

Abstract

Click chemistry has been established rapidly as one of the most valuable methods for the chemical transformation of complex molecules. Due to the rapid rates, clean conversions to the products, and compatibility of the reagents and reaction conditions even in complex settings, it has found applications in many molecule-oriented disciplines. From the vast landscape of click reactions, approaches have emerged in the past decade centered around oxidative processes to generate in situ highly reactive synthons from dormant functionalities. These approaches have led to some of the fastest click reactions know to date. Here, we review the various methods that can be used for such oxidation-induced “one-pot” click chemistry for the transformation of small molecules, materials, and biomolecules. A comprehensive overview is provided of oxidation conditions that induce a click reaction, and oxidation conditions are orthogonal to other click reactions so that sequential “click-oxidation-click” derivatization of molecules can be performed in one pot. Our review of the relevant literature shows that this strategy is emerging as a powerful approach for the preparation of high-performance materials and the generation of complex biomolecules. As such, we expect that oxidation-induced “one-pot” click chemistry will widen in scope substantially in the forthcoming years.
1. Introduction
Click chemistry is now a preferred approach for many bond-forming reactions. This is especially the case when large molecules have to be joined or when molecules have to be connected in heterogeneous systems such as the modification of surfaces. In the seminal Angewandte Chemie paper of Sharpless and co-workers the concept of “click chemistry” was illustrated by the relative energy levels of chemical components that participate in representative chemical transformations (Figure 1A).1 In this scheme, the process of petrochemical cracking leads to an increase in the oxidation state of the carbon atoms involved. The resulting raised molecular energy levels pave the way for additional chemical transformations, often aided by catalysts, to install heteroatoms or heteroatom-containing functional groups. These functional groups serve as an anchor point for follow-up conversions to generate fine chemicals and other compounds of interest. In many cases, the oxidation of organic molecules is allowing a variety of chemical reactions, especially with electron-rich species. This is exemplified by the oxygenation of a hydrocarbon followed by stepwise oxidation to its carboxylic acid (Figure 1B). In this process, the sp2-hybridized carbon atoms that are formed in the higher oxidation states, i.e. oxidation state +1 (in a carbonyl) and +3 (in a carboxylic acid), under suitable conditions become susceptible to nucleophilic attack, leading to the formation of imines or amides, respectively. Besides the increased reactivity of the atoms with higher oxidation state, the electron-withdrawing nature of the introduced heteroatoms may activate neighboring proton-containing (carbon) atoms for deprotonation (Figure 1C), thereby generating electron-rich species that can connect to electrophilic species such as the electron-poor carbon atoms in carbonyl compounds, as for example applied in aldol condensations. This chemistry is, interestingly so, also extensively used by natural biosynthesis pathways.2 For example, both lipid and polyketide biosyntheses rely on Claisen condensation of carbonyl groups (C=O) (Figure 1C). As such, Nature uses oxidation to generate more reactive carbon atoms to be used for the construction of larger molecules and as such has been a tremendous source of inspiration for the development of artificial oxidation-assisted ligation methods.
Figure 1.

Schematic depiction of the changes in oxidation states of carbon atoms during various chemical transformations (formal oxidation states are indicated in red). (A) Schematic depiction of the conversion of hydrocarbons to a representative set of fine chemicals via a process of cracking and catalysis, and three representative transformations of the heteroatom-containing derivatives. (B) Oxidation ladder of 3-hybridized a carbon atom when oxidized from sp3-hybridization in an alkane and alcohol to sp2-hybridization in an aldehyde and carboxylic acid. (C) Reaction equations of two classical C–C bond-forming reactions (aldol condensation and Claisen condensation) enabled by the oxidation state of α-carbon atoms.
1.1. Click Chemistry
The concept of “click chemistry” was launched with a set of criteria and characteristics for the process (Table 1). Nevertheless, approximately 20 years after the formal definition of click chemistry, multiple click reactions fail to match all qualifications. For example, despite being a game-changer in the field of chemistry, the extensive review on copper-catalyzed alkyne–azide cycloaddition (CuAAC) reactions by Christensen and Meldal3 teaches that the simplicity of the reaction can be obscured by tedious optimization procedures that are often required to obtain full conversion. Similarly, few click reactions are stereospecific. Therefore, the list provided in Table 1 is nowadays regarded as an ideal set of parameters of which most but not all are met by procedures generally referred to as “click reactions”.
Table 1. Summary of the Most Important Criteria and Characteristics of Click Chemistry.
| Criteria | Characteristics |
|---|---|
| Modular | Simple reaction conditions |
| Wide in scope | Readily available starting materials and reagents |
| High yielding | No or benign solvent (e.g. water) that is easily removed |
| Stereospecific | Simple product isolation |
| Inoffensive byproducts | High thermodynamic driving force (>20 kcal/mol) |
1.2. Oxidation-Induced Click Reactions
Oxidation of an organic substrate in order to induce click chemistry has emerged as an appealing approach. Although many click chemistry building blocks can be generated in situ by means of addition of heteroatoms along the pathways described (e.g. when the Nu: is an azide anion (N3–) in Figure 1A), the ability to “activate” one of the substrates by means of oxidation in the presence of the other substrate is an attractive alternative, especially if isolation of intermediates is not required. If the oxidation is sufficiently chemoselective, a one-pot procedure can be designed based on click reactions that are orthogonal to the oxidation step, i.e. oxidation-orthogonal click reactions (cf. bio-orthogonal click reactions). Since oxidation often leads to functional groups with enhanced reactivity, the rate of the induced click reaction needs to outcompete potential undesired side reactions. For example, for a relatively stable moiety such as a nitrile oxide, a rate constant k > 1 M–1·s–1 will still lead to efficient click conversion to the targeted product. However, for the more reactive ortho-quinones, the rate constants for any click reaction need to be at least 2 orders of magnitude higher to effectively suppress competing Michael addition. Fortunately, these high reaction rates also pave the way for the application under high dilution in heterogeneous media (e.g. in serum or inside cells).4 Therefore, the oxidation of certain organic substrates to higher energy levels enables unique “click-type” reaction pathways that rapidly lead to desired products without competing side reactions. Figure 2A provides an overview of organic molecules used in one-pot oxidation-induced click-chemistry approaches by change of oxidation state. It also shows how oxidation changes the shape of the HOMO and LUMO orbitals of some of these organic molecules (Figure 2B).
Figure 2.

(A) Concept of oxidation of organic substrates into clickable moieties. (B) Changes in shapes of the HOMO and LUMO orbitals as a result of oxidation, based on wb97xd/6-311+G(d,p) calculations of energy-minimized structures.
1.3. Scope of This Review
This review focuses on one-pot oxidation and click approaches that have been developed in the past decade (i.e., from 2010–2020). As such, the concept distinguishes itself from approaches involving prior oxidation of one of the substrates before click reaction, as for example to ensure sulfur(VI)-fluoride exchange (SuFEx): despite being directly connected to the sulfur atom in its highest oxidation state, procedures in which oxidation and SuFEx occur in one pot have yet not been reported. For the interested reader we refer to the following papers on the growing applications of SuFEx chemistry5 for the preparation of advanced materials,6,7 in drug discovery,8,9 and chemical biology10−13 or the latest discoveries that reveal stereospecific conversions14,15 and inverted drug discovery.16 Similarly, in situ activation methods for click reactions different from oxidation are not included.17−23
The outline of this review is as follows. We first cover various oxidation-induced one-pot click reactions that have been described for small molecules. Due to the broad compatibility of most small molecules to harsh reaction conditions, a wide range of oxidation conditions is tolerated. Next, we will provide an overview of conditions that have been established for the modification of polymers and of surfaces, and we close this review with an analysis of the modification of large biomolecules (proteins).
2. Small Molecules
In situ oxidation of small molecules in order to induce a click reaction is often applied to uncover parameters specifically associated with the intended transformation (i.e., kinetics, solvent compatibility, scope, etc.). The resulting proof-of-concept conditions can consequently also be applied to larger molecules as well as complex biomolecules.
2.1. Oxidative Activation of the Alkyne
As a framework for our review we consider the strain-promoted alkyne–azide cycloaddition (SPAAC).24 For this [3 + 2] cycloaddition, wherein the azide (R–N3) and the alkyne react as a 1,3-dipole and a dipolarophile, respectively, we have compared the rate constants for two of the most used cyclooctynes, i.e., DBCO 1 (also known as DIBAC)25 and BCN 2,26 with two different azides (Figure 3A and B). The reaction rates between benzyl azide (Bn–N3) and DBCO or BCN to produce product 3a or 4a are 0.24 M–1·s–1 and 0.07 M–1·s–1, respectively, revealing a 3.4 times higher rate of DBCO over BCN in this reaction. Interestingly, an inverse order of reactivity is apparent for the aromatic phenyl azide (Ph–N3); that is, in that case BCN reacts 6 times faster than DBCO.27 Specifically, the rates between Ph–N3 and DBCO or BCN to produce SPAAC product 3b or 4b are 0.033 M–1·s–1 or 0.2 M–1·s–1, respectively (in CH3CN:H2O = 3:1).27 DFT calculations indicated that Ph–N3 and BCN react via an inverse-electron demand (IED) mechanism controlled by HOMOBCN/LUMOazide. Based on this, an electron-poor azide was designed that resulted in one of the fastest SPAAC reactions: the reaction between BCN-OH 2 and 4-azido-1-methylpyridinium iodide, producing product 4c, proceeds with a rate constant of 2–2.9 M–1·s–1.27 This has been the upper limit of SPAAC for a while.
Figure 3.

SPAAC between an aliphatic or aromatic azide and DBCO (A; R′ = -CH2CH2NH2) or BCN (B). Panel C points to a significant change of the relevant energy levels (by extension of the π-system) by oxidation and thereby to the observed increased rate of SPAAC (DMP = Dess–Martin periodinane).
Very recently, it was shown that the SPAAC rate can be further increased by a delicate combination of substituents on the aromatic rings of DIBO derivative 5. Oxidation of the alcohol moiety in DIBO to its corresponding carbonyl in 6 (a.k.a. keto-DIBO) led to a faster formation of 7 than of 8 from 5 (Figure 3C).28 As a result of this oxidation, the reaction rate increased 3.5–7-fold, to a rate constant of 3.5 M–1·s–1. This enhancement is analogous to the differences in the rate constants between benzyl azide and BARAC and DBCO (k = 0.96 M–1·s–1 and 0.31 M–1·s–1, respectively,29 a 3-fold difference); these two cyclooctynes also differ from BCN by the presence of sp2-hybridized C atoms in the 8-membered ring. Interestingly, the fluorescence of the hydrophobic cell-permeable cyclooctyne 5 increased as a result of its oxidation to 6 (Φ = 0.13) and decreased again after SPAAC, which allowed for its use for imaging in protozoan parasites.28
Whereas above-mentioned nonoxidized alkynes are stable enough to allow prolonged storage, more reactive and intrinsically unstable alkynes can be obtained by in situ activation methods from oxidized species. By clever design of a synthetic procedure to oxidize substituents on the benzene ring of 9 to diazobenzene 10, which was converted in a series of steps to benzoic acid derivative 11, a product was obtained that could be photoactivated (Figure 4).30 Irradiation of 11 led to disintegration and produced benzyne 12, which is an oxidized form of benzene. In the presence of benzylazide, benzyne 12 underwent SPAAC efficiently, and conversions were typically complete within 3–15 min. This method complements alternative methods developed by Larock,31 Moses,32 Feringa,33 and Reddy,34 in which click chemistry is induced by in situ benzyne generation from 2-(trimethylsilyl)phenyl trifluoromethanesulfonate and cesium fluoride.
Figure 4.

Photoinduced preparation of benzyne 12, an in situ generated oxidized version of benzene, and its SPAAC with benzylazide.
A direct method for the oxidative activation of a precursor molecule to a strained alkyne involves protection of the acetylene moiety as a cobalt hexacarbonyl complex (Figure 5). As such, orthogonality between the reactivity of a terminal alkyne and a strained alkyne was obtained that allowed the combination of CuAAC with SPAAC in a two-step approach.35 After CuAAC derivatization of the terminal alkyne in 12, oxidative conditions were applied to remove the cobalt protecting group from the strained internal alkyne. This deprotection procedure could be performed in the presence of an azide, resulting in a one-pot oxidation-induced derivatization of the cycloheptyne to form SPAAC product 13 (Figure 5A). A similar approach was developed for BCN, but now having both the azide and the alkyne-protected BCN in one molecule 14, leading to the tandem CuAAC-SPAAC product 15 (Figure 5B).36 As such, the protection of strained alkynes as metal complexes has emerged as an appealing strategy to increase orthogonality in the reactivity of alkynes.37
Figure 5.

Oxidative removal of a dicobalt hexacarbonyl complex from a strained alkyne to facilitate CuAAC conjugation on a distal alkyne (A) or azide (B). In blue, the one-pot oxidation-induced click reaction is shown, leading to SPAAC and CuAAC products 14 and 16.
2.2. Oxidative Activation of 1,3-Dipoles and Dienes
Whereas the N–N–N 1,3-dipole of azides is usually not obtained by oxidation,38 some other 1,3-dipoles that react with strained unsaturated C–C bonds can be accessed via this route. This is especially the case for the C–N–O 1,3-dipole found in nitrile oxides and nitrones.
Due to the high reactive nature, nitrile oxides are generally prepared by in situ oxidation (Figure 6A). When this is done with a mild reagent such as a hypervalent iodine species, oxime 17 can be converted to nitrile oxide 18 to produce R–C≡N+–O– as a C–N–O 1,3-dipole that reacts with unsaturated C–C bonds in a reaction called strain-promoted alkyne–nitrile oxide cycloaddition (SPANOC).39,40 In the presence of an olefin, the oxidation with phenyliodine bis(acetate) (PIDA) provided isoxazoline 19.41 Analogously, the presence of a terminal alkyne in combination with phenyliodine bis(trifluoroacetate) (PIFA) provided isoxazole 20.42 The PIDA-induced oxidation activation was also compatible with click chemistry of DIBO-functionalized molecules.40 Even the oxidative cycloaddition of oximes to maleimides in the presence of a catalytic amount of iodonium species has been described.43 Interestingly, the reaction rate of SPAAC could be increased in this manner for the strained alkyne BCN-OH 2: complete conversion to tricyclic construct 21 was observed within 2–5 min. The rate constant 1.8 M–1·s–1 is approximately 10 times higher than the reaction of BCN 2 with benzyl azide under identical conditions (k ∼ 0.18 M–1·s–1). The application to DIBO led to even higher rate constants, of up to 3.9 M–1·s–1, which is ∼60 times higher than the reaction between DIBO and Bn-N3 under the same conditions.
Figure 6.

Click reactions of 1,3-dipoles with unsaturated C–C bonds. (A) One-pot oxidation of oximes 17 to nitrile oxide 18 and subsequent SPANOC with olefins (to form isooxazoline 19), with terminal alkynes or internal alkynes to form isoxazoles (20 and 21). (B) SPANC of nitrone 22, which can be obtained by means of in situ oxidation, with DBCO derivative 23 to form isoxazoline 24.
Although nitrones are typically not prepared in situ, and hence would fall outside the scope of this review, the in situ preparation of nitrones at the N-terminally positioned serine residue in proteins that is mentioned toward the end of this review is facilitated by oxidation (vide infra). Therefore, we include this short treatment of the use of nitrones in click reactions. When the nitrogen atom of the C–N–O-based 1,3-dipole contains a carbon substituent, a nitrone moiety (i.e., the (R–C=N+(R′)–O–) is obtained that also undergoes click chemistry (Figure 6B). Nitrones are not only of biomedical relevance,44 they can conveniently be obtained by means of oxidation of amines or imines45 and can be readily introduced on nanoparticles46 or in proteins47 (See section 4.6.). Furthermore, nitrones can be used in the Kinugasa reaction to form β-lactams.48 However, a variety of nitrones such as 22 has been subjected to strain-promoted alkyne–nitrone cycloaddition (SPANC) with DBCO 23, yielding the formation of isoxazoline 24. Rate constants of the reaction of strained alkynes with nitrones are 1–60 M–1·s–1, which are substantially higher than with nitrile oxides and azides.49,50 Also, they tend to be higher than the reaction between BCN-OH 2 and a cyclic nitrone, which is in the lower single-digit range.51 The fixed E-configuration of the nitrone double bond, the electronic influence of the substituents, and the additional strain exerted by the five-membered ring were mentioned as probable causes for the enhanced rates, and the term “doubly strain-promoted” was introduced.49
For many chemical transformations the rates of unactivated 1,3-dipolar cycloaddition reactions are sufficient. However, restrictions imposed on conjugation reactions in complex or stringent settings usually demand higher rates. For example, the concentrations of biomolecules of interest in cytoplasm are in the order of μM–nM, and in order to achieve full conversion bimolecular rate constants of at least 104 M–1·s–1 are required.52 Only such rate constants provide the ability to compete with enzymatic rate constants (103–106 M–1·s–1).53 Up to now rates based on 1,3-dipolar cycloaddition reactions fall short of this demand (vide supra), and conjugation methods were pursued that are associated with high kinetics while still leading to clean conversions. In principle, already adding one extra atom to the number of three that form 1,3-dipoles, thereby moving to dienes that can be formed by four atoms, makes available additional means to pursue oxidation-induced click chemistry. These are described below.
One set of reactions that filled the need for rapid click conjugations is [4 + 2] cycloadditions of unsaturated C=C and C≡C bonds on 1,2,4,5-tetrazines (Tz, a.k.a. s-tetrazines).54,55 These inverse electron-demand Diels–Alder (IEDDA) cycloadditions are among the fastest man-made ligation reactions known to date, with rates that outcompete those for many enzymes. Specifically, the reaction of trans-cyclooctene sTCO (a.k.a. cpTCO) and dipyridyl-tetrazine (dipyTz) proceeds with a second order rate constant of 3.3 × 106 M–1·s–1,56 which was only surpassed recently by reaction of dipyTz with trans-1-sila-4-cycloheptene (k = 1.14 × 107 M–1·s–1).57
Many current synthetic pathways include as a final step the oxidation of dihydrotetrazine (e.g. DHTz 25) to its corresponding Tz 26 (Figure 7A).58 In most cases the Tz is isolated before it is subjected to a click reaction. However, the group of Fox recently showed that this oxidation can also be performed in situ, resulting in a one-pot oxidation-induced click reaction.59 Using a clever combination of starting materials, a DHTz/Tz pair was selected that is stable in the reduced and in the oxidized state. For example, when DHTz was flanked with alkyl or phenyl groups, it had poor stability toward O2, but when Tz was flanked with carboxylic esters, it displayed poor stability toward H2O and other nucleophiles. A suitable balance was found in Tz that was flanked by 2-pyridine substituents: both the DHTz 27 and Tz 28 were stable enough in various organic solvents but also aqueous buffer conditions that contained serum. As such, the in situ oxidation of DHTz 27 involving a variety of photosensitizer dyes was studied. Of these, 4 μM of methylene blue catalyzed the complete oxidation of 21 μM DHTz to Tz in 200 s upon irradiation with 660 nm light, whereas under ambient light, the conversion was substantially slower and only half of DHTz was oxidized to 29 after 2 h. Instead of methylene blue (λmax 665 nm), Rose bengal (λmax = 550 nm) could also be used, for example to avoid spectral overlap with a sTCO-functionalized dye 29. As an alternative to light-induced oxidation, complete oxidation of DHTz 27 to Tz 28 was observed in 10 min by means of horseradish peroxidase (HRP) in the presence of 2 mM H2O2. Interestingly, other heme enzymes were not able to catalyze this oxidation, and without hydrogen peroxide, the oxidation by HRP occurred even faster (KM = 100 μM and kcat = 27 s–1). We note that in this case the enzyme-catalyzed reaction is the rate-determining step. Other chemical60−63 and electrochemical64,65 methods for oxidation have also been applied for the in situ formation of Tz but not in combination with a one-pot click reaction. For example, 3,6-dichlorotetrazine was used as an organocatalyst in an aerobic oxidative-catalyzed nitroso-Diels–Alder reaction between arylhydroxylamines and dienecarbamates, but a detailed treatment thereof is outside the scope of this review.66
Figure 7.

(A) Classical synthesis of 1,2,4,5-tetrazine (Tz, 26) by means of oxidation of dihydrotetrazine (DHTz, 25). (B) One-pot in situ oxidation-induced click chemistry activates DHTz 27 by forming Tz 28, which rapidly undergoes an IEEDA cycloaddition reaction with sTCO-dye 29 to form adduct 30.
Another [4 + 2] IEEDA cycloaddition reaction is the reaction between an ortho-quinone (e.g. 31) and a cyclooctyne (32) under formation of diketone 33 (Figure 8A) or between ortho-quinone 34 and olefin 35a/alkyne 35b to form bicyclo[2.2.2]octadiene diketone 36a or 36b (Figure 8B).67,68 Applications of this reaction in more complex settings will be treated below; here we focus on the model studies that were used in order to determine the rates, scope, and limitations of this conversion. Specifically, the model substrate 4-tert-butyl-1,2-benzoquinone 34 was used to determine the reaction rate constants with various strained alkenes and alkynes (Figure 8C). The difference for IEDDA of these two dienophiles with 4-tert-butyl-1,2-benzoquinone 34 is 1:110. In contrast, the difference in reactivity between TCO 37 and BCN 2 when it comes to IEEDA with tetrazines is 440:1. The reaction of quinone 34 with endo-BCN 38 is 2–3 times faster than exo-BCN 2 and is the fastest recorded reaction of a quinone with alkynes. As such, it can compete with nucleophiles such as Ac-Cys-OH, which reacts with 34 with rates that are in the same order of magnitude as BCN (unpublished results). However, by isomerization of the Z-bicyclo[6.1.0]nonene to E-alkene, under formation of exo-cpTCO 39, a highly strained system was obtained that resulted in an even faster strain-promoted oxidation-controlled cycloalkyne–quinone (SPOCQ), with rates constants of 2900 ± 115 M–1·s–1. An in-depth study on the mechanism of SPOCQ revealed that the observed differences in rates could be explained by the differences in ΔH‡ values for the reaction between 4-tert-butyl-1,2-benzoquinone and BCN 2, TCO 37, and DBCO 1 or 4.5, 7.3, and 12.2 kcal/mol, respectively.69 Computational investigations revealed that secondary orbital interactions enhance the reactivity of alkynes in IEDDA with ortho-quinones.70
Figure 8.

Strain-promoted oxidation-controlled quinone cycloaddition reactions (first example from 1979 in panel A, and more recent example from 2010 in panel B, including various strained olefins or acetylenes that have been used in recent years (C). Note: DCE = 1,2-dichloroethane, MeOH = methanol.
3. Materials
The observation that many click reactions proceed with near-complete conversion has triggered the application of this type of reaction in demanding settings. For the preparation or functionalization of advanced materials, two specific examples will be treated in detail: the preparation of hydrogels and polymers and the chemical modification of hard surfaces.
3.1. Hydrogels
The water-swollen networks of cross-linked hydrophilic polymers have given such hydrogels advantageous properties for (injectable) drug delivery, for the immobilization of biomolecules, and as a matrix for regenerative medicine. Most hydrogels are prepared prior to their application, although trigger-responsive elements have been incorporated for controlled release.71,72 However, for several applications such as injectables a spatiotemporal control of hydrogel formation is highly beneficial.
In order to work reliably, the formation kinetics of the polymers have to be very fast. This inspired the group of Van Hest to develop oxidation-induced click chemistry recently emerged as an approach to prepare activatable cross-linking hydrogels.73 For this, star-shaped PEG units (10 kDa) were functionalized with 3,4-dihydroxyphenylacetic acid (DHPA, in 40) or with BCN (in 41) (Figure 9). Upon in situ oxidation of the catechol to its ortho-quinone (2-(3,4-dioxocyclohexa-1,5-dien-1-yl)acetic acid, DOCA), a highly reactive diene was generated that rapidly performed a cycloaddition reaction to BCN-functionalized PEG units (Figure 9). In the presence of NaIO4 gel formation was almost instantaneous, and in the presence of mushroom tyrosinase (mTyr) the gelation time was 65 ± 8 s for 2000 units·mL–1 and increased with decreasing enzyme concentration to 1147 ± 100 s with 100 units·mL–1 (based on rheological measurements). The mechanical properties of the formed gel were comparable to other PEG-based hydrogels. That gelation was caused by the cross-coupling of BCN to DOCA was confirmed by very slow gelation when only DHPA-star-PEG was present (130 mg·mL–1, 18 min for NaIO4 and 2 days for 25000 units·mL–1 of mTyr). For comparison, gelation was much slower when the SPAAC reaction was used: when the DHPA-star-PEG was replaced with N3-star-PEG, gelation was slower with 20 min for 30 mg·mL–1 and 6 h for 10 mg·mL–1. Interestingly, when nonequimolar amounts of functionalized star-PEG units were applied, i.e., 0.8:1.0 of DHPA-star-PEG:BCN-star-PEG, an off-stoichiometric hydrogel was formed that contained remaining BCN functionalities (Figure 9, inset lower right). These could be derivatized with azide-functionalized dyes. Due to the substantially higher reaction rate for the SPOCQ reaction when compared to the SPAAC reaction, even a one-pot procedure could be applied in which BCN-star-PEG (1.0 equiv) and DHPA-star-PEG (0.8 equiv) were mixed, after which the N3-functionalized dye was added together with the oxidizing agent.
Figure 9.

Hydrogel formation as a result of the oxidation-induced SPOCQ reaction between DHPA-functionalized (red, 40), BCN-functionalized (black, 41), and star-PEG units. The bicyclo[2,2,2]octadienenone that connects the hydrogel monomers is highlighted. The inset (right bottom) shows the hydrogel that contains a SPAAC-linked dye on the BCN-functionalized ends that remained using nonequimolar amounts of BCN-star-PEG and DHPA-star-PEG.
3.2. Polymers
The group of Fox explored how their light or enzyme-activated tetrazine-based click reactions could be applied in polymeric materials (Figure 10).59 In a biphasic setup hydrophobic trifunctional molecule 42 that contains a latent dihydrotetrazine (DHTz) functionality and two sTCO moieties reacted at the biphasic interface with water-soluble bis-tetrazine monomer 43 under formation of a DHTz-functionalized polymer fiber. Upon long-wavelength photocatalytic generation of reactive Tz moieties with either methylene blue or rose bengal, sTCO-functionalized derivatives (44) containing fluorophores, peptides, and even entire proteins could be attached postsynthetically to the meter-long polymer fiber (45). The authors note that the HRP-induced conversion of DHTz to Tz also works, but with limited efficiency, which was most likely due to the heterogeneous system and the steric difficulty of the enzyme active site to interact with DHTz groups on the fiber. Functionalization via this click reaction of the fibers with RGD peptides stimulated adhesion of fibroblast cells that maintained a healthy morphology.59
Figure 10.
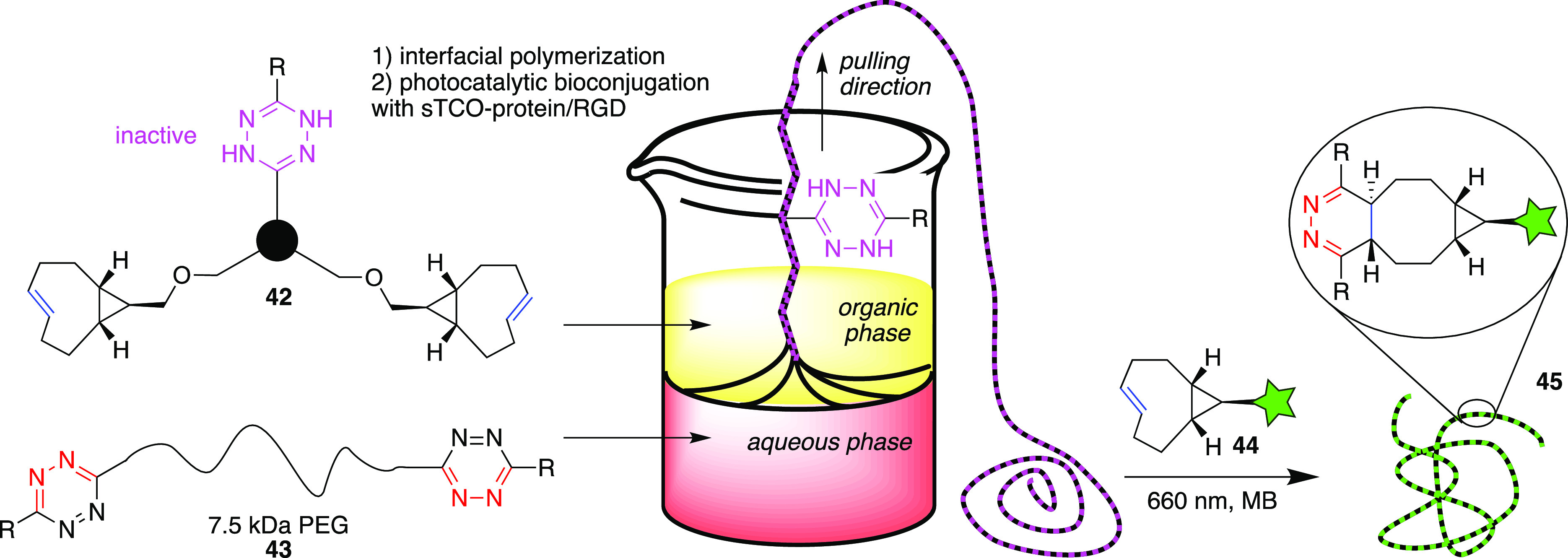
Schematic representation of interfacial polymerization with a dhTz-functionalized monomer with diTz-functionalized PEG and subsequent photocatalytic modification of the fiber with cpTCO-functionalized derivative 44.
Apart from hydrogel-forming and soft polymers, oxidation-induced click chemistry has also been applied for the synthesis of harder polymers such as polystyrene. By means of an appropriate design in the synthetic route, Boons et al. have prepared copolymers that contained three clickable functional groups: oxime, azide, and nitrones.74 All of these could be derivatized with DIBO derivatives, and of these, the oximes could be subjected to in situ oxidation with PIDA, after which these could be clicked to DIBO moieties by means of SPANOC. Kinetic analysis revealed that the SPANOC reaction was 20 times faster than the SPAAC reaction. Even more, a block copolymer that contained both azide and oxime groups in segregated blocks could be subjected to sequential SPAAC and SPANOC functionalization, resulting in polymers that self-assembled into well-defined nanostructures. In a later study, the same authors described how this in situ oxidative installation of a 1,3-dipole followed by the SPANOC reaction to a set of DIBO derivatives could be applied to end functionalization of polymers.75 A more in-depth treatment of the role of nitrile oxides and nitrones in polymer science can be found elsewhere.76
3.3. Surfaces
The heterogeneous nature that is associated with surface modification has made it difficult to achieve complete surface coverage. For example, most SPAAC reactions stall at 80%.77−79 For many applications, a high-yielding surface modification is required as unreacted surface-bound sites cannot be removed from the surface and can also not always be “neutralized” so that they do not interfere with the application of the surface. In pursuit of a method that led to complete surface coverage, Zuilhof et al. studied the applicability of the oxidation-induced SPOCQ click reaction (Figure 11).65 For this, a surface was coated with catechol moieties and subsequently oxidized to their corresponding ortho-quinones in order to perform the SPOCQ reaction.80 Whereas the rate constant for the solution-phase SPOCQ reaction is typically in the order of 500–1000 M–1·s–1, for this reaction at the interface a rate of 10–4 s–1 was measured, which was still 2-fold faster than the rate for the interfacial SPAAC reaction. It turned out that the ∼6 kcal mol–1 higher activation enthalpy for the SPOCQ reaction on the monolayer surface when compared to the SPAAC reaction was compensated by the entropy of activation. Furthermore, it was shown that the rates were influenced by the level of exposure of the quinone, with very exposed quinones reaction 3-times faster than more buried ones.
Figure 11.

Conversion of a catechol-functionalized surface by means of oxidation-induced SPOCQ reaction with BCN 46 (left) and cyclopropene 47 (right).
Quantitative modification of the monolayer was also obtained when BCN was replaced with a cyclopropene (cProp) moiety.81 It deserves to be mentioned that comparison of the rates of the SPOCQ reactions of BCN 46 or cProp 47 with immobilized quinones with their soluble counterparts revealed striking differences in the extent of reduction of the reaction. Whereas the cProp–quinone combination displayed a 4-fold lower reaction rate of the heterogeneous system when compared to the homogeneous reaction, this difference was 150-fold for the BCN–quinone combination. Apparently, the small size of the cProp moiety is advantageous in crowded environments as present on modified surfaces or polymer brushes and is better able to keep up with the rate of the reaction between soluble partners. Compatibility of this click reaction with other surface-bound click reactions, such as CuAAC and SuFEx, was also demonstrated.7
4. Bioconjugation Chemistry
Proteins are complex biopolymers that play pivotal roles in all living organisms. In order to unravel the role of proteins in relation to the inner workings of living systems, chemically modified proteins have proven particularly useful.82 In order to prepare the desired chemically modified proteins, site-specific modification of proteins has been developed so that uniform bioconjugates are obtained.
At the moment, many methods exist that provide access to the chemical modification of proteins with high precision. Many of these rely on the installation of a uniquely reactive amino acid residue in the protein structure, most notably on its surface.83 However, when it comes to the precision modification of native proteins for which bulk production has been established, e.g. enzymes and antibodies, direct derivatization of those biomolecules with moieties of interest is much more demanding.
Whichever route is taken, one general restriction applies to all proteins: their modification is performed under dilute conditions, usually in the mM−μM range, and performed at ambient conditions (pH 6–8, ≤40 °C, aqueous media). Therefore, fast kinetics are required to obtain high conversions and yields of bioconjugate products. This is even more so for the modification of endogenous proteins in biological systems.
Whereas uncontrolled oxidation of biomolecules such as proteins84,85 and DNA86 is undesired in most cases, a few residues can be oxidized in a more controlled manner,87 leading to derivatives that can be subjected to click chemistry modification. The methods that have been developed for proteinogenic amino acids, especially the sulfhydryl-containing cysteine (Cys, C) and the phenol-containing tyrosine (Tyr, Y), will be reviewed below. A few examples also emerged targeting the indole ring of tryptophan (Trp, W) and the thioether moiety of methionine (Met, M).88−91
4.1. Tyrosine
The ionizable phenol ring of the tyrosine (Tyr) side chain (pKa 9.7–10.1) in proteins (e.g. 48) has become a valued partner for tyrosine ligation reactions (TLRs) and oxidation-induced click chemistry approaches in the past decades.92,93 Not only is Tyr sufficiently exposed to allow chemical modification,94 its pH-dependent reduction potential facilitates modification with redox-active molecules (Figure 12). On top of this, the availability of chemical reagents and enzymes that can oxygenate one of the two ortho positions of Tyr allows it to be converted to the catechol functional group (as in 49) that has the ability to undergo additional electrochemical transformations (Figure 12).95,96 Specifically, this oxygenation of Tyr gives access to an energy level that facilitates further oxidation to an ortho-quinone (e.g. 50) that enables Michael addition and cycloaddition chemistry.
Figure 12.

Schematic depiction of the various oxidation pathways for protein-bound Tyr and its oxygenated DOPA derivative.
4.1.1. Single Electron-Transfer Oxidation of Substrates for Click Chemistry on Tyr
One of the earliest strategies to utilize Tyr redox chemistry for protein functionalization was the interior chemical modification of the MS2 viral capsid using a hetero-Diels–Alder cycloaddition.97 Using a site-selective diazonium coupling reaction with diazonium salt 51, the azo conjugate 52 of Tyr85 was prepared (Figure 13). This could be converted into ortho-iminoquinone 53 by a reduction–oxidation combination. After this, a hetero-Diels–Alder cycloaddition occurred in the presence of N-(4-aminophenyl)acrylamide 54, and only product 55 was observed. The authors specifically mentioned that the (4 + 2) cycloaddition product 56 was not identified. By controlling the pH of the various steps in the procedure, >60% functionalization of the capsid protein with the spontaneously oxidized product 57 was achieved in 4 h.
Figure 13.

Chemical modification of Tyr85 by means of diazonium reaction, followed by a reduction–oxidation conversion to iminoquinone 53, after which a hetero-Diels–Alder cycloaddition with acrylamide resulted in product 57.
A few years later, Barbas and co-workers introduced an aqueous ene-type conjugation procedure for Tyr residues.98 The resulting conjugate displayed a higher stability than Tyr-based conjugates that were obtained by earlier methods. As such, its applicability in the generation of more complex bioconjugates was demonstrated.99 Inspired by the high reactivity of diazodicarboxylate reagents toward substituted phenols,100 the redox-active cyclic diazodicarboxamide PTAD101 was developed for Tyr-specific bioconjugation reactions. Exposure of an equimolar mixture of N-acyl methyl amides of His, Trp, Ser, Cys, Lys, and Tyr revealed exclusive reactivity of Tyr, thereby revealing the chemoselectivity of the reaction for phenol residues.102 Interestingly, this ligation method for Tyr could be used in concert with SPAAC conjugation for the preparation of densely cross-linked protein-based hydrogels.103 Unfortunately, the highly reactive PTAD moiety decomposes to isocyanate byproducts that react with nucleophilic amines (e.g. Lys residues and N-termini in proteins), thereby restricting its scope.104 To counter this, the reduced form of PTAD (i.e., its triazolidine-3,5-dione derivative, PTAID) was applied. This nonreactive moiety enabled functionalization with the integrin αVβ3-binding cyclic RGD peptide, yielding latent construct 58. For conjugation to trastuzumab the PTAID-functionalized RGD peptide was subjected to in situ oxidation by means of NBS/pyridine and then applied to a phosphate-buffered solution of the antibody (Figure 14A). This resulted in clean conjugation product 59 where the RGD peptide was linked to exposed Tyr residues of the antibody. Alternatively, oxidation of PTAID could also be performed by 1,3-dibromo-5,5-dimethylhydantoin (DBH) and applied as a Tyr modification reagent.103
Figure 14.

(A) In situ oxidation of PTAID 58 and its subsequent conjugation to Tyr residues in trastuzumab. (B) Hemin-catalyzed oxidative coupling of N-methyl phthalic hydrazide 60 to angiotensin II (top) and photocatalytic oxidation of a peptidic Tyr residue, followed by its cross-coupling to the dimethylaniline-based radical transfer reagent (RTA, 62) (bottom).
Another means to overcome the instability of PTAD was developed by Sato and Nakamura. Instead of using PTAD, they resorted to the application of N-methyl derivatized PTAD and a range of luminol derivatives. Especially this latter set of derivatives proved useful. In the presence of hemin as a catalyst and H2O2 as oxidant, they found that N-methylated phthalic hydrazide (NMPH, 60) gave >95% conversion for the modification of the Tyr residue in angiotensin II (Figure 14B).105 For comparison, phthalic hydrazide itself modified 61% of the same peptide, and luminol modified even less, with only 21%. Apparently, the generation of a soluble radical that cross-couples to Tyr residues on proteins already facilitates a significant level of control over the precision of the conjugation chemistry. However, better control was achieved when the radical was generated on the protein surface. By using photocatalyst 61 the oxidation of exposed Tyr residues to their corresponding tyrosyl radicals (see Figure 12) resulted in short-lived active species (20 ms) that rapidly cross-linked to a radical transfer agent (RTA, 62), also known as tyrosyl radical trapping (TRT) agents (Figure 14B). Especially when the ruthenium-based photocatalyst was bound to a protein ligand, the modification was restricted to the periphery of the protein–ligand interface106,107 and not dictated by the reactivity of the most accessible residues toward a soluble radical. As such, this ligand-directed protein modification offered specific advantages over methods that activate the soluble label to initiate the oxidative cross-coupling. Due to the cell permeability of a Ru(bipy)3 complex and TRT reagent, the method was tested in intact erythrocytes, and intracellular target-selective protein modification was shown. In a recent study this chemistry was extended to immunohistochemistry applications.108
After the basic chemistry was established using peptides, the shift to proteins was made. Sato and Nakamura also showed for these much more complex biomolecules that in situ activation of functionalized N-methyl luminol (NML) derivatives by means of hemin (redox potential: −0.35 V) and H2O2 led to substantially cleaner modification of Tyr residues when compared to the application of PTAD.105 When hemin was replaced with the heme-containing enzyme horseradish peroxidase (HRP; oxidation potential 1.1 V109), Tyr modification efficiencies were boosted substantially so that the amount of catalyst that was needed to obtain >95% conversion could be decreased from 10% (for hemin) to 0.1% for HRP (Figure 15).110 Importantly, self-modification of HRP was not observed, probably due to the hidden nature of the Tyr residues that are present in this enzyme. Also, (uncontrolled) polymerization by competing Tyr-Tyr cross-coupling reactions that were previously observed111−115 was not observed in these experiments. Experiments with NADH as activating agent revealed that HRP activates NML and not protein-bound Tyr residues. Whereas the method based on hemin required an excess of H2O2, which caused substantial unwanted Cys and Met oxidation and protein aggregation, the application of HRP allowed for a reduction of the concentration of H2O2 and resulted in cleaner conversions. As a result, more complex proteins such as antibodies were successfully subjected to the optimized modification conditions.116 Indeed, also for these large proteins, modification of only the most exposed residues was observed. Even though modification was observed for a Tyr residue in the complementarity-determining region (CDR) (i.e., Tyr57) of the antibody trastuzumab, this did not hamper antigen-binding nor its biological activity. When an antibody that contained more Tyr residues in the CDR was taken, i.e. rituximab (contains five Tyr residues in the CDR), four of these were modified. Remarkably, also this did not hamper its CD20 affinity.
Figure 15.

Enzymatic oxidation-induced click reaction of NML-derivatives to protein Tyr residues.
Very recently the nonheme copper enzyme laccase (oxidation potential: 0.8–1.0 V109) was discovered as a powerful catalyst for the tyrosine click reaction (Figure 15).117 The most important feature of this reaction is its use of O2 as oxidizing agent and that efficient Tyr modification was detected and no modification of other residues could be detected. In a one-on-one comparison to HRP, conversions with laccase were substantially better (i.e., 75% vs 95%), although more double-modified Tyr was detected when laccase was used (i.e., ratio of mono:bis = 1:0.06 vs 1:0.4, respectively).
Apart from these biocatalytic methods for the activation SET-mediated Tyr modification, electrochemical methods were also described. For example, a so-called “e-Y-click” method using urazoles and a potential of 0.36 V led to near complete modification of multiple Tyr residues in peptides and proteins, for example with carbohydrates (Figure 16).118 This method was also compatible with a two-step labeling procedure in which the PTAD derivative contained an azide that could be conjugated to a fluorescent dye by means of SPAAC.
Figure 16.

Electrochemical activation of urazole species in situ (e-Y-click). Reproduced from the Journal of the American Chemical Society.118 Copyright 2018 American Chemical Society.
Although enzymes are usually unmatched in their catalytic potency, some of their functions can be imitated successfully with artificial constructs. One of the most studied mimics of horseradish peroxidase (HRP) is the hemin/G-quadruplex (hGQ) DNAzyme complex (Figure 17A).119 Although the redox potential hemin in a hGQ complex is similar to that of hemin (i.e., −0.35 V, for hemin bound to the telomeric G-quadruplex [AGGG(TTAGGG)3] repeat), the electrocatalytic ability of hemin toward H2O2 is significantly enhanced by the presence of the GQ nanostructure.120 This is most likely due to the participation of neighboring groups in the formation of the catalytically active complex (i.e., compound I, Figure 17B).121 The many GQ structures that are available and the ease by which they can be modified make this an ideal platform to be applied in demanding protein modification settings.122 Indeed, Keijzer123 and Masuzawa122 recently showed that hGQs have the ability to efficiently modify proteins with an NML derivative 63. Whereas others have shown that, in the absence of labels that can be cross-coupled to Tyr, Tyr-Tyr cross-coupling124 or self-labeling of DNA can occur,125 this was not seen when NML derivatives were applied. In the presence of NML and H2O2 most hGQ structures reached full conversion in 15–30 min, and depending on the type of GQ different site-specificity of the modification was observed. Lastly, a trigger-responsive element was introduced, so that the protein modifying ability could repeatedly be switched ON and OFF.
Figure 17.

(A) Schematic depiction of the protein modification by means of hGQ DNAzymes and NML derivatives, including a model of the hemin/G-quadruplex DNAzyme derived from PDB-code 6PNK. (B) Activation of the hGQ DNAzyme by means of H2O2 and a neighboring adenine base.
Whereas these SET approaches allow radical-based oxidation-induced click reactions on Tyr, two-electron transfer and four-electron transfer oxidations facilitate alternative bioconjugation reactions. Most notably, depending on the substrate that is subjected to these conditions, access is obtained to very fast IEEDA cycloaddition reactions.
4.1.2. Click Reactions Induced by Two-Electron Transfer Oxidations
The two-electron oxidation of exogenous catechol derivatives to yield ortho-quinones facilitates a click reaction to BCN-functionalized proteins. This was first described using 3,4-dihydroxyphenylacetic acid (DHPA) containing peptide 64 (Figure 18).126 This oxygenated Tyr derivative can be converted into its corresponding ortho-quinone by means of a two-electron transfer oxidation. The reaction with BCN-functionalized biotin 65 formed two isomers of the product 66 and even a small amount of the decarbonylated retro-Diels–Alder aromatic product. The observed efficiency of the conjugation reaction opens it up for in-depth bioconjugation studies.
Figure 18.

Two-electron oxidation of catechol 64 to its corresponding quinone, and its SPOCQ with BCN-functionalized biotin derivative 65, resulting in product 66.
Under appropriate conditions the oxygenation of a phenol can be halted at the stage of the catechol. In this, only the oxidation state of one carbon atom next to the aromatic OH group is increased (Figure 2). Interestingly, L-DOPA derivatives can be generated on protein structures by means of enzymatic oxidation of Tyr by tyrosinase or by means of genetic incorporation of L-DOPA, although this last method may suffer from protein misfolding and lower titers.127 When proteins that contain an exposed Tyr residue are treated with tyrosinase in the presence of ascorbic acid (HAsc), the oxidation is halted at the L-DOPA state (these conditions limit the diphenolase tyrosinase to its monophenolase activity). The generated catechol moiety can be subjected to “click chemistry” to boronic acids, although we apply a stricter definition in this review.128
The high rates for this reaction also made it possible to expose a BCN-functionalized protein 68 to the 1,2-quinone-functionalized dye 69 (Figure 19), even though ortho-quinones such as 70 readily react with naturally occurring nucleophiles such as amines (Lys, N-terminus) and thiols (Cys). Nevertheless, full and clean labeling of BCN-protein 67 to the SPOCQ product 71 was obtained (Figure 19A).126 The time-resolved SPOCQ conjugation with the oxidized lissamine rhodamine B label 72 is shown in Figure 19B. It also shows that the reduced form of this label, i.e. 73, does not perform the labeling reaction and that labeling with azide-functionalized lissamine 74 is not only substantially slower but also fails to lead to completion, even though a catechol-azide containing bifunctional linker 75 could be used for the preparation of dimers (Figure 19C).
Figure 19.

(A) NaIO4 oxidation-induced activation of a substrate (10 mM, 4 equiv) and its subsequent SPOCQ to a BCN-functionalized protein (20 μM). (B) SPOCQ conjugation of the BCN-protein with the oxidatively activated lissamine rhodamine B fluorophore. (C) Dimerization of the BCN-functionalized protein with bifunctional linker 75. Reproduced from Bioconjugate Chemistry.126 Copyright 2015 American Chemical Society.
Genetic incorporation of L-DOPA into the protein provides the protein with a selectively oxidizable handle, effectively inverting the reaction partners shown in Figure 19. In a method developed by Lee et al. in 2018, L-DOPA 76 is biosynthesized in E. coli from catechol 77 (8–10 mM), pyruvate 78 (100 mM), and ammonia (25 mM) and directly incorporated into their target protein (Figure 20).129 Mild oxidation by NaIO4 in the presence of DBCO-Cy5.5 dye 79 resulted in the generation of a protein–Cy5.5 conjugate 80 by means of SPOCQ. Apparently, the competing DOPA oligomerization observed by others130 is outcompeted by the high rate of SPOCQ.
Figure 20.

Biosynthesis of L-DOPA 76 by tyrosine phenol-lyases (TPL) from catechol 77, pyruvate 78, and ammonia, followed by its genetic incorporation by means of an evolved aatRNA and aaRS pair. The DOPA-derivatized protein was subjected to a SPOCQ with Cy5.5-DBCO 79 in the presence of sodium periodate.
4.1.3. Click Reactions Induced by Four-Electron Transfer Oxidations
In the presence of tyrosinase alone, i.e. without the reducing agent ascorbic acid, phenol derivatives are oxidized to their corresponding quinone, an increase in the oxidation state of two neighboring oxygen-containing carbon atoms (Figure 2). Alternatively, DOPA derivatives can be oxidized by catechol oxidase (diphenolase activity)131 or by mild oxidizing agents such as NaIO4 (vide supra).129 The resulting ortho-quinone derivatives can be applied for a variety of bioconjugation chemistries.132−134 The most notable of these are the Michael addition reaction of most nucleophiles to the carbon atom at the δ-position of the amino acid,135−137 although a recent example emerged where it was shown that Cys residues attach to the carbon atoms at the ε-position (the resulting Cys-Tyr protein–protein conjugates were quantitatively formed in 2 h).138
Alternative to the classic Michael-addition type of conjugation reactions, the tyrosine-derived ortho-quinone was recently uncovered as a potent partner in cycloaddition chemistry. Due to the significantly lower orbital energy levels of the quinone when compared to the phenol group (see Figure 2), reaction rates are achieved that are 3 orders of magnitude higher compared to SPAAC (k < 2 M–1·s–1).
The concept of switching SPOCQ partners between the protein and the functional label is shown in Figure 21, i.e. by generating the ortho-quinone on the protein and having the dienophile on the label.139 In the presence of tyrosinase, a deliberately positioned genetically encoded exposed Tyr residue was oxidized to the corresponding ortho-quinone to create an electron-deficient diene on the surface of the protein, thereby facilitating the IEDDA cycloaddition with an electron-rich alkyne. Using a hyperthermostable endo-β-1,3-glucanase laminarinase A (LamA, 32 kDa) that contained a C-terminal tetraglycyltyrosine extension, a one-pot procedure was developed in which treatment with BCN-lissamine 81 (4 equiv) in the presence of mushroom tyrosinase (mTyr, 7.5%) resulted in full protein labeling after 30 min. In the absence of the G4Y-tag, no modification was observed.
Figure 21.

SPOCQ labeling of G4Y-tagged laminarinase A by reaction of BCN-modified reagent 81 with in situ generated 1,2-quinone on the protein laminarinase A. Reproduced from Bioconjugate Chemistry.139 Copyright 2017 American Chemical Society.
For a much larger protein, trastuzumab (Tras, 150 kDa) was selected and oxidation-induced SPOCQ was performed on the antibody that contained a G4Y-tag on the C-terminus of the light chain (i.e., Tras[LC]G4Y). In this case, however, some nonlabeled product remained, due to thwarted accessibility by the strained dienophile, allowing competing Michael addition from a neighboring nucleophilic residue. More recently, this chemistry was extended to prepare monofunctionalized knob-in-hole antibodies and antibody dimers.140 The application of asymmetric antibodies facilitated efficient derivatization of the C-terminus of the heavy chain with large structures such as proteins by means of SPOCQ chemistry.
To counter the competition of the Michael addition, Bruins et al. applied cpTCO-functionalized dyes. In a head-on comparison between exo-cpTCO–OH 39 and endo-BCN–OH 2, the first reacted three times faster with a model quinone than the latter (2900 M–1·s–1 vs 1112 M–1·s–1; see Figure 8).141 With this reactive moiety in hand, it was possible to reduce the quantity of oxidative undesired cross-linked products by replacing BCN-lissamine with cpTCO-lissamine and it was concluded that the oxidation of the exposed Tyr by means of mTyr was the rate-limiting step. We note that, even though the SPOCQ cycloaddition is among the most rapid click reactions that can be performed on enzymatically generated dienes on proteins, the presence of nucleophilic functionalities inside a cell prevents in vitro applications of this ligation chemistry.
Alternative to the application of NaIO4 or the enzyme tyrosinase, Tyr can also be oxidized to its corresponding ortho-quinone by means of Fremy’s salt (i.e., ON(SO3K)2, 82).142,143 Recently, this chemo-oxidation reaction was described in the context of protein bioconjugation chemistry to perform strain-promoted oxidation-controlled quinone click reactions with BCN and DBCO, such as 83 (Figure 22).144 For the first step, the oxidation of Tyr to its quinone, the chemical oxidation was 6-fold faster than the enzymatic version (i.e., 0.57 M–1·s–1 vs 3.16 M–1·s–1). Since oxidation is the rate-limiting step in the overall SPOCQ process, it might be beneficial in certain cases to apply this chemical method for Tyr oxygenation and oxidation, although undesirable oxidation of other amino acid side chains could be an issue.
Figure 22.

Schematic representation of SPOCQ on protein tyrosine residues by means of Fremy’s salt 82 and DBCO-functionalized dye 83.
It is clear that oxidation-induced click reactions on tyrosine have become a powerful and multifaceted tool in the field of bioconjugation chemistry. These methods complement approaches that utilize the unique nucleophilic reactivity of the Tyr side chain for protein conjugation chemistry: sulfur-fluoride (and its derivatives) exchange (SuFEx),145 Mannich-type electrophilic aromatic substitution,146,147 alkylation with π-allyl complexes,148 and covalent complexation with metal complexes.149,150 Altogether, Tyr has emerged besides Cys and Lys as the third-most important proteinogenic residue for controlled chemical modification.
4.2. Tryptophan
Apart from the oxidation-mediated functionalization of Tyr, of the other three aromatic residues (i.e., His, Phe, and Trp) only Trp has been subjected to oxidative modification. Specifically, the chemoselective oxidative Trp modification has been achieved with 9-azabicyclo[3.3.1]nonane-3-one-N-oxyl (keto-ABNO).151 As this method obeys many of the characteristics associated with oxidation-induced click chemistry, we include it in this review. Following a serendipitous finding that showed modification of Trp with keto-ABNO in the presence of CuI/NOx,152 optimized conditions that contained 1 equiv of keto-ABNO 85, 0.6 equiv of NaNO2, and 0.1% AcOH resulted in 95% modification of Trp residue 84 in 30 min under ambient conditions (Figure 23). Under these conditions keto-ABNO 85 was oxidized to its corresponding oxoammonium cation 86, which reacted as an electrophile with carbon-3 of the indole ring of Trp 84. Hydration resulted in a stable end-product 86 that was observed by LC-MS, as confirmed by the crystal structure of the Trp-modified lysozyme. The keto group in the conjugated product was successfully subjected to a second modification reaction with hydroxylamine. Other elegant methods for the modification of Trp, but that do not classify as click chemistry, can be found elsewhere.153−158
Figure 23.

Metal-free oxidation-induced modification of Trp 84 with keto-ABNO 86.
4.3. Thiol-Containing Residues
Both sulfur-containing canonical amino acid residues can be used for oxidation-induced chemical modification. Of these two, cysteine (Cys, C) is the most-applied residue for protein conjugation chemistry. It is mostly modified via classical alkylation or disulfide bond-forming strategies or via desulfurization approaches that yield dehydroalanines that have unique reactivity for cross-coupling conjugation chemistry, but some methods rely on the oxidative modification of Cys. The other sulfur-containing residue, methionine (Met, M), has recently also been subjected to oxidative modification that has been referred to as click chemistry.
4.4. Cysteine
The sulfhydryl group of cysteine can easily be oxidized. Methods to study the rich chemical biology that is associated with the eight distinct oxidation states (from −2 to +6) of sulfur is extensively reviewed elsewhere.159,160 In short, the oxidative post-translational modifications (oxPTMs) of Cys involve reactions with reactive sulfur species to yield persulfides and disulfides, with reactive oxygen species to form sulfenic acid, sulfenic acid, and sulfonic acid, and with reactive nitrogen species to form S-nitrosthiols and sulfonamides. The difference in the biochemistry of these various stages of Cys oxidation has been the driving force behind the development of probes that, ideally, react with only one of those oxPTMs in order to study their biological function (i.e., sulfenomics161). Alternatively, the specific oxidation states of Cys are harnessed for immunological techniques.162 Of these various oxidized Cys residues sulfenic acid has proven to be the most suitable residue for oxidation-induced click chemistry ligation approaches, and it complements the ligation techniques that use the sulfhydryl of Cys.163
The mono-oxidized form of Cys, its sulfenic acid (see Figure 2), is a key regulator for protein function, catalysis, and signaling. Often, Cys sulfenic acid represents an early oxidation product of the reaction of Cys with reactive oxygen species and reactive nitrogen species and can be considered as a biomarker for oxidative stress. These residues are formed through the oxidation of the thiolate side chain by H2O2.164 Apart from the importance of the compounds acidity for this reaction, evidence suggests that a suitable microenvironment can enhance the reactivity 106-fold.165 Sulfenic acid exists in two tautomeric forms: proton on sulfur [R-SH(=O)] or on oxygen [R-S-OH], the latter of which is favored (Figure 24).166 In Cys sulfenic acid, the S-atom exhibits both electrophilic and weak nucleophilic character and it can react with a variety of probe molecules. Also, it can react with the backbone amide nitrogen to form a cyclic sulfonamide167 or with an adjacent thiol to form an intramolecular disulfide,168 thereby preventing further oxidation to sulfinic and sulfonic acid.
Figure 24.

Oxidation of Cys residues to its corresponding sulfenic, sulfinic, and sulfonic acids, with emphasis on the derivatization of the sulfenic acid moieties by means of nucleophilic attack or cycloaddition reactions.
Many sulfenic acids are thought to be short-lived species, thereby imposing serious demands on the reactivity of the probe molecules. The earliest 1,3-diketone probes (e.g. dimedone or 5,5-dimethyl-1,3-cyclohexanedione)169 displayed reasonable reactivity (k ∼ 0.8 min–1) despite being more reactive at lower pH values. At physiological pH values, linear 1,3-diketone probes display an improved reactivity (k ∼ 3.8 min–1), even though this is still slow compared to the rate by which sulfenic acids are sometimes formed (depending on the protein microenvironment the rate of oxidation by H2O2 can vary from 1–108 M–1·s–1).170 A quest for more reactive probes that are still chemoselective for sulfenic acids was initiated by several researchers.
The dual, tautomer-induced reactivity of sulfenic acids with on the one hand nucleophiles and on the other hand unsaturated systems via a concerted cycloaddition mechanism to give sulfoxide adducts171 yielded probe molecules that displayed a high reactivity toward strained alkenes and alkynes (Figure 25).172 Specifically, the reaction between Cys sulfenic acid and BCN proceeded with a rate that is 2 orders of magnitude higher than with dimedone (k ∼ 28.7 ± 0.6 M–1·s–1 for BCN-OH 2 and the fluorogenic sulfenic acid derivative of anthraquinone Fries acid, vs 0.05 M–1·s–1 for a dimedone-based probe with model protein sulfenic acids) (Figure 25A). In comparison, trans-cyclooctene 88 is five times less reactive than dimedone (k ∼ 0.01 M–1·s–1) (Figure 25B). The reactivity of BCN to azides and sulfenic acids has inspired the application of bifunctional BCN derivative 89 for the imaging and enrichment of Cys sulfenic acids in biological samples (Figure 25C). One bifunctional linker 89 has been reacted with one equivalent of sulfenic acid, allowing the other strained alkyne on 90 to be subjected to a subsequent SPAAC conjugation. A major disadvantage of the application of BCN, however, is that it also reacts with nucleophilic thiols,173 although this depends on the reactivity of the Cys and is not as rapid as the reaction of Cys with DBCO174 or the reaction of BCN with persulfides (k ∼ 19 M–1·s–1).175 One major benefit of strained alkenes/alkynes over dimedone probes is that the former do not react with sulfenamides.176 By applying moderately strained alkenes such as norbornenes (91) under formation of sulfoxide adduct 92, a balance between high reactivity and high selectivity was struck that allowed bypass of off-target reactions (Figure 25D).177 More recent developments were focused on the application of norbornene probes such as 93 and 94 in cells (Figure 25E).178
Figure 25.

(A and B) Strained alkyne 2 and alkene 88 reacting with sulfenic acids under formation of covalent adducts. (C) Biscyclooctyne probe 89 reacting with protein sulfenic acids to install BCN moieties on Cys-OH in the form of 90. (D) Norbornene derivatives 91 reacting with short-lived sulfenic acids to form adduct 92. (E) Two norbornene probes for cysteine sulfenic acid detection, one containing an alkyne (93) and the other a biotin (94). (F) Application of SAM-TCO probe 95 in the labeling of biological sulfenic acid derivatives via the highly reactive thiiranium ion 96.
For a long time, dimedone-derived probes offered the best selectivity for studying cysteine sulfenic acids in cells. By means of a systematic variation of dimedone-inspired carbon-centered nucleophiles, highly reactive probes were identified that displayed interesting cellular labeling.179 In this search, it was found that with less strain, norbornene was more selective than cyclooctynes.177,179 The straightforward synthesis and modification of the norbornene core facilitates additional functionalization with handles that enable follow-up experimentation. This enabled cellular evaluation of the occurrence of sulfenic acid in cell lysates and cells.178 In a head-to-head comparison between norbornene and dimedone different labeling profiles indicated a higher selectivity of the former. Tests in living HeLa cells that were treated with norbornene probe 94 for 2 h at 37 °C, after which the cells were incubated for 2 h with H2O2, showed that labeling increased with increasing concentrations of H2O2. Proteomic analysis of the labeled proteins revealed 148 new protein members of the sulfenome, evidently showing that these norbornene-based probes are complementary to previously reported probes.180
One of the latest developments utilizes the reactivity of sulfenic acid modifying trans-cyclooctenol (SAM-TCO) derivative (such as 95) that (i) contains a strained E-alkene that reacts with sulfenic acid to form [3.3.1]oxabicycle 97 after intramolecular nucleophilic attack and ring opening of 96, (ii) facilitates in situ quench of the remaining SAM-TCO 95 with a methyltetrazine (MeTz) derivative, and (iii) contains an alkyne as handle for secondary labeling of the reacted sulfenic acid-containing proteins by means of CuAAC to form biotin-labeled product 97, for example.181 The combination of the high in vitro reaction rate of 95 and sulfenic acids of 750 M–1·s–1 and the even higher reaction rate constants of the IEEDA between 95 and MeTz enable time-resolved chemoproteomic profiling of sulfenylation in cell lysates.
The higher oxidized forms of Cys, i.e. its sulfinic acid (pKa ∼ 2) and sulfonic acid (pKa < 2), exhibit decreased reactivity in comparison to the lower oxidation states, and only a handful of chemical probes have been designed for conjugation to these moieties.158 Although electrophilic aryl nitroso compounds reacted with sulfinic acids—and the labile N-sulfonyl hydroxylamine could be intramolecularly trapped as N-sulfonylbenzisoxazolone—their high reactivity to thiols limits their applicability to samples in which sulfhydryl groups are first blocked with alkylating agents such as iodoacetamide or N-ethyl maleimide. Better selectivity was obtained by reaction with S-nitrosothiols, but since the thiosulfonates that are formed readily exchange, free thiols must first be alkylated with iodoacetamide.182 Sulfinic acids also react with N-ethyl maleimide, and the products are only stable at pH < 6, which limits practical applications for bioconjugation purposes.
4.5. Methionine
Although methionine can be oxidized to its sulfoxide, this product is rather inert and has not been used for click reactions. Inspired by the reaction of Met with cyanogen bromide, alkylation of Met was tuned so it could be applied on polypeptides.183,184 Alternatively, a hypervalent iodine reagent was developed by the group of Gaunt and was found to yield fast, selective, and click-like bioconjugation products that enabled secondary protein functionalization strategies.185
Recently, a robust method for the selective modification of Met by means of an oxidative sulfur imidation reaction with oxaziridine derivatives 98 was reported (Figure 26).186 The method not only worked for the modification of Met residues in isolated proteins (with k = 18.0 ± 0.6 M–1·s–1) but also facilitated the identification of hyperreactive Met residues in whole proteomes. Depending on the presence of water, preferential formation of the N-transfer product 99 was observed over O-transfer-product 100. Recently, this chemistry was also employed for the functionalization of peptides and proteins with 18F.187
Figure 26.

Oxidative Met derivatization with oxaziridine 98 leading to functionally derivatized N-transfer product 99.
4.6. Oxidative Installation of Click 1,3-Dipoles
So far, we have covered approaches in which oxidation activates one of the two reaction partners for the click reaction. Below, we briefly mention two approaches in which mild oxidation is used to introduce a moiety in proteins that facilitates introduction of a clickable nitrone moiety in a one-pot procedure.
4.6.1. N-Terminal Serine
In the absence of a functional proteinogenic side chain, an N-terminus-positioned serine (Ser) or threonine (Thr) residue can be used for the oxidative modification of proteins.188,189 Alternatively, (protein-bound) sialic acids have been used for oxidative modification (vide infra).190 When an N-terminal Ser or Thr is not present, pyridoxal phosphate (PLP) can be used to oxidize the Cα atom of the terminal amino acid to its corresponding aldehyde. This moiety possesses unique reactivity toward nucleophiles and allows the introduction of clickable moieties. However, this approach is more abundantly applied for the installation of additional functionalities by means of oxime or hydrazone ligation (k < 101 M–1·s–1 and k < 102 M–1·s–1, respectively), using the small family of Pictet–Spengler ligation reactions (i.e., normal Pictet–Spengler reaction,191 the iso-Pictet–Spengler reaction with k ∼ 10 M–1·s–1 at pH 4.5,192 and the hydrazino-iso-Pictet–Spengler ligation with k ∼ 4.2 M–1·s–1 at pH 6193), and the trapped Knoevenagel ligation (k = 0.39 M–1·s–1).194 As such, the pyrodoxal phosphate method resembles the application of formylglycine (fGly)—which is generated by the chemoenzymatic oxidation and hydrolysis of a Cys residue in the consensus sequence Cys-Xaa-Pro-Xaa-Arg (where Xaa is a random amino acid residues) that is targeted by the Formyglycine Generating Enzyme (FGE)—as a handle for protein modification.195
The glyoxyl-terminated protein that is generated by the chemical oxidation of an N-terminal Ser residue has provided direct access to SPANC modification. In a one-pot, three-step procedure, the N-terminal serine residue of interleukin-8 (IL-8, 101) was oxidized by NaIO4 to its aldehyde, which then could be converted into a nitrone by treatment with a cocktail of p-MeOC6H4SH, MeNHOH·HCl, and p-anisidine (Figure 27A).196 This introduced a SPANC handle into the protein, which then could be derivatized with a moiety that contained DBCO 102, yielding PEGylated IL-8 103.179 This method also worked in a heterogeneous system where anti-HER2 scFv 104 was conjugated to nanoparticles (NPs) that were coated with a BDCO-functionalized polymer, yielding functionalized hybrid materials that were able to target HER2-positive breast-cancer cells (Figure 27B).197 Interestingly, the original procedure caused unwanted nanoparticle agglomeration, which was avoided by adjusting the reaction conditions slightly. By adding the DBCO-functionalized NPs to in situ-formed nitrone, NP aggregation was avoided and the coated NPs 105 were obtained. Due to the mild oxidation conditions that were used, SPANC could be combined with subsequent azide–alkyne click chemistry in order to facilitate dual functionalization.198
Figure 27.

SPANC derivatization of proteins by attachment of a PEG unit (A) or to a nanoparticle (B).
4.6.2. Protein-Bound Sialic Acids
Although proteinogenic amino acids offer various entries into oxidation-induced click reactions (vide supra), other protein-bound functionalities can be utilized for this chemistry as well. Inspired by the application of sialic acids on glycoproteins,199 Boons et al. developed a method for the one-pot oxidation-induced SPANOC reaction to derivatize these carbohydrates.40 Specifically, the sialylated protein fetuin was treated with NaIO4 for 5 min, after which the resulting aldehyde was converted into an oxime by means of exposure to hydroxyl amine. Following a short oxidation treatment with PIDA, the resulting nitrile oxide was clicked to a DIBO-functionalized biotin.
5. Conclusions and Outlook
The applicability of click reactions is largely determined by the combination of scope, efficiency, ease of use, and the potential to avoid competing chemistry. In this regard, a particularly appealing chemical approach involves the augmentation of reactivity, which is induced within a mixture of chemical components. By careful design of reagents, such activation can be readily achieved via the mild and controlled in situ oxidation of one of the components. This review has outlined the rapid rise of such oxidation-induced “one-pot” click reactions and its enormous potential to be applied in fields as diverse as bioconjugation, hydrogel formation, and surface modification. As the field has not yet reached a mature status, its true value is expected to become more apparent in forthcoming studies, both at a fundamental level and in further applications. In particular, the identification of oxidative conditions that are fully compatible with the integrity of the various components that are involved in the click reaction, i.e. develop “oxidation-orthogonal click reactions”, is most important to bring this field to an even further fruition.
Acknowledgments
This work is funded by the ECHO grant from the Dutch Organization for Scientific research (NWO) (project number 711.017.004).
Biographies
Bauke Albada obtained his Ph.D. degree from the Utrecht University (2009) after which he performed postdoctoral studies at the Ruhr University of Bochum (Germany) and The Hebrew University of Jerusalem (Israel). Since 2016, he is an assistant professor in the Laboratory of Organic Chemistry (Wageningen University & Research, The Netherlands), where his group works on various projects in the bio-organic chemistry domain and primarily focuses on the development of new methods for the site-specific modification of proteins and approaches to perform unclicking reactions. He is (co)author on >60 peer-reviewed publications, and his research has been awarded with several fellowships and grants.
Jordi Keijzer was born in Zwijndrecht, The Netherlands, in 1993. He graduated from Leiden University and TU Delft in 2014, earning a bachelor/s degree in Molecular Science and Technology. Continuing at Leiden University, he received his master’s degree in Chemistry in 2016, specializing in Chemical Biology under the supervision of Prof. Mario van der Stelt and Prof. Hermen Overkleeft. Currently, he is employed as a Ph.D. candidate at the Organic Chemistry Group of Prof. Han Zuilhof at Wageningen University & Research Centre. He works with Dr. Bauke Albada and Prof. Floris van Delft in the field of bioconjugation to develop novel methods for selective modification of native proteins using trigger-activated catalytic DNAzyme nanostructures.
Han Zuilhof is the Chair of Organic Chemistry at Wageningen University (Wageningen, The Netherlands), Perennial Distinguished Guest Professor of Molecular Science at Tianjin University (Tianjin, China), and Distinguished Adjunct Professor of Materials Chemistry at the King Abdulaziz University (Jeddah, Saudi Arabia). He is a Senior Editor of Langmuir, an Editorial Advisory Board member of Advanced Materials Interfaces, Applied Surface Science, and Cell Reports, Physical Science. His interests focus on surface-bound organic chemistry and the accompanying analytical chemistry to properly characterize that and on novel supramolecular materials. He likes all of China, but Guizhou is his favorite province.
Floris van Delft graduated in organic chemistry from Leiden University (1996, cum laude) in The Netherlands. After a postdoctoral stay at the Scripps Research Institute (San Diego, USA), he has held assistant/associate professorship positions at the University of Amsterdam and Radboud University Nijmegen, as well as a special professorship at Wageningen University in The Netherlands (2015–2020). During his academic career, his research was focused primarily on click chemistry, carbohydrate chemistry, and protein conjugation technologies. In 2010, Floris was a cofounder of Synaffix, of which he became the full-time chief scientific officer (CSO) in 2014. He has been the driving force behind the invention of the proprietary technologies that now form the basis of Synaffix’s best-in-class antibody-drug conjugates (ADCs) for targeted cancer therapy. Floris has (co)authored >150 peer-reviewed publications and is the (co)inventor of >25 patent applications.
The authors declare the following competing financial interest(s): FVD is an employee of Synaffix BV.
References
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Kalgutkar A. S.; Gardner I.; Obach R. S.; Shaffer C. L.; Callegari E.; Henne K. R.; Mutlib A. E.; Dalvie D. K.; Lee J. S.; Nakari Y.; O’Donnell J. P.; Boer J.; Harriman S. P. A comprehensive listing of bioactivation pathways of organic functional groups. Curr. Drug Metab. 2005, 6, 161–225. 10.2174/1389200054021799. [DOI] [PubMed] [Google Scholar]
- Meldal M.; Tornøe C. W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 2008, 108, 2952–3015. 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46, 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Barrow A. S.; Smedley C. J.; Zheng Q.; Li S.; Dong J.; Moses J. E. The growing applications of SuFEx click chemistry. Chem. Soc. Rev. 2019, 48, 4731–4758. 10.1039/C8CS00960K. [DOI] [PubMed] [Google Scholar]
- Gao B.; Zhang L.; Zheng Q.; Zhou F.; Klivansky L. M.; Lu J.; Liu Y.; Dong J.; Wu G.; Sharpless K. B. Bifluoride-catalysed suflur(VI) fluoride exchange reaction for the synthesis of polysulfates and polysulfonates. Nat. Chem. 2017, 9, 1083–1088. 10.1038/nchem.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahtory D.; Sen R.; Pujari S.; Li S.; Zheng Q.; Moses J. E.; Sharpless K. B.; Zuilhof H. Quantitative and orthogonal formation and reactivity of SuFEx platforms. Chem. - Eur. J. 2018, 24, 10550–10556. 10.1002/chem.201802356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hett E. C.; Xu H.; Geoghegan K. F.; Gopalsamy A.; Kyne R. E. Jr; Menard C. A.; Narayanan A.; Parikh M. D.; Liu S.; Roberts L.; Robinson R. P.; Tones M. A.; Jones L. H. Rational targeting of active-site tyrosine residues using sulfonyl fluoride probes. ACS Chem. Biol. 2015, 10, 1094–1098. 10.1021/cb5009475. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Li J.; Li S.; Li G.; Sharpless K. B.; Wu P. SuFEx click chemistry enabled late-stage drug functionalization. J. Am. Chem. Soc. 2018, 140, 2919–2925. 10.1021/jacs.7b12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan A.; Jones L. H. Sulfonyl fluorides as privileged warheads in chemical biology. 2015, 6, 2650–2659. 10.1039/C5SC00408J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattah T. A.; Saeed A.; Albericio F. Recent advances towards sulfur (VI) fluoride exchange (SuFEx) click chemistry. J. Fluorine Chem. 2018, 213, 87–112. 10.1016/j.jfluchem.2018.07.008. [DOI] [Google Scholar]
- Zheng Q.; Woehl J. L.; Kitamura S.; Santos-Martins D.; Smedley C. J.; Li G.; Forli S.; Moses J. E.; Wolan D. W.; Sharpless K. B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 18808–18814. 10.1073/pnas.1909972116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondoni A.; Marra A. SuFEx: a metal-free click ligation for multivalent biomolecules. Org. Biomol. Chem. 2017, 15, 1549–1553. 10.1039/C6OB02458K. [DOI] [PubMed] [Google Scholar]
- Liang D.-D.; Streefkerk D. E.; Jordaan D.; Wagemakers J.; Baggerman J.; Zuilhof H. Silicon-free SuFEx reactions of sulfonimidoyl fluorides: scope: enantioselectivity, and mechanism. Angew. Chem., Int. Ed. 2020, 59, 7494–7500. 10.1002/anie.201915519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greed S.; Briggs E. L.; Idiris F. I. M.; White A. J. P.; Lücking U.; Bull J. A. Synthesis of highly enantioenriched sulfonimidoyl fluorides and sulfonimidamides by stereospecific sulfur-fluorine exchange (SuFEx) reaction. Chem. - Eur. J. 2020, 26, 12533–12538. 10.1002/chem.202002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brighty G. J.; Botham R. C.; Li S.; Nelson L.; Mortenson D. E.; Li G.; Morisseau C.; Wang H.; Hammock B. D.; Sharpless K. B.; Kelly J. W. Using sulfuramidimidoyl fluorides that undergo sulfur(VI) fluoride exchange for inverse drug discovery. Nat. Chem. 2020, 12, 906–913. 10.1038/s41557-020-0530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Laughlin S. T. Modular activatable bioorthogonal reagents. Methods Enzymol. 2019, 622, 153–182. 10.1016/bs.mie.2019.02.007. [DOI] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. Photoclick chemistry: a fluorogenic light-triggered in vivo ligation reaction. Curr. Opin. Chem. Biol. 2014, 21, 89–95. 10.1016/j.cbpa.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam S.; Orski S. V.; Mbua N. E.; McNitt C.; Boons G.-J.; Locklin J.; Popik V. V. Photo-click chemistry strategies for spatiotemporal control of metal-free ligation, labeling, and surface derivatization. Pure Appl. Chem. 2013, 85, 1499–1513. 10.1351/PAC-CON-13-01-08. [DOI] [Google Scholar]
- Yu Z.; Lin Q. Design of spiro[2.3]hex-1-ene, a genetically encodable double-strained alkene for superfast photoclick chemistry. J. Am. Chem. Soc. 2014, 136, 4153–4156. 10.1021/ja5012542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Ohulchanskyy T. Y.; An P.; Prasad P. N.; Lin Q. Fluorogenic, two-photon-triggered photoclick chemistry in live mammalian cells. J. Am. Chem. Soc. 2013, 135, 16766–16769. 10.1021/ja407867a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloukhtine A. A.; Mbua N. E.; Wolfert M. A.; Boons G.-J.; Popik V. V. Selective labeling of living cells by a photo-triggered click reaction. J. Am. Chem. Soc. 2009, 131, 15769–15776. 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z.; Pan Y.; Wang Z.; Wang J.; Lin Q. Genetically encoded cyclopropene directs rapid, photoclick-chemistry-mediated protein labeling in mammalian cells. Angew. Chem., Int. Ed. 2012, 51, 10600–10604. 10.1002/anie.201205352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debets M. F.; Van Berkel S. S.; Dommerholt J.; Dirks A. J.; Rutjes F. P. J. T.; Van Delft F. L. Bioconjugation with strained alkenes and alkynes. Acc. Chem. Res. 2011, 44, 805–815. 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]
- Debets M. F.; Van Berkel S. S.; Schoffelen S.; Rutjes F. P. J. T.; Van Hest J. C. M.; Van Delft F. L. Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3 + 2) cycloaddition. Chem. Commun. 2010, 46, 97–99. 10.1039/B917797C. [DOI] [PubMed] [Google Scholar]
- Dommerholt J.; Schmidt S.; Temming R.; Hendriks L. J. A.; Rutjes F. P. J. T.; Van Hest J. C. M.; Lefeber D. J.; Friedl P.; Van Delft F. L. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem., Int. Ed. 2010, 49, 9422–9425. 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dommerholt J.; Van Rooijen O.; Borrmann A.; Guerra C. F.; Bickelhaupt F. M.; Van Delft F. L. Highly accelerated inverse electron-demand cycloaddition of electron-deficient azides with aliphatic cyclooctynes. Nat. Commun. 2014, 5, 5378. 10.1038/ncomms6378. [DOI] [PubMed] [Google Scholar]
- Terzic V.; Pousse G.; Méallet-Renault R.; Grellier P.; Dubois J. Dibenzocyclooctynes: effect of aryl substitution on their reactivity toward strain-promoted alkyne-azide cycloaddition. J. Org. Chem. 2019, 84, 8542–8551. 10.1021/acs.joc.9b00895. [DOI] [PubMed] [Google Scholar]
- Dommerholt J.; Rutjes F. P. J. T.; Van Delft F. L. Strain-promoted 1,3-dipolar cycloaddition of cycloalkynes and organic azides. Top. Curr. Chem. (Z) 2016, 374, 16. 10.1007/s41061-016-0016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gann A. W.; Amoroso J. W.; Einck V. J.; Rice W. P.; Chambers J. J.; Schnarr N. A. A photoinduced, benzyme click reaction. Org. Lett. 2014, 16, 2003–2005. 10.1021/ol500389t. [DOI] [PubMed] [Google Scholar]
- Shi F.; Waldo J. P.; Larock R. C. Benzyne click chemistry: synthesis of benzotriazole from benzynes and azides. Org. Lett. 2008, 10, 2409–2412. 10.1021/ol800675u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Moses J. E. Benzyne click chemistry with in situ generated aromatic azides. Org. Lett. 2009, 11, 1587–1590. 10.1021/ol9002338. [DOI] [PubMed] [Google Scholar]
- Campbell-Verduyn L.; Elsinga P. H.; Mirfeizi L.; Dierckx R. A.; Feringa B. L. Copper-free ‘click’: 1,3-dipolar cycloaddition of azides and arynes. Org. Biomol. Chem. 2008, 6, 3461–3463. 10.1039/b812403e. [DOI] [PubMed] [Google Scholar]
- Chandrasekhar S.; Seenaiah M.; Rao Ch. L.; Reddy R. Ch A smooth access to benzotriazoles via azide-benzyne cycloaddition. Tetrahedron 2008, 64, 11325–11327. 10.1016/j.tet.2008.08.115. [DOI] [Google Scholar]
- Cormier M.; Fouquet E.; Hermange P. Expedient synthesis of a symmetric cycloheptyne-Co2(CO)6 complex for orthogonal Huisgen cycloadditions. Org. Chem. Front. 2019, 6, 1114–1117. 10.1039/C9QO00086K. [DOI] [Google Scholar]
- Gobbo P.; Romagnoli T.; Barbon S. M.; Price J. T.; Keir J.; Gilroy J. B.; Workentin M. S. Expanding the scope of strained-alkyne chemistry: a protection-deprotection strategy via the formation of a dicobalt-hexacarbonyl complex. Chem. Commun. 2015, 51, 6647–6650. 10.1039/C5CC01522G. [DOI] [PubMed] [Google Scholar]
- Yoshida S.; Hatakeyama Y.; Johmoto K.; Uekusa H.; Hosoya T. Transient protection of strained alkynes from click reaction via complexation with copper. J. Am. Chem. Soc. 2014, 136, 13590–13593. 10.1021/ja507660x. [DOI] [PubMed] [Google Scholar]
- Although it can be triggered to click to electron-deficient olefin by means of oxidation, see:; Gangaprasad D.; Raj J. P.; Kiranmye T.; Karthikeyan K.; Elangovan J. Another example of organo-click reactions: TEMPO-promoted oxidative azide-olefin cycloaddition for the synthesis of 1,2,3-triazoles in water. Chem. Eur. J. Org. 2016, 2016, 5642–5646. 10.1002/ejoc.201601121. [DOI] [Google Scholar]
- Singh I.; Heaney F. Solid phase strain promoted “click” modification of DNA via [3 + 2]-nitrile oxide-cyclooctyne cycloadditions. Chem. Commun. 2011, 47, 2706–2708. 10.1039/C0CC03985C. [DOI] [PubMed] [Google Scholar]
- Sanders B. C.; Friscourt F.; Ledin P. A.; Mbua N. E.; Arumugam S.; Guo J.; Boltje T. J.; Popik V. V.; Boons G.-J. Metal-free sequential [3 + 2]-dipolar cycloadditions using cyclooctynes and 1,3-dipoles of different reactivity. J. Am. Chem. Soc. 2011, 133, 949–957. 10.1021/ja1081519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn B. A.; Lee S.; Kim S.; Teyssier F.; Aulakh V. S.; Ciufolini M. A. Oxidation of oximes to nitrile oxides with hypervalent iodine reagents. Org. Lett. 2009, 11, 1539–1542. 10.1021/ol900194v. [DOI] [PubMed] [Google Scholar]
- Jawalekar A. M.; Reubsaet E.; Rutjes F. P. J. T.; Van Delft F. L. Synthesis of isoxazoles by hypervalent-induced cycloaddition of nitrile oxides to alkynes. Chem. Commun. 2011, 47, 3198–3200. 10.1039/c0cc04646a. [DOI] [PubMed] [Google Scholar]
- Yoshimura A.; Nguyen K. C.; Rohde G. T.; Saito A.; Yusubov M. S.; Zhdakin V. V. Oxidative cycloaddition of aldoximes with maleimides using catalytic hydroxy(aryl)iodonium species. Adv. Synth. Catal. 2016, 358, 2340–2344. 10.1002/adsc.201600331. [DOI] [Google Scholar]
- Marco-Contelles J. Recent advances on nitrones design for stroke treatment. J. Med. Chem. 2020, 63, 13413–1427. 10.1021/acs.jmedchem.0c00976. [DOI] [PubMed] [Google Scholar]
- Murahashi S.-I.; Imada Y. Synthesis and transformations of nitrones for organic synthesis. Chem. Rev. 2019, 119, 4684–4716. 10.1021/acs.chemrev.8b00476. [DOI] [PubMed] [Google Scholar]
- Ghiassian S.; Yu L.; Gobbo P.; Nazemi A.; Romagnoli T.; Luo W.; Luyt L. G.; Workentin M. S. Nitron-modified gold nanoparticles: synthesis, characterization, and their potential as 18F-labeled positron emission tomography probes via I-SPANC. ACS Omega 2019, 4, 19106–19115. 10.1021/acsomega.9b02322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay C. S.; Blake J. A.; Cheng J.; Danielson D. C.; Pezacki J. P. Strain-promoted cycloadditions of cyclic nitrones with cyclooctynes for labeling human cancer cells. Chem. Commun. 2011, 47, 10040–10042. 10.1039/c1cc13808a. [DOI] [PubMed] [Google Scholar]
- Kinugasa M.; Hashimoto S. The reactions of copper(I) phenylacetylide with nitrones. J. Chem. Soc., Chem. Commun. 1972, 466–467. 10.1039/c39720000466. [DOI] [Google Scholar]
- McKay C. S.; Moran J.; Pezacki J. P. Nitrones as dipoles for rapid strain-promoted 1,3-dipolar cycloadditions with cyclooctynes. Chem. Commun. 2010, 46, 931–933. 10.1039/B921630H. [DOI] [PubMed] [Google Scholar]
- MacKenzie D. A.; Sherratt A. R.; Chigrinova M.; Cheung L. L. W.; Pezacki J. P. Strain-promoted cycloadditions involving nitrones and alkynes - rapid tunable reactions for bioorthogonal labeling. Curr. Opin. Chem. Biol. 2014, 21, 81–88. 10.1016/j.cbpa.2014.05.023. [DOI] [PubMed] [Google Scholar]
- MacKenzie D. A.; Pezacki J. P. Kinetics studies of rapid strain-promoted [3 + 2] cycloadditions of nitrones with bicyclo[6.1.0]nonyne. Can. J. Chem. 2014, 92, 337–340. 10.1139/cjc-2013-0577. [DOI] [Google Scholar]
- Blizzard R. J.; Backus D. R.; Brown W.; Bazewicz C. G.; Li Y.; Mehl R. A. Ideal bioorthogonal reactions using a site-specifically encoded tetrazine amino acid. J. Am. Chem. Soc. 2015, 137, 10044–10047. 10.1021/jacs.5b03275. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Bioorthogonal reactions for labeling proteins. ACS Chem. Biol. 2014, 9, 16–20. 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130, 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W.; Wang D.; Dai C.; Hamelberg D.; Wang B. Clicking 1,2,4,5-tetrazine and cyclooctynes with tunable reaction rates. Chem. Commun. 2012, 48, 1736–1738. 10.1039/C2CC16716F. [DOI] [PubMed] [Google Scholar]
- Darko A.; Wallace S.; Dmitrenko O.; Machovina M. M.; Mehl R. A.; Chin J. W.; Fox J. M. Conformationally strained trans-cyclooctene with improved stability and excellent reactivity in tetrazine ligation. Chem. Sci. 2014, 5, 3770–3776. 10.1039/C4SC01348D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasco J. M. J. M.; Coelho J. A. S. Predictive multivariate models for bioorthogonal inverse-electron demand Diels-Alder reactions. J. Am. Chem. Soc. 2020, 142, 4235–4241. 10.1021/jacs.9b11948. [DOI] [PubMed] [Google Scholar]
- Wu H.; Devaraj N. K. Advances in tetrazine bioorthogonal chemistry driven by the synthesis of novel tetrazines and dienophiles. Acc. Chem. Res. 2018, 51, 1249–1259. 10.1021/acs.accounts.8b00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Trout W. S.; Liu S.; Andrade G. A.; Hudson D. A.; Scinto S. L.; Dicker K. T.; Li Y.; Lazouski N.; Rosenthal J.; Thorpe C.; Jia X.; Fox J. Rapid bioorthogonal chemistry turn-on through enzymatic or long wavelength photocatalytic activation of tetrazine ligation. J. Am. Chem. Soc. 2016, 138, 5978–5983. 10.1021/jacs.6b02168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj R.; Fox J. M. An efficient and mild oxidant for the synthesis of s-tetrazines. Tetrahedron Lett. 2014, 55, 4795–4797. 10.1016/j.tetlet.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerl G.; Senkovska I.; Kaskel S. Tetrazine functionalized zirconium MOF as an optical sensor for oxidizing gases. Chem. Commun. 2015, 51, 2280–2282. 10.1039/C4CC08136F. [DOI] [PubMed] [Google Scholar]
- Dzandzi J. P. K.; Beckford Vera D. R.; Genady A. R.; Albu S. A.; Eltringham-Smith L. J.; Capretta A.; Sheffield W.; Valliant J. F. Fluorous analogue of chloramine-T: preparation, X-ray structure determination, and use as an oxidant for radioiodination and s-tetrazine synthesis. J. Org. Chem. 2015, 80, 7117–7125. 10.1021/acs.joc.5b00988. [DOI] [PubMed] [Google Scholar]
- Albu S. A.; Al-Karmi S. A.; Vito A.; Dzandzi J. P. K.; Zlitni A.; Beckford-Vera D.; Blacker M.; Janzen N.; Patel R. M.; Capretta A.; Valliant J. F. 125I-tetrazines and inverse-electron-demand Diels-Alder chemistry: a convenient radioiodination strategy for biomolecule labeling, screening, and biodistribution studies. Bioconjugate Chem. 2016, 27, 207–216. 10.1021/acs.bioconjchem.5b00609. [DOI] [PubMed] [Google Scholar]
- Ehret F.; Wu H.; Alexander S. C.; Devaraj N. K. Electrochemical control of rapid bioorthogonal tetrazine ligations for selective functionalization of microelectrodes. J. Am. Chem. Soc. 2015, 137, 8876–8879. 10.1021/jacs.5b03371. [DOI] [PubMed] [Google Scholar]
- Clavier G.; Audebert P. s-Tetrazines as building blocks for new functional molecules and molecular materials. Chem. Rev. 2010, 110, 3299–3314. 10.1021/cr900357e. [DOI] [PubMed] [Google Scholar]
- Le T.; Courant T.; Merad J.; Allain C.; Audebert P.; Masson G. Aerobic tetrazine-catalyzed oxidative nitroso-Diels-Alder reaction of N-arylhydroxylamines with dienecarbamates: access to functionalized 1,6-dihydro-1,2-oxazines. ChemCatChem 2019, 11, 5282–5286. 10.1002/cctc.201901373. [DOI] [Google Scholar]
- Verboom W.; Bos H. J. T. Cycloaddition reactions of cyclooctyne with 9,10-phenanthrenequinone and 3,5-di-tert-butyl-o-benzoquinone. Recl. Trav. Chim. Pays-Bas 1981, 100, 207–213. 10.1002/recl.19811000509. [DOI] [Google Scholar]
- Meier H.; Molz T.; Merkle U.; Echter T.; Lorch M. Synthesis of aromatic-hydrocarbons from 1,2,3-selenadiazoles and -pyrones. Liebigs Ann. Chem. 1982, 1982, 914–923. 10.1002/jlac.198219820511. [DOI] [Google Scholar]
- Escorihuela J.; Das A.; Looijen W. J. E.; Van Delft F. L.; Aquino A. J. A.; Lischka H.; Zuilhof H. Kinetics of the strain-promoted oxidation-controlled cycloalkyne-1,2-quinone cycloaddition: experimental and theoretical studies. J. Org. Chem. 2018, 83, 244–252. 10.1021/acs.joc.7b02614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levandowski B. J.; Svatunek D.; Sohr B.; Mikula H.; Houk K. N. Secondary orbital interactions enhance the reactivity of alkynes in Diels-Alder cycloadditions. J. Am. Chem. Soc. 2019, 141, 2224–2227. 10.1021/jacs.8b13088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn J. S.; Hu Y.; Willner I. Stimuli-responsive DNA-based hydrogels: from basic principles to applications. Acc. Chem. Res. 2017, 50, 680–690. 10.1021/acs.accounts.6b00542. [DOI] [PubMed] [Google Scholar]
- Grim J. C.; Marozas I. A.; Anseth K. S. Thiol-ene and photo-cleavage chemistry for controlled presentation of biomolecules in hudrogels. J. Controlled Release 2015, 219, 95–106. 10.1016/j.jconrel.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker A. M.; Borrmann A.; Van Eck E. R. H.; Van Delft F. L.; Löwik D. W. P. M.; Van Hest J. C. M. A fast and activatable cross-linking strategy for hydrogel formation. Adv. Mater. 2015, 27, 1235–1240. 10.1002/adma.201404448. [DOI] [PubMed] [Google Scholar]
- Ledin P. A.; Kolishetti N.; Boons G.-J. Multifunctionalization of polymers by strain-promoted cycloadditions. Macromolecules 2013, 46, 7759–7768. 10.1021/ma400913a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledin P. A.; Kolishetti N.; Hudlikar M. S.; Boons G.-J. Exploring strain-promoted 1,3-dipolar cycloadditions of end functionalized polymers. Chem. - Eur. J. 2014, 20, 8753–8760. 10.1002/chem.201402225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J.; Xiao Z.; Müllner M.; Connal L. A. The emergence of oxime click chemistry and its utility in polymer science. Polym. Chem. 2016, 7, 3812–3826. 10.1039/C6PY00635C. [DOI] [Google Scholar]
- Kuzmin A.; Poloukhtine A.; Wolfert M. A.; Popik V. V. Surface functionalization using catalyst-free azide-alkyne cycloaddition. Bioconjugate Chem. 2010, 21, 2076–2085. 10.1021/bc100306u. [DOI] [PubMed] [Google Scholar]
- Guo J.; Chen G.; Ning X.; Wolfert M. A.; Li X.; Xu B.; Boons G.-J. Surface modification of polymeric micelles by strain-promoted alkyne-azide cycloadditions. Chem. - Eur. J. 2010, 16, 13360–13366. 10.1002/chem.201002532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen R.; Escorihuela J.; Smulders M. M. J.; Zuilhof H. Use of ambient ionization high-resolution mass spectrometry for the kinetic analysis of organic surface reactions. Langmuir 2016, 32, 3412–3419. 10.1021/acs.langmuir.6b00427. [DOI] [PubMed] [Google Scholar]
- Sen R.; Gahtory D.; Carvalho R. R.; Albada B.; Van Delft F. L.; Zuilhof H. Ultrathin covalently bound organic layers on mica: formation of atomically flat biofunctionalizable surfaces. Angew. Chem., Int. Ed. 2017, 56, 4130–4134. 10.1002/anie.201701301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahtory D.; Sen R.; Kuzmyn A. R.; Escorihuela J.; Zuilhof H. Strain-promoted cycloaddition of cyclopropenes with o-quinones: a rapid click reaction. Angew. Chem., Int. Ed. 2018, 57, 10118–10122. 10.1002/anie.201800937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadish J. A.; DeForest C. A. Site-selective protein modification: from functionalized proteins to functional biomaterials. Matter 2020, 2, 50–77. 10.1016/j.matt.2019.11.011. [DOI] [Google Scholar]
- Tamura T.; Hamachi I. Chemistry for covalent modification of endogenous/native proteins: from test tubes to complex biological systems. J. Am. Chem. Soc. 2019, 141, 2787–2799. [DOI] [PubMed] [Google Scholar]
- Li S.; Cai H.; He J.; Chen H.; Lam S.; Cai T.; Zhu Z.; Bark S. K.; Cai C. Extent of the oxidative side reactions to peptides and proteins during the CuAAC reaction. Bioconjugate Chem. 2016, 27, 2315–2322. 10.1021/acs.bioconjchem.6b00267. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Li K.; Cai C.-Z. Anaerobic conditions to reduce oxidation of proteins and to accelerate the copper-catalyzed “clicl” reaction with a water-soluble bis(triazole) ligand. Chem. Commun. 2011, 47, 3186–3188. 10.1039/c0cc05376g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel G. R.; Calabrese Z. A.; Ayco J.; Hein J. E.; Ye T. Measuring and suppressing the oxidative damage to DNA during Cu(I)-catalyzed azide-alkyne cycloaddition. Bioconjugate Chem. 2016, 27, 698–704. 10.1021/acs.bioconjchem.5b00665. [DOI] [PubMed] [Google Scholar]
- Osberger T. J.; Rogness D. C.; Kohrt J. T.; Stepan A. F.; White M. C. Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. 10.1038/nature18941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudi L.; Kissner R.; Nauser T.; Koppenol W. H. Electrode potentials of L-tryptophan, L-tyrosine, 3-nitro-L-tyrosine, 2,3-difluoro-L-tyrosine, and 2,3,5-trifluoro-L-tyrosine. Biochemistry 2016, 55, 2849–2856. 10.1021/acs.biochem.6b00019. [DOI] [PubMed] [Google Scholar]
- DeFelippis M. R.; Murthy C. P.; Broitman F.; Weinraub D.; Faraggi M.; Klapper M. H. Electrochemical properties of tyrosine phenoxy and tryptophan indolyl radicals in peptides and amino acid analogues. J. Phys. Chem. 1991, 95, 3416–3419. 10.1021/j100161a081. [DOI] [Google Scholar]
- Glover S. D.; Tyburski R.; Liang L.; Tommos C.; Hammarström L. Pourbaix diagram, proton-coupled electron transfer, and decay kinetics of a protein tryptophan radical: comparing the redox properties of W32• and Y32• generated inside the structurally characterized α3W and α3Y proteins. J. Am. Chem. Soc. 2018, 140, 185–192. 10.1021/jacs.7b08032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close D. M.; Wardman P. Calculation of standard reduction potentials of amino acid radicals and the effects of water and incorporation into peptides. J. Phys. Chem. A 2018, 122, 439–445. 10.1021/acs.jpca.7b10766. [DOI] [PubMed] [Google Scholar]
- Patureau F. W. The phenol-phenothiazine coupling: an oxidative click concept. ChemCatChem 2019, 11, 5227–7231. 10.1002/cctc.201900152. [DOI] [Google Scholar]
- Alvarez-Dorta D.; Deniaud D.; Mével M.; Gouin S. G. Tyrosine conjugation methods for protein labelling. Chem. - Eur. J. 2020, 26, 14257–14269. 10.1002/chem.202001992. [DOI] [PubMed] [Google Scholar]
- Koide S.; Sidhu S. S. The importance of being tyrosine: lessons in molecular recognition from minimalist synthetic binding proteins. ACS Chem. Biol. 2009, 4, 325–334. 10.1021/cb800314v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomäki M.; Marttila L.; Kivelä H.; Ouvinen T.; Lukkari J. Effects of pH and oxidants on the first steps op polydopamine formation: a thermodynamic apporach. J. Phys. Chem. B 2018, 122, 6314–6327. 10.1021/acs.jpcb.8b02304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier G. P.; Bernt C. M.; Butler A. Catechol oxidation: considerations in the design of wet adhesive materials. Biomater. Sci. 2018, 6, 332–339. 10.1039/C7BM00884H. [DOI] [PubMed] [Google Scholar]
- Hooker J. M.; Kovacs E. W.; Francis M. B. Interior surface modification of bacteriophage MS2. J. Am. Chem. Soc. 2004, 126, 3718–3719. 10.1021/ja031790q. [DOI] [PubMed] [Google Scholar]
- Ban H.; Gavrilyuk J.; Barbas C. F. Tyrosine bioconjugation through aqueous ene-type reactions: a click-like reaction for tyrosine. J. Am. Chem. Soc. 2010, 132, 1523–1525. 10.1021/ja909062q. [DOI] [PubMed] [Google Scholar]
- Ban H.; Nagano M.; Gavrilyuk J.; Hakamata W.; Inokuma T.; Barbas C. F. III Facile and stabile linkages through tyrosine: bioconjugation strategies with the tyrosine-click reaction. Bioconjugate Chem. 2013, 24, 520–532. 10.1021/bc300665t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter S. H. The reaction of phenols with ethyl azodicarboxylate. J. Org. Chem. 1969, 34, 4012–4015. 10.1021/jo01264a056. [DOI] [Google Scholar]
- Alstanei A.-M.; Hornoiu C.; Aycard J.-P.; Carles M.; Volanschi E. Electrochemical behaviour and redox reactivity of some 4-R-1,2,4-triazolin-3,5-diones. J. Electroanal. Chem. 2003, 542, 13–21. 10.1016/S0022-0728(02)01441-9. [DOI] [Google Scholar]
- Although a recent example showed that PTAD can be used for click conjugation to indole rings:; Billiet S.; De Bruycker K.; Driessen F.; Goossens H.; Van Speybroeck V.; Winne J. M.; Du Prez F. E. Triazolinediones enable ultrafast and reversible click chemistry for the design of dynamic polymer systems. Nat. Chem. 2014, 6, 815–821. 10.1038/nchem.2023. [DOI] [PubMed] [Google Scholar]
- Madl C. M.; Heilshorn S. C. Tyrosine-selective functionalization for bio-orthogonal cross-linking of engineered protein hydrogels. Bioconjugate Chem. 2017, 28, 724–730. 10.1021/acs.bioconjchem.6b00720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Q.-Y.; Allan M.; Adamo R.; Quinn D.; Zhai H.; Wu G.; Clark K.; Zhou J.; Ortiz S.; Wang B.; Danieli E.; Crotti S.; Tontini M.; Brogioni G.; Berti F. Synthesis of a well-defined glycoconjugate vaccine by a tyrosine-selective conjugation strategy. Chem. Sci. 2013, 4, 3827–3832. 10.1039/c3sc51694f. [DOI] [Google Scholar]
- Sato S.; Nakamura K.; Nakamura H. Tyrosine-specific chemical modification with in situ hemin-activated luminol derivatives. ACS Chem. Biol. 2015, 10, 2633–2640. 10.1021/acschembio.5b00440. [DOI] [PubMed] [Google Scholar]
- Sato S.; Nakamura H. Ligand-directed selective protein modification based on local single-electron-transfer catalysis. Angew. Chem., Int. Ed. 2013, 52, 8681–8684. 10.1002/anie.201303831. [DOI] [PubMed] [Google Scholar]
- Sato S.; Hatano K.; Tsushima M.; Nakamura H. 1-methyl-4-aryl-urazole (MAUra) labels tyrosine in proximity to ruthenium photocatalysts. Chem. Commun. 2018, 54, 5871–5874. 10.1039/C8CC02891E. [DOI] [PubMed] [Google Scholar]
- Sato S.; Yoshida M.; Hatano K.; Matsumura M.; Nakamura H. N’-acyl-N-methylphenylenediamine as a novel proximity labeling agent for signal amplification in immunohistochemistry. Bioorg. Med. Chem. 2019, 27, 1110–1118. 10.1016/j.bmc.2019.01.036. [DOI] [PubMed] [Google Scholar]
- Kersten P. J.; Kalyanaraman B.; Hammel K. E.; Reinhammar B.; Kirk T. K. Comparison of lignin peroxidase, horseradisch peroxidase and laccase in the oxidation of methoxybenzenes. Biochem. J. 1990, 268, 475–480. 10.1042/bj2680475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S.; Nakamura K.; Nakamura H. Horseradish-peroxidase-catalyzed tyrosine click reaction. ChemBioChem 2017, 18, 475–478. 10.1002/cbic.201600649. [DOI] [PubMed] [Google Scholar]
- Gross A. J.; Sizer I. W. The oxidation of tyramine, tyrosine, and related compounds by peroxidase. J. Biol. Chem. 1959, 234, 1611–1614. 10.1016/S0021-9258(18)70059-8. [DOI] [PubMed] [Google Scholar]
- Minamihata K.; Goto M.; Kamiya N. Site-specific protein cross-linking by peroxidase-catalyzed activation of a tyrosine-containing peptide tag. Bioconjugate Chem. 2011, 22, 74–81. 10.1021/bc1003982. [DOI] [PubMed] [Google Scholar]
- Minamihata K.; Goto M.; Kamiya N. Site-specific conjugation of an antibody-binding protein catalyzed by horseradish peroxidase creates a multivalent protein conjugate with high affinity to IgG. Biotechnol. J. 2015, 10, 222–226. 10.1002/biot.201400512. [DOI] [PubMed] [Google Scholar]
- Kodadek T.; Duroux-Richard I.; Bonnafous J.-C. Techniques: oxidative cross-linking as an emergent tool for the analysis of receptor-mediated signalling events. Trends Pharmacol. Sci. 2005, 26, 210–217. 10.1016/j.tips.2005.02.010. [DOI] [PubMed] [Google Scholar]
- Meunier S.; Strable E.; Finn M. G. Crosslinking of a coupling to viral capsid proteins by tyrosine oxidation. Chem. Biol. 2004, 11, 319–326. 10.1016/j.chembiol.2004.02.019. [DOI] [PubMed] [Google Scholar]
- Sato S.; Matsumura M.; Kadonosono T.; Abe S.; Ueno T.; Ueda H.; Nakamura H. Site-selective protein chemical modification of exposed tyrosine residues using tyrosine click reaction. Bioconjugate Chem. 2020, 31, 1417–1424. 10.1021/acs.bioconjchem.0c00120. [DOI] [PubMed] [Google Scholar]
- Sato S.; Nakane K.; Nakamura H. A laccase-catalysed tyrosine click reaction. Org. Biomol. Chem. 2020, 18, 3664–3668. 10.1039/D0OB00650E. [DOI] [PubMed] [Google Scholar]
- Alvarez-Dorta D.; Thobie-Gautier C.; Croyal M.; Bouzelha M.; Mevel M.; Deniaud D.; Boujtita M.; Gouin S. G. Electrochemically promoted tyrosine-click-chemistry for protein labeling. J. Am. Chem. Soc. 2018, 140, 17120–17126. 10.1021/jacs.8b09372. [DOI] [PubMed] [Google Scholar]
- Travascio P.; Li Y.; Sen D. DNA-enhanced peroxidase activity of a DNA aptamer-hemin complex. Chem. Biol. 1998, 5, 505–517. 10.1016/S1074-5521(98)90006-0. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Nie Y.; Zhu X.; Liu X.; Li G. Study on the electrocatalytic activity of human telomere G-quadruplex-hemin complex and its interaction with small molecular ligands. Electrochim. Acta 2009, 55, 276–280. 10.1016/j.electacta.2009.08.050. [DOI] [Google Scholar]
- Li W.; Li Y.; Liu Z.; Lin B.; Yi H.; Xu F.; Nie Z. Insight into G-quadruplex-hemin DNAzyme/RNAzyme: adjacent adenine as the intramolecular species for remarkable enhancement of enzymatic activity. Nucleic Acids Res. 2016, 44, 7373–7384. 10.1093/nar/gkw634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuzawa M.; Sato S.; Niwa T.; Taguchi H.; Nakamura H.; Oyoshi T. G-quadruplex-proximity protein labeling based on peroxidase activity. Chem. Commun. 2020, 56, 11641–11644. 10.1039/D0CC02571B. [DOI] [PubMed] [Google Scholar]
- Keijzer J. F.; Albada B. Site-specific and trigger-activated modification of proteins by means of catalytic hemin/G-quadruplex DNAzyme nanostructures. Bioconjugate Chem. 2020, 31, 2283–2287. 10.1021/acs.bioconjchem.0c00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler T.; Patsis P. A.; Hahn D.; Ruland A.; Naas C.; Muller M.; Thiele J. DNAzymes as catalysts for L-tyrosine and amyloid b oxidation. ACS Omega 2020, 5, 7059–7064. 10.1021/acsomega.9b02645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einarson O. J.; Sen D. Self-biotinylation of DNA G-quadruplexes via intrinsic peroxidase activity. Nucleic Acids Res. 2017, 45, 9813–9822. 10.1093/nar/gkx765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann A.; Fatunsin O.; Dommerholt J.; Jonker A. M.; Löwik D. W. P. M.; Van Hest J. C. M.; Van Delft F. L. Strain-promoted oxidation-controlled cyclooctyne-1,2-quinone cycloaddition (SPOCQ) for fast and activatable protein conjugation. Bioconjugate Chem. 2015, 26, 257–261. 10.1021/bc500534d. [DOI] [PubMed] [Google Scholar]
- Lepthien S.; Merkel L.; Budisa N. In vivo double and triple labeling of proteins using synthetic amino acids. Angew. Chem., Int. Ed. 2010, 49, 5446–5450. 10.1002/anie.201000439. [DOI] [PubMed] [Google Scholar]
- Montanari E.; Gennari A.; Pelliccia M.; Manzi L.; Donno R.; Oldham N. J.; MacDonald A.; Tirelli N. Tyrosinase-mediated bioconjugation. A versatile approach to chimeric macromolecules. Bioconjugate Chem. 2018, 29, 2550–2560. 10.1021/acs.bioconjchem.8b00227. [DOI] [PubMed] [Google Scholar]
- Kim S.; Sung B. H.; Kim S. C.; Lee H. S. Genetic incorporation of L-dihydroxyphenylalanine (DOPA) biosynthesized by a tyrosine phenol-lyase. Chem. Commun. 2018, 54, 3002–3005. 10.1039/C8CC00281A. [DOI] [PubMed] [Google Scholar]
- Ayyadurai N.; Prabhu N. S.; Deepankumar K.; Jang Y. J.; Chitrapriya N.; Song E.; Lee N.; Kim S. K.; Kim S.-G.; Soundrarajan N.; Lee S.; Cha H. J.; Budisa N.; Yun H. Bioconjugation of L-3,4-dihydroxyphenylalanine containing protein with a polysaccharide. Bioconjugate Chem. 2011, 22, 551–555. 10.1021/bc2000066. [DOI] [PubMed] [Google Scholar]
- Panis F.; Kampatsikas I.; Bijelic A.; Rompel A. Conversion of walnut tyrosinase into a catechol oxidase by site directed mutagenesis. Sci. Rep. 2020, 10, 1659. 10.1038/s41598-020-57671-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins J. J.; Albada B.; Van Delft F. Ortho-quinones and analogues thereof: highly reactive intermediates for fast and selective biofunctionalization. Chem. - Eur. J. 2018, 24, 4749–4756. 10.1002/chem.201703919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollett A.; Thallinger B.; Ohradanova-Repic A.; Machacek C.; Walenta E.; Cavaco-Paulo A.; Birner-Gruenberger R.; Bogner-Strauss J. G.; Stockinger H.; Guebitz G. M. Enzymatic synthesis of antibody-human serum albumin conjugate for targeted drug delivery using tyrosinase from Agaricus bisporus. RSC Adv. 2013, 3, 1460–1467. 10.1039/C2RA22560C. [DOI] [Google Scholar]
- For the application of protein-bound para-quinone in oxime ligations, see:; Park S.; Westcott N. P.; Luo W.; Dutta D.; Yousaf M. N. General chemoselective and redox-responsive ligation and release strategy. Bioconjugate Chem. 2014, 25, 543–551. 10.1021/bc400565y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long M. J. C.; Hedstrom L. Mushroom tyrosinase oxidizes tyrosine-rich sequences to allow selective protein functionalization. ChemBioChem 2012, 13, 1818–1825. 10.1002/cbic.201100792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmelstein A. M.; Lobba M. J.; Mogilevsky C. S.; Maza J. C.; Brauer D. D.; Francis M. B. Tyrosinase-mediated oxidative coupling of tyrosine tags on peptides and proteins. J. Am. Chem. Soc. 2020, 142, 5078–5086. 10.1021/jacs.9b12002. [DOI] [PubMed] [Google Scholar]
- Maza J. C.; Bader D. L. V.; Xiao L.; Marmelstein A. M.; Brauer D. D.; ElSohly A. M.; Smith M. J.; Krska S. W.; Parish C. A.; Francis M. B. Enzymatic modification of N-terminal proline residues using phenol derivatives. J. Am. Chem. Soc. 2019, 141, 3885–3892. 10.1021/jacs.8b10845. [DOI] [PubMed] [Google Scholar]
- Lobba M. J.; Fellmann C.; Marmelstein A. M.; Maza J. C.; Kissman E. N.; Robinson S. A.; Staahl B. T.; Urnes C.; Lew R. J.; Mogilevsky C. S.; Doudna J. A.; Francis M. B. Site-specific bioconjugation through enzyme-catalyzed tyrosine-cysteine bond formation. ACS Cent. Sci. 2020, 6, 1564–1571. 10.1021/acscentsci.0c00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins J. J.; Westphal A. H.; Albada B.; Wagner K.; Bartels K.; Spits H.; Van Berkel W. J. H.; Van Delft F. L. Inducible, site-specific protein labeling by tyrosine oxidation-strain-promoted (4 + 2) cycloaddition. Bioconjugate Chem. 2017, 28, 1189–1193. 10.1021/acs.bioconjchem.7b00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins J. J.; Van de Wouw C.; Wagner K.; Bartels L.; Albada B.; Van Delft F. L. Highly efficient mono-functionalization of knob-in-hole antibodies with strain-promoted click chemistry. ACS Omega 2019, 4, 11801–11807. 10.1021/acsomega.9b01727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruins J. J.; Blanco-Ania D.; Van der Doef V.; Van Delft F. L.; Albada B. Orthogonal, dual protein labelling by tandem cycloaddition of strained alkenes and alkynes to ortho-quinones and azides. Chem. Commun. 2018, 54, 7338–7341. 10.1039/C8CC02638F. [DOI] [PubMed] [Google Scholar]
- Dukler S.; Wilchek M.; Lavie D. Oxidation of tyrosine and its peptides with potassium nitrosodisulphonate. Tetrahedron 1971, 27, 607–614. 10.1016/S0040-4020(01)90729-X. [DOI] [Google Scholar]
- Wilchek M.; Miron T. Mussel-Inspired New approach for polymerization and cross-linking of peptides and proteins containing tyrosines by Fremy’s salt oxidation. Bioconjugate Chem. 2015, 26, 502–510. 10.1021/bc5006152. [DOI] [PubMed] [Google Scholar]
- George A.; Priya G. K.; Ilamaran M.; Kamini N. R.; Ganesh S.; Easwaramoorthi S.; Ayyadurai N. Accelerated Strain-promoted and oxidation-controlled cyclooctyne-quinone cycloaddition for cell labeling. Chem. Select 2017, 2, 7117–7122. [Google Scholar]
- Brulet J. W.; Borne A. L.; Yuan K.; Libby A. H.; Hsu K.-L. Liganding functional tyrosine sites on proteins using sulfur-triazole exchange chemistry. J. Am. Chem. Soc. 2020, 142, 8270–8280. 10.1021/jacs.0c00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi N. S.; Whitaker L. R.; Francis M. B. A three-component Mannich-type reaction for selective tyrosine bioconjugation. J. Am. Chem. Soc. 2004, 126, 15942–15943. 10.1021/ja0439017. [DOI] [PubMed] [Google Scholar]
- Romanini D. W.; Francis M. B. Attachment of peptide building blocks to proteins through tyrosine bioconjugation. Bioconjugate Chem. 2008, 19, 153–157. 10.1021/bc700231v. [DOI] [PubMed] [Google Scholar]
- Tilley S. D.; Francis M. B. Tyrosine-selective protein alkylation using π-allylpalladium complexes. J. Am. Chem. Soc. 2006, 128, 1080–1081. 10.1021/ja057106k. [DOI] [PubMed] [Google Scholar]
- Ohata J.; Miller M. K.; Mountain C. M.; Vohidov F.; Ball Z. T. A three-component organometallic tyrosine bioconjugation. Angew. Chem., Int. Ed. 2018, 57, 2827–2830. 10.1002/anie.201711868. [DOI] [PubMed] [Google Scholar]
- Albada H. B.; Wieberneit F.; Dijkgraaf I.; Harvey J. H.; Whistler J. L.; Stoll R.; Metzler-Nolte N.; Fish R. H. The chemoselective reactions of tyrosine-containing G-protein-coupled receptor peptides with [Cp*Rh(H2O)3](OTf)2, including 2D NMR structures and the biological consequences. J. Am. Chem. Soc. 2012, 134, 10321–10324. 10.1021/ja303010k. [DOI] [PubMed] [Google Scholar]
- Seki Y.; Ishiyama T.; Sasaki D.; Abe J.; Sohma Y.; Oisaki K.; Kanai M. Transition metal-free tryptophan-selective bioconjugation of proteins. J. Am. Chem. Soc. 2016, 138, 10798–10801. 10.1021/jacs.6b06692. [DOI] [PubMed] [Google Scholar]
- Seki Y.; Tanabe K.; Sasaki D.; Sohma Y.; Oisaki K.; Kanai M. Serine-selective aerobic cleavage of peptides and a protein using a water-soluble copper-organoradical conjugate. Angew. Chem., Int. Ed. 2014, 53, 6501–6505. 10.1002/anie.201402618. [DOI] [PubMed] [Google Scholar]
- Antos J. M.; Francis M. B. Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc. 2004, 126, 10256–10257. 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- Ruiz-Rodriguez J.; Albericio F.; Lavilla R. Postsynthetic modification of peptides: chemoselective C-arylation of tryptophan residues. Chem. - Eur. J. 2010, 16, 1124–1127. 10.1002/chem.200902676. [DOI] [PubMed] [Google Scholar]
- Popp B. V.; Ball Z. T. Proximity-driven metallopeptide catalysis: remarkable side-chain scope enables modification of the Fos BZip domain. Chem. Sci. 2011, 2, 690–695. 10.1039/c0sc00564a. [DOI] [Google Scholar]
- Seim K. L.; Obermeyer A. C.; Francis M. B. Oxidative modification of native protein residues using cerium(IV) ammonium nitrate. J. Am. Chem. Soc. 2011, 133, 16970–16976. 10.1021/ja206324q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams T. J.; Reay A. J.; Whitwood A. C.; Fairlamb I. J. S. A mild and selective Pd-mediated methodology for the synthesis of highly fluorescent 2-arylated tryptophans and tryptophan-containing peptides: a catalytic role for Pd0 nanoparticles?. Chem. Commun. 2014, 50, 3052–3054. 10.1039/C3CC48481E. [DOI] [PubMed] [Google Scholar]
- Hansen M. B.; Hubálek F.; Skrydstrup T.; Hoeg-Jensen T. Chemo- and regioselective ethynylation of tryptophan-containing peptides and proteins. Chem. - Eur. J. 2016, 22, 1572–1576. 10.1002/chem.201504462. [DOI] [PubMed] [Google Scholar]
- Alcock L. J.; Perkins M. V.; Chalker J. M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2018, 47, 231–268. 10.1039/C7CS00607A. [DOI] [PubMed] [Google Scholar]
- Couvertier S. M.; Zhou Y.; Weerapana E. Chemical-proteomic strategies to investigate cysteine posttranslational modifications. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844, 2315–2330. 10.1016/j.bbapap.2014.09.024. [DOI] [PubMed] [Google Scholar]
- Akter S.; Huang J.; Waszczak C.; Jacques S.; Gevaert K.; Van Breusegem F.; Messens J. Cysteines under ROS attack in plants: a proteomics view. J. Exp. Bot. 2015, 66, 2935–2944. 10.1093/jxb/erv044. [DOI] [PubMed] [Google Scholar]
- Cobley J. N.; Husi H. Immunological techniques to assess protein thiol redox state: opportunities, challenges and solutions. Antioxidants 2020, 9, 315. 10.3390/antiox9040315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright T. H.; Bower B. J.; Chalker J. M.; Bernardes G. J. L.; Wiewiora R.; Ng W.-L.; Raj R.; Faulkner S.; Vallée M. R. J.; Phanumartwiwath A.; et al. Posttranslational mutagenesis: a chemical strategy for exploring protein side-chain diversity. Science 2016, 354, aag1465-1. [DOI] [PubMed] [Google Scholar]
- Hugo M.; Turell L.; Manta B.; Botti H.; Monteiro G.; Netto L. E. S.; Alvarez B.; Radi R.; Trujillo M. Thiol and sulfenic acid oxidation of AphE, the one-cysteine peroxiredoxin from Mycobacterium tuberculosis: kinetics, acidity constants, and conformational dynamics. Biochemistry 2009, 48, 9416–9426. 10.1021/bi901221s. [DOI] [PubMed] [Google Scholar]
- Peskin A. V.; Low F. M.; Paton L. N.; Maghzal G. J.; Hampton M. B.; Winterbourn C. C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- Bruice T. C.; Sayigh A. B. The structure of anthraquinone-1-sulfenic acid (Fries’ acid) and related compounds. J. Am. Chem. Soc. 1959, 81, 3416–3420. 10.1021/ja01522a066. [DOI] [Google Scholar]
- Salmeen A.; Andersen J.; Myers M.; Meng T.-C.; Hinks J. A.; Tonks N. K.; Barford D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. 10.1038/nature01680. [DOI] [PubMed] [Google Scholar]
- Sohn J.; Rudolph J. Catalytic and chemical competence of regulation of Cdc25 phosphatase by oxidation/reduction. Biochemistry 2003, 42, 10060–10070. 10.1021/bi0345081. [DOI] [PubMed] [Google Scholar]
- Benitez L. V.; Allison W. S. The inactivation of the acyl phosphatase activity catalyzed by the sulfenic acid form of glyceraldehyde 3-phosphate dehydrogenase by dimedone and olefins. J. Biol. Chem. 1974, 249, 6234–6243. 10.1016/S0021-9258(19)42244-8. [DOI] [PubMed] [Google Scholar]
- Gupta V.; Carroll K. S. Profiling the reactivity of cyclic C-nucleophiles towrds electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 2016, 7, 400–415. 10.1039/C5SC02569A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison W. S. Formation and reactions of sulfenic acids in proteins. Acc. Chem. Res. 1976, 9, 293–299. 10.1021/ar50104a003. [DOI] [Google Scholar]
- Poole T. H.; Reisz J. A.; Zhao W.; Poole L. B.; Furdui C. M.; King S. B. Strained cycloalkynes as new protein sulfenic acid traps. J. Am. Chem. Soc. 2014, 136, 6167–6170. 10.1021/ja500364r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Geel R.; Pruijn G. J. M.; Van Delft F. L.; Boelens W. C. Preventing thiol-yne addition improves the specificity of strain-promoted azide-alkyne cycloaddition. Bioconjugate Chem. 2012, 23, 392–398. 10.1021/bc200365k. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Dai P.; Vinogradov A. A.; Gates Z. P.; Pentelute B. L. Site-selective cysteine-cyclooctyne conjugation. Angew. Chem., Int. Ed. 2018, 57, 6459–6463. 10.1002/anie.201800860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galardon E.; Padovani D. Reactivity of persulfides toward strained bicyclo[6.1.0]nonyne derivatives: relevance to chemical tagging of proteins. Bioconjugate Chem. 2015, 26, 1013–1016. 10.1021/acs.bioconjchem.5b00243. [DOI] [PubMed] [Google Scholar]
- Forman H. J.; Davies M. J.; Krämer A. C.; Miotto G.; Zaccarin M.; Zhang H.; Ursini F. Protein cysteine oxidation in redox signalling: caveats on sulfenic acid detection and quantification. Arch. Biochem. Biophys. 2017, 617, 26–37. 10.1016/j.abb.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcock L. J.; Farrell K. D.; Akol M. T.; Jones G. H.; Tierney M. M.; Kramer H. B.; Pukala T. L.; Bernardes G. J. L.; Perkins M. V.; Chalker J. M. Norbornene probes for the study of cysteine oxidation. Tetrahedron 2018, 74, 1220–1228. 10.1016/j.tet.2017.11.011. [DOI] [Google Scholar]
- Alcock L. J.; Oliveira B. L.; Deery M. J.; Pukala T. L.; Perkins M. V.; Bernardes G. J. L.; Chalker J. M. Norbornene probes for the detection of cysteine sulfenic acid in cells. ACS Chem. Biol. 2019, 14, 594–598. 10.1021/acschembio.8b01104. [DOI] [PubMed] [Google Scholar]
- Gupta V.; Carroll K. S. Profiling the reactivity of cyclic C-nucleophiles towards electrophilic sulfur in cysteine sulfenic acid. Chem. Sci. 2016, 7, 400–412. 10.1039/C5SC02569A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcock L. J.; Langini M.; Stühler K.; Remke M.; Perkins M. V.; Bernardes G. J. L.; Chalker J. M. Proteome-wide survey of cysteine oxidation by using a norbornene probe. ChemBioChem 2020, 21, 1329–1334. 10.1002/cbic.201900729. [DOI] [PubMed] [Google Scholar]
- Scinto S. L.; Ekanayake O.; Seneviratne U.; Pigga J. E.; Boyd S. J.; Taylor M. T.; Liu J.; am Ende C. W.; Rozovsky S.; Fox J. M. Dual-reactivity trans-cyclooctenol probes for sulfenylation in live cells enable temporal control via bioorthogonal quenching. J. Am. Chem. Soc. 2019, 141, 10932–10937. 10.1021/jacs.9b01164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmudar J. D.; Konopko A. M.; Labby K. J.; Tom C. T. M. B.; Crellin J. E.; Prakash A.; Martin B. R. Harnessing redox cross-reactivity to profile distinct cysteine modifications. J. Am. Chem. Soc. 2016, 138, 1852–1859. 10.1021/jacs.5b06806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer J. R.; Deming T. J. Preparation of multifunctional and multireactive polypeptides via methionine alkylation. Biomacromolecules 2012, 13, 1719–1723. 10.1021/bm300807b. [DOI] [PubMed] [Google Scholar]
- Kramer J. R.; Deming T. J. Reversible chemoselective tagging and functionalization of methionine containing peptides. Chem. Commun. 2013, 49, 5144–5146. 10.1039/c3cc42214c. [DOI] [PubMed] [Google Scholar]
- Taylor M. T.; Nelson J. E.; Suero M. G.; Gaunt M. J. A protein functionalization platform based on selective reactions at methionine residues. Nature 2018, 562, 563–568. 10.1038/s41586-018-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S.; Yang X.; Jia S.; Weeks A. M.; Hornsby M.; Lee P. S.; Nichiporuk R. V.; Iavarone A. T.; Wells J. A.; Toste F. D.; Chang C. J. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355, 597–602. 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin D.; Wallace M.; Allentoff A. J.; Donnelly D. J.; Gomes E.; Voronin K.; Gong S.; Huang R. Y.-C.; Kim H.; Caceres-Cortes J.; Bonacorsi S. Jr Chemoselective methionine bioconjugation: site-selective fluorine-18 labeling of proteins and peptides. Bioconjugate Chem. 2020, 31, 1908–1916. 10.1021/acs.bioconjchem.0c00256. [DOI] [PubMed] [Google Scholar]
- Geoghegan K. F.; Stroh J. G. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjugate Chem. 1992, 3, 138–146. 10.1021/bc00014a008. [DOI] [PubMed] [Google Scholar]
- Spears R. J.; Fascione M. A. Site-selective incorporation and ligation of protein aldehydes. Org. Biomol. Chem. 2016, 14, 7622–7638. 10.1039/C6OB00778C. [DOI] [PubMed] [Google Scholar]
- Hage D. S.; Wolfe C. A. C.; Oates M. R. Development of a kinetic model to describe the effective rate of antibody oxidation by periodate. Bioconjugate Chem. 1997, 8, 914–920. 10.1021/bc970112o. [DOI] [PubMed] [Google Scholar]
- Sasaki T.; Kodama K.; Suzuki H.; Fukuzawa S.; Tachibana K. N-terminal labeling of proteins by the Pictet-Spengler reaction. Bioorg. Med. Chem. Lett. 2008, 18, 4550–4553. 10.1016/j.bmcl.2008.07.033. [DOI] [PubMed] [Google Scholar]
- Agarwal P.; Van der Weijden J.; Sletten E. M.; Rabuka D.; Bertozzi C. R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 46–51. 10.1073/pnas.1213186110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Kudirka R.; Albers A. E.; Barfield R. M.; de Hart G. W.; Drake P. M.; Jones L. C.; Rabuka D. Hydrazino-Pictet-Spengler ligation as a biocompatible method fort he generation of stable protein conjugates. Bioconjugate Chem. 2013, 24, 846–851. 10.1021/bc400042a. [DOI] [PubMed] [Google Scholar]
- Kudirka R.; Barfield R. M.; McFarland J.; Albers A. E.; de Hart G. W.; Drake P. M.; Holder P. G.; Banas S.; Jones L. C.; Garofalo A. W.; Rabuka D. Generating site-specifically modified proteins via a versatile and stable nucleophilic carbon ligation. Chem. Biol. 2015, 22, 293–298. 10.1016/j.chembiol.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Appel M. J.; Bertozzi C. R. Formylglycine, a post-translationally generated residue with unique catalytic capabilities and biotechnology applications. ACS Chem. Biol. 2015, 10, 72–84. 10.1021/cb500897w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning X.; Temming R. P.; Dommerholt J.; Guo J.; Ania D. B.; Debets M. F.; Wolfert M. A.; Boons G. J.; Van Delft F. L. Protein modification by strain-promoted alkyne-nitrone cycloaddition. Angew. Chem., Int. Ed. 2010, 49, 3065–3068. 10.1002/anie.201000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M.; Sommaruga S.; Mazzucchelli S.; Polito L.; Verderio P.; Galeffi P.; Corsi F.; Tortora P.; Prosperi D. Site-specific conjugation of scFv antibodies to nanoparticles by bioorthogonal strain-promoted alkyne-nitrone cycloaddition. Angew. Chem., Int. Ed. 2012, 51, 496–499. 10.1002/anie.201106775. [DOI] [PubMed] [Google Scholar]
- Temming R. P.; Eggermont L.; Van Eldijk M. B.; Van Hest J. C. M.; Van Delft F. L. N-terminal dual protein functionalization by strain-promoted alkyne-nitrone cycloaddition. Org. Biomol. Chem. 2013, 11, 2772–2779. 10.1039/c3ob00043e. [DOI] [PubMed] [Google Scholar]
- Zeng Y.; Ramya T. N. C.; Dirksen A.; Dawson P. E.; Paulson J. C. High-efficiency labeling of sialylated glycoproteins on living cells. Nat. Methods 2009, 6, 207–209. 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]