

Abstract
Induced pluripotent stem cells (iPSCs) provide a unique platform for individualized cell therapy approaches. Currently, episomal DNA, mRNA, and Sendai virus-based RNA reprogramming systems are widely used to generate iPSCs. However, they all rely on the use of multiple (three to six) components (vectors/plasmids/mRNAs) leading to the production of partially reprogrammed cells, reducing the efficiency of the systems. We produced a one-cycle measles virus (MV) vector by substituting the viral attachment protein gene with the green fluorescent protein (GFP) gene. Here, we present a highly efficient multi-transgene delivery system based on a vaccine strain of MV, a non-integrating RNA virus that has a long-standing safety record in humans. Introduction of the four reprogramming factors OCT4, SOX2, KLF4, and cMYC via a single, “one-cycle” MV vector efficiently reprogrammed human somatic cells into iPSCs, whereas MV vector genomes are rapidly eliminated in derived iPSCs. Our MV vector system offers a new reprogramming platform for genomic modification-free iPSCs amenable for clinical translation.
Introduction
Since the first reprogramming of somatic cells into induced pluripotent stem cells (iPSCs) in 2006 [1–3], the fields of stem cell biology and regenerative medicine have entered a new exiting era. Patient-derived iPSC and iPSC-derived progenies are increasingly utilized for human “disease in a dish” modeling, as well as the testing of patient-specific drug efficacy/toxicity [4]. Numerous vehicles were developed to deliver the reprogramming genes, from integrating lentiviral and retroviral vectors, RNAs, proteins to plasmids [5], which reprogrammed somatic cells into iPSCs. For clinical iPSC applications, however, the use of integrating vectors adds concern over increased tumorigenicity due to insertional mutagenesis [6], whereas non-viral reprogramming systems typically show low reprogramming efficiency of patient-derived cells [5]. The recent first-in-human iPSCs clinical trial used autologous iPSCs made by an episomal DNA reprogramming vector system [7–10]. However, this trial was suspended due to two genetic variants found in the iPSCs from the second patient [8, 9]. Thus, for widespread autologous iPSC-based therapies, it is critical to develop a clinically applicable, genomic modification-free reprogramming system.
Non-integrating reprogramming system based on episomal plasmid [11, 12], Sendai virus (SeV) [13, 14], or mRNA transduction [15] have emerged as powerful nuclear reprogramming systems [16]. Episomal plasmid-based reprogramming allow derivation of integration-free iPSCs, but the efficiency is low and the aneuploidy rate is elevated compare with other system. Additionally, the vectors persist in iPSC over 12 passages [17]. SeV-based reprogramming vectors allow efficient derivation of integration-free iPSCs through robust and sustained expression of reprogramming factors (RFs) [13, 16, 18], and remains for up to 12 passages in the iPSC after reprogramming. In contrast, after mRNA-based reprogramming, all iPSC lines are free of reprogramming agents by passage 5, but this system requires repetitive transduction of the somatic cells in the early stage of reprogramming.
In 2015, we published the proof of principle that measles virus (MV), a human negative strand RNA Paramyxovirus, with a proven safety vaccination record in nearly a billion children [19] can be used for nuclear reprogramming [20]. We had generated a MV virus identical to the Food and Drug Administration-approved Moraten vaccine strain and generated a vector deficient in its attachment protein expressing the RF OCT4 (MV(OCT4)). Using this vector, and three lentiviral vectors (LVs), each expressing SOX2, KLF4, or cMYC, we showed that MV(OCT4) could lead to the production of iPSC with identic characteristic than iPSCs produced with four LV vectors system. This MV(OCT4)-derived iPSC were able to re-differentiate into the three germ lineages, and we also demonstrate that the MV(OCT4) was eliminated quickly from the established iPSC after reprogramming, suggesting a great potential for the MV reprogramming platform [20]. MV is a negative strand RNA human Paramyxovirus, with its life cycle/RNA replication occurring in the cytoplasm without DNA intermediates [21]. This property rules out the risk of the reprogramming vector genome integration into the host genome, as shown for SeV [22, 23]. Moreover, genetically engineered, replication-competent MV vaccine lineage-based viruses are safe even when delivered at very high doses in humans [24].
We present here the production of a one-cycle MV vector expressing the four RFs, OCT4, SOX2, KLF4, and cMYC, in one single measles genome and the first application of this vector system for nuclear reprogramming. We reprogrammed human fibroblasts into iPSCs and characterized these MV-derived iPSCs for expression of stem cell makers, induction of endogenous pluripotency-associated genes, and pluripotency. In summary, we have developed a new vector system based on the MV vaccine platform, which could accommodate a large amount of genetic material demonstrating the extreme tolerance and flexibility of the MV vector delivery system. This new reprogramming system would provide a safe and efficient platform for the production of human iPSCs.
Materials and methods
Human cell line used in this study was purchased from the American Type Culture Collection and did not require consent or the need for Mayo Institutional Review Board approval. The Mayo Clinic Institutional Biosafety Committee has approved all vectors, viruses, and experiments performed in this study.
Maintenance of cell culture
All Vero (ATCC #CCL81), helper 293–3-46-H2 cells [20] and Vero-H2 [20] cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (GE Life Sciences HyClone, SH30022.01) containing 10% fetal calf serum (FCS, Life Technologies, #10437–028), 1% penicillin/streptomycin (P/S, Corning Mediatech, 30–002-Cl) (DMEM-10). Helper 293–3-46-H2 cells were cultured with 1.2 mg/ml G418 (Cardinal Healthcare, MT61234RG) in addition to DMEM-10. Human BJ cells, neonatal foreskin human cells (ATCC #CRL 2522) were maintained in DMEM containing 10% embryonic stem cell qualified FCS (ES-FCS, Life Technologies, #16141–079) containing 0.1 mM non-essential amino acids (Corning Mediatech, 25–025-Cl) and 1% P/S (media 1). iPSCs were maintained in 80% Pluriton (Stemgent, #00–0070), 20% mTeSR1 (STEMCELL Technologies, #05851), and 1% P/S (media 2). When 80–90% confluency was reached, iPSC were splitted using ReLSR, following manufacturer’s instructions, and propagated as clumps in new matrigel-coated plates. iPSC were frozen at passages 2, 5, 10, and 20 in CryoStor CS10 (BioLife Solutions, 210102). All cell lines stated above were cultured in humidified atmosphere with 5% CO2 at 37 °C under atmospheric oxygen conditions.
Full-length MV cDNA production
In this study, the complementary DNAs (cDNAs) used to produced the MV vectors were previously produced with the coding capacity identical to that of a MV Moraten/Schwartz vaccine strain (GenBank accession numbers AF266287 and AF266291 [25–27]). Full-length cDNA vector p( + ) MVvac2ΔH(OSK)(GFP)H (MV3F, Fig. 1a) was produced by inserting the codon optimized sequence encoding OCT4, SOX2, KLF4 (OSK, sequence provided in supplementary data S1) instead of the H gene using the EcoRV and SmaI restriction site. The OSK fragment (Supplement Fig. 1) was PCR amplified using the forward 5ʹ-ATGATTACGCGTGATATCGGATCCGCCACCATGG-3ʹ and reverse 5ʹ-AATCATACTAGTTCGAGACGTCTGCGCGCGATATCTATCAGAAGTGCCTTTTCATG-3ʹ primers to introduce two EcoRV sites. A EcoRV fragment was cloned into a EcoRV–SmaI digested intermediate vector pCG containing the PacI–SpeI fragment from full-length pB( + )MVvac2(GFP)H [28] (MV, Fig. 1a). The sequence encoding the cMYC gene (Supplement Fig. 1) was PCR amplified using the forward 5ʹ-GCCCATCAACGCGTCTGGATTTTTTTCGGGTAG-3ʹ and reverse 5ʹ-ACTAGTTCGAGACGTCGCGCGCTTACGCACAAGAGTTCCG-3ʹ primers to introduce the MluI and BssHII restriction sites, then cloned in the ATU in H position, replacing the GFP. The PacI–SpeI fragment was then cloned back into the MV full-length genome containing a GFP in an additional transcription unit (ATU) in front of the N gene. The ATU in N position is identical to the one from the original MVeGFP described previously [29]. The resulting full-length vector was called p( + ) MVvac2 (GFP)uNΔH(OSK)(cMYC)H (MV4FΝ, Fig. 1a). All cloning were performed in accord with the “rule of six” and verified by sequencing.
Fig. 1.
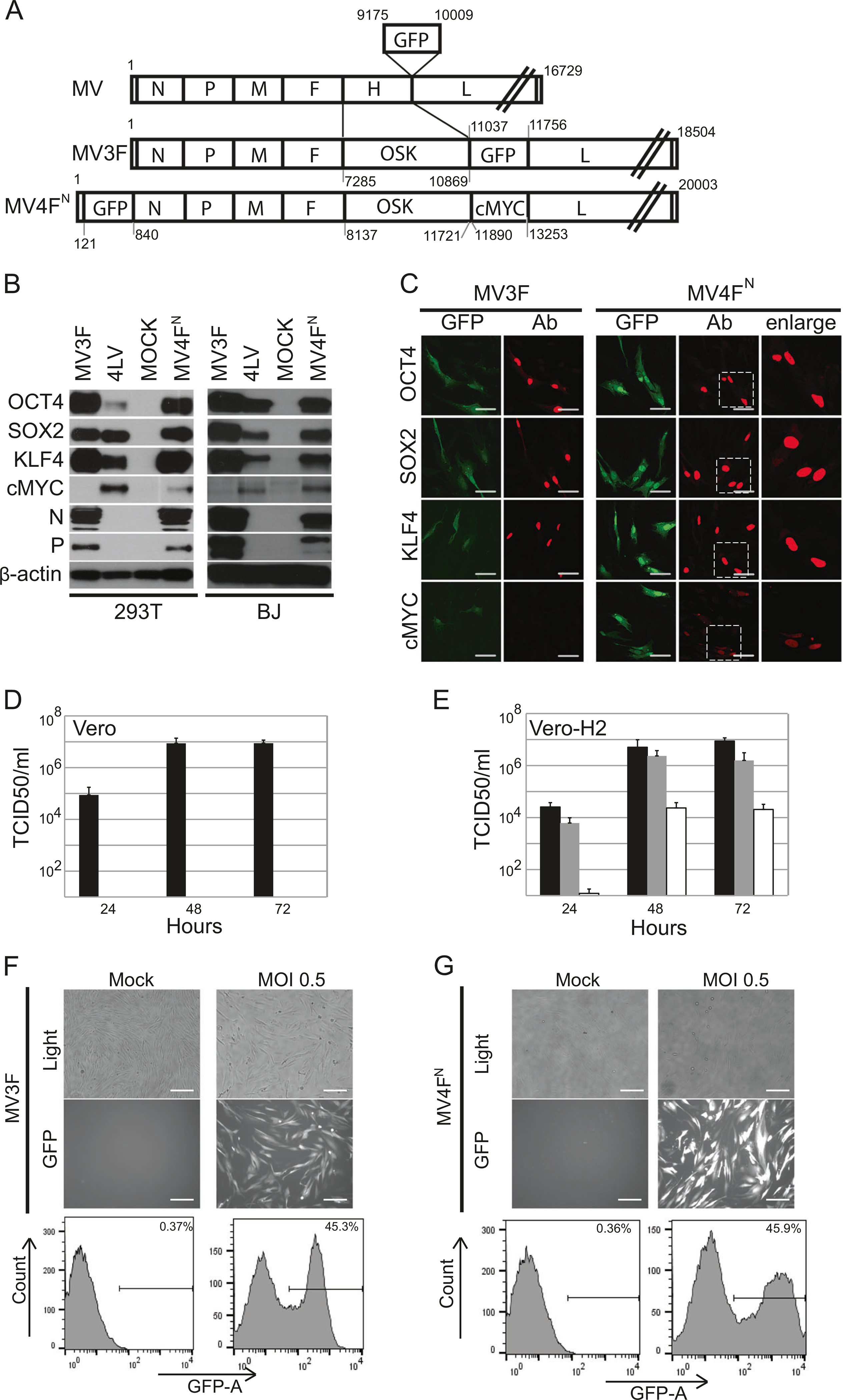
Generation and characterization of a one-cycle measles virus vector expressing OCT4, SOX2, KLF4, cMYC, and GFP. a Schematic representation of MV(GFP)H (top), MV3F (middle), and MV4FΝ (bottom). The measles virus (MV) antigenome (plus strand) is represented with its 5′ end on the left; the six genes are indicated by capital letters, numbers represent nucleotides position with number 1 being the first nucleotide of measles genome. b Immunoblot analysis of OCT4, SOX2, KLF4, cMYC expression in 293T and human fibroblast (BJ) transduced cells with the indicated vector. Antibodies against the indicated proteins were used. Uninfected BJ and 293T cells (MOCK) were used as controls. Cells transduced with LVOCT4, LVSOX2, LVKLF4, and LVcMYC (4LV) were used as positive control. β-Actin was used as loading control. c Immunofluorescence analysis of OCT4, SOX2, KLF4, cMYC expression in transduced human fibroblast (BJ) cells with the indicated vector. Cells were stained with indicated antibodies (red). GFP (green) was expressed during infection. Scale bars represent 50 μm. d, e Titers of cell-associated and released virus produced upon infection of Vero (d) and Vero-H2 (e) cells with MV3F (gray columns), MV4FN (white columns) or MV (black columns), determined at 24 h, 48 h or 72 h post-infection. Values and error bars reflect the mean and standard deviation of at least two biological replicates. f Level of transduction of human fibroblasts with MV3F. Cells (BJ) were infected with MV3F or mock infected. Forty-eight hours post-infection, pictures were taken under phase contrast (top panels), fluorescence (bottom panels) or quantified by flow cytometry (bottom panel). g Level of transduction of human fibroblasts with MV4FN. Cells (BJ) were infected with MV4FN or mock infected. Forty-eight hours post-infection, pictures were taken under phase contrast (top panels), fluorescence (middle panels) or quantified by flow cytometry (bottom panel). Scale bars represent 0.2 mm
Cells and viruses production
Vero-H2 and helper 293–3-46-H2 cell lines were previously described [20]. Recombinant MVs were produced according to previously published procedures [20]. In brief, helper 293–3-46-H2 cells were transfected using calcium phosphate precipitation (ProFection kit, Promega, E1200) with two plasmids encoding for the MV genome and MV polymerase (pEMCLa). Three days after transfection, the helper cells were overlaid on Vero-H2 cells. Appearance of infectious centers was monitored by observing green fluorescent protein (GFP) expression under fluorescence microscope. Single viruses were then picked and propagated on Vero-H2 cells.
For virus stock preparation, Vero-H2 cells were infected at a multiplicity of infection (MOI) of 0.05 in OptiMEM (Life Technologies, 221705) for 2 h at 37 °C. DMEM-10 medium was then added on top and transferred to 32 °C until 95% of the cells expressed GFP. Cell culture media were removed and cells were scraped in OptiMEM. Viral particles were released by two freeze–thaw cycles. Titers of virus stocks were determined by 50% end-point dilution (tissue culture infectious dose 50, or TCID50) on Vero-H2 cells using the Spearman–Kärber method [30].
Infection efficiency quantification by flow cytometry
BJ cells (7 × 104) were infected with MV3F or MV4FN at MOI of 0.25 or 0.5 dilutes in OptiMEM or mock infected and submitted to spinoculation at 1100 rpm for 1 h at room temperature. The inoculum was removed, the cells washed with PBS and 1 ml of media 1 was added. Cells were collected after 48 h post-infection by washing once with PBS and detached with trypsin (Corning Mediatech, 25–053-Cl). After an additional wash with Phosphate Buffer Saline (PBS), cells were fixed with 0.5 ml of PBS-2% paraformaldehyde (PFA). Percentage of GFP expression was determined using flow cytometry performed on Becton Dickinson FACS Calibur (Becton Dickinson) and analyzed by FlowJo_V10 software.
MV vector infectivity
Vero or Vero-H2 cells (4 × 105) were infected with MV3F, MV4FN, or MV control virus at a MOI of 0.05 for 2 h at 37 °C in OptiMEM. After 2 h, the inoculum was removed and cells were washed once with PBS and 1 ml of medium was added to the cells. Cells were then incubated at 37 °C until time of collection. Cells were collected in the 1 ml of supernatant at 24, 48, and 72 h post-infection. Samples were freeze–thawed once to release cell-associated virus. Virus titers were determined using Speaman–Kärber method.
Immunostaining and confocal microscopy
BJ cells (2 × 104) were seeded in matrigel (Corning, #354277) coated chamber slides (Lab Tek II) and infected with MV3F or MV4FN at 0.5 MOI. Thirty-six hours post-transduction, cells were washed with PBS and fixed in PBS-2% PFA for 15 min. Cells were then permeabilized in PBS–2% PFA–0.1% Trition X-100 for 15 min, washed three times with PBS and incubated overnight in blocking solution (PBS-5% FCS). Overnight incubation with primary antibodies: rabbit anti-OCT4 (Cell Signaling Technology, #2750, 1:200), rabbit anti-SOX2 (Cell Signaling Technology, #2748, 1:200), mouse anti-KLF4 (Stemgent, 09–0021, 1:200), mouse anti-cMYC (Santa Cruz Biotechnology, sc-40, 1:200) were performed, followed by three PBS washes and corresponding Alexa 594 secondary antibodies, anti-rabbit or anti-mouse (Life Technologies, A21207 or A21203, 1:1000), incubation for 1 h. All antibodies were diluted in blocking solution. After five washes with PBS, cells were mounted with Prolong Gold Antifade reagent containing DAPI (4’,6-diamidino-2-phenylindole; Life Technologies, P36930) and analysis were performed using Zeiss LSM 780 confocal microscope followed by analysis with Zen black software (Zeiss). Further processing was performed with Adobe Photoshop CS6 (Adobe Systems) for color contrast.
For analysis of undifferentiated iPSCs, iPSCs were seeded on matrigel-coated chamber slides. Fixation, permeabilization, washing, mounting, and analysis were done as stated above. Primary antibodies staining were performed with mouse anti-SSEA-1, SSEA-4, TRA1–60, TRA1–81 (Millipore, #SCR001, 1:100), rabbit anti-OCT4 (1:200), rabbit anti-SOX2 (1:200), and NANOG (Abcam, #ab21624, 1:100) diluted in blocking solution. Corresponding AlexaFluor 488 anti-mouse or anti-rabbit (Life Technologies, A21202 or A21206, 1:1000) were used for secondary antibody staining. Confocal microscopy analysis was performed as described above.
Immunoblot analysis of cell extracts
BJ (2.1 × 105) and 293 T (3 × 105) were transduced with 4LV (LV OCT4, LV SOX2, LV KLF4, and LV cMYC), MV3F or MV4FN vectors at MOI 0.5. After 36 h, cells were processed according to previously described procedures [25]. Samples were fractionated on 4–15% sodium dodecyl sulfate-polyacrylamide gels (Bio-Rad Laboratories, 345–0028, 345–0027) and transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore, IPVH00010). Membranes were blocked for 2 h in 10% dry milk and incubated with the following primary antibodies diluted in 5% milk overnight at 4 °C: rabbit anti-OCT4 (1:1000), rabbit anti-SOX2 (1:1000), goat anti-KLF4 (R&D Systems, AF3640, 1:2000), mouse anti-cMYC (ThermoFisher Scientific, MA1–980, 1:1000), mouse anti-N (Cl25 [20], 1:5000), rabbit anti-P254 (P254 [31], 1:5000). After three washes in TBS-Tween 1% buffer, membranes were incubated with peroxidase-conjugated anti-mouse (Calbiochem, 401215, 1:2500), peroxidase-conjugated anti-rabbit (Jackson ImmunoResearch, 111–035-003, 1:7500), peroxidase-conjugated anti-goat (Jackson ImmunoResearch, 1:5000), or loading control peroxidase-conjugated anti-β-actin (Sigma-Aldrich, A3854 1:10000) for 2 h at room temperature. After three washes in TBS-Tween 1%, ECL2 substrate (Thermo Pierce, PI80196) was then used to detect presence of specific antibody binding.
Reprogramming of human foreskin fibroblasts
BJ foreskin cells (7 × 104) were seeded on matrigel-coated plates. Cells were either transduced with MV3F or MV4FN at 0.5 or 0.25 MOI in OptiMEM. Cells transduced with MV3F were co-transduced with LV cMYC (50 μl). The amount of LV cMYC was determined after reprogramming of BJ cells using a 4LV system, using different volume of each single LV vectors in order to get the best reprogramming efficiency. Cell and virus were subjected to spinoculation at 1100 rpm for 1 h at 25 °C, then the inoculum was left overnight on top of the cells. The day after, cells were washed once with PBS, and medium 1 added and was changed every 2 days until day 7 for MV3F and day 8 for MV4FN. Feeding media 1 was changed to media 2 and was changed every day thereafter. For reprogramming with small molecules (sm); SB431542 (Stemgent, 04–0010-05, 5 μM), PD0325901 (Stemgent, 04–0006-02, 0.2 μM), and Thiazovivin (Stemgent, 04–0017, 0.5 μM), small molecules were added to media 2 from day 7 to 14 according to concentration provided in previous studies [32]. iPSCs clones were picked on the basis of size and morphology, around days 25–35 and transferred individually on a matrigel-coated 12-well for further expansion and study. For each conditions, a minimum of two to five iPSCs from three independent reprogramming were analyzed.
Cellular and viral gene transcription
Confluent well (80%) of 12-well plate iPSCs were detached with versene (Life Technologies, 15040–066) and pelleted. Total RNA was isolated with Trizol reagent (Life Technologies, 221705) according to manufacturer’s protocols. RNA to cDNA reverse transcription was performed with EcoDry TM Premix (Oligo dT) kit (Takara Bio, 639543) using 1 μg of RNA. Semiquantitative PCR was done using Platinum Taq DNA polymerase (Life Technologies, 10966–026). Reaction mixture was made according to manufacturer’s protocols and all PCRs were performed using 1 μl of cDNA at 55 °C for 40 cycles. Primers for OCT4, SOX2, KLF4, NANOG, GDF3, hTERT, and cMYC pluripotency markers sequence were previously described [18]. OSK (Fwd: 5ʹAACTAGCATCGAGAACAG-3ʹ; Rev: 5ʹ-GAGTAGTCACGGACACG-3ʹ), viral cMYC (Fwd: 5ʹ-GTCGTACGTCGCGACTGG-3ʹ; Rev: 5ʹ-GTGACCGCAACGTAGG-3ʹ), N (Fwd: 5ʹ-CGGAGCTAAGAAGGTGGATAA A-3ʹ; Rev: 5ʹ-CAGTCCAAGAGCAGGATACATAG-3ʹ) and P (Fwd: 5ʹ-AGCTGCTGAAGGAATTTC-3ʹ; Rev: 5ʹ-CTACTTCATTATTATCTTCATC-3ʹ) primers were used to detect viral mRNA.
Quantitative PCR were performed with N (Fwd: 5ʹ-CGGAGCTAAGAAGGTGGATAAA-3ʹ’; Rev: 5ʹ-CAGTCCAAGAGCAGGATACATAG-3ʹ) and GAPDH (IDT PrimeTime Predesigned Primers, Hs.PT.58.40035104, NM_002046) using Bullseye EvaGreen qPCR Master Mix (MidSci, BEQPCR-R) with 1 μl of cDNA final reaction volume of 25 μl. As positive control, we performed a quantitative PCR on dilution of different amounts of MV vector in 1 μl of cDNA from 4LV-iPSC clone (4000, 40, 0.4 molecule of vector were used). The negative control was 1 μl of cDNA from 4LV-iPSC. Two technical replicates were performed for each sample. Measurements were collected using Applied Biosystems 7300 Real-Time PCR systems at 55 °C annealing temperature for 40 cycles. Relative gene expression of N mRNA and GAPDH were compared using 2^(−ΔCT) method.
Spontaneous differentiation assay
MV- and 4LV-derived iPSC clones were detached using ReLeSR™ (Stemcell Technologies, #05872) and cultured on low-adhesion plates in iPSC medium for 10 days to form embryoid bodies (EBs). After culturing for 10 days in suspension, EBs were moved to matrigel-coated chamber slides (LABTECKR II, 154526) and cultured in DMEM 20% ES-FCS for an additional 10 days to allow for differentiation. Differentiated cells were fixed, permeabilized, blocked and immunostained with either rabbit anti-FOXA2 (Millipore, #07–633, 1:100) for endoderm, chicken anti-β-III tubulin (Abcam, ab41489, 1:1000) for ectoderm, and goat anti-CD31 (Santa Cruz Biotechnology, sc-1506, 1:50) for mesoderm diluted in blocking solution overnight at 4 C. After three PBS washes, secondary antibodies AlexaFluor 594 anti-rabbit (1:1000), FITC anti-chicken (Jackson ImmunoResearch, 703–095-155, 1:500), Alexa Flour 488 anti-goat (Jackson ImmunoResearch, 705–546-147, 1:1000) were used on corresponding primary antibodies. Confocal microscopy analysis was performed as described above.
Guided differentiation assay
Guided differentiation assay were performed using STEMdiff™ Trilineage Differentiation Kit (Stemcell Technologies, #05230) according to manufacturer’s directions. iPSCs obtained through 4LVs and MV4FN reprogramming were seeded at full or half the recommended seeding density on matrigel-coated four-well chamber slides according to manufacturer’s protocol. Differentiation was carried out for 5 days for mesoderm and endoderm, and 7 days for ectoderm. Differentiated cells were analyzed by immunostaining using markers suggested by manufacturer’s protocol: rabbit anti-Nestin (Millipore, ABD69, 1:500) and mouse anti-Pax-6 (Abcam, ab78545, 1:100) for Ectoderm, rabbit anti-FoxA2 (1:100) and mouse anti-SOX17 (R&D Systems, MAB1924, 1:100) for Endoderm and mouse anti-CD56 (NCAM) (Stemcell Technologies, #60021, 1:100) and rabbit anti-Brachyury (R&D Systems, MAB20851, 1:75) for Mesoderm. AlexaFluor 488 anti-mouse (1:1000) and AlexaFluor 594 anti-rabbit (1:1000) secondary antibodies were used. Undifferentiated iPSCs were stained using the same antibodies and used as control. Staining and confocal microscopy analysis is performed as described above.
Gene expression analysis
Microarray analysis was conducted according to manufacturer’s instructions for the Affymetrix 3ʹ IVT Plus kit (Thermofisher Scientific, 902416). Briefly, RNA quality was assessed by Agilent Bioanalyzer (Agilent Technologies). Reverse transcription to second-strand cDNA was generated from 100 ng of high-quality total RNA. Subsequently, the products were in vitro transcribed to generate biotin-labeled cRNA. The IVT products were then bead-purified (Affymetrix), fragmented, and hybridized onto Affymetrix U133Plus 2 GeneChips® at 45 °C for 16 h. Subsequent to hybridization, the arrays were washed and stained with streptavidin–phycoerythrin, then scanned in an Affymetrix GeneChip® Scanner 3000 (Santa Clara, CA). All Affymetrix array experiments were carried out at the Mayo Clinic Genome Analysis Core. Control parameters were confirmed to be within normal ranges before normalization and data reduction was initiated using the GeneChipTM Command ConsoleTM Software. The .cel files were processed using Partek Genomics Suite software, version 6.6 Copyright ©; 2017 Partek Inc., St. Louis, MO, USA. The files were normalized using quantile normalization with a log probes using base 2. The differential expression was performed using the analysis of variance method in Partek. The scatter plots was generate using R script version 3.1.1. The heat map was generated using Excel software.
Cytogenetic analysis
Conventional cytogenetic analysis was performed on 20 metaphase cells of three iPSC clones and parental BJ cells. Chromosomes were banded following standard methods for high-resolution G-banding. Cells were captured and karyotyped using a CytoVision Karyotyping System (Genetix, New Milton, UK).
Statistical analysis
Reprogramming efficiency: results are expressed as mean of number clones per reprogramming (n = 3). Comparisons of repeated values between groups were performed using Student’s t-test. Significance was determined as *P ≤ 0.05.
Results
Generation of “one-cycle” MV vector expressing three or four RFs from a single genome
We generated two full-length cDNA genomes from MV (GFP)H ([28] and Fig. 1a, top genome, MV). MV3F encodes three RFs as a polycistronic gene OCT4-SOX2-KLF4 (OSK), and MV4FN encodes all four RFs with cMYC in the ATU located just downstream of OSK in the MV genome (Fig. 1a, middle and bottom genomes, MV3F and MV4FN, respectively). In both MV3F and MV4FN, the attachment protein H gene was substituted by OSK, to produce a replication-defective, “one cycle” vector. Vector particles were rescued and propagated in a helper/packaging cell line expressing the MV H protein [20].
We first verified expression and correct processing of OSK in 293T and human fibroblast. All three proteins OCT4, SOX2, and KLF4 were expressed and had the same apparent molecular weight as the proteins expressed in cells infected by corresponding LVs (Fig. 1b). We took advantage of the MV transcription gradient [33] to express cMYC at lower levels than the other three RFs in MV4FN-infected cells (Fig. 1b). All factors were expressed in the nuclei (Fig. 1c). We documented high viral titers in Vero cells infected with the replication-competent virus MV(GFP)H (Fig. 1d, black histograms), but no virus was produced in cells infected with the “single-cycle” vector MV3F or MV4FN (Fig. 1d, gray and white histograms). In contrast, cells expressing MV-H produced high titers of MV3F or MV4FN (Fig. 1e, gray and white histograms), confirming that these are “single-cycle” vectors in the absence of trans-supplementation of MV-H. Viral stocks of MV3F and MV4FN reached an average of 1 to 3 × 106 TCID50/ml. Efficiency of transduction was assessed and spinoculation of MV3F and MV4FN at MOI of 0.5 efficiently transduced 45 and 46% of human fibroblast 48 h post-transduction, respectively (Figs. 1f, g).
MV4FN vector can efficiently reprogram human fibroblasts into iPSC-like cells and is eliminated rapidly from the established iPSC
We first tested the reprogramming capacity of the OSK gene. Human fibroblasts were transduced with MV3F in the presence of a LV vector encoding cMYC following the protocol described in Fig. 2a. Three weeks post-transduction, GFP positive, sharp-edge, flat, and tightly packed iPSC-like colonies were found in the infected cells (Fig. 2b). Single cell-derived clones were picked and expanded. All MV3F-derived iPSC clones expressed human pluripotency-associated markers, SSEA-4, TRA-1–60, TRA-1–81, OCT4, SOX2, and NANOG, at passage 2 and after prolonged culture (at passage 25), indicating stability of the derived clones (Fig. 2c). Multi-lineages propensity of all MV3F-derived clones was confirmed by formation of EBs and spontaneous differentiation into mesoderm (CD31) endoderm (FOXA2), or ectoderm (β-III tubulin) (Fig. 2d, left, middle, and right panel, respectively). Thus, MV expressing a polycistronic OSK gene can reprogram fibroblasts into iPSCs when cMYC was trans-supplemented by a LV-cMYC co-infection.
Fig. 2.
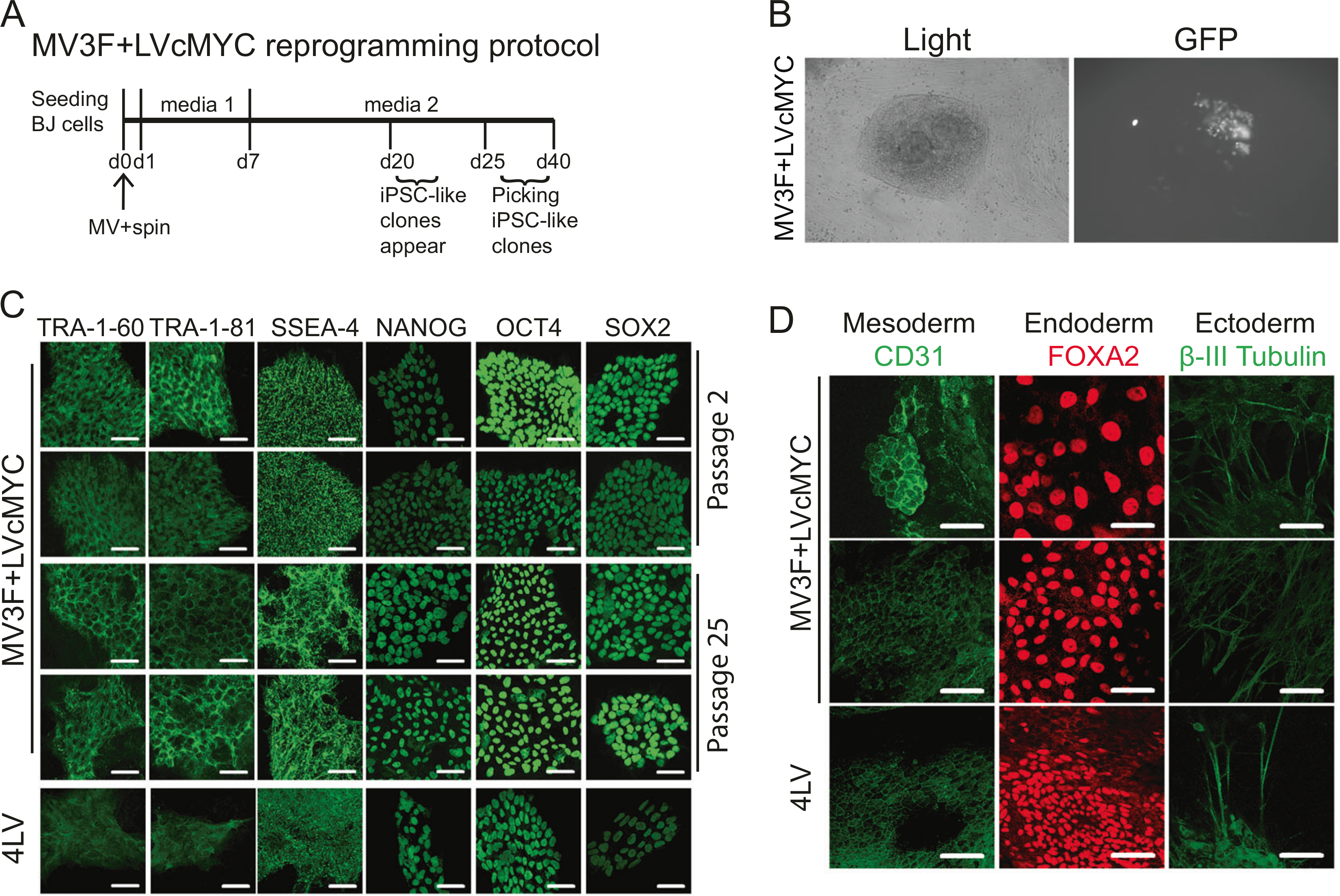
Generation of induced pluripotent stem cell (iPSC) using MV3F and elimination of the vector after reprogramming (a). Reprogramming schedule of human fibroblasts (BJ) transduced with MV3F + LVcMYC (MV3F). b Representative iPSC-like clone obtained 20 days post-transduction with MV3F + LVcMYC under light and fluorescence microscopy (left and right panel, respectively). c Two representative MV3F + LVcMYC-derived iPSC clones and one 4LV-derived iPSC control (4LV) were cultured under feeder-free conditions on a matrigel-based slide and examined for expression of human pluripotent stem cell markers by immunofluorescence. Passages 2 and 25 were analyzed. Scale bars represent 50 μm. d The same iPSC clones were analyzed by immunofluorescence for lineage markers for three germ layers (endoderm, mesoderm, and ectoderm). iPSC clones were spontaneously differentiated through embryoid body formation. Pluripotency of derived iPSC clones was verified by generation of cells of ectoderm (β-III tubulin, green, top row), endoderm (FOXA2, red, second row), and mesoderm (CD31, green, bottom row) upon spontaneous differentiation. Clones were tested at passages 4 and 20. Scale bars indicate 50 μm
Next, we assessed the ability of MV4FN to reprogram human fibroblasts. We also tested the impact of supplementation of small molecules, SB431542, PD0325901, and Thiazovivin, for improved nuclear reprogramming [32] (Fig. 3a). Although reprogramming at MOI 0.5 with MV4FN (or MV3F), with or without small molecules, led to noticeable toxicity over the first week, GFP-expressing cells remained visible over 30 days. Around 2 weeks post-transduction, GFP-positive, iPSC-like clones emerged in the fibroblast monolayer (Fig. 3b). The number of collectable clones for MV4FN ranged from 1 to 3 clones per 2.1 × 105 cells, whereas the number of clones observed for MV4FN with small molecules ranged from 2 to 4 times higher (Fig. 3c). The effect of the three small molecules was the same on the two MOI tested (Fig. 3c). These results indicate that MV4FN vector alone is sufficient to reprogram human fibroblasts, and that the addition of the small molecules improves reprogramming efficiency.
Fig. 3.
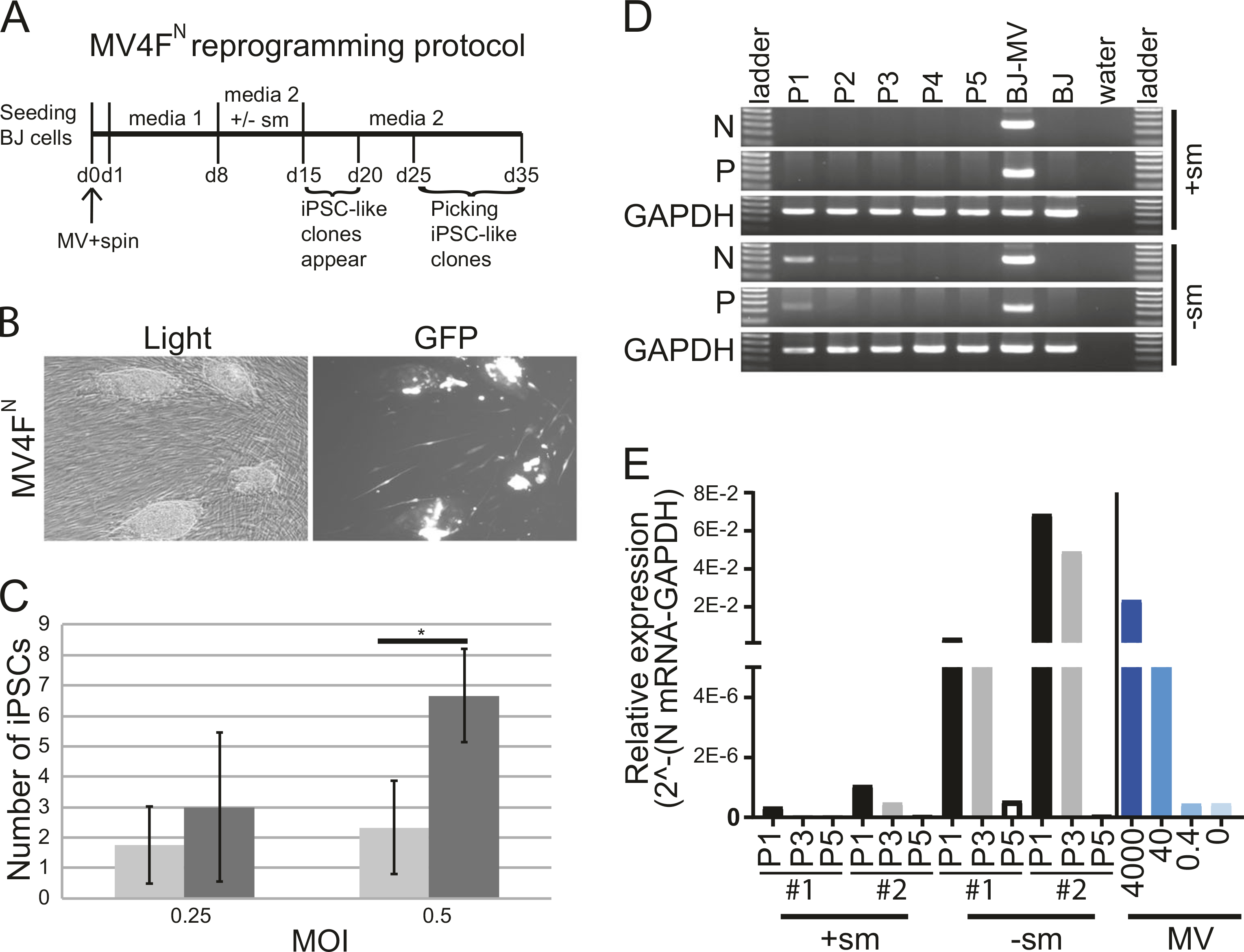
Generation of induced pluripotent stem cell (iPSC) using MV4FN and elimination of the vector after reprogramming (a). Reprogramming schedule of human fibroblasts (BJ) transduced with MV4FN. b Representative pictures of iPSC-like clones obtained ~15 days post-transduction with MV4FN under light and fluorescence microscopy. c Reprogramming efficiency: average number of iPSC clones produced after transduction of 2.1 × 105 BJ cells with MV4FN, with (black columns) or without (gray columns) small molecules. Values and error bars reflect the mean and standard deviation of three biological replicates. *P < 0.05 with small molecules versus without small molecules, Student’s t-test. d Loss of viral gene expression after passaging. Nucleoprotein (N) and Phosphoprotein (P) mRNA expression levels were analyzed in MV4FN -derived iPSC clones by semiquantitative RT-PCR at passages 1, 2, 3, 4, and 5. GAPDH is the cellular internal control, and water is the negative control. Controls: (BJ-MV) BJ cells infected with MV4FN, (BJ) BJ mock infected. e Elimination of the vector analysis. Quantitative RT-PCR analysis of the relative expression of the N mRNA in four iPSC clones obtained in presence ( + sm) or absence (−sm) of small molecules at passage 1, 3, and 5 (P1, P3, and P5; black, gray, and white columns, respectively). The right part of the graph (blue histograms) represents a quantitative PCR from 4000 to 0 molecules vector cDNA genome
We determined the persistence of the MV4FN vector genomes in derived iPSC clones after reprogramming. The two most abundant MV mRNAs, nucleoprotein (N) and phosphoprotein (P) were detected in all iPSC clones produced without small molecules up to passages P3 to P5 (Figs. 3d, e, −sm). All clones produced with small molecules had detectable mRNA only at P1 (Figs. 3d, e, +sm), indicating fast clearance of MV vector after reprogramming in presence of the small molecules (Fig. 3d). Quantitative analysis of four independent iPSC clones indicate that MV vector is eliminated by passage 5 in the iPSC after reprogramming (Fig. 3e). The fast clearance could be explained by the initial low MOI of transduction and by the fact that the small molecules favor fast multiplication of the iPSCs therefore diluting the vector, as shown by the loss of GFP expression in the iPSCs (Fig. 3b).
The MV4FN-derived iPSC-like clones displayed characteristics of human iPSC
The MV4FN-derived clones were characterized for expression of markers of pluripotency. Over 20 clones in total were tested (6 without small molecules and 14 with small molecules). All clones expressed pluripotency-associated markers, SSEA-4, TRA-1–60, TRA-1–81, OCT4, SOX2, and NANOG, at low passage, and remained stable over 20 passages (Fig. 4a). Induction of endogenous pluripotency-associated genes including OCT4, SOX2, KLF4, NANOG, GDF3, hTERT, and cMYC was comparable to those observed in the control 4LV clone (Fig. 4b). Their expression remained unchanged from passage 3 to passage 22 (Fig. 4b). Using specific primers for the viral and cellular RFs, we confirmed the reactivation of endogenous pluripotency genes, OCT4 and cMYC (Fig. 4c).
Fig. 4.
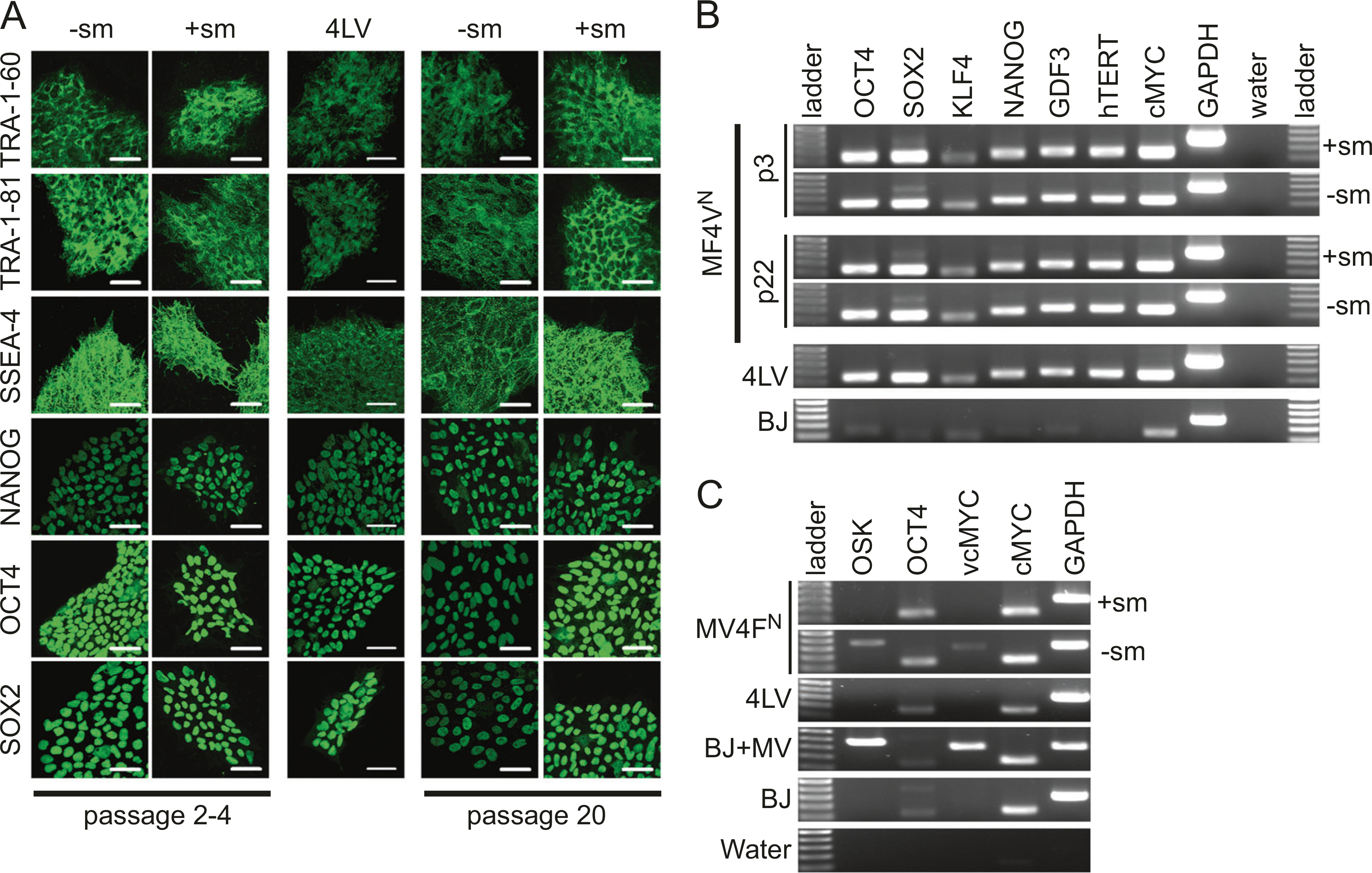
Characterization of MV4FN-derived induced pluripotent stem cell (iPSC) clones for pluripotency markers. a One representative MV4FN-derived iPSC clones obtained with or without small molecules ( + sm, −sm) and one 4LV-derived iPSC control (4LV) were cultured under feeder-free conditions on a matrigel-based slide and examined for expression of human pluripotent stem cell markers by immunofluorescence. Passages 2–4 and 20 were analyzed. Scale bars represent 50 μm. b RT-PCR analysis assessing transcription of key pluripotency-associated genes (OCT4, SOX2, KLF4, NANOG, GDF3, hTERT, cMYC) using total cellular RNA of two representative iPSC clones obtained with or without small molecules at passages 3 and 22. GAPDH is the cellular internal control, and water is the negative control. c RT-PCR analysis assessing transcription of viral and cellular genes OCT4, and cMYC were analyzed using total cellular RNA of one representative iPSC clones obtained with or without small molecules at passages 1. GAPDH is the cellular internal control, and water is the negative control. BJ cells and measles virus (MV)-infected BJ cells were used as negative and positive control of viral OCT4 and cMYC
To confirm that the MV-derived iPSC-like clones are pluripotent, we further characterized MV4FN-derived clones for their propensity to differentiate into the three germ layers: endoderm, ectoderm, and mesoderm. All MV4FN-derived iPSCs could form EBs when cultured in suspension in vitro and differentiate spontaneously into ectoderm, endoderm, or mesoderm (Fig. 5a, left two rows). They maintained their differentiation propensity over 20 passages, indicating stability of the pluripotency state (Fig. 5a, middle two rows). Following guided differentiation, all iPSCs tested differentiated into the ectodermal, endodermal, or mesodermal pathways (Fig. 5b) further confirming multi-lineage differentiation propensity of all MV4FN-derived iPSCs.
Fig. 5.
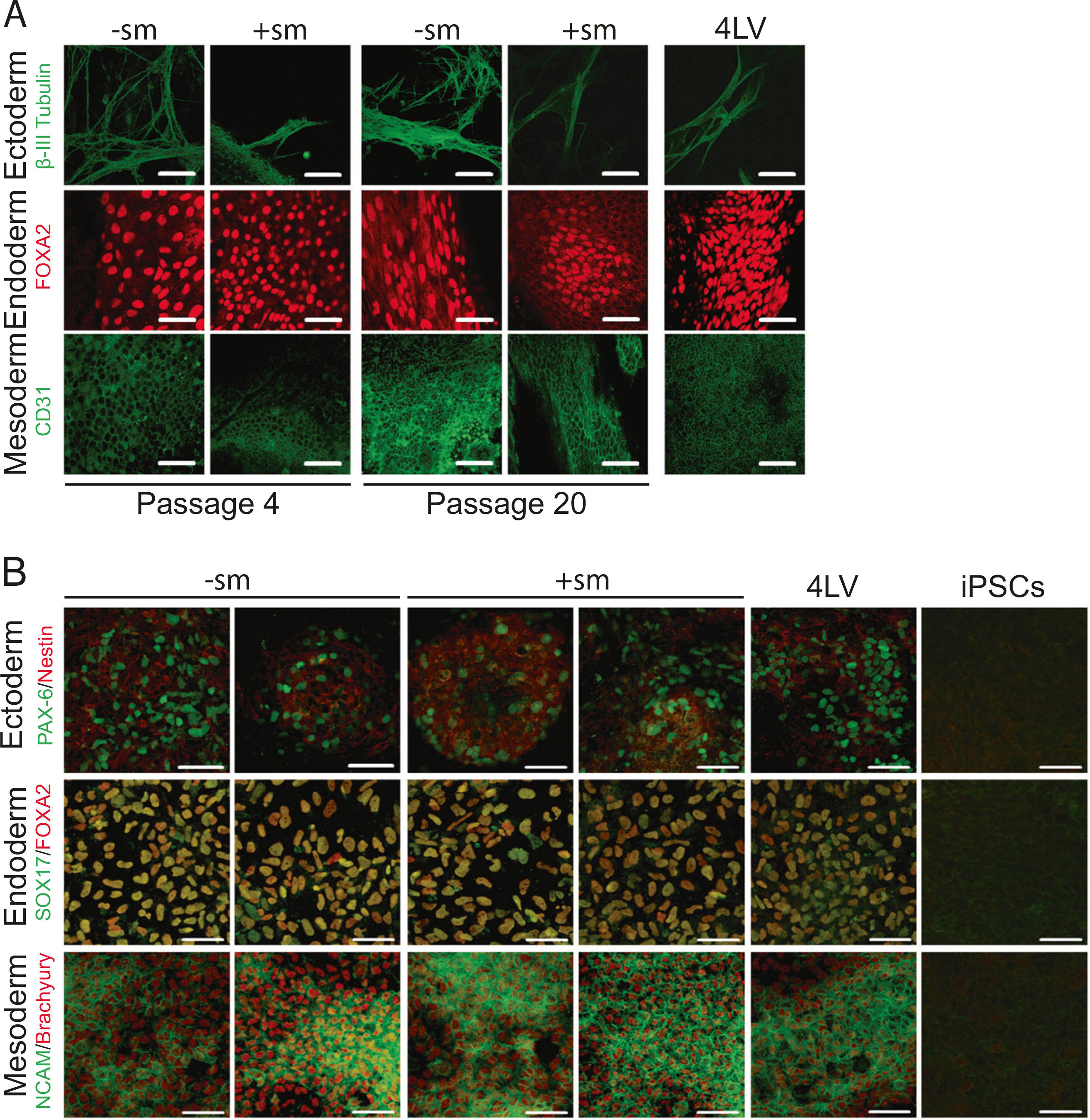
Spontaneous and guided differentiation of the MV4FN-derived induced pluripotent stem cell (iPSC) clones. a Representative MV4FN-derived iPSC clones obtained with or without small molecules (+sm, −sm) and one 4LV-derived iPSC control (4LV) were analyzed by immunofluorescence for lineage markers for three germ layers (endoderm, mesoderm, and ectoderm). iPSC clones were spontaneously differentiated through embryoid body formation. Pluripotency of derived iPSC clones was verified by generation of cells of ectoderm (β-III tubulin, green, top row), endoderm (FOXA2, red, second row), and mesoderm (CD31, green, bottom row) upon spontaneous differentiation. Clones were tested at passages 4 and 20. Scale bars indicate 50 μm. b Representative MV4FN-derived iPSC clones obtained with or without small molecules (+sm, −sm), and one 4LV-derived iPSC control (4LV) were analyzed by immunofluorescence for lineage markers for three germ layers (endoderm, mesoderm, and ectoderm). iPSC clones were differentiated through guided differentiation using the STEMdiff™ Trilineage Differentiation kit. Pluripotency of derived iPSC clones was verified by generation of cells of ectoderm (Nestin [red] and PAX-6 [green], top row), endoderm (FOXA2 [red] and SOX17 [green], second row), and mesoderm (NCAM [green] and Brachyury [red], bottom row) upon guided differentiation. Clones were tested at passage 4. Control staining was done on not differentiated iPSCs (right three panels, iPSCs). Scale bars indicate 50 μm
Finally, we determined the global gene expression profiles of MV4FN clones (GSE122790, GEO DataSets, Boston, MA) and compared it against parental fibroblast cells and H9 human ES cells (GSM551202; GEO DataSets, Boston, MA). Scatter plot analysis demonstrated that the transcriptome of MV4FN-derived clones showed higher similarity to those of ES than parental fibroblast cells (Figs. 6a, b). Heat map analysis of differentially expressed genes further confirmed that gene expression patterns of derived iPSC clones were similar to those of human ESCs, but highly divergent from control human fibroblast cells (Fig. 6c). The transcriptome of three independent MV4FN-derived iPSC clones showed striking similarity to each other. Thus, global gene expression profiles support a high degree of similarity in transcriptome between MV4FN-iPSCs and human ES cells. Karyotyping analysis of derived iPSC clones was performed at passages 5–7. All iPSC clones tested have normal diploid karyotypes (Fig. 6d).
Fig. 6.
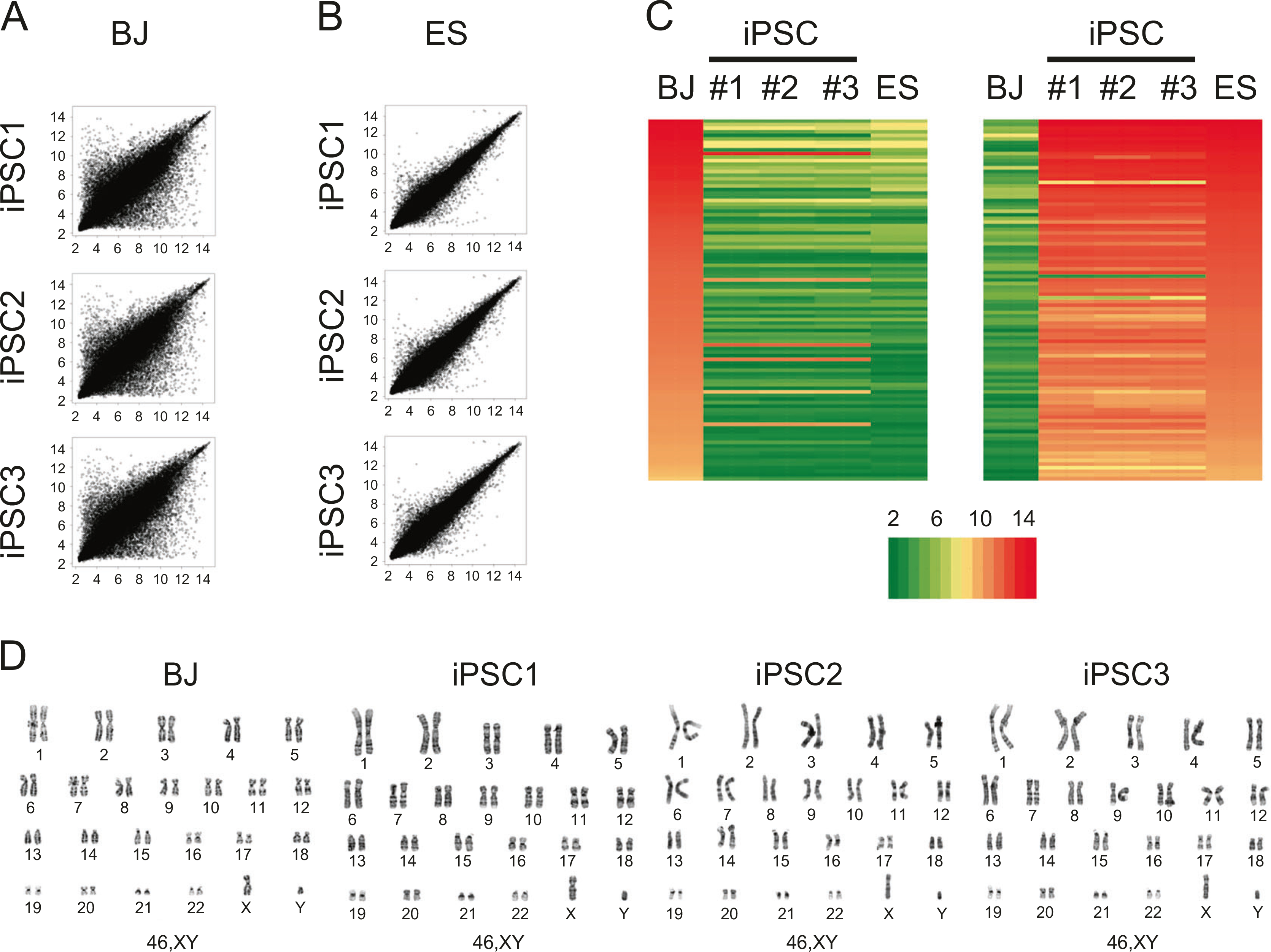
Global gene expression comparison between measles virus (MV)-derived induced pluripotent stem cell (iPSC) and human ES cells. a Global gene expression patterns were compared between human fibroblast (BJ) and three MV4FN-iPSC clones. b Global gene expression patterns were compared between human embryonic stem (ES) cells (H9, GSM551202) and the same three MV4FN-iPSC clones (GSE122790). c Heat map analysis of human fibroblast (BJ), MV4FN-iPSC clones (iPSC1, iPSC2, and iPSC3) and human ES cells (ES). Expression of genes that are differentially expressed between BJ and iPSC clones. (Right) Top hundred human ES cell-enriched probe sets; (left) top hundred fibroblast enriched probe sets. The color key indicates the color code gene expression in log2 scale. d G-banding chromosome analysis of parental BJ cells and the three same MV4FN-iPSC clones, iPSC1, iPSC2, and iPSC3
Discussion
Our results show that a MV vector can express the four RFs and a reporter gene from a single genome and can efficiently reprogram human fibroblasts into iPSCs. The reprogramming of somatic cells requires a low MOI and the vector genome is quickly eliminated from the established iPSCs. The MV4FN-derived iPSCs have similar characteristic than LV-derived iPSCs. Our MV4FN system provides a novel, safe reprogramming platform for efficient transgene-free iPSC derivation, amenable for timely clinical applications.
Viral vectors are typically engineered to express a transgene of interest. They are structurally similar to their wild-type counterparts but lack some or all of the viral genes impeding or obliterating their ability to replicate. There are two major considerations for the use of viral vector in humans. The first of which is safety. Some platforms, such as Ad vectors, have been tested extensively in human clinical trials for many years and are accepted as being safe for human use [34]. Other platforms, such as Vesicular Stomatitis Virus or SeV, are still in their infancy and the safety in humans remains to be determined [34]. As more clinical trials take place, we will gain a better understanding of these vectors and their performance in humans. The second consideration is the vector’s genomic capacity for a transgene insert. Depending on designs of viral vectors, their packaging capacity can range from <1 kb to 35 kb [34]. These must all be factored together to determine the ideal virus vector that is suitable for use in humans, but also with reasonable transgene delivery efficacies. Thus, we need to select appropriate viral vector systems depending on applications.
The MV4FN, used in this study, is based on the MV vaccine stran moraten/Schwartz and has a wide tropism, as it can use all three identified receptors, CD46, CD150, and Nectin-4 to enter the cells, conferring the vector a wide range of potential targets. Additionally, it was widely reported that the hemagglutinin of MV could be detargeted from its natural receptor and retargeted to a different protein leading to a retargeted vector for specific delivery. Recombinant MV vaccine has been used since several years as backbone for the development of oncolytic vector or prophylactic vaccine. MV has been shown to stably express up to 6 kb long heterologous antigen-coding sequence [35]. Here in MV4FN, we report the introduction of ~5.2 kb genetic sequence (OSK, cMYC, and GFP), however, the total increase of genome size is only 4 kb, as we deleted the H gene. In contrast, the reported cargo size of the SeV vector system is currently limited for reprogramming into iPSC is ~3.59 kb of additional genetic material (KSO polycistron) [36] introduced into a F-deficient SeV vector and to up to ~4.5 kb in the SeV Cas9 virus [37].
A wild-type strain of MV-ICB was modified with a segmented genome in one viral particle. The authors showed that such a virus could express up to four foreign genes from two independent segments [38]. Based on this virus, Hiramoto et al. reported the use of a segmented measles vector deficient in F gene to transfer multiple genes (OCT4, SOX2, KLF4, L-MYC PIN1, and GFP) into human cells [39, 40]. Efficient transduction of hematopoietic cells (HSC), T cells were reported. Moreover, they claims the successful generation of iPSC from human fibroblasts, T cells and HSC [39, 40]. While we have not yet tested MV4FN in adult fibroblasts or Peripheral Blood Mononuclear Cell (HSC or T cells), in view of the results from the Hiramoto et al. studies, we are confident that reprogramming will be successful with adult cells and these studies are underway.
MV vector as a reprogramming platform
One of the main advantages of negative strand RNA viral vectors over most other reprogramming systems is their replication in the cytoplasm without DNA intermediates [21], ruling out the potential risk of viral vector integration into host genome. Although SeV-based reprogramming system has been widely used for transgene-free iPSCs generation in basic and pre-clinical studies [13, 14], the popular, commercially available SeV reprogramming kit (ThermoFisher, #A16517) requires transduction with three independent SeV vectors at high MOIs of three to five for each vector and it can take >11 passages to eliminate the vector [17]. The MV-based reprogramming system offers efficient, integration-free reprogramming and as the vector expresses the four RFs from a single genome, and its reprogramming efficiency allows reduction of the vector dose to below 1 TCID50/cell. Importantly, iPSCs made with a clinically applicable MV platform would facilitate timely clinical translation of iPSC products in humans. Furthermore, our single MV4FN vector system would reduce the cost for clinical grade vector production. Since SeV vectors persistently infect iPSCs upon iPSC derivation, the current SeV vector system employs a temperature-sensitive SeV strain, allowing elimination of SeV genomes in SeV-derived iPSCs after cultivation at 38–39 °C for 5–7 days [13]. The rapid elimination of the MV vector genome from MV-generated and dividing iPSCs, without additional modifications to the vector or iPSCs, is an additional safety property that would facilitate the generation of clinical grade iPSCs. We noticed some toxicity associated with transduction of the cells with MV4FN and MV3F, most likely due to a high uptake of the vector and apoptosis. Toxicity related to reprogramming viral vector is not uncommon, as similar effects can occur with Sendai reprogramming and is mentioned in the reprogramming protocol of Cytotune 2.0. MV4FN is the first generation of the MV reprogramming vectors, and its reprogramming efficiency (~0.0006%) can seem to be low compare with more established and commercialized technologies, such as Episomal vector (0.0013%), SeV (0.077%), or mRNA (2.1%) [17]. However, we are currently working on the next generation of vectors and have already been able to increase reprogramming efficiency by a factor of at least 10–20 (Wang et al. unplublished).
The iPSC technology is facilitating autologous pluripotent stem cell-based regenerative medicine approaches. However, the first-in-human clinical trial with autologous iPSC-derived cells was suspended, at least in part, due to identification of mutations in the second patient’s iPSCs including two single-nucleotide variations and three copy-number variants, which are not detectable in the patient’s original fibroblast [41]. Since iPSCs frequently show chromosomal instability in culture [42], we show that MV reprogramming or iPSC culturing process does not appear to induced mutations. Nevertheless, the trial is now modified to test partially matched, well-characterized allogeneic iPSCs, instead of autologous iPSCs [43]. In addition to the potential safety concern, creating and characterizing autologous iPSCs can be costly, as nearly $900,000 was spent to develop and test the iPSCs for the first autologous iPSC trial [43], highlighting the challenge of autologous iPSC approaches and the requirement of a safe and reliable reprogramming platform for clinical autologous iPSC derivation. Our single MV reprogramming system offers efficient, integration-free iPSC reprogramming, via the pediatric vaccine platform with an over 30 years safety record [44]. Genetically modified MVs are also used in several cancer clinical trials, which have established the safety of genetically modified MVs in humans [24]. Thus, along with the reduced cost for manufacturing the single reprogramming vector, our novel cellular reprogramming system could support accelerated clinical applications of autologous iPSCs.
MV vector as a gene delivery platform
Although we found rapid elimination of the MV vector genome from derived iPSCs, without additional modifications to the vector or iPSCs, it is important to note that the MV vector supported the marker gene expression in non-reprogrammed human fibroblasts over 30 days post-transduction. This observation suggests that MV vector with the large packaging capacity could provide an unique RNA delivery platform, facilitating sustained multi-transgene delivery. We showed that the expression of four or five genes expressed from two or three position in measles genome, ATU before N gene (GFP), instead of the H gene (OSK) and ATU after H gene (GFP or cMYC) does not affect virus recovery and that viral propagation is only slightly decreased with the addition of fourth factor cMYC. When we produced a second MV4F vector, where the GFP was moved to the ATU after the P gene, MV4FP, the propagation of this vector was similar to MV3F or MV control, indicating that decrease in viral propagation is likely due to the position of GFP, but not to the addition of cMYC (Wang, Rallanbandi et al., unpublished). Measles transcription, like all Paramyxoviruses, follows a transcription gradient that can be useful to control the level of expression of the transgene of interest [21, 28]. In the case of vector with the GFP positioned in front of N, it could reduced the level of the N and P viral proteins, two important components for viral transcription and replication, which led to the reduction in viral propagation of this vector. Despite the positive of MV as a vector, there is one cavea that need to be addressed to improve MV as a vector for gene delivery, this is the cells toxicity that we observed in the 4–6 days post-transduction. The cells toxicity might be related to an accumulation of viral proteins leading cell death. While the toxicity might not affect the reprogramming, it will affect its use as efficient delivery vector and the addition of a mechanism to control viral protein production in the cells using microRNA might allow longer and sustained expression of the vector in cells.
In conclusion, we established a novel MV vector platform and used it for transgene-free iPSC derivation. This single MV reprogramming vector allows expression of four RFs, facilitating efficient iPSC derivation by a low dose vector transduction. Based on the long-standing safety record of the vaccine strain of MV, along with recent clinical experiences of genetically modified MV applications, in humans, this MV vector system offers a safe new vector platform for transient transgene expression.
Supplementary Material
Acknowledgements
We thank Andrew Badley, Roberto Cattaneo, Eva Galanis, and Jason Tonne for reading the manuscript. We thank Megan Rasmussen, Christopher Driscoll, Jason Tonne for technical assistance. We thank Debra Schultz from the Mayo Clinic Medical Genome Facility Gene Expression Core for performing the microarray and Vivekananda Sarangi from the Mayo Clinic Bioinformatics Core for helping with the gene expression analysis. We thank the Mayo Clinic Cancer Center for the use of the Cytogenetics Core, which provided karyotyping analysis services. Mayo Clinic Cancer Center is supported in part by an NCI Cancer Center Support Grant 5P30 CA15083-45. We also thank Todd DanDeWalker, Tony Goble, and Patricia T Greipp for cytogenetic analysis.
Funding This work was supported by Mayo Center for Regenerative Medicine (PD), Mayo Graduate School, and the National Institutes of Health (R21AI105233 to PD). This publication was also made possible by CTSA Grant Number UL1TR000135 and UL1TR002377 from the National Center for Advancing Translational Sciences (NCATS), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIH.
Footnotes
Conflict of interest PD and YI are inventors on a patent application (WO2018064460A1) for the content of the manuscript. The remaining authors declare that they have no conflict of interest.
Compliance with ethical standards
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information The online version of this article (https://doi.org/10.1038/s41434-019-0058-7) contains supplementary material, which is available to authorized users.
Data availability
The datasets used and/or analyzed during the current study are available from GEO DataSets, Boston, MA: GSE122790.
References
- 1.Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–72. [DOI] [PubMed] [Google Scholar]
- 2.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. [DOI] [PubMed] [Google Scholar]
- 3.Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–20. [DOI] [PubMed] [Google Scholar]
- 4.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2017;16:115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu C, Li L. Current reprogramming systems in regenerative medicine: from somatic cells to induced pluripotent stem cells. Regen Med. 2016;11:105–32. [DOI] [PubMed] [Google Scholar]
- 6.El-Khatib M, Ohmine S, Jacobus EJ, Tonne JM, Morsy SG, Holditch SJ, et al. Tumor-free transplantation of patient-derived induced pluripotent stem cells progeny for customized islet regeneration. Stem Cells Transl Med. 2016;5:694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyodo. First iPS cell transplant patient makes progress one yearon. In: Japan Times; [online], 2015. https://www.japantimes.co.jp/news/2015/10/02/national/science-health/first-ips-cell-transplant-patient-makes-progress-one-year/#.XE-WSc9Khgg [Google Scholar]
- 8.Kimbrel EA, Lanza R. Current status of pluripotent stem cells: moving the first therapies to the clinic. Nat Rev Drug Discov. 2015;14:681–92. [DOI] [PubMed] [Google Scholar]
- 9.Trounson A, DeWitt ND. Pluripotent stem cells progressing to the clinic. Nat Rev Mol Cell Biol. 2016;17:194–200. [DOI] [PubMed] [Google Scholar]
- 10.Mandai M, Watanabe A, Kurimoto Y, Hirami Y, Morinaga C, Daimon T, et al. Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med. 2017;376:1038–46. [DOI] [PubMed] [Google Scholar]
- 11.Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–12. [DOI] [PubMed] [Google Scholar]
- 12.Yu J, Hu K, Smuga-Otto K, Tian S, Stewart R, Slukvin II, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ban H, Nishishita N, Fusaki N, Tabata T, Saeki K, Shikamura M, et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc Natl Acad Sci USA. 2011;108:14234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fusaki N, Ban H, Nishiyama A, Saeki K, Hasegawa M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci. 2009;85:348–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren L, Manos PD, Ahfeldt T, Loh YH, Li H, Lau F, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7:618–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kudva YC, Ohmine S, Greder LV, Dutton JR, Armstrong AS, Genebriera De Lamo J. et al. Transgene-free Disease-specific induced pluripotent stem cells from patients with type 1 and type 2 diabetes. Stem Cell Transl Med. 2012;1:451–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlaeger TM, Daheron L, Brickler TR, Entwisle S, Chan K, Cianci A, et al. A comparison of non-integrating reprogramming methods. Nat Biotechnol. 2015;33:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thatava T, Kudva YC, Edukulla R, Squillace K, De Lamo JG, Khan YK, et al. Intrapatient variations in type 1 diabetes-specific iPS cell differentiation into insulin-producing cells. Mol Ther. 2013;21:228–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moszynski P Measles campaign’s “historic victory” for global public health. BMJ. 2007;334:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Driscoll CB, Tonne JM, El Khatib M, Cattaneo R, Ikeda Y, Devaux P. Nuclear reprogramming with a non-integrating human RNA virus. Stem Cell Res Ther. 2015;6:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lamb RA, Parks GD. Paramyxoviridae: the viruses and their replication. In: Fields BN, Knipe DM, Howley PM editors. Fields virology, 5th ed. Lippincott-Raven: Philadelphia; 2007. p. 1305–40. [Google Scholar]
- 22.Bitzer M, Armeanu S, Lauer UM, Neubert WJ. Sendai virus vectors as an emerging negative-strand RNA viral vector system. J Gene Med. 2003;5:543–53. [DOI] [PubMed] [Google Scholar]
- 23.Li HO, Zhu YF, Asakawa M, Kuma H, Hirata T, Ueda Y, et al. A cytoplasmic RNA vector derived from nontransmissible Sendai virus with efficient gene transfer and expression. J Virol. 2000;74:6564–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robinson S, Galanis E. Potential and clinical translation of oncolytic measles viruses. Expert Opin Biol Ther. 2017; 17:353–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Devaux P, von Messling V, Songsungthong W, Springfeld C, Cattaneo R. Tyrosine 110 in the measles virus phosphoprotein is required to block STAT1 protein phosphorylation. Virology. 2007;360:72–83. [DOI] [PubMed] [Google Scholar]
- 26.Parks CL, Lerch RA, Walpita P, Wang HP, Sidhu MS, Udem SA. Analysis of the noncoding regions of measles virus strains in the Edmonston vaccine lineage. J Virol. 2001;75:921–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parks CL, Lerch RA, Walpita P, Wang HP, Sidhu MS, Udem SA. Comparison of predicted amino acid sequences of measles virus strains in the Edmonston vaccine lineage. J Virol. 2001; 75:910–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.del Valle JR, Devaux P, Hodge G, Wegner NJ, McChesney MB, Cattaneo R. A vectored measles virus induces hepatitis B surface antigen antibodies while protecting macaques against measles virus challenge. J Virol. 2007;81:10597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duprex WP, McQuaid S, Hangartner L, Billeter MA, Rima BK. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol. 1999;73:9568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kärber G Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch Exp Pathol Pharmakol. 1931;162:480–3. [Google Scholar]
- 31.Toth AM, Devaux P, Cattaneo R, Samuel CE. Protein kinase PKR mediates the apoptosis induction and growth restriction phenotypes of C protein-deficient measles virus. J Virol. 2009;83:961–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lin T, Ambasudhan R, Yuan X, Li W, Hilcove S, Abujarour R, et al. A chemical platform for improved induction of human iPSCs. Nat Methods. 2009;6:805–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cattaneo R, Rebmann G, Schmid A, Baczko K, ter Meulen V, Billeter MA. Altered transcription of a defective measles virus genome derived from a diseased human brain. EMBO J. 1987;6:681–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lundstrom K Viral vectors in gene therapy. Diseases 2018;6:42–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Billeter MA, Naim HY, Udem SA. Reverse genetics of measles virus and resulting multivalent recombinant vaccines: applications of recombinant measles viruses. Curr Top Microbiol Immunol. 2009;329:129–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujie Y, Fusaki N, Katayama T, Hamasaki M, Soejima Y, Soga M, et al. New type of Sendai virus vector provides transgene-free iPS cells derived from chimpanzee blood. PLoS ONE. 2014;9: e113052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park A, Hong P, Won ST, Thibault PA, Vigant F, Oguntuyo KY, et al. Sendai virus, an RNA virus with no risk of genomic integration, delivers CRISPR/Cas9 for efficient gene editing. Mol Ther Methods Clin Dev. 2016;3:16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takeda M, Nakatsu Y, Ohno S, Seki F, Tahara M, Hashiguchi T, et al. Generation of measles virus with a segmented RNA genome. J Virol. 2006;80:4242–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiramoto T, Tahara M, Miura Y, Nakatsu Y, Kubota T, Kurita R, et al. Newly developed measles viral vector can efficiently transduce multiple genes into naïve T cells. Blood. 2014;124:4798. [Google Scholar]
- 40.Hiramoto T, Tahara M, Sakamoto C, Nakatsu Y, Kubota T, Ono H, et al. 5. Newly developed measles virus vector can simultaneously transfer multiple genes into human hematopoietic cells and induce ground state like pluripotent stem cells. In: The American society of gene & cell therapy. New Orleans, LA: Elsevier Inc.; 2015. p. pS2–S3. [Google Scholar]
- 41.Garber K RIKEN suspends first clinical trial involving induced pluripotent stem cells. Nat Biotechnol. 2015;33:890–1. [DOI] [PubMed] [Google Scholar]
- 42.Pera MF. Stem cells: the dark side of induced pluripotency. Nature. 2011;471:46–7. [DOI] [PubMed] [Google Scholar]
- 43.Normile D iPS cell therapy reported safe. Science. 2017;355: 1109–10. [DOI] [PubMed] [Google Scholar]
- 44.Pless RP, Bentsi-Enchill AD, Duclos P. Monitoring vaccine safety during measles mass immunization campaigns: clinical and programmatic issues. J Infect Dis. 2003;187(Suppl 1):S291–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used and/or analyzed during the current study are available from GEO DataSets, Boston, MA: GSE122790.