

Abstract
The 1,4-conjugate addition reaction between activated alkynes or acetylenic Michael acceptors and nucleophiles (i.e., the nucleophilic Michael reaction) is a historically useful organic transformation. Despite its general utility, the efficiency and outcomes can vary widely and are often closely dependent upon specific reaction conditions. Nevertheless, with improvements in reaction design, including catalyst development and an expansion of the substrate scope to feature more electrophilic alkynes, many examples now present with features that are congruent with Click chemistry. Although several nucleophilic species can participate in these conjugate additions, ubiquitous nucleophiles such as thiols, amines, and alcohols are commonly employed and, consequently, among the most well developed. For many years, these conjugate additions were largely relegated to organic chemistry, but in the last few decades their use has expanded into other spheres such as bioorganic chemistry and polymer chemistry. Within these fields, they have been particularly useful for bioconjugation reactions and step-growth polymerizations, respectively, due to their excellent efficiency, orthogonality, and ambient reactivity. The reaction is expected to feature in increasingly divergent application settings as it continues to emerge as a Click reaction.
1. Introduction
Alkynes that have electron withdrawing substituents in conjugation with the acetylenic moiety (i.e., activated alkynes) are tremendously useful synthetic intermediates as a result of their combination of good bench stability with high reactivity. Although these compounds can react via numerous pathways, including as dienophiles in cycloaddition reactions, they are most renowned for their interaction with nucleophiles in conjugate addition reactions. For alkynes in immediate conjugation with carbonyl moieties such as ketones (ynones), esters (propiolates), or amides (propiolamides), nucleophilic attack commonly furnishes a 1,2-addition product (at the carbonyl carbon) or the 1,4-addition product (at the acetylenic moiety) in addition to other more esoteric products (Figure 1).1 The former reaction is usually induced by hard or strong nucleophiles, including organometallic reagents such as organolithiums or Grignard reagents, while the latter is famously more efficient and achieved using softer nucleophiles.
Figure 1.

Nucleophilic addition reactions to activated alkynes
The 1,4-conjugate addition reaction, which is colloquially referred to as the “Michael addition” or “Michael reaction”, remains an extremely useful synthetic transformation. Although it was first established using carbon-centered nucleophiles in carbon–carbon bond forming reactions,2 the use of heteronucleophiles has continued to gain prominence as a consequence of their superior reaction efficacy and the general ubiquity of heteroatoms in organic compounds. Specifically, the heteronucleophilic 1,4-conjugate addition has been employed extensively for the construction of heterocycles and for highly efficient conjugation to biological compounds for bioorganic and medicinal chemistry applications. This especially applies to amine- and thiol-based nucleophiles which are abundant functionalities in many naturally occurring compounds, including amino acid residues of proteins or biopolymers such as polysaccharides. The high performance of nucleophilic conjugate additions has translated beyond organic transformations and into other disciplines, most notably polymer chemistry. In particular, it has stimulated increased interest in step-growth polymerizations to afford heteroatom-containing polymers of novel compositions, replacing inefficient condensation chemistries (such as acid-catalyzed esterification) which struggle to reach full conversion and lower the molecular weight of the resultant polymers. However, heteronucleophilic Michael addition reactions often afford very high molecular weight, well-defined step-growth polymers under comparatively mild reaction conditions and in some cases the polymerizations may proceed spontaneously at ambient temperature. These considerations are significant because polymer molecular weight is intimately connected to the performance of the end-material properties. More recently, Michael addition reactions have featured in postpolymerization conjugation reactions to afford functional materials and they have also been used to create high performance network polymers such as hydrogels as a consequence of excellent compatibility with aqueous environments.
Regardless of application setting and the overall utility of the transformation, reaction outcomes have historically been divergent, ranging from poor yields even at high temperatures to quantitative conversion at ambient temperatures without the use of a catalyst; the eventual outcome of the reaction is strongly reliant upon several aspects including electrophilicity of the activated alkyne, the strength of the nucleophile, solvent polarity, and the nature of the catalytic species in many cases. Even though some reactions are uncatalyzed, the majority are not, and judicious catalyst choice often has significant bearing on reaction efficiency and selectivity. In this Review, we have focused on reactions that are most congruent with classical Click chemistry criteria that were first outlined nearly 20 years ago.3 While some reported examples satisfy all (or most) Click requirements, others are less “Click-like” but are still highlighted because of either their historical significance in the development of this chemistry or to show where improvements are required to make the reactions more efficient and/or selective. Moreover, we have narrowed our focus to reactions that employ the most abundant and versatile nucleophiles, which includes thiols (thiol-yne), amines (amino-yne), and alcohols (hydroxyl-yne) because these have most successfully translated to other chemistry spheres outside of organic chemistry. Other heteroatom-centered nucleophiles (including boron, phosphorus, selenium, and tellurium) or dinucleophilic species (such as hydrazines, amidines, or hydroxylamines) are beyond the scope of this review because these are comparatively inefficient and/or require inert atmospheres, thus largely precluding them from being reasonably termed as Click reactions and hence inhibiting their widespread use beyond organic synthesis. In addition, they have been well documented in other recent reviews.4,5 Finally, we made an effort to emphasize studies relating to polymeric materials and bioconjugate chemistry as these are rapidly expanding areas in which the heteronucleophilic Michael reaction is envisioned to be increasingly impactful as a consequence of its orthogonality, modularity, simple reaction requirements, and overall efficiency.
1.1. Substrate Overview
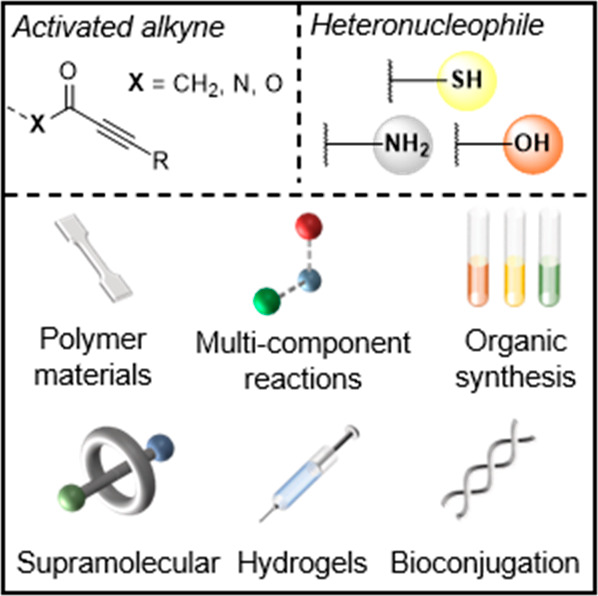
In this brief section, we will provide some context on the availability of substrates (nucleophiles and electrophilic alkynes) that commonly react in Michael reactions. It is by no means exhaustive, rather it is intended to provide a sense of the accessibility of frequently encountered substrates (Figure 2).
Figure 2.

Commonly employed activated alkynes and heteronucleophiles for Click Michael additions.
Because of their potent nucleophilicity, thiols are the most commonly used heteroatomic substrate in Click Michael reactions. Unfortunately, they lack relative abundance in commodity compounds and only naturally occur in a few amino acids. As a result, commercially sourced thiol-containing compounds can be relatively expensive in comparison to alcohols or amines. Still, many desired thiol substrates can be produced from alcohols, albeit in multistep schemes typically via a thioester intermediate6,7 or disulfide,8 but more direct catalytic pathways9,10 are also available. On the other hand, amines are abundant building blocks, particularly diamines due to their commercial importance in polyamides and polyurethanes. Finally, the hydroxyl group is among the most central functionalities in organic chemistry. However, alcohols are generally poorer nucleophiles compared to thiols and amines, often requiring unusual catalysts and/or conditions to react efficiently in conjugate additions.
In general, many of the electron deficient alkynes that are presented in this Review are isolated from readily accessible precursors using convenient synthetic techniques. Propiolates are arguably the most abundant substrate in associated literature. They are commonly synthesized using straightforward acid-catalyzed esterification11 (or analogous protocols such as the Steglich esterification12 for more challenging substrates13) of the commodity chemical propiolic acid or via simple O-alkylation of the carboxylate salt.14 It should be noted that the more reactive propiolyl chloride15 can also be esterified, however it is only transiently stable and can quickly decompose to hazardous byproducts. The synthesis of ynones is also very versatile, with many synthetic pathways available. Transition metal-catalyzed coupling between terminal alkynes and functional acyl chlorides enables a modular route to an expansive library of aromatic or alkyl ynones.16−19 Another prevalent method is the reaction between metalated alkynes and Weinreb–Nahm20 amides to yield desired ynone structures.21,22 However, a more exhaustive account of ynone synthesis is well described in another review.4 The synthesis of propiolamides can often be more challenging than propiolates because the condensation reaction between amines and propiolic acid derivatives competes with the amine conjugate addition (i.e., amino-yne reaction) to the electron deficient alkyne. However, reasonable yields can be obtained via carefully controlled condensation reactions between propiolates and amines.23,24 Additionally, there are other methods that afford propiolamides such as acylation reactions employing carbamoyl chlorides,16,25,26 photocatalyzed decarboxylative ynonylation,27 dehydrogenative coupling of terminal alkynes with formamides,28 or palladium-catalyzed multicomponent reactions among alkynes, isonitriles, and carboxylates.29 More esoteric electron deficient alkynes such as ethynylsulfones are conveniently isolated from sulfonyl chlorides,30,31 sulfonyl hydrazides,32 sulfinates,33,34 or via multicomponent reactions, for example, photocatalyzed coupling of sulfur dioxide and alkynyl bromides.35,36 While their use as a substrate in Michael reactions is comparatively rare next to other activated alkynes, they are still very reactive in reported examples30,31 and this provides ample opportunity for further work. Propiolonitriles are also susceptible to 1,4-conjugate additions37 from heteronucleophiles, however, they are not as common in this context due to a lack of functional modularity.
2. Nucleophilic Thiol-yne Reactions
2.1. Overview and Historical Development
Soon after Arthur Michael’s seminal report on the conjugate addition of carbon nucleophiles to unsaturated substrates,2 Ruhemann et al. discovered the heteronucleophilic conjugate addition reaction between sodium thiophenolate and propiolates to furnish α,β-unsaturated esters (Scheme 1).38 These initial explorations were conducted at ambient temperature with yields in excess of 80% using the sodium thiophenolate nucleophile, but the reaction produced large exotherms that led to side products. Despite the use of strong bases needed to activate the nucleophiles, this study would lay the groundwork to eventually enable the nucleophilic thiol-yne Michael addition to become a versatile Click reaction in modern chemistry.
Scheme 1. First Instance of Heteronuclear 1,4-Conjugate Addition Reaction between Sodium Thiophenolate and Ethyl Phenylpropiolate.38.

In the last several decades, the nucleophilic thiol-yne addition has advanced to a point where most, if not all, Click criteria are fulfilled. Quantitative conversions can be observed under ambient reaction conditions, which is undoubtedly aided by the high nucleophilicity of the thiolate anion and/or the use of highly electrophilic alkynes. Base catalysis has also been a key development in this area and further increased the already large thermodynamic driving force of the thiol-yne reaction, consequently producing exotherms which are frequently reported in the literature.39 But perhaps the most attractive Click feature is the expansive modularity of the thiol-yne conjugate addition. With numerous electron-withdrawing groups, β-carbon substituents, and building blocks containing thiols, the reaction can be exquisitely tailored for a desired outcome.
Stereospecificity is another key aspect of the Click umbrella and is pertinent to the thiol-yne nucleophilic reaction and its unsaturated products. Ruhemann’s initial reports did not indicate the stereochemical configuration of the reaction product from sodium thiophenolate and phenylpropiolate, but a later study suggested the products to be primarily Z-isomers.40 A firmer understanding for the observed reaction stereochemistries arrived many decades later when the anti-addition rule (which largely applies for most conjugate additions) to the electron-deficient alkyne was formulated from a detailed study into the thiol-yne conjugate addition reaction by Truce et al.(41) In this report, the addition of sodium p-toluenethiolate to numerous electrophilic alkynes in methanol established the Z-isomer as the dominant product, but occasionally the E-isomer can be prevalent in small quantities (Table 1). The authors initially assumed that isomerization of the product was responsible for the observed small quantity of the E-isomer (and this is still sometimes a concern that can obfuscate modern reaction outcomes). Heating the thiol-yne Michael adducts to 50 °C in the presence of catalytic amounts of HCl or sodium p-toluenethiolate in methanol afforded higher quantities of the more stable E-isomer under these conditions. However, control experiments performed on the adduct formed from methyl propiolate and p-toluenethiolate at a range of temperatures (0–35 °C) with and without radical inhibitor indicated that both radical and thermal isomerization would not proceed under the initial reaction conditions for the thiol-yne Michael addition. Interestingly, the E-isomer was observed only in adducts containing a carbonyl moiety; it was postulated that the carbonyl group enabled the formation of a stable enol intermediate enabling small quantities of the more thermodynamically stable E-isomer to form directly.
Table 1. Reaction between Sodium p-Toluenethiolate and Various Electrophilic Alkynes in Methanol and Subsequent Isomerization of Unsaturated Product with Sodium p-Toluenethiolate Unless Stated Otherwise41.

| R1 | conversion (%) | initial E/Z | final E/Z |
|---|---|---|---|
| –CN | 100 | 0/100 | 67/33 |
| –SO2C7H7-p | 65 | 0/100 | 100/0 |
| –C6H4NO2-p | 98 | 0/100 | 100/0 |
| –CO2Me | 93 | 8/92 | 78/22 |
| –COMea | 93 | 18/82 | 78/22 |
| –CONH2 | 97 | 13/93 | 77/23 |
Dilute HCl used as isomerization catalyst.
Nevertheless, analysis of other historical references indicating E/Z stereochemistry should be treated with caution as the prevalence of isomerization (resulting from temperatures greater than 50 °C, UV light or base catalysis) can obfuscate the actual obtained stereochemistry in the reaction products.42,43 Since the seminal works by Truce et al., the importance of controlling E/Z stereochemistry in unsaturated reaction products has become a central research theme. Through variation of base catalyst, temperature, and solvent conditions, E/Z isomerization can be effectively tuned, which will be highlighted in many forthcoming examples. Finally, thiol-yne Michael adducts, much like the well-known thiol-ene counterparts,44 are also reversible, undergoing retro-Michael addition reactions45 under various conditions such as high temperatures or alkaline environments. This topic has been a rising research focus in recent years as interest in dynamic covalent chemistries46,47 has significantly grown.
In addition to the nucleophilic pathway for the thiol-yne reaction, radical or thermally driven reactions can also ensue and complicate reaction selectivity and products.48−50 However, it should be noted that the thermally initiated thiol additions were also found to be inhibited in the presence of a radical trapping agent, which suggests that the thermal mechanism is likely facilitated via radical pathways.51,52 As it stands, the radical variant of the thiol-yne reaction has traditionally featured high conversions and good functional group tolerance which led to its growth in both organic and material chemistry alike.53 However, this method is inherently less controllable than the nucleophilic mechanism, primarily due to the facile thiol conjugate addition (higher rate than the initial addition) to the α,β-unsaturated product when in the presence of radicals.54,55 As well as double addition side products, the regio- and stereochemistry of resulting adducts increases the number of possible combinations, and a simple radical-mediated thiol-yne addition may yield up to six distinct reaction products (Scheme 2).56,57 Together, these factors exclude the radical-based thiol-yne reaction from Click categorization, unlike the nucleophilic variant that is a focus of this Review.
Scheme 2. Radical Mediated Reaction of Terminal Alkynes and Monofunctional Thiyl Radicals with the Six Observable Products.

2.2. Trends in Reactivity
This straightforward conjugate addition reaction is now a multifaceted tool that has been utilized in a variety of settings, however, understanding the effects of reaction conditions (including solvent), additives, or catalysts and the nature of the reactants have been key to recent advances. In this section, we have classified each sub-section according to these primary reaction conditions, with the aim of demonstrating how these parameters can influence the reaction outcomes for the thiol-yne reaction and whether Click criteria (related to scope, efficiency, stereospecificity, and orthogonality) can be satisfied.
2.2.1. Substrate Effects
Thiols, and corresponding thiolate anions, are excellent nucleophiles as a consequence of the large atomic radius of sulfur that is able to stabilize charge effectively. With that said, the high nucleophilicity can lead to competing reactions with other functional groups present in the electrophile. Historic reactions of carbonyl-based electrophiles (propiolates and ynones) with thiolate anions often led to competing 1,2-addition reactions.58 In modern examples, these byproducts are rarely reported due to milder conditions that frequently employ only catalytic amounts of base to enhance the nucleophilicity of the thiol. Thiols are notorious for their oxidation to form disulfide species, however, this formation is not kinetically competitive with the reaction rates for conjugate addition reactions and thus does significantly limit their utility in the presence of the open atmosphere which is an important requisite for Click reactions. It has also been noted that some phosphine catalysts (such as dimethylphenylphosphine) can even effectively reduce disulfide bonds under ambient conditions, thus further supporting excellent reaction efficacy under ambient atmosphere.59
A survey of the literature reveals there is a lack of fundamental studies that detail the effect of thiol structure (such as acidity or sterics) on the outcome of nucleophilic thiol-yne reactions. Nevertheless, some comparisons can be drawn from the analogous nucleophilic thiol-ene reaction which has been investigated in greater detail. A detailed mechanistic study of the nucleophilic thiol-ene reaction of hexyl acrylate with various thiols reported that the reaction rate (and overall conversion) was dependent upon the acidity of the thiol (Table 2).60 The addition with ethyl thioglycolate reached near quantitative conversion after 120 s, but the corresponding reaction with hexanethiol only achieved 19% conversion after 500 s. This relationship could be reasonably expected to hold with increasingly acidic thiols (thiophenols and thioacids) as well as correlate to the thiol-yne Michael addition. As it stands, no systemic study details the correlation between the thiol steric environment and the reaction efficiency, however, it was recently investigated for radical-based addition reactions.61
Table 2. Relationship of Thiol Acidity to Reaction Conversion and Rate for the Thiol-ene Michael Addition of Various Thiols to Hexylacrylate60.


There are a variety of acetylenic Michael acceptors that have been investigated for thiol-yne additions which have included the following electron withdrawing groups (including ester,62 ketone,63 amide,64 sulfone,65 and cyano66) in conjugation with the alkyne unit. A noteworthy study investigated the reactivity differences (reaction rates and product stereochemistry) between propiolamides, propiolates, and ynones in base-catalyzed conjugate addition reactions with thiol terminated peptides in aqueous acetonitrile solvent mixtures at ambient temperature (Table 3).67 Quantitative yields were observed for the alkyl and aryl terminal ynones, while methylpropiolate (30%) and phenylpropiolamide (<5%) gave poorer yields. The rate of reaction followed the same trend and correlated to the electron-withdrawing ability of the functional group in conjugation with the alkyne, where ketone > ester > amide. It was also noted that use of excess electrophile led to competing addition to the primary amine of alanine also present in the peptide substrate; this was only observed when using terminal ynones (where R1 = Ph, R2 = H, and R1 = CH2CH2Ph, and R2 = H). In addition to studying the effect of the withdrawing group, the influence of β-carbon substitution was also investigated (Table 3). A lower yield is observed for ynones that possess β-substituted n-butyl groups in neutral water/acetonitrile mixtures (<5%), however, quantitative yields are observed in pH = 8 buffered mixtures. The n-butyl groups seem to have little effect on the electronic nature of the ynone as it relates to reactivity and appear to only present a steric barrier for the thiolate addition.
Table 3. Conversion between a Thiol-Containing Peptide and Various Electrophilic Alkynes in Aqueous Acetonitrile Mixtures67.

| conversion (%) |
|||
|---|---|---|---|
| R1 | R2 | H2O | pH = 8 buffer |
| –NHPh | H | <5 | 60 |
| –OMe | H | 30 | 100 |
| –Ph | H | 100 | 100 |
| –CH2CH2Ph | H | 100 | 100 |
| –Ph | –C4H9 | <5 | 100 |
| –CH2CH2Ph | –C4H9 | <5 | 100 |
The E/Z stereochemical outcome of the unsaturated product is also affected by the electronics of the Michael acceptor. Shiu et al. reacted a protected cysteine with various electrophilic alkynes in aqueous acetonitrile conditions (Table 4).67 Quantitative functionalization of the thiol moiety in buffered water:acetonitrile mixture (pH = 8) was achieved with all electrophiles, while the propiolamide failed to reach full conversion in unbuffered solution. Interestingly, the E/Z stereoselectivity was strongly correlated to the electron density of the acceptor (when the solvent was buffered to pH = 8), with the propiolamide affording the highest Z-content (∼95%) followed by the propiolate (∼89%) and the aryl ynone (83%), however, the alkyl ynone substrate yielded a more mixed stereochemical content (E/Z = 34/66). The same ratio (E/Z = 34/66) was observed for ynones with β-substituted n-butyl, possibly indicating an equilibrium position for the stereochemistry. The overall trend in stereochemistry may be impacted by isomerization reactions, but it is more likely dependent upon stabilization of the enolate intermediates, where bond rotation to the more thermodynamically stable trans-isomer becomes more favorable as the electron withdrawing ability increases (Scheme 3).68
Table 4. Stereochemistry of Reaction Products between Protected Cysteine and Various Electrophilic Alkynes in pH Buffer and Aqueous Acetonitrile67.

|
E/Z |
|||
|---|---|---|---|
| R1 | R2 | H2O | pH = 8 buffer |
| –NHPh | H | 20/80 | 5/95 |
| –OMe | H | 66/34 | 11/89 |
| –Ph | H | 25/75 | 17/83 |
| –CH2CH2Ph | H | 34/66 | 34/66 |
| –Ph | –C4H9 | 20/80 | 34/66 |
| –CH2CH2Ph | –C4H9 | 66/34 | 34/66 |
Scheme 3. Proposed Mechanism for an Allene Intermediate That Dictates the Stereochemical Outcome68.

The influence of the electron withdrawing group is not limited to the initial addition reaction but also to the stability of the Michael adduct itself. Thermally induced reversibility of the reaction has been reported for ynone45,67 and propiolate67 substrates as well as network materials using ynone monomers,30 but propiolamide–thiol adducts are irreversible under similar reaction conditions. The relative electron withdrawing strength of the functionalities can account for the discrepancy, at first glance, but further study is certainly warranted to wholly elucidate the relative reaction barriers and to screen other conceivable electron withdrawing groups (such as sulfones) for their reversibility.
Since the earliest studies, the efficiency of the reaction has shown little sensitivity to β-carbon substituents on the Michael acceptor. Acceptors bearing aromatic69,70 and alkyl71 groups have previously been shown to still react efficiently despite the steric barrier. For example, an n-butyl β-substituted substrate still reached near quantitative conversion, albeit at a notably slower rate than the unsubstituted analogue.67 Another study performed a N-methylmorpholine-catalyzed thiol conjugate addition to ethyl propiolate with varying β-functionality (Scheme 4).72 Using the terminal alkyne afforded the highest yield (91%) with the ethyl ester β-substituted substrate producing slightly lower yield (86%), followed by the n-butyl derivative (yield = 62%). Any form of substitution on the β-position presents a steric barrier, which explains the lower yield in both latter cases. However, the additional ester group also activates the alkyne, making it “doubly” activated and thus more susceptible to nucleophilic attack, which can explain the increased yield for the ester substituent compared to the electron donating butyl group.
Scheme 4. Reaction of 4-Pentyn-1-thiol with Various β-Substituted Propiolates in Dichloromethane and Catalysed by N-Methylmorpholine72.

2.2.2. Solvent Effects
One of the greatest hallmarks of the thiol-yne Michael addition is that the suitable solvent scope is enormous and no additional solvent purification (e.g., drying or degassing) is usually required. Even in the presence of potentially nucleophilic hydroxy groups, such as in aqueous conditions or alcoholic solvents, the thiol-yne reaction can proceed cleanly with no hydroxyl-yne products.73 In one study, water was proposed to facilitate the reaction via hydrogen bonding activation of both carbonyl and thiol substrates because yields are significantly higher in water compared to neat conditions. The analogous thiol-ene reaction is also efficient in water without the need of an added catalyst species.74 The vast range of compatible solvents has led to several studies explicitly examining solvent effects on the Michael reaction. In most cases, solvent choice leads to a rather pronounced effect on both the rate of reaction and the stereochemistry of the α,β-unsaturated reaction products.
Generally, solvents with high polarity (i.e., large dielectric constant, including water) enhance the reaction rate and overall conversion, while less polar solvents retard the reactivity. A detailed investigation of solvent effects on the uncatalyzed, equimolar addition of thiophenol to ethyl propiolate was reported by Randive et al. (Table 5).75 Polar solvents such as methanol, acetonitrile, or dimethylformamide, delivered high yields (>80%) in less than 30 min, and reactions performed in water approached quantitative yields (98%) in just 5 min. Reactions conducted in relatively nonpolar solvents (including toluene and n-hexane) proceed much slower and reached lower overall conversions. A moderate range of E/Z stereoconfigurations (53–97% Z-isomer) were also accessible by changing the reaction solvent; high polarity solvents (water, acetonitrile, and methanol) typically yielded products with high Z-isomer content with the notable exception of dimethylformamide.
Table 5. Reaction between Thiophenol and Ethyl Propiolate in Various Solvents at 0.2 M and Ambient Temperature75.

| solvent | time (min) | yield (%) | E/Z |
|---|---|---|---|
| H2O | 5 | 98 | 3/97 |
| MeCNa | 15 | 90 | 13/87 |
| MeOH | 25 | 85 | 15/85 |
| DMFb | 20 | 80 | 47/53 |
| CH2Cl2 | 20 | 44 | 26/74 |
| THFc | 30 | 30 | 32/68 |
| EtOAc | 25 | 12 | 17/83 |
| acetone | 20 | 74 | 20/80 |
| toluene | 40 | 18 | 25/75 |
| n-C6H13 | 40 | 19 | 33/67 |
MeCN = acetonitrile.
DMF = dimethylformamide.
THF = tetrahydrofuran.
Another study investigated solvent effects of the triethylamine-catalyzed addition of dodecanethiol to ethyl propiolate (Table 6).76 As previously noted, both the conversion and reaction rate were positively correlated to the dielectric constant of the solvent where dimethyl sulfoxide, acetonitrile, and acetone afforded quantitative conversions within 1 h. The stereochemistry of the reaction product was also discovered to be intimately tied to the solvent dielectric with product E/Z content ranging from 98/2 (benzene) to 22/78 (dimethyl sulfoxide). The authors posited that the higher polarity of the reaction medium more effectively stabilized the enolate intermediate to enable anti- over syn-addition during the proton transfer step. On the other hand, the combination of a low polarity solvent with triethylamine could possibly form a hydrogen-bonded pair, so the syn-attack would be more favorable under those conditions.
Table 6. Reaction between Ethyl Propiolate and Dodecanethiol Catalyzed by Triethylamine in Various Solvents at 0.1 M and Ambient Temperature76.

| solvent | dielectric constant | conversion (%) | E/Z |
|---|---|---|---|
| C6H6 | 2.27 | 50 | 98/2 |
| CHCl3 | 4.81 | 96 | 97/3 |
| acetone | 20.7 | 100 | 52/48 |
| MeCNa | 37.5 | 100 | 34/66 |
| DMSOb | 46.7 | 100 | 22/78 |
MeCN = acetonitrile.
DMSO = dimethylsulfoxide.
As previously disclosed, the thiol-yne reaction can be conducted efficiently in water73,75 and/or aqueous mixtures, which is another hallmark of a Click reaction. Moreover, the pH of the aqueous reaction solvent can have a pronounced effect on the reaction rate and overall conversion (Table 3).67 “Greener” recyclable solvents such as polyethylene glycol (PEG)77 have also shown promise for the thiol-yne Michael addition. For this uncatalyzed addition of thiophenol to ethyl 3-phenylpropiolate, yields near 90% were achieved at 60 °C, which falls short of the Click definition but could still be greatly improved by simply employing organobase catalysts. The steadfast efficiency under a wide range of reaction conditions, especially tolerance to aqueous environments, has vaulted the reaction into a similar class as its more popular thiol-ene counterpart and propelled it closer to biorthogonality.78,79
2.2.3. Catalyst Effects
Although there are several examples of the thiol-yne Michael addition proceeding spontaneously, this is only achievable under narrowly defined reaction conditions such as in highly polar solvents and/or alkaline pH or by employing highly acidic thiols.75 However, employing a catalyst greatly broadens the scope of both operative Michael acceptors and thiols, which serves to improve the rate of reaction, total conversion, and stereoselectivity in many cases.
The rudimentary use of stoichiometric sodium to generate an isolated thiolate anion is effective,80 but it offers poor atom economy, more reaction steps, and ultimately does not fulfill Click criteria. However, the use of a catalytic quantity of amine was first described more than a century ago by Ruhemann. In this early report, the reaction of thiophenol with 1,3-diphenylprop-2-yn-1-one was found to be extremely exothermic upon addition of piperidine to only produce the desired unsaturated Michael adduct in quantitative yield at ambient temperature.39 Nevertheless, the mechanism of the amine base-catalyzed thiol-yne Michael addition was not fully elucidated until decades later. These base-catalyzed processes are believed to proceed via two primary pathways, sometimes simultaneously competing in the same reaction, according to the nucleophilicity of the catalyst species. Strongly basic amines can act as a Brønsted–Lowry base to deprotonate the thiol, or it may attack the electron deficient alkyne as a nucleophile to form an allene enolate that can serve to deprotonate the thiol (Scheme 5). For some time, there were concerns that the use of these “nucleophilic” catalysts could form stable catalyst–Michael acceptor adducts to inhibit reactivity.81 However, this matter was largely settled in a thorough study that reported negligible adduct formation for phosphine- or tertiary amine-catalyzed reactions, even when the catalyst was employed in stoichiometric quantities.59
Scheme 5. Organocatalyzed Thiol-yne Pathway Where the Base May Serve As the Initial Nucleophile.

There are several examples that compare different organobases as thiol-yne catalysts; one notable study investigated a range of amine and phosphine catalysts for the reaction between ethyl propiolate and 1-dodecanethiol in order to establish comparative catalyst performance on reaction rate, yield, and stereochemistry (Table 7).76 Products of this reaction could be retrieved in high yields from simple acid washing procedures to remove residual base catalyst. When primary amines such as hexylamine or isopropylamine were used, no conversion was observed because the catalyst was inactivated from competing addition of the amine to the electrophilic alkyne. Only trace amounts of products (reaction conversion <1%) were observed for the secondary amines (diisopropylamine and dicyclohexylamine), presumably as a consequence of similar addition byproducts. The relatively non-nucleophilic tertiary amine (triethylamine) offered a much-improved outcome (85% conversion in 60 min). Finally, the stronger amidine and guanidine bases, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), were far more active, achieving quantitative conversions at low catalyst loadings (1 and 0.1 mol %, respectively) in just 10 min.
Table 7. Reaction of Ethyl Propiolate with Dodecanethiol Employing Different Organocatalysts at 0.1 M and Ambient Temperature76.

| catalyst | loading (mol %) | time (min) | conversion (%) | E/Z |
|---|---|---|---|---|
| n-hexylamine | 10 | 60 | 0 | |
| isopropylamine | 10 | 60 | 0 | |
| diisopropylamine | 10 | 60 | <1 | 50/50 |
| dicyclohexylamine | 10 | 60 | <1 | 50/50 |
| NEt3 | 10 | 60 | 85 | 97/3 |
| DIPEAa | 10 | 60 | <1 | 50/50 |
| DBUb | 1 | 10 | 100 | 34/66 |
| TBDc | 0.1 | 10 | 100 | 10/90 |
| PPhMe2 | 1 | 10 | 100 | 50/50 |
| PPh3 | 1 | 60 | 20 | 30/70 |
DIPEA = diisopropylethylamine.
DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene.
TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene
Trivalent phosphines have also shown promise for the nucleophilic thiol-yne reaction,82−84 shortly after they were discovered to be highly active catalysts in the thiol-ene reaction.85,86 Phosphine catalysts are widely assumed to follow the nucleophilic catalyst pathway (Scheme 5) on account that they lack the requisite basicity to effectively deprotonate many thiols; as such, their nucleophilicity is very important to overall catalytic efficiency.82,83 Hence, dimethylphenylphosphine was found to possess similar activity to the strong amidine bases, such as DBU, reaching quantitative conversion in 10 min.76 However, in the same study, triphenylphosphine was relatively ineffective as a catalyst (only 20% conversion in 60 min), likely as a consequence of its poor nucleophilicity. These results were corroborated in another report where tributylphosphine afforded quantitative yields in 15 min, but the triphenylphosphine-catalyzed reaction proceeded to only 17% conversion after 3 d using the same reaction conditions (ambient temperature in dichloromethane solvent).84
Similar to the observed solvent effects on stereochemistry, the catalyst choice can also play an important role.76 All organobase-catalyzed thiol-yne reactions produced products with mixed stereochemistry or high Z-isomer content (up to 90%) from the thiol anti-addition across an alkyne, with the exception of triethylamine, which afforded high E selectivity (97%). It was postulated that these stark differences could be explained by relative basicity differences where the triethylamine could form an ion-pair with the thiol that would then add via a syn-addition to the alkyne moiety.87 Differences in the stereochemical outcome among other catalysts in the study could be explained by varying nucleophilicity and/or basicity that influenced the relative rates between both mechanistic pathways (e.g., anti- vssyn-addition).
Double thiol addition to an alkyne to form the thioacetal species can also be observed when suitably strong bases are used in sufficient catalyst loadings and the stoichiometry of the thiol is adjusted. For example, employing TBD at 1 mol % loading in conjunction with 2 equiv of dodecanethiol (relative to ethyl propiolate) yields the corresponding thioacetal in near quantitative yield (>97%) at ambient temperature in just 10 min (Scheme 6).76 A similar reaction using tributylphosphine (at 20 mol % loading) with 2 equiv of benzyl mercaptan (relative to methyl propiolate) produces the thioacetal in 80% yield after 24 h at ambient temperature.88 In this example, the second addition of thiol proceeded approximately 1000 times slower, which implies that judicious choice of catalyst loading and/or reaction time can yield exclusively single addition products even when employing highly active nucleophilic catalysts. Finally, increasing the reaction temperature can also distinguish selectivity between single and double addition products. For example, the thiol-yne reaction in water at ambient temperature afforded single-substituted products, but increasing the reaction temperature to 80 °C yielded thioacetal products in suitable yields (67–81%).73
Scheme 6. TBD-Catalyzed Reaction of Ethyl Propiolate with 2 mol equiv of 1-Dodecanethiol to Exclusively Afford the Dithioacetal Product76.

Inorganic-based reagents, including BF389 or NaH,90 have also been investigated to catalyze the thiol-yne reaction, however, the catalysts that arguably show greater promise are heterogeneous species that allow simple separation and potential recyclability. A study into the influence of common lab inorganic solids on the addition of 2-napthalenethiol to methyl propiolate was undertaken by Kodomari et al. (Table 8).91 Neutral alumina achieved a far higher conversion (96%) compared to reactions at all other conditions, including a control without any additive (19%), silica (20%), and even triethylamine (85%). Furthermore, when employing neutral alumina the Z-isomer was heavily preferred, while other catalysts were less stereoselective and afforded a mixture of E/Z isomers. However, although alumina is abundant, a vast quantity (>500 wt %) was employed, which falls short of the high atom economy requirement of a Click reaction. Finally, the reactions were efficient when using aromatic thiols, but alkyl thiols were not as reactive, only reaching <90% conversion after 48 h. Nevertheless, the use of a heterogeneous catalyst like alumina could still be attractive if it retained catalytic activity for batch recycling or flow chemistry processes.
Table 8. Reaction of Methyl Propiolate with 2-Napthalenethiol Catalyzed by Various Inorganic Solids and Triethylamine in Diethyl Ether91.

| catalyst | conversion (%) | E/Z |
|---|---|---|
| none | 19 | 50/50 |
| Al2O3 | 96 | 10/90 |
| SiO2 | 20 | 45/55 |
| NEt3 | 85 | 58/42 |
The synthesis of dithioacetals have also been reported using heterogeneous catalysts.92 A more recent investigation found high surface area magnesium oxide could achieve near quantitative yields of the double addition product (98%) from the reaction of 1,3-propanedithiol to 4-phenyl-3-butyn-2-one.93 The reaction also showed a good tolerance when acceptors with β-subtitutents were employed, such as −SiMe3 (yield = 95%, 1 h) or −C5H11 (yield = 98%, 3 h). As it stands, inorganic catalysts are less efficient than organic counterparts (often employed in at least stoichiometric quantities) and their future role in advancing the thiol-yne reaction will be contingent on the ability to leverage their heterogeneity, i.e., by physically recycling them.
2.3. Applications in Organic Synthesis
In addition to its high efficiency and good orthogonality, the ability to furnish multiple reactive functionalities (especially an electron deficient alkene) in the reaction products has allowed for widespread utility of the nucleophilic thiol-yne reaction in organic synthesis. This has most commonly been exploited in cascade heterocyclic syntheses, but it has also played a role in bioconjugate chemistry where orthogonal reactions can be utilized in tandem.
2.3.1. Heterocycle Synthesis
Versatile functionality, selectivity, and a large thermodynamic driving force are crucial to the synthesis of heterocyclic compounds, making the mild nucleophilic thiol-yne addition an attractive reaction in the synthesis of functional heterocycles. In particular the use of commercially available “doubly activated” alkynes (e.g., diethyl acetylenedicarboxylate), which have gained increasing attention in organic chemistry, especially for the synthesis of sulfur-containing heterocycles.94−96 Additionally, the α,β-unsaturated products from the thiol-yne Michael addition have also been effectively exploited in subsequent intramolecular cyclization reactions to afford heterocyclic compounds.97 However, many of these heterocycle syntheses do not exhibit Click features, and thus this section will not cover every example; instead, the focus will be directed to more efficient and/or informative reports.
An early interesting reaction that demonstrated the versatility of the thiol-yne Michael addition centered on the attempted synthesis of functional benzothiazines from 2-aminothiophenol. Initially, the reaction was investigated using a doubly activated alkyne (dimethyl acetylenedicarboxylate) as the Michael acceptor, where the thiol Michael addition occurs before the in situ intramolecular amidation (Scheme 7-top).94 However, when using a singular activated alkyne (3-phenylpropiolonitrile), the Michael addition is selective for the thiol addition without the intramolecular amino Michael addition side reaction (Scheme 7, bottom).98 After this step, the substrate could be cyclized to yield benzothiazine and benzothiazole depending on the reaction conditions. This example highlights the orthogonal reactivity of the thiol over the amine in the Michael addition and should be an important consideration for reactions between complex substrates that may feature several functionalities.
Scheme 7. Synthesis of Benzothiazines and Benzothiazoles Using Tandem Michael Addition-Cyclization Reactions94,98.

Thiophenes are important building blocks in conjugated materials and medicinal chemistry, but the well-controlled synthesis of functionalized thiophene rings is a historical challenge. Variations of Fiesselmann’s99 thiophene synthesis exploit the functionality control of the Michael acceptor β-carbon substituent, the electronic nature of the alkyne activating group, and thiol nucleophile to effectively yield highly substituted thiophenes. In the seminal report, a propiolate or ynone was reacted with thiolglycolate to form a Michael adduct which then undergoes an intramolecular cyclization.99 Although the Michael addition reaction is high yielding when employing DBU as the catalyst, the subsequent cyclization is not, but it can be improved by changing the base catalyst from DBU to CsCO3 to afford substituted thiophenes in good yields (Table 9).100
Table 9. Fiesselmann’s Thiophene Synthesis Using CsCO3/MgSO4 (1/2) in Methanol at Ambient Temperature100.

| R1 | R2 | yield (%) |
|---|---|---|
| Ph | CH(OEt)2 | 83 |
| Ph | CH2NHBoc | 89 |
| CF3 | CH(OEt)2 | 79 |
| C5H11 | CH(OEt)2 | 84 |
The reaction is also famously insensitive to β-ynone substituents, and careful selection of the electron withdrawing groups has been exploited to afford thiophenes flanked by notoriously difficult-to-access functional groups. For example, amino-functionalized thiophene rings have been synthesized using Fiesselmann’s method by employing propiolonitrile acceptors, however, isolated yields are typically moderate (∼70%) and sodium thiolate nucleophiles are generated instead of using catalytic bases.101 There has been a long-standing interest in synthesizing thiophene derivatives containing organoboron groups for further modification in cross-coupling applications. By installing a trifluoroborate salt on the β-position of an ynone reactant, the Fiesselmann route can afford a borate-substituted thiophene (Scheme 8).102 The initial Michael addition of the thioglycolate to the phenylpropynone borate salt proceeded quantitatively and an efficient subsequent cyclization afforded the thiophene compound in high yield (95%) under ambient conditions. However, the yield of this reaction is heavily contingent on reagents and conditions; for example, without K2CO3 or MeOH the reaction barely proceeds (<5% yield) and the yield fluctuates according to the nature of the ynone. Another representative example for the utility of the thiol-yne reaction is the synthesis of fluorinated thiophenes, which are challenging synthetic targets, but the Fiesselmann reaction can be employed to furnish heavily fluorinated thiophenes.103,104
Scheme 8. Synthesis of a Borate-Substituted Thiophenes102.

Longstanding limitations of the Fiesselmann synthetic pathway included the inability to functionalize each position independently and/or synthesize fully (tetra-) substituted thiophenes. However, these issues were simultaneously addressed by using pyridinium 1,4-zwitterionic thiolates as nucleophiles in the Fiesselmann thiophene synthesis.105 Nevertheless, these reactions do not fully satisfy Click requirements, as exemplified by their modest yields (60–75%) at 85 °C and the overall poor atom economy (due to the pyridine leaving group). Another important hallmark of Click chemistry is employing readily available reagents; a recent example sourced the low-cost and abundant mercaptoacetaldehyde dimer to synthesize thiophene (Table 10).106 Treating a functional ynone species with dimeric mercaptoacetaldehyde and stoichiometric triethylamine leads to a cyclic thioether intermediate possessing a hydroxyl group that can undergo subsequent dehydration via treatment with SiO2 or HCl. Another advantage is the broad range of suitable ynones, however, the use of stoichiometric additives is not ideal, but further exploration of other organobases could optimize reaction conditions to be more compatible with Click requirements.
Table 10. Organobase-Catalyzed Intermolecular Heterocyclization of Ynones with Dimeric 2-Mercaptoacetaldehyde106.

| R1 | R2 | yield (%) |
|---|---|---|
| Me | Ph | 75 |
| Ph | Ph | 91 |
| Ph | n-butyl | 91 |
| Ph | SiMe3 | 80 |
The combination of good orthogonality and rapid reaction rates has made the thiol-yne a popular choice for one-pot multistep syntheses. In one example, the ynone Michael acceptor was synthesized via Sonogashira coupling followed by in situ Michael addition and cyclization to afford a functional thiophene substrate from acyl chlorides (Table 11).107 The reaction demonstrated a good functional group tolerance for the α- and β-carbon substituents on the ynone, as exemplified by yields in excess of 90%. The same approach has also been used to make oligomeric thiophenes, which are known to exhibit aggregation induced emission (AIE).108,109 Despite good reaction orthogonality in this study, as evidenced by tolerance of metal species and diverse functionalities, the use of large quantities (up to equimolar amounts) of organobase catalysts fall short of the high atom economy that is crucial to satisfy Click criteria.
Table 11. Tandem Reaction of Sonogashira Coupling and Thiol-yne Addition to Afford Trisubstituted Thiophenes107.

| R1 | R2 | yield (%) |
|---|---|---|
| p-MeOC6H4 | Ph | 97 |
| p-NCC6H4 | p-(CH3)3C6H4 | 91 |
| 2-thienyl | Ph | 90 |
| p-MeC6H4 | ferrocenyl | 91 |
2.3.2. Bioconjugation Reactions
Thiols are exemplary targeting moieties for bioconjugation reactions as a result of their high nucleophilicity compared to primary or secondary amino groups in peptides and related biomolecules. For example, the thiol of cysteine was determined to be 4 orders of magnitude more reactive as a nucleophile than any other amino acid functionality.110 It should be no surprise that the thiols in biological systems have been exploited via Michael addition reactions with electrophilic alkynes. An interesting application includes functionalizing free cysteine thiol groups in cotton with alkyne-containing reactive dyes to improve the staining efficiency.111 The reaction has even been successfully demonstrated in an analytical assay to quantify sulfur content in certain wines.112
Historically, maleimide was the preferred Michael acceptor used for antibody–drug conjugates113,114 due to its superior reaction rate,60,86 however, stability issues arose due to the reversibility of the resulting Michael adduct because irreversible conjugation was required along with good targeting selectivity of the thiol. Wagner and co-workers screened multiple acetylenic electrophiles for rapid cysteine tagging in aqueous conditions (PBS:DMSO mixture) and observed good yields for 3-phenylpropiolonitrile (94%), N-phenylmaleimide (99%), N-phenylpropiolamide (72%), and phenylpropynone (99%).115 Unlike the adducts formed from the ynone or maleimides, products from propiolonitrile substrates were indefinitely stable under the reaction conditions (Scheme 9).115,116 Another investigation highlighted that 3-phenylpropiolonitrile was far more selective for cysteine conjugation compared to the maleimide substrate, which was found to couple with multiple other amino acids (histidine, glycine, and serine) in non-negligible amounts (≥2.8%).116
Scheme 9. Irreversible Cysteine Tagging Using 3-Phenylpropiolonitrile115.

In medicinal applications, alkyne Michael acceptors have shown great promise in irreversible inhibition of protein targets.117−119 They show particular promise in the inhibition of Bruton’s tyrosine kinase (BTK): a protein linked to non-Hodgkin’s lymphoma. Acalabrutinib is an FDA approved drug for the inhibition of BTK that features a targeting propiolamide Michael acceptor that can bind with the protein substrate.120 Importantly, the drug has shown better targeting selectivity than the analogous drug candidate that contained an alkene Michael acceptor moiety, thus demonstrating the superior chemoselectivity of using acetylenic substrates.
Thiol-containing amino acids (such as cysteine or homocysteine) and small peptides featuring these units (such as glutathione) are important indicator molecules for assays related to disease diagnostics. However, it is crucial to be able to orthogonally discern among different thiol containing biomolecules, and this can be achieved by judicious choice of the alkyne Michael acceptor. In a particularly elegant example, a fluorophore with a flanking ynone functionality was first reacted with a thiol to generate a nonfluorescent complex. Cysteine or homocysteine was then reacted with the complex to release the thiol quenching molecule and generate a new fluorescent adduct that could be accurately quantified by fluorescence spectroscopy (Figure 3).121 This reaction relied on the reversible displacement of the thiol-ynone Michael adduct, which can be dynamic under alkaline pH so in this example the amino acid conjugation was performed at pH = 8.1 to ensure the efficient thiol displacement.
Figure 3.

Fluorescent probe assay based on reversible thiol-yne Michael reaction to quantify cysteine and homocysteine content.121 Reproduced with permission from ref (121). Copyright 2010 John Wiley and Sons.
More recently, Cheng et al. designed a very simple and selective fluorescent tag based on a propiolamide electrophile that reacted preferentially with thiol nucleophiles over amines.122 The resultant thiol-Michael adduct itself is not fluorescent, however, upon an intramolecular cyclization with the neighboring amino group (which is not present in peptide-based thiols such as glutathione), the adduct becomes fluorescent (Figure 4).
Figure 4.
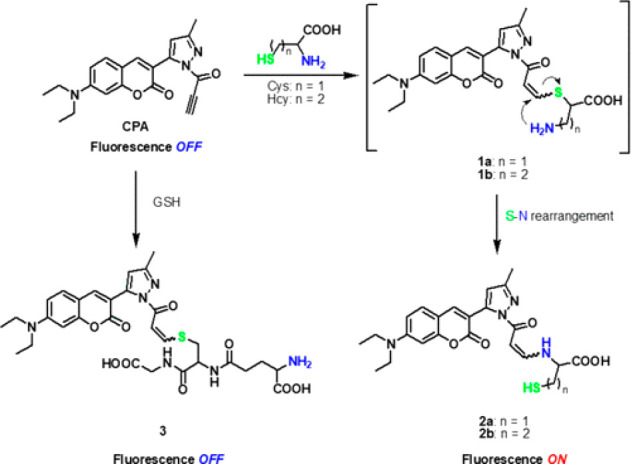
Fluorescent probe assay based on the intramolecular (S–N) exchange of the Michael adduct to quantify cysteine and homocysteine content.122 Reproduced with permission from ref (122). Copyright 2019 American Chemical Society.
2.3.3. Intermediates or Reagents
The thiol-yne Michael reaction has also been featured as a means to furnish valuable synthetic reagents and/or intermediates. For example, phosphonium ylides can be synthesized through the thiol-yne Michael addition of 2-aminothiophenol and di(methyl/ethyl) acetylene carboxylate in the presence of triphenylphosphine.123 Additionally, cyclic thioacetals are versatile umpolungs for carbonyl synthons,124 and they are smoothly produced from successive conjugate additions of propanedithiol or ethanedithiol.92,125,126 Efficient pathways for selective protection/deprotection of thiols remains challenging in organic synthesis. Vinylic Michael acceptors had previously been investigated as thiol protecting groups but did not perform well, especially during the deprotection step, however, the sulfone-based alkyne Michael acceptor, tosylacetylene, was demonstrated as a useful protecting group.127 Specifically, the Michael addition of tosylacetylene to thiols could be achieved in good to excellent yields (80–100%) at ambient temperature. The protecting group was also shown to have excellent reaction selectivity for thiols in the presence of alcohols, and the resulting adduct was stable in acidic and alkaline media, thus highlighting its utility.128 Finally, deprotection to regenerate the thiol was quantitatively achieved by reacting with excess pyrrolidine.
The deprotection of a thioacetic acid-propiolate Michael adduct with CsCO3 has also been used as an efficient route to vinylic thioethers including cysteine and homocysteine derivatives (Scheme 10).129 It is interesting to note that subsequent intermolecular reactions were not reported despite the nucleophilicity of the thiolate species. The thiol-yne reaction was also not quantitative (yield = 79%) in this example despite the low pKa of the thiol, but this can be rationalized by the fact that it is a secondary thiol and imidazole is a relatively poor base. Finally, the addition reactions of pyrrolecarbodithioates to activated alkenes and alkynes was reported as a path to divinylsulfanes, albeit in low yields.130
Scheme 10. Formation of Vinylic Thiols from the Stepwise Thiol-yne Michael Addition and Base-Catalyzed Alkylation129.

In a final example, the thiol-yne reaction between thiols and propiolates or propiolamides was employed for the synthesis of rotaxanes in chloroform using K2CO3 as an additive (150 mol %) (Figure 5).131 The addition reaction using only the acetylenic “axle” Michael acceptors (i.e., without the macrocycle) were successful for both the propiolate (24%) and propiolamide (53%). This was further translated to the reaction with the “wheel” to form the rotaxane in similar yields (19% for propiolate and 35% for propiolamide). Interestingly, the analogous thiol-ene reaction (i.e., employing an acrylate or acrylamide substrate) failed to form the targeted compounds. This example is also interesting because the propiolamide substrates afforded higher reaction yields even though the propiolate is normally accepted as the more reactive of the two moieties.
Figure 5.

Synthesis of rotaxanes using the thiol-yne reaction.131 Reproduced with permission from ref (131). Copyright 2000 American Chemical Society.
2.4. Applications in Polymer Chemistry
Soon after Click reactions gained prominence in organic chemistry, their use was investigated in polymer synthesis. One of the greatest success stories has been the thiol-ene reaction, including both nucleophilic and radical mechanisms, which has been used to synthesize step-growth polymers in high conversion with rapid kinetics and good orthogonality.132,133 Although the thiol-yne reaction is comparatively less developed in polymer chemistry, it offers not only many of the same advantages attributed to thiol-ene polymerizations but a few others as well. Principally, if the thiol-yne reaction proceeds via a nucleophilic pathway the product is an unsaturated polymer which provides opportunities for both postpolymerization functionalization as well as stereochemical structure–property manipulation. To date, much of the limited work on thiol-yne polymerizations has featured radical-based processes but these are inherently less selective (e.g. competition between single and double addition).53 Nevertheless, there are a few examples of nucleophilic thiol-yne polymerizations and its application in polymer chemistry is expanding rapidly.134,135
2.4.1. Step-Growth Polymers
As a consequence of their exceptional reactivity, ynones were the first Michael acceptors to be polymerized with dithiols in a thiol-yne polyaddition.136−138 Bass and co-workers synthesized a series of disubstituted aryl internal ynones from the addition of lithium phenylacetylide to aryl dialdehydes.139 These diynone species were then reacted with dithiophenols in the presence of methylmorpholine to furnish unsaturated step-growth polymers with pendant phenyl groups. The synthesis of conjugated polymers can be challenging and is often restricted to organometallic cross-coupling reactions with a limited number of accessible building blocks.140 However, the straightforward synthesis of the diynone monomers and dithiophenols opens a library of conjugated structures where various stereochemistry, functionality, and diverse heterocycles can easily be incorporated. Unfortunately, these aromatic thiol-yne polymers were difficult to fully characterize as a result of their limited solubility, however, the polymers could be retrieved in high yields (>90%) by a simple precipitation procedure in methanol (Scheme 11).136 Poor overall solubility made typical characterization techniques challenging, nevertheless, the aromatic backbones resulted in thermally robust polymers as evidenced by a high decomposition temperature (Td,10% ≥ 330 °C) with glass transition temperatures (Tg) above 100 °C that were also dependent on regiochemistry, where an increasing quantity of 1,4-aryl substitution over 1,3-substitution in the polymer backbone led to a ∼20 °C increase in the Tg.
Scheme 11. Unsaturated Step-Growth Thiol-yne Polymers Formed between Thiophenols and Internal Diynones136.

Terminal aromatic ynones have also been polymerized with dithiols and these reactions proceeded spontaneously at low temperatures (0–5 °C), thus highlighting the significant difference in reactivity between the internal and terminal ynones. Both types of aromatic polymers (from both internal and terminal ynones) were thermally stable and could be fabricated into useable materials using compression molding at high temperatures but this is somewhat surprising since terminal ynone–thiol adducts have been investigated for their dynamic behavior.45 Finally, another early study investigated the reaction of dithiothreitol with an aromatic ynone monomer using a catalytic quantity of triethylamine to afford unsaturated polymers with a greater solubility than previously reported polymers from aliphatic dithiols.138
More recently, propiolates have emerged as excellent monomers for thiol-yne step-growth polymerizations. The reaction of aromatic dithiols with aromatic dipropiolates (internal and terminal) proceeded to high conversion using a range of amine organobase catalysts (Scheme 12).141 Diphenylamine proved to be an active catalyst to afford a high molecular weight polymer (weight-average molar mass, Mw = 30 kDa) in 98% yield with simple isolation via precipitation. Very recently, it was discovered that the same reaction (varying the monomer only with an additional β-substituted methyl group) can proceed uncatalyzed in dimethylformamide at 60 °C to produce polymers with comparable molecular weights and isolated yields.142 In addition to their high thermal stability, these polymers possess exceptional optical transparency and refractive index (resulting from their high sulfur content), which makes them potentially useful in optics applications, however, there lacks critical analysis of the structure–property relationships. Some of the polymers also exhibit aggregation induced emission (AIE), where the polymers in the solution state are weakly emissive, but as they become less soluble, they aggregate and emit at a higher intensity. These AIE aromatic polymers have potential value for the detection of explosives.142
Scheme 12. Base-Catalyzed Step-Growth Polyaddition of Aromatic Dithiol and Aromatic Dipropiolate141.
Stereochemistry has been a key consideration for the thiol-yne Michael addition for decades, however, its importance was largely unheeded in polymer chemistry. More recently, there has been a push to rationally manipulate stereochemistry in order to influence bulk properties of polymers and materials.143 A series of dipropiolate monomers were synthesized from the Fischer esterification of propiolic acid and commercially available diols. These monomers were polymerized with commercial aliphatic dithiols using various solvents and organobases to modulate the E/Z-content in the polymer backbone (Figure 6a).144 Linear unsaturated polyesters were attained at very high molecular weight (>100 kDa) with excellent control of Z-isomer content (32–80%). The stereochemistry dictated the bulk mechanical properties where the high Z-content polymer was significantly tougher and stronger than the high E polymer. This trend in mechanical strength was attributed to greater polymer chain entanglement and alignment of the Z unit that led to increased crystallization; this is illustrated by wide-angle X-ray scattering (WAXS) (Figure 6b) and endothermic transitions in the differential scanning calorimetry (DSC) thermograms (Figure 6c). The alkenes remaining in the unsaturated polymer were also reacted with sulfur (i.e., vulcanized) to create an elastic material which further exemplifies the utility of the thiol-yne reaction in polymer synthesis. An analogous series of thiol-yne polyester biomaterials also displayed independently tunable mechanical and degradation properties, which addresses a long-standing challenge in synthetic biomaterials, by simultaneously manipulating the Z-isomer content (stereocontrol) and succinate amount (compositional control) of the polymer.145 Previously, controlling one property involved complex changes to the material formulation that would then affect the former property. However, this approach could conveniently afford two materials with similar mechanical strength, but different degradation rates and vice versa. In general, thiol-yne polyesters display excellent mechanical strength, in addition to facile manipulation of bulk physical properties, which can be finely tuned according to alkene stereochemistry and/or monomer choice.
Figure 6.

Thiol-yne polymerization to afford stereocontrolled polyesters.144 (a) Reaction of dipropiolate with 1,6-hexanedithiol to afford unsaturated polyesters. (b) Wide-angle X-ray scattering (WAXS) indicating more order in higher cis content materials. (c) DSC thermograms showing crystallinity in high cis polymers. Reproduced under Creative Commons Attribution License from ref (144).
Another report described the synthesis of high molecular weight (>100 kDa) stereocontrolled unsaturated polyamides from dipropiolamide monomers (Figure 7a).24 The polyamides were extremely processable and malleable, relative to conventional nylons, and the stereochemistry once again influenced thermomechanical properties where the greater Z-content polymers displayed higher glass transition temperatures and greater physical stiffness than their isomeric analogues (Figure 7b-c). Although these trends corroborated the structure–property relationships observed for analogous polyesters, the differences among the properties were significantly less pronounced. This is likely due to the fact that the polyamides were amorphous at all E/Z ratios, whereas crystallinity was found to drive the changes to the bulk mechanical properties for the polyester materials. Nevertheless, the lack of crystallinity in the thiol-yne polyamides was beneficial to processing the materials, especially compared to conventional semicrystalline nylons that require extremely high temperatures to effectively process. Efforts to further improve stereoselectivity in this area toward 100% E or Z polymers continues to be a major aim and should afford even more divergent physical properties within respective polymer classes.
Figure 7.

Thiol-yne polymerization to afford stereocontrolled polyamides.24 (a) Reaction of dipropiolamide with 1,6-hexanedithiol to afford unsaturated polyamide. (b) Plot of Young’s modulus and Glass transition temperature against the E/Z content of the unsaturated polyamides. (c) Physical demonstration of excellent moldability compared to conventional nylons. Reproduced under Creative Commons Attribution License from ref (24).
The nucleophilic thiol-yne Michael reaction predominantly results in a single thiol-addition rather than a second conjugate addition to the unsaturated product, unless a strong base or excessive catalyst loadings are employed. This is often regarded as one of its prime advantages over the radical thiol-yne reaction, however, recent studies have exploited this second conjugate addition to synthesize linear thioacetal polymers.146 Endo and co-workers disclosed the efficient polyaddition of 1,4-bis(mercaptomethyl)benzene and methyl propiolate catalyzed by large quantities of tributylphosphine (50 mol %), but the barrier for second addition led to slow kinetics and poor overall conversions, only affording low molecular weight polythioacetals (<10 kDa).146 Nevertheless, thioacetal polymers have the potential to offer unique degradation pathways and optical properties due to their susceptibility to hydrolysis and high sulfur content. A similar system exploited β-substitution of ethyl propiolate to furnish a hydrophobic fluorinated polythioether.147 In another example, a “doubly” activated alkyne (dimethyl acetylenedicarboxylate) was reacted with several dithiols using the strong organobase, TBD, as the catalyst in chloroform (Figure 8).148 High molecular weight (>30 kDa) linear polymers were afforded in under 60 min and isolated by simple precipitation in acidified methanol. As a consequence of the high electrophilicity of the Michael acceptor and the strength of the organobase, the double addition was favored, resulting in linear polythioethers. A few other acetylenedicarboxylate esters (ethyl and tert-butyl) were also successfully polymerized with the potential to use other moieties to expand possible pendant functionality. Among the dithiols that were tested, a striking discovery found that both 1,2-ethanedithiol and 1,3-propanedithiol both favored intramolecular cyclization over polymerization to afford the dithiane product. Importantly, these polymers were found to be dynamic and could be effectively depolymerized by the subsequent addition of monofunctional thiol and TBD. These studies perfectly highlight the scope of the thiol-yne Michael addition, and specifically how modular substrate design can provide access to a broad range of versatile polymers for different applications.
Figure 8.

Polyaddition of dithiol with doubly activated alkyne with dithiol using a TBD catalyst.148 Reproduced with permission from ref (148). Copyright 2019 American Chemical Society.
The thiol-yne Michael addition is also particularly suited for multicomponent tandem reactions because of its orthogonality. This strategy has been utilized by Tang and co-workers to create functional linear polymers (Scheme 13).149 Tandem Sonogashira coupling and conjugate addition reactions employing various combinations of acyl chlorides, phenylacetylenes, and thiols (where at least two of the species were disubstituted) formed linear polymers in a one-pot reaction. The high molecular weight linear polymers (>30 kDa) were retrieved by a filtration/precipitation protocol and isolated in near quantitative yields (>95%). The polymers showed varying UV absorption behavior and different refractive indexes, and this is likely to be related simply to the specific aryl functionalities that were employed. Nevertheless, the reaction illustrates the orthogonality of the Michael reaction with other catalysts and functionalities present. However, it should be noted that the Sonogashira coupling reaction is sensitive to atmospheric oxygen and therefore deviates from Click criteria.
Scheme 13. Multicomponent Reactions Combining Sonogashira Coupling and Thiol-yne Addition to Afford Various Linear Polymers149.

The rapid reaction rates and high efficiency of the thiol-yne Michael addition is also represented by its use in postpolymerization functionalization reactions. A polymer with propiolate-containing pendant groups was smoothly reacted with thiol in the presence of NEt3 to afford the single addition thiol-Michael adduct.150 A subsequent second addition with a different thiol can be achieved using 1 equiv of monofunctional thiol and employing a stronger base such as TBD. Both reactions are observed to proceed to completion and are purified by precipitation to isolate the functionalized polymer in high yields (>90%). The same principle of stepwise thiol additions has also been applied to polyesters containing an activated internal alkyne along the polymer backbone achieving quantitative conversions when using TBD at 25 mol % loading.151
2.4.2. Network Polymers
Hydrogels are important materials comprised of cross-linked (network) polymers that are swollen with aqueous media, with critical applications ranging from food to healthcare. Hydrogel synthesis has historically turned to natural polymers (such as alginates), but hydrophilic synthetic polymers are increasingly being used. In particular, the thiol-yne Michael reaction has proven to be highly useful for hydrogel synthesis due to its great efficiency and tolerance to aqueous environments.152−158 Multiarm polyethylene glycols (PEG) were functionalized with thiol and propiolate moieties via straightforward Fischer esterification and then reacted with one another in an aqueous solution to form robust hydrogels in under 30 min. This simple approach enabled tunable mechanical properties and mesh sizes by selecting three- or four-arm PEG units and varying the length of two-arm substrates (Figure 9).155 The same thiol-yne system has been exploited in conjunction with tetrazine–norbornene adducts to form interpenetrating dual network hydrogels.158 Both Click reactions proceeded orthogonally to form independent networks that were considerably tougher than either single network system.
Figure 9.

A thiol-yne hydrogel system from multiarm PEG-thiols and PEG-propiolates.155 Reproduced with permission from ref (155). Copyright 2017 American Chemical Society.
Using a similar system, nonswellable hydrogels153 were also obtained, which is important for tissue engineering because uncontrolled swelling behavior can diminish mechanical performance of the material. A hydrogel based on the thiol-yne system still has residual electron deficient alkenes within the network; these can be further functionalized. As an example, an antimicrobial PEG-based thiol-yne hydrogel was formed via postsynthetic conjugation with a thiol-terminated antimicrobial peptide.157 Compatibility with biological systems has already been highlighted for the thiol-yne reaction, but this also enables the versatility of incorporating numerous natural polymers (such as alginate, chitosan, or hyaluronic acid) into synthetic hydrogels.152,156 Thiol-yne hydrogels that incorporate these natural polymers have exhibited self-healing behavior and high stretchability rendering them superior to hydrogels of similar composition (i.e., the same biopolymer) but not cross-linked with thiol-yne chemistry.152
The reversibility of the second thiol addition in the thiol-ynone reaction has been used to create reprocessable cross-linked materials.30,159 Similar to other dynamic Michael addition systems, the reversibility of the reaction is triggered by base or heat. This feature has been exploited to deliver self-healing injectable hydrogels by cross-linking multiarm PEG thiols with a small molecule alkyl ynone.159 The dynamic nature of the thiol-ynone reaction was triggered by high pH buffer solution, enabling the hydrogels to undergo shear-thinning under stress, meaning they could be quickly injected using a syringe only to reform into a “solid” material after delivery (i.e., when the stress was removed). A similar thiol-ynone double addition reaction was also employed to afford covalent adaptable networks (CANs) based on reversible cross-linking. By varying the substituents on the ynone monomers, the exchange rates of the second thiol addition were tuned by several orders of magnitude to create materials with diverse rheological properties that were important for their reprocessing behavior.30 Model reactions of the thiol exchange process showed that the presence of electron withdrawing groups on the aromatic unit increased the exchange rate of the thiols and ultimately reduced the requisite reprocessing temperature for the bulk materials (Figure 10).
Figure 10.
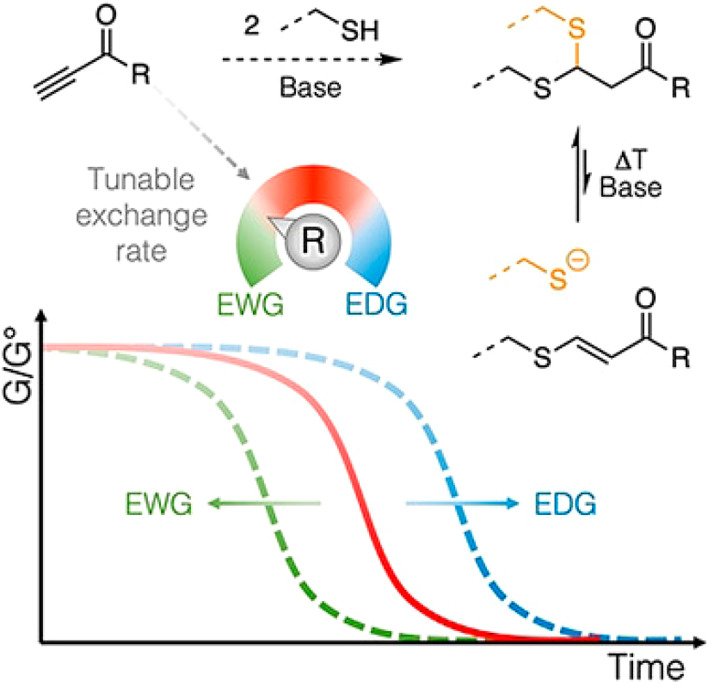
Dynamic exchange of dithioacetals influenced by electronic differences among ynone substrates.30 Reproduced with permission from ref (30). Copyright 2020 John Wiley and Sons.
3. Amino-yne Reactions
3.1. Overview and Historical Development
The amino-yne Michael reaction is broadly similar to the ubiquitous thiol-yne motif, where a primary or secondary amine attacks the β-carbon on an electron deficient alkyne, although differences lie in the catalysis because many amino-yne reactions proceed spontaneously (i.e., without an added catalyst) at ambient temperatures. As such, the efficiency of the reaction rivals, and in some cases surpasses, the thiol-yne Michael addition as a result of the excellent nucleophilicity of the amine functionality.160 Consequently, the reaction has been adopted in numerous materials and biological applications. Furthermore, the reactivity and versatility of the enamine Michael adduct itself has found good utility in organic synthesis.
The first amino-yne nucleophilic addition was reported in 1899 by Ruhemann and co-workers where the reactions of diethylamine (or piperidine) with diethylacetylene dicarboxylate and ethyl phenylpropiolate were reported.161 The reactions were described as “violently exothermic” and proceeded in the absence of a catalyst at ambient temperature. This early work originally focused on employing secondary amine nucleophiles to avoid a second addition of the nucleophilic enamine Michael adduct. Subsequent work featured primary and multifunctional amines to induce intramolecular condensation reactions after the initial conjugate addition to furnish heterocyclic compounds such as pyrones, pyridines, and cyclic amides.162,163 Other early work in the area also expanded the scope of Michael acceptors to propiolonitriles164 and ynones165 in equally high efficiency to the propiolates. It should be noted that nucleophilic amino-yne additions can also proceed without the requisite of an electron withdrawing functionality in conjugation with the alkyne, i.e., electronically neutral alkynes are sometimes reactive as well. The addition of amines and ammonia to nonactivated alkyl alkynes has been known for decades but typically requires harsh reaction conditions including high temperatures (>300 °C) and pressures or heavy metal catalysts.166,167 As such, these transformations are not discussed in further detail.
Modern organic synthesis has continued to focus on transition metal-catalyzed intramolecular (and intermolecular168) cyclization reactions of amines to electron deficient alkynes (primarily ynones), and this area has been critically reviewed in recent years.4,5 Even though this section will feature overlap with some of this content, it will ultimately focus on highly efficient reactions presenting with Click features, emphasizing those with (near) quantitative yields, high selectivity, and/or simple reaction conditions. Finally, the section will be presented slightly differently than the thiol-yne reaction because there are not many conclusions to be made about reactivity trends according to reaction conditions and/or substrate scope. There will be a brief overview on reactivity, but subsequent parts of the section will be divided by catalyst class for organic transformations and then conclude with emerging applications in polymer and materials chemistry.
3.2. Trends in Reactivity
This convenient reaction has become a mainstay in modern organic chemistry, however, a detailed understanding of the effects of reaction conditions (including solvent, additives, and/or catalysts) and the structure of the reactants on reaction outcomes has not been systematically investigated. This is problematic because the efficiency of the reaction varies widely, sometimes proceeding spontaneously at ambient temperature or instead requiring the use of metal catalysts at elevated temperatures. Nevertheless, we will attempt to draw meaningful comparisons within and among reports, especially as related to the aforementioned reaction conditions.
The description of alkene stereochemistry in the amino-yne Michael adducts is notably less discussed in the literature because it is heavily contingent on internal hydrogen bonding and steric considerations rather than charge stabilization of the intermediate species as in the thiol-yne analogue. This means that there is often less opportunity to manipulate the stereochemical outcomes unless the amino nucleophile is substituted. Nevertheless, many conjugate additions of primary amines to acetylenic Michael acceptors typically select for the Z-isomer, especially when propiolates, propiolamides, and ynones are employed due to intramolecular hydrogen bonding with the adjacent carbonyl moiety, while secondary amines tend to be less stereoselective and sometimes favor the E-isomer (Scheme 14).169−171 This of course is not a universal characteristic, as the influence of the Michael acceptor can also change the stereochemistry and isomerization of the Michael adducts is possible, both of which can obfuscate stereochemical trends.
Scheme 14. Generic Scheme Showing the 1,4-Conjugate Addition of 1° and 2° Amine Nucleophiles to Carbonyl-Containing Michael Acceptors.

3.3. Applications in Organic Synthesis
3.3.1. Metal-Catalyzed Reactions
The metal-catalyzed amino-yne reaction has been used to a great extent to synthesize N-containing heterocycles, primarily substituted pyrroles or indoles, from precursors containing acetylenic units and amines. The first report of pyrrole synthesis via conjugate amino-yne addition was reported by Nozaki and co-workers in 1981 using a palladium catalyst to synthesize pyrroles in quantitative yields.172 Gabriele et al. expanded on this motif to form highly substituted pyrroles and quinolines at similar efficiency using a variety of palladium(II) and copper(II) catalysts (Scheme 15).173−177 The use of precious metal gold- and silver-based catalysts have also been employed with analogous reaction outcomes.178,179
Scheme 15. Transition Metal-Catalyzed Intramolecular Amino-yne Cyclization to Form Pyrroles173.

In 2010, Kievanloo et al. developed a one-pot quinoxaline synthesis (in yields of up to 92%) by Sonogashira coupling and subsequent intramolecular amino-yne coupling using a Pd/Cu catalyst at 70 °C.180,181 A palladium-catalyzed synthesis of isoindolines using Sonogashira coupling has also been reported with up to 94% yield in water at 50 °C.182 The copper(I)/triethylamine-catalyzed synthesis of pyrazoles in yields up to 99% was accomplished from the reaction of various ynones and substituted hydrazines (Scheme 16a).183 Copper(II) triflate without a cocatalyst was also employed to catalyze the conjugate addition of anilines and nitro-substituted alkynes at ambient temperature with similar efficiencies (Scheme 16b).184 These examples illustrate some of the more moderate reaction conditions (such as water as a solvent and/or low reaction temperatures) in the area and thus align more closely with Click criteria.
Scheme 16. Synthesis of 5-Membered Nitrogen Heterocycles from Copper-Catalyzed Intermolecular Amino-yne Cyclizations.

(a) Pyrazoles via formation and isolation of hydrazone intermediate;183 (b) pyrrole via a one-pot method.184.
Six-membered nitrogen-containing heterocycles have also been intensely investigated using the amino-yne reaction, with a few examples reporting good efficiency. A one-pot strontium- and barium-catalyzed synthesis of isoquinolines by cascading amino-yne and amino-ene reactions was reported, achieving yields of up to 99% but at a higher temperature (≥130 °C) which decreases its compatibility with Click criteria (Scheme 17a).185 The Gouault group described the total synthesis of (−)-epimyrtine, wherein one of the critical steps was a highly selective gold-catalyzed intramolecular amino-yne cyclization of an ynone to form a six-membered pyridone from a secondary amine (Scheme 17b).186 Although this reaction step was not quantitative, this is likely due to steric considerations from both the amino functionality and the ynone moiety.
Scheme 17. Intramolecular Amino-yne Cyclization Reactions to Form Six-Membered Nitrogen-Containing Heterocycles.

(a) Isoquinolines from cascading amino-yne/amino-ene reactions;185 (b) pyridone via gold-catalyzed reaction.186
Ribecai and co-workers used alkali metal salts or organobases to produce indoles via intramolecular cyclization with yields of up to 99% and 90%, respectively, however microwave heating at 200 °C diminishes the experimental simplicity.187 The Verma group later reported KOH to catalyze the conjugate addition of imidazole with electron deficient alkynes at yields of up to 84%.188 This example is particularly interesting because imidazole is sometimes employed as an organobase catalyst for other conjugate addition reactions and its nucleophilicity is comparatively low compared to other amine nucleophiles.
3.3.2. Organocatalyzed Reactions
Transition metal complexes are undoubtedly the catalyst of choice for amino-yne reactions, but organocatalyzed reactions have started to appear in the previous decade. Svete and co-workers developed a series of peptide syntheses employing N-methylmorpholine to catalyze the reaction of primary amines with ynone-terminated peptides at low temperatures (from 0–25 °C), affording vinylogous peptides in up to 92% isolated yields and with 100% E-selectivity or 87% Z-selectivity depending on the functional group.189N-Methylimidazole has also been used to catalyze the synthesis of carboxylated pyrroles from propiolates, achieving yields of up to 87% (Scheme 18).190 Proline-catalyzed activation of Michael acceptors is commonly encountered in organic synthesis, and this was employed to facilitate the reaction between an ynal (the aldehyde equivalent of an ynone) and an aniline derivative in quantitative yields at ambient conditions.191
Scheme 18. N-Methylimidazole Catalyzed Pyrrole Synthesis in Water from Two Activated Alkynes and an Amine Nucleophile190.

Analogous to thiol-yne reactions, trivalent phosphines have also shown efficacy as catalysts for the amino-yne Michael reaction. Kwon and co-workers have reported several phosphine-catalyzed nitrogen-containing heterocycles, synthesized in acetonitrile at ambient temperature and often in quantitative yields.192−194 Numerous benzannulated nitrogen heterocycles (such as indoles or benzimidazolines) were synthesized from simple dinucleophiles and acetylenic Michael acceptors by employing 1,3-bis(diphenylphosphino)propane at 20 mol % (Scheme 19a).192 Generally, the reactions were quite efficient (yields ranging from 82–96%), but trends regarding reaction outcomes according to electrophile (i.e., ynone vs propiolate) did not emerge. A one-pot synthesis of quinolines, with near quantitative yields observed for some substrates, was reported from the triphenylphosphine-catalyzed conjugate addition between o-tosylamidobenzaldehydes with several classes of activated acetylenes (Scheme 19b).194 Generally, the aryl ynones afforded the best yields (86–99%) followed by the alkyl ynone (88%) and alkyl propiolate (61%), which is not unexpected considering their relative electrophilicities. The efficiency is quite remarkable considering that triphenylphosphine is relatively non-nucleophilic, compared to trialkylphosphines, and a catalytic nucleophilic mechanism is presumably invoked for this case.
Scheme 19. Phosphine-Catalyzed Intramolecular Amino-yne Reaction to Form Nitrogen Heterocycles.

(a) Synthesis of saturated heterocycles via a double Michael addition using a diphosphine catalyst;192 (b) synthesis of quinolines using triphenyl phosphine.194
3.3.3. Uncatalyzed Reactions
Spontaneous amino-yne reactions are also documented in the literature, especially for the highly nucleophilic primary or secondary alkyl amines. However, the extreme reactivity can sometimes lead to deleterious side reactions (such as “over-addition”), especially for primary amines that form a nucleophilic enamine Michael adduct. Together, these features detract from Click characteristics. Anilines are less nucleophilic than alkyl amines, but they have also been reported in spontaneous Michael addition reactions. En route to quinolines, an uncatalyzed, solvent-free conjugate addition of 2-aminobenzaldehyde with an alkyl ynone proceeded in 61% yield with predominantly Z-isomer formation (Scheme 20a).195 An iron-catalyzed one-pot method was further employed without isolating the intermediate enamine Michael adduct (or vinylogous amide) and afforded substituted quinolines in moderate to excellent yields ranging from 52–94% with aryl ynones being more efficient (Scheme 20b).195
Scheme 20. Reaction of o-Aminoarylaldehydes and Ynones195.

(a) Spontaneous amino-yne reaction to form a vinylogous amide with mixture of stereochemistry; (b) iron-catalyzed synthesis of quinolines via a vinylogous amide intermediate.
Recently, Tang and co-workers reported the uncatalyzed reaction of primary amines with a dual-activated alkyne to form maleimides in ethanol at ambient temperature with isolated yields up to 88%.196 This result is particularly encouraging because maleimides are usually formed from harsh condensation reactions requiring high temperatures and/or the use of catalysts. In 2010, the Sekiguchi group reported an uncatalyzed amino-disilyne reaction at cryogenic temperatures with a yield of 78%.197,198 The Michael addition reaction of various nucleophiles (anilines, amines, thiols, or thiophenol) and 4-hydroxy-2-alkynoates were investigated in water at ambient temperature under ultrasound sonication (Scheme 21).70 When amines were employed instead of thiols, a cascade conjugate addition/lactonization process afforded 4-amino-furan-2-one derivatives. The initial conjugate addition step was also most efficient when employing thiophenol derivatives, however primary amines also afforded good yields compared to alkyl thiols and anilines. Finally, the importance of water as the solvent was demonstrated and reactions conducted in acetonitrile required heating to 80 °C in order to obtain satisfactory yields.
Scheme 21. Conjugate Addition of Thiols or Amines to Alkyl 4-Hydroxy-2-alkynoates to Afford Michael Adducts or Substituted Lactones from a Conjugate Addition–Cyclization Reaction70.

A particularly powerful example of the amino-yne reaction is the efficient bioconjugation to native molecules without tedious prefunctionalization of the targets.199−201 An ynone substrate was quantitatively tagged onto several different biomolecules (including peptides, proteins, and polysaccharides) via the amino-yne reaction. Importantly, the reactions reached completion within minutes at ambient temperatures and also proceeded with high stereoselectivity (∼92% Z-isomer formation). In this study, the amino-yne reaction was more efficient than the analogous thiol-yne or hydroxyl-yne reactions, from reaction with thiols or alcohols, respectively, with the targeted biomolecules (Figure 11).199 The same group also reported the use of amino-yne Click reactions to efficiently immobilize proteins onto cell surfaces, representing a forward step toward biorthogonality.200 In another contemporary example, pectin (which is an enzyme) was conjugated to an amino-functionalized polymer with up to 90% efficiency under ambient conditions and in the absence of a catalyst.201 Immobilization of the enzyme greatly improved its shelf life with 70% activity still observed after 60 d in addition to increased thermal and pH tolerances.
Figure 11.

Quantitative bioconjugation using the amino-yne reaction between a fluorescent ynone substrate and amino groups on native molecules.199 Reproduced under Creative Commons Attribution License from ref (199).
3.4. Applications in Polymer Chemistry
3.4.1. Step-Growth Polymers
Just over 30 years ago, Bass and co-workers reported a new uncatalyzed step-growth polymerization between aromatic diynones and diamines to afford unsaturated nitrogen containing polymers.139,202 Although the polymers were isolated in near-quantitative yields, the reaction conditions required high temperatures of 100 °C. Soon after, Wilbur and Bonner extended this motif to afford aliphatic poly(enamine ketone)s with ≥100% Z-isomer selectivity at ambient temperatures, which brought the reaction more in line with Click criteria.137 More than two decades later, the polyaddition reaction was again investigated by Tang and co-workers, where they presented a copper-catalyzed amino-yne Click reaction using internal dipropiolate substrates to form a small library of moderate molecular weight (up to 12.8 kDa) polymers under a variety of reaction conditions (Scheme 22a).203 The use of CuCl was found to be catalytically superior to other salts, and interestingly, the Z-content decreased as the size of the halide increased (CuCl, E/Z = 12/88; CuBr, E/Z = 18/82; CuI, E/Z = 61/39). For the copper-catalyzed reaction: solvent effects on stereochemistry were also scrutinized where the Z-isomer within the unsaturated polymer ranged from 50% (dimethylformamide or DMSO) to 88% (tetrahydrofuran), however, the stereochemistry was not strongly correlated to solvent polarity, and the bulk reaction produced the highest stereoselectivity (E/Z = 6/94). The polymers were found to have good UV shielding efficiency below 400 nm and nearly perfect transmittance of visible light. The same group subsequently developed a room temperature, spontaneous version of this reaction in dichloromethane to afford high molecular weight polymers (up to 64 kDa) with quantitative conversion after only several hours (Scheme 22b).204 Dissimilar to the metal-catalyzed protocol, this reaction was predominantly selective for the E-isomer, which better satisfies Click requirements. Interestingly, the addition of triethylamine afforded a polymer with more mixed stereochemistry, although isomerization to 100% E-content was observed when the reaction mixture was left at ambient temperatures for prolonged periods of time. Finally, these two examples also superbly demonstrate the importance of the Michael acceptor on reaction efficiency: the terminal propiolate proceeds without a catalyst at ambient temperature, whereas the former requires high temperatures and the use of a catalyst to yield only modest molecular weight polymers.
Scheme 22. Amino-yne Polyadditions Using Dipropiolate Monomers.

(a) Copper-catalyzed reaction at elevated temperature;203 (b) polymerization without an added catalyst at ambient temperature.204
A variety of different polymer systems were reported soon after, including high molecular weight poly(β-aminovinylsulfone)s with perfect stereoregularity (100% E) from ethynylsulfone monomers.31 This study is also interesting because it appears that the reaction is stereoselective for the E-isomer even when employing primary amine monomers, however, a postpolymerization isomerization to the thermodynamic product cannot be ruled out as a contributing factor. Another recent report described the spontaneous polymerization between aryl ynones and various 1° or 2° diamines205 and is similar to the substrates employed by Bass and co-workers in their seminal studies.202 Nevertheless, this work did improve the molecular weight of the resulting polymers (up to 49 kDa) and investigated the polymer stereochemistry where Z-isomeric polymers were obtained when employing 2° amines and E-isomers for 1° amines, in addition to high thermal stabilities (Tds > 300 °C). In a related example, a multicomponent tandem polymerization employing hydrazines, ynones, and terephthaloyl chloride yielded conjugated polypyrazoles in high yields, all of which had high decomposition temperatures (Tds > 350 °C) and possessed photoluminescent properties.206 Ni and co-workers have also used an amino-functionalized doxorubicin prodrug to end-cap an amino-yne polymer for drug delivery applications which proceeded at 35 °C in dichloromethane (Scheme 23).207 Recent trends in the field have pivoted away from the use of chlorinated solvents to more benign solvents such as tetrahydrofuran205 or water (in vivo)208 and even solvent-free,209 which are more congruent with Click reaction principles.
Scheme 23. Amino-yne Polymers End-Capped with Doxorubicin for Drug Delivery Applications207.

Finally, an interesting example featured a sequence-controlled polymer by employing sequential tandem amino-yne/thiol-ene reactions (Figure 12).210 Amine-terminated sequence-controlled oligomers were first obtained by exploiting the orthogonality between the two nucleophiles, i.e., amine vs thiol monomers, before a subsequent step-growth polymerization with a dipropiolate monomer to afford high molecular weight (up to ∼60 kDa) materials in dimethylformamide at ambient temperature (up to 82% yield). Although this example loses some Click characteristics, it does showcase good modularity and applicability for the Michael reaction.
Figure 12.

Sequenced controlled polyesters produced by sequential amino-yne/thiol-ene reactions.210 Reproduced with permission from ref (210). Copyright 2018 John Wiley and Sons.
3.4.2. Network Polymers
As a consequence of its exceptional efficiency and compatibility with aqueous environments, the amino-yne reaction has recently been employed for the synthesis of hydrogels.211,212 Injectable and pH responsive hydrogels based on cross-linking the free amino groups within the biopolymer chitosan provided a platform for tunable drug release behavior (Figure 13).211 Importantly, the amino-yne cross-linking reaction proceeded spontaneously under physiological conditions which is critical for tissue engineering applications. Langer and co-workers also utilized the amino-yne reaction to afford synthetic hydrogels based on propiolate terminated multiarm PEG precursors.212 The study was also thorough in its investigation of the substitution pattern for the multifunctional amine nucleophiles. Interestingly, the use of 2° amines efficiently formed gels under all conditions, while 1° amines resulted in gelation at only a high solids content (30 wt %). The greater efficiency of 2° amines vs 1° amines is similar to the trend observed in previously referenced reports for step-growth polymers.203,204 Nevertheless, the gels displayed good physical properties that could enhance their biocompatibility including fracture stresses ranging from 16 to 65 kPa, compressive modulus of 6 to 27 kPa, and the ability to swell up to 300%.
Figure 13.
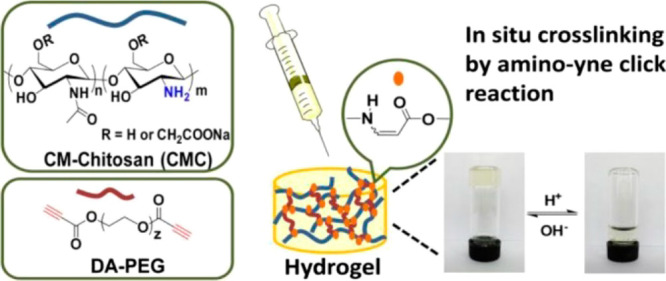
Hydrogels formed from the spontaneous amino-yne reaction of chitosan and PEG-propiolates.211 Reproduced with permission from ref (211). Copyright 2018 American Chemical Society.
The reaction has also been employed to form hyperbranched polymer architectures.213−215 In one report, the polymerization of a fluorescent amine monomer was used to probe the aggregation behavior of amphiphilic amino-yne polymers as they self-assembled into nanoparticles.214 In another interesting study, the amino-yne reaction was used as a junction point to create a miktoarm copolymer.213 A secondary amine terminated with two alcohols was quantitatively reacted with a propiolate-PEG substrate to afford a difunctional PEG macromonomer. This species was then used to initiate the ring-opening polymerization (ROP) of caprolactone to afford an amphiphilic copolymer that formed nanostructured materials with various morphologies that depended on the polymer arm lengths and reaction conditions. The biorthogonality of the amino-yne reaction, combined with its impressive versatility, should enable innovative biomaterial applications.
4. Hydroxyl-yne Reactions
4.1. Overview and Historical Development
Alcohols have also been widely employed as nucleophiles for 1,4-conjugate addition reactions with activated alkynes in what is now commonly known as the hydroxyl-yne reaction. In fact, some of the first heteronuclear conjugate addition reactions by Ruhemann in 1900 also screened phenolates to furnish α,β-unsaturated esters (Scheme 24).38,216
Scheme 24. Seminal Report of Hydroxyl-yne Reaction between Sodium Phenolate and Ethyl Phenylpropiolate216.

At first glance, the hydroxyl-yne reaction appears principally analogous to the thiol or amine variants, however, a distinguishing feature is the reduced acidity (relative to the thiol) and nucleophilicity (compared to either) of the hydroxyl species. The effect of this characteristic is that hydroxyl-yne reactions do not proceed spontaneously and demand the addition of a catalyst and/or base to suitably activate the hydroxyl moiety. The net effect of these harsher reaction requirements means that the orthogonality of the hydroxyl-yne reaction can also suffer. Nevertheless, the reaction can still proceed efficiently under certain conditions and there are examples that satisfy Click characteristics. Finally, alcohols are one of the most ubiquitous functional groups in organic chemistry so this balances the comparative inefficiency relative to less abundant heteronucleophiles and also provides palpable motivation for continued progress in the field. However, as a result of the aforementioned drawback of hydroxyl-yne reactions the overall lesser number of Click reactions is still perceptible, especially compared to thiol-yne or amino-yne reactions. Similar to the amino-yne section, we will start with a brief overview on reactivity and then categorize the reactions according to catalyst choice and conclude with examples in polymer chemistry.
4.2. Trends in Reactivity
There are just a handful of studies that investigate structure-reactivity relationships for hydroxyl-yne reactions. The most noteworthy study concluded that the organobase-catalyzed reaction of primary alcohols and propiolates can proceed efficiently (i.e., quantitative conversion in <2 h at ambient temperature), however when secondary and tertiary alcohols are employed as nucleophiles, the reactions are comparatively sluggish and rarely reach completion.217 Unfortunately, other reaction factors such as the nature of the acetylenic acceptor or solvent effects were not examined, although both are known to be important factors related to reaction outcomes in other conjugate addition reactions. Finally, the authors described the products as primarily Z-isomers, although specific values were not reported in this example.
In general, the hydroxyl-yne reaction is often reported to generally favor the E-isomer over the Z-isomer in many cases. Similarly to the thiol-yne variant, the stereochemical reaction outcome shows dependence on both the solvent and catalyst choice. A detailed study by Inanaga et al. investigated the effects of catalyst and solvent on the addition reaction between benzyl alcohol and methyl propiolate (Table 12).82 When the reaction was catalyzed by triphenylphosphine, the quantity of E-isomer in the product only fluctuates slightly (75–83%) despite large differences in solvent polarity (e.g., benzene vs acetonitrile). These solvent effects are dramatically smaller than those reported for the thiol-yne reaction.76 Moreover, when the reaction is catalyzed by tributylphosphine, the solvent effect is nullified and reaction is stereoselective for the E-isomer. Together the surveyed reaction conditions only provide products ranging from 75–100% E-content. Although this demonstrates a smaller range compared to the thiol-yne reaction, some examples are perfectly stereoselective which satisfies an important Click requirement.
Table 12. Screening the Hydroxyl-yne Reaction between Benzyl Alcohol and Methyl Propiolate Using Various Solvents and Different Trivalent Phosphine Catalysts82.

| catalyst | solvent | time (min) | conversion (%) | E/Z |
|---|---|---|---|---|
| PPh3 | C6H6 | 480 | 86 | 75/25 |
| PPh3 | CH2Cl2 | 480 | 85 | 83/17 |
| PPh3 | THFa | 480 | 62 | 75/25 |
| PPh3 | MeCNb | 480 | >98 | 83/17 |
| PBu3 | C6H6 | 10 | 83 | 100/0 |
| PBu3 | CH2Cl2 | 10 | >98 | 100/0 |
| PBu3 | THF | 10 | >98 | 100/0 |
| PBu3 | MeCN | 10 | >98 | 99/1 |
| P(c-Hex)3 | MeCN | 120 | 66 | 88/12 |
| P(OMe)3 | MeCN | 480 |
THF = tetrahydrofuran.
MeCN = acetonitrile.
4.3. Applications in Organic Synthesis
The most popular use of the hydroxyl-yne reaction has been to synthesize heterocyclic compounds, notably furans. Nozaki and co-workers reported the first furan syntheses via intramolecular hydroxyl-yne reactions,218 and this has been extensively reviewed in the 1990s.219 There are a few recent, excellent summaries that describe the chemistry of ynones and reports therein have suitably captured work in this area from the last few decades.4,5 Instead, we will largely spotlight reactions that avoid ynone substrates. For example, ethynylbenzene (or phenylacetylene) derivatives have been successfully employed and we will cover these among others that stand out for their efficiency.
4.3.1. Metal-Catalyzed Reactions
Since the earlier furan studies, other efficient transformations to oxygen-containing heterocycles have appeared. Gabriele et al. reported the palladium-catalyzed synthesis of benzofuran and isochromene compounds reaching quantitative yields for some substrates, albeit at elevated temperatures (Scheme 25).220,221 The Nishibayashi group reported the ruthenium-catalyzed intramolecular cyclization of 3-butyne-1,2-diols into substituted furans with yields up to 70%.222 Although the reaction was not nearly quantitative, the yields were surprisingly good considering the alkyne was actually deactivated with a neighboring alcohol moiety. In similar intramolecular cyclizations involving unactivated alkynes and diols, a heterogeneous AgNO3-SiO2 catalyst afforded near quantitative yields at ambient temperature.223 Additionally, the AgNO3 supported catalyst displayed good recyclability. Another example used a nonsupported AgNO3 catalyst to accomplish a similar transformation in only 87% yield on the way to a natural product synthesis.224
Scheme 25. Palladium-Catalyzed Intramolecular Hydroxyl-yne Cycloadditions to Form Either 1,3-Dihydroisobenzofurans or Isochromenes According to Solvent Selection221.

The pioneering work on the gold-catalyzed hydroxyl-yne addition was accomplished by Larock and co-workers with a tandem conjugate addition reaction where the intermediate enolate attacked the internal alkyne as the nucleophile using AuCl3 as the catalyst, affording heavily substituted furans (in up to 90% isolated yields) under ambient conditions in only 1 h (Scheme 26).225 Many comparable gold-catalyzed furan syntheses have been reported using this general approach and are excellently summarized in several reviews226−229 so they will not be discussed further.
Scheme 26. Gold-Catalyzed Tandem Conjugate Additions to Form Substituted Furans225.

The nucleophilic hydroxylation of nonactivated acetylenes has also afforded various nitrogen- or oxygen-based heterocycles using gold-230−234 and copper-catalyzed176,177,235 approaches. In particular, the reactions catalyzed by gold species have primarily involved intramolecular cyclization of enyne alcohols and can often be high yielding depending on the functional groups (Scheme 27).
Scheme 27. Metal-Catalyzed Intramolecular Hydroxyl-yne Cycloadditions Form Unsaturated Heterocycles.

(a) Gold-catalyzed cascade reactions to form bridged heterocycles;233 (b) copper-catalyzed synthesis of pyrroles and furans.235
4.3.2. Organocatalyzed Reactions
Organocatalysts have become increasingly popular for hydroxyl-yne transformations in recent years. Nucleophilic phosphine catalysts were already mentioned as efficient catalysts for the hydroxyl-yne reaction, however, amine catalysts have also been screened.217 At 20 mol % catalyst loading, 1,4-diazabicyclo[2.2.2]octane (DABCO) afforded quantitative conversion (along with tributylphosphine) for the 1,4-conjugate addition of octanol to methyl propiolate in just 2 h. Interestingly, other amines performed more poorly, including 4-dimethylaminopyridine (DMAP) (26%), triethylamine (38%), and N-methylmorpholine (85%). Regardless of the catalyst and overall reaction efficiency, the E-isomer was still formed exclusively.
Proline- and pyrrolidine-based catalysts have become very useful after their first report in 2006 by Gouverneur and co-workers, affording yields of up to 90% and E/Z ratios of >99/1.236 The Taran group later reported use of a trivalent phosphine to catalyze the ambient temperature intermolecular reaction between an azido-alcohol and an ynone to afford 1,3-oxazines or 1,4-oxazepines, dependent upon the substituents (aryl vs alkyl) of the alcohol substrate, in up to 98% yield (Scheme 28).237
Scheme 28. Phosphine-Catalyzed Intermolecular Hydroxyl-yne Cycloadditions Yielding Unsaturated Oxygen-Containing Heterocycles237.

Several studies have reported proline-catalyzed reaction of ynones and ynals with phenols in quantitative yields as a room-temperature synthesis of chromene derivatives (Scheme 29).238−240 Pyrrolidine was employed in a more recent report to catalyze the tandem Michael addition of ortho-substituted aminophenols to ynals at yields of up to 95%.241 Another study also described the efficient bioconjugation of polysaccharides with ynone substrates by employing DMAP as a catalyst. The authors also examined the stereochemistry of the conjugates, reporting 100% E-isomer stereoconfiguration for the reaction products.199
Scheme 29. Enantioselective Organocatalytic Cascade oxa-Michael Reactions to Afford Chromenes239,240.

4.4. Applications in Polymer Chemistry
In the 1990s, Endo and co-workers disclosed several reports describing polyaddition reactions that satisfy most Click criteria by reacting a variety of diols and electron-deficient diacetylenes.71,242,243 Specifically, the authors employed tributylphosphine to catalyze the polymerizations at ambient temperature in tetrahydrofuran, affording a large library of moderate molecular weight (up to 20 kDa) polymers in excellent yields (up to 94%). More recently, the Tang group244 reported DMAP as a catalyst for the step-growth polymerization between diphenols (such as bisphenol A) and diynones to produce stereoregular E-isomeric polymers with high molecular weights (up to 35 kDa). On the other hand, the same group also reported polymers with mixed stereoconfigurations (∼50/50 E/Z) and high thermal stability (Td > 320 °C) when hydroxyl-yne polymerizations were catalyzed by a phosphazene species.245 However, because different alkyne monomers (aryl acetylenes vs ynones) were employed, it is difficult to draw meaningful conclusions between the studies. Very recently, 1,4-diazabicyclo[2.2.2]octane (DABCO) was used to catalyze hydroxyl-yne polymerizations at ambient temperature using propiolate monomers and various diols producing polymers with Td > 300 °C and Tg = 15–50 °C, indicating most are glassy plastics at ambient temperature.246 When aliphatic diols were reacted, the resulting polymers primarily contained E-isomeric units but stereoselectivity diminished when employing phenolic monomers. Although there are only a handful of reports that feature alcohol nucleophiles in Michael addition polymerizations, the monomer scope is already extensive and exemplifies its versatility (Scheme 30). There is also one example of a network polymer where a 4-armed hydroxy-terminated polycaprolactone macromonomer was reacted with bis-propiolate monomers at ambient temperature to afford luminescent gels (Figure 14).247 A functionalization reaction of hydroxypropylcellulose (HPC) with an ynone substrate catalyzed by DMAP proceeded at ambient temperature in good yield (Scheme 31).199
Scheme 30. Representative Substrate Scope of Organocatalyzed Hydroxyl-yne Polyaddition Reactions244−246.
Figure 14.

Highly cross-linked network polymers from hydroxyl-yne click polymerizations between an alcohol-terminated four-armed macromonomer and bis-propiolates.247 (a) Synthesis of PCL3600 network from alkyl propiolate and (b) luminescent PCL5800 network from tetraphenylethylene bis-propiolate. Reproduced with permission from ref (247). Copyright 2020 Royal Society of Chemistry.
Scheme 31. Functionalization of Polysaccharide Using the Hydroxyl-yne Reaction199.

5. Summary and Outlook
Nucleophilic conjugate addition reactions of heteronucleophiles to activated acetylenes, chiefly the thiol-yne, amino-yne, and hydroxyl-yne reactions, have long served as efficient transformations in organic synthesis and more recently within polymer science where their usage has prompted the development of many emergent and innovative materials. In many settings, the Michael reactions can satisfy Click criteria by exhibiting wide scope and good selectivity (orthogonal chemical reactivity, regioselectivity, and stereoselectivity) under mild temperatures and ambient atmosphere. The ability to perform efficient reactions under ambient conditions is one of the greatest hallmarks of the reaction and truly distinguishes it from metal-catalyzed cross-coupling transformations in organic synthesis and the inefficient condensation chemistries currently employed for the synthesis of some commodity polymers.
Nevertheless, reaction requirements and outcomes can vary widely among similar reactions even by simply changing the nucleophile, alkyne, solvent, or catalyst. To date, the field is lacking more fundamental studies that could help establish universal concepts, especially those related to the influence of reaction conditions on stereochemistry, kinetics, and reversibility. By undertaking systematic studies to assess structure–reactivity relationships, this should unlock important insights in order to establish more uniform design principles for the Michael reaction. This could be especially beneficial to applications in bioconjugate chemistry, where reliable protocols are critical to the clinical translation of technologies. It is envisioned that future developments in the field will ultimately seek to optimize conditions (especially relating to catalyst development) to enable a more generalized approach and rationally obtain Click reactivity on demand.
Selective modulation of product stereochemistry by judicious choice of catalyst is a unique advantage compared to many other Click reactions, especially the corresponding thiol-ene reaction. However, although the stereoselectivity can be rationally tuned through careful choice of base or solvent, thus far stereopure reaction products have remained rare. This presents the opportunity to develop more stereoselective catalysts, including other organocatalysts or single-site transition metal catalysts, which are both areas in need of development. Achieving improvements to stereoselectivity will push this family of reactions closer to satisfying complete Click criteria. Stereocontrol will also be critical to the effective translation to a broader range of applications. This is especially important for polymeric materials, where thiol-yne polymers have demonstrated superb structure–property manipulation just by simply changing the stereochemistry of the alkene in the polymer backbone. From a fundamental viewpoint, this is very exciting because it could enable the creation of a material portfolio where polymers are compositionally similar but feature a range of divergent physical properties. By reacting the same monomers and only adjusting the stereochemical content of the polymer, the material behavior could vary between hard/soft or elastic/plastic, which provides an exceptionally powerful approach to synthesizing next-generation, functional materials.
Acknowledgments
A.P.D., C.J.S. and J.C.W. acknowledge funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 681559. M.J.P. thanks The University of Birmingham Global PhD Scholarship for financial support.
Biographies
Joshua C. Worch is the Group Leader for Professor Andrew Dove in the School of Chemistry at the University of Birmingham. Josh completed a Bachelor of Arts degree in Chemistry from Manchester University (Indiana, USA) in 2011 and a Ph.D. in Chemistry from Carnegie Mellon University (Pennsylvania, USA) in 2016, where he developed organic semiconductors for energy applications under the supervision of Prof. Kevin Noonan. Josh then moved across the Atlantic to the UK and joined the research group of Prof. Andrew Dove, where he was supported by a Marie Skłodowska-Curie Fellowship from 2017 to 2019 to investigate stereochemical structure–property relationships in polymers. His current research interests are centered on sustainable polymers, encompassing degradable materials for engineering applications and biomedicine to the chemical recycling of plastics.
Connor J. Stubbs is currently studying his Ph.D. under the supervision of Prof. Andrew Dove at the University of Birmingham. Previously, he graduated with a MChem degree from the University of Hull in 2016. His research interests range from leveraging backbone stereochemistry to manipulate bulk polymer properties and designing chemically recyclable polymeric materials.
Matthew J. Price is a postgraduate researcher under Professor Andrew Dove in the School of Chemistry at the University of Birmingham. Prior to this, he studied at the University of Cambridge, completing a Master’s in Natural Science including research work on polymer postfunctionalization and supramolecular cage synthesis. His research now focuses on the depolymerization of “real-world” polymer wastes.
Andrew P. Dove is a Professor of Chemistry in the School of Chemistry at the University of Birmingham. Andrew graduated from the University of York with an MChem degree in 1999. His subsequent Ph.D. studies were conducted under the supervision of Prof. Vernon C. Gibson FRS at Imperial College, London, focused on metal catalyzed coordination insertion polymerization. Andrew undertook postdoctoral research first under the guidance of Prof. Robert M. Waymouth at Stanford University, California, and then as a CIPMA postdoctoral fellow at IBM, San Jose, California, under the supervision of Dr. James L. Hedrick and Prof. Robert M. Waymouth. Andrew returned to the UK to take up a RCUK Fellowship in Nanotechnology at the University of Warwick in 2005 and moved to the University of Birmingham in 2018. His research focusses on the development and application of sustainable polymers and degradable polymeric biomaterials.
The authors declare no competing financial interest.
References
- Fraile A.; Parra A.; Tortosa M.; Alemán J. Organocatalytic Transformations of Alkynals, Alkynones, Propriolates, and Related Electron-deficient Alkynes. Tetrahedron 2014, 70, 9145–9173. 10.1016/j.tet.2014.07.023. [DOI] [Google Scholar]
- Michael A. Ueber die Addition von Natriumacetessig- und Natriummalonsäureäthern zu den Aethern ungesättigter Säuren. J. Prakt. Chem. 1887, 35, 349–356. 10.1002/prac.18870350136. [DOI] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Nájera C.; Sydnes L. K.; Yus M. Conjugated Ynones in Organic Synthesis. Chem. Rev. 2019, 119, 11110–11244. 10.1021/acs.chemrev.9b00277. [DOI] [PubMed] [Google Scholar]
- Li Y.; Yu J.; Bi Y.; Yan G.; Huang D. Tandem Reactions of Ynones: Via Conjugate Addition of Nitrogen-, Carbon-, Oxygen-, Boron-, Silicon-, Phosphorus-, and Sulfur-Containing Nucleophiles. Adv. Synth. Catal. 2019, 361, 4839–4881. 10.1002/adsc.201900611. [DOI] [Google Scholar]
- Keddie D. J.; Grande J. B.; Gonzaga F.; Brook M. A.; Dargaville T. R. Amphiphilic Silicone Architectures via Anaerobic Thiol-Ene Chemistry. Org. Lett. 2011, 13, 6006–6009. 10.1021/ol202051y. [DOI] [PubMed] [Google Scholar]
- Shu P.; Zeng J.; Tao J.; Zhao Y.; Yao G.; Wan Q. Selective S-deacetylation inspired by native chemical ligation: practical syntheses of glycosyl thiols and drug mercapto-analogues. Green Chem. 2015, 17, 2545–2551. 10.1039/C5GC00084J. [DOI] [Google Scholar]
- Wessig P.; Gerngroß M.; Freyse D.; Bruhns P.; Przezdziak M.; Schilde U.; Kelling A. Molecular Rods Based on Oligo-spiro-thioketals. J. Org. Chem. 2016, 81, 1125–1136. 10.1021/acs.joc.5b02670. [DOI] [PubMed] [Google Scholar]
- Bandgar B. P.; Sadavarte V. S.; Uppalla L. S. Remarkably Fast Direct Synthesis of Thiols from Alcohols under Mild Conditions. Chem. Lett. 2000, 29, 1304–1305. 10.1246/cl.2000.1304. [DOI] [Google Scholar]
- Monaco M. R.; Prévost S.; List B. Catalytic Asymmetric Synthesis of Thiols. J. Am. Chem. Soc. 2014, 136, 16982–16985. 10.1021/ja510069w. [DOI] [PubMed] [Google Scholar]
- Rhinesmith H. S. Action of Grignard Reagents on the Esters of Propiolic Acid. J. Org. Chem. 1975, 40, 1773–1776. 10.1021/jo00900a021. [DOI] [Google Scholar]
- Neises B.; Steglich W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem., Int. Ed. Engl. 1978, 17, 522–524. 10.1002/anie.197805221. [DOI] [Google Scholar]
- Balas L.; Jousseaume B.; Langwost B. Improved Preparation of Aliphatic Propynoic Esters. Tetrahedron Lett. 1989, 30, 4525–4526. 10.1016/S0040-4039(01)80735-8. [DOI] [Google Scholar]
- Shen C. C.; Ainsworth C. I. Selective Alkylation of 2-Butynoic Acid. Tetrahedron Lett. 1979, 20, 83–86. 10.1016/S0040-4039(01)85888-3. [DOI] [Google Scholar]
- Balfour W. J.; Greig C. C.; Visaisouk S. Preparation and characterization of propiolyl chloride. J. Org. Chem. 1974, 39, 725–726. 10.1021/jo00919a037. [DOI] [Google Scholar]
- Tohda Y.; Sonogashira K.; Hagihara N. A Convenient Synthesis of 1-Alkynyl Ketones and 2-Alkynamides. Synthesis 1977, 1977, 777–778. 10.1055/s-1977-24574. [DOI] [Google Scholar]
- Chowdhury C.; Kundu N. G. Copper(I)-catalysed Acylation of Terminal Alkynes. Tetrahedron Lett. 1996, 37, 7323–7324. 10.1016/0040-4039(96)01598-5. [DOI] [Google Scholar]
- Cox R. J.; Ritson D. J.; Dane T. A.; Berge J.; Charmant J. P. H.; Kantacha A. Room Temperature Palladium Catalysed Coupling of Acyl Chlorides with Terminal Alkynes. Chem. Commun. 2005, 1037–1039. 10.1039/b414826f. [DOI] [PubMed] [Google Scholar]
- Alonso D. A.; Nájera C.; Pacheco M. C. Synthesis of Ynones by Palladium-Catalyzed Acylation of Terminal Alkynes with Acid Chlorides. J. Org. Chem. 2004, 69, 1615–1619. 10.1021/jo035761+. [DOI] [PubMed] [Google Scholar]
- Nahm S.; Weinreb S. M. N-Methoxy-N-Methylamides as Effective Acylating Agents. Tetrahedron Lett. 1981, 22, 3815–3818. 10.1016/S0040-4039(01)91316-4. [DOI] [Google Scholar]
- Peil S.; Bistoni G.; Goddard R.; Fürstner A. Hydrogenative Metathesis of Enynes via Piano-Stool Ruthenium Carbene Complexes Formed by Alkyne gem-Hydrogenation. J. Am. Chem. Soc. 2020, 142, 18541–18553. 10.1021/jacs.0c07808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost B. M.; Hung C.-I. J.; Gnanamani E. Tuning the Reactivity of Ketones through Unsaturation: Construction of Cyclic and Acyclic Quaternary Stereocenters via Zn-ProPhenol Catalyzed Mannich Reactions. ACS Catal. 2019, 9, 1549–1557. 10.1021/acscatal.8b04685. [DOI] [Google Scholar]
- Ferris T. D.; Lee P. T.; Farrar T. C. Synthesis of Propiolamide and 1H, 13C and 15N NMR Spectra of Formamide, Acetamide and Propiolamide. Magn. Reson. Chem. 1997, 35, 571–576. . [DOI] [Google Scholar]
- Worch J. C.; Weems A. C.; Yu J.; Arno M. C.; Wilks T. R.; Huckstepp R. T. R.; O’Reilly R. K.; Becker M. L.; Dove A. P. Elastomeric Polyamide Biomaterials with Stereochemically Tuneable Mechanical Properties and Shape Memory. Nat. Commun. 2020, 11, 3250. 10.1038/s41467-020-16945-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Motoyama Y.; Tsuji K.; Yoon S.-H.; Mochida I.; Nagashima H. (Z)-Selective Partial Hydrogenation of Internal Alkynes by Using Palladium Nanoparticles Supported on Nitrogen-Doped Carbon Nanofiber. ChemCatChem 2012, 4, 778–781. 10.1002/cctc.201200058. [DOI] [Google Scholar]
- Suda T.; Noguchi K.; Hirano M.; Tanaka K. Highly Enantioselective Synthesis of N,N-Dialkylbenzamides with Aryl-Carbonyl Axial Chirality by Rhodium-Catalyzed [2 + 2+2] Cycloaddition. Chem. - Eur. J. 2008, 14, 6593–6596. 10.1002/chem.200800953. [DOI] [PubMed] [Google Scholar]
- Huang H.; Zhang G.; Chen Y. Dual Hypervalent Iodine(III) Reagents and Photoredox Catalysis Enable Decarboxylative Ynonylation under Mild Conditions. Angew. Chem., Int. Ed. 2015, 54, 7872–7876. 10.1002/anie.201502369. [DOI] [PubMed] [Google Scholar]
- Wu J.-J.; Li Y.; Zhou H.-Y.; Wen A. H.; Lun C.-C.; Yao S.-Y.; Ke Z.; Ye B.-H. Copper-Catalyzed Carbamoylation of Terminal Alkynes with Formamides via Cross-Dehydrogenative Coupling. ACS Catal. 2016, 6, 1263–1267. 10.1021/acscatal.5b02881. [DOI] [Google Scholar]
- He Y.; Wang Y.; Liang X.; Huang B.; Wang H.; Pan Y.-M. Palladium-Catalyzed Three-Component Reaction: A Novel Method for the Synthesis of N-Acyl Propiolamides. Org. Lett. 2018, 20, 7117–7120. 10.1021/acs.orglett.8b03068. [DOI] [PubMed] [Google Scholar]
- Van Herck N.; Maes D.; Unal K.; Guerre M.; Winne J. M.; Du Prez F. E. Covalent Adaptable Networks with Tunable Exchange Rates Based on Reversible Thiol-yne Cross-linking. Angew. Chem., Int. Ed. 2020, 59, 3609–3617. 10.1002/anie.201912902. [DOI] [PubMed] [Google Scholar]
- Chen X.; Hu R.; Qi C.; Fu X.; Wang J.; He B.; Huang D.; Qin A.; Tang B. Z. Ethynylsulfone-Based Spontaneous Amino-yne Click Polymerization: A Facile Tool toward Regio- and Stereoregular Dynamic Polymers. Macromolecules 2019, 52, 4526–4533. 10.1021/acs.macromol.9b00670. [DOI] [Google Scholar]
- Mo Z.-Y.; Zhang Y.-Z.; Huang G.-B.; Wang X.-Y.; Pan Y.-M.; Tang H.-T. Electrochemical Sulfonylation of Alkynes with Sulfonyl Hydrazides: A Metal- and Oxidant-free Protocol for the Synthesis of Alkynyl Sulfones. Adv. Synth. Catal. 2020, 362, 2160–2167. 10.1002/adsc.201901607. [DOI] [Google Scholar]
- Meng X.; Xu H.; Cao X.; Cai X.-M.; Luo J.; Wang F.; Huang S. Electrochemically Enabled Sulfonylation of Alkynes with Sodium Sulfinates. Org. Lett. 2020, 22, 6827–6831. 10.1021/acs.orglett.0c02341. [DOI] [PubMed] [Google Scholar]
- Kim D.-K.; Um H.-S.; Park H.; Kim S.; Choi J.; Lee C. Silyloxymethanesulfinate as a sulfoxylate equivalent for the modular synthesis of sulfones and sulfonyl derivatives. Chem. Sci. 2020, 11, 13071–13078. 10.1039/D0SC02947E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X.; Yang M.; Liu J.-B.; He F.-S.; Wu J. Photoinduced Synthesis of Alkylalkynyl Sulfones through a Reaction of Potassium Alkyltrifluoroborates, Sulfur Dioxide, and Alkynyl Bromides. Org. Chem. Front. 2020, 7, 938–943. 10.1039/D0QO00100G. [DOI] [Google Scholar]
- Gong X.; Yang M.; Liu J.-B.; He F.-S.; Fan X.; Wu J. A Metal-free Route to Alkynyl Sulfones Under Photoinduced Conditions with the Insertion of Sulfur Dioxide. Green Chem. 2020, 22, 1906–1910. 10.1039/D0GC00332H. [DOI] [Google Scholar]
- Bhatt D.; Kumari C.; Goswami A. Syntheses and Applications of 2-Alkynylnitriles. Asian J. Org. Chem. 2019, 8, 1985–2001. 10.1002/ajoc.201900463. [DOI] [Google Scholar]
- Ruhemann S.; Stapleton H. E. CIX.—Condensation of Phenols with Esters of the Acetylene Series. Part III. Synthesis of Benzo-γ-pyrone. J. Chem. Soc., Trans. 1900, 77, 1179–1185. 10.1039/CT9007701179. [DOI] [Google Scholar]
- Ruhemann S. LVII.—The Combination of Mercaptans with Unsaturated Ketonic Compounds. J. Chem. Soc., Trans. 1905, 87, 461–468. 10.1039/CT9058700461. [DOI] [Google Scholar]
- Truce W. E.; Heine R. F. The Stereochemistry of Base-catalyzed Additions of p-Toluenethiol to Several Negatively-substituted Acetylenes. An Exception to the Rule of Trans-nucleophilic Addition. J. Am. Chem. Soc. 1957, 79, 5311–5313. 10.1021/ja01576a062. [DOI] [Google Scholar]
- Truce W. E.; Tichenor G. J. W. Effect of Activating Group on Trans-stereoselectivity of Thiolate Additions to Activated Acetylenes. J. Org. Chem. 1972, 37, 2391–2396. 10.1021/jo00980a007. [DOI] [Google Scholar]
- Bowden K.; Braude E. A.; Jones E. R. H. 206. Researches on Acetylenic Compounds. Part VIII. Miscellaneous Addition Reactions of Ethynyl Ketones. J. Chem. Soc. 1946, 945–948. 10.1039/jr9460000945. [DOI] [Google Scholar]
- Selling H. A. Additions of Thiols to Acetylenic Sulfones. Tetrahedron 1975, 31, 2381–2390. 10.1016/0040-4020(75)80244-4. [DOI] [Google Scholar]
- Zhang B.; Chakma P.; Shulman M. P.; Ke J.; Digby Z. A.; Konkolewicz D. Probing the mechanism of thermally driven thiol-Michael dynamic covalent chemistry. Org. Biomol. Chem. 2018, 16, 2725–2734. 10.1039/C8OB00397A. [DOI] [PubMed] [Google Scholar]
- Joshi G.; Anslyn E. V. Dynamic Thiol Exchange with β-Sulfido-α,β-Unsaturated Carbonyl Compounds and Dithianes. Org. Lett. 2012, 14, 4714–4717. 10.1021/ol301781u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S. J.; Cantrill S. J.; Cousins G. R. L.; Sanders J. K. M.; Stoddart J. F. Dynamic Covalent Chemistry. Angew. Chem., Int. Ed. 2002, 41, 898–952. . [DOI] [PubMed] [Google Scholar]
- Jin Y.; Yu C.; Denman R. J.; Zhang W. Recent Advances in Dynamic Covalent Chemistry. Chem. Soc. Rev. 2013, 42, 6634–6654. 10.1039/c3cs60044k. [DOI] [PubMed] [Google Scholar]
- Kohler E. P.; Potter H. The Properties of Unsaturated Sulfur Compounds. I. Alpha Beta Unsaturated Sulfones. J. Am. Chem. Soc. 1935, 57, 1316–1321. 10.1021/ja01310a045. [DOI] [Google Scholar]
- Bader H.; Cross L. C.; Heilbron I.; Jones E. R. H. 132. Researches on acetylenic compounds. Part XVIII. The addition of thiolacetic acid to acetylenic hydrocarbons. The conversion of monosubstituted acetylenes into aldehydes and 1:2-dithiols. J. Chem. Soc. 1949, 619–623. 10.1039/jr9490000619. [DOI] [Google Scholar]
- Oswald A. A.; Griesbaum K.; Hudson B. E.; Bregman J. M. Organic Sulfur Compounds. XIII. Free-Radical Addition of Thiols to Phenylacetylene. J. Am. Chem. Soc. 1964, 86, 2877–2884. 10.1021/ja01068a023. [DOI] [Google Scholar]
- Kondoh A.; Takami K.; Yorimitsu H.; Oshima K. Stereoselective Hydrothiolation of Alkynes Catalyzed by Cesium Base: Facile Access to (Z)-1-Alkenyl Sulfides. J. Org. Chem. 2005, 70, 6468–6473. 10.1021/jo050931z. [DOI] [PubMed] [Google Scholar]
- Yao B.; Mei J.; Li J.; Wang J.; Wu H.; Sun J. Z.; Qin A.; Tang B. Z. Catalyst-Free Thiol-Yne Click Polymerization: A Powerful and Facile Tool for Preparation of Functional Poly(vinylene sulfide)s. Macromolecules 2014, 47, 1325–1333. 10.1021/ma402559a. [DOI] [Google Scholar]
- Lowe A. B.; Hoyle C. E.; Bowman C. N. Thiol-yne Click Chemistry: A Powerful and Versatile Methodology for Materials Synthesis. J. Mater. Chem. 2010, 20, 4745–4750. 10.1039/b917102a. [DOI] [Google Scholar]
- Blomquist A. T.; Wolinsky J. Addition of Ethyl Mercaptan to Acetylenic Compounds. J. Org. Chem. 1958, 23, 551–554. 10.1021/jo01098a014. [DOI] [Google Scholar]
- Fairbanks B. D.; Scott T. F.; Kloxin C. J.; Anseth K. S.; Bowman C. N. Thiol-Yne Photopolymerizations: Novel Mechanism, Kinetics, and Step-Growth Formation of Highly Cross-Linked Networks. Macromolecules 2009, 42, 211–217. 10.1021/ma801903w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe A. B. Thiol-yne ‘Click’/coupling Chemistry and Recent Applications in Polymer and Materials Synthesis and Modification. Polymer 2014, 55, 5517–5549. 10.1016/j.polymer.2014.08.015. [DOI] [Google Scholar]
- Truce W. E.; Simms J. A. Stereospecific Reactions of Nucleophilic Agents with Acetylenes and Vinyl-type Halides. IV. The Stereochemistry of Nucleophilic Additions of Thiols to Acetylenic Hydrocarbons. J. Am. Chem. Soc. 1956, 78, 2756–2759. 10.1021/ja01593a029. [DOI] [Google Scholar]
- Posner T. Zur Kenntniss der disulfone. IX. Weitere Mittheilungen über schwefelhaltige derivate ungesättigter Ketone. Ber. Dtsch. Chem. Ges. 1902, 35, 799–816. 10.1002/cber.190203501127. [DOI] [Google Scholar]
- Frayne S. H.; Northrop B. H. Evaluating Nucleophile Byproduct Formation during Phosphine- and Amine-Promoted Thiol-Methyl Acrylate Reactions. J. Org. Chem. 2018, 83, 10370–10382. 10.1021/acs.joc.8b01471. [DOI] [PubMed] [Google Scholar]
- Chan J. W.; Hoyle C. E.; Lowe A. B.; Bowman M. Nucleophile-Initiated Thiol-Michael Reactions: Effect of Organocatalyst, Thiol, and Ene. Macromolecules 2010, 43, 6381–6388. 10.1021/ma101069c. [DOI] [Google Scholar]
- Long K. F.; Bongiardina N. J.; Mayordomo P.; Olin M. J.; Ortega A. D.; Bowman C. N. Effects of 1°, 2°, and 3° Thiols on Thiol-Ene Reactions: Polymerization Kinetics and Mechanical Behavior. Macromolecules 2020, 53, 5805–5815. 10.1021/acs.macromol.0c00369. [DOI] [Google Scholar]
- Kobayashi S.; Mukaiyama T. The Sterospecific Preparation of Methyl Farnesoate and Synthetic Precursors of C18- and C17 Juvenile Hormones. Chem. Lett. 1974, 3, 1425–1428. 10.1246/cl.1974.1425. [DOI] [Google Scholar]
- Omar M. T.; Basyouni M. N. Stereochemistry of Ionic Thiol Addition to Acetylenic Ketones. Bull. Chem. Soc. Jpn. 1974, 47, 2325–2326. 10.1246/bcsj.47.2325. [DOI] [Google Scholar]
- Tsou H.-R.; et al. 6-Substituted-4-(3-bromophenylamino)quinazolines as Putative Irreversible Inhibitors of the Epidermal Growth Factor Receptor (EGFR) and Human Epidermal Growth Factor Receptor (HER-2) Tyrosine Kinases with Enhanced Antitumor Activity. J. Med. Chem. 2001, 44, 2719–2734. 10.1021/jm0005555. [DOI] [PubMed] [Google Scholar]
- De Lucchi O.; Lucchini V.; Marchioro C.; Valle G.; Modena G. Self-induced Diastereoselective Oxidation of Vinyl Sulfoxides Bearing a Chiral Hydroxy Group as a way of Preparation of Optically Active Sulfinyl Dienophiles and Their Use in the Asymmetric Diels-Alder Reactions to Cyclopentadiene. J. Org. Chem. 1986, 51, 1457–1466. 10.1021/jo00359a014. [DOI] [Google Scholar]
- Trofimov B. A.; Mal’kina A. G.; Shemyakina O. A.; Nosyreva V. V.; Borisova A. P.; Khutsishvili S. S.; Krivdin L. B. Synthesis of Functionalized L-Cysteine and L-Methionine by Reaction with Electron-Deficient Acetylenes. Synthesis 2009, 2009, 3136–3142. 10.1055/s-0029-1217602. [DOI] [Google Scholar]
- Shiu H.-Y.; Chan T.-C.; Ho C.-M.; Liu Y.; Wong M.-K.; Che C.-M. Electron-Deficient Alkynes as Cleavable Reagents for the Modification of Cysteine-Containing Peptides in Aqueous Medium. Chem. - Eur. J. 2009, 15, 3839–3850. 10.1002/chem.200800669. [DOI] [PubMed] [Google Scholar]
- Caramella P.; Houk K. N. The Influence of Electron-withdrawing Substituents on the Geometries and Barriers to Inversion of Vinyl Anions. Tetrahedron Lett. 1981, 22, 819–822. 10.1016/0040-4039(81)80005-6. [DOI] [Google Scholar]
- Basyouni M. N.; Omar M. T.; Ghali E. A. Stereochemistry of Base-catalysed Addition of Methyl Mercaptoacetate to Acetylenic Ketones and Esters. Effects of Activating Groups and Solvents. Bull. Chem. Soc. Jpn. 1980, 53, 1739–1742. 10.1246/bcsj.53.1739. [DOI] [Google Scholar]
- Arcadi A.; Alfonsi M.; Marinelli F. Facile Reaction of Thiols and Amines with Alkyl 4-hydroxy-2-alkynoates in Water Under Neutral Conditions and Ultrasound Irradiation. Tetrahedron Lett. 2009, 50, 2060–2064. 10.1016/j.tetlet.2009.02.106. [DOI] [Google Scholar]
- Kuroda H.; Tomita I.; Endo T. A Novel Polyaddition of Bifunctional Acetylenes Containing Electron-withdrawing Groups: 4. Synthesis of Polymers having Enone Moieties by the Reaction of Ynones with Bifunctional Heteronucleophiles. Polymer 1997, 38, 3655–3662. 10.1016/S0032-3861(96)00901-9. [DOI] [Google Scholar]
- Journet M.; Rouillard A.; Cai D.; Larsen R. D. Double Radical Cyclization/β-Fragmentation of Acyclic ω-Yne Vinyl Sulfides. Synthesis of 3-Vinyldihydrothiophene and Dihydrothiopyran Derivatives. A New Example of a 5-Endo-trig Radical Cyclization. J. Org. Chem. 1997, 62, 8630–8631. 10.1021/jo971787n. [DOI] [Google Scholar]
- Bhadra S.; Ranu B. C. Water-promoted regioselective hydrothiolation of alkynes. Can. J. Chem. 2009, 87, 1605–1609. 10.1139/V09-130. [DOI] [Google Scholar]
- Khatik G. L.; Kumar R.; Chakraborti A. K. Catalyst-Free Conjugated Addition of Thiols to α,β-Unsaturated Carbonyl Compounds in Water. Org. Lett. 2006, 8, 2433–2436. 10.1021/ol060846t. [DOI] [PubMed] [Google Scholar]
- Randive N. A.; Kumar V.; Nair V. A. A Facile Approach to Substituted Acrylates by Regioselective and Stereoselective Addition of Thiols and Amines to an Alkynyl Ester in Water. Monatsh. Chem. 2010, 141, 1329–1332. 10.1007/s00706-010-0399-9. [DOI] [Google Scholar]
- Truong V. X.; Dove A. P. Organocatalytic, Regioselective Nucleophilic “Click” Addition of Thiols to Propiolic Acid Esters for Polymer-Polymer Coupling. Angew. Chem., Int. Ed. 2013, 52, 4132–4136. 10.1002/anie.201209239. [DOI] [PubMed] [Google Scholar]
- de Lima Borges E.; Carvalho Nobre P.; Silva M. S. D.; Jacob R. G.; Lenardão E. J.; Perin G. PEG-400 as a Recyclable Solvent in the Synthesis of β-Arylthio-α,β-unsaturated Esters, Ketone and Aldehyde Under Base and Catalyst-free Conditions. J. Environ. Chem. Eng. 2016, 4, 2004–2007. 10.1016/j.jece.2016.03.027. [DOI] [Google Scholar]
- Nguyen S. S.; Prescher J. A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat. Rev. Chem. 2020, 4, 476–489. 10.1038/s41570-020-0205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem., Int. Ed. 2009, 48, 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truce W. E.; Goldhamer D. L. The Stereochemistry of the Base-catalyzed Addition of p-Toluenethiol to Sodium and Ethyl Phenylpropiolate. J. Am. Chem. Soc. 1959, 81, 5795–5798. 10.1021/ja01530a060. [DOI] [Google Scholar]
- Li G.-Z.; Randev R. K.; Soeriyadi A. H.; Rees G.; Boyer C.; Tong Z.; Davis T. P.; Becer C. R.; Haddleton D. M. Investigation into Thiol-(meth)acrylate Michael Addition Reactions using Amine and Phosphine Catalysts. Polym. Chem. 2010, 1, 1196–1204. 10.1039/c0py00100g. [DOI] [Google Scholar]
- Inanaga J.; Baba Y.; Hanamoto T. Organic Synthesis with Trialkylphosphine Catalysts. Conjugate Addition of Alcohols to α,β-Unsaturated Alkynic Acid Esters. Chem. Lett. 1993, 22, 241–244. 10.1246/cl.1993.241. [DOI] [Google Scholar]
- White D. A.; Baizer M. M. Catalysis of the Michael Reaction by Tertiary Phosphines. Tetrahedron Lett. 1973, 14, 3597–3600. 10.1016/S0040-4039(01)86980-X. [DOI] [Google Scholar]
- Zhao S.; Zhou Q. F.; Liu J. Z.; Tang W. F.; Lu T. Tributylphosphine-catalyzed Reaction of Ethanethiol with Alkynyl Ketones. Chin. Chem. Lett. 2011, 22, 397–400. 10.1016/j.cclet.2010.11.007. [DOI] [Google Scholar]
- Chan J. W.; Hoyle C. E.; Lowe A. B. Sequential Phosphine-Catalyzed, Nucleophilic Thiol-Ene/Radical-Mediated Thiol-Yne Reactions and the Facile Orthogonal Synthesis of Polyfunctional Materials. J. Am. Chem. Soc. 2009, 131, 5751–5753. 10.1021/ja8099135. [DOI] [PubMed] [Google Scholar]
- Nguyen L.-T. T.; Gokmen M. T.; Du Prez F. E. Kinetic Comparison of 13 Homogeneous Thiol-X Reactions. Polym. Chem. 2013, 4, 5527–5536. 10.1039/c3py00743j. [DOI] [Google Scholar]
- Halphen P. D.; Owen T. C. Carboxyalthioacrylates. J. Org. Chem. 1973, 38, 3507–3510. 10.1021/jo00960a015. [DOI] [Google Scholar]
- Kuroda H.; Tomita I.; Endo T. Facile Synthetic Method for the Preparation of Dithioacetals by the Double Conjugate Addition of Acetylenes Bearing Electron-Withdrawing Groups with Thiols. Synth. Commun. 1996, 26, 1539–1543. 10.1080/00397919608003520. [DOI] [Google Scholar]
- Zhou Q. F.; Chu X. P.; Zhao S.; Lu T.; Tang W. F. BF3·t2O Promoted Conjugate Addition of Ethanethiol to Electron-deficient Alkynes. Chin. Chem. Lett. 2012, 23, 639–642. 10.1016/j.cclet.2012.04.008. [DOI] [Google Scholar]
- Medel R.; Monterde M. I.; Plumet J.; Rojas J. K. Total Stereochemical Control in the Addition of Thiols to p-Toluenesulfonylacetylene. Synthesis of Z- and E-2-Sulfanylvinylsulfonyl Derivatives. J. Org. Chem. 2005, 70, 735–738. 10.1021/jo0402576. [DOI] [PubMed] [Google Scholar]
- Kodomari M.; Saitoh G.; Yoshitomi S. Alumina-Catalyzed Addition of Thiols to Methyl Propiolate. Bull. Chem. Soc. Jpn. 1991, 64, 3485–3487. 10.1246/bcsj.64.3485. [DOI] [Google Scholar]
- Ranu B. C.; Bhar S.; Chakraborti R. Synthesis of β-keto 1,3-Dithianes from Acetylenic Ketones. J. Org. Chem. 1992, 57, 7349–7352. 10.1021/jo00052a064. [DOI] [Google Scholar]
- Xu C.; Bartley J. K.; Enache D. I.; Knight D. W.; Lunn M.; Lok M.; Hutchings G. J. On the Synthesis of β-Keto-1,3-dithianes from Conjugated Ynones Catalyzed by Magnesium Oxide. Tetrahedron Lett. 2008, 49, 2454–2456. 10.1016/j.tetlet.2008.02.030. [DOI] [Google Scholar]
- Kalbag S. M.; Nair M. D.; Rajagopalan P.; Talaty C. N. On the Structure of the Product of the Reaction of 2-Aminothiophenol with Diethyl Acetylene Dicarboxylate. Tetrahedron 1967, 23, 1911–1914. 10.1016/S0040-4020(01)82593-X. [DOI] [Google Scholar]
- Ziyaei-Halimehjani A.; Saidi M. R. Synthesis of Aza-Henry Products and Enamines in Water by Michael Addition of Amines or Thiols to Activated Unsaturated Compounds. Tetrahedron Lett. 2008, 49, 1244–1248. 10.1016/j.tetlet.2007.12.042. [DOI] [Google Scholar]
- Howe R. K.; Gruner T. A.; Carter L. G.; Black L. L.; Franz J. E. Cycloaddition Reactions of Nitrile Sulfides with Acetylenic Esters. Synthesis of Isothiazolecarboxylates. J. Org. Chem. 1978, 43, 3736–3742. 10.1021/jo00413a024. [DOI] [Google Scholar]
- Wadsworth D. H.; Detty M. R. Regiochemical Control of the Addition of Aryl Selenols and Aryl Thiols to the Triple Bond of Arylpropiolates. Synthesis of Seleno- and Thioflavones and Seleno- and Thioaurones. J. Org. Chem. 1980, 45, 4611–4615. 10.1021/jo01311a013. [DOI] [Google Scholar]
- Liso G.; Trapani G.; Berardi V.; Latrofa A.; Marchini P. Adducts of 2-Aminothiophenol with Acetylenic Nitriles or Esters and Their Conversion into Benzothiazoles and/or 1,4-Benzothiazines. J. Heterocycl. Chem. 1980, 17, 793–796. 10.1002/jhet.5570170435. [DOI] [Google Scholar]
- Fiesselmann H.; Schipprak P.; Zeitler L. Über Oxythiophen-carbonsäureester, II. Mitteil.): Synthese und Reaktionen von 3-Oxy-thiophen-carbonsäure-(2)-estern. Chem. Ber. 1954, 87, 841–848. 10.1002/cber.19540870609. [DOI] [Google Scholar]
- Obrecht D.; Gerber F.; Sprengeir D.; Masquelin T. A Novel Approach towards 2,3,5-Trisubstituted Thiophenes via tandem Michael Addition/intramolecular Knoevenagel Condensation. Helv. Chim. Acta 1997, 80, 531–537. 10.1002/hlca.19970800217. [DOI] [Google Scholar]
- Ren W.-Y.; Rao K. V. B.; Klein R. S. Convenient Synthesis of Substituted 3-Aminothiophene-2-carbonitriles from α-Acetylenic Nitriles and Their Conversion to Thieno[3,2-d]pyrimidines. J. Heterocycl. Chem. 1986, 23, 1757–1763. 10.1002/jhet.5570230631. [DOI] [Google Scholar]
- Fricero P.; Bialy L.; Czechtizky W.; Méndez M.; Harrity J. P. A. Synthesis of Bifunctional Thiophenes via Fiesselmann Condensation of Ynone Trifluoroborate Salts. Org. Lett. 2018, 20, 198–200. 10.1021/acs.orglett.7b03558. [DOI] [PubMed] [Google Scholar]
- Muzalevskiy V. M.; Iskandarov A. A.; Nenajdenko V. G. Reaction of CF3-ynones with Methyl Thioglycolate. Regioselective Synthesis of 3-CF3-Thiophene Derivatives. J. Fluorine Chem. 2018, 214, 13–16. 10.1016/j.jfluchem.2018.07.013. [DOI] [Google Scholar]
- Chauvin A.; Greiner J.; Pastor R.; Cambon A. Synthese et Reactivite des Perfluoroalkyl-3 Alkylthio-3 Propenoates d’Ethyle: Thiophenes et β-Thioxoesters Perfluoroalkyles. Tetrahedron 1986, 42, 663–668. 10.1016/S0040-4020(01)87467-6. [DOI] [Google Scholar]
- Cheng B.; Duan X.; Li Y.; Zhang X.; Li H.; Wu F.; Li Y.; Wang T.; Zhai H. Development and Application of Pyridinium 1,4-Zwitterionic Thiolates: Synthesis of Polysubstituted Thiophenes. Eur. J. Org. Chem. 2020, 2020, 1896–1906. 10.1002/ejoc.202000165. [DOI] [Google Scholar]
- Vatansever E. C.; Kılıç K.; Özer M. S.; Koza G.; Menges N.; Balci M. Intermolecular Heterocyclization of Alkynones with 2-Mercaptoacetaldehyde under Metal-free Conditions: Synthesis of 2,3-Disubstituted Thiophenes. Tetrahedron Lett. 2015, 56, 5386–5389. 10.1016/j.tetlet.2015.07.090. [DOI] [Google Scholar]
- Teiber M.; Müller T. J. J. Rapid Consecutive Three-component Coupling-Fiesselmann Synthesis of Luminescent 2,4-disubstituted Thiophenes and Oligothiophenes. Chem. Commun. 2012, 48, 2080–2082. 10.1039/c2cc17548g. [DOI] [PubMed] [Google Scholar]
- Merkt F. K.; Müller T. J. J. Solid State and Aggregation Induced Emissive Chromophores by Multi-component Syntheses. Isr. J. Chem. 2018, 58, 889–900. 10.1002/ijch.201800058. [DOI] [Google Scholar]
- Teiber M.; Giebeler S.; Lessing T.; Müller T. J. J. Efficient Pseudo-five-component Coupling-Fiesselmann Synthesis of Luminescent Oligothiophenes and Their Modification. Org. Biomol. Chem. 2013, 11, 3541–3552. 10.1039/c3ob40124c. [DOI] [PubMed] [Google Scholar]
- Brotzel F.; Mayr H. Nucleophilicities of Amino Acids and Peptides. Org. Biomol. Chem. 2007, 5, 3814–3820. 10.1039/b713778h. [DOI] [PubMed] [Google Scholar]
- Gordon P. F.; Gregory P., Application and Fastness Properties of Dyes. In Organic Chemistry in Colour; Gordon P. F., Gregory P., Eds.; Springee: Berlin, Heidelberg, 1987; pp 262–304. [Google Scholar]
- Herbst-Johnstone M.; Piano F.; Duhamel N.; Barker D.; Fedrizzi B. Ethyl Propiolate Derivatisation for the Analysis of Varietal Thiols in Wine. J. Chromatogr. A 2013, 1312, 104–110. 10.1016/j.chroma.2013.08.066. [DOI] [PubMed] [Google Scholar]
- Alley S. C.; Benjamin D. R.; Jeffrey S. C.; Okeley N. M.; Meyer D. L.; Sanderson R. J.; Senter P. D. Contribution of Linker Stability to the Activities of Anticancer Immunoconjugates. Bioconjugate Chem. 2008, 19, 759–765. 10.1021/bc7004329. [DOI] [PubMed] [Google Scholar]
- Sun Y.; Liu H.; Cheng L.; Zhu S.; Cai C.; Yang T.; Yang L.; Ding P. Thiol Michael Addition Reaction: A Facile Tool for Introducing Peptides into Polymer-Based Gene Delivery Systems. Polym. Int. 2018, 67, 25–31. 10.1002/pi.5490. [DOI] [Google Scholar]
- Koniev O.; Leriche G.; Nothisen M.; Remy J.-S.; Strub J.-M.; Schaeffer-Reiss C.; Van Dorsselaer A.; Baati R.; Wagner A. Selective Irreversible Chemical Tagging of Cysteine with 3-Arylpropiolonitriles. Bioconjugate Chem. 2014, 25, 202–206. 10.1021/bc400469d. [DOI] [PubMed] [Google Scholar]
- Kolodych S.; Koniev O.; Baatarkhuu Z.; Bonnefoy J.-Y.; Debaene F.; Cianférani S.; Van Dorsselaer A.; Wagner A. CBTF: New Amine-to-thiol Coupling Reagent for Preparation of Antibody Conjugates with Increased Plasma Stability. Bioconjugate Chem. 2015, 26, 197–200. 10.1021/bc500610g. [DOI] [PubMed] [Google Scholar]
- Li H.-Q.; Li D.-D.; Lu X.; Xu Y.-Y.; Zhu H.-L. Design and Synthesis of 4,6-Substituted-(diaphenylamino)quinazolines as Potent EGFR Inhibitors with Antitumor Activity. Bioorg. Med. Chem. 2012, 20, 317–323. 10.1016/j.bmc.2011.10.085. [DOI] [PubMed] [Google Scholar]
- Discafani C. M.; et al. Irreversible Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase with In Vivo Activity by N-[4-[(3-bromophenyl)amino]-6-quinazolinyl]-2-butynamide (CL-387,785). Biochem. Pharmacol. 1999, 57, 917–925. 10.1016/S0006-2952(98)00356-6. [DOI] [PubMed] [Google Scholar]
- Pan Z.; et al. Discovery of Selective Irreversible Inhibitors for Bruton’s Tyrosine Kinase. ChemMedChem 2007, 2, 58–61. 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- Byrd J. C.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. 10.1056/NEJMoa1509981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiu H.-Y.; Chong H.-C.; Leung Y.-C.; Wong M.-K.; Che C.-M. A Highly Selective FRET-Based Fluorescent Probe for Detection of Cysteine and Homocysteine. Chem. - Eur. J. 2010, 16, 3308–3313. 10.1002/chem.200903121. [DOI] [PubMed] [Google Scholar]
- Cheng T.; Huang W.; Gao D.; Yang Z.; Zhang C.; Zhang H.; Zhang J.; Li H.; Yang X.-F. Michael Addition/S,N-Intramolecular Rearrangement Sequence Enables Selective Fluorescence Detection of Cysteine and Homocysteine. Anal. Chem. 2019, 91, 10894–10900. 10.1021/acs.analchem.9b02814. [DOI] [PubMed] [Google Scholar]
- Esmaili A. A.; Ghereghloo M.; Islami M. R.; Bijanzadeh H. R. One-pot Synthesis of Stable Phosphonium Ylides using 2-Aminothiophenol. Tetrahedron 2003, 59, 4785–4788. 10.1016/S0040-4020(03)00693-8. [DOI] [Google Scholar]
- Seebach D.; Corey E. J. Generation and Synthetic Applications of 2-Lithio-1,3-dithianes. J. Org. Chem. 1975, 40, 231–237. 10.1021/jo00890a018. [DOI] [Google Scholar]
- Gaunt M. J.; Sneddon H. F.; Hewitt P. R.; Orsini P.; Hook D. F.; Ley S. V. Development of β-Keto 1,3-Dithianes as Versatile Intermediates for Organic Synthesis. Org. Biomol. Chem. 2003, 1, 15–16. 10.1039/B208982C. [DOI] [PubMed] [Google Scholar]
- Smith A. B.; Adams C. M.; Lodise Barbosa S. A.; Degnan A. P. Total Synthesis of (+)-13-Deoxytedanolide. J. Am. Chem. Soc. 2003, 125, 350–351. 10.1021/ja0289649. [DOI] [PubMed] [Google Scholar]
- Arjona O.; Iradier F.; Medel R.; Plumet J. p-Toluenesulfonylacetylene as Thiol Protecting Group. J. Org. Chem. 1999, 64, 6090–6093. 10.1021/jo990308c. [DOI] [Google Scholar]
- Arjona O.; Medel R. O.; Rojas J.; Costa A. M.; Vilarrasa J. Chemoselective Protection of Thiols Versus Alcohols and Phenols. The Tosvinyl Group. Tetrahedron Lett. 2003, 44, 6369–6373. 10.1016/S0040-4039(03)01614-9. [DOI] [Google Scholar]
- O’Donnell J. S.; Singh S. P.; Metcalf T. A.; Schwan A. L. Cesium (Z)-2-Carbomethoxyethenethiolate: A Reagent for the Preparation of (Z)-2-Carbomethoxyethenyl Thioethers Including Selected Cysteine and Homocysteine Derivatives. Eur. J. Org. Chem. 2009, 2009, 547–553. 10.1002/ejoc.200801025. [DOI] [Google Scholar]
- Sobenina L. N.; Demenev A. P.; Mikhaleva A. B. I.; Elokhina V. N.; Mal’kina A. G.; Tarasova O. G. A.; Ushakov I. A.; Trofimov B. A. The Addition Reactions of Pyrrolecarbodithioates to Activated Alkenes and Alkynes. Synthesis 2001, 2001, 0293–0299. 10.1055/s-2001-10819. [DOI] [Google Scholar]
- Reuter C.; Vögtle F. Rotaxanes via Michael Addition. Org. Lett. 2000, 2, 593–595. 10.1021/ol990350u. [DOI] [PubMed] [Google Scholar]
- Mather B. D.; Viswanathan K.; Miller K. M.; Long T. E. Michael Addition Reactions in Macromolecular Design for Emerging Technologies. Prog. Polym. Sci. 2006, 31, 487–531. 10.1016/j.progpolymsci.2006.03.001. [DOI] [Google Scholar]
- Hoyle C. E.; Bowman C. N. Thiol-Ene Click Chemistry. Angew. Chem., Int. Ed. 2010, 49, 1540–1573. 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- Huang D.; Liu Y.; Qin A.; Tang B. Z. Recent Advances in Alkyne-based Click Polymerizations. Polym. Chem. 2018, 9, 2853–2867. 10.1039/C7PY02047C. [DOI] [Google Scholar]
- Daglar O.; Luleburgaz S.; Baysak E.; Gunay U. S.; Hizal G.; Tunca U.; Durmaz H. Nucleophilic Thiol-yne reaction in Macromolecular Engineering: From synthesis to applications. Eur. Polym. J. 2020, 137, 109926. 10.1016/j.eurpolymj.2020.109926. [DOI] [Google Scholar]
- Bass R. G.; Cooper E.; Hergenrother P. M.; Connell J. W. Poly(enonsulfides) from the Addition of Aromatic Dithiols to Aromatic Dipropynones. J. Polym. Sci., Part A: Polym. Chem. 1987, 25, 2395–2407. 10.1002/pola.1987.080250907. [DOI] [Google Scholar]
- Wilbur J. M. Jr; Bonner B. A. Synthesis of Hydrogen-terminated Aliphatic Bis(ethynyl ketone)s and Aliphatic Poly(enamine-ketone)s and Poly(enonesulfide)s. J. Polym. Sci., Part A: Polym. Chem. 1990, 28, 3747–3759. 10.1002/pola.1990.080281318. [DOI] [Google Scholar]
- Dix L. R.; Ebdon J. R.; Hodge P. Chain Extension and Crosslinking of Telechelic Oligomers—II. Michael Additions of Bisthiols to Bismaleimides, Bismaleates and Bis(acetylene ketone)s to Give Linear and Crosslinked Polymers. Eur. Polym. J. 1995, 31, 653–658. 10.1016/0014-3057(95)00006-2. [DOI] [Google Scholar]
- Sinsky M. S.; Bass R. G.; Connell J. W.; Hergenrother P. M. Poly(enamine-ketones) from Aromatic Diacetylenic Diketones and Aromatic Diamines. J. Polym. Sci., Part A: Polym. Chem. 1986, 24, 2279–2295. 10.1002/pola.1986.080240922. [DOI] [Google Scholar]
- Chujo Y.Conjugated Polymer Synthesis: Methods and Reactions; Wiley-VCH Verlag, 2010. [Google Scholar]
- Jim C. K. W.; Qin A.; Lam J. W. Y.; Mahtab F.; Yu Y.; Tang B. Z. Metal-Free Alkyne Polyhydrothiolation: Synthesis of Functional Poly(vinylenesulfide)s with High Stereoregularity by Regioselective Thioclick Polymerization. Adv. Funct. Mater. 2010, 20, 1319–1328. 10.1002/adfm.200901943. [DOI] [Google Scholar]
- Du J.; Huang D.; Li H.; Qin A.; Tang B. Z.; Li Y. Catalyst-Free Click Polymerization of Thiol and Activated Internal Alkynes: A Facile Strategy toward Functional Poly(β-thioacrylate)s. Macromolecules 2020, 53, 4932–4941. 10.1021/acs.macromol.0c00311. [DOI] [Google Scholar]
- Worch J. C.; Prydderch H.; Jimaja S.; Bexis P.; Becker M. L.; Dove A. P. Stereochemical Enhancement of Polymer Properties. Nat. Rev. Chem. 2019, 3, 514–535. 10.1038/s41570-019-0117-z. [DOI] [Google Scholar]
- Bell C. A.; Yu J.; Barker I. A.; Truong V. X.; Cao Z.; Dobrinyin A. V.; Becker M. L.; Dove A. P. Independent Control of Elastomer Properties through Stereocontrolled Synthesis. Angew. Chem., Int. Ed. 2016, 55, 13076–13080. 10.1002/anie.201606750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wandel M. B.; Bell C. A.; Yu J.; Arno M. C.; Dreger N. Z.; Hsu Y.-H.; Pitto-Barry A.; Worch J. C.; Dove A. P.; Becker M. L.; et al. Concomitant control of mechanical properties and degradation in resorbable elastomer-like materials using stereochemistry and stoichiometry for soft tissue engineering. Nat. Commun. 2021, 12, 446. 10.1038/s41467-020-20610-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda H.; Tomita I.; Endo T. A novel phosphine-catalysed polyaddition of terminal acetylenes bearing electron-withdrawing groups with dithiols. Synthesis of polymers having dithioacetal moieties in the main chain. Polymer 1997, 38, 6049–6054. 10.1016/S0032-3861(97)00152-3. [DOI] [Google Scholar]
- Daglar O.; Cakmakci E.; Gunay U. S.; Hizal G.; Tunca U.; Durmaz H. A Straightforward Method for Fluorinated Polythioether Synthesis. Macromolecules 2020, 53, 2965–2975. 10.1021/acs.macromol.0c00548. [DOI] [Google Scholar]
- Daglar O.; Gunay U. S.; Hizal G.; Tunca U.; Durmaz H. Extremely Rapid Polythioether Synthesis in the Presence of TBD. Macromolecules 2019, 52, 3558–3572. 10.1021/acs.macromol.9b00293. [DOI] [Google Scholar]
- Zheng C.; Deng H.; Zhao Z.; Qin A.; Hu R.; Tang B. Z. Multicomponent Tandem Reactions and Polymerizations of Alkynes, Carbonyl Chlorides, and Thiols. Macromolecules 2015, 48, 1941–1951. 10.1021/acs.macromol.5b00175. [DOI] [Google Scholar]
- He H.; Liu B.; Wang M.; Vachet R.; Thayumanavan S. Sequential Nucleophilic “Click” Reactions for Functional Amphiphilic Homopolymers. Polym. Chem. 2018, 10, 187–193. 10.1039/C8PY01341A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunay U. S.; Cetin M.; Daglar O.; Hizal G.; Tunca U.; Durmaz H. Ultrafast and Efficient Aza- and Thiol-Michael Reactions on a Polyester Scaffold with Internal Electron Deficient Trriple Bonds. Polym. Chem. 2018, 9, 3037–3054. 10.1039/C8PY00485D. [DOI] [Google Scholar]
- Pérez-Madrigal M. M.; Shaw J. E.; Arno M. C.; Hoyland J. A.; Richardson S. M.; Dove A. P. Robust Alginate/hyaluronic Acid Thiol-yne Click-hydrogel Scaffolds with Superior Mechanical Performance and Stability for Load-bearing Soft Tissue Engineering. Biomater. Sci. 2020, 8, 405–412. 10.1039/C9BM01494B. [DOI] [PubMed] [Google Scholar]
- Macdougall L. J.; Pérez-Madrigal M. M.; Arno M. C.; Dove A. P. Nonswelling Thiol-Yne Cross-Linked Hydrogel Materials as Cytocompatible Soft Tissue Scaffolds. Biomacromolecules 2018, 19, 1378–1388. 10.1021/acs.biomac.7b01204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdougall L. J.; Pérez-Madrigal M. M.; Shaw J. E.; Inam M.; Hoyland J. A.; O’Reilly R.; Richardson S. M.; Dove A. P. Self-healing, Stretchable and Robust Interpenetrating Network Hydrogels. Biomater. Sci. 2018, 6, 2932–2937. 10.1039/C8BM00872H. [DOI] [PubMed] [Google Scholar]
- Macdougall L. J.; Truong V. X.; Dove A. P. Efficient In Situ Nucleophilic Thiol-yne Click Chemistry for the Synthesis of Strong Hydrogel Materials with Tunable Properties. ACS Macro Lett. 2017, 6, 93–97. 10.1021/acsmacrolett.6b00857. [DOI] [PubMed] [Google Scholar]
- Racine L.; Costa G.; Bayma-Pecit E.; Texier I.; Auzély-Velty R. Design of Interpenetrating Chitosan and Poly(ethylene glycol) Sponges for Potential Drug Delivery Applications. Carbohydr. Polym. 2017, 170, 166–175. 10.1016/j.carbpol.2017.04.060. [DOI] [PubMed] [Google Scholar]
- Cai X. Y.; Li J. Z.; Li N. N.; Chen J. C.; Kang E.-T.; Xu L. Q. PEG-based Hydrogels Prepared by Catalyst-free Thiol-yne Addition and Their Post-Antibacterial Modification. Biomater. Sci. 2016, 4, 1663–1672. 10.1039/C6BM00395H. [DOI] [PubMed] [Google Scholar]
- Truong V. X.; Ablett M. P.; Richardson S. M.; Hoyland J. A.; Dove A. P. Simultaneous Orthogonal Dual-Click Approach to Tough, in-Situ-Forming Hydrogels for Cell Encapsulation. J. Am. Chem. Soc. 2015, 137, 1618–1622. 10.1021/ja511681s. [DOI] [PubMed] [Google Scholar]
- Fan B.; Zhang K.; Liu Q.; Eelkema R. Self-Healing Injectable Polymer Hydrogel via Dynamic Thiol-Alkynone Double Addition Cross-Links. ACS Macro Lett. 2020, 9, 776–780. 10.1021/acsmacrolett.0c00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-I.; Hwang S.-J.; Park Y.-M.; Um I.-H. Michael-type Reactions of 1-(X-substituted phenyl)-2-propyn-1-ones with Alicyclic Secondary Amines in MeCN and H2O: Effect of Medium on Reactivity and Transition-State Structure. Bull. Korean Chem. Soc. 2010, 31, 1199–1203. 10.5012/bkcs.2010.31.5.1199. [DOI] [Google Scholar]
- Ruhemann S.; Cunnington A. V. XCII.—Studies of the acids of the acetylene series. J. Chem. Soc., Trans. 1899, 75, 954–963. 10.1039/CT8997500954. [DOI] [Google Scholar]
- Ruhemann S.; Stapleton H. E. LXXII.—Condensation of ethyl acetylenedicarboxylate with bases and β-ketonic esters. J. Chem. Soc., Trans. 1900, 77, 804–810. 10.1039/CT9007700804. [DOI] [Google Scholar]
- Ruhemann S. XXXIX.—Formation of α-pyrone compounds and their transformation into pyridine derivatives. J. Chem. Soc., Trans. 1899, 75, 411–416. 10.1039/CT8997500411. [DOI] [Google Scholar]
- Moureu C.; Lazennec I. Bull. Soc. Chim. Fr. 1906, 1, 1179–1190. [Google Scholar]
- Andre M. E. Ann. Chim. 1913, 29, 540. [Google Scholar]
- Barluenga J.; Aznar F.; Liz R.; Rodes R. Catalytic and Non-catalytic Addition of Aromatic Amines to Terminal Acetylenes in the Presence of Mercury(II) Chloride and Acetate. J. Chem. Soc., Perkin Trans. 1 1980, 2732–2737. 10.1039/p19800002732. [DOI] [Google Scholar]
- Severin R.; Doye S. The Catalytic Hydroamination of Alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. 10.1039/b600981f. [DOI] [PubMed] [Google Scholar]
- Yim J. C. H.; Schafer L. L. Efficient Anti-Markovnikov-Selective Catalysts for Intermolecular Alkyne Hydroamination: Recent Advances and Synthetic Applications. Eur. J. Org. Chem. 2014, 2014, 6825–6840. 10.1002/ejoc.201402300. [DOI] [Google Scholar]
- Chisholm D. R.; Valentine R.; Pohl E.; Whiting A. Conjugate Addition of 3-Buytn-2-one to Anilines in Ethanol: Alkene Geometric Insights through In Situ FTIR Monitoring. J. Org. Chem. 2016, 81, 7557–7565. 10.1021/acs.joc.6b01110. [DOI] [PubMed] [Google Scholar]
- Sengee M.; Sydnes L. K. Michael Addition of Various Nitrogen and Oxygen Nucleophiles to 1,1-Diethoxybut-3-yn-2-one. Synthesis 2011, 2011, 3899–3907. 10.1055/s-0031-1289294. [DOI] [Google Scholar]
- Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]
- Utimoto K.; Miwa H.; Nozaki H. Palladium-catalyzed Synthesis of Pyrroles. Tetrahedron Lett. 1981, 22, 4277–4278. 10.1016/S0040-4039(01)82932-4. [DOI] [Google Scholar]
- Gabriele B.; Salerno G.; Fazio A.; Bossio M. R. Palladium-catalyzed Cycloisomerization of (Z)-(2-En-4-ynyl)amines: A New Synthesis of Substituted Pyrroles. Tetrahedron Lett. 2001, 42, 1339–1341. 10.1016/S0040-4039(00)02206-1. [DOI] [Google Scholar]
- Gabriele B.; Salerno G.; Fazio A. General and Regioselective Synthesis of Substituted Pyrroles by Metal-Catalyzed or Spontaneous Cycloisomerization of (Z)-(2-En-4-ynyl)amines. J. Org. Chem. 2003, 68, 7853–7861. 10.1021/jo034850j. [DOI] [PubMed] [Google Scholar]
- Gabriele B.; Mancuso R.; Salerno G.; Ruffolo G.; Plastina P. Novel and Convenient Synthesis of Substituted Quinolines by Copper- or Palladium-Catalyzed Cyclodehydration of 1-(2-Aminoaryl)-2-yn-1-ols. J. Org. Chem. 2007, 72, 6873–6877. 10.1021/jo071094z. [DOI] [PubMed] [Google Scholar]
- Gabriele B.; Mancuso R.; Lupinacci E.; Spina R.; Salerno G.; Veltri L.; Dibenedetto A. Recyclable Catalytic Synthesis of Substituted Quinolines: Copper-catalyzed Heterocyclization of 1-(2-Aminoaryl)-2-yn-1-ols in Ionic Liquids. Tetrahedron 2009, 65, 8507–8512. 10.1016/j.tet.2009.08.017. [DOI] [Google Scholar]
- Gabriele B.; Veltri L.; Plastina P.; Mancuso R.; Vetere M. V.; Maltese V. Copper-Catalyzed Synthesis of Substituted Furans and Pyrroles by Heterocyclodehydration and Tandem Heterocyclodehydration-Hydration of 3-Yne-1,2-diols and 1-Amino-3-yn-2-ol Derivatives. J. Org. Chem. 2013, 78, 4919–4928. 10.1021/jo400533j. [DOI] [PubMed] [Google Scholar]
- Egi M.; Azechi K.; Akai S. Cationic Gold(I)-Mediated Intramolecular Cyclization of 3-Alkyne-1,2-diols and 1-Amino-3-alkyn-2-ols: A Practical Route to Furans and Pyrroles. Org. Lett. 2009, 11, 5002–5005. 10.1021/ol901942t. [DOI] [PubMed] [Google Scholar]
- Aponick A.; Li C.-Y.; Malinge J.; Marques E. F. An Extremely Facile Synthesis of Furans, Pyrroles, and Thiophenes by the Dehydrative Cyclization of Propargyl Alcohols. Org. Lett. 2009, 11, 4624–4627. 10.1021/ol901901m. [DOI] [PubMed] [Google Scholar]
- Keivanloo A.; Bakherad M.; Rahimi A.; Taheri S. A. N. One-pot Synthesis of 1,2-Disubstituted Pyrrolo[2,3-b]quinoxalines via Palladium-catalyzed Heteroannulation in Water. Tetrahedron Lett. 2010, 51, 2409–2412. 10.1016/j.tetlet.2010.02.123. [DOI] [Google Scholar]
- Keivanloo A.; Bakherad M.; Nasr-Isfahani H.; Esmaily S. Highly Efficient Synthesis of 5,6-Disubstituted-5H-pyrrolo[2,3-b]pyrazine-2,3-Dicarbonitriles through a One-pot Palladium-catalyzed Coupling Reaction/cyclization in Water. Tetrahedron Lett. 2012, 53, 3126–3130. 10.1016/j.tetlet.2012.04.016. [DOI] [Google Scholar]
- Chatterjee N.; Sarkar S.; Pal R.; Sen A. K. A Green Approach for the Regio- and Stereo-selective Syntheses of (Z)-3-Methyleneisoindoline-1-ones in Aqueous Medium. Tetrahedron Lett. 2013, 54, 3748–3751. 10.1016/j.tetlet.2013.05.004. [DOI] [Google Scholar]
- Zora M.; Kivrak A. Synthesis of Pyrazoles via CuI-Mediated Electrophilic Cyclizations of α,β-Alkynic Hydrazones. J. Org. Chem. 2011, 76, 9379–9390. 10.1021/jo201685p. [DOI] [PubMed] [Google Scholar]
- Bharathiraja G.; Sengoden M.; Kannan M.; Punniyamurthy T. Expedient Synthesis of Tetrasubstituted Pyrroles via a Copper-catalyzed Cascade Inter-/Intramolecular Cyclization of 1,3-Enynes Carry a Nitro Group with Amines. Org. Biomol. Chem. 2015, 13, 2786–2792. 10.1039/C4OB02508C. [DOI] [PubMed] [Google Scholar]
- Reid S.; Barrett A. G. M.; Hill M. S.; Procopiou P. A. Heavier Alkaline Earth Catalyzed Ene-yne Cyclizations: Atom-Efficient Access to Tetrahydroisoquinoline Frameworks. Org. Lett. 2014, 16, 6016–6019. 10.1021/ol502600g. [DOI] [PubMed] [Google Scholar]
- Trinh T. T. H.; Nguyen K. H.; de Aguiar Amaral P.; Gouault N. Total Synthesis of (−)-Epimyrtine by a Gold-catalyzed Hydroamination Approach. Beilstein J. Org. Chem. 2013, 9, 2042–2047. 10.3762/bjoc.9.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpita A.; Ribecai A.; Stabile P. Microwave-assisted Synthesis of Indole- and Azaindole-derivatives in Water via Cycloisomerization of 2-Alkynylanilines and Alkynylpyridinamines Promoted by Amines or Catalytic Amounts of Neutral or Basic Salts. Tetrahedron 2010, 66, 7169–7178. 10.1016/j.tet.2010.06.083. [DOI] [Google Scholar]
- Patel M.; Saunthawal R. K.; Verma A. K. Base-Catalyzed Stereoselective Intermolecular Addition of Imidazoles onto Alkynes: An Easy Access to Imidazolyl Enamines. Tetrahedron Lett. 2014, 55, 1310–1315. 10.1016/j.tetlet.2013.12.100. [DOI] [Google Scholar]
- Šenica L.; Grošelj U.; Kasunič M.; Kočar D.; Stanovnik B.; Svete J. Synthesis of Enaminone-Based Vinylogous Peptides. Eur. J. Org. Chem. 2014, 2014, 3067–3071. 10.1002/ejoc.201402033. [DOI] [Google Scholar]
- Sayyed-Alangi S. Z.; Hossaini Z.; Rostami-Charati F. Synthesis of Highly Functionalized Pyrroles from Primary Amines and Activated Acetylenes in Water. Chin. Chem. Lett. 2012, 23, 1119–1121. 10.1016/j.cclet.2012.06.042. [DOI] [Google Scholar]
- Zhang X.; Song X.; Li H.; Zhang S.; Chen X.; Yu X.; Wang W. An Organocatalytic Cascade Approach toward Polysubstituted Quinolines and Chiral 1,4-Dihydroquinolines-Unanticipated Effect of N-Protecting Groups. Angew. Chem., Int. Ed. 2012, 51, 7282–7286. 10.1002/anie.201202161. [DOI] [PubMed] [Google Scholar]
- Sriramurthy V.; Kwon O. Diphosphine-Catalyzed Mixed Double-Michael Reaction: A Unified Synthesis of Indolines, Dihydropyrrolopyridines, Benzimidazolines, Tetrahydroquinolines, Tetrahydroisoquinolines, Dihydrobenzo-1,4-oxazines, and Dihydrobenzo-3,1-oxazines. Org. Lett. 2010, 12, 1084–1087. 10.1021/ol100078w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y. C.; Kwon O. Diversity-Oriented Synthesis Based on the DPPP-Catalyzed Mixed Double-Michael Reactions of Electron-Deficient Acetylenes and β-Amino Alcohols. Molecules 2011, 16, 3802–3825. 10.3390/molecules16053802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khong S.; Kwon O. One-Pot Phosphine-Catalyzed Syntheses of Quinolines. J. Org. Chem. 2012, 77, 8257–8267. 10.1021/jo3015825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Xu X.; Yang J.; Xie X.; Huang H.; Li Y. Iron-catalyzed Cascade Reaction of Ynone with o-Aminoaryl Compounds: A Michael Addition-cyclization Approach to 3-Carbonyl Quinolines. Tetrahedron Lett. 2011, 52, 530–533. 10.1016/j.tetlet.2010.11.106. [DOI] [Google Scholar]
- Zhu Q.; Ye Z.; Yang W.; Cai X.; Tang B. Z. One-Pot Synthesis and Structure-Property Relationship of Aminomaleimides: Fluorescence Efficiencies in Monomers and Aggregates Easily Tuned by Switch of Aryl and Alkyl. J. Org. Chem. 2017, 82, 1096–1104. 10.1021/acs.joc.6b02706. [DOI] [PubMed] [Google Scholar]
- Takeuchi K.; Ikoshi M.; Ichinohe M.; Sekiguchi A. Addition of Amines and Hydroborane to the Disilyne RSi≡SiR (R = SiiPr[CH(SiMe3)2]2) giving Amino- and Boryl-substituted Disilenes. J. Am. Chem. Soc. 2010, 132, 930–931. 10.1021/ja910157h. [DOI] [PubMed] [Google Scholar]
- Takeuchi K.; Ikoshi M.; Ichinohe M.; Sekiguchi A. Silicon Version of Enamines: Amino-substituted Disilenes by the Reactions of the Disilyne RSiSiR (R = SiiPr[CH(SiMe3)2]2) with Amines. J. Organomet. Chem. 2011, 696, 1156–1162. 10.1016/j.jorganchem.2010.07.008. [DOI] [Google Scholar]
- Hu X.; Zhao X.; He B.; Zhao Z.; Zheng Z.; Zhang P.; Shi X.; Kwok R. T. K.; Lam J. W. Y.; Qin A.; Tang B. Z. A Simple Approach to Bioconjugation at Diverse Levels: Metal-Free Click Reactions of Activated Alkynes with Native Groups of Biotargets without Prefunctionalization. Research 2018, 2018, 3152870. 10.1155/2018/3152870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Shen J.; Hu R.; Shi X.; Hu X.; He B.; Qin A.; Tang B. Z. Fast Surface Immobilization of Native Proteins through Catalyst-free Amino-yne Click Bioconjugation. Chem. Sci. 2020, 11, 3931–3935. 10.1039/D0SC00062K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oktay B.; Demir S.; Kayaman-Apohan N. Immobilization of Pectinase on Polyethyleneimine Based Support via Spontaneous Amino-yne Click Reaction. Food Bioprod. Process. 2020, 122, 159–168. 10.1016/j.fbp.2020.04.010. [DOI] [Google Scholar]
- Bass R. G.; Sinsky M. S.; Connell J. W.; Waldbauer R. O.; Hergenrother P. M. Poly(enamine-ketones) from Hydrogen-terminated Aromatic Dipropynones and Aromatic Diamines. J. Polym. Sci., Part A: Polym. Chem. 1989, 27, 171–183. 10.1002/pola.1989.080270115. [DOI] [Google Scholar]
- He B.; Zhen S.; Wu Y.; Hu R.; Zhao Z.; Qin A.; Tang B. Z. Cu(I)-Catalyzed Amino-yne Click Polymerization. Polym. Chem. 2016, 7, 7375–7382. 10.1039/C6PY01501H. [DOI] [Google Scholar]
- He B.; et al. Spontaneous Amino-yne Click Polymerization: A Powerful Tool toward Regio- and Stereospecific Poly(β-aminoacrylate)s. J. Am. Chem. Soc. 2017, 139, 5437–5443. 10.1021/jacs.7b00929. [DOI] [PubMed] [Google Scholar]
- Chen X.; Bai T.; Hu R.; Song B.; Lu L.; Ling J.; Qin A.; Tang B. Z. Aroylacetylene-based Amino-yne Click Polymerization toward Nitrogen-containing Polymers. Macromolecules 2020, 53, 2516–2525. 10.1021/acs.macromol.9b02747. [DOI] [Google Scholar]
- Tang X.; Zheng C.; Chen Y.; Zhao Z.; Qin A.; Hu R.; Tang B. Z. Multicomponent Tandem Polymerizations of Aromatic Diynes, Terephthaloyl Chloride, and Hydrazines toward Functional Conjugated Polypyrazoles. Macromolecules 2016, 49, 9291–9300. 10.1021/acs.macromol.6b02192. [DOI] [Google Scholar]
- Li D.; Song Y.; He J.; Zhang M.; Ni P. Polymer-doxorubicin Prodrug with Biocompatibility, pH Response, and Main Chain Breakability Prepared by Catalyst-free Click Reaction. ACS Biomater. Sci. Eng. 2019, 5, 2307–2315. 10.1021/acsbiomaterials.9b00301. [DOI] [PubMed] [Google Scholar]
- Hu R.; Chen X.; Zhou T.; Si H.; He B.; Kwok R. T. K.; Qin A.; Tang B. Z. Lab-in-cell Based on Spontaneous Amino-yne Click Polymerization. Sci. China: Chem. 2019, 62, 1198–1203. 10.1007/s11426-019-9517-9. [DOI] [Google Scholar]
- Qi C.; Zheng C.; Hu R.; Tang B. Z. Direct Construction of Acid-responsive Poly(indolone)s through Multicomponent Tandem Polymerizations. ACS Macro Lett. 2019, 569–575. 10.1021/acsmacrolett.9b00297. [DOI] [PubMed] [Google Scholar]
- Yu L.; Zhang Z.; You Y.-Z.; Hong C.-Y. Synthesis of Sequence-controlled Polymers via Sequential Thiol-ene and Amino-yne Click Reactions in One Pot. Eur. Polym. J. 2018, 103, 80–87. 10.1016/j.eurpolymj.2018.02.006. [DOI] [Google Scholar]
- Huang J.; Jiang X. Injectable and Degradable pH-Responsive Hydrogels via Spontaneous Amino-Yne Click Reaction. ACS Appl. Mater. Interfaces 2018, 10, 361–370. 10.1021/acsami.7b18141. [DOI] [PubMed] [Google Scholar]
- Fenton O. S.; Andresen J. L.; Paolini M.; Langer R. β-Aminoacrylate Synthetic Hydrogels: Easily Accessible and Operationally Simple Biomaterials Networks. Angew. Chem., Int. Ed. 2018, 57, 16026–16029. 10.1002/anie.201808452. [DOI] [PubMed] [Google Scholar]
- Saravanakumar G.; Park H.; Kim J.; Park D.; Pramanick S.; Kim D. H.; Kim W. J. Miktoarm Amphiphilic Block Copolymer with Singlet Oxygen-Labile Stereospecific β-Aminoacrylate Junction: Synthesis, Self-Assembly, and Photodynamically Triggered Drug Release. Biomacromolecules 2018, 19, 2202–2213. 10.1021/acs.biomac.8b00290. [DOI] [PubMed] [Google Scholar]
- Jiang R.; et al. Fabrication of AIE-active Fluorescent Polymeric Nanoparticles with Red Emission through a Facile Catalyst-free Amino-yne Click Polymerization. Dyes Pigm. 2018, 151, 123–129. 10.1016/j.dyepig.2017.12.046. [DOI] [Google Scholar]
- He B.; Zhang J.; Wang J.; Wu Y.; Qin A.; Tang B. Z. Preparation of Multifunctional Hyperbranched Poly(β-aminoacrylate)s by Spontaneous Amino-yne Click Polymerization. Macromolecules 2020, 53, 5248–5254. 10.1021/acs.macromol.0c00813. [DOI] [Google Scholar]
- Ruhemann S.; Beddow F. LXXXVIII.—Condensation of Phenols with Esters of the Acetylene Series. Part I. Action of Phenols on Ethyl Phenylpropiolate. J. Chem. Soc., Trans. 1900, 77, 984–990. 10.1039/CT9007700984. [DOI] [Google Scholar]
- Tejedor D.; Álvarez-Méndez S. J.; López-Soria J. M.; Martín V. S.; García-Tellado F. A Robust and General Protocol for the Lewis-Base-Catalysed Reaction of Alcohols and Alkyl Propiolates. Eur. J. Org. Chem. 2014, 2014, 198–205. 10.1002/ejoc.201301303. [DOI] [Google Scholar]
- Wakabayashi Y.; Fukuda Y.; Shiragami H.; Utimoto K.; Nozaki H. Preparation of Furans from Alkynols Utilizing Palladium Catalyzed Intramolecular Addition of Alcohol to Acetylene as a Key Reaction. Tetrahedron 1985, 41, 3655–3661. 10.1016/S0040-4020(01)91384-5. [DOI] [Google Scholar]
- Hou X. L.; Cheung H. Y.; Hon T. Y.; Kwan P. L.; Lo T. H.; Tong S. Y.; Wong H. N. C. Regioselective Syntheses of Substituted Furans. Tetrahedron 1998, 54, 1955–2020. 10.1016/S0040-4020(97)10303-9. [DOI] [Google Scholar]
- Gabriele B.; Salerno G.; Lauria E. A General and Facile Synthesis of Substituted Furans by Palladium-Catalyzed Cycloisomerization of (Z)-2-En-4-yn-1-ols. J. Org. Chem. 1999, 64, 7687–7692. 10.1021/jo990847h. [DOI] [PubMed] [Google Scholar]
- Gabriele B.; Salerno G.; Fazio A.; Pittelli R. Versatile Synthesis of (Z)-1-Alkylidene-1,3-dihydroisobenzofurans and 1H-Isochromenes by Palladium-catalyzed Cycloisomerization of 2-Alkynylbenzyl Alcohols. Tetrahedron 2003, 59, 6251–6259. 10.1016/S0040-4020(03)01019-6. [DOI] [Google Scholar]
- Yada Y.; Miyake Y.; Nishibayashi Y. Ruthenium-Catalyzed Intramolecular Cyclization of 3-Butyne-1,2-diols into Furans. Organometallics 2008, 27, 3614–3617. 10.1021/om800298r. [DOI] [Google Scholar]
- Hayes S. J.; Knight D. W.; Menzies M. D.; O’Halloran M.; Tan W.-F. An Efficient Furan Synthesis using Heterogeneous Catalysis. Tetrahedron Lett. 2007, 48, 7709–7712. 10.1016/j.tetlet.2007.08.102. [DOI] [Google Scholar]
- Sakai M.; Sasaki M.; Tanino K.; Miyashita M. Synthetic Studies of Zoanthamine Alkaloids. Stereoselective Synthesis of the ABC Ring System of Norzoanthamine by an Intramolecular Diels-Alder reaction. Tetrahedron Lett. 2002, 43, 1705–1708. 10.1016/S0040-4039(02)00079-5. [DOI] [Google Scholar]
- Yao T.; Zhang X.; Larock R. C. AuCl3-Catalyzed Synthesis of Highly Substituted Furans from 2-(1-Alkynyl)-2-alken-1-ones. J. Am. Chem. Soc. 2004, 126, 11164–11165. 10.1021/ja0466964. [DOI] [PubMed] [Google Scholar]
- Ma J.; Zhang L.; Zhu S. Enynal/Enynone: A Safe and Practical Carbenoid Precursor. Curr. Org. Chem. 2015, 20, 102–118. 10.2174/1385272819666150608222517. [DOI] [Google Scholar]
- Siva Kumari A. L.; Siva Reddy A.; Swamy K. C. K. Exploring the Gold Mine: [Au]-Catalysed Transformations of Enynals, Enynones and Enynols. Org. Biomol. Chem. 2016, 14, 6651–6671. 10.1039/C6OB00698A. [DOI] [PubMed] [Google Scholar]
- Chen L.; Liu Z.; Zhu S. Donor- and Acceptor-enynals/enynones. Org. Biomol. Chem. 2018, 16, 8884–8898. 10.1039/C8OB02348D. [DOI] [PubMed] [Google Scholar]
- Chen L.; Chen K.; Zhu S. Transition-Metal-Catalyzed Intramolecular Nucleophilic Addition of Carbonyl Groups to Alkynes. Chem. 2018, 4, 1208–1262. 10.1016/j.chempr.2018.02.001. [DOI] [Google Scholar]
- Barluenga J.; Calleja J.; Mendoza A.; Rodríguez F.; Fañanás F. J. Synthesis of Polycyclic Compounds by a Cascade Cycloisomerisation/Diels-Alder Reaction. Chem. - Eur. J. 2010, 16, 7110–7112. 10.1002/chem.201000515. [DOI] [PubMed] [Google Scholar]
- Yeh M.-C.; Liang C.-J.; Jiang X.-Y. Synthesis of 3-exo-Aroylhexahydroindoles via Sequential Gold(I)-Catalyzed Claisen-Type Rearrangement-Epimerization Reactions of cis-4-[N-Tosyl-N-(3-arylprop-2-ynyl)amino]cyclohex-2-en-1-ols. Synthesis 2014, 46, 2220–2224. 10.1055/s-0033-1339129. [DOI] [Google Scholar]
- Guo R.; Li K.-N.; Gong L.-Z. Catalytic Cascade Hydroalkoxylation/isomerization/[4 + 2] Cycloaddition using Enyne Alcohols as Latent Dienes or Dienophiles. Org. Biomol. Chem. 2013, 11, 6707. 10.1039/c3ob41611a. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; Fernández A.; Diéguez A.; Rodríguez F.; Fañanás F. J. Gold- or Platinum-Catalyzed Cascade Processes of Alkynol Derivatives Involving Hydroalkoxylation Reactions Followed by Prins-Type Cyclizations. Chem. - Eur. J. 2009, 15, 11660–11667. 10.1002/chem.200900856. [DOI] [PubMed] [Google Scholar]
- Barluenga J.; Fernández A.; Satrústegui A.; Diéguez A.; Rodríguez F.; Fañanás F. J. Tandem Intramolecular Hydroalkoxylation-Hydroarylation Reactions: Synthesis of Enantiopure Benzofused Cyclic Ethers from the Chiral Pool. Chem. - Eur. J. 2008, 14, 4153–4156. 10.1002/chem.200800312. [DOI] [PubMed] [Google Scholar]
- Gabriele B.; Plastina P.; Vetere M. V.; Veltri L.; Mancuso R.; Salerno G. A Simple and Convenient Synthesis of Substituted Furans and Pyrroles by CuCl2-catalyzed Heterocyclodehydration of 3-Yne-1,2-diols and N-Boc- or N-Tosyl-1-amino-3-yn-2-ols. Tetrahedron Lett. 2010, 51, 3565–3567. 10.1016/j.tetlet.2010.05.001. [DOI] [Google Scholar]
- Silva F.; Sawicki M.; Gouverneur V. Enantioselective Organocatalytic Aldol Reaction of Ynones and Its Synthetic Applications. Org. Lett. 2006, 8, 5417–5419. 10.1021/ol0624225. [DOI] [PubMed] [Google Scholar]
- Frančois-Endelmond C. L.; Carlin T.; Thuery P.; Loreau O.; Taran F. D. R. A Phosphine-Mediated Construction of 1,4-Oxazepines and 1,3-Oxazines. Org. Lett. 2010, 12, 40–42. 10.1021/ol9024309. [DOI] [PubMed] [Google Scholar]
- Alemán J.; Núñez A.; Marzo L.; Marcos V.; Alvarado C.; García Ruano J. L. Asymmetric Synthesis of 4-Amino-4H-Chromenes by Organocatalytic Oxa-Michael/Aza-Baylis-Hillman Tandem Reactions. Chem. - Eur. J. 2010, 16, 9453–9456. 10.1002/chem.201001293. [DOI] [PubMed] [Google Scholar]
- Liu C.; Zhang X.; Wang R.; Wang W. One-Pot” Access to 4 H -Chromenes with Formation of a Chiral Quaternary Stereogenic Center by a Highly Enantioselective Iminium-allenamine Involved Oxa-Michael-Aldol Cascade. Org. Lett. 2010, 12, 4948–4951. 10.1021/ol102096s. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Zhang S.; Wang W. Iminium-Allenamine Cascade Catalysis: One-Pot Access to Chiral 4H-Chromenes by a Highly Enantioselective Michael-Michael Sequence. Angew. Chem., Int. Ed. 2010, 49, 1481–1484. 10.1002/anie.200906050. [DOI] [PubMed] [Google Scholar]
- Song A.; Chen X.; Song X.; Zhang X.; Zhang S.; Wang W. Synthesis of Benzoxazoles via an Amine-Catalyzed [4 + 1] Annulation. Org. Lett. 2013, 15, 2510–2513. 10.1021/ol400988e. [DOI] [PubMed] [Google Scholar]
- Kuroda H.; Tomita I.; Endo T. A Novel Polyaddition of Bifunctional Acetylenes Containing Electron-withdrawing Groups. III. Synthesis of Polymers having β-Alkoxyenoate Moieties by the Reaction of Internal Diynes with Diols. J. Polym. Sci., Part A: Polym. Chem. 1996, 34, 1597–1604. . [DOI] [Google Scholar]
- Kuroda H.; Tomita I.; Endo T. A Novel Polyaddition of Diols with Bifunctional Acetylenes Having Electron-Withdrawing Groups. Macromolecules 1995, 28, 433–436. 10.1021/ma00106a005. [DOI] [Google Scholar]
- Shi Y.; et al. Phenol-yne Click Polymerization: An Efficient Technique to Facilely Access Regio- and Stereoregular Poly(vinylene ether ketone)s. Chem. - Eur. J. 2017, 23, 10725–10731. 10.1002/chem.201702966. [DOI] [PubMed] [Google Scholar]
- Wang J.; Li B.; Xin D.; Hu R.; Zhao Z.; Qin A.; Tang B. Z. Superbase Catalyzed Regio-selective Polyhydroalkoxylation of Alkynes: A Facile Route Towards Functional Poly(vinyl ether)s. Polym. Chem. 2017, 8, 2713–2722. 10.1039/C7PY00363C. [DOI] [Google Scholar]
- Si H.; Wang K.; Song B.; Qin A.; Tang B. Z. Organobase-catalysed Hydroxyl-yne Click Polymerization. Polym. Chem. 2020, 11, 2568–2575. 10.1039/D0PY00095G. [DOI] [Google Scholar]
- Wang K.; Si H.; Wan Q.; Wang Z.; Qin A.; Tang B. Z. Luminescent Two-way Reversible Shape Memory Polymers Prepared by Hydroxyl-yne Click Polymerization. J. Mater. Chem. C 2020, 8, 16121–16128. 10.1039/D0TC03888A. [DOI] [Google Scholar]