

Abstract
Bioorthogonal cleavage chemistry has been rapidly emerging as a powerful tool for manipulation and gain-of-function studies of biomolecules in living systems. While the initial bond formation-centered bioorthogonal reactions have been widely adopted for labeling, tracing, and capturing biomolecules, the newly developed bond cleavage-enabled bioorthogonal reactions have opened new possibilities for rescuing small molecules as well as biomacromolecules in living systems, allowing multidimensional controls over biological processes in vitro and in vivo. In this Outlook, we first summarized the development and applications of bioorthogonal cleavage reactions (BCRs) that restore the functions of chemical structures as well as more complex networks, including the liberation of prodrugs, release of bioconjugates, and in situ reactivation of intracellular proteins. As we embarked on this fruitful progress, we outlined the unmet scientific needs and future directions along this exciting avenue. We believe that the potential of BCRs will be further unleashed when combined with other frontier technologies, such as genetic code expansion and proximity-enabled chemical labeling.
Short abstract
Bioorthogonal cleavage chemistry has been rapidly emerging as a powerful tool for manipulation and gain-of-function studies of biomolecules under living conditions.
Introduction
Chemical reactions that are orthogonal to endogenous molecules and reactions would provide a powerful tool to dissect and manipulate biological processes within sophisticated living systems. Since the concept of bioorthogonal reactions was proposed almost two decades ago,1 we have witnessed an explosion of interest in developing and optimizing bioorthogonal reactions such as Staudinger ligation,2,3 Cu-catalyzed azide–alkyne cycloaddition (CuAAC),4 strain-promoted azide–alkyne cycloaddition (SPAAC), and inverse-electron-demand Diels–Alder (IDA) cycloaddition.5 These reactions have enabled the labeling, tracing, or enriching of biomolecules in living cells and have found exciting applications in areas such as biocompatible materials, biomedical engineering, and nanomedicine.6−11 The biocompatibility of bioorthogonal reactions has been improved from cell lysate to live cells and even live animals.1,12,13 For example, in 2008, the SPAAC reaction was applied to label cell surface glycans in a time-lapse manner to visualize the dynamic change in zebrafish embryos during their development, which demonstrated the high bioorthogonality of this exogenous chemical reaction to monitor delicate biological processes.14 More representative work in this field has been comprehensively reviewed elsewhere.9,13,15−30
The content and boundaries of bioorthogonal reactions are also continuously expanding. Early examples of bioorthogonal chemistry mainly focused on ligation-type reactions between two biologically inert but mutually reactive components. In recent years, bond breakage-enabled bioorthogonal reactions, namely, bioorthogonal cleavage reactions (BCRs), have rapidly emerged.31 Unlike bioorthogonal ligation reactions, BCRs utilize bond cleavage chemistry to achieve the spatially and/or temporally controlled breakage of different components on a target of interest22 (Figure 1a). Relying on this new reaction type, the functions of target molecules can be temporarily masked by a protecting group (e.g., a chemical cage) that can be subsequently deprotected by the corresponding cleavage trigger (e.g., chemical decaging), leading to restored activity. A collection of BCRs have since been rapidly developed and improved in terms of their reaction scopes, biocompatibilities, and efficiencies, which enables the on-demand activation of small molecules, biomolecules, and bioconjugates inside living cells, multicellular organisms, and even animals24,25 (Figure 1). Encouragingly, some of these reactions hold promise for clinical applications.13,32−34 For example, the IDA reaction-triggered decaging strategy was recently applied to activate caged doxorubicin (Dox)-SQP33 for solid tumor therapy in human patients.35 As a new direction in bioorthogonal chemistry, BCRs have been greatly inspired by deprotection reactions in organic chemistry, which were initially not often compatible with living conditions. Extensive efforts have thus focused on reinventing such reactions as biocompatible reactions that can be performed in living systems. A panel of BCRs with distinct reaction mechanisms, tunable reaction rates, and different biocompatibilities with various in vivo settings have now been reported. Herein, we aim to offer an overview of this exciting new avenue and introduce the scope, biocompatibility, and applications of different BCRs with a particular focus on the type of molecules and functions that could be manipulated by each reaction.
Figure 1.
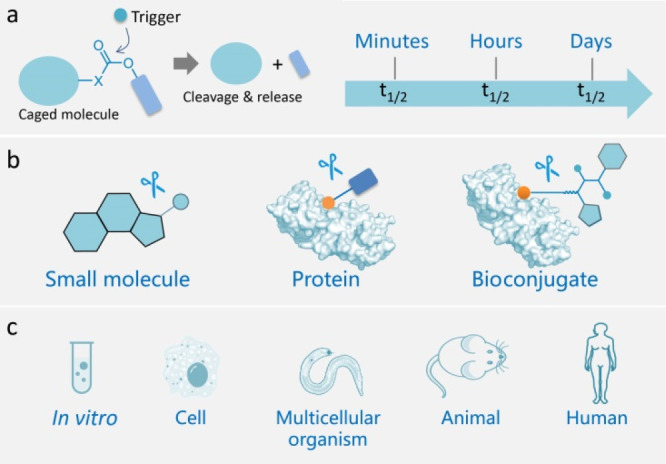
Landscape of bioorthogonal cleavage reactions (BCRs). (a) Concept of BCRs and the usual reaction kinetics. (b) Controllable release of functional biomolecules by BCRs. (c) Biocompatibility evolution of BCRs. The rich repertoire of deprotection reactions in chemical synthesis has greatly facilitated the development of BCRs. However, the reaction conditions are usually too harsh for biological settings, and reinventing these reactions for living systems is not a trivial task. Extensive efforts have thus been devoted to developing BCRs with high biocompatibility and a broad range of utility in various living systems, opening a new avenue in bioorthogonal chemistry.
Overview of Current BCRs
Traditionally, ultraviolet (UV) light-mediated deprotection reactions have been employed for the controllable release of small-molecule drugs, nucleotides, and proteins. These excellent works have been comprehensively reviewed and discussed previously.36 In contrast, metal- or small-molecule-triggered BCRs hold great potential due to their versatility, tunability, and better penetration and spatial targeting abilities in living tissues and animals.31 As shown in Figure 2, a collection of small-molecule-triggered cleavage reactions have been developed in recent years.
Figure 2.

State-of-the-art bioorthogonal cleavage reactions. BCRs can be classified as transition-metal-triggered cleavage reactions (entries 1–11), cycloaddition-mediated cleavage reactions (entries 12–25 and 31), and redox-based cleavage reactions (entries 26–30), all of which have found an array of exciting applications with excellent biocompatibility both in vitro and in living cells or animals. Reported pseudo-second-order constants (k2) are listed in the Figure; the k2 values of entries 4, 7, and 30 are calculated by the increasing signal of products, and the k2 values of entries 12, 14–22, 24, and 31 are calculated by the decreasing signals of reactants.
Transition-metal-catalyzed deprotection reactions were tested in living systems more than a decade ago (Figure 2, entries 1–11). The catalytic cycle of the cleavage reaction is often initiated via coordination between metals and alkene/alkyne moieties, followed by nucleophilic substitution to release the final products. In 2006, Meggers et al. reported the first Ru-mediated deallyloxycarbonyl (allyl carbamate) reaction in live cells for the activation of a fluorescent dye.37 The efficiency of this reaction was further improved by optimization of the catalyst ligands, allowing catalyst loading as low as 5 μM in living cells.38 Since then, additional catalysts formed by metals such as palladium,39 ruthenium,21,40 copper,41,42 gold,43 and platinum44 have been applied to trigger allyl or propargyl deprotection under physiological conditions.45,46
Pd is one of the most widely explored transition metals for BCRs, with catalysts containing Pd(0), Pd(II), and Pd(IV) species or Pd nanoparticles.17,34,39,47−56 For instance, Koide et al. reported the Pd-triggered deallylation of fluorescent probes in 2007,47 which was later evaluated in zebrafish by using 5 μM PdCl2.48 Our group extended this reaction to biomacromolecules for a gain-of-function study in proteins in 2014.49 In this study, we exploited the Pd-triggered depropargylation reaction to rescue a premasked catalytic Lys residue on a protein of interest (POI) using 10 μM Pd(dba)2 or allyl2Pd2Cl2, which enabled metal-triggered bioorthogonal protein activation in living cells.49
As one of the most widely used metals for catalyzing bioorthogonal ligation reactions, such as CuAAC, Cu has recently been used to trigger BCRs41,57−59 (Figure 2, entries 5, 9, and 11). Inspired by the in vitro deprotection of the dimethyl propargyl group using Cu(I) by Finn et al. in 2013,42 we employed this bioorthogonal cleavage pair for the manipulation of cell surface interactions.41 In addition, Chang et al. remodeled the Cu(I)-chelating ligand as a hydroxyl protecting group, which could be oxidatively cleaved upon coordination with Cu(I) (Figure 2, entry 11).57,58
BCRs triggered by other metals have also been investigated. For instance, Ward et al. designed a Ru-triggered olefin metathesis reaction for the controlled release of a hydroxyl group21 (Figure 2, entry 6). Bernardes et al. reported the Pt-triggered deprotection of N-propargyls. In the latter work, the authors discovered that pentynoyl amides can be cleaved by the Pt catalyst, which added a new bioorthogonal caging group on the amide other than the carbamate group44 (Figure 2, entry 4). Finally, Huang et al. used the Pt(IV) complex as a precatalyst, which can be selectively converted into Pt(II) in tumor cells, triggering the cleavage of O/N propargyl-caged prodrugs.60
In general, an advantage of transition-metal-triggered BCRs is that their reactivity/selectivity can be finely tuned by the metals or ligands utilized, which provides exciting chemical space for further exploration. Currently, although more than a stoichiometric amount of metal is often used, catalytic amounts of metal reagents may eventually be used to trigger BCRs in living systems. Recent progress reported by Koide et al. conceptually demonstrated this potential. The authors used the cleavage of allylic and propargylic ethers as platforms to systematically screen metals and ligands to discover fast and efficient BCRs.59 After examining over 800 reactions, several previously unknown metal–ligand pairs were disclosed, indicating that the selectivity/reactivity of the metals can be tuned by specific phosphine ligands and that the cleavage reaction can be efficiently conducted using a catalytic amount of the metal catalyst.59 Nevertheless, the potential toxicity of metal ions to living systems needs to be considered. In addition, these transition-metal-triggered BCRs are often sensitive to the reaction conditions, rendering it difficult to adopt the optimized reaction to a more complex environment.61
Small-molecule-triggered BCRs have attracted much attention due to their high biocompatibility, fast reaction rate, and robustness (Figure 2, entries 11–31). In 2008, Fox et al. reported bioorthogonal ligation between trans-cyclooctene (TCO) and tetrazine (Tz) with excellent reaction kinetics.62 This IDA reaction has since been intensively studied and adopted in various settings. In particular, Robillard et al. discovered that when a carbamate group was anchored to the allylic position of TCO, liberation of free amines would occur via a series of intramolecular rearrangements of the [4 + 2] cycloaddition adducts.63,64 We are the first group to employ this “click-to-release” reaction on proteins in living cells as well as in animals.65 Notably, although the kinetics of Tz/TCO conjugation are very fast,62 the overall reaction rate of this two-step reaction is slow due to the rate-determining release process.63,64 As shown in Figure 2, the k2 of the cycloaddition-based BCRs (entries 12, 14–22, 24, and 31) is calculated by measuring the decreasing rate of reactants (usually Tz), indicating that mostly the k2 value reflects the rate of the cycloaddition step instead of the whole cleavage reaction. Usually, the following releasing step is much slower than the typical IEDDA cycloaddition reaction. Encouraged by the high reactivity and bioorthogonality, IDA-based BCRs have been intensively studied. Based on the study of the reaction mechanism and substitution effects of Tz, IDA-based BCRs have been well optimized in terms of their reaction rate, efficiency, and stability.66,67 In addition to the liberation of an amine, the IDA reaction was further expanded to release alcohol, phenol, and carboxyl groups.68,69 It was even possible to release H2S by replacing the carbamate group with a thiocarbamate70 (Figure 2, entry 13). Whereas the reaction features fast kinetics, challenges still exist due to the possible instability of the highly strained TCO ring in living systems. Other than TCO, dienophiles, including benzonorbornene,71,72 vinyl esters,73,74 vinylboronic acid,75 and isocyanide,76 have also been developed as Tz-responsive caging groups (Figure 2, entries 12–19). In particular, Tz itself can serve as a protecting group, which can be decaged by several triggers, including TCO,77 cyclooctyne,78 and isonitrile79 (Figure 2, entries 20–22). Additional cycloaddition-based BCRs have also been developed. Gamble et al. reported a TCO-triggered BCR in which the para-azido benzyl group could be deprotected by TCO to liberate a free amine group from its carbamate precursor80 (Figure 2, entry 23). Similarly, the deprotection reaction occurs due to the instability of both the [3 + 2] cycloadducts of TCO and phenyl azides. A series of intramolecular rearrangements would generate an imine intermediate that would be hydrolyzed in living systems to give an unstable para-aminobenzyl structure, and subsequent 1,6-elimination releases the final amine products. This unstable and self-rearrangement structure is usually termed self-immolative linker, which is widely used in the BCRs. Taran et al. reported iminosydnones as caging groups cleavable by cyclooctyne81,82 (Figure 2, entry 24). Based on the intramolecular cycloaddition between the alkyne and cycloketones as well as the alkyne and sulfone derivatives, Wang et al. designed BCRs to release SO2 and CO, respectively83,84 (Figure 2, entry 25).
In addition to cycloaddition chemistry, BCR redox chemistry has also found broad applications.85−87 For instance, azide reduction followed by 1,4- or 1,6-elimination could lead to the controllable release of functional groups in living systems85 (Figure 2, entries 26–28). In 2016, Pluth et al. designed a “self-promoting” strategy based on the H2S reduction of azides in living cells.70 The 1,6-elimination of the thiocarbamate-containing linker generates more H2S, further promoting the initial azide reduction.70 In 2019, Peng et al. reported a formaldehyde-triggered 2-aza-cope reaction to release the Lys side chain from a POI88 (Figure 2, entry 29). Moreover, boronic acid or boronic acid ester oxidation reactions can also be used to restore the hydroxyl or amine group; thus, the decaging of a boronic-acid-caged fluorophore can be used for the detection of various reactive oxygen species.89 In 2015, Bertozzi et al. designed a “conditional bioorthogonal” reaction pair between an amine oxide and a diboronic acid pinacol ester90 (Figure 2, entry 30). Amine oxides are generally considered unstable but were creatively used here as a bioorthogonal tertiary amine protecting group. Moreover, oxidation of the diboronic acid pinacol ester enabled the transfer of oxygen atoms from the amine oxide to the boronic reagent, allowing the release of a tertiary amine.90
While BCRs initially focused on the release of amine groups, reports on the release of other functional groups have also emerged. In 2009, Koide et al. and Ahn et al. reported the Pd-mediated removal of allyl and propargyl groups to liberate a phenolic hydroxyl moiety on fluorescent probes.47,48 In 2016, we reported the Pd-mediated removal of allenes to release a phenolic hydroxyl group.55 In 2017, Devaraj et al. reported Tz-mediated vinyl removal to release a phenolic hydroxyl group.74 A subsequent study in 2019 demonstrated that vinylboronic acid is superior to vinyl groups in terms of release kinetics.75 Furthermore, IDA BCR-mediated chemistry has recently been reported to liberate a carboxyl group69 (Figure 2, entry 31).
The fast emergence of BCRs has greatly expanded the scope of the decaging reactions of different functional groups in complex biological systems. It is expected that each type of biomolecule of interest may find a suitable BCR for gain-of-function studies within the native cellular context. In addition to the expansion of reaction types and molecule types, BCRs have also significantly expanded in terms of their biological applications, which will be discussed in the following sections.
BCRs for Prodrug Activation
One motivation for the development of BCRs is the on-demand activation of prodrugs, such as anticancer agents, at the tumor site. Although various prodrug strategies have previously been developed to circumvent the toxicity of anticancer drugs to normal cells, such methods typically rely on the tumor-specific microenvironment or enzymatic activity for prodrug activation. Alternatively, BCR-enabled prodrugs are more stable than traditional prodrugs due to the bioorthogonality of the installed caging group and can thus be further reactivated via a bioorthogonal trigger. Such bioorthogonal prodrugs could be selectively activated at the tumor site, further minimizing the side effects to normal cells. To date, a series of bioorthogonal prodrugs have been developed, and representative work will be discussed below.24
Staudinger chemistry between azide and phosphine compounds was among the first examples of BCRs being used for prodrug activation. In 2006, Florent et al. reported the design of a prodrug by remodeling the Staudinger ligation reaction. Triarylphosphine was modified as an amine protecting group to inactivate Dox, which could be liberated upon the addition of an azide reagent. Over 90% of Dox was rescued within 3 h at 37 °C in a H2O–THF solution (Figure 3a, entry 1).86 Conversely, Robillard et al. used p-azidobenzyl (PAB) as the protecting group, which is self-immolative upon azide reduction by a triarylphosphine compound (Figure 3a, entry 2). Full conversion of PAB-protected Dox was observed under physiological conditions after 20 h of treatment with triarylphosphine. The cell proliferation assay showed that Dox and PAB–Dox had comparable IC50 values after multiple treatments with triarylphosphine.91 However, notable rapid quenching of the trigger triarylphosphine in the cell growth medium was observed due to its oxidation, requiring repeated addition of the trigger. Moreover, the potential toxicity of phosphine to animals would be another challenge for the in vivo applications of Staudinger chemistry-based drug release. In 2015, Gamble et al. demonstrated TCO-triggered PAB deprotection and PAB–Dox activation (Figure 3a, entry 2). Unlike the Staudinger reduction reaction, the 1,3-dipolar cycloaddition between TCO and the azides gave unstable 1,2,3-triazoline adducts, which further underwent intramolecular rearrangement, hydrolysis, and self-immolation to liberate a free amine or hydroxyl group. The advantages of TCO over triarylphosphine reagents in terms of its serum stability and low cytotoxicity make the TCO/PAB cleavage pair a more promising BCR for living systems.80 However, a limitation remains: its relatively slow kinetics (k2 = 0.027 M–1 s–1). The relationship between the substitution on PAB and the reaction kinetics was further studied by the same group in 2018.92 Although fluorine-substituted PAB showed faster 1,3-dipolar cycloaddition with TCO, slower hydrolysis and self-immolation in the subsequent processes were observed,92 indicating the complexity and difficulty in optimizing this multistep BCR.66
Figure 3.

BCR-enabled prodrug activation. (a) Representative prodrugs are summarized in which Dox and MMAE are the two drugs that have attracted dominant interest for prodrug design. (b) The triggers that have been used to trigger prodrug activation are listed and include transition metals and small molecules.
The IDA reaction between strained olefins and Tz is one of the most attractive bioorthogonal reactions due to its high bioorthogonality and fast reaction rate. In 2013, Robillard et al. reported their elegant work on the use of the IDA reaction for prodrug activation93 (Figure 3a, entry 3). The release of Dox from TCO–Dox by 3,6-dimethyltetrazine was tested in PBS/MeCN, with 79% yield obtained in 20 min. In the cytotoxicity analysis, Dox and TCO–Dox treated with 10 μM 3,6-dimethyltetrazine gave comparable EC50 values (0.037 μM vs 0.049 μM). This same research group further developed releasable antibody–drug conjugates (ADCs) by transforming the TCO group from a terminal to an internal cleavable linker.63,64,94,95 In 2016, Royzen and Yigit et al. prepared multifunctionalized magnetic nanoparticles with both Tz and the Cy5.5 fluorophore for image-guided TCO–Dox activation in situ.96 Additionally, Oneto and Royzen et al. functionalized an alginate hydrogel with Tz, which was used as a preimplanted biomaterial for TCO–Dox activation at the tumor site.32 This approach was evaluated to treat soft tissue sarcoma in mouse xenograft models. A better suppression of tumor growth and fewer side effects, including bone marrow suppression and a decrease in body weight, were observed in the prodrug group as compared to the control group.32 While this approach showed promising results, the triggers were consumed during prodrug activation, requiring repeated injection of the hydrogel. Our group and collaborators developed a phosphatase-instructed self-assembly consisting of a Tz-containing peptide enriched inside cancer cells, which allowed the selective activation of TCO-caged Dox at the tumor site by this pretargeted Tz trigger.97 Bernardes et al. also reported a Tz pretargeting strategy via single-walled carbon nanotubes (SWCNTs) for selective doxorubicin prodrug activation and instantaneous fluorescence imaging in living cells.98
Interestingly, a recent report demonstrated that TCO itself can serve as a trigger to deprotect the Tz-caged molecule, which can be used for prodrug activation77 (Figure 3a, entry 4). Notably, the Tz-carbamate group reacts with TCO faster in the conjugation step (k2 = 287 ± 10 to 23 800 ± 400 M–1 s–1)77 as compared to the Tz-involved reaction with TCO-carbamate (k2 = 57.7 ± 5.0 M–1 s–1).93 Actually, the Tz-caged prodrug exhibited a slower reaction rate in the releasing step, which finally resulted in a similar total reaction rate. Nevertheless, this improvement enables a lesser usage of activator reagents to afford considerable release yields.77 This reaction was applied to release monomethyl auristatin E (MMAE) from Tz-protected MMAE efficiently (>80%) at nanomolar concentrations under complex reaction conditions.77
IDA reactions between simple olefins and Tz have also been used for prodrug activation. In 2016, Deveraj et al. reported the BCR between a vinyl ester and Tz for mRNA detection via a proximity-enabled decaging reaction.74 In 2017, this reaction was used for prodrug activation by Bernardes et al. The authors developed a vinyl-protected duocarmycin analogue as an inactive prodrug, which can be reactivated through Tz-mediated Winstein spirocyclization to liberate the hydroxyl group to regenerate the duocarmycin analogue73 (Figure 3a, entry 5). However, a major limit for these BCRs is the slow reaction kinetics. Rate enhancement could be partly achieved by attaching a boronic acid group onto the terminal vinyl via a B–N coordination effect between boronic acid and 3,6-pyridyltetrazine71,75 (Figure 3a, entry 6).
The product of Tz in this IDA reaction is pyridazine, a core miR21 inhibitor structure, which makes dual prodrug activation from one reaction possible. Bradley et al. designed vinyl-protected camptothecin (CPT, Figure 3a, entry 7) and 3-(3-nitrophenyl)-6-methylthiotetrazine as prodrugs of camptothecin and pyridazine, respectively (Figure 3a, entry 8). The IDA reaction between these two prodrugs simultaneously liberated CPT and pyridazine, offering an elegant approach for dual prodrug activation.99 Finally, additional groups such as benzonorbornadienes have also been used as Tz-triggered deprotecting groups for bioorthogonal prodrug development71 (Figure 3a, entry 9).
The Tz-triggered deprotection of the 3-isocyanopropyl group (ICP) has also been applied for prodrug activation, diversifying the BCRs for prodrug activation. In 2018, Frazini et al. reported the protection of different functional groups using ICP to generate their corresponding prodrugs, including the amine group of Dox (Figure 3a, entry 10), the phenol group of SN-38 (Figure 3a, entry 11), the sulfonyl group of 6-mercaptopurine (Figure 3a, entry 12), and the aziridine group of mitomycin C100 (Figure 3a, entry 13). A rescue rate greater than 75% was observed for these prodrugs upon the addition of 3,6-dipyridyl tetrazine.100 Recently, the same group reported the converse of this Tz/ICP pair: a tetrazylmethyl protecting group was designed that could be deprotected by ICP to liberate a free amine. Accordingly, tetrazylmethyl–Dox was synthesized as a prodrug that could be rescued by the ICP reagent (Figure 3a, entry 14). Although the reaction kinetics is moderate (Figure 2, entry 22, k2 = 0.05–0.38 M–1 s–1), the unique feature of this BCR is that Tz and ICP can deprotect each other, making it straightforward to design a dual prodrug activation system.79
In 2018, Wang et al. reported a different strategy for designing prodrugs in which reaction kinetics were employed for the control of drug release in a concentration-sensitive manner78 (Figure 3a, entry 15). Specifically, Dox was protected with Tz as a prodrug via an amide bond, which was anchored to a triphenylphosphonium (TPP) group for mitochondrial accumulation, and 3-hydroxycyclooctyne was used as the trigger. When the trigger was anchored to TPP, a significant improvement in the prodrug activation in living cells was observed compared to that without TPP, indicating the concentration effect on prodrug activation through organelle-specific accumulation. The effectiveness of this strategy was further demonstrated by the release of the gaseous drug CO using cyclopentadienone as the CO precursor and cyclooctyne as the trigger.101,102
While small-molecule-triggered BCRs have been extensively employed in bioorthogonal prodrug activation, transition-metal-mediated decaging chemistry has also been used, and the catalytic nature of such reactions holds unique advantages in reducing the loading dosage and frequency of the trigger.
In 2014, Unciti-Broceta and Bradley et al. reported the activation of 5-fluorouracil prodrugs (Figure 3a, entry 16) and gemcitabine prodrugs (Figure 3a, entry 17) by using a nano-palladium-loaded resin. In this work, the authors proposed that local implantation of resin containing the Pd catalyst was surgically feasible and could achieve localized prodrug activation. For these homogeneous metal catalysts, nanoencapsulation and passive targeting strategies could be used to achieve tumor selectivity.50,51 They also expanded the Pd-triggered depropargylation for the controllable release of vorinostat, a histone deacetylase inhibitor.51,54 In 2018, Bernardes et al. reported a thioether-directed Pd-cleavable linker to release small-molecule drugs39 (Figure 3a, entry 18).
In addition, transition-metal-mediated prodrug activation has been extended to other metals, including Ru, Au, Cu, Pt, and others. For instance, Ru-triggered deprotection of an allyl group has been intensively studied for prodrug activation in living systems by Meggers et al.38 (Figure 3a, entry 19). The Unciti-Broceta team verified that Au-triggered deprotection of the propargyl group could also be utilized for the design of prodrugs.43 They further used immobilized Au catalysts for the removal of propargyl groups in zebrafish.
Our group has been interested in Cu due to the wide availability of biocompatible ligands owing to the broad applications of the CuAAC reaction in living systems. A systematic survey of the ligands and metals identified the Cu-2-[4-[[bis[(1-tertbutyltriazol-4-yl)methyl]amino]methyl]triazol-1-yl] acetic acid (BTTAA)/disubstituted propargyl group as a new bioorthogonal cleavage pair, which facilitated the further extension of this chemistry from terminal to internal bond cleavage of the substituted propargyl scaffold41 (Figure 3a, entries 20 and 21). In addition, a recent report by Bernardes et al. utilized the Pt-triggered deprotection of pentynoyl or propargyl groups on amines for the release of MMAE and 5-FU44 (Figure 3a, entry 22). Remarkably, certain Pt compounds, such as cisplatin, are anticancer agents that could synergize with the prodrug activation strategy as a unique dual drug system in cancer therapy.44 Combined with targeting methods, bioorthogonal prodrug activation via BCRs will unleash power in clinical applications.
BCRs for Protein Activation in Living Systems
Beyond small molecules, BCRs have also been extensively employed to manipulate biomolecules. In particular, here, we would like to focus on the precise on-demand activation of proteins, which represents one of the most exciting playgrounds for BCRs. The activity of a POI can be premasked by replacing the key residue with chemically caged or photocaged unnatural amino acids (UAAs) via the genetic code expansion technique, which could later be rescued by a BCR in living systems. As an alternative approach to extensive protein engineering methods, this genetic-based strategy (switchable proteins103,104 and split proteins105) has shown superior spatial and/or temporal specificity on POIs in situ(31) (Figure 4).
Figure 4.

Activation of BCR-enabled proteins. (a) Structures of caged amino acids and their corresponding applied proteins. (b) Protein functions that can be activated by chemical decaging as well as the corresponding caged amino acids. Structures of OspF, Fluc, MEK1, TnaA, Cre, Cas9, Taq, LF, and affibody were regenerated from PDB files 2Q8Y, 4D36, 1S9I, 5W1B, 1CRX, 4CMP, 1BGX, 1PWV, and 2B89, respectively.
In this regard, the photoactivation of proteins was initially studied, but attention has been given to chemical activation strategies due to concerns about cytotoxicity, permeability, and chemical diversity. In 2014, we applied Pd-triggered depropargylation chemistry for POI activation in living cells. In this study, we applied our genetically encoded chemical decaging strategy to OspF (Figure 4, entry 1), a bacterial effector protein containing a catalytic Lys residue (K134) to irreversibly dephosphorylate the T–E–Y motif on phosphorylated extracellular signal regulated kinase (p-Erk), resulting in the downregulation of the mitogen-activated protein kinase (MAPK) signaling cascade. By using the genetic code expansion technique, a premasked OspF variant bearing the propargyl-caged Lys at K134 (OspF-K134ProK) was expressed in living cells, which could be turned on via the Pd-triggered depropargylation reaction that converts ProK back into free Lys. Upon Pd addition, significant dephosphorylation of p-Erk was observed in living cells.49 In the same year, we also expanded this strategy to employ IDA-mediated BCRs for rapid protein activation in living cells. Our systematic screening of the TCO stereoposition as well as the 3,6-substitution of Tz showed that dimethyl-Tz (DM-Tz) and ax-TCO have the fastest decaging rates. We then site-specifically incorporated TCO-caged Lys (ax-TCOK) into proteins, which can be rapidly decaged by DM-Tz. Since firefly luciferase (Fluc) contains a key Lys residue that catalyzes the luciferin oxidation reaction, it was chosen as the model enzyme to verify this reaction in living cells and animals (Figure 4, entry 2). Indeed, the premasked Fluc variant Fluc-K529-TCOK can be converted back into wild-type Fluc in 10 min in living cells.65 Our continuous efforts to optimize the IDA reaction by systematically investigating the structure–activity relationships of Tz have yielded a rationally designed unsymmetric Tz with a significantly improved reaction rate, which has led to the complete activation of Fluc within 4 min.66 In addition, Weissleder et al. found that this reaction is sensitive to pH, and rationally designed acid-functionalized Tz both enhances and accelerates the release of the caged small molecules.67 Based on this discovery, we envision that carboxylic-modified Tz could be used to improve the temporal resolution of protein activation. Since many POIs contain key Lys residue(s), we have demonstrated the generality of our genetically encoded Lys decaging strategy for the activation of a range of proteins involved in signal transduction (kinases, e.g., MEK, FAK, Src),33 amino acid metabolism in bacteria (tryptophanase),106 and proximal labeling enzymes (biotin ligase, unpublished results).
In 2016, Dieters et al. reported Staudinger reduction-mediated protein activation based on ortho-azido benzyl carbamate-caged Lys (OABK) and a trisubstituted phosphine trigger.87 This decaging reaction was further used for the functional manipulation of a fluorescent protein, nuclear localization sequence, recombinase (Cre), and gene editing enzyme (CRISPR/Cas9). We also showed that TCO itself can serve as a bioorthogonal cleavage trigger that is able to rescue para-azido benzyl carbamate-caged Lys (PABK) for protein activation (Figure 4, entry 3). In addition, by taking advantage of the unique chemistry of the aryl azide group, we further demonstrated the diverse utility of PABK, including its use in CuAAC-mediated protein labeling as well as phototriggered protein cross-linking107 (Figure 4, entry 4). Recently, Peng et al. designed another chemically caged Lys analogue, PrAK, which could be decaged by formaldehyde (FA) via a 2-aza-Cope rearrangement. Replacing the key Lys residue on EGFP or Fluc with PrAK was used as a protein-based sensor for FA detection in vitro and in live cells88 (Figure 4, entry 5).
In addition to Lys, this decaging strategy was also expanded to include other amino acid side chains. For example, we developed an allenyl-caged Tyr that can be decaged by Pd to regenerate free Tyr in situ (Figure 4, entry 6). This genetically encoded Tyr decaging strategy has been applied to regulate posttranslational modifications on kinases (Src-Y416), activate DNA polymerase (Taq-Y671), and activate bacterial toxins (e.g., lethal factor (LF) from Bacillus anthracis).55 We also found that Cu(I)-BTTAA was able to catalyze the cleavage of a disubstituted propargyl group caged Tyr (EMPY) to rescue the function of a POI. The binding of affibody–ZHER can be blocked and rescued before and after Cu(I)-triggered decaging (Figure 4, entry 7). In addition, this disubstituted propargyl group can be used as a Lys caging group to control ZHER–Her2 binding (Figure 4, entry 8).
Site-specific caging and decaging of a catalytic residue on a protein are a one-stop shop for a category of POIs that share the same catalytic residue. It can be achieved via a genetic incorporation of a caged-UAA for blockage, subsequently being precisely decaged for selective on-demand protein activation (Figure 5a). However, currently the bioorthogonal rescue strategy is only achievable on a few kinds of amino acids. Expanding the BCRs from Lys (chemical- and photo-decaging), Tyr (chemical- and photo-decaging), Ser (photo-decaging), and Cys (photo-decaging) to other residues, such as His, Trp, Glu, and Arg, among others, will increase the protein coverage and fulfill the toolbox.
Figure 5.

Proximal decaging (CAGE-prox) as a universal protein activation strategy. (a) Schematic illustration of the proximal decaging strategy in comparison with active site decaging. The active site decaging strategy is highly dependent on the active site residue, while the unified CAGE-prox strategy relies on a single BCR that can be utilized in the proximal decaging and activation of various POIs. (b) Protein families that can be activated by CAGE-prox. The proximal sites for incorporation and decaging of the unified UAA are recommended after computational design. The structures of MEK1, KRAS, FTO, caspase-3, Fluc, and LF were regenerated from the PDB files 1S9I, 4IE5, 4EPY, 4DCO, 4D36, and 1PWV, respectively.
In addition, a unified BCR that can be applied for a broad range of proteins, even proteins with unknown catalytic residues, is a more exciting approach to explore. To expand the generality of this approach, we and our collaborators developed a proximal decaging strategy that may activate any POI in living cells.108 This strategy, termed CAGE-prox, relies on the basic concept that a universal “proximal cage” (caged UAA) can be introduced by the genetic code expansion technique in close proximity with the functional site of a POI for temporal blockage of its activity until it is rescued by a cleavage trigger (Figure 5a). To identify such a proximal “anchor site” in a POI and avoid exhaustive experimental screening, we first developed a computer-aided program to facilitate the identification of such positions that can be further validated experimentally. Since the computational method does not require information about the mechanism of action of a POI, the CAGE-prox strategy can therefore be applied to a wide range of proteins (Figure 5b). Notably, the proximal-decaged POI differs from its native counterpart by at most a single amino acid residue, allowing preservation of the structure, folding, and protein–protein interactions of the wild-type protein.108
Embarking on this CAGE-prox strategy, we created a time-resolved proteomics technique that can capture and identify substrates during caspase hydrolysis, providing valuable resources for understanding dynamic biological processes such as cell apoptosis.108 Additionally, spatiotemporally controlled activation of B. anthracis LF as a protein-based prodrug was also achieved in living mice.108
BCR-Enabled Intramolecular Cleavage of Bifunctional Molecules
While BCRs used for prodrug activation are mainly terminal decaging reactions, there is a growing demand for internal bond cleavage, for which bioorthogonal-cleavable linkers can be utilized in various applications, such as cleavable ADCs. There have been a growing number of ADCs with cleavable linkers that depend on overexpressed enzymes or the tumor microenvironment (e.g., acidic or hypoxic conditions) to release the toxic payload from the conjugated antibody at the tumor site.109 Alternatively, BCR-enabled cleavable ADCs may release anticancer agents by a bioorthogonal trigger administered at the tumor site on demand.
Although many internal cleavable linkers have been remodeled from the terminal decaging chemistry by introducing an extra arm for attaching targeting molecules (e.g., antibody, affibody, or organelle targeting groups; Figure 4), only those protecting groups with an extra attachment site can be considered. In particular, introduction of a linking arm should not affect the efficiency of the cleavage reaction. For example, the Tz/TCO bioorthogonal cleavage pair has attracted great attention because, in addition to its excellent bioorthogonality, stability, and fast kinetics, the TCO group can be further modified as an internal linker rather than a terminal cage. By introducing an extra carboxylic acid group into the TCO ring to attach additional molecules, Robillard et al. transformed TCO to an internal cleavable linker for ADC conjugation. The resulting TAG72-targeting ADC was able to release Dox or MMAE on the cancer cell surface upon the addition of the Tz trigger94,95 (Figure 6, entries 1–2). Interestingly, the Robillard group recently remodeled Tz as a TCO-responsive cleavable linker,77 affording better reaction kinetics and diversified internal cleavable linkers. Additionally, this approach makes the TCO/Tz pair a more convenient internal cleavable linker choice since the synthesis of the Tz-based linker is less complicated and time-consuming than the TCO-based linker (Figure 6, entry 3). In addition, Wang et al. reported the breakage of a Tz-bearing linker by 3-hydroxyl cyclooctyne with moderate cleavage kinetics. The Tz-linked Dox and tetraphenylphosphine (mitochondria-targeting molecule) are released in situ in mitochondria78 (Figure 6, entry 4), which provides the enrichment-triggered subcellular release of functional molecules with fast (i.e., seconds to minutes) and complete (i.e., >99%) cleavage. More recently, Mikula et al. designed a C2-symmetric trans-cyclooctene as a cleavable linker to link functional molecules at two allylic positions. This design avoids the unexpected cycloaddition product (head-to-tail addition) and increased the yield and rate of cleavage. By further optimization of the Tz trigger structure, the authors achieved the complete release (i.e., >99%) of functional small molecules within minutes.110
Figure 6.
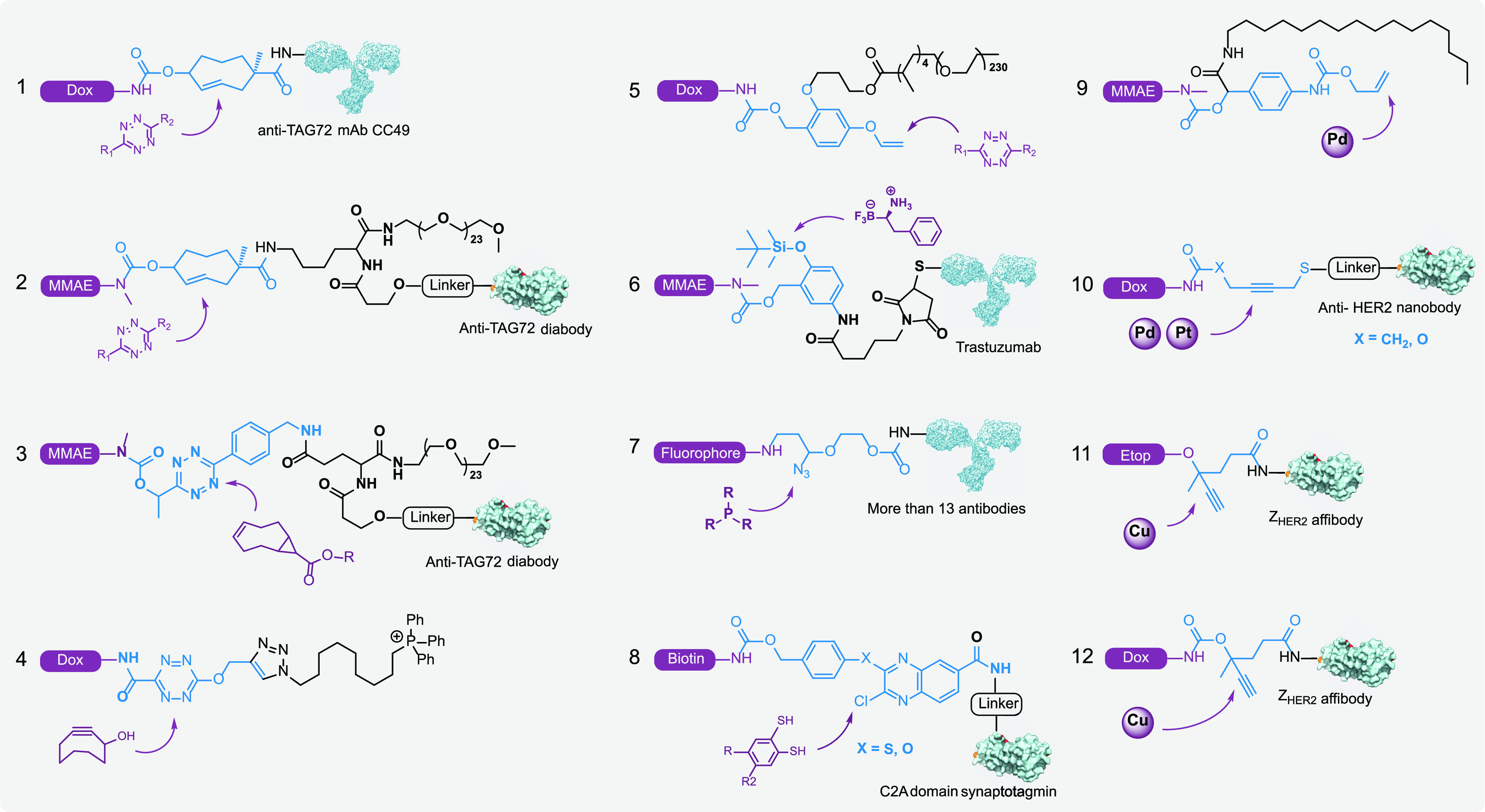
BCR-enabled intramolecular bioconjugate cleavage. Representative bifunctional molecules that can be cleaved by BCRs, including ADCs (entries 1, 6, and 7), nanobody/affibody-drug conjugates (entries 2–3, 8, and 10–12), and subcellular targeted drugs (entries 2, 4, and 9). In the designed bioconjugates, the modified/substituted caging group could be utilized as a cleavable linker for the on-demand release of prodrugs.
The vinyl group has also been employed in a Tz-sensitive, self-immolative linker on molecules containing the 4-hydroxybenzylic alcohol moiety111 (Figure 6, entry 5). The vinyl group reacts with Tz to release a phenol group, which initiates 1,6-elimination for cleavage of the linker. A similar strategy was recently used by Liu and Shao et al. to develop a fluoro reagent-sensitive linker to release the gasdermin family protein GSDMA3 from nanoparticles. As the gasdermin family of pore-forming proteins has been shown to directly trigger pyroptosis by Shao et al.,112,113 this study was the first to demonstrate that delivery of GSDMA3 into tumor cells will cause pyroptotic cell death.114 Additionally, by controlling the release of the protein gasdermin, the resulting pyroptosis and inflammation boosted the antitumor immune response and increased the efficacy of the checkpoint blockade (PD-1 antibody).114 The bioorthogonal cleavage chemistry used in this work was based on the deprotection of silyl groups from phenols with a fluoro reagent114 (Figure 6, entry 6). In addition, other BCRs, such as Staudinger reduction115 and nucleophilic substitution reactions,116 may also hold the potential for developing internal cleavable linkers for the on-demand release of bioconjugates (Figure 6, entries 7 and 8).
Since transition metals have mainly been applied on smaller protecting groups (e.g., allylic or propargyl moieties), there are limited sites that are able to be remodeled as internal cleavable linkers. By employing the 4-amino benzylic alcohol-derived self-immolative linker, Weissleder et al. developed the Pd-triggered release of MMAE based on the deprotection of the allylic group from aniline117 (Figure 6, entry 9). Bernardes et al. attached a methylene thioether group to the terminal carbon of propargyl, making it an internally cleavable linker to release Dox from the anti-HER2 nanobody by Pd or Pt triggers39,44 (Figure 6, entry 10).
By surveying a range of commonly used transition metal catalysts, we found that Cu(I)-BTTAA was able to catalyze the cleavage of disubstituted propargyl groups to release free amine or hydroxyl groups. Such a disubstituted propargyl moiety can be further extended by the conjugation of one arm with the ZHER affibody, which converts the propargyl moiety from a terminal decaging group to a traceless internal linker for releasing bioconjugates41 (Figure 6, entries 11 and 12). Both Dox and etoposide (Etop) could be efficiently released from the cleavable ADCs by Cu-BTTAA41 (Figure 6, entries 11 and 12). Additionally, reversible cell surface remodeling was achieved by this traceless linker and the corresponding Cu(I)-BTTAA-mediated internal cleavage.41 Given that the second substitution at the α position of the propargyl moiety (e.g., the methyl group) can also be transformed into a conjugation arm, more diversifications and applications of this internal cleavable linker can be further explored.
BCRs in Animals
Expanding BCRs into animals is another active frontier that has been increasingly pursued. Reactions in animals can be used to establish preclinical models to dissect clinical problems and obtain evidence to support the potential clinical applications of BCRs, such as prodrug activation.13 In fact, BCRs have already been conducted in several animal models. Robillard et al. reported the activation of TCO-caged Dox in mice by conjugating this prodrug with a tumor-targeting antibody to achieve the selective killing of cancer cells.94 In 2017, Weissleder et al.34 and Wu et al.118 reported liposome- and hydrogel-encapsulated Pd catalysts for tumor targeting and the activation of allyloxycarbonyl-caged prodrugs, respectively. Oneto et al. designed a Tz-modified hydrogel that can be implanted into tumor loci in mice enabling local activation of the TCO-caged prodrug inside the tumor.32,119 In 2016, our group reported for the first time the IDA-based BCR for protein activation in mice, which offered a facial tool for time-resolved protein study and protein-based prodrug development in animals.33 By using the circulatory system, small-molecule triggers can be injected into the caudal vein and activate a POI in target cells or tissues, enabling on-demand and remote-controlled activation. We and our collaborators developed a pretargeting strategy for Tz triggers in mice, which enabled the selective activation of TCO-caged Dox at the tumor site.97 We also developed a prodrug–antibody conjugate (Pro–ADC) for on-demand tumor chemotherapy.120 The conjugated prodrug can be released first via the acidic tumor microenvironment and then further activated within the tumor cell.120
In addition to small-molecule prodrugs, BCRs also allow the controlled activation of protein-based toxins. For example, we used both active site-decaging and CAGE-prox methods to activate LF from B. anthracis, allowing remote activation of this bacterial toxin inside tumor cells, as demonstrated in a xenograft animal model. In addition, we and our collaborators used a cationic lipid to deliver the bacterial type III secretion toxin OspF into mice. This, in conjunction with the BCR-enabled activation of LF, holds the potential for exploring the rich repertoire of bacterial effectors as bioorthogonally activatable protein prodrugs with improved efficacy and safety in vivo or in clinical settings.121 Recently, Liu and Shao et al. codeveloped an in vivo BCR between Phe-BF3 and a silyl ether, which was applied for the controllable release of toxin from an ADC and release of the GSDMA3 protein from a nanoparticle conjugate114 in mice. As demonstrated by this work, BCR-triggered pyroptosis and a further immune response hold immunotherapeutic potential for enhancing the efficacy of the immune checkpoint blockade.
Outlook
The development of bioorthogonal chemistry has enabled the in situ manipulation of functional molecules within living cells and animals. Bioorthogonal ligation reactions are continuously thriving with exciting applications. For example, multiple reactions have been designed with mutual orthogonality and can be conducted simultaneously.27,122,123 In addition, reactions that are specific to native protein side chains, which can be coupled with a small-molecule ligand or proteins as a warhead for residue-specific or site-specific labeling, have also drawn great attention.18,124−126 Moreover, the rapid emergence of BCRs has greatly leveraged the strength of bond cleavage chemistry in living systems. It is conceivable that new BCRs with improved reaction kinetics and biocompatibility will be continuously developed by expanding the boundary and utility of this powerful chemical biology toolkit. Finally, we hope to outline the future perspectives along this exciting avenue.
Expanding Reaction Types and Applications
Despite the fast growth of BCRs, unmet biological needs remain. As the functional groups of biomolecules are varied, expanding the BCRs for releasing diverse chemical groups is one of the most urgent directions to explore. In particular, rather than being derived from previous deprotection reactions in organic chemistry, completely new BCRs that are developed from scratch are highly desirable. There is usually a trade-off between bioorthogonality and reactivity for BCR development. We envision that new approaches could be used to create new BCRs and promote the chemical reactivities, including (1) physical approaches, such as ultrasound or magnetic fields, etc.; (2) bioorthogonal enzymatic BCRs; and (3) BCRs triggered by the sub-microenvironment of living systems. In addition to the increased types of caging groups and uncaging triggers, functional manipulation of biomolecules should also be expanded to move from being protein-centered to including other biomolecules such as nucleic acids, glycans, and lipids. For example, BCRs represent an efficient approach in cell engineering, such as in remodeling cell surface glycans,127 unmasking the epitope of major histocompatibility complex class I (MHC-I),128,129 and removing chemically labeled molecular probes.41 More innovative applications in this direction will likely emerge in the future.
Remote Control beyond Small-Molecule Triggers
Cleavage triggers that are currently used in BCRs are not optimal for in vivo or clinical settings. For example, although photoinduced decaging by UV or visible light offers a noninvasive approach for precise activation with spatial resolution, poor tissue penetration limits their in vivo applications. Small-molecule-mediated chemical decaging, as well as two-photon130 or upconversion nanoparticle-triggered decaging,131 may partly address this issue, but additional noninvasive methods with an excellent remote-control ability are highly desired. Recently, Liu et al. reported the radiation-triggered unmasking of functional molecules, which provided a proof-of-concept study of remote activation in vivo.132 Beyond this proof-of-concept study, new methods for remote decaging/release that are suitable not only for living animals but also for humans may appear in the near future.
In Vivo Chemistry Holds Potential for Clinical Applications
Bioorthogonal prodrugs enabled by BCRs have emerged as a valuable approach for the on-target and on-demand activation of small-molecule- or protein-based toxins that otherwise would have very narrow therapeutic windows due to toxicity to normal tissues. However, so far, only a few BCRs can be carried out in vivo, and intensive efforts have been made to optimize BCRs for mammals. The main reason is that the reaction between the caging group and decaging trigger must proceed with very fast kinetics due to their low concentrations in vivo. Furthermore, optimization of the reaction pair to meet the desired pharmacokinetic properties and safety standards is highly challenging. Fortunately, some reported BCRs have demonstrated satisfying results in animals and hold promise for potential use in humans, although systematic evaluation of their safety and efficacy is required. Indeed, the activation of TCO-caged Dox by Tz in humans has recently entered a phase 1 clinical trial, raising the high expectation of its clinical utility.35 We have reasons to believe that more in vivo BCRs may be launched for clinical studies in the near future.
Acknowledgments
This work was supported by the National Key Research and Development Program of China (2016YFA0501500 and 2019YFA0904201), the National Natural Science Foundation of China (21521003, 21740001, and 21937001 for P.R.C.; 91957101 and 22077004 for X.F.; and 22077059 for J.W.), and Beijing National Laboratory for Molecular Sciences (BNLMS202012).
Author Contributions
∥ J.W. and X.W. contributed equally.
The authors declare no competing financial interest.
Originally published ASAP on April 30, 2021; References 108−110 replaced May 7, 2021.
References
- Prescher J. A.; Bertozzi C. R. Chemistry in Living Systems. Nat. Chem. Biol. 2005, 1 (1), 13–21. 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- Saxon E.; Armstrong J. I.; Bertozzi C. R. A ″traceless″ Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2000, 2 (14), 2141–2143. 10.1021/ol006054v. [DOI] [PubMed] [Google Scholar]
- Lemieux G. A.; De Graffenried C. L.; Bertozzi C. R. A fluorogenic dye activated by the Staudinger ligation. J. Am. Chem. Soc. 2003, 125 (16), 4708–4709. 10.1021/ja029013y. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40 (11), 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 2004, 126 (46), 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Borrmann A.; Van Hest J. C. M. Bioorthogonal chemistry in living organisms. Chem. Sci. 2014, 5 (6), 2123–2134. 10.1039/c3sc52768a. [DOI] [Google Scholar]
- Haun J. B.; Devaraj N. K.; Hilderbrand S. A.; Lee H.; Weissleder R. Bioorthogonal chemistry amplifies nanoparticle binding and enhances the sensitivity of cell detection. Nat. Nanotechnol. 2010, 5 (9), 660–665. 10.1038/nnano.2010.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. From mechanism to mouse: A tale of two bioorthogonal reactions. Acc. Chem. Res. 2011, 44 (9), 666–676. 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj N. K. The Future of Bioorthogonal Chemistry. ACS Cent. Sci. 2018, 4 (8), 952–959. 10.1021/acscentsci.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azagarsamy M. A.; Anseth K. S. Bioorthogonal Click Chemistry: An Indispensable Tool to Create Multifaceted Cell Culture Scaffolds. ACS Macro Lett. 2013, 2 (1), 5–9. 10.1021/mz300585q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Z.; Hong S.; Chen X.; Chen P. R. Introducing Bioorthogonal Functionalities into Proteins in Living Cells. Acc. Chem. Res. 2011, 44 (9), 742–751. 10.1021/ar200067r. [DOI] [PubMed] [Google Scholar]
- He C. Chemical decaging in living systems. Natl. Sci. Rev. 2015, 2 (3), 250–251. 10.1093/nsr/nwv030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Li J.; Chen P. R. Bioorthogonal chemistry in living animals. Natl. Sci. Rev. 2017, 4 (3), 300–302. 10.1093/nsr/nwx010. [DOI] [Google Scholar]
- Laughlin S. T.; Baskin J. M.; Amacher S. L.; Bertozzi C. R. In vivo imaging of membrane-associated glycans in developing zebrafish. Science 2008, 320 (5876), 664–667. 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertozzi C. R. A Decade of Bioorthogonal Chemistry. Acc. Chem. Res. 2011, 44 (9), 651–653. 10.1021/ar200193f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem., Int. Ed. 2009, 48 (38), 6974–6998. 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilyard M. K.; Bailey H. J.; Raich L.; Gafitescu M. A.; Machida T.; Iglésias-Fernández J.; Lee S. S.; Spicer C. D.; Rovira C.; Yue W. W.; Davis B. G. Palladium-mediated enzyme activation suggests multiphase initiation of glycogenesis. Nature 2018, 563 (7730), 235–240. 10.1038/s41586-018-0644-7. [DOI] [PubMed] [Google Scholar]
- Wright T. H.; Bower B. J.; Chalker J. M.; Bernardes G. J. L.; Wiewiora R.; Ng W.-L.; Raj R.; Faulkner S.; Vallée M. R. J.; Phanumartwiwath A.; Coleman O. D.; Thézénas M.-L.; Khan M.; Galan S. R. G.; Lercher L.; Schombs M. W.; Gerstberger S.; Palm-Espling M. E.; Baldwin A. J.; Kessler B. M.; Claridge T. D. W.; Mohammed S.; Davis B. G. Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity. Science 2016, 354 (6312), aag1465. 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Prescher J. A. Finding the right (bioorthogonal) chemistry. ACS Chem. Biol. 2014, 9 (3), 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Ramil C. P.; Lin Q. Bioorthogonal chemistry: Strategies and recent developments. Chem. Commun. 2013, 49 (94), 11007–11022. 10.1039/c3cc44272a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino V.; Rebelein J. G.; Ward T. R. ″Close-to-Release″: Spontaneous Bioorthogonal Uncaging Resulting from Ring-Closing Metathesis. J. Am. Chem. Soc. 2019, 141 (43), 17048–17052. 10.1021/jacs.9b07193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J.; Xu M.; Franzini R. M. Dissociative Bioorthogonal Reactions. ChemBioChem 2019, 20 (13), 1615–1627. 10.1002/cbic.201800810. [DOI] [PubMed] [Google Scholar]
- Yang M.; Li J.; Chen P. R. Transition metal-mediated bioorthogonal protein chemistry in living cells. Chem. Soc. Rev. 2014, 43 (18), 6511–6526. 10.1039/C4CS00117F. [DOI] [PubMed] [Google Scholar]
- Ji X.; Pan Z.; Yu B.; De La Cruz L. K.; Zheng Y.; Ke B.; Wang B. Click and release: bioorthogonal approaches to ″on-demand″ activation of prodrugs. Chem. Soc. Rev. 2019, 48 (4), 1077–1094. 10.1039/C8CS00395E. [DOI] [PubMed] [Google Scholar]
- Oliveira B. L.; Guo Z.; Bernardes G. J. L. Inverse electron demand Diels-Alder reactions in chemical biology. Chem. Soc. Rev. 2017, 46 (16), 4895–4950. 10.1039/C7CS00184C. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem. Rev. 2014, 114 (9), 4764–4806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Nguyen S. S.; Prescher J. A. Developing bioorthogonal probes to span a spectrum of reactivities. Nat. Rev. Chem. 2020, 4 (9), 476–489. 10.1038/s41570-020-0205-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q.; Dong T.; Liu X.; Lei X. A Bioorthogonal Ligation Enabled by Click Cycloaddition of o-Quinolinone Quinone Methide and Vinyl Thioether. J. Am. Chem. Soc. 2013, 135 (13), 4996–4999. 10.1021/ja401989p. [DOI] [PubMed] [Google Scholar]
- Lim R. K. V.; Lin Q. Bioorthogonal chemistry: recent progress and future directions. Chem. Commun. 2010, 46 (10), 1589–1600. 10.1039/b925931g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Robillard M. S. Pretargeted imaging using bioorthogonal chemistry in mice. Curr. Opin. Chem. Biol. 2014, 21, 161–9. 10.1016/j.cbpa.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Li J.; Chen P. R. Development and application of bond cleavage reactions in bioorthogonal chemistry. Nat. Chem. Biol. 2016, 12 (3), 129–137. 10.1038/nchembio.2024. [DOI] [PubMed] [Google Scholar]
- Mejia Oneto J. M.; Khan I.; Seebald L.; Royzen M. In Vivo Bioorthogonal Chemistry Enables Local Hydrogel and Systemic Pro-Drug To Treat Soft Tissue Sarcoma. ACS Cent. Sci. 2016, 2 (7), 476–482. 10.1021/acscentsci.6b00150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G.; Li J.; Xie R.; Fan X.; Liu Y.; Zheng S.; Ge Y.; Chen P. R. Bioorthogonal chemical activation of kinases in living systems. ACS Cent. Sci. 2016, 2 (5), 325–331. 10.1021/acscentsci.6b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. A.; Askevold B.; Mikula H.; Kohler R. H.; Pirovich D.; Weissleder R. Nano-palladium is a cellular catalyst for in vivo chemistry. Nat. Commun. 2017, 8, 15906. 10.1038/ncomms15906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee N. A.; Srinivasan S.; Royzen M.; Oneto J. M. M. Abstract LB-002: SQ3370 enhances the safety of chemotherapeutics via local activation therapy. Cancer Res. 2019, 79, LB-002. 10.1158/1538-7445.SABCS18-LB-002. [DOI] [Google Scholar]
- Klán P.; Šolomek T.; Bochet C. G.; Blanc A.; Givens R.; Rubina M.; Popik V.; Kostikov A.; Wirz J. Photoremovable protecting groups in chemistry and biology: Reaction mechanisms and efficacy. Chem. Rev. 2013, 113 (1), 119–191. 10.1021/cr300177k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streu C.; Meggers E. Ruthenium-induced allylcarbamate cleavage in living cells. Angew. Chem., Int. Ed. 2006, 45 (34), 5645–5648. 10.1002/anie.200601752. [DOI] [PubMed] [Google Scholar]
- Völker T.; Dempwolff F.; Graumann P. L.; Meggers E. Progress towards Bioorthogonal Catalysis with Organometallic Compounds. Angew. Chem., Int. Ed. 2014, 53 (39), 10536–10540. 10.1002/anie.201404547. [DOI] [PubMed] [Google Scholar]
- Stenton B. J.; Oliveira B. L.; Matos M. J.; Sinatra L.; Bernardes G. J. L. A thioether-directed palladium-cleavable linker for targeted bioorthogonal drug decaging. Chem. Sci. 2018, 9 (17), 4185–4189. 10.1039/C8SC00256H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. A.; Chalker J. M.; Floyd N.; Bernardes G. J. L.; Davis B. G. Allyl sulfides are privileged substrates in aqueous cross-metathesis: Application to site-selective protein modification. J. Am. Chem. Soc. 2008, 130 (30), 9642–9643. 10.1021/ja8026168. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu Y.; Fan X.; Wang J.; Ngai W. S. C.; Zhang H.; Li J.; Zhang G.; Lin J.; Chen P. R. Copper-Triggered Bioorthogonal Cleavage Reactions for Reversible Protein and Cell Surface Modifications. J. Am. Chem. Soc. 2019, 141 (43), 17133–17141. 10.1021/jacs.9b05833. [DOI] [PubMed] [Google Scholar]
- Kislukhin A. A.; Hong V. P.; Breitenkamp K. E.; Finn M. G. Relative performance of alkynes in copper-catalyzed azide-alkyne cycloaddition. Bioconjugate Chem. 2013, 24 (4), 684–689. 10.1021/bc300672b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-López A. M.; Rubio-Ruiz B.; Sebastián V.; Hamilton L.; Adam C.; Bray T. L.; Irusta S.; Brennan P. M.; Lloyd-Jones G. C.; Sieger D.; Santamaría J.; Unciti-Broceta A. Gold-Triggered Uncaging Chemistry in Living Systems. Angew. Chem., Int. Ed. 2017, 56 (41), 12548–12552. 10.1002/anie.201705609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira B. L.; Stenton B. J.; Unnikrishnan V. B.; de Almeida C. R.; Conde J.; Negrão M.; Schneider F. S. S.; Cordeiro C.; Ferreira M. G.; Caramori G. F.; Domingos J. B.; Fior R.; Bernardes G. J. L. Platinum-Triggered Bond-Cleavage of Pentynoyl Amide and N-Propargyl Handles for Drug-Activation. J. Am. Chem. Soc. 2020, 142 (24), 10869–10880. 10.1021/jacs.0c01622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Völker T.; Meggers E. Transition-metal-mediated uncaging in living human cells-an emerging alternative to photolabile protecting groups. Curr. Opin. Chem. Biol. 2015, 25, 48–54. 10.1016/j.cbpa.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Völker T.; Meggers E. Chemical Activation in Blood Serum and Human Cell Culture: Improved Ruthenium Complex for Catalytic Uncaging of Alloc-Protected Amines. ChemBioChem 2017, 18 (12), 1083–1086. 10.1002/cbic.201700168. [DOI] [PubMed] [Google Scholar]
- Song F.; Garner A. L.; Koide K. A highly sensitive fluorescent sensor for palladium based on the allylic oxidative insertion mechanism. J. Am. Chem. Soc. 2007, 129 (41), 12354–12355. 10.1021/ja073910q. [DOI] [PubMed] [Google Scholar]
- Santra M.; Ko S. K.; Shin I.; Ahn K. H. Fluorescent detection of palladium species with an O-propargylated fluorescein. Chem. Commun. 2010, 46 (22), 3964–3966. 10.1039/c001922d. [DOI] [PubMed] [Google Scholar]
- Li J.; Yu J.; Zhao J.; Wang J.; Zheng S.; Lin S.; Chen L.; Yang M.; Jia S.; Zhang X.; Chen P. R. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6 (4), 352–361. 10.1038/nchem.1887. [DOI] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Macleod K. G.; Rybski W.; Fraser C.; Torres-Sánchez C.; Patton E. E.; Bradley M.; Carragher N. O.; Unciti-Broceta A. Extracellular palladium-catalysed dealkylation of 5-fluoro-1-propargyl-uracil as a bioorthogonally activated prodrug approach. Nat. Commun. 2014, 5, 3277. 10.1038/ncomms4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Dawson J. C.; Fraser C.; Rybski W.; Torres-Sánchez C.; Bradley M.; Patton E. E.; Carragher N. O.; Unciti-Broceta A. Development and bioorthogonal activation of palladium-labile prodrugs of gemcitabine. J. Med. Chem. 2014, 57 (12), 5395–5404. 10.1021/jm500531z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. T.; Carragher N. O.; Unciti-Broceta A. Palladium-mediated dealkylation of N-propargyl-floxuridine as a bioorthogonal oxygen-independent prodrug strategy. Sci. Rep. 2015, 5, 9329. 10.1038/srep09329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman Ohana R.; Levin S.; Wood M. G.; Zimmerman K.; Dart M. L.; Schwinn M. K.; Kirkland T. A.; Hurst R.; Uyeda H. T.; Encell L. P.; Wood K. V. Improved Deconvolution of Protein Targets for Bioactive Compounds Using a Palladium Cleavable Chloroalkane Capture Tag. ACS Chem. Biol. 2016, 11 (9), 2608–2617. 10.1021/acschembio.6b00408. [DOI] [PubMed] [Google Scholar]
- Rubio-Ruiz B.; Weiss J. T.; Unciti-Broceta A. Efficient Palladium-Triggered Release of Vorinostat from a Bioorthogonal Precursor. J. Med. Chem. 2016, 59 (21), 9974–9980. 10.1021/acs.jmedchem.6b01426. [DOI] [PubMed] [Google Scholar]
- Wang J.; Zheng S.; Liu Y.; Zhang Z.; Lin Z.; Li J.; Zhang G.; Wang X.; Li J.; Chen P. R. Palladium-Triggered Chemical Rescue of Intracellular Proteins via Genetically Encoded Allene-Caged Tyrosine. J. Am. Chem. Soc. 2016, 138 (46), 15118–15121. 10.1021/jacs.6b08933. [DOI] [PubMed] [Google Scholar]
- Adam C.; Pérez-López A. M.; Hamilton L.; Rubio-Ruiz B.; Bray T. L.; Sieger D.; Brennan P. M.; Unciti-Broceta A. Bioorthogonal Uncaging of the Active Metabolite of Irinotecan by Palladium-Functionalized Microdevices. Chem. - Eur. J. 2018, 24 (63), 16783–16790. 10.1002/chem.201803725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heffern M. C.; Park H. M.; Au-Yeung H. Y.; Van de Bittner G. C.; Ackerman C. M.; Stahl A.; Chang C. J. In vivo bioluminescence imaging reveals copper deficiency in a murine model of nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (50), 14219–14224. 10.1073/pnas.1613628113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. Y.; Posimo J. M.; Lee S.; Tsang T.; Davis J. M.; Brady D. C.; Chang C. J. Activity-based ratiometric FRET probe reveals oncogene-driven changes in labile copper pools induced by altered glutathione metabolism. Proc. Natl. Acad. Sci. U. S. A. 2019, 116 (37), 18285–18294. 10.1073/pnas.1904610116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham D.; Deter C. J.; Reinard M. C.; Gibson G. A.; Kiselyov K.; Yu W.; Sandulache V. C.; St Croix C. M.; Koide K. Using Ligand-Accelerated Catalysis to Repurpose Fluorogenic Reactions for Platinum or Copper. ACS Cent. Sci. 2020, 6 (10), 1772–1788. 10.1021/acscentsci.0c00676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T.; Lv T.; Wu J.; Zhu M.; Fei Y.; Zhu J.; Zhang Y.; Huang Z. General Strategy for Integrated Bioorthogonal Prodrugs: Pt(II)-Triggered Depropargylation Enables Controllable Drug Activation In Vivo. J. Med. Chem. 2020, 63 (22), 13899–13912. 10.1021/acs.jmedchem.0c01435. [DOI] [PubMed] [Google Scholar]
- Latocheski E.; Dal Forno G. M.; Ferreira T. M.; Oliveira B. L.; Bernardes G. J. L.; Domingos J. B. Mechanistic insights into transition metal-mediated bioorthogonal uncaging reactions. Chem. Soc. Rev. 2020, 49 (21), 7710–7729. 10.1039/D0CS00630K. [DOI] [PubMed] [Google Scholar]
- Blackman M. L.; Royzen M.; Fox J. M. Tetrazine ligation: Fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 2008, 130 (41), 13518–13519. 10.1021/ja8053805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin R.; Van Den Bosch S. M.; Ten Hoeve W.; Carvelli M.; Versteegen R. M.; Lub J.; Robillard M. S. Highly reactive trans-cyclooctene tags with improved stability for diels-alder chemistry in living systems. Bioconjugate Chem. 2013, 24 (7), 1210–1217. 10.1021/bc400153y. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Läppchen T.; Van Den Bosch S. M.; Laforest R.; Robillard M. S. Diels-alder reaction for tumor pretargeting: In vivo chemistry can boost tumor radiation dose compared with directly labeled antibody. J. Nucl. Med. 2013, 54 (11), 1989–1995. 10.2967/jnumed.113.123745. [DOI] [PubMed] [Google Scholar]
- Li J.; Jia S.; Chen P. R. Diels-Alder reaction–triggered bioorthogonal protein decaging in living cells. Nat. Chem. Biol. 2014, 10 (12), 1003–1005. 10.1038/nchembio.1656. [DOI] [PubMed] [Google Scholar]
- Fan X.; Ge Y.; Lin F.; Yang Y.; Zhang G.; Ngai W. S. C.; Lin Z.; Zheng S.; Wang J.; Zhao J.; Li J.; Chen P. R. Optimized Tetrazine Derivatives for Rapid Bioorthogonal Decaging in Living Cells. Angew. Chem., Int. Ed. 2016, 55 (45), 14046–14050. 10.1002/anie.201608009. [DOI] [PubMed] [Google Scholar]
- Carlson J. C. T.; Mikula H.; Weissleder R. Unraveling Tetrazine-Triggered Bioorthogonal Elimination Enables Chemical Tools for Ultrafast Release and Universal Cleavage. J. Am. Chem. Soc. 2018, 140 (10), 3603–3612. 10.1021/jacs.7b11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies S.; Qiao L.; Oliveira B. L.; Navo C. D.; Jiménez-Osés G.; Bernardes G. J. L. Tetrazine-Triggered Release of Carboxylic-Acid-Containing Molecules for Activation of an Anti-inflammatory Drug. ChemBioChem 2019, 20 (12), 1541–1546. 10.1002/cbic.201900098. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; ten Hoeve W.; Rossin R.; de Geus M. A. R.; Janssen H. M.; Robillard M. S. Click-to-Release from trans-Cyclooctenes: Mechanistic Insights and Expansion of Scope from Established Carbamate to Remarkable Ether Cleavage. Angew. Chem., Int. Ed. 2018, 57 (33), 10494–10499. 10.1002/anie.201800402. [DOI] [PubMed] [Google Scholar]
- Steiger A. K.; Yang Y.; Royzen M.; Pluth M. D. Bio-orthogonal ″click-and-release″ donation of caged carbonyl sulfide (COS) and hydrogen sulfide (H2S). Chem. Commun. 2017, 53 (8), 1378–1380. 10.1039/C6CC09547J. [DOI] [PubMed] [Google Scholar]
- Xu M.; Tu J.; Franzini R. M. Rapid and efficient tetrazine-induced drug release from highly stable benzonorbornadiene derivatives. Chem. Commun. 2017, 53 (46), 6271–6274. 10.1039/C7CC03477F. [DOI] [PubMed] [Google Scholar]
- Xu M.; Galindo-Murillo R.; Cheatham T. E.; Franzini R. M. Dissociative reactions of benzonorbornadienes with tetrazines: Scope of leaving groups and mechanistic insights. Org. Biomol. Chem. 2017, 15 (46), 9855–9865. 10.1039/C7OB02191G. [DOI] [PubMed] [Google Scholar]
- Jiménez-Moreno E.; Guo Z.; Oliveira B. L.; Albuquerque I. S.; Kitowski A.; Guerreiro A.; Boutureira O.; Rodrigues T.; Jiménez-Osés G.; Bernardes G. J. L. Vinyl Ether/Tetrazine Pair for the Traceless Release of Alcohols in Cells. Angew. Chem., Int. Ed. 2017, 56 (1), 243–247. 10.1002/anie.201609607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Alexander S. C.; Jin S.; Devaraj N. K. A Bioorthogonal Near-Infrared Fluorogenic Probe for mRNA Detection. J. Am. Chem. Soc. 2016, 138 (36), 11429–11432. 10.1021/jacs.6b01625. [DOI] [PubMed] [Google Scholar]
- Lelieveldt L.; Eising S.; Wijen A.; Bonger K. M. Vinylboronic acid-caged prodrug activation using click-to-release tetrazine ligation. Org. Biomol. Chem. 2019, 17 (39), 8816–8821. 10.1039/C9OB01881F. [DOI] [PubMed] [Google Scholar]
- Kaur T.; Wadhwa P.; Bagchi S.; Sharma A. Isocyanide based [4 + 1] cycloaddition reactions: An indispensable tool in multi-component reactions (MCRs). Chem. Commun. 2016, 52 (43), 6958–6976. 10.1039/C6CC01562J. [DOI] [PubMed] [Google Scholar]
- van Onzen A.; Versteegen R. M.; Hoeben F. J. M.; Filot I. A. W.; Rossin R.; Zhu T.; Wu J.; Hudson P. J.; Janssen H. M.; Ten Hoeve W.; Robillard M. S. Bioorthogonal Tetrazine Carbamate Cleavage by Highly Reactive trans-Cyclooctene. J. Am. Chem. Soc. 2020, 142 (25), 10955–10963. 10.1021/jacs.0c00531. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Ji X.; Yu B.; Ji K.; Gallo D.; Csizmadia E.; Zhu M.; Choudhury M. R.; De La Cruz L. K. C.; Chittavong V.; Pan Z.; Yuan Z.; Otterbein L. E.; Wang B. Enrichment-Triggered prodrug activation demonstrated through mitochondria-Targeted delivery of doxorubicin and carbon monoxide. Nat. Chem. 2018, 10 (7), 787–794. 10.1038/s41557-018-0055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J.; Svatunek D.; Parvez S.; Eckvahl H. J.; Xu M.; Peterson R. T.; Houk K. N.; Franzini R. M. Isonitrile-responsive and bioorthogonally removable tetrazine protecting groups. Chem. Sci. 2020, 11 (1), 169–179. 10.1039/C9SC04649F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikonda S. S.; Orsi D. L.; Staudacher V.; Jenkins I. A.; Fiedler F.; Chen J.; Gamble A. B. Bioorthogonal prodrug activation driven by a strain-promoted 1,3-dipolar cycloaddition. Chem. Sci. 2015, 6 (2), 1212–1218. 10.1039/C4SC02574A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard S.; Audisio D.; Riomet M.; Bregant S.; Sallustrau A.; Plougastel L.; Decuypere E.; Gabillet S.; Kumar R. A.; Elyian J.; Trinh M. N.; Koniev O.; Wagner A.; Kolodych S.; Taran F. Bioorthogonal Click and Release Reaction of Iminosydnones with Cycloalkynes. Angew. Chem., Int. Ed. 2017, 56 (49), 15612–15616. 10.1002/anie.201708790. [DOI] [PubMed] [Google Scholar]
- Riomet M.; Decuypere E.; Porte K.; Bernard S.; Plougastel L.; Kolodych S.; Audisio D.; Taran F. Design and Synthesis of Iminosydnones for Fast Click and Release Reactions with Cycloalkynes. Chem. - Eur. J. 2018, 24 (34), 8535–8541. 10.1002/chem.201801163. [DOI] [PubMed] [Google Scholar]
- Wang W.; Ji X.; Du Z.; Wang B. Sulfur dioxide prodrugs: Triggered release of SO2 via a click reaction. Chem. Commun. 2017, 53 (8), 1370–1373. 10.1039/C6CC08844A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Viennois E.; Ji K.; Damera K.; Draganov A.; Zheng Y.; Dai C.; Merlin D.; Wang B. A click-and-release approach to CO prodrugs. Chem. Commun. 2014, 50 (100), 15890–15893. 10.1039/C4CC07748B. [DOI] [PubMed] [Google Scholar]
- van Brakel R.; Vulders R. C. M.; Bokdam R. J.; Grüll H.; Robillard M. S. A doxorubicin prodrug activated by the staudinger reaction. Bioconjugate Chem. 2008, 19 (3), 714–718. 10.1021/bc700394s. [DOI] [PubMed] [Google Scholar]
- Azoulay M.; Tuffin G.; Sallem W.; Florent J. C. A new drug-release method using the Staudinger ligation. Bioorg. Med. Chem. Lett. 2006, 16 (12), 3147–3149. 10.1016/j.bmcl.2006.03.073. [DOI] [PubMed] [Google Scholar]
- Luo J.; Liu Q.; Morihiro K.; Deiters A. Small-molecule control of protein function through Staudinger reduction. Nat. Chem. 2016, 8 (11), 1027–1034. 10.1038/nchem.2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Du Y.; Li M.; Zhang D.; Xiang Z.; Peng T. Activity-Based Genetically Encoded Fluorescent and Luminescent Probes for Detecting Formaldehyde in Living Cells. Angew. Chem., Int. Ed. 2020, 59 (38), 16352–16356. 10.1002/anie.202001425. [DOI] [PubMed] [Google Scholar]
- Chan J.; Dodani S. C.; Chang C. J. Reaction-based small-molecule fluorescent probes for chemoselective bioimaging. Nat. Chem. 2012, 4 (12), 973–984. 10.1038/nchem.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Bertozzi C. R. A Bioorthogonal Reaction of N-Oxide and Boron Reagents. Angew. Chem., Int. Ed. 2015, 54 (52), 15777–15781. 10.1002/anie.201508861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brakel R. v.; Vulders R. C. M.; Bokdam R. J.; Grüll H.; Robillard M. S. A Doxorubicin Prodrug Activated by the Staudinger Reaction. Bioconjugate Chem. 2008, 19 (3), 714–718. 10.1021/bc700394s. [DOI] [PubMed] [Google Scholar]
- Matikonda S. S.; Fairhall J. M.; Fiedler F.; Sanhajariya S.; Tucker R. A. J.; Hook S.; Garden A. L.; Gamble A. B. Mechanistic Evaluation of Bioorthogonal Decaging with trans-Cyclooctene: The Effect of Fluorine Substituents on Aryl Azide Reactivity and Decaging from the 1,2,3-Triazoline. Bioconjugate Chem. 2018, 29 (2), 324–334. 10.1021/acs.bioconjchem.7b00665. [DOI] [PubMed] [Google Scholar]
- Versteegen R. M.; Rossin R.; Ten Hoeve W.; Janssen H. M.; Robillard M. S. Click to release: Instantaneous doxorubicin elimination upon tetrazine ligation. Angew. Chem., Int. Ed. 2013, 52 (52), 14112–14116. 10.1002/anie.201305969. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Van Duijnhoven S. M. J.; Ten Hoeve W.; Janssen H. M.; Kleijn L. H. J.; Hoeben F. J. M.; Versteegen R. M.; Robillard M. S. Triggered Drug Release from an Antibody-Drug Conjugate Using Fast ″click-to-Release″ Chemistry in Mice. Bioconjugate Chem. 2016, 27 (7), 1697–1706. 10.1021/acs.bioconjchem.6b00231. [DOI] [PubMed] [Google Scholar]
- Rossin R.; Versteegen R. M.; Wu J.; Khasanov A.; Wessels H. J.; Steenbergen E. J.; Ten Hoeve W.; Janssen H. M.; Van Onzen A. H. A. M.; Hudson P. J.; Robillard M. S. Chemically triggered drug release from an antibody-drug conjugate leads to potent antitumour activity in mice. Nat. Commun. 2018, 9 (1), 1484. 10.1038/s41467-018-03880-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I.; Agris P. F.; Yigit M. V.; Royzen M. In situ activation of a doxorubicin prodrug using imaging-capable nanoparticles. Chem. Commun. 2016, 52 (36), 6174–6177. 10.1039/C6CC01024E. [DOI] [PubMed] [Google Scholar]
- Yao Q.; Lin F.; Fan X.; Wang Y.; Liu Y.; Liu Z.; Jiang X.; Chen P. R.; Gao Y. Synergistic enzymatic and bioorthogonal reactions for selective prodrug activation in living systems. Nat. Commun. 2018, 9 (1), 5032. 10.1038/s41467-018-07490-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Conde J.; Guerreiro A.; Bernardes G. J. L. Tetrazine Carbon Nanotubes for Pretargeted In Vivo ″Click-to-Release″ Bioorthogonal Tumour Imaging. Angew. Chem., Int. Ed. 2020, 59 (37), 16023–16032. 10.1002/anie.202008012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann K.; Gambardella A.; Lilienkampf A.; Bradley M. Tetrazine-mediated bioorthogonal prodrug–prodrug activation. Chem. Sci. 2018, 9 (36), 7198–7203. 10.1039/C8SC02610F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu J.; Xu M.; Parvez S.; Peterson R. T.; Franzini R. M. Bioorthogonal Removal of 3-Isocyanopropyl Groups Enables the Controlled Release of Fluorophores and Drugs in Vivo. J. Am. Chem. Soc. 2018, 140 (27), 8410–8414. 10.1021/jacs.8b05093. [DOI] [PubMed] [Google Scholar]
- Ji X.; Wang B. Strategies toward Organic Carbon Monoxide Prodrugs. Acc. Chem. Res. 2018, 51 (6), 1377–1385. 10.1021/acs.accounts.8b00019. [DOI] [PubMed] [Google Scholar]
- Pan Z.; Chittavong V.; Li W.; Zhang J.; Ji K.; Zhu M.; Ji X.; Wang B. Organic CO Prodrugs: Structure–CO-Release Rate Relationship Studies. Chem. - Eur. J. 2017, 23 (41), 9838–9845. 10.1002/chem.201700936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop A. C.; Ubersax J. A.; Petsch D. T.; Matheos D. P.; Gray N. S.; Blethrow J.; Shimizu E.; Tsien J. Z.; Schultz P. G.; Rose M. D. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 2000, 407 (6802), 395–401. 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Dagliyan O.; Tarnawski M.; Chu P. H.; Shirvanyants D.; Schlichting I.; Dokholyan N. V.; Hahn K. M. Engineering extrinsic disorder to control protein activity in living cells. Science 2016, 354 (6318), 1441–1444. 10.1126/science.aah3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt M. R.; Schwartz E. C.; Muir T. W. Small-molecule-mediated rescue of protein function by an inducible proteolytic shunt. Proc. Natl. Acad. Sci. U. S. A. 2007, 104 (27), 11209. 10.1073/pnas.0700816104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L.; Liu Y.; Zhang G.; Ge Y.; Fan X.; Lin F.; Wang J.; Zheng H.; Xie X.; Zeng X.; Chen P. R. Genetically Encoded Chemical Decaging in Living Bacteria. Biochemistry 2018, 57 (4), 446–450. 10.1021/acs.biochem.7b01017. [DOI] [PubMed] [Google Scholar]
- Ge Y.; Fan X.; Chen P. R. A genetically encoded multifunctional unnatural amino acid for versatile protein manipulations in living cells. Chem. Sci. 2016, 7 (12), 7055–7060. 10.1039/C6SC02615J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Liu Y.; Liu Y.; Zheng S.; Wang X.; Zhao J.; Yang F.; Zhang G.; Wang C.; Chen P. R. Time-resolved protein activation by proximal decaging in living systems. Nature 2019, 569 (7757), 509–513. 10.1038/s41586-019-1188-1. [DOI] [PubMed] [Google Scholar]
- Beck A.; Goetsch L.; Dumontet C.; Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discovery 2017, 16 (5), 315–337. 10.1038/nrd.2016.268. [DOI] [PubMed] [Google Scholar]
- Wilkovitsch M.; Haider M.; Sohr B.; Herrmann B.; Klubnick J.; Weissleder R.; Carlson J. C. T.; Mikula H. Cleavable C2-Symmetric trans-Cyclooctene Enables Fast and Complete Bioorthogonal Disassembly of Molecular Probes. J. Am. Chem. Soc. 2020, 142 (45), 19132–19141. 10.1021/jacs.0c07922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann K.; Jain S.; Gambardella A.; Walker S. E.; Valero E.; Lilienkampf A.; Bradley M. Tetrazine-Responsive Self-immolative Linkers. ChemBioChem 2017, 18 (1), 91–95. 10.1002/cbic.201600560. [DOI] [PubMed] [Google Scholar]
- Shi J.; Zhao Y.; Wang K.; Shi X.; Wang Y.; Huang H.; Zhuang Y.; Cai T.; Wang F.; Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526 (7575), 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Ding J.; Wang K.; Liu W.; She Y.; Sun Q.; Shi J.; Sun H.; Wang D.-C.; Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535 (7610), 111–116. 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Wang Y.; Ding J.; Wang C.; Zhou X.; Gao W.; Huang H.; Shao F.; Liu Z. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 2020, 579 (7799), 421–426. 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- Mondal M.; Liao R.; Xiao L.; Eno T.; Guo J. Highly Multiplexed Single-Cell In Situ Protein Analysis with Cleavable Fluorescent Antibodies. Angew. Chem., Int. Ed. 2017, 56 (10), 2636–2639. 10.1002/anie.201611641. [DOI] [PubMed] [Google Scholar]
- Li Y.; Lou Z.; Li H.; Yang H.; Zhao Y.; Fu H. Bioorthogonal Ligation and Cleavage by Reactions of Chloroquinoxalines with ortho-Dithiophenols. Angew. Chem., Int. Ed. 2020, 59 (9), 3671–3677. 10.1002/anie.201913620. [DOI] [PubMed] [Google Scholar]
- Miller M. A.; Mikula H.; Luthria G.; Li R.; Kronister S.; Prytyskach M.; Kohler R. H.; Mitchison T.; Weissleder R. Modular Nanoparticulate Prodrug Design Enables Efficient Treatment of Solid Tumors Using Bioorthogonal Activation. ACS Nano 2018, 12 (12), 12814–12826. 10.1021/acsnano.8b07954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Liu P.; Wu H.; Xie X.; Chen Z.; Zeng F.; Wu S. A bioorthogonal nanosystem for imaging and in vivo tumor inhibition. Biomaterials 2017, 138, 57–68. 10.1016/j.biomaterials.2017.05.036. [DOI] [PubMed] [Google Scholar]
- Czuban M.; Srinivasan S.; Yee N. A.; Agustin E.; Koliszak A.; Miller E.; Khan I.; Quinones I.; Noory H.; Motola C.; Volkmer R.; Di Luca M.; Trampuz A.; Royzen M.; Mejia Oneto J. M. Bio-Orthogonal Chemistry and Reloadable Biomaterial Enable Local Activation of Antibiotic Prodrugs and Enhance Treatments against Staphylococcus aureus Infections. ACS Cent. Sci. 2018, 4 (12), 1624–1632. 10.1021/acscentsci.8b00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F.; Chen L.; Zhang H.; Ching Ngai W. S.; Zeng X.; Lin J.; Chen P. R. Bioorthogonal Prodrug–Antibody Conjugates for On-Target and On-Demand Chemotherapy. CCS Chemistry 2019, 1 (2), 226–236. 10.31635/ccschem.019.20180038. [DOI] [Google Scholar]
- Yang S.; Tang Q.; Chen L.; Chang J.; Jiang T.; Zhao J.; Wang M.; Chen P. R. Cationic Lipid-based Intracellular Delivery of Bacterial Effectors for Rewiring Malignant Cell Signaling. Angew. Chem., Int. Ed. 2020, 59, 18087. 10.1002/anie.202009572. [DOI] [PubMed] [Google Scholar]
- Tu J.; Svatunek D.; Parvez S.; Liu A. C.; Levandowski B. J.; Eckvahl H. J.; Peterson R. T.; Houk K. N.; Franzini R. M. Stable, Reactive, and Orthogonal Tetrazines: Dispersion Forces Promote the Cycloaddition with Isonitriles. Angew. Chem., Int. Ed. 2019, 58 (27), 9043–9048. 10.1002/anie.201903877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamber D. N.; Nazarova L. A.; Liang Y.; Lopez S. A.; Patterson D. M.; Shih H.-W.; Houk K. N.; Prescher J. A. Isomeric Cyclopropenes Exhibit Unique Bioorthogonal Reactivities. J. Am. Chem. Soc. 2013, 135 (37), 13680–13683. 10.1021/ja407737d. [DOI] [PubMed] [Google Scholar]
- Lin S.; Yang X.; Jia S.; Weeks A. M.; Hornsby M.; Lee P. S.; Nichiporuk R. V.; Iavarone A. T.; Wells J. A.; Toste F. D.; Chang C. J. Redox-based reagents for chemoselective methionine bioconjugation. Science 2017, 355 (6325), 597. 10.1126/science.aal3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. T.; Nelson J. E.; Suero M. G.; Gaunt M. J. A protein functionalization platform based on selective reactions at methionine residues. Nature 2018, 562 (7728), 563–568. 10.1038/s41586-018-0608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall N.; Da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Site-selective protein-modification chemistry for basic biology and drug development. Nat. Chem. 2016, 8 (2), 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Wang J.; Cheng B.; Li J.; Zhang Z.; Hong W.; Chen X.; Chen P. R. Chemical remodeling of cell-surface sialic acids through a palladium-triggered bioorthogonal elimination reaction. Angew. Chem., Int. Ed. 2015, 54 (18), 5364–5368. 10.1002/anie.201409145. [DOI] [PubMed] [Google Scholar]
- Pawlak J. B.; Gential G. P. P.; Ruckwardt T. J.; Bremmers J. S.; Meeuwenoord N. J.; Ossendorp F. A.; Overkleeft H. S.; Filippov D. V.; Van Kasteren S. I. Bioorthogonal deprotection on the dendritic cell surface for chemical control of antigen cross-presentation. Angew. Chem., Int. Ed. 2015, 54 (19), 5628–5631. 10.1002/anie.201500301. [DOI] [PubMed] [Google Scholar]
- Van Der Gracht A. M. F.; De Geus M. A. R.; Camps M. G. M.; Ruckwardt T. J.; Sarris A. J. C.; Bremmers J.; Maurits E.; Pawlak J. B.; Posthoorn M. M.; Bonger K. M.; Filippov D. V.; Overkleeft H. S.; Robillard M. S.; Ossendorp F.; Van Kasteren S. I. Chemical Control over T-Cell Activation in Vivo Using Deprotection of trans-Cyclooctene-Modified Epitopes. ACS Chem. Biol. 2018, 13 (6), 1569–1576. 10.1021/acschembio.8b00155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Tang S.; Zheng J.-S.; Zhao R.; Wang Z.-P.; Shao W.; Chang H.-N.; Cheng J.-Y.; Zhao H.; Liu L.; Qi H. Chemical synthesis of a two-photon-activatable chemokine and photon-guided lymphocyte migration in vivo.Nat. Nat. Commun. 2015, 6, 7220. 10.1038/ncomms8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar M. K. G.; Idris N. M.; Zhang Y. Remote activation of biomolecules in deep tissues using near-infrared-to-UV upconversion nanotransducers. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (22), 8483–8488. 10.1073/pnas.1114551109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q.; Li H.; Wang C.; Shen S.; Ma H.; Liu Z. External Radiation-Induced Local Hydroxylation Enables Remote Release of Functional Molecules in Tumors. Angew. Chem., Int. Ed. 2020, 59, 2–8. 10.1002/anie.202084862. [DOI] [PubMed] [Google Scholar]