypes were generated for each expressed gene and then plotted as a ratio distribution. The distributions for the disomies were partitioned into genes that are present on the varied chromosome (cis) versus those that are in the remainder of the genome (trans) according to the cis gene list in Table 1. For cis genes, a dosage effect was designated if the ratio is 2.0, while no relative change in expression (ratio = 1.0) was designated as dosage compensation. For trans genes, the ratios below 1.0 or above 1.0 were designated as inverse effect or direct effect, respectively.
The mean, median, and standard deviation (sd) for each distribution were computed (Supplemental Data Set 2). We also performed several statistical tests including a normality test (Lilliefors test), Kolmogorov–Smirnov (K–S), and Bartlett’s test. The normality test was used to determine the deviation of the distributions from normal (Supplemental Data Set 3). The K–S test was performed to test for distribution differences (Supplemental Data Set 4) and Bartlett’s test was used to compare the variances across distributions (Supplemental Data Set 5).
The cis genes in all disomies show a spread across a landscape of effects with each being distinct (Figure 5). The expression levels of many cis genes in disomies show a dosage effect (peak near 2.0), while a notable subset of cis genes show dosage compensation (ratio ∼1.0) but also a spread that extends above (ratio >2.0) and below (ratio <1.0) these levels. The major peaks in cis for disomies of TB-1Sb, TB-1La, TB-2Sa, TB-3La, TB-4Lb, TB-5Sc, TB-5Lb, TB-6Lc, TB-7Sc, TB-7Lb, TB-9Lc, and TB-10L19 are between 1.0 and 2.0, which suggest the expression of most genes in these disomies are compensated to some degree. For a few regions there are minor peaks of cis genes at the inverse level even below the compensated mark, most notably for TB-2Sa, TB-3Sb, TB-4Lb, TB-5Lb, TB-8Lc, and TB-10L19, suggesting that a subset of genes are doubly inversely affected independently. Also, when gene expression approaches dosage compensation in cis, the trans genes skew more towards the inverse effect (major peaks < 1.0). Pearson correlation analysis revealed that the medians of cis and trans distributions in each comparison are highly positively correlated across all disomies (Supplemental Figure 7), indicating cis and trans peaks in each comparison were shifting in the same direction, which suggests a related mechanistic basis. Besides the inverse effect, TB-1La, TB-2Sa, TB-3La, TB-4Lb, TB-6Lc, TB-7Lb, TB-9Lc, and TB-10L19 displayed some direct effects in trans, but to a lesser degree. In the TB-2Sa-deletion, TB3-Sb, TB-4Sa, TB-8Lc, and TB-9Sd, more genes are inversely modulated than directly in trans even though the major peak is near 1. Taken together, all disomies showed an inverse effect in trans, with direct effects to a lesser degree. It should be noted that the ratio distributions reflect the relative expression per transcriptome and should be considered in the context of the transcriptome size measurements described above. Scatter plots testing the significance of the cis and trans differential gene expression complement the ratio distributions (Figure 6) and illustrate these trends.
Figure 5.

Ratio distributions of gene expression in each disomy compared with haploids. The normalized counts from RNA-seq were averaged for the biological replicates. Those genes with a sum of averaged counts of disomy and haploid <1 are regarded as lowly expressed genes and were filtered out. For each expressed gene, a ratio of the averaged normalized value in the disomy was made over the normalized counts in the haploid control. These ratios were plotted in bins of 0.05. The x-axis notes the value for each bin, and the y axis notes the number of genes per bin (frequency). For each disomy, genes were partitioned into those encoded on the varied chromosome (cis, orange) versus those encoded on the remainder of the genome that were not varied in dosage (trans, blue). A ratio of 1.0 represents no change in the experimental genotype versus the haploid. For cis genes, a ratio of 2.0 represents a gene-dosage effect, whereas 1.0 represents dosage compensation. A ratio of 0.5 represents the inverse ratio of gene expression in trans. These ratio values are demarcated with labeled vertical lines in purple (2.0) and red (0.5).
Figure 6.

Scatter plots of differential gene expression. Scatter plots of significant differential expression (using Cuffdiff) for each gene comparing disomic to haploid are shown. The scatter plot analysis was used as a complement to ratio distributions to illustrate both the magnitude of deviation from the haploid and the magnitude of expression difference. Lowly expressed genes were filtered using the same criteria as in the ratio distributions. The x-axis represents log2-fold change of the disomy to the haploid control and the y-axis shows the mean of normalized counts of the disomy and haploid. Data points with a q-value (adjusted P-value) <0.05 and a corresponding log-fold change of disomy to haploid more than 0 were depicted in magenta, while points with a q value <0.05 and a corresponding log-fold change of disomy to haploid <0 were depicted in green. Otherwise, they were designated in black. For the respective disomies, genes were partitioned into those encoded on the varied chromosome (cis) and those in the remainder of the genome (trans). Three designated ratio values, 0.5, 1.0, and 2.0, were depicted in red, black and purple vertical dashed lines, respectively.
Alternative validation
Because the RNA-seq data for this study removed ribosomal RNA (rRNA) before sequencing (in order to eventually examine the effect on organelle expression), we selected six disomies of different sizes and effects, namely, TB-1La, TB-3Sb, TB-4Sa, TB-6Lc, TB-7Lb, and TB-10L19, for polyA mRNA selection RNA-sequencing to compare to the RNA-seq data from the rRNA removal method (rRNA depletion). The results showed that both methods exhibit similar patterns of ratio distributions (Supplemental Figure 8). Indeed, there was no significant difference between the distributions both for cis and trans genes with these two methods (Supplemental Data Set 4).
B chromosome analysis
The disomies were generated from B-A translocations and thus contain varying amounts of the B chromosome. We examined whether the B portion in disomies might have an effect on the genes from the A chromosomes. The ratio distributions of haploid lines with one or two copies of the B chromosome compared to the lines without the B chromosome showed a sharp major peak around 1 (Supplemental Figure 9). Differential gene expression analysis showed that there are a few differentially expressed genes in the lines with the B chromosomes (Supplemental Figure 9; Supplemental Data Set 6). The distribution difference (K-S) and variance (Bartlett’s) tests showed that the A chromosome disomy groups are significantly different from the group comparison of haploid with one or two B chromosomes (Supplemental Data Sets 4 and 5). The few differentially expressed genes between the B chromosome versus haploid comparisons show a dosage effect with the B chromosome number and likely result from B encoded genes (Huang et al., 2016). These results indicate that the effects observed in A chromosome disomies, not only for the phenotype but also for modulations in trans, are due the copy number variation from the cis genes on the A chromosome portion.
Aneuploidy versus Polyploidy
The companion paper (Shi et al., 2021) examined gene expression in a ploidy series as well as in diploid aneuploidy. The haploid disomy collection studied here routinely shows greater modulation than the haploid/diploid comparison (Supplemental Data Sets 4 and 5). At both haploid and diploid ploidies, aneuploidy produces a greater spread of gene expression than balanced ploidies but the haploid disomy effects are to a greater degree than the trisomy effects at the diploid level, as a general rule.
Functional groups of genes and maize subgenomes show different patterns of expression
Genome imbalance studies in Arabidopsis and maize suggest that genes with diverse functions may respond differently in aneuploids (Hou et al., 2018; Johnson et al., 2020). Functional gene lists (generated as described in Johnson et al., 2020 and Shi et al., 2021) were retrieved and subjected to the ratio distribution and differential gene expression analysis (Supplemental Figures 10 and 11; Supplemental Data Set 7).
From our results, signal and stress genes in cis showed similar patterns with all genes from K-S tests (Supplemental Data Set 4). However, in some disomies, both functional groups showed significant differences from all genes (e.g. TB-2Sa; Supplemental Data Set 4). Furthermore, the distributions of TFs in trans display significant differences with all genes in most disomies even though no difference appears in cis for most disomies (Supplemental Data Set 4). TFs appear to be less inversely modulated in trans compared with all genes after analyzing the mean and median of the distributions in trans within the two groups (Supplemental Data Set 2; Supplemental Figure 12).
Interestingly, however, ribosomal genes were much more modulated than the pattern of all genes both in cis and trans (Supplemental Figures 10 − 12). In all disomies except TB-3Sb, TB-4Sa, and TB-9Lc, the ribosomal genes were even more inversely affected in trans with the major peaks of some disomies, such as TB-1Sb, TB-1La, TB-2Sa, TB-2Sa deletion and TB5Sc, shifted close to 0.5. On the other hand, the ribosomal genes in TB-4Sa and TB-9Lc displayed much stronger direct effects in trans (major peak close to 1.5) than the distribution of all genes. Only TB-3Sb showed a major peak close to 1, which is similar to the pattern of all genes. The K-S test showed that the distributions of the trans ribosomal genes in all disomies are significantly different from the distribution of all genes (Supplemental Data Set 4).
For nuclear-encoded genes whose products are targeted to the organelles including chloroplast, mitochondrion, and peroxisome, there are different responses compared to all genes. The nuclear chloroplast and peroxisome-targeted genes are modulated to a wide range with the distribution of nuclear chloroplast genes being significantly different from all genes in trans in all disomies and in cis for some disomies. There are few disomies showing a significant difference between peroxisome-targeted and all genes both in cis and trans, probably due to the limited gene number in this functional group. The trans nuclear mitochondrial genes distribute differently from all genes in most disomies and only few show differences in cis (Supplemental Data Set 4; Supplemental Figures 10, 11, and 13).
Maize descended from an allopolyploid event such that the remnants of the two founding genomes remain (Gaut et al., 2000) but have different reactions to fractionation, namely maize1 and maize2 (Schnable et al., 2011). We sought to examine whether there was a difference between the two genomes to imbalance. The effects on the two genomes only show differences for some disomies in trans, suggesting that target genes respond similarly (Supplemental Data Set 4; Supplemental Figures 10−12).
Discussion
The phenotypic effect of genomic imbalance was observed a century ago but there is still little knowledge about the molecular aspects. Here, we first identified 17 distal segmental disomies that cover about 80% of the maize genome. All the disomies showed a typical aneuploid phenotype of decreased plant stature. Examining the modulation of gene expression in disomies by utilizing RNA-seq data revealed dosage compensation and dosage effects in cis and an obvious trans-modulation on the unaltered chromosomal segments. An inverse effect is the most prevalent one. Functional groups of genes respond differently to the disomies, with ribosomal components and nuclear chloroplast genes showing distinct effects compared with TFs, signaling components, and stress-related genes, as examples.
Greater magnitude of dosage change in disomies causes severe phenotypic effects
Throughout various aneuploid studies there is documentation of aneuploid phenotypes (Sinnott et al., 1934; Satina et al., 1937; Lee et al., 1996a, 1996b; Sheridan and Auger, 2008; Brunelle and Sheridan, 2014). Maize is excellent material to produce aneuploids with varied chromosomal segments by utilizing the non-disjunction of the B chromosome coupled with the ability to generate haploids at reasonable frequencies. The evolutionary history of maize allows this ancient allotetraploid (Gaut et al., 2000) to have more tolerance in response to the aneuploids than other organisms with no readily discernible recent WGD. Previous morphological investigations of the maize monosomies and trisomies revealed that the severity of the dosage responses characteristically varied with the chromosome segment involved, rather than being a general consequence of aneuploidy (Lee et al., 1996a, 1996b). In this study, phenotypes observed in haploid disomies with greater imbalance are generally more severe. The severity is greater for larger aneuploid regions. Morphological observations of different stages of disomies (14 days and 45 days) in the present study revealed that the phenotypic differences between the disomies and control were evident at an early stage.
Transcriptome size change in some disomies
Across several biological scales, from cellular to organismal, there is a trend of a generalized linear correlation between cell size, transcriptome size, and whole-organism ploidy (Sinnott and Blakeslee, 1922; Steinitz-Sears, 1963; Loven et al., 2012; Robinson et al., 2018; Song et al., 2020). By contrast, there is little knowledge about the change of transcriptome size with regard to the impact of copy number variants on the phenotype. Cell size measurements in Datura trisomies decades ago showed that some specific cell types exhibit a decreased or increased cell size (Sinnott and Blakeslee, 1922; Sinnott et al., 1934). Given the correlation between cell size and transcriptome size noted with ploidy, the question arises if there are transcriptome size changes in aneuploidy, which has not previously been examined. In general, in our study, there was some variation in transcriptome size but four regions in particular had an obvious reduction surrounding an inverse modulation of the expression of the assayed genes. The chromosomal regions with this effect were understandably quite detrimentally affected by disomy. Aneuploidy effects are probably cell and developmentally distinct to some degree so these results raise the possibility that there are transcriptome size changes in individual cell types. Those regions identified in this study with the most obvious effects have a whole transcriptome modulation that is apparently uniform across cell types. Indeed, a PCA analysis of the ddPCR results (relative expression per cell) found these four disomies with inversely modulated transcriptome sizes clustered differently from the remainder (Supplemental Figure 13). Nevertheless, it is unlikely that the aneuploids have any major differences in the developmental types of cells compared to the haploid. The ratio distribution analysis (Figure 5) would reveal if that were the case by a large increase in the “outlier” peaks that would contain more cell type specifically expressed genes with many fold greater or lesser expression levels between the two genotypes if there were major differences.
The possibility that the reductions of gene expression in trans are a reflection of a sequencing bias due to gene dosage effects in cis occupying a greater amount of the sequencing aliquot is discounted by several observations. First, measurement of the expression of selected genes was validated against genomic DNA, which revealed a similar expression relationship to the RNA-seq data (Supplemental Figure 6). Indeed, for some regions, there is an inverse effect on the whole transcriptome (Figure 4), which cannot be explained by this hypothesis. Further, different functional types of genes have complex landscapes of trans modulations (Supplemental Figure 10) whereas a sequencing bias would predict a uniform and consistent effect. Lastly, the inverse effect can be observed phenotypically in Datura trisomies (Blakeslee, 1921), in Drosophila segmental aneuploids (Muller, 1950; Birchler et al., 1990; Sabl and Birchler, 1993; Sun et al., 2013a), and even in single gene dosage series (Rabinow et al., 1991; Birchler et al., 2001)—none of which can be explained by sequencing bias. It should be noted that a strong inverse effect could potentially cause a sequencing bias in the absence of controls to detect it and that would give the appearance of a stronger cis dosage effect than actually occurs, which would be exacerbated further by normalizing cis values by trans.
Implications for imbalance in haploid phases of the life cycle
The finding that haploid disomies have much more extreme effects than diploid trisomies has implications for the effects of copy number variation of dosage-sensitive regulatory genes in the haploid gametophyte generations. Classical studies have regularly found that unbalanced pollen tubes are at a great disadvantage in achieving fertilization in competition with balanced ones (Carlson, 1988). Obtaining pure samples of balanced and unbalanced pollen to address gene expression is not possible with this system. Thus, our results can serve as a surrogate example. In haploid pollen, regulatory genes for which the relative stoichiometry has an effect on target gene expression will have an extremely strong selection against them with regard to pollen tube growth when compared to the alternative if varied in dosage. Thus, selection against dosage-sensitive regulatory copy number variation in populations is likely to occur unless there is a strong counterselection for increased expression of the included genes. Fractionation after WGDs has usually retained TFs and signal transduction components in duplicate for longer periods of evolutionary time than other types of genes, as noted above. While divergence in expression levels between the duplicates might occur in sporophytic generations with lesser effects, the haploid gametophyte generation is likely to be a more stringent stage involved in maintaining the stoichiometry of interacting genes than other life history stages and thus would have a strong impact on the evolution of dosage-sensitive regulatory genes after WGD or segmental duplication.
Comparison to haploid yeast disomies
Disomic yeast has been examined with regard to gene expression. These aneuploids typically grow more slowly than normal haploid yeast in laboratory strains (Torres et al., 2007; Chen et al., 2019). Yet, there is extensive aneuploidy in natural isolates that tolerate genetic imbalance with genetic variants affecting this difference (Hose et al., 2020). The disomics in maize analyzed here are comparable to the division of the genome by individual yeast chromosomes, although each division carries many more genes in maize and the phenotypes are quite detrimental. A group of stress-associated genes is typically activated in yeast aneuploids (Terhorst et al., 2020) but this response is not seen in Arabidopsis (Hou et al., 2018) or in maize (present work). The stoichiometric genomic changes in maize modulate gene expression across the genome, which affects the expression of the varied chromosome similarly. While there are both positive and negative responses, the inverse effect is more common and therefore will mediate dosage compensation of a subset of genes on the varied region. Dosage compensation likewise has been observed for some genes on varied chromosomes in yeast (Hose et al., 2015). The kinetics of regulatory interactions is expected to be species blind and indeed there is evidence for the inverse effect in yeast as well (Hou et al., 2018). There is also a correlation between the amount of dosage compensation in cis and the amount of inverse effects in trans (Hou et al., 2018). However, it is possible that selection has occurred in unicellular yeast via various homeostatic mechanisms (Dephoure et al., 2014) to ameliorate the impact of regulatory imbalance to some degree as evidenced by the amount of aneuploidy in wild strains. This might be the case to capitalize on any advantage of increased absolute dosage of metabolic genes (Conant and Wolfe, 2007; Conant, 2014) in disomies in various environmental circumstances encountered, which the unicellular nature of yeast could accommodate. Just as various sex chromosomal dosage compensation mechanisms have evolved to ameliorate the impact of genomic imbalance caused by two-fold differences when they evolve from a homologous pair of chromosomes, yeast might have evolved mechanisms to facilitate a trade-off between genomic imbalance and any advantages of absolute dosage. This does not appear to be the case for aneuploidy in multicellular organisms.
Global inverse modulation
The most common effect on gene expression is a generalized inverse modulation on the unvaried chromosomes in aneuploids throughout various organisms (Sun et al., 2013a, 2013b; Hou et al., 2018; Johnson, et al., 2020). Our disomic data compared to trisomies of the same regions (Shi et al., 2021) show there is generally more decreased than increased modulation of trans genes, with larger segmental disomies being more pronounced in trans. The ratio distributions of TFs and signal transduction component genes display inverse modulations in trans compared to all genes. Given that these classes of genes have previously and generally been found to be dosage-sensitive on their targets, including other TFs (e.g. Birchler et al., 2001; Hou et al., 2018), the results suggest a cascade of effects from genomic imbalance. The fact that every region regardless of size produced recognizable effects is consistent with this view. However, the larger disomies in general have more affected genes suggesting cumulative effects. The change of stoichiometry of dosage-sensitive regulatory genes is postulated to cause a dominant negative effect on target gene expression particularly with hyperploidy, as is the case here, and as discussed in the companion paper (Shi et al., 2021). The dosage sensitivity of gene expression suggests that there is a rheostat of possible quantitative levels of expression with innumerable possibilities. The results presented here illustrate the need to incorporate genome balance principles into an understanding of gene expression, the control of quantitative traits, and the evolution of duplicate genes in populations and following WGDs.
Materials and methods
Materials and haploid induction
Hyperploid heterozygotes (A, A-B, B-A, B-A) from each translocation line except TB-7Sc and TB-2Sa were selected by a kernel phenotype showing full purple (TB-1La, TB-3La, TB-4Sa, TB-4Lb, TB-5Sc, TB-5Lc, TB-8Lc, TB-9Sd, TB-9Lc, and TB-10L19) or dotted purple (TB-1Sb, TB-3Sb, TB-6Lc, and TB-7Lb) embryo and yellow endosperm from crosses between hyperploid heterozygotes and their tester lines. Hyperploid heterozygotes of TB-7Sc show opaque endosperm and the kernel size is small compared to euploid (A, A-B, B-A) or monosomy (A, A-B). TB-2Sa tetrasomic plants (2, 2, B-2Sa, B-2Sa plus B chromosomes) were chosen from kernels showing purple embryos and yellow endosperm. All materials and their respective tester stocks have been converged to the W22 inbred line carrying the R-scm2 allele, which conditions anthocyanin pigment in both the embryo and endosperm, for a minimum of seven backcrosses. Two haploid inducer lines were used, namely RWSK; R1-navajo (R1-nj); c1-I or RWS; GFP; R1-nj (Rober et al., 2005; Yu and Birchler, 2016). RWSK (developed in the Birchler lab from a hybrid of RWS and RWK76 from the University of Hohenheim) has the anthocyanin inhibitor gene c1-I; thus, the haploid kernels produced from this line have pigmented embryos (purple) and colorless (yellow) endosperms because of the maternal inheritance of R-scm2 and the respective dominant anthocyanin marker on the B-A chromosome. Hyperploid heterozygotes (TB-1La, TB-3La, TB-4Sa, TB-4Lb, TB-5Sc, TB-5Lc, TB-8Lc, TB-9Sd, TB-9Lc, and TB-10L19) or tetrasomy from TB-2Sa were crossed as female by the haploid inducer line RWSK; R1-nj; C1-I. For some translocations (TB-1-Sb, TB-3Sb, TB-6Lc, and TB-7Lb) that use transposable elements (e.g. Dotted or Activator) to activate anthocyanin gene expression of reporter alleles or TB-7Sc with no anthocyanin marker, RWS; GFP; R1-nj was used instead. Roots of haploid seedlings show an absence of GFP signal when using a NIGHTSEA BLUESTAR flashlight. For the haploids with one or two B chromosomes, W22 diploids plus various B chromosomes were crossed by RWSK and the kernels showing purple embryo and yellow endosperm were chosen. Fluorescence in situ hybridization (FISH) identification confirmed the chromosome constitution of all seedlings. Haploid seedlings were grown in the Sears greenhouse at the University of Missouri-Columbia under the following conditions: 16 h light, 25°C day/20°C night.
FISH identification
Kernels were germinated at 28°C for 2–3 days in moist vermiculite; the primary root tip was excised and then the seedlings were transplanted to the greenhouse. Probe preparation, somatic chromosome spreading, FISH, image capture, and processing were performed as described previously with minor modification (Kato et al., 2004; Lamb et al., 2007). Generally, excised root tips were treated with gaseous nitrous oxide (Kato, 1999) for 2–3 h. Treated root tips were fixed in ice-cold acetic acid-ethanol (9:1 dilution) for 10 min and stored in 70% ethanol at −20°C until use. After washing in water on ice for 10 min, the root tip containing dividing cells was dissected and digested in 1% pectolyase and 3% cellulase solution for 30–50 min at 37°C depending on the excised root tip size. After digestion, the root sections were briefly washed in 100% ethanol twice. The root sections were carefully broken using a needle in acetic acid methanol (9:1 dilution) solution. The cell suspension was dropped onto glass slides in a box lined with wet paper towels and dried slowly. After the cell spreads were dried on slides, they were UV-crosslinked for 1 min (total energy, 120 mJ/cm2). At the center of the cell spreads, 10 μl of 2× SSC solution containing probe mixture (36 ng CentC, 10 ng TAG, 6 ng B-repeat, 10 ng Telo, and 50 ng NOR if applicable) was dropped. After the application of a mineral-oil-coated plastic coverslip, the slide preparation was denatured by placing onto a wet paper towel in an aluminum tray floating in boiling water (100°C) for 5 min. The slides were transferred immediately to a 55°C preheated humidity chamber containing water-soaked paper towelling and incubated at 55°C oven overnight. Slides were submerged vertically into a Coplin jar filled with room temperature 2× SSC to remove coverslips and then washed in 2× SSC for 15 min at 55°C. Slides were removed from 2× SSC and shaken dry by hand. A drop of Vectashield (with DAPI) was added to the center of the probed cells and a 22 × 50 mm2 glass coverslip was placed on the slide. After at least 20 min, spreads were screened using a fluorescence Olympus (BX61) microscope.
Genomic extraction and DNA sequencing
Genomic DNA extraction of the disomic lines was performed using DNeasy Plant Mini Kit (QIAGEN, 69104). Approximately 100 ng of genomic DNA were subjected to agarose gel electrophoresis to examine DNA integrity before being sent to the DNA Core at the University of Missouri in Columbia for DNA sequencing. The DNA Core performed TruSeq genomic library preparation. The library insert size was selected for 350bp. The DNA library was sequenced in NextSeq 1x75 flow cells. A coverage of ∼12Gb per sample was produced.
Copy number variation analysis
After removal of low quality sequences from the raw dataset with the FASTX toolkit (Hannon, 2010) using parameter -Q 33 -q 20 -p 80, ∼6× coverage of maize genome single reads were aligned to the maize W22 reference genome (Zm-W22-REFERENCE-NRGENE-2.0; Springer et al., 2018) or B73 reference genome plus mitochondrion and chloroplast genomes (Jiao et al., 2017) using Bowtie2 (Langmead and Salzberg, 2012). The threshold of the maximum number of distinct alignments was set to 10. MULTICOM-MAP (Li et al., 2015) was used to remove the reads mapped to multiple locations on the maize W22 or B73 sequences with more than two mismatches. The first step was to count the aligned read numbers using 1 Mb resolution along the maize chromosomes and compare the read numbers from aneuploid to the haploid control. This analysis allows one to find the copy number change for the varied chromosome arm (e.g. chromosome 1L in TB-1La) and also can determine whether there is copy number variation on other chromosomes (all chromosomes except chromosome 1 in TB-1La in this example). Also, by examining the copy number change with a single gene window, the genes on the varied chromosome arm (cis genes) and those in the remainder of the genome can be determined.
ddPCR
The ddPCR reaction mixture was prepared from a 2× ddPCR EvaGreen mix (1864033, Bio-Rad), primers (250 nM primers) and template (50 ng cDNA+gDNA mix) in a final volume of 22 μL; 20 μl of each assembled ddPCR reaction mixture was then loaded into the sample well of an eight-channel disposable droplet generator cartridge (1864008, Bio-Rad). A volume of 70 μL of droplet generation oil (1864006, Bio-Rad) was loaded into the oil well for each channel. The cartridge was placed into the droplet generator (1864002, Bio-Rad) to generate droplets. The droplets were collected and transferred to a 96-well PCR plate (12001925, Bio-Rad). The plate was heat-sealed with a foil seal (1814040, Bio-Rad) and then placed on a conventional thermal cycler with deep wells (e.g. C1000, Bio-Rad). For ddPCR technology, the post-cycling protocol was in accordance with the kit instructions (186-4033, Bio-Rad). Generally, it contains a 95°C enzyme activation step for 5 min followed by 40 cycles of a two-step cycling protocol (95°C for 30 s and 58°C for 1 min). Post cycling, a signal stabilization protocol was applied (a single step of 4°C for 5 min followed by 90°C for 5 min). The ramp rate between these steps was set to 2 C/second. After PCR, the 96-well PCR plate was loaded on the droplet reader (1864003, Bio-Rad), which automatically reads the droplets from each well of the plate. Analysis of the ddPCR data was performed with QuantaSoft analysis software (Bio-Rad) that accompanied the droplet reader.
Genome normalized expression
We used a genome normalized expression method (Coate and Doyle, 2010) in our ddPCR assay. The key step is co-extracting both RNA and gDNA from the same tissue using Dr. P Isolation Kit (BioChain) so that in vivo RNA/gDNA ratios are preserved. About 1µg of RNA and gDNA mixture (TNA) was subjected to reverse transcription using iScript™ Reverse Transcription Supermix [oligo(dT) and random primer] (1708840, Bio-Rad). Primers were designed to specifically amplify either the cDNA or gDNA. For designing the cDNA specific primers, at least one primer from each pair should be designed across exon-exon junctions to avoid non-specific binding to gDNA. Genome specific primers were designed from regions that locate upstream of the 5′ UTR or downstream of the 3′ UTR to avoid binding on mature or precursor mRNA. Primers for the amplicons range from 70 bp to 150 bp were blasted to NCBI or MaizeGDB to insure the specificity. All cDNA primers were tested for specificity by amplifying amplicons when using a cDNA template and not the gDNA. The gDNA primers can only produce products using gDNA and not the cDNA template.
The absolute value of the cDNA amplicon of each gene was compared to the corresponding genome amplicon to obtain the relative genome normalized expression values. The genome normalized expression of the disomy was compared to the haploid to obtain the relative expression per genome. For genes on the varied chromosome arm (cis), the disomy has twice as many copies as the haploid control; thus, relative expression per cell in disomy for cis genes was obtained by multiplying relative expression per genome by two. For the genes in the remainder of the genome (trans), the relative expression per cell is equal to the relative expression per genome.
RNA extraction and RNA-seq
At least three different plants (biological replicates) were identified by FISH for each disomy or haploid control. At 45 days after germination, the fifth and sixth leaf blades were collected and the central portion without the mid-rib was kept for RNA extraction. Total RNA was extracted using mirVana™ miRNA Isolation Kit (AM1560, Ambion) plus plant isolation aid (AM9690, ambion). External RNA Control Consortium (ERCC) Spike-in RNA (4456740) was added to the total RNA. There are two kinds of RNA selection, rRNA depletion (rRNA removal) and poly-A selection (mRNA selection). All samples (total 113) were subjected to stranded RNA-Seq libraries (TruSeq) with rRNA depletion libraries sequenced in NextSeq 1x75 flow cells. For the mRNA selection, stranded RNA-Seq libraries were constructed and sequenced on a NovaSeq S2 SE100 flow cell. An average read count of approximately 50 million reads per sample was obtained.
RNA-seq data and PCA plot analysis
The RNA-seq data and PCA plot analyses were described (Shi et al., 2021). Specifically, low quality reads, ERCC reads, and reads mapped to organelle genomes (mitochondrion and chloroplast) were filtered. Finally, the reads were mapped to the maize W22 genome using Tophat2 and differential expression analysis was performed using Cuffdiff. PCA was performed using normalized read counts (deposited at GEO under the accession number GSE156986) to determine the similarity of gene expression levels among biological replicates.
Relative transcriptome size
We used the Coate and Doyle (2010) method to calculate the transcriptome size. To estimate the size of the disomy transcriptome relative to each haploid control, we divided the per cell expression ratios (disomy/haploid) from the ddPCR assay by the per transcriptome expression ratios (disomy/haploid) from the RNA-Seq data set. Eight genes were used for the relative transcriptome size calculation. For each gene, the expression per cell (ddPCR) was divided by expression per transcriptome (RNA-Seq). The mean of these eight independent estimates (and associated sd) was taken as the estimate of relative transcriptome size.
Functional group gene lists
Functional gene lists were retrieved from Johnson et al. (2020). These gene lists include TFs, nuclear-encoded genes but targeted to the chloroplast (chloroplast targeted), mitochondrion (mitochondrial targeted), and peroxisome (peroxisomal-targeted), as well as stress related genes (stress), signal transduction pathway genes (signaling), structural components of the ribosome (ribosomal) and the proteasome (proteasomal).
Ratio distributions and scatter plots
Ratio distributions and scatter plots were performed using the same methods as described (Shi et al., 2021). The ratio was generated by dividing the mean of experimental counts (disomies) by the mean of control counts (W22 haploid, except with TB-5Sc, for which the euploid controls were used).
Statistical analysis
Statistical tests were performed using R with extreme values of ratio (ratio >6 or <1/6) excluded. Details of the statistical tests were described in the companion study (Shi et al., 2021). Mean, median and sd of each comparison were computed. Lilliefors test was used to check a normality assumption. The K–S test was used to check for differences between two ratio distributions. The Bartlett’s test was used to examine if variances are equal across different groups.
Data availability
All sequencing data were deposited at the Gene Expression Omnibus (GEO) repository under the accession number GSE156986.
Supplemental data
The following materials are available in the online version of this article.
Supplemental Figure 1. Haploid induction using GFP.
Supplemental Figure 2. FISH identification.
Supplemental Figure 3. Phenotypes of disomic seedlings (2–3 weeks) and disomic plants (45 days).
Supplemental Figure 4. Copy number analysis.
Supplemental Figure 5. PCA plots.
Supplemental Figure 6. ddPCR versus RNA-seq comparisons.
Supplemental Figure 7. Relationship between expression of cis and trans genes in each disomy comparison.
Supplemental Figure 8. rRNA removal and mRNA-selection comparison.
Supplemental Figure 9. Ratio distributions and scatter plots of haploids with B chromosomes.
Supplemental Figure 10. Ratio distributions of different functional groups and maize subgenomes.
Supplemental Figure 11. Scatter plots of differential gene expression of different functional groups and maize subgenomes.
Supplemental Figure 12. Comparison of the median of all trans gene expression to those of different functional groups.
Supplemental Figure 13. PCA analysis of ddPCR results in all disomies.
Supplemental Table 1. Statistics of the DNA sequencing data that were used to determine the cis gene number.
Supplemental Table 2. Sequence for primer sets used in ddPCR analysis.
Supplemental Data Set 1. Read mapping statistics and group information of each RNA-seq experiment.
Supplemental Data Set 2. Mean, median, standard deviation (sd) for each distribution.
Supplemental Data Set 3. Normality test for each distribution.
Supplemental Data Set 4. Distribution comparisons with K–S tests of significance.
Supplemental Data Set 5. Comparisons of distribution variances with Bartlett’s test.
Supplemental Data Set 6. DEGs statistics in each disomy. Lowly expressed genes were excluded for this analysis.
Supplemental Data Set 7. Functional group gene lists.
Funding
This work was supported by National Science Foundation grants (IOS-1545780, IOS-1444514, NSF 1615789, and NSF 1853556).
Conflict of interest statement. None declared.
Supplementary Material
Contributor Information
Hua Yang, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Xiaowen Shi, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
Chen Chen, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
Jie Hou, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
Tieming Ji, Department of Statistics, University of Missouri, Columbia, Missouri 65211, USA.
Jianlin Cheng, Department of Electrical Engineering and Computer Science, University of Missouri, Columbia, Missouri 65211, USA.
James A Birchler, Division of Biological Sciences, University of Missouri, Columbia, Missouri 65211, USA.
J.B., J.C., H.Y., X.S. designed the research; H.Y., X.S., C.C., J.H. performed research; C.C., J.H., T.J., H.Y., X.S., J.C. contributed analytical/computational tools; H.Y., X.S., C.C., J.H., T.J., J.B. analyzed data; H.Y., X.S., J.B. wrote the paper.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (https://academic.oup.com/plcell) is: James A. Birchler (BirchlerJ@Missouri.edu).
References
- Alfenito MR, Birchler JA (1993) Characterization of a maize B chromosome centric sequence. Genetics 135: 589–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aury JM, Jaillon O, Duret L, Noel B, Jubin C, Porcel BM, Ségurens B, Daubin V, Anthouard V, Aiach N, et al. (2006) Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 444: 171–178 [DOI] [PubMed] [Google Scholar]
- Bastiannse H, Zinkgraf M, Canning C, Tsai H, Lieberman M, Comai L, Henry I, Groover A (2019) A comprehensive genomic scan reveals gene dosage balance impacts on quantitative traits in Populus trees. Proc Natl Acad Sci U S A 116: 13690–13699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett JB (1991) Cytogenetic, genetic and plant breeding applications of B–A translocations in maize. In Gupta PK, Tsuchiya T, eds, Chromosome Engineering in Plants: Genetics, Breeding, Evolution (Part A). Elsevier, Amsterdam, pp 493–529 [Google Scholar]
- Birchler JA (1979) A study of enzyme activities in a dosage series of the long arm of chromosome one in maize. Genetics 92: 1211–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA (1981) The genetic basis of dosage compensation of alcohol dehydrogenase-1 in maize. Genetics 97: 625–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Alfenito MR (1993) Marker systems for B-A translocations in maize. J Hered 84: 135–138 [Google Scholar]
- Birchler JA, Bhadra U, Bhadra MP, Auger DL (2001) Dosage-dependent gene regulation in multicellular eukaryotes: implications for dosage compensation, aneuploid syndromes, and quantitative traits. Dev Biol 234: 275–288 [DOI] [PubMed] [Google Scholar]
- Birchler JA, Hiebert JC, Paigen K (1990) Analysis of autosomal dosage compensation involving the alcohol dehydrogenase locus in Drosophila melanogaster. Genetics 124: 679–686 [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Newton KJ (1981) Modulation of protein levels in chromosomal dosage series of maize: The biochemical basis of aneuploid syndromes. Genetics 99: 247–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA, Riddle NC, Auger DL, Veitia RA (2005) Dosage balance in gene regulation: Biological implications. Trends Genet 21: 219–226 [DOI] [PubMed] [Google Scholar]
- Birchler JA,, Veitia RA (2007) The gene balance hypothesis: From classical genetics to modern genomics. Plant Cell 19: 395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA,, Veitia RA (2010) The gene balance hypothesis: Implications for gene regulation, quantitative traits and evolution. New Phytol 186: 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birchler JA,, Veitia RA (2012) Gene balance hypothesis: Connecting issues of dosage sensitivity across biological disciplines. Proc Natl Acad Sci U S A 109: 14746–14753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakeslee AF (1934) New jimson weeds from old chromosomes. J Hered 25: 81–108 [Google Scholar]
- Blakeslee AF (1921) Types of mutations and their possible significance in evolution. Am Nat 55: 254–267 [Google Scholar]
- Blakeslee AF, Belling J, Farnham ME (1920) Chromosomal duplication and Mendelian phenomena in Datura mutants. Science 52: 388–390 [DOI] [PubMed] [Google Scholar]
- Blanc G, Wolfe KH (2004) Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell 16: 1679–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomme T, Vandepoele K, Simillion SD, Maere C, Van de Peer S, , Bodt Y (2006) The gain and loss of genes during 600 million years of vertebrate evolution. Genome Biol 7: R43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boell L, Pallares LF, Chen C, Christian Y, Kousa JL, Kuss YA, Nelsen P, Novikov S, Schutte O, , Brodski BC, et al. (2013) Exploring the effects of gene dosage on mandible shape in mice as a model for studying the genetic basis of natural variation. Dev Genes Evol 223: 279–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridges CB (1925) Sex in relation to chromosomes and genes. Am Nat 59: 127–137 [Google Scholar]
- Brunelle DC, Sheridan WF (2014) The effect of varying chromosome arm dosage on maize plant morphogenesis. Genetics 198: 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson PS (1972) Locating genetic loci with aneuploids. Mol General Genet 114: 273–280 [DOI] [PubMed] [Google Scholar]
- Carlson WR (1988) The cytogenetics of corn. Corn Corn Improv 18: 259–343. [Google Scholar]
- Chen Y, Chen S, Li K, Zhang Y, Huang X, Li T, Wu S,, Wang Y, Carey LB, Qian W (2019) Overdosage of balanced protein complexes reduces proliferation rate in aneuploid cells. Cell Systems 9: 129–142 [DOI] [PubMed] [Google Scholar]
- Clark NM, Fisher AP, Berckmans B, Van den Broek L, Nelson EC, Nguyen TT, Bustillo-Avendano E, Zebell SG, Moreno-Risueno MA, Simon R, et al. (2020) Protein complex stoichiometry and expression dynamics of transcription factors modulate stem cell division. Proc Natl Acad Sci U S A 117: 15332–15342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coate JE, Doyle JJ (2010) Quantifying whole transcriptome size, a prerequisite for understanding transcriptome evolution across species: An example from a plant allopolyploid . Genome Biol Evol 2: 534.– [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant GC (2014) Comparative genomics as a time machine: How relative gene dosage and metabolic requirements shaped the time-dependent resolution of yeast polyploidy. Mol Biol Evol 31: 3184–3193 [DOI] [PubMed] [Google Scholar]
- Conant GC, Wolfe KH (2007) Increased glycolytic flux as an outcome of whole-genome duplication in yeast. Mol Syst Biol 3: 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defoort J, Van de Peer Y, Carretero-Paulet L (2019) The evolution of gene duplicates in angiosperms and the impact of protein-protein interactions and the mechanism of duplication. Genome Biol Evol 11: 2292–2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Hwang S, O’Sullivan C, Dodgson SE, Gygi SP, Amon A, Torres EM (2014) Quantitative proteomic analysis reveals posttranslational responses to aneuploidy in yeast. eLife 3: e03023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deimling S, Röber F, Geiger HH (1997) Methodik und Genetik der in-vivo-Haploideninduktion bei Mais. Vortr Pflanzenzüchtg 38: 203–224 [Google Scholar]
- Freeling M, Lyons E, Pedersen B, Alam M, Ming R, Lisch D (2008) Many or most genes in Arabidopsis transposed after the origin of the order Brassicales. Genome Res 18: 1924–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeling M,, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16: 805–814 [DOI] [PubMed] [Google Scholar]
- Gaut BS, Le Thierry d'Ennequin M, Andrew S,, Peek AS, Mark C, Sawkins MC (2000) Maize as a model for the evolution of plant nuclear genomes. Proc Natl Acad Sci U S A 97: 7008–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grell EH (1962) The dose effect of ma-l+ and ry+ on xanthine dehydrogenase activity in Drosophila melanogaster. Zeitschrift für Vererbungslehre 93: 371–377 [Google Scholar]
- Guo M, Birchler JA (1994) Trans-acting dosage effects on the expression of model gene systems in maize aneuploids. Science 266: 1999–2002 [DOI] [PubMed] [Google Scholar]
- Gout J-F,, Lynch M (2015) Maintenance and loss of duplicated genes by dosage subfunctionalization. Mol Biol Evol 32: 2141–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannon GJ (2010) FASTX-Toolkit. http://hannonlab.cshl.edu/fastx_toolkit
- Hose J, Escalante LE, Clowers KJ, Dutcher HA, Robinson D, Bouriakov V, Coon JJ, Shishkova E, Gasch AP (2020) The genetic basis of aneuploidy tolerance in wild yeast. eLife 9: e52063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hose J, Yong CM, Sardi M, Wang Z, Newton MA, Gasch AP (2015) Dosage compensation can buffer copy-number variation in wild yeast. eLife 4: e05462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Shi X, Chen C, Islam MS, Johnson AF, Kanno T, Huettel B, Yen M-R, Hsu F-M, Ji T, et al. (2018) Global impacts of chromosomal imbalance on gene expression in Arabidopsis and other taxa. Proc Natl Acad Sci U S A 115, E11321–E11330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Du Y, Zhao X, Jin W (2016) B chromosome contains active genes and impacts the transcription of A chromosomes in maize (Zea mays L.). BMC Plant Biol 16: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Peluso P, Shi J, Liang T, Stitzer MC, Wang B, Campbell MS, Stein JC, Wei X, Chin CS, et al. (2017) Improved maize reference genome with single-molecule technologies. Nature 546: 524–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AF, Hou J, Yang H, Shi X, Chen C, Islam MS, Ji T, Cheng J, Birchler JA (2020) Magnitude of modulation of gene expression in aneuploid maize. J Genet Genomics 47: 93–103 [DOI] [PubMed] [Google Scholar]
- Kato A (1999) Air drying method using nitrous oxide for chromosome counting in maize. Biotech Histochem 74: 160–166 [DOI] [PubMed] [Google Scholar]
- Kato A, Lamb JC, Birchler JA (2004) Chromosome paintingusing repetitive DNA sequences as probes for somatic chromosome identification in maize. Proc Natl Acad Sci 101: 13554–13559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov FA, Koonin EV (2004) A common framework for understanding the origin of genetic dominance and evolutionary fates of gene duplications. Trends Genet 20: 287–290 [DOI] [PubMed] [Google Scholar]
- Lamb JC, Riddle NC, Cheng YM, Theuri J, Birchler JA (2007) Localization and transcription of a retrotransposon-derived element on the maize B chromosome. Chromosome Res 15: 383–398 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EA, Darrah LL,, Coe EH (1996a) Genetic variation in dosage effects in maize aneuploids. Genome 39: 711–721 [DOI] [PubMed] [Google Scholar]
- Lee EA, Darrah LL, Coe EH (1996b) Dosage effects on morphological and quantitative traits in maize aneuploids. Genome 39: 898–908 [DOI] [PubMed] [Google Scholar]
- Li J, HouJ, SunL, WilkinsJM, LuY, Niederhuth CE, MeridethBR, Mawhinney TP, Mossine VV, Greenlief CM, et al . (2015) From Gigabyte to Kilobyte: A Bioinformatics Protocol for Mining Large RNA-Seq Transcriptomics Data. PLOS ONE 10 (4): e0125000. 10.1371/journal.pone.0125000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA (2012) Revisiting global gene expression analysis. Cell 151: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Bodt SD, Raes J, Casneuf T, Montagu MV, Kuiper M, de Peer YV (2005) Modelling gene and genome duplications in eukaryotes. Proc Natl Acad Sci U S A 102: 5454–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y (2008) RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath CL, Gout J-F, Johri P, Doak TG, Lynch M (2014) Differential retention and divergent resolution of duplicate genes following whole-genome duplication. Genome Res 24: 1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B (2008) Mapping and quantifying mammalian transcriptomes by RNA-seq. Nat Methods 5: 621–628 [DOI] [PubMed] [Google Scholar]
- Muller HJ (1950) The Harvey Lectures, Series XLIII. Chas. C. Thomas, Springfield, IL, pp. 165–229 [Google Scholar]
- O’Brien SJ, Gethmann RC (1973) Segmental aneuploidy as a probe for structural genes in Drosophila: mitochondrial membrane enzymes. Genetics 75: 155–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pál C, Hurst LD (2003) Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197 [DOI] [PubMed] [Google Scholar]
- Rabinow L, Nguyen-Huynh AT, Birchler JA (1991) A trans-acting regulatory gene that inversely affects the expression of the white, brown and scarlet loci in Drosophila. Genetics 129: 463–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raznahan A, Parikshak NN, Chandran V, Blumenthal JD, Clasen LS, Alexander-Bloch AF, Zinn AR, Wangsa D, Wise J, Murphy DGM, et al. (2018) Sex-chromosome dosage effects on gene expression in humans. Proc Natl Acad Sci U S A 115: 7398–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rober FK, Gordillo GA, Geiger HH (2005) In vivo haploid induction in maize - performance of new inducers and significance of doubled haploid lines in hybrid breeding. Maydica 50: 275–e283. [Google Scholar]
- Robinson DO, Coate JE, Singh A, Hong L, Bush M, Doyle JJ, Roeder AHK (2018) Ploidy and size at multiple scales in the Arabidopsis sepal. Plant Cell 30: 2308–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabl JF, Birchler JA (1993) Dosage dependent modifiers of white alleles in Drosophila melanogaster. Genet Res 62: 15–22 [DOI] [PubMed] [Google Scholar]
- Satina S, Blakeslee AF, Avery AG (1937) Balanced and unbalanced haploids in Datura. J Heredity 28: 193–202 [Google Scholar]
- Schinzel A (2001) Catalogue of Unbalanced Chromosome Aberrations in Man. Walter de Gruyter, Berlin, New York [Google Scholar]
- Schnable JC, Springer NM, Freeling M (2011) Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss. Proc Natl Acad Sci U S A 108: 4069–4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman JG, Seidman C (2002) Transcription factor haploinsufficiency: When half a loaf is not enough. J Clin Invest 109: 451–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheridan WF, Auger DL (2008) Chromosome segmental dosage analysis of maize morphogenesis using B-A-A translocations. Genetics 180: 755–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Rahmani RS, Gugger PF, Wang M, Li H, Zhang Y, Li Z, Wang Q, Van de Peer Y, Marchal K, et al. (2020) Distinct expression and methylation patterns for genes with different fates following a single whole-genome duplication in flowering plants. Mol Biol Evol 37: 2394–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Yang H, Chen C, Hou J, Hanson KM, Albert PS, Ji T, Cheng J, Birchler JA (2021) Genomic imbalance determines positive and negative modulation of gene expression in diploid maize. The Plant Cell 33: 917–939 [DOI] [PMC free article] [PubMed]
- Simillion C, Vandepoele K, Van Montagu MC, Zabeau M, Van de Peer Y (2002) The hidden duplication past of Arabidopsis thaliana. Proc Natl Acad Sci U S A 99: 13627–13632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott EW, Blakeslee AF (1922) Structural changes associated with factor mutations and with chromosome mutations in Datura. Proc Natl Acad Sci U S A 8: 17–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnott EW, Houghtaling H, Blakeslee AF (1934) The Comparative Anatomy of ExtraChromosomal Types in Datura stramonium. Carnegie Institution of Washington, Washington, DC [Google Scholar]
- Song MJ, Potter B, Doyle JJ, Coate JE (2020) Gene balance predicts transcriptional responses immediately following ploidy change in Arabidopsis thaliana. Plant Cell 32: 1434–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben-Zvi G, et al. (2018) The maize W22 genome provides a foundation for functional genomics and transposon biology. Nat Genet 50: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Stahl Y, Grabowski S, Bleckmann A, Kuhnemuth R, Weldtkamp-Peters S, Pinto KG, Kirschner GK, Schmid JB, Wink RH, Hulsewede A, et al. (2013) Moderation of Arabidopsis root stemness by CLAVATA1 and ARABIDOPSIS CRINKLY4 receptor kinase complexes. Curr Biol 23: 362–371 [DOI] [PubMed] [Google Scholar]
- Steinitz-Sears LM (1963) Chromosome studies in Arabidopsis thaliana. Genetics 48: 483–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, et al. (2007) Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315: 848–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Johnson AF, Donohue RC, Li J, Cheng J, Birchler JA (2013a) Dosage compensation and inverse effects in triple X metafemales of Drosophila. Proc Natl Acad Sci U S A 110: 7383–7388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Johnson AF, Li J, Lambdin AS, Cheng J, Birchler JA (2013b) Differential effect of aneuploidy on the X chromosome and genes with sex-biased expression in Drosophila. Proc Natl Acad Sci U S A 110: 16514–16519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdighian S, Van Bel M, Li Z, Van de Peer Y, Carretero-Paulet L, Maere S (2017) Reciprocally retained genes in the angiosperm lineage show the hallmarks of dosage balance sensitivity. Plant Cell 29: 2766–2785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terhorst A, Sandikci A, Keller A, Whittaker CA, Dunham MJ, Amon A (2020) The environmental stress response causes ribosome loss in aneuploidy yeast cells. Proc Natl Acad Sci U S A 117: 17031–17040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas BC, Pedersen B, Freeling M (2006) Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res 16: 934–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A (2007) Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 317: 916–924 [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A (2008) Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science 322: 703–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH, Shields DC (1997) Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708–713 [DOI] [PubMed] [Google Scholar]
- Yu W, Birchler JA (2016) A green fluorescent protein-engineered haploid inducer line facilitates haploid mutant screens and doubled haploid breeding in maize. Mol Breed 36:1–12 [Google Scholar]
- Zhang X, Hong D, Ma S, Ward T, Ho M, Pattni R, Duren Z, Stankov A, Shrestha SB, Hallmayer J, et al. (2020) Integrated functional genomic analyses of Klinefelter and Turner syndromes reveal global network effects of altered X chromosome dosage. Proc Natl Acad Sci U S A 117: 4864–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data were deposited at the Gene Expression Omnibus (GEO) repository under the accession number GSE156986.