

SUMMARY
Natural killer (NK) cell effector functions are dependent on metabolic regulation of cellular function; however, less is known about in vivo metabolic pathways required for NK cell antiviral function. Mice with an inducible NK-specific deletion of Cox10, which encodes a component of electron transport chain complex IV, were generated to investigate the role of oxidative phosphorylation in NK cells during murine cytomegalovirus (MCMV) infection. Ncr1-Cox10Δ/Δ mice had normal numbers of NK cells but impaired expansion of antigen-specific Ly49H+ NK cells and impaired NK cell memory formation. Proliferation in vitro and homeostatic expansion were intact, indicating a specific metabolic requirement for antigen-driven proliferation. Cox10-deficient NK cells upregulated glycolysis, associated with increased AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) activation, although this was insufficient to protect the host. These data demonstrate that oxidative metabolism is required for NK cell antiviral responses in vivo.
Graphical Abstract
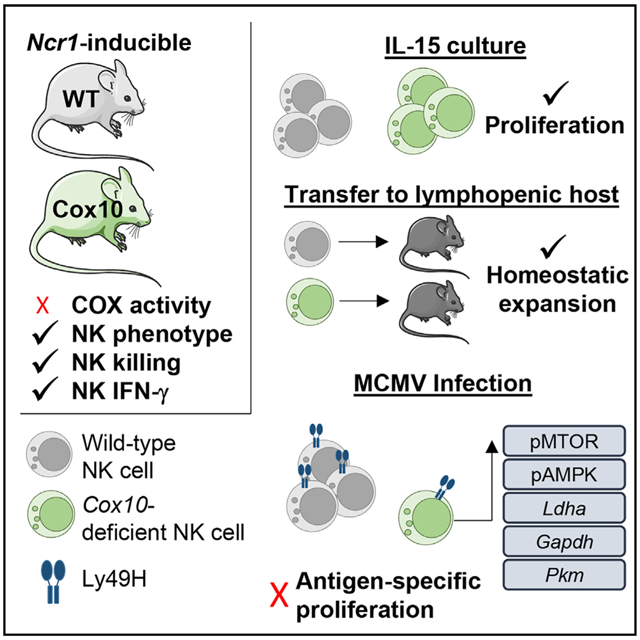
In brief
Cox10 encodes a key component of complex IV in the mitochondrial electron transport chain. Mah-Som et al. use an inducible model of Cox10 deletion in type I ILCs to demonstrate that COX10 is required for antigen-specific expansion of NK cells during viral infection but not cytokine-driven proliferation.
INTRODUCTION
Natural killer (NK) cells are cytotoxic innate lymphocytes important for the early response to viral infection and malignant transformation. NK cell activation is stimulated through inflammatory cytokines and activation of germline-encoded activating receptors on their cell surface (Yokoyama, 2013). NK cells are of increasing translational interest, and they are being used clinically in the context of cancer immunotherapy. There is also the potential to harness anti-viral properties of NK cells for treatment of infection and vaccination (Fehniger and Cooper, 2016; Liu et al., 2020; Myers and Miller, 2021; Sun and Lanier, 2018). While important triggers of NK cell activation and relevant intracellular signaling pathways are well understood, less clear are the metabolic regulators of NK cell function, particularly in the context of viral infection.
Immune cell metabolism and availability of metabolic fuels is increasingly recognized as essential for immune cell regulation and function (Buck et al., 2017; Kelly and Pearce, 2020). Oxidative phosphorylation (OXPHOS) and glycolysis, the breakdown of glucose, are two major metabolic pathways required for generation of energy and building blocks for cellular functions, including proliferation and expansion of antigen-specific cells. In immune cells, regulation of these and other metabolic pathways can dictate function, including memory formation and pro- and anti-inflammatory responses; for example, a switch to primarily OXPHOS and fatty acid oxidation associated with memory T cell formation (Buck et al., 2016).
Investigation of NK cell responses to murine cytomegalovirus (MCMV) has proven to be a useful tool for interrogation of NK cell anti-viral effectors functions in vivo. In the C57BL/6 genetic background, approximately 50% of NK cells stochastically express the germline-encoded Ly49H activating receptor, which recognizes the MCMV-encoded ligand m157. This recognition strategy allows for a robust NK cell response to infection, and in the absence of either Ly49H or m157 hosts are highly susceptible to MCMV infection (Arase et al., 2002; Cheng et al., 2008; Smith et al., 2002). Following infection with MCMV, there is expansion and contraction of Ly49H-expressing NK cells leading to a pool of innate “memory” cells that have enhanced recall response to MCMV and other stimuli (O’Sullivan et al., 2015b; Sun et al., 2009). In humans, a similar population of NK cells emerges following human CMV (HCMV) infection, frequently associated with the germline-encoded NKG2C activating receptor that can recognize HCMV peptides (Hammer et al., 2018). These human adaptive NK cells have enhanced function, particularly when triggered by antibodies, and they also expand and respond to other infections (Fehniger and Cooper, 2016).
Previous studies have suggested that metabolism is important for NK function during MCMV infection. For example, inhibition of mammalian target of rapamycin (mTOR), a regulator of metabolism, impairs multiple NK cell functions during MCMV infection in vivo (Donnelly et al., 2014; Nandagopal et al., 2014), while genetic deletion of mTOR inhibits NK cell differentiation (Marçais et al., 2014). Administration of glucose metabolism inhibitor during MCMV infection inhibited NK cell killing and led to susceptibility to infection (Mah et al., 2017). Previous in vitro work demonstrated that murine NK cells primarily use glucose-driven OXPHOS at rest (Keating et al., 2016; Keppel et al., 2015), and both murine and human NK cells require OXPHOS to produce the cytokine interferon (IFN)-γ in response to certain stimuli (Keating et al., 2016; Keppel et al., 2015).
To investigate the effects of oxidative metabolism on NK cell function, we generated mice with tamoxifen-inducible NK-specific deletion of Cox10 (Ncr1-Cox10Δ/Δ). The Cox10 gene encodes a farnesyltransferase involved in the synthesis of heme a, part of the catalytic core of cytochrome c oxidase (COX). COX, also known as complex IV, is a key component of the mitochondrial electron transport chain (ETC) and is a rate-limiting step of OXPHOS (Diaz, 2010). Defects in complex IV, including Cox10 deficiency, result in mitochondrial disease in humans (Antonicka et al., 2003; Valnot et al., 2000). Pediatric patients with mitochondrial disease have increased susceptibility to infection, in particular respiratory viral infections (Tarasenko et al., 2017). Cox10 was required for maximal T cell proliferation during viral infection in a mouse model of T cell-specific deletion of Cox10 (Tarasenko et al., 2017). These clinical observations suggest that defects in oxidative metabolism may alter the immune response to pathogens.
Antiviral responses of mice with NK-specific Cox10 deficiency were investigated during MCMV infection. Cox10-deficient NK cells developed normally, and in vitro functions are largely intact; however, we observed an NK cell-intrinsic impaired expansion of Ly49H+ MCMV-specific NK cells lacking Cox10.
RESULTS
Inducible NK cell-specific deletion of Cox10
An NK cell and ILC1-specific inducible model of Cox10 deletion, Ncr1-Cox10Δ/Δ, was generated by crossing mice with tamoxifen-inducible iCre recombinase driven by Ncr1 (Ncr1-iCreERT2) (Wagner et al., 2020) with mice carrying loxP sites flanking exon 6 of Cox10 (Diaz et al., 2005) and yellow fluorescent protein (YFP) Cre-reporter mice. Ncr1 encodes the cell surface receptor NKp46 and was chosen, as its expression early during NK cell development defines a murine NK cell compared to an NK precursor. Control mice with tamoxifen-inducible Ncr1-driven Cre and the same YFP reporter were used as controls (Ncr1-wild-type [WT]). For all flow cytometry-based experiments, NK cells from Ncr1-Cox10Δ/Δ and Ncr1-WT mice were gated on YFP+ NK cells to identify cells with Cre-recombinase activity (Figure 1A and representative flow gating in Figure S1). Following 2 days of tamoxifen chow, Cre activity was observed in 60%–80% of NK cells based on YFP expression (Figure 1A). Cox10 transcript was undetectable 3 days after induction in YFP-positive cells from Ncr1-Cox10Δ/Δ mice (Figure 1B). YFP-negative cells also had lower expression of Cox10 transcript, suggesting that there may have been Cre-mediated excision in some cells that had not yet expressed YFP protein.
Figure 1. Phenotype of NK cells in inducible Ncr1-Cox10Δ/Δ mice.

(A) Mice were fed tamoxifen-containing chow for 2 days and then analyzed after 3 or 7 days. Representative flow of Cre induction by the YFP reporter (day 3) is shown.
(B) Cox10 transcript (day 3), normalized to YFP− cells from Ncr1-Cox10Δ/Δ mice (Mann-Whitney test, n = 2–5 mice/group, two independent experiments, error bars represent SEM).
(C) Absolute numbers of YFP+ NK cells in the spleen, bone marrow (BM), and liver (two-way ANOVA, n = 8 mice/group, three independent experiments, pooled data shown, error bars represent SD).
(D) NK maturation of YFP+ cells by CD11b and CD27; stage 2 = CD27+CD11b−, stage 3 = CD27+CD11b+, stage 4 = CD27−CD11b+ (two-way ANOVA, n = 8 mice/group, three independent experiments with all p > 0.1000, except as shown, error bars represent SD).
(E) Normal expression of Ly49H (day 3, n = 4 mice/group, paired t test, error bars represent SEM).
(F) Degranulation of NK cells (CD107a) was similar (n = 9–10 mice/group, three independent experiments, two-way ANOVA, error bars represent SD).
(G) Killing of YAC-1 targets demonstrates similar cytotoxic capacity (day 3, n = 3 mice/group, two-way ANOVA, one experiment shown representative of three independent experiments, error bars represent SD).
(H) Percentage of IFN-γ+ NK cells following stimulation with cytokines (IL12+IL-15 or IL-12+IL-18) or activating receptors (anti-Ly49H, anti-Ly49H+IL-15, and anti-NK1.1) (mixed effects analysis with Sidak’s multiple comparisons test, n = 5–6 mice/group, two independent experiments, pooled data shown, error bars represent SD).
(I–M) Extracellular flux assay of YFP+ NK cells cultured with IL-2.
(I and J) Oxygen consumption rate (OCR; I) and OCR/ECAR (extracellular acidification rate) ratio (J) during Mito Stress test (Agilent Technologies).
(K) Cox10-deficient NK cells had a lower baseline OCR and higher ECAR, with a decreased OCR/ECAR ratio.
(L and M) Spare respiratory capacity (SRC; L) and maximal OCR (M) after carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) were similar (n = 3-5 technical replicates using n = 5–8 pooled mice, unpaired Student’s t test, error bars show SEM).
NK cells from Ncr1-Cox10Δ/Δ and Ncr1-WT mice were present in similar numbers in the spleen, bone marrow, and liver at days 3 and 7 after Cre induction (Figure 1C). However, total numbers of NK cells were increased in the bone marrow of Ncr1-Cox10Δ/Δ mice at day 7 (Figure 1C), suggesting increased production of Cox10-sufficient NK cells at this time point. Maturation of Cox10-deficient NK cells measured by CD11b and CD27 (Chiossone et al., 2009) demonstrated the presence of mature stage 4 (CD11b+CD27−) and intermediate stage 3 (CD11b+CD27+) and stage 2 (CD11b−CD27+) populations, but no stage 1 immature (CD27−CD11b−) cells. There were no differences in maturation stage of Cox10-deficient or WT NK cells, with the exception of increased stage 4 cells in the liver at day 7 (Figure 1D). Expression of common activating and inhibitory receptors, including Ly49H (Figure 1E), Ly49C/I, NKG2A/C/E, and NKG2D (data not shown), was also similar. To test cytotoxic capacity, degranulation was measured in response to YAC-1 tumor targets, and it was similar between Cox10-deficient and WT NK cells (Figure 1F). Killing of YAC targets was also intact in Cox10-deficient NK cells (Figure 1G). Cells from both models produced similar IFN-γ in response to cytokine (interleukin [IL]-12+IL-15, IL-12+IL-18) and receptor (anti-Ly49H oranti-NK1.1) stimulation (Figure 1H). These findings demonstrate that deletion of Cox10 in NK cells at a time when the Ncr1 gene is expressed does not alter NK cell maturation or ex vivo cytokine and cytotoxic function.
Extracellular flux assays were performed on purified YFP+ cells cultured with IL-2 for 3 days, and they identified a baseline decreased oxygen consumption rate (OCR) with a higher rate of aerobic glycolysis (extracellular acidification rate [ECAR]) and lower OCR/ECAR ratio (Figures 1I-1K) in Cox10-deficient NK cells. However, their ability to upregulate OXPHOS (spare respiratory capacity [SRC]) remained intact (Figure 1L), and maximal OCR rates were not significantly different compared to WT (Figure 1M). A COX activity assay demonstrated decreased activity in NK cells purified from Ncr1-Cox10Δ/Δ mice compared to WT mice (Figure S1C). Overall, these findings demonstrate that Cox10-deficient NK cells have impaired complex IV activity and following IL-2 activation have decreased baseline OXHOS and increased glycolysis.
Mice with constitutive Ncr-1-driven Cre were also generated, and they showed percentages and numbers of NK cells similar to controls in different organs (Figures S2A and S2B) with normal maturation (Figure S2C) and expression of NK receptors (Figure S2D). Similar to inducible Ncr1 deletion, NK cells from the constitutive model also had intact IFN-γ production (Figure S3A).
Taken together, these findings suggest that acute deletion of Cox10 in NK cells at the time of Ncr1 expression does not significantly alter NK cell maturation, activation, IFN-γ production, or target killing at baseline. Inducible deletion of Cox10 results in an increased baseline glycolytic rate, potentially as a compensatory mechanism for loss of this gene encoding an important component of complex IV.
Defect in Cox10-deficient NK cell antigen-induced proliferation
Infection of C57BL/6 mice with MCMV leads to well-characterized NK cell responses, including expansion of cells expressing the activating receptor Ly49H, which recognizes the MCMV-encoded ligand m157 (Yokoyama, 2013). To determine whether Cox10 was important for this antiviral function of NK cells, mice were infected with MCMV (Figure 2A). At day 4, the peak time for proliferation of Ly49H+ NK cells (Dokun et al., 2001), Ncr1-Cox10Δ/Δ mice had significantly fewer splenic Ly49H+ NK cells than did control mice (Figure 2A). Similar observations were made in mice with constitutive Ncr1-Cre driving Cox10 deficiency (Figures S3B and S3C), suggesting that the effect on proliferation was not due to the abrupt deletion of Cox10 in NK cells that developed with intact OXPHOS. Analysis of NK cells at an earlier time point in the constitutive model also demonstrated intact IFN-γ production following MCMV infection (Figure S3D), an important NK cell function during the first 1–2 days of infection driven by inflammatory cytokines.
Figure 2. Cox10-deficient NK cells have impaired proliferation in response to activating ligand stimulation during MCMV infection.

(A–C) Female Ncr1-Cox10Δ/Δ and Ncr1-WT mice were infected with 5 × 104 plaque-forming units (PFU) of MCMV and analyzed at day 4.
(A) The percentage and number of YFP+Ly49H+ NK cells were significantly lower in Ncr1-Cox10Δ/Δ mice.
(B) BrdU incorporation was lower in Ly49H+ NK cells lacking Cox10 (YFP+ Ly49H cells from Ncr1-Cox10Δ/Δ mice), demonstrating a defect in proliferation in antigen-specific NK cells.
(C) MCMV copy number normalized to β-actin was higher in Ncr1-Cox10Δ/Δ mice.
Data in (A)–(C) represent four separate experiments (individual mice are shown), analyzed using an unpaired t test for (A), a two-way ANOVA for (B), and an unpaired t test of log-transformed data for (C).
(D) NK cell proliferation following 3 days of IL-15 culture. Representative CTV histograms are shown for WT (blue) or Cox10-deficient (green) NK cells with low-dose IL-15 (LD, 5–10 ng/mL) or high-dose IL-15 (HD, 100 ng/mL). The percentage of proliferated cells was the same between the LD and HD groups. The proliferation index was the same between WT and Cox10-deficient cells and was higher with HD IL-15 compared to LD IL-15 (n = 6–14/group in four independent experiments, pooled data shown, two-way ANOVA).
(E) WT and Cox10-deficient NK cells were co-transferred into Rag2−/− γc−/− host mice, with similar ratios of cells recovered. (n = 2 hosts/group in three independent experiments, pooled data analyzed by two-way ANOVA).
(F) Ncr1-Cox10Δ/Δ and Ncr1-WT splenocytes cultured with IL-15 for 2 days followed by addition of Ba/F3-m157 cells for 3 days. Representative flow histograms of CTV for Ly49H+ and Ly49H− NK cells (all YFP+) are shown.
(G–I) There was no difference in percentage of cells that divided (G), but proliferation analysis demonstrated significantly reduced (H) proliferation index and (I) replication index in Ly49H+ Cox10-deficient NK cells compared to WT (two-way ANOVA, n = 13 mice/group in five independent experiments).
(J) Annexin V staining was similar, representative flow plots with WT on the left and Cox10-deficient on the right and pooled data shown (two-way ANOVA, n = 7–9 mice/group, three independent experiments). For all experiments, error bars represent mean and SD.
Both Cox10-deficient and WT NK cells displayed preferential proliferation of splenic Ly49H+ cells, measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation into replicating DNA (Figure 2B). However, significantly fewer Cox10-deficient Ly49H+ NK cells from Ncr1-Cox10Δ/Δ mice (YFP+Ly49H+) proliferated compared to WT-YFP+ cells. There was no defect in proliferation of YFP-negative cells from Ncr1-Cox10Δ/Δ mice (Figure 2B, YFP−), further supporting a specific role for Cox10 deletion. This proliferative defect in splenic NK cells was associated with impaired viral control at that site, as evidenced by increased MCMV copy number in the spleen (Figure 2C). There were no differences in MCMV copy number in the liver, suggesting that there may be local differences in viral control. Interestingly, ILC1s, the other major population affected by Cox10 deletion in this Ncr1-driven system, had normal proliferation measured by BrdU incorporation (Figure S4), suggesting differences in metabolic regulation of innate lymphocyte proliferation.
To determine whether defects in NK cell proliferation were specific to viral infection, the proliferative capacity of Cox10-deficient NK cells to other stimuli was tested. Stimulation with IL-15 demonstrated normal proliferation by Cox10-deficient NK cells in vitro (Figure 2D; Figure S3E). To test NK cell proliferation in vivo, NK cells were transferred into Rag2−/−γc−/− mice to examine homeostatic proliferation, a process driven largely by cytokines (Prlic et al., 2003). Congenically marked splenic Ncr1-Cox10Δ/Δ and Ncr1-WT cells were co-transferred at similar ratios into Rag2−/−γc−/− hosts, and analysis of spleens at days 3 and 5 showed similar percentages of each population (Figure 3E).
Figure 3. Single-cell RNA-seq and evaluation of mTOR and AMPK activation in NK cells during MCMV infection.

(A and B) Ncr1-Cox10Δ/Δ and Ncr1-WT NK cells were examined by single-cell sequencing 4 days after MCMV infection with uninfected controls.
(A) UMAP clustering of expression data using 20 principal components. Clusters 8 and 9 were expanded in response to MCMV infection in both groups, but cluster 10 was primarily expanded only with Cox10-deficient infected mice, accounting for ~20% of NK cells.
(B) Dot plot showing percentage of cells per sample expressing gene of interest (size) and average expression of gene (color) of transcriptionally regulated glycolytic genes upregulated during MCMV infection.
(C and D) NK cells from Ncr1-Cox10Δ/Δ and Ncr1-WT mice 4 days after MCMV expressing phosphorylated (C) mTOR and (D) AMPK in Ly49H− and Ly49H+ populations (mixed effects model with Tukey’s multiple comparison test, three independent experiments, n = 10–12 mice/group, pooled data, error bars represent SD). pAMPK was upregulated specifically in Ly49H+ Cox10-deficient NK cells.
These findings suggested that the proliferative defect seen in vivo was due to Ly49H stimulation. To investigate this, we used an in vitro system to simulate both cytokine and Ly49H stimulation (Figure 3F) to model stimuli seen during MCMV, since early NK cell activation is driven by cytokines followed by antigen-specific Ly49H stimulation (Dokun et al., 2001). Splenocytes from Ncr1-Cox10Δ/Δ and Ncr1-WT mice were labeled with CellTrace Violet (CTV) and cultured in 20 ng/mL IL-15 for 2 days, followed by addition of m157-expressing Ba/F3 cells. There was preferential expansion of Ly49H+ NK cells (Figure 2F, histograms). While the absolute percentage of Ly49H− and Ly49H+ NK cells that divided was the same (Figure 2G), the total number of divisions (proliferative index, Figure 2H) and fold expansion (replication index, Figure 2I) of dividing cells was significantly higher in Ly49H+ cells in both Cox10-deficient and WT mice, similar to that seen with Ly49H-specific proliferation during MCMV. Cox10-deficient Ly49H+ NK cells had an overall decreased proliferation compared to WT Ly49H+ cells, based on proliferation and the replication index of dividing cells (Figures 2H and 2I). Annexin V staining of cells to measure apoptosis did not show any differences, suggesting that the defect observed with Ly49H stimulation is proliferative and not induction of apoptosis (Figure 2J).
These results suggest that Cox10-deficient NK cells have a specific defect in activation receptor-induced proliferation, and that this effect is likely amplified in vivo in the context of viral infection.
Single-cell RNA sequencing identifies viral-specific NK cells
To investigate possible mechanisms behind this defect in virus-driven proliferation, single cell RNA-sequencing (scRNA-seq) was performed on YFP+ NK cells from Ncr1-Cox10Δ/Δ and Ncr1-WT mice 4 days after MCMV infection and compared to uninfected cells. Both Ncr1-Cox10Δ/Δ and Ncr1-WT NK cells demonstrated a strong MCMV-induced transcriptional signature with differential expression of >500 genes that was generally similar between the two groups (Figure S5A). Analysis by UMAP clustering identified two clusters of cells (clusters 8 and 9) that were expanded with MCMV infection in both WT and Cox10-deficient mice (Figure 3A). These clusters consisted largely of Ly49H+ cells (Figure S5B). There was also a distinct population (cluster 10) present primarily in Cox10-deficient MCMV-infected NK cells that accounted for 19% of cells in those cells (Figure 3A). Cluster 10 was comprised mostly of Ly49H+ cells largely negative for the NK transcription factors Eomes and T-bet (Figure S5B). Among the most highly upregulated genes in this cluster were Spp1, Gzmb, and Ly6a, all of which are known to increase in NK cells during MCMV infection (Fogel et al., 2013; Leavenworth et al., 2015). The presence of granzyme B transcript, which encodes a major protein in NK cell cytotoxic granules, supports that these are bona fide cytotoxic NK cells and not ILC1 cells, which express some NK cell receptors but do not produce cytotoxic granules (Figure S5C). No major differences in pro- or anti-apoptotic gene expression were seen in this cluster (Figures S6A and 6SB). Rather, cells in cluster 10 were best classified as in the G2/M (mitotic) phase of the cell cycle based on transcriptional analysis (Figure S6C). However, many genes responsible for triggering mitosis (Shen et al., 2018), including cyclin B (Ccnb), Cdk1, and Cdc25c, were expressed by few cells and at lower levels in this cluster (Figure S6D), suggesting that these NK cells may be in G2 arrest. By comparison, cells in cluster 9, also in the G2/M phase, had upregulated expression of these genes, suggesting active cycling (Figure S6D). Consistent with cell cycle arrest, the most transcriptionally downregulated processes were mitosis and DNA synthesis/repair (Figure S6E). Overall, the signature of cluster 10 suggests that this population was responding to MCMV but may be experiencing cell cycle arrest at the G2/M checkpoint due to Cox10 deficiency, suggesting a mechanism whereby Cox10-deficient Ly49H+ NK cells fail to expand appropriately during MCMV infection.
Cox10 deficiency leads to upregulation of mTOR and AMP-activated protein kinase (AMPK) activation in NK cells
Focusing on metabolic genes revealed that MCMV infection led to upregulation of transcriptionally regulated glycolytic genes (Ldha, Gapdh, Pkm, Eno1, Pgk1, and Aldoa) with infection, which was increased in Cox10-deficient cells (Figure 3B). Two major regulators of glycolysis in immune cells are mTOR and AMPK. We hypothesized that either of these pathways may be associated with increased glycolysis in cells. Generally, mTOR is downregulated by AMPK; however, Cox10-deficient NK cells had increased expression of phosphorylated mTOR (pmTOR) and AMPK (pAMPK) compared to WT during MCMV infection (Figures 3C and 3D). Increased pmTOR and pAMPK was seen in both Ly49H-negative and -positive Cox10-deficient NK cells, suggesting it was not limited to the Ly49H compartment. However, pAMPK was significantly higher in Ly49H+ cells from Cox10-deficient NK cells and not WT cells, suggesting that the compensatory increase in pmTOR with Cox10 deficiency was not sufficient for the metabolic stress in those cells.
NK cells lacking Cox10 do not expand as required for induction of an MCMV memory population
Expansion of Ly49H+ NK cells during MCMV infection leads to the induction of a group of cells with adaptive features, including increased effector functions in the context of subsequent MCMV infection (Sun et al., 2009). The expansion and contraction of these cells are required for formation of such MCMV memory NK cells (Adams et al., 2020). Based on impaired proliferation of Ly49H+ cells we hypothesized that formation of such memory would be defective. Indeed, a switch from glycolysis to OXPHOS is associated with T cell memory formation (Geltink et al., 2018). The potential for expansion of NK cells, and thus the potential to form memory, can be directly measured in a competitive transfer system in which the input cells are the only ones expressing Ly49H (O’Sullivan et al., 2015a; Sun et al., 2009). To determine the intrinsic effects of Cox10 deficiency on expansion capacity, we adoptively transferred YFP+ cells from Ncr1-Cox10Δ/Δ (Ly5.2+) and Ncr1-WT (Ly5.1+) into the same Ly49H-deficient host (B6.BXD8) followed by infection with MCMV (Figure 4A). This transfer model also addresses confounding factors, for example if Cox10 deficiency changed the cytokine milieu or viral titer. YFP+ NK cells from Ncr1-Cox10Δ/Δ (Ly5.2+) and Ncr1-WT (Ly5.1+) mice were co-transferred at similar ratios into Ly49H-deficient hosts (B6.BXD8) followed by MCMV infection (Figure 4A). NK cell numbers in the blood were measured by serial bleeds of animals. The peak numbers of Ly49H+ NK cells occur at day 7 post-infection, after which they contract to form a memory population (Sun et al., 2009). After 7 days, there were very few detectable Cox10-deficient NK cells, while there was robust expansion and contraction of WT Ly49H+ NK cells as measured by the percentage of NK cells that were Ly49H+ in both inducible and constitutive models (Figure 4B; Figures S3F and S3G). NK cells contracted over 28 days, and at all time points there were significantly more WT than Cox10-deficient NK cells, measured as the percentage of all NK cells that were Ly49H+ (Figure 4B) or by comparing the percentage of donor cells that were Cox10-deficient or WT (Figure 4C). At day 28, the absolute numbers of cells present in the spleen were similar to those in uninfected hosts, while only WT NK cells expanded following MCMV infection (Figure 4D). These experiments demonstrate an NK-intrinsic defect in MCMV-driven expansion of Ly49H+ NK cell required for memory formation.
Figure 4. Cox10 deficiency impairs MCMV-induced expansion required for memory formation.

(A) Congenic Ncr1-WT and Ncr1-Cox10Δ/Δ mice NK cells were co-transferred at a similar ratio into Ly49H-deficient hosts, and infected with MCMV. The proportion of each cell type was tracked in the blood weekly and in the spleen at day 28.
(B) Percentage of transferred Ly49H+ cells present among all NK cells in the host.
(C) Ratio of WT to Cox10-deficient NK cells in the blood and spleen (two-way ANOVA for blood, and one-way ANOVA for the spleen, ***p < 0.0001).
(D) Absolute number of transferred NK cells in the spleen at day 28 (two-way ANOVA). Pooled data are from three separate experiments; n = 11 recipient infected, 4 uninfected mice, error bars represent SD.
DISCUSSION
In this study, we demonstrate that Cox10, a gene encoding a component of electron transport chain complex IV of the mitochondrial electron transport chain, is required for NK cell antigen-specific expansion and control of MCMV infection in both inducible and constitutive NK-specific genetic models. Interestingly, acute or chronic deletion of Cox10 in mature NK cells with the inducible or constitutive Cre models did not significantly alter NKphenotype, homeostasis, cytotoxic function, or cytokine production. This somewhat surprising finding demonstrates that robust OXPFIOS is not required for NK cell development, at least after the stage at which NKp46 (Ncr1) is expressed. However, during MCMV infection, there was a clear requirement for Cox10 for expansion of Ly49H+ NK cells, a requisite step for generation of NK cell memory. This effect was cell-intrinsic, as demonstrated by co-transfer experiments. Impaired antigen-specific proliferation could be recapitulated in vitro in the presence of cytokine followed by co-culture with antigen-bearing target cells; however, proliferation in response to cytokines alone or homeostatic proliferation in vivo was normal. The fact that in vitro function for the most part was normal, while in vivo effector function (proliferation) was impaired, also highlights the challenges in re-capitulating metabolic microenvironments in vitro and the need to study immune cell metabolism in vivo.
NK cells primarily rely on OXPHOS for energy generation and IFN-γ production downstream of activating receptor stimulation at baseline (Keppel et al., 2015; Marçais et al., 2014). Following prolonged cytokine activation, NK cells upregulate glycolysis and OXPHOS (O’Brien and Finlay, 2019). A genetic model of disrupted Srepb signaling and the citrate-malate shuttle led to impaired OXPHOS and glycolysis, with defects in NK cell proliferation in response to cytokines (Assmann et al., 2017). In the present study, Cox10 deficiency partially impaired OXPHOS with increased glycolysis with IL-2 stimulation or MCMV infection. However, Cox10-deficient NK cells had normal proliferation in response to IL-15 and homeostatic proliferation, which is largely driven by cytokines. This highlights metabolic flexibility in NK cells and distinct requirements for different effector functions depending on the context in which NK cells are activated.
Deficiency of Cox10 led to increased activation of both mTOR and AMPK during MCMV infection. mTOR is important for NK cell maturation and function, in part through upregulation of glycolysis (Donnelly et al., 2014; Marçais et al., 2014; Nandagopal et al., 2014). Deficiencies in complex IV activate AMPK, a major energy sensor in cells (Kogot-Levin et al., 2016), and AMPK is phosphorylated in response to energetic stress to increase production of ATP by upregulating glycolysis and oxidative metabolism (Garcia and Shaw, 2017). Increased phosphorylation of mTOR in Cox10-deficient NK cells was not dependent on antigen-specific responses, and it was similarly increased in both Ly49H+ and Ly49H− Cox10-deficient NK cells. We hypothesize that this is due to the energetic stress of infection, including cytokines associated with infection, and is consistent with the known role of mTOR in upregulating glycolysis in NK cells (Donnelly et al., 2014; Marçais et al., 2014). While mTOR generally inhibits AMPK activation, there was significantly more pAMPK in antigen-specific, Ly49H+, Cox10-deficient NK cells as compared to Ly49H− cells, suggesting an effect of activating receptor engagement here. AMPK is a critical sensor for low energy (AMP/ATP ratio) and responds by activating catabolic processes, including glycolysis and fatty acid oxidation, while inhibiting protein and macromolecule synthesis (Garcia and Shaw, 2017), thus preserving cellular fitness at the expense of cellular replication. Thus, a possible explanation for activation of both mTOR and AMPK is the need to upregulate glycolysis to ensure cell survival during infection. AMPK has also been shown to cause G2 arrest (Shen et al., 2018). We hypothesize that in the setting of MCMV infection, Cox10 deficiency leads to activation of AMPK and subsequent G2 arrest, as suggested by scRNA-seq data, with dampening of antigen-specific NK cell expansion.
Interestingly, upregulation of AMPK at the peak of NK cell expansion has been shown to promote NK cell memory formation during MCMV infection through induction of autophagy (O’Sullivan et al., 2015a). Here, Cox10 deficiency was associated with upregulated phosphorylation of AMPK in antigen-specific Ly49H+ cells 4 days after infection. Failed expansion of Cox10-deficient NK cells following MCMV infection in a competitive transfer model supports the lack of induction of memory response due to Cox10 deficiency (Sun et al., 2009). These seemingly contrasting findings highlight the importance of the timing of metabolic changes. In the present study, failure to induce the initial expansion of antigen-specific cells was associated with upregulation of AMPK, whereas O’Sullivan et al. (2015a) demonstrated that activation of AMPK during the contraction phase of NK cells, after peak expansion, is important for memory formation.
Work by Sheppard et al. (2021, this issue of Cell Reports) using a genetic model of NK-specific lactate dehydrogenase A (Ldha) deficiency suggests that limiting fermentation and aerobic glycolysis impairs NK cells and also impairs proliferative capacity and memory formation, as well as cytotoxicity. Interestingly, these NK Ldha-deficient mice also had normal numbers and phenotype of NK cells at baseline, and the requirement for glycolysis only became apparent when the NK cells encountered challenges, viral infection, and tumor (Sheppard et al., 2021). We observed increased Ldha expression associated with an increased glycolytic signature in Cox10-deficient NK cells; however, this was not sufficient to allow for normal proliferation and host viral control. This supports that both aerobic glycolysis and OXPHOS must be intact for normal NK cell responses to MCMV. Collectively, these studies of genetic manipulation of OXPHOS and glycolysis in NK cells demonstrate that while some NK cell functions have metabolic flexibility, for example development and response to cytokines, antigen-driven proliferation during viral infection has absolute metabolic requirements for both glycolysis and OXPHOS.
In summary, deficiency in the mitochondrial respiratory chain gene Cox10 led to impaired antigen-specific expansion of NK cells during MCMV infection. This model also demonstrates that NK cells have metabolic flexibility that can compensate for defects in OXPHOS, but that these mechanisms fail during antigen-driven proliferation in vitro and in vivo. NK cells are important for host control of viruses and tumors, and identification of relevant metabolic pathways for their function in these contexts is important for considering therapeutic approaches targeting NK cells, as well as for a better understanding of the immune response in patients with metabolic defects.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Megan Cooper (cooper_m@wustl.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
The single cell RNA sequencing data generated during this study is available at Gene Expression Omnibus with accession number GSE149659.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal studies were performed at Washington University using an Animal Studies Committee (ASC) approved protocol and conducted in accordance with ASC regulations. All mice were maintained in specific pathogen-free conditions in group housing, bred inhouse, used between 8-14 weeks of age, and euthanized in accordance with institutional guidelines. Mice with tamoxifen-inducible Ncr1-specific Cre expression, Ncr1-iCreERT2, were recently published (Wagner et al., 2020). Transgenic Ncr1-Cre mice with constitutive Ncr1-driven Cre were previously published (Eckelhart et al., 2011), and used for experiments shown in supplemental figures. Mice with Cre-inducible Cox10 deletion (JAX 024697) (Diaz et al., 2005) and YFP reporter (JAX 006148) (Srinivas et al., 2001), were purchased from Jackson Laboratories. Mice were bred to generate either tamoxifen-inducible or constitutive deletion of Cox10, referred to here as Ncr1-Cox10Δ/Δ or cNcr1-Cox10Δ/Δ respectively. Control mice with tamoxifen-inducible or constitutive Ncr1-driven Cre also crossed to YFP-reporter mice were used for all experiments. Some experiments were performed with congenic CD45.1+ mice. B6.BXD8 mice were previously described and obtained from W. Yokoyama (Cheng et al., 2008). All mice were on the C57BL/6 background. For Cre induction, mice were fed 250 mg/kg tamoxifen-containing Teklad chow (Envigo) which gives a daily dose of 40 mg/kg. Both sexes were used, and sex-matched to Ncr1-Cre (Ncr1-WT) controls for experiments.
Cell lines
Ba/F3-m157 cells [from W. Yokoyama, (Smith et al., 2002)] were cultured in R10 supplemented with puromycin for selection of m157 expression and 5% IL-3- containing supernatant from X63 cells. YAC1 cells (CVCL_2244, ATCC TIB-160) were grown in R10. All cells were kept in a 37°C 5% CO2 incubators.
METHOD DETAILS
Cell culture
Primary splenocytes from mice were cultured in “R10” RPMI 1640 (Mediatech) supplemented with 10% FBS (Sigma-Aldrich), L-glutamine to a final concentration of 4 mM (Sigma), 50 μM 2-ME (Sigma), and antibiotics (penicillin/streptomycin).
NK purification
Murine NK cells were first enriched from spleens by negative selection (EasySep, StemCell Technologies) and then FACS-purified by sorting cells based on CD3−NK1.1+ with or without YFP as indicated (FACSAria Fusion sorter, BD).
Flow cytometry
Alive/dead stain was included to measure viability (Zombie Yellow, BioLegend). Cells were blocked with anti-FcγRIII (2.4G2) prior to surface staining. Cells were fixed with 1% paraformaldehyde for surface analysis or fixed with Cytofix/Cytoperm (BD Bioscience) for intracellular staining. For intranuclear staining, cells were fixed in Cytofix/Cytoperm and Cytoperm Plus buffer (BD) and DNase treated. For phospho mTOR staining, cells were fixed with 2% paraformaldehyde and permeabilized with methanol. For phospho AMPK staining, cells were fixed with Cytofix/Cytoperm (BD Bioscience), and stained overnight at 4C with anti-pAMPK. Data was acquired on a Cytek-modified FACScan (BD and Cytek) or LSRFortessa (BD); data was analyzed using FlowJo 7.6.5. Geometric mean fluorescence intensity and % proliferated were calculated using FlowJo proliferation analysis (see below). The following fluorochrome-conjugated antibodies were used: BrdU (BrdU kit, BD), CD107a (1D4B, BD), CD3ε (145-2C11, BD/BioLegend), CD11b (M1/70, BD), CD226/DNAM-1 (10E5, BioLegend), CD27 (LG.3A10, BD), CD45.1 (A20 Biolegend), anti-human IFN-γ (XMG1.2, BioLegend), Ly49C/I (5E6, BD), Ly49H (3D10, BD/BioLegend), NK1.1 (PK136, BD), NKG2A/C/E (20d5, BD), NKG2D (CX5, Biolegend), NKp46 (29A1.4, BD), pAMPKa, (40H9, Cell Signaling), and pMTOR (MRRBY, Invitrogen).
NK cell functional assays
To assess intracellular IFN-γ production splenocytes were stimulated with 10 ng/ml murine IL-12 (PeproTech) and 50 ng/mL murine IL-15 (StemCell), 10ng/mL of IL-12 and 50ng/mL of murine IL-18, or in plates coated with 20 μg/ml purified anti-Ly49H (with or without 10ng/mL of IL-15), anti-NK1.1 (PK136; BioXcell) or control antibody for a total of 6 hours. Brefeldin A was added to cells after 1h of culture and intracellular IFN-γ production was assayed by flow cytometry using BD Cytofix/Cytoperm (BD Biosciences). CD107a degranulation assays and killing assays were performed with splenocytes incubated with YAC-1 target cells at E:T ratios of 10:1 or 25:1 (splenocytes:target) in a 96 well round bottom plate as previously described (Keppel et al., 2013). For killing assays, cells were incubated with 7-aminoactinomycin D (7AAD, Calbiochem) after 4 hours of co-culture. CD107a was stained by incubating splenocytes with anti-CD107a (1D4B, BD) and GolgiStop (monensin, BD) for 4 hours at 37°C. For analysis of IFN-γ production, proliferation, and CD107a, NK cells (CD3−NK1.1+) were gated on YFP+ cells to identify NK cells with Cre activity.
Cox10 quantitative PCR
Enriched NK cells were sorted for YFP expression, and RNA was extracted using the RNA Easy kit (QIAGEN) according to the manufacturer’s protocol. cDNA from RNA was reverse transcribed using random hexamers (Promega). Both Cox10 primer/probe and Beta-actin primer/probe set was from ThermoFisher. Fold change was calculated using the 2^-ΔΔCt method normalized to YFP-NK cells from Ncr1-Cox10Δ/Δ mice.
Extracellular flux assays
FACS-purified YFP+ NK cells were cultured with 1000 IU/mL of murine IL-2 for 72hrs. Extracellular flux assays were performed using a XF96 Analyzer (Seahorse Bioscience/Agilent) as previously described (Keppel et al., 2015). Cell were washed and plated 4 × 105 cells per well on poly-L-lysine-coated plates in at least triplicate for extracellular flux analysis of oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). Mitochondrial stress tests were performed in non-buffered, phenol red-free RPMI media with glucose, L-glutamine, sodium pyruvate, and 1% FBS (pH 7.4). Drug concentrations during the assay were: 10 μM oligomycin, 10 μM FCCP, 10 μM rotenone, and 10 μM antimycin A.
Cox10 activity assay
Cytochrome C Oxidase (COX) activity was measured using the Cytochrome C Oxidase Assay Kit (Sigma Aldrich). NK cells were sorted for YFP expression, and cell pellets were snap-frozen at −80°C in 1-2 × 105 cells/vial. Samples were thawed on ice and incubated for 30-45 minutes in 20 μL enzyme dilution buffer (10 mM Tris-HCl, 250 mM Sucrose, 1 mM N-dodecyl-β-D-maltoside, pH = 7), then mixed with 200 μL assay buffer (10 mM Tris-HCl, 120 mM KCl). Cytochrome c was dissolved in molecular-grade H2O at 2.7 mg/mL and reduced in a final concentration of 2 mM dithiothreitol (DTT) for 2 hours at room temperature. 20 μL of reduced cytochrome c solution was added to the sample. Absorbance at 550 nm (A500 nm) was measured in a 96-well plate every minute for 10 minutes with intermittent shaking on a Biotek Gen5 Microplate Reader. Control samples of Assay Buffer only, fully reduced cytochrome c, and fully oxidized cytochrome c were also measured. COX activity was determined by the slope of the ΔA550 nm from minute 0 to minute 10.
MCMV infection
8-12 week old mice were injected intraperitoneally (i.p.) with 0.5 × 104 - 2 × 105 PFU of Smith strain salivary gland MCMV (ATCC, prepared in BALB/c mice), or media as a control. To assess proliferation during MCMV infection, mice were injected with 2 mg BrdU (Sigma) i.p. 3 hours before harvest. For memory experiments with constitutive Cre mice, 20-30 × 106 splenocytes from cNcr1-Cox10Δ/Δ and WT-Cox10Δ/Δ littermates were adoptively transferred by tail vein injection at a 1:1 ratio into Ly49H-deficient Bxd8 mice. For competitive expansion/memory experiments with inducible Cre mice, YFP+ FACS-purified NK cells from CD45.2+ Ncr1- Cox10Δ/Δ and CD45.15 Ncr1-WT mice were mixed and 1-2 × 105 cells transferred into Ly49H-deficient mice (B6.BXD8). Male recipients were infected with 2.5 × 103 PFU MCMV 18-24 hours post transfer. Females were infected with 2.0 × 103 PFU MCMV. Ly49H+ expansion was tracked through bleeding from the cheek vein once per week for 4 weeks. On day 28, spleen and blood were harvested. MCMV copy number was measured as previously described (Mah et al., 2017). The limit of detection was 100 copies of MCMV, and all values below 100 were rounded up. Graphs represent [(copies MCMV ie1)/(copies Actb)] x 1000. Statistics were performed on log-transformed data.
Proliferation assays
For IL-15-induced proliferation, splenocytes were stained with 2.5-5 μM Tag-it/CellTrace Violet (Biolegend) and cultured with either 5-10ng/ml (low-dose) or 100ng/mL (high-dose) murine IL-15 for 72hrs. Cells were analyzed for proliferation by CTV using FlowJo Proliferation platform (see below).
For homeostatic proliferation, NK cells were sorted for YFP expression from CD45.2+ Ncr1-Cox10Δ/Δ and CD45.1+ Ncr1-Cre YFP-reporter mice (1-2 × 105 cells per mouse) and labeled with Tag-it/CellTrace Violet (CTV). They were mixed 1:1 and adoptively transferred by tail vein injection into congenic CD45.2+Rag-2−/−γc−/− hosts and the spleens were harvested 3 and 5 days later. The ratio of transferred cells was calculated as %recovered/%inputed, e.g. % of NK cells that were CD45.1+ NK cells in spleen/% of CD45.1+ NK cells injected.
For Ba/F3-m157 assays, splenocytes were labeled with CTV and cultured in 20ng/mL murine IL-15 for two days, followed by addition of Ba/F3-m157 cells at a 10:1 splenocyte:Ba/F3-m157 ratio with fresh 20ng/mL IL-15 in the media for three additional days. Cells were analyzed for proliferation by CTV using FlowJo Proliferation platform.
Proliferation using the FlowJo platform was assessed by identifying and setting an undivided peak for all samples from the same experiment (generation 0 peak). Percentage of divided cells was calculated based on this gate. Following assignment of proliferating cells to different generations, the following measurements of proliferative capacity of dividing cells were determined in FlowJo and are presented here: Proliferation index (PI) = , Replication Index (RI) = , and Percent Divided = , where i is the generation number and Ni is the number of cells per generation I (Roederer, 2011)
Single-cell RNA sequencing and analysis
YFP+ NK cells were sorted from MCMV-infected mice at day 4 for single cell RNA-seq (10X Chromium platform, Illumina Hi-Seq). Sequencing was performed by the McDonnell Genomics Institute at Washington University in St. Louis. Raw FASTQ files were aligned to the mm10 reference using CellRanger (v3.1.0) count algorithm, and the barcode and UMI count files were aggregated and normalized using CellRanger (v.3.1.0) aggr default options (Zheng et al., 2017). Quality control and analysis of the matrix file was performed using Seurat V3 software for R (Stuart et al., 2019). Cells that expressed less than 200 genes and cells that expressed more than 10% of mitochondrial associated genes were removed, remaining a total of 46,129 cells and 19,436 genes across all samples.
Comparative analysis was done by performing an integrated analysis on all cells using Seurat software. For cluster visualization, data was scaled and the RunUMAP tool was used to map cells, using a PCA reduction with 20 principal components. Differentially regulated genes for each cluster and comparison between samples were determined using the FindMarkers function. Heatmaps were made using the Heatmap function. Cell cycle analysis was performed using CellCycleScoring in the Seurat package using genes from Nestorowa et al. (2016) and Tirosh et al. (2016). Pathway analysis was performed using Metascape (Zhou et al., 2019). Only genes with adjusted p value < 0.05 were retained for pathway analysis. Cell identity analysis was performed using the Immunological Genome Project (ImmGen)’s My Geneset tool with ultra-low input (ULI) RNA-Seq data (Heng et al., 2008). ScRNA-Seq data is available at the Gene Expression Omnibus with accession number GSE149659.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
Statistical details of experiments can be found in figure legends. “n” represents number of experimental replicates and/or number of animals in an experiment. Numbers of independent experiments performed are in the figure legends. Statistics calculated in Prism 9 with specific tests discussed in figure legends and p values noted in the figure or legend. All treatment groups used in an experiment were included in the ANOVA analysis, including controls. For MCMV copy number, t tests were performed on log-transformed data.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Alexa-647 rat anti-mouse NKp46 (clone 29A1.4) | BD Biosciences | Cat# 560755; RRID:AB_1727464 |
| APC mouse anti-mouse NK1.1 (clone PK136) | BD Biosciences | Cat#550627; RRID:AB_398463 |
| APC/Cyanine7 anti-mouse CD19 (clone 6D5) | Biolegend | Cat# 115530; RRID:AB_830707 |
| APC-Cy7 Hamster Anti-Mouse CD11c (Clone HL3) | BD Biosciences | Cat# 561241; RRID:AB_10611727 |
| APC-Cy7 hamster anti-mouse CD3epsilon (clone 145-2C11) | Biolegend | Cat# 100330; RRID:AB_1877170 |
| BV421 hamster anti-mouse CD3epsilon (clone 145-2C11) | Biolegend | Cat# 100336; RRID:AB_11203705 |
| BV421 mouse anti-mouse CD45.1 (clone A20) | BioLegend | Cat#110732; RRID:AB_2562563 |
| BV421 mouse anti-mouse NK1.1 (clone PK136) | BD Biosciences | Cat# 562921; RRID:AB_2728688 |
| BV421 rat anti-mouse Ly49H (clone 3D10) | BD Biosciences | 744260; RRID:AB_2742099 |
| BV421 hamster anti-mouse CD49a (clone Ha31/8) | BD Biosciences | Cat# 740046; RRID:AB_2739815 |
| BV605 NKG2A/C/E (clone 20d5) | BD Biosciences | Cat#564382; RRID:AB_2738782 |
| Mouse anti-mouse Ly49C+I (clone 5e6) | BD Biosciences | Cat#553277; RRID:AB_394751 |
| PE goat anti-rabbit F(ab’)2 (polyclonal) | Cell Signaling Technologies | Cat# 8885; RRID:AB_2797677 |
| PE hamster anti-mouse CD3epsilon (clone 145-2C11) | Biolegend | Cat# 100308; RRID:AB_312673 |
| PE mouse anti-mouse BRDU (clone 3D4) | BD Biosciences | Cat# 556029; RRID:AB_396305 |
| PE mouse anti-mouse phospho-MTOR (clone MRBBY) | ThermoFisher | Cat# 12-9718-42; RRID:AB_2572724 |
| PE rat anti-mouse (clone XMG1.2) | BioLegend | Cat#505808; RRID:AB_315402 |
| PE rat anti-mouse CD107a (clone 1D4B) | BD Biosciences | Cat# 558661; RRID:AB_1645247 |
| PE rat anti-mouse CD11b (clone M1/70) | BD Biosciences | Cat#557397; RRID:AB_396680 |
| PE rat anti-mouse Ly49H (clone 3D10) | BioLegend | Cat#144705; RRID AB_2561673 |
| PE Annexin V (Ca2+ dependent phospholipidbinding protein) | BD Biosciences | Cat# 556421; RRID:AB_2869071 |
| PE-Cy7 hamster anti-mouse CD27 (clone LG.3A10) | BD Biosciences | Cat#563604; RRID:AB_2738309 |
| PE-Cy7 mouse anti-mouse CD45.1 (clone A20) | BioLegend | Cat# 110730; RRID:AB_1134168 |
| PE-Cy7 rat anti-mouse Ly49H (clone 3D10) | Biolegend | 144714; RRID:AB_2783113 |
| PE-Cy7 rat anti-mouse CD127 (clone A7R34) | Biolegend | Cat# 135014; RRID:AB_1937265 |
| PerCP/Cyanine5.5 anti-mouse DX5 (clone CD49b) | Biolegend | Cat# 108916; RRID:AB_2129358 |
| PerCP-Cy5.5 hamster anti-mouse CD3epsilon (clone 145-2C11) | Biolegend | Cat# 100328; RRID:AB_893318 |
| Rat anti mouse CD226/DNAM-1 (clone 10E5) | BioLegend | Cat#128806; RRID:AB_1186119 |
| Rat anti-mouse NKG2D (clone CX5) | BioLegend | Cat# 130212; RRID:AB_1236372 |
| Unconjugated mouse anti-mouse NK1.1 (PK136) | BioXcell | Cat# BE0036; RRID:AB_1107737 |
| Unconjugated rabbit anti-human phospho-AMPK-alpha (clone 40H9) | Cell Signaling Technologies | Cat# 2535; RRID:AB_331250 |
| Unconjugated rat anti-mouse CD16 (clone 2.4g2) | ATCC | ATCC Cat# HB-197; RRID:CVCL_9148 |
| Unconjugated anti-mouse Ly49H (clone 3D10) | Biolegend | Cat# 144702; RRID:AB_2561549 |
| Bacterial and virus strains | ||
| Murine cytomegalovirus (MCMV) | ATCC | Cat#ATCC VR-1399 |
| Chemicals, peptides, and recombinant proteins | ||
| 7-AAD (7-Amino Actinomycin D) | Millipore-Sigma | Cat# 129935 |
| BD Cytofix/cytoperm | BD Biosciences | Cat# 554714 |
| BrdU | MilliporeSigma | Cat# B5002 |
| Brefeldin A | BD Biosciences | Cat# 555029 |
| Draq5 | Cell Signaling | Cat# 4084S |
| GolgiStop (monensin) | BD Biosciences | Cat #: 554724 |
| Mouse recombinant IL-12 | PeproTech | Cat# 210-12 |
| Mouse recombinant IL-15 | Stem Cell | Cat# 78080.1 |
| Mouse recombinant IL-18 | MBL | Cat#B002-5 |
| Tag-it Violet | BioLegend | Cat# 425101 |
| Tamoxifen Chow | Envigo | Cat# TD.130856 |
| Zombie Yellow | Biolegend | Cat# 423104 |
| Critical commercial assays | ||
| ATPLite Luminescence Assay | Perkin Elmer | Cat# 6016941 |
| BrdU Assay | BD Biosciences | Cat# 552598 |
| COX assay | MilliporeSigma | Cat# CYTOCOX1 |
| cytochrome c | MilliporeSigma | Cat #: C3131 |
| Dithiothreitol (DTT) | MilliporeSigma | Cat #: D0632 |
| Gentra Puregene Tissue Kit | QIAGEN | Cat# 158667 |
| Mouse NK cell Enrichment (EasySep) | StemCell | Cat# 19855 |
| N-dodecyl-B-D-Maltoside | MilliporeSigma | Cat #: D4641 |
| RNA Easy kit | QIAGEN | Cat# 74104 |
| Seahorse XF Cell Mito Stress Test Kit | Agilent | Cat #: 103015-100 |
| Deposited data | ||
| Single cell RNA sequencing raw data | This paper | GSE149659 |
| Experimental models: Cell lines | ||
| YAC1 | ATCC | ATCC TIB-160; RRID CVCL_2244 |
| Ba/F3-m157 cells | W. Yokoyama | Smith et al., 2002 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Cox10Δ/Δ; B6.129X1-Cox10tm1Ctm/J | The Jackson Laboratory | Cat# 024697; RRID IMSR_JAX:024697 |
| Mouse: YFP reporter; B6.129X1-Gt(ROSA) 26Sortm1(EFYP)Cos/J | The Jackson Laboratory | Cat# 006148; RRID IMSR_JAX:006148 |
| Mouse: B6.Bxd8; B6.BXD8-Klra8Cmv1-del/WumJ | The Jackson Laboratory | Cat# 008633; RRID IMSR_JAX:008633 |
| Mouse: Rag2−/−γc−/−; B10;B6-Rag2tm1Fwa Il2rgtm1Wjl | Taconic | Cat# 4111; RRID IMSR_TAC:4111 |
| Mouse: cNcr1; Tg(Ncr1-icre)265Sxl | Eckelhart et al., 2011 | MGI ID: 4941472 |
| Mouse: Ly5.1; B6.SJL-PtprcaPepcb/BoyCrCrl | Charles River | Cat# 564, RRID IMSR_CRL:564 |
| Mouse: Ncr1-iCreERT2 | Wagner et al., 2020 | N/A |
| Oligonucleotides | ||
| Beta actin primer/probe set | Thermofisher | Cat# 4352341E |
| COX10 primer/probe set | Thermofisher | Cat# Mm00617695_m1 |
| MCMV (IE1) primer/probe set Primer 1 CCCTCTCCTAACTCTCCCTTT | IDT Custom PrimeTime Probe Parikh et al., 2015 | N/A |
| MCMV (IE1) primer/probe set Primer 2 TGGTGCTCTTTTCCCGTG | IDT Custom PrimeTime Probe Parikh et al., 2015 | N/A |
| MCMV (IE1) primer/probe set Probe FAM TCTCTTGCCCCGTCCTGAAAACC | IDT Custom PrimeTime Probe Parikh et al., 2015 | N/A |
| Software and algorithms | ||
| Flowjo 10.7.1 | Flowjo | https://www.flowjo.com/solutions/flowjo/downloads |
| ImmGen | Heng et al., 2008 | https://www.immgen.org |
| Metascape | Zhou et al., 2019 | http://metascape.org |
| Prism 9 | Graphpad | N/A |
| QuantStudios3 | Thermofisher | Cat# A28136 |
| Seurat 3.0 for R | Stuart et al., 2019 | https://satijalab.org/seurat/ |
Highlights.
Inducible loss of Cox10, a component of complex IV, does not alter NK cell homeostasis
Cox10-deficient NK cells proliferate normally in response to IL-15
Antigen-specific proliferation of NK cells is impaired in Cox10-deficient NK cells
Induced Cox10 deletion inhibits the ability of NK cells to expand during MCMV infection
ACKNOWLEDGMENTS
This work was supported by NIH grant R01AI127752 (to M.A.C.); the Children’s Discovery Institute (M.A.C.); the Rheumatology Research Foundation (to M.A.C.); NIH grants F30AI129110 (to A.Y.M.-S.), T32GM007200 (to A.Y.M.-S. and J.M.T.), R01AI078994 (to A.R.F.), and R01 CA205239 and P50CA171963 (to T.A.F.); and by Austrian Science Foundation (FWF) grant P28571 (to V.S.). We thank the Genome Technology Access Center and McDonnell Genome Institute Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center support grant P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA grant UL1TR002345 from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH). This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. We thank the Tissue Culture Support Center at Washington University in St. Louis for technical assistance. This work used data and tools assembled by the ImmGen Consortium and Metascape, and programming support from the Satija lab.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109209.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams NM, Grassmann S, and Sun JC (2020). Clonal expansion of innate and adaptive lymphocytes. Nat. Rev. Immunol 20, 694–707. [DOI] [PubMed] [Google Scholar]
- Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, and Shoubridge EA (2003). Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum. Mol. Genet 12, 2693–2702. [DOI] [PubMed] [Google Scholar]
- Arase H, Mocarski ES, Campbell AE, Hill AB, and Lanier LL (2002). Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 296, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, Heinrich P, Oefner PJ, Lynch L, Gardiner CM, et al. (2017). Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol 18, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial dynamics controls T cell fate through metabolic programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic Instruction of Immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng TP, French AR, Plougastel BF, Pingel JT, Orihuela MM, Buller ML, and Yokoyama WM (2008). Ly49h is necessary for genetic resistance to murine cytomegalovirus. Immunogenetics 60, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, and Walzer T (2009). Maturation of mouse NK cells is a 4-stage developmental program. Blood 113, 5488–5496. [DOI] [PubMed] [Google Scholar]
- Diaz F (2010). Cytochrome c oxidase deficiency: Patients and animal models. Biochim. Biophys. Acta 1802, 100–110. [DOI] [PubMed] [Google Scholar]
- Diaz F, Thomas CK, Garcia S, Hernandez D, and Moraes CT (2005). Mice lacking COX10 in skeletal muscle recapitulate the phenotype of progressive mitochondrial myopathies associated with cytochrome c oxidase deficiency. Hum. Mol. Genet 14, 2737–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, and Yokoyama WM (2001). Specific and nonspecific NK cell activation during virus infection. Nat. Immunol 2, 951–956. [DOI] [PubMed] [Google Scholar]
- Donnelly RP, Loftus RM, Keating SE, Liou KT, Biron CA, Gardiner CM, and Finlay DK (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol 193, 4477–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckelhart E, Warsch W, Zebedin E, Simma O, Stoiber D, Kolbe T, Rülicke T, Mueller M, Casanova E, and Sexl V (2011). A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK-cell survival and development. Blood 117, 1565–1573. [DOI] [PubMed] [Google Scholar]
- Fehniger TA, and Cooper MA (2016). Harnessing NK cell memory for cancer immunotherapy. Trends Immunol. 37, 877–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogel LA, Sun MM, Geurs TL, Carayannopoulos LN, and French AR (2013). Markers of nonselective and specific NK cell activation. J. Immunol 190, 6269–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geltink RIK, Kyle RL, and Pearce EL (2018). Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol 36, 461–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer Q, Rückert T, Borst EM, Dunst J, Haubner A, Durek P, Heinrich F, Gasparoni G, Babic M, Tomic A, et al. (2018). Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol 19, 453–463. [DOI] [PubMed] [Google Scholar]
- Heng TS, and Painter MW; Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, and Gardiner CM (2016). Metabolic reprogramming supports IFN-γ production by CD56bright NK cells. J. Immunol 196, 2552–2560. [DOI] [PubMed] [Google Scholar]
- Kelly B, and Pearce EL (2020). Amino assets: How amino acids support immunity. Cell Metab. 32, 154–175. [DOI] [PubMed] [Google Scholar]
- Keppel MP, Yang L, and Cooper MA (2013). Murine NK cell intrinsic cytokine-induced memory-like responses are maintained following homeostatic proliferation. J. Immunol 190, 4754–4762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keppel MP, Saucier N, Mah AY, Vogel TP, and Cooper MA (2015). Activation-specific metabolic requirements for NK cell IFN-γ production. J. Immunol 194, 1954–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogot-Levin A, Saada A, Leibowitz G, Soiferman D, Douiev L, Raz I, and Weksler-Zangen S (2016). Upregulation of mitochondrial content in cytochrome c oxidase deficient fibroblasts. PLoS ONE 11, e0165417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavenworth JW, Verbinnen B, Wang Q, Shen E, and Cantor H (2015). Intracellular osteopontin regulates homeostasis and function of natural killer cells. Proc. Natl. Acad. Sci. USA 112, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, et al. (2020). Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med 382, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, Tripathy SK, Agarwal SK, Jeng EK, Wong HC, et al. (2017). Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight 2, e95128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabilloud J, Mayol K, Tavares A, Bienvenu J, et al. (2014). The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol 15, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers JA, and Miller JS (2021). Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol 18, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandagopal N, Ali AK, Komal AK, and Lee SH (2014). The critical role of IL-15-PI3K-mTOR pathway in natural killer cell effector functions. Front. Immunol 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Göttgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien KL, and Finlay DK (2019). Immunometabolism and natural killer cell responses. Nat. Rev. Immunol 19, 282–290. [DOI] [PubMed] [Google Scholar]
- O’Sullivan TE, Johnson LR, Kang HH, and Sun JC (2015a). BNIP3- and BNIP3L-mediated mitophagy promotes the generation of natural killer cell memory. Immunity 43, 331–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan TE, Sun JC, and Lanier LL (2015b). Natural killer cell memory. Immunity 43, 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh BA, Piersma SJ, Pak-Wittel MA, Yang L, Schreiber RD, and Yokoyama WM (2015). Dual Requirement of Cytokine and Activation Receptor Triggering for Cytotoxic Control of Murine Cytomegalovirus by NK Cells. PLoS Pathog 11, e1005323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prlic M, Blazar BR, Farrar MA, and Jameson SC (2003). In vivo survival and homeostatic proliferation of natural killer cells. J. Exp. Med 197, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roederer M (2011). Interpretation of cellular proliferation data: avoid the panglossian. Cytometry A 79, 95–101. [DOI] [PubMed] [Google Scholar]
- Shen Y, Sherman JW, Chen X, and Wang R (2018). Phosphorylation of CDC25C by AMP-activated protein kinase mediates a metabolic checkpoint during cell-cycle G2/M-phase transition. J. Biol. Chem 293, 5185–5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard S, Santosa EK, Lau CM, Violante S, Giovanelli P, Kim H, Cross JR, Li MO, and Sun JC (2021). Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 35, this issue, 109210-1–109210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, et al. (2002). Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 99, 8826–8831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, and Costantini F (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol 1, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, and Lanier LL (2018). Is there natural killer cell memory and can it be harnessed by vaccination? NK cell memory and immunization strategies against infectious diseases and cancer. Cold Spring Harb. Perspect. Biol 10, a029538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, and Lanier LL (2009). Adaptive immune features of natural killer cells. Nature 457, 557–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarasenko TN, Pacheco SE, Koenig MK, Gomez-Rodriguez J, Kapnick SM, Diaz F, Zerfas PM, Barca E, Sudderth J, DeBerardinis RJ, et al. (2017). Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 25, 1254–1268.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valnot I, von Kleist-Retzow JC, Barrientos A, Gorbatyuk M, Taanman JW, Mehaye B, Rustin P, Tzagoloff A, Munnich A, and Rötig A (2000). A mutation in the human heme A:farnesyltransferase gene (COX10 ) causes cytochrome c oxidase deficiency. Hum. Mol. Genet 9, 1245–1249. [DOI] [PubMed] [Google Scholar]
- Wagner JA, Wong P, Schappe T, Berrien-Elliott MM, Cubitt C, Jaeger N, Lee M, Keppel CR, Marin ND, Foltz JA, et al. (2020). Stage-specific requirement for Eomes in mature NK cell homeostasis and cytotoxicity. Cell Rep. 31, 107720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama WM (2013). Natural killer cells. In Fundamental Immunology, Paul W, ed. (Lippincott, Williams &Wilkins; ), pp. 395–430. [Google Scholar]
- Zheng GX, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The single cell RNA sequencing data generated during this study is available at Gene Expression Omnibus with accession number GSE149659.