

Abstract
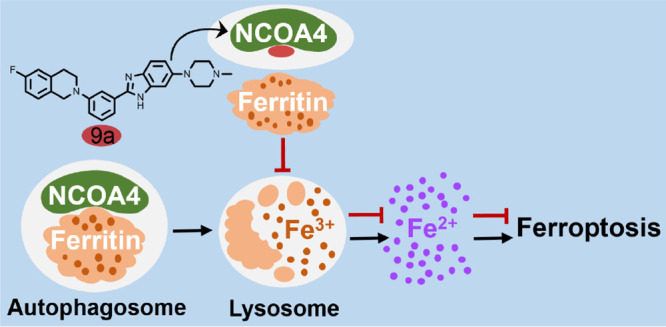
Ferroptosis is an iron-dependent form of oxidative cell death, and the inhibition of ferroptosis is a promising strategy with which to prevent and treat neurological diseases. Herein we report a new ferroptosis inhibitor 9a with a novel mechanism of action. It is demonstrated that nuclear receptor coactivator 4 (NCOA4), a cargo receptor for ferritinophagy, is the target of 9a. Compound 9a blocks ferroptosis by reducing the amount of bioavailable intracellular ferrous iron through disrupting the NCOA4–FTH1 protein–protein interaction. Further studies indicate that 9a directly binds to recombinant protein NCOA4383–522 and effectively blocks the NCOA4383–522–FTH1 interaction. In a rat model of ischemic stroke, 9a significantly ameliorates the ischemic-refusion injury. With the first ligand 9a, this work reveals that NCOA4 is a promising drug target. Additionally, 9a is the first NCOA4–FTH1 interaction inhibitor. This work paves a new road to the development of ferroptosis inhibitors against neurological diseases.
Short abstract
New ferroptosis inhibitory mechanism is described. A novel ferroptosis inhibitor 9a disrupts the protein−protein interaction of NCOA4 and ferritin to inhibit ferroptosis.
Introduction
Ferroptosis, a new type of programmed cell death, is characterized by excessive iron-dependent lipid peroxidation.1,2 In the presence of ferrous iron, the abnormal accumulation of lipid hydroperoxides results in membrane destruction and irreversible cell death.3 Glutathione peroxidase 4 (GPX4), a glutathione (GSH)-dependent selenoenzyme, plays a crucial role in preventing ferroptosis by reducing toxic lipid hydroperoxides to nontoxic lipid alcohols.4,5 When the enzymatic activity of GPX4 is inhibited, lipid hydroperoxides accumulate significantly, leading to ferroptosis. Cysteine, the rate-limiting substrate for GSH synthesis, is obtained primarily from extracellular cystine through the cystine–glutamate antiporter system xc–-mediated transport.6 Consequently, the inhibition of system xc– or the inactivation or depletion of GPX4 can induce ferroptosis.4,7,8
Intracellular redox-active iron can promote the production of lipid reactive oxygen species (ROS), and the presence of iron is indispensable for the execution of ferroptosis.7,9 Iron chelators such as deferoxamine (DFO) suppress ferroptosis by reducing the availability of iron; exogenous iron makes cells more sensitive to this cell death.1,3 Moreover, genes and proteins that regulate iron metabolism, including its import, export, utilization, and storage, can modulate the ferroptosis sensitivity.1−3,10−14 The process of selective autophagic turnover of ferritin (termed ferritinophagy) plays a crucial role in controlling iron homeostasis and is intimately involved in ferroptosis.
Ferritinophagy is mediated by nuclear receptor coactivator 4 (NCOA4), a recently identified autophagic cargo receptor.15,16 Ferritin, consisting of 24 subunits of heavy (FTH1) and light (FTL) isoforms, is a cytosolic iron storage protein complex capable of chelating as many as 4500 iron atoms. By sequestering redox-active iron, ferritin plays an important antioxidant role in cells.17,18 Recent studies demonstrate that ferritin is a key ferroptotic regulator, and its level affects the susceptibility to ferroptosis in vitro and in vivo.19−21 In ferritinophagy, the cargo receptor NCOA4 directly recognizes and binds FTH1, then delivers iron-bound ferritin to autophagosomes for lysosomal degradation and iron release. It has been reported that ferritinophagy participates in ferroptosis. The knockdown of NCOA4 can inhibit ferritinophagy and block lipid peroxidation and ferroptosis by reducing the amount of bioavailable intracellular labile iron pools.22,23
Increasing evidence demonstrates that ferroptosis is implicated in the pathological cell death associated with many neurological diseases, including neurodegenerative disorders and strokes.24−32 Blocking ferroptosis is considered to be a promising therapeutic strategy for these devastating diseases and has exhibited positive outcomes.24−26
Previously, we reported that benzimidazole derivatives inhibited the system xc– inhibitor glutamate-induced ferroptosis in HT22 mouse hippocampal cells.33 To further explore more potent ferroptosis inhibitors and the mechanism of action, we designed and synthesized a library of benzimidazole derivatives, which were assayed with Erastin-induced ferroptosis experiments. This resulted in the discovery of compound 9a, a submicromolar ferroptosis inhibitor. In this work, 9a is identified as the first ligand for NCOA4 and an inhibitor of the NCOA4–FTH1 interaction. It is demonstrated that developing inhibitors for the NCOA4–FTH1 interaction is a new strategy to halt ferroptosis and develop novel therapeutic solutions to combat neurological diseases, such as ischemic stroke.
Results and Discussion
Discovery of a Potent Ferroptosis Inhibitor
In our earlier work, we identified ten benzimidazole derivatives with inhibitory activity against glutamate-induced ferroptosis in HT22 cells.33 In this study, we further confirmed that these compounds also inhibited ferroptosis of HT22 cells induced by Erastin (another inhibitor of system xc–), as shown in Figure S1.
To explore the ferroptosis inhibitory activity space, we designed and synthesized a new library of benzimidazole derivatives (Figure 1A) by introducing lipophilic groups at R1. The substituents at R1 are cyclic amines (N-methylpiperazine, methylpiperazine, morpholine, and piperidine) to improve the solubility of the compound; the central aromatic ring is benzene or pyridine. This led to the first-generation library with eight compounds (4a–h). These compounds were tested for their inhibitory activities against Erastin-induced ferroptosis in HT22 cells, and the results are listed in Table 1.
Figure 1.

Structures of synthesized benzimidazole derivatives.
Table 1. Inhibitory Activities of Synthesized Compounds against Erastin-Induced Ferroptosis in HT22 Cellsa.
| ID | R | EC50 (μM) | ID | R | EC50 (μM) |
|---|---|---|---|---|---|
| 4a | 0.50 | 4b | 0.64 | ||
| 4c | 1.21 | 4d | 1.70 | ||
| 4e | 0.78 | 4f | 0.67 | ||
| 4g | n.d. | 4h | n.d. | ||
| 9a | 6-F | 0.29 | 9b | 6-Cl | 0.51 |
| 9c | 6-OCH3 | 1.01 | 9d | 6-OH | n.d. |
| 9e | 7-CH3 | 0.77 | 9f | 8-OCH3 | 0.33 |
| 9g | 8-OH | n.d. | 9h | 6,8-di-OCH3 | n.d. |
| 9i | 6,7-di-OCH3 | 1.44 | 9j | 6,8-di-OH | n.d. |
| 9k | 6,7-di-OH | n.d. | 14a | 6-OCH2CH3 | 0.51 |
| 14b | 6-OCH2CH2CH3 | 0.39 | 14c | 6-OCH(CH3)2 | 0.53 |
| 14d | 6-OCH2C(CH) | 0.88 | |||
| 17a | n.d. | 17b | n.d. | ||
| 17c | n.d. | ||||
| Fer-1 | 0.082 | DFO | 5.75 |
n.d., cell viability was lower than 50% at 1.0 μM in cells cotreated with Erastin.
In the first-generation library, 4a was the most potent compound (EC50 = 0.50 μM). Therefore, a second-generation library was designed and synthesized based on 4a. This resulted in 18 compounds (Figure 1B), and we discovered more compounds (9a–b, 9e–f, and 14a–d) whose activities were comparable to that of 4a. Their antiferroptosis EC50 values are in the submicromolar range. The most potent compound, 9a, was chosen for further antiferroptosis evaluation.
Cysteine depletion using a culture medium free of cystine can also induce ferroptosis. We found that the cotreatment of HT22 cells with 9a, the classic ferroptosis inhibitor Fer-1, or DFO significantly reduced the level of cell death from cysteine depletion (Figure S2A). l-Buthionine sulfoximine (BSO), an inhibitor of the GSH synthetic enzyme γ-glutamate-cysteine ligase, depletes intracellular GSH, triggering ferroptosis.2,7,8 With the assay of BSO-induced ferroptosis in HT22 cells, we found that 9a dose-dependently prevented cell death (Figure S2B). In addition, using either the assay of the GPX4 covalent inhibitor (1S,3R)-RSL3 (hereafter RSL3) or ML210-induced ferroptosis in HT22 cells, we demonstrated that 9a had a ferroptosis inhibitory behavior similar to those of Fer-1 and DFO (Figures 2A and S2C, respectively). Morphologically, 9a also protected HT22 cells from the damage triggered by RSL3 (Figure S2D). Furthermore, 9a inhibited ferroptosis induced by Erastin, RSL3, or ML210 in HT-1080 human fibrosarcoma cells, another ferroptosis-sensitive cell line (Figure S2E). As shown in Figure 2B, 9a also significantly suppressed ferroptosis induced by knockdown of GPX4 in HT-1080 cells.
Figure 2.

Compound 9a inhibits ferroptosis. (A) Dose–response relationship for the inhibition of RSL3 (1 μM, 24 h)-induced death in HT22 cells by 9a (red), Fer-1 (blue), and DFO (yellow). Cell viability was assayed using a cell counting kit-8 (CCK-8) kit. (B) Inhibitory activity of 9a against ferroptosis induced by the knockdown of GPX4 in HT-1080 cells. Cells were transfected with scrambled siRNA (si-NC, negative control) or a GPX4-targeting siRNA (si-GPX4) for 12 h, then treated with or without 0.5 μM 9a, 0.5 μM Fer-1, or 20 μM DFO for 36 h. Cell viability was assayed using a CCK-8 kit. Data shown represent the mean ± SEM from three independent experiments; ***p < 0.001. (C) Confocal imaging of oxidized BODIPY-C11 in HT22 cells treated with 1 μM RSL3 in the absence or presence of 0.5 μM 9a or 0.5 μM Fer-1 for 3 h. Nuclei were stained with Hoechst 33342 (blue). Flow cytometry analyses of (D) lipid ROS levels and (E) cytosolic ROS levels in HT22 cells treated with the indicated stimuli for 3 h. (F) HT22 cells were treated as indicated for 12 h. PTGS2 mRNA was measured by quantitative PCR (qPCR) with reverse transcription. Data shown represent the mean ± SEM from three independent experiments; ##p < 0.01 compared with control cells, and **p < 0.01 and ***p < 0.001 compared with cells treated with RSL3 alone.
Lipid peroxidation is the hallmark of ferroptosis. The levels of lipid ROS were measured with BODIPY 581/591 C11 (BODIPY-C11), a lipid peroxidation-sensitive dye, using confocal laser scanning microscopy and flow cytometry. In HT22 cells, RSL3 induced significant lipid ROS accumulation, which was indicated by the increase in the amount of oxidized BODIPY-C11 (green fluorescence). When cells were coincubated with RSL3 and either 9a or Fer-1, the fluorescence intensity was remarkably decreased (Figures 2C and D). In addition, 9a inhibited lipid ROS accumulation induced by the knockdown of GPX4 in HT-1080 cells (Figure S2F). The RSL3-induced cytosolic ROS accumulation was also suppressed by 9a in a dose-dependent manner in HT22 cells. This was detected by flow cytometry with 5- (and 6-) carboxy-2′,7′-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA), a cytosolic ROS sensor (Figure 2E). The increased level of PTGS2 mRNA expression induced by RSL3 is a marker for the lipid peroxidation that occurs during ferroptosis.4,34 As shown in Figure 2F, 9a dose-dependently inhibited the upregulation of PTGS2 expression.
It should be noted that 9a cannot inhibit staurosporine (STS)-induced apoptosis, hydrogen peroxide (H2O2)-induced necrosis, or TNF-α/SM-164/zVAD-fmk-induced necroptosis (Figure S2G). Together, these results demonstrate that 9a is a selective ferroptosis inhibitor that effectively suppresses lipid peroxidation and rescues cells from ferroptosis.
Compound 9a Inhibits Ferroptosis by Decreasing the Amount of Intracellular Free Fe2+
The aforementioned experiments revealed that 9a suppressed ferroptosis induced by directly inactivating or depleting GPX4. Additionally, 9a did not change the intracellular GSH level (Figure S3A) or GPX4 expression (Figures S3B and C). Therefore, 9a probably inhibits ferroptosis downstream of or in parallel to GPX4. It has been reported that Fer-1 and liproxstatin-1 (Lip-1), the most commonly used ferroptosis inhibitors, suppress ferroptosis by acting as radical-trapping antioxidants (RTAs).1,27,35 The 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay under cell-free conditions, however, demonstrated that 9a cannot directly scavenge free radicals (Figure 3A), indicating that the compound was devoid of antioxidant activity. Since the iron chelator DFO is also a classic ferroptosis inhibitor, we tested the iron chelating properties of 9a using UV–vis spectroscopy. As shown in Figure 3B, DFO displays a characteristic peak with a maximum absorption at ∼430 nm in the presence of irons.36,37 However, no new peak was observed when adding irons to the solution of 9a, suggesting that 9a cannot chelate Fe3+ or Fe2+. In spite of this, given the crucial role of ferrous iron in promoting lipid ROS formation and ferroptosis, we explored whether 9a can alter the levels of available Fe2+ in live cells using the Fe2+-selective fluorescent probe FerroOrange. Similar to that treated with DFO, the fluorescence intensity of FerroOrange in HT22 cells significantly decreased upon treatment with 9a (Figures 3C and D), suggesting that 9a reduced the level of free Fe2+. Interestingly, it was also observed that the large quantity of Fe2+ in lysosomes (colocalization of FerroOrange and LysoTracker Green) was diminished when HT22 cells were treated with 9a (Figures 3E and F). The number of lysosomes was not changed by 9a (Figures S4A and B), precluding the possibility that the decrease in the amount of lysosomal Fe2+ was due to the decrease in the number of lysosomes. The reduction of the Fe2+ level induced by 9a was also observed in HT-1080 cells, again without altering the number of lysosomes (Figures S4C–H).
Figure 3.

Compound 9a reduces the amount of bioavailable ferrous iron to block ferroptosis. (A) Cell-free antioxidant potential monitored by changes in the absorbance at 517 nm of the stable radical DPPH. The concentration of all tested compounds was 50 μM. Data shown represent the mean ± SEM from three independent experiments; ns, not significant; ***p < 0.001. (B) UV–vis absorption spectra of DFO and 9a in the absence or presence of Fe3+ or Fe2+. (C) Confocal imaging of FerroOrange (red) in HT22 cells treated with 0.5 μM 9a or 50 μM DFO for 6 h. Nuclei were stained with Hoechst 33342 (blue). (D) Quantification of the fluorescence intensity of FerroOrange. (E) Confocal imaging of FerroOrange (red) and LysoTracker Green (green) in HT22 cells treated with 0.5 μM 9a for 6 h. The images show the subcellular localization of Fe2+ in live cells. LysoTracker Green stains the lysosomes. (F) Quantification of the fluorescence intensity of FerroOrange colocalized with LysoTracker Green per cell. Approximately 100 cells were measured per condition. (G) Cell viability of HT22 cells treated as indicated. In the absence or presence of 0.5 μM 9a, cells were treated with 1 μM RSL3 ± 10 μg/mL ferric ammonium citrate (FAC), 25 μM ferric citrate (FC), or 20 μM iron chloride hexahydrate (IHC) for 12 h. (D, F, and G) Data shown represent the mean ± SEM from three independent experiments; ***p < 0.001.
To further confirm that 9a suppresses ferroptosis by lowering the amount of bioavailable intracellular Fe2+, we examined the effects of iron addition on the inhibitory activity of 9a against ferroptosis. We found that treatment of HT22 cells with three different exogenous sources of iron (ferric ammonium citrate, ferric citrate, and iron chloride hexahydrate) abolished or significantly mitigated the ability of 9a to rescue cells from RSL3-induced ferroptosis (Figure 3G). Because 9a cannot bind irons, the induced decrease in Fe2+ level is most likely a result of the regulation of 9a on iron metabolism.
Compound 9a Inhibits the Degradation of Ferritin Complexes in Lysosomes (Ferritinophagy) and Localizes in Autophagosomes
As the major iron storage protein complex in mammals, ferritin plays a vital role in iron metabolism and protecting against ferroptosis. Large amounts of sequestered iron can be liberated from the ferritin complex via ferritinophagy, the process of the autophagic turnover of ferritin.15,16,38 Because 9a diminished the level of Fe2+ in lysosomes, we assumed that 9a inhibits ferritinophagy. To prove this hypothesis, we first determined the levels of ferritin by Western blotting, and found that upon treatment with 9a, the protein level of ferritin increased in a dose-dependent manner (Figures 4A and B). Additionally, using qPCR experiments, we revealed that 9a did not change the mRNA expressions of FTH1 and FTL (Figure 4C). Therefore, 9a induced an increase in the amount of ferritin by inhibiting protein degradation, not by upregulating gene transcription.
Figure 4.

Compound 9a inhibits lysosomal ferritin degradation and localizes in autophagosomes. (A) Immunoblot analysis of HT22 cells treated with the indicated concentrations of 9a for 6 h. (B) Quantification of the immunoblot analysis in panel A. Data shown represent the mean ± SEM from three independent experiments; *p < 0.05. (C) Effects of 9a on the mRNA levels of FTH1 and FTL. HT22 cells were treated with 0.5 μM 9a for 6 h. Data shown represent the mean ± SEM from three independent experiments. (D) Immunofluorescence imaging of ferritin (red) in HT-1080 cells treated with 9a. LAMP2 (green) stains lysosomes. (E) Confocal imaging of 9a (magenta) in A549 cells that stably expressed GFP-LC3 and were treated for 2 h with the compound at 10 μM. The colocalized foci are indicated by white arrows and shown in an enlarged image of the yellow box.
To further validate that 9a suppressed lysosomal ferritin degradation, we performed immunofluorescence experiments to observe the ferritin lysosomal localization. To promote ferritin accumulation in lysosomes, HT-1080 cells were loaded with FAC for 12 h, then incubated with DFO for 6 h in the presence of lysosomal protease inhibitors (E64-d and Pepstatin A). As previously reported, cells exhibited punctate ferritin lysosomal localization,15,39 whereas the presence of 9a led to diffuse ferritin staining (Figure 4D). This suggests that 9a indeed blocks ferritin colocalization with lysosomes and causes impaired ferritinophagy.
Compound 9a has a conjugated system with fluorescent emission (Figure S5A), and this characteristic was used to investigate its distribution in live cells. With confocal laser scanning microscopy, we observed that 9a accumulated as punctate vesicles in the cytoplasm in both HT22 and HT-1080 cells, regardless of the presence of RSL3 (Figure S5B). Interestingly, the 9a foci (magenta staining) well colocalized with the green fluorescent protein (GFP)-LC3 puncta (merged white staining, Figures 4E and S5C) in A549 human lung adenocarcinoma cells stably expressing GFP-fused LC3 protein (a marker of autophagosomes), suggesting that 9a accumulated in autophagosomes. To further confirm this, the alkyne compound 14d (a derivative of 9a) with antiferroptosis activity was labeled by means of in situ click chemistry, and immunofluorescence staining was carried out. As shown in Figure S5F, 14d also gave punctate cell labeling, and these foci colocalized with LC3. Again, this result supports that 9a targets autophagosomes. In contrast, 9a did not accumulate in the endoplasmic reticulum (ER) or mitochondria (Figure S6). To some extent, the localization of 9a in autophagosomes indicates that 9a interferes with ferritinophagy.
Compound 9a Inhibits Ferritinophagy by Disrupting the NCOA4–FTH1 Interaction through Binding to NCOA4 in Cells
Ferritinophagy is mediated by the cargo receptor NCOA4, which directly recognizes FTH1 and then transports the ferritin complex to autophagosomes for lysosomal degradation and iron release.15,16 It is clear that the NCOA4–FTH1 protein–protein interaction (PPI) is essential for ferritinophagy. Therefore, with coimmunoprecipitation (co-IP) assays, we investigated whether 9a abolished the NCOA4–FTH1 interaction to inhibit ferritinophagy. HEK-293T cells were transfected with Myc- and His-tagged FTH1 and Flag-tagged NCOA4, followed by DMSO or 9a treatment for 16 h, and then a co-IP assay was performed. As shown in Figure 5A, the NCOA4–FTH1 interaction was substantially impaired by 9a. In addition, we detected the endogenous interaction between the two proteins in HT-1080 cells. Consistently, 9a also inhibited the endogenous NCOA4 interaction with FTH1 (Figure 5B). These results demonstrate that 9a is a potent inhibitor of the NCOA4–FTH1 interaction in cells. Furthermore, we examined the impact of 9a on autophagy. In A549 cell line stably expressing GFP-LC3, the autophagy inhibitor chloroquine (CQ) significantly enhanced the number of GFP-LC3 puncta. While these A549 cells were treated with 9a, the number of GFP-LC3 puncta remained nearly the same as that in the control cells even at a high concentration (Figures S7A and B), indicating that 9a cannot promote the formation of GFP-LC3 puncta. In line with this, the Western blotting analysis revealed that, unlike CQ, 9a had no ability to induce an increase in the LC3-II level in HT22 cells (Figures S7C and D). Taken together, these data indicate that 9a is not an autophagy inhibitor. Additionally, these results exclude the possibility that 9a inhibits ferritinophagy by affecting autophagy.
Figure 5.

Compound 9a disrupts the NCOA4–FTH1 interaction and binds to NCOA4. (A) Immunoprecipitation (IP) and immunoblot analysis of the interaction of FTH1-Myc-His and NCOA4-Flag in HEK-293T cells. 9a was added at 8 h post-transfection. The lysates from HEK-293T cells expressing the indicated proteins were immunoprecipitated with anti-Flag (Flag-IP) and immunoblotted with the indicated antibodies. (B) IP and immunoblot analysis of the interaction of endogenous FTH1 and NCOA4 in HT-1080 cells treated with 9a for 6 h. IgG was used as a negative control. (C) Live-cell imaging of 9a (magenta) in HT-1080 cells expressing EGFP-NCOA4; Nuc, nucleus. (D) Structures of Biotin-PEG3-N3 and Biotin-14d. (E) Specific binding of 9a to NCOA4. The lysates from HEK-293T cells expressing FTH1-Myc-His and NCOA4-Flag were pretreated with either 9a or DMSO at 4 °C for 6 h and then incubated with increasing concentrations of Biotin-14d (precoupled with streptavidin magnetic beads) at 4 °C overnight, followed by pull-down with streptavidin magnetic beads. The precipitates were immunoblotted with the indicated antibodies. Biotin-PEG3-N3 was used as a negative control for Biotin-14d.
Since NCOA4 is an autophagic cargo receptor, we hypothesized that 9a directly binds to NCOA4 instead of FTH1. To prove this, we tested whether 9a colocalized with NCOA4 in live cells. HT-1080 cells were transfected with a plasmid expressing enhanced GFP (EGFP)-tagged NCOA4, incubated with 9a, and then imaged directly. As shown in Figure 5C, the 9a foci were almost completely located in EGFP-NCOA4 puncta, demonstrating that 9a targets NCOA4 in live cells. Given the autophagosomal distribution of NCOA4 in the cytoplasm, this result also supports the localization of 9a in autophagosomes, as demonstrated above.
To further confirm the binding of 9a to NCOA4, we synthesized a biotin-tagged chemical probe by conjugating biotin-PEG3-N3 with the active alkyne compound 14d. We found that the probe (named Biotin-14d) effectively blocks RSL3-induced ferroptosis in HT22 cells (EC50 = 3.66 μM). The activity is comparable to the activity of its parent 14d (EC50 = 0.69 μM, Figure S7E). Therefore, biotin–streptavidin pull-down experiments were carried out using Biotin-14d. Cell lysates from HEK-293T cells expressing FTH1-Myc-His and NCOA4-Flag were incubated with different concentrations of Biotin-14d or free biotin-PEG3-N3. The mixture was precipitated with streptavidin magnetic beads, followed by immunoblotting with the corresponding antitag antibody. As shown in Figure 5E, only NCOA4 was specifically pulled down by Biotin-14d in a dose-dependent manner, and the binding of Biotin-14d and NCOA4 was competitively inhibited by 9a. Overall, these results demonstrate that 9a binds to NCOA4 and blocks its interaction with FTH1 in live cells, thereby inhibiting ferritinophagy.
Compound 9a Directly Binds NCOA4383–522 Protein and Inhibits the NCOA4383–522–FTH1 Interaction
It has been reported that NCOA4 associates with FTH1 via the fragment containing amino acids 383–522, and this region of NCOA4 (NCOA4383–522) is also required for ferritinophagy in live cells.39 Hence, we prepared and purified the recombinant human NCOA4383–522 protein for further analysis. As shown in Figure 6A, the in vitro recombinant NCOA4383–522 protein was pulled down by the synthesized biotin-tagged probe Biotin-14d in a dose-dependent manner, which was detected by silver staining and immunoblotting. Additionally, the binding of NCOA4383–522 and Biotin-14d was blocked when the protein was preincubated with 9a. Meanwhile, we did not detect the binding of Biotin-14d to recombinant human FTH1 protein. This result further confirms that 9a specifically interacts with NCOA4383–522. The binding affinity of 9a to NCOA4383–522 was measured with surface plasmon resonance (SPR). The equilibrium dissociation constant (KD) of the binding of 9a to NCOA4383–522 is 7.48 μM. As a negative control, the inactive compound 9h has no binding to the recombinant protein (Figure 6B). Moreover, to confirm that 9a directly inhibits the NCOA4383–522–FTH1 interaction, we developed an enzyme-linked immunosorbent assay (ELISA) to detect NCOA4383–522 binding to FTH1 protein (a schematic is shown in Figure 6C). In this assay, 9a significantly disrupted the NCOA4–FTH1 interaction (IC50 = 3.26 μM), as shown in Figure 6D.
Figure 6.

Compound 9a directly binds NCOA4383–522 and inhibits the NCOA4383–522–FTH1 interaction. (A) Specific binding of 9a to NCOA4383–522. The recombinant NCOA4383–522 or FTH1 protein was pretreated with 9a or DMSO for 1 h and then incubated with increasing concentrations of Biotin-14d (precoupled with streptavidin magnetic beads) for 1 h. The proteins bound to the beads were detected by silver staining and Western blotting. (B) SPR sensorgrams for the binding of 9a and the inactive compound 9h to NCOA4383–522. (C) Schematic of the ELISA assay used to detect the NCOA4383–522–FTH1 interaction and its disruption by 9a. (D) Dose–response relationship for the inhibition of the NCOA4383–522–FTH1 interaction by 9a. The indicated concentration of 9a was preincubated with NCOA4383–522, and the binding of NCOA4383–522 to FTH1 was monitored by ELISA. Data shown represent the mean ± SEM from three independent experiments.
Compound 9a Alleviates Cerebral Ischemic Injury in Rats Subjected to Transient Middle Cerebral Artery Occlusion (MCAO)
Ischemic stroke occurs as a result of vascular occlusion and is the most common form of stroke, which is a leading cause of death and permanent disability. To date, there is no effective therapy for this disease.26,40 Current studies indicate that ferroptotic pathways have been involved in the pathological process of ischemic stroke, and the inhibition of ferroptosis can ameliorate ischemic brain injury in vivo.1,24,26,41,42
Since 9a has a pronounced inhibitory activity against ferroptosis in cells, we evaluated whether 9a can be protective in vivo in a MCAO model of ischemic stroke. In this model, rats were given 9a (10 mg/kg), Fer-1 (10 mg/kg), or vehicle by intraperitoneal injection. A filament suture was used to occlude the middle cerebral artery for 1.5 h and then withdrawn for reperfusion. The results from brain sections stained with 2,3,5-tripenyltetrazolium chloride (TTC) showed that, similar to Fer-1, 9a also substantially reduced infarct volume at 24 h post reperfusion (Figures 7A and B). Additionally, the Longa test indicated that 9a administration improved neurological deficits following focal ischemia (Figure 7C). The protection of 9a against cerebral ischemic injury was also confirmed by histological observations, including H&E and Nissl staining (Figure 7D). For example, compared to normal cells with a regular shape and even staining, many neurons in the cortex of the lesioned hemisphere from MCAO-treated rats were shrunken with aberrant morphology, as indicated by Nissl staining. Strikingly, 9a treatment maintained most neuronal cells in a regular shape and reduced the percentage of injured neurons in the cortex (Figure 7E).
Figure 7.

The administration of 9a ameliorates ischemic reperfusion injury in the MCAO model. (A) Representative images of TTC staining in brain sections at 24 h after MCAO and reperfusion. The infarction area is indicated by the white color with a black outline. Either 9a or Fer-1 was injected into rats 0.5 h before and 2 h after the MCAO occlusion. (B) Quantification of the infarction volume (corrected by the contralateral structure) as indicated by TTC staining using ImageJ (n = 8–9). (C) Statistical analysis of the neurological score (higher numbers indicate more severe impairment) in the Longa test performed at 24 h after MCAO and reperfusion (n = 12). (D) Representative images of H&E (hematoxylin and eosin) and Nissl staining of the cortex of the lesioned hemisphere. The scale bar is 100 μm. (E) Quantification of the percentage of injured neurons in the cortex of the lesioned hemisphere (n = 6). (F) Relative PTGS2 mRNA, (G) MDA, and 4-HNE levels in the lesioned hemisphere (n = 6). Data shown represent the mean ± SEM; *p < 0.05, **p < 0.01, and ***p < 0.001 compared with the group treated with vehicle.
In this model, we also detected the expression of PTGS2 mRNA, a marker for the assessment of ferroptosis in vivo.4 As shown in Figure 7F, 9a significantly decreased the gene expression of PTGS2 in the lesioned hemisphere. Moreover, 9a reduced the levels of malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE), two characteristic products of lipid peroxidation (Figure 7G). Together, in vivo data demonstrate that 9a effectively alleviates cerebral ischemic damage through inhibiting ferroptosis.
Conclusions
In this work, a new and potent ferroptosis inhibitor 9a was identified through phenotypic screening and structural modification. Systematic investigations reveal that through inhibiting ferritinophagy, 9a lowers the level of intracellular labile Fe2+, leading to ferroptosis suppression. This mechanism is distinct from that of the existing ferroptosis inhibitors Fer-1 and DFO. It was observed that 9a accumulates in autophagosomes and colocalizes with the autophagic receptor NCOA4 in live cells. With the synthesized probe Biotin-14d, NCOA4 was further identified as the target of 9a. Through binding to NCOA4, compound 9a disrupts the NCOA4–FTH1 interaction and then blocks NCOA4-dependent ferritinophagy. We also demonstrate that 9a directly binds to NCOA4 in vitro with the purified ferritin-binding domain of human NCOA4, fragment 383–522 (NCOA4383–522). Moreover, the inhibition of 9a in regard to the NCOA4383–522–FTH1 interaction was confirmed in a developed ELISA assay. As an effective ferroptosis inhibitor, 9a protects brain from ischemic reperfusion injury in rats subjected to MCAO.
With the first ligand 9a, this study uncovers that NCOA4 is a potential drug target. Moreover, this work demonstrates that the interaction of NCOA4 and iron-bound ferritin is important for ferroptosis. Gene-depleting NCOA4 can inhibit ferritinophagy and ferroptosis, as previously reported;22,23 blocking the NCOA4–FTH1 interaction with small molecules can also hinder this selective autophagy process, leading to a decrease in the amount of bioavailable Fe2+ as well as ferroptosis suppression, as revealed in this work. To our best knowledge, 9a is the first NCOA4–FTH1 protein–protein interaction inhibitor. This study indicates that except for certain enzymes associated with lipid metabolism, such as GPX4, acyl-CoA synthetase long-chain family member 4 (ACSL4), and lipoxygenases (LOXs), the NCOA4–FTH1 interaction is also a key target when developing new ferroptosis inhibitors.
Acknowledgments
This work was supported by the National Key R&D Program of China (2017YFB02034043) and the Science and Technology Planning Project of Guangdong Province (2016A020217005). We thank Dr. Min Li (School of Pharmaceutical Sciences, Sun Yat-sen University) for kindly providing A549 cells that stably expressed GFP-LC3 and the Jianwen Chen team (New Drug R&D Technology Center, School of Pharmaceutical Science, Sun Yat-sen University) for assistance during the in vivo surgery.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01592.
Experimental details and NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Dixon S. J.; Lemberg K. M.; Lamprecht M. R.; Skouta R.; Zaitsev E. M.; Gleason C. E.; Patel D. N.; Bauer A. J.; Cantley A. M.; Yang W. S.; Morrison B.; Stockwell B. R. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell B. R.; Friedmann Angeli J. P.; Bayir H.; Bush A. I.; Conrad M.; Dixon S. J.; Fulda S.; Gascon S.; Hatzios S. K.; Kagan V. E.; Noel K.; Jiang X.; Linkermann A.; Murphy M. E.; Overholtzer M.; Oyagi A.; Pagnussat G. C.; Park J.; Ran Q.; Rosenfeld C. S.; Salnikow K.; Tang D.; Torti F. M.; Torti S. V.; Toyokuni S.; Woerpel K. A.; Zhang D. D. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassannia B.; Vandenabeele P.; Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019, 35, 830–849. 10.1016/j.ccell.2019.04.002. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; SriRamaratnam R.; Welsch M. E.; Shimada K.; Skouta R.; Viswanathan V. S.; Cheah J. H.; Clemons P. A.; Shamji A. F.; Clish C. B.; Brown L. M.; Girotti A. W.; Cornish V. W.; Schreiber S. L.; Stockwell B. R. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedmann Angeli J. P.; Schneider M.; Proneth B.; Tyurina Y. Y.; Tyurin V. A.; Hammond V. J.; Herbach N.; Aichler M.; Walch A.; Eggenhofer E.; Basavarajappa D.; Radmark O.; Kobayashi S.; Seibt T.; Beck H.; Neff F.; Esposito I.; Wanke R.; Forster H.; Yefremova O.; Heinrichmeyer M.; Bornkamm G. W.; Geissler E. K.; Thomas S. B.; Stockwell B. R.; O’Donnell V. B.; Kagan V. E.; Schick J. A.; Conrad M. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppula P.; Zhang Y.; Zhuang L.; Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer commun (London, England) 2018, 38, 12–24. 10.1186/s40880-018-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon S. J.; Stockwell B. R. The hallmarks of ferroptosis. Annu. Rev. Cancer Biol. 2019, 3, 35–54. 10.1146/annurev-cancerbio-030518-055844. [DOI] [Google Scholar]
- Cao J. Y.; Dixon S. J. Mechanisms of ferroptosis. Cell. Mol. Life Sci. 2016, 73, 2195–2209. 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell B. R.; Jiang X. A physiological function for ferroptosis in tumor suppression by the immune system. Cell Metab. 2019, 30, 14–15. 10.1016/j.cmet.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez S. W.; Sviderskiy V. O.; Terzi E. M.; Papagiannakopoulos T.; Moreira A. L.; Adams S.; Sabatini D. M.; Birsoy K.; Possemato R. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017, 551, 639–643. 10.1038/nature24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassannia B.; Wiernicki B.; Ingold I.; Qu F.; Van Herck S.; Tyurina Y. Y.; Bayir H.; Abhari B. A.; Angeli J. P. F.; Choi S. M.; Meul E.; Heyninck K.; Declerck K.; Chirumamilla C. S.; Lahtela-Kakkonen M.; Van Camp G.; Krysko D. V.; Ekert P. G.; Fulda S.; De Geest B. G.; Conrad M.; Kagan V. E.; Vanden Berghe W.; Vandenabeele P.; Vanden Berghe T. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J. Clin. Invest. 2018, 128, 3341–3355. 10.1172/JCI99032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S.; Henson E. S.; Chen Y.; Gibson S. B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016, 7, e2307–e2317. 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X.; Ou Z.; Xie M.; Kang R.; Fan Y.; Niu X.; Wang H.; Cao L.; Tang D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625. 10.1038/onc.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Li X.; Zhang X.; Kang R.; Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844. 10.1016/j.bbrc.2016.08.034. [DOI] [PubMed] [Google Scholar]
- Mancias J. D.; Wang X.; Gygi S. P.; Harper J. W.; Kimmelman A. C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. 10.1038/nature13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowdle W. E.; Nyfeler B.; Nagel J.; Elling R. A.; Liu S.; Triantafellow E.; Menon S.; Wang Z.; Honda A.; Pardee G.; Cantwell J.; Luu C.; Cornella-Taracido I.; Harrington E.; Fekkes P.; Lei H.; Fang Q.; Digan M. E.; Burdick D.; Powers A. F.; Helliwell S. B.; D’Aquin S.; Bastien J.; Wang H.; Wiederschain D.; Kuerth J.; Bergman P.; Schwalb D.; Thomas J.; Ugwonali S.; Harbinski F.; Tallarico J.; Wilson C. J.; Myer V. E.; Porter J. A.; Bussiere D. E.; Finan P. M.; Labow M. A.; Mao X.; Hamann L. G.; Manning B. D.; Valdez R. A.; Nicholson T.; Schirle M.; Knapp M. S.; Keaney E. P.; Murphy L. O. Selective VPS34 inhibitor blocks autophagy and uncovers a role for NCOA4 in ferritin degradation and iron homeostasis in vivo. Nat. Cell Biol. 2014, 16, 1069–1079. 10.1038/ncb3053. [DOI] [PubMed] [Google Scholar]
- Pantopoulos K.; Porwal S. K.; Tartakoff A.; Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry 2012, 51, 5705–5724. 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaldan S.; Bush A. I.; Devos D.; Rolland A. S.; Moreau C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radical Biol. Med. 2019, 133, 221–233. 10.1016/j.freeradbiomed.2018.09.033. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Lu J.; Hao X.; Li H.; Zhang G.; Liu X.; Li X.; Zhao C.; Kuang W.; Chen D.; Zhu M. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of Parkinson’s disease. Neurotherapeutics 2020, 17, 1796–1812. 10.1007/s13311-020-00929-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui T.; Wang H.; Li Q.; Cheng Y.; Gao Y.; Fang X.; Ma X.; Chen G.; Gao C.; Gu Z.; Song S.; Zhang J.; Wang C.; Wang Z.; Wang T.; Zhang M.; Min J.; Chen X.; Tao L.; Wang F.; Luo C. Deletion of ferritin H in neurons counteracts the protective effect of melatonin against traumatic brain injury-induced ferroptosis. J. Pineal Res. 2021, 70, e12704. 10.1111/jpi.12704. [DOI] [PubMed] [Google Scholar]
- Fang X.; Cai Z.; Wang H.; Han D.; Cheng Q.; Zhang P.; Gao F.; Yu Y.; Song Z.; Wu Q.; An P.; Huang S.; Pan J.; Chen H. Z.; Chen J.; Linkermann A.; Min J.; Wang F. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ. Res. 2020, 127, 486–501. 10.1161/CIRCRESAHA.120.316509. [DOI] [PubMed] [Google Scholar]
- Hou W.; Xie Y.; Song X.; Sun X.; Lotze M. T.; Zeh H. J. 3rd; Kang R.; Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Monian P.; Pan Q.; Zhang W.; Xiang J.; Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. 2016, 26, 1021–1032. 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim I.; Caulfield J. T.; Chen Y.; Swarup V.; Geschwind D. H.; Ivanova E.; Seravalli J.; Ai Y.; Sansing L. H.; Ste.Marie E. J.; Hondal R. J.; Mukherjee S.; Cave J. W.; Sagdullaev B. T.; Karuppagounder S. S.; Ratan R. R. Selenium drives a transcriptional adaptive program to block ferroptosis and treat stroke. Cell 2019, 177, 1262–1279. 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- Skouta R.; Dixon S. J.; Wang J.; Dunn D. E.; Orman M.; Shimada K.; Rosenberg P. A.; Lo D. C.; Weinberg J. M.; Linkermann A.; Stockwell B. R. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo Q. Z.; Lei P.; Jackman K. A.; Li X. L.; Xiong H.; Li X. L.; Liuyang Z. Y.; Roisman L.; Zhang S. T.; Ayton S.; Wang Q.; Crouch P. J.; Ganio K.; Wang X. C.; Pei L.; Adlard P. A.; Lu Y. M.; Cappai R.; Wang J. Z.; Liu R.; Bush A. I. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530. 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- Angeli J. P. F.; Shah R.; Pratt D. A.; Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498. 10.1016/j.tips.2017.02.005. [DOI] [PubMed] [Google Scholar]
- Li Q.; Han X.; Lan X.; Gao Y.; Wan J.; Durham F.; Cheng T.; Yang J.; Wang Z.; Jiang C.; Ying M.; Koehler R. C.; Stockwell B. R.; Wang J. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. 10.1172/jci.insight.90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M.; Karuppagounder S. S.; Chen Y.; Gough P. J.; Bertin J.; Finger J.; Milner T. A.; Jonas E. A.; Ratan R. R. Neuronal death after hemorrhagic stroke in vitro and in vivo shares features of ferroptosis and necroptosis. Stroke 2017, 48, 1033–1043. 10.1161/STROKEAHA.116.015609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L.; Hambright W. S.; Na R.; Ran Q. Ablation of the ferroptosis inhibitor glutathione peroxidase 4 in neurons results in rapid motor neuron degeneration and paralysis. J. Biol. Chem. 2015, 290, 28097–28106. 10.1074/jbc.M115.680090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland A.; Wang Y.; Wu W.; Lan X.; Han X.; Li Q.; Wang J. Ferroptosis and its role in diverse brain diseases. Mol. Neurobiol. 2019, 56, 4880–4893. 10.1007/s12035-018-1403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do Van B.; Gouel F.; Jonneaux A.; Timmerman K.; Gele P.; Petrault M.; Bastide M.; Laloux C.; Moreau C.; Bordet R.; Devos D.; Devedjian J. C. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. 10.1016/j.nbd.2016.05.011. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Zhou H.; Gu Q.; Xu J. Synthesis and evaluation of tetrahydroisoquinoline-benzimidazole hybrids as multifunctional agents for the treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2019, 167, 133–145. 10.1016/j.ejmech.2019.02.008. [DOI] [PubMed] [Google Scholar]
- Fang X.; Wang H.; Han D.; Xie E.; Yang X.; Wei J.; Gu S.; Gao F.; Zhu N.; Yin X.; Cheng Q.; Zhang P.; Dai W.; Chen J.; Yang F.; Yang H. T.; Linkermann A.; Gu W.; Min J.; Wang F. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 2672–2680. 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka O.; Shah R.; Li B.; Friedmann Angeli J. P.; Griesser M.; Conrad M.; Pratt D. A. On the mechanism of cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent. Sci. 2017, 3, 232–243. 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z.; Qiao J.; Nagy T.; Xiong M. P. ROS-triggered degradable iron-chelating nanogels: Safely improving iron elimination in vivo. J. Controlled Release 2018, 283, 84–93. 10.1016/j.jconrel.2018.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Yang Y.; Liu Q.; Zhan A.; Yang Y.; Liu H. A novel star like eight-arm polyethylene glycol-deferoxamine conjugate for iron overload therapy. Pharmaceutics 2020, 12, 329–341. 10.3390/pharmaceutics12040329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Liu J.; Kang R.; Klionsky D. J.; Kroemer G.; Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. 10.1016/j.semcancer.2019.03.002. [DOI] [PubMed] [Google Scholar]
- Mancias J. D.; Pontano Vaites L.; Nissim S.; Biancur D. E.; Kim A. J.; Wang X.; Liu Y.; Goessling W.; Kimmelman A. C.; Harper J. W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. 10.7554/eLife.10308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhorne P.; Bernhardt J.; Kwakkel G. Stroke rehabilitation. Lancet 2011, 377, 1693–1702. 10.1016/S0140-6736(11)60325-5. [DOI] [PubMed] [Google Scholar]
- Yigitkanli K.; Pekcec A.; Karatas H.; Pallast S.; Mandeville E.; Joshi N.; Smirnova N.; Gazaryan I.; Ratan R. R.; Witztum J. L.; Montaner J.; Holman T. R.; Lo E. H.; van Leyen K. Inhibition of 12/15-lipoxygenase as therapeutic strategy to treat stroke. Ann. Neurol. 2013, 73, 129–135. 10.1002/ana.23734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Liu X.; Song C.; Ji S.; Yang J.; Liu Y.; You J.; Zhang J.; Huang S.; Cheng W.; Shao Z.; Li L.; Yang S. Structure-activity relationship studies of phenothiazine derivatives as a new class of ferroptosis inhibitors together with the therapeutic effect in an ischemic stroke model. Eur. J. Med. Chem. 2021, 209, 112842–112862. 10.1016/j.ejmech.2020.112842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.