

Abstract
Trichloroethene (TCE) exposure is associated with the induction of autoimmune diseases (ADs). Although oxidative stress plays a major role in TCE-mediated autoimmunity, the underlying molecular mechanisms still need to be delineated. Dysregulation of redox-sensitive nuclear factor (erythroid-derived 2)-like2 (Nrf2), resulting in uncontrolled antioxidant and cytoprotective genes, and pro-inflammatory MAPK signaling pathways could be critical in TCE-mediated disease progression. This study was, therefore, focused on establishing status and contribution of Nrf2 and MAPK signaling in TCE-mediated inflammatory and autoimmune responses, especially during disease progression. To achieve these objectives, time-response studies were conducted by treating female MRL+/+ mice with TCE (0.5 mg/ml, a dose relevant to human exposure) for 24, 36 and 52 wks. TCE exposure led to reduction in Nrf2 expression, but increased phos-NF-κB (p65) and iNOS along with increased phosphorylation of MAPKs (p38, ERK and JNK) and downstream pro-inflammatory cytokines IL-12, TNF-α and RANTES in the livers in a time-dependent manner. These changes were also associated with time-dependent increases in liver protein carbonyls and induction of serum anti-dsDNA antibodies (marker of systemic lupus erythematosus disease), further supporting the role of oxidative stress and Nrf2/MAPK signaling in TCE-mediated autoimmune response progression. The mechanistic role of MAPK in TCE-mediated autoimmunity was further established by treating MRL+/+ mice with sulforaphane (SFN; 8 mg/kg, i.p., every other day) along with TCE (10 mmol/kg, i.p., every 4th day) for 6 wks using an established protocol, and by in vitro treatment of T cells with dichloroacetyl chloride (a TCE metabolite) with/without p38 MAPK inhibitor. SFN treatment attenuated the TCE-mediated phosphorylation of p38 MAPK. More importantly, treatment with SFN or p38 inhibitor led to suppression of downstream pro-inflammatory cytokines IL-12 and TNF-α. These findings thus support the contribution of Nrf2 and MAPK signaling pathways and help in delineating novel potential therapeutic targets against TCE-mediated autoimmunity.
Keywords: Nrf2, MAPK, trichloroethene, autoimmunity, sulforaphane
1. Introduction
Autoimmune diseases (ADs) are the third most common type of diseases in the United States after cardiovascular disease and cancer (Desai et al., 2019). In the United States, the estimated prevalence of ADs is between 14.7 to 23.5 million people (Desai et al., 2019). ADs, such as systematic lupus erythematosus (SLE), scleroderma, rheumatoid arthritis (RA) and autoimmune hepatitis (AIH), are chronic pathologies mediated by the loss of immune tolerance to self-antigens, increased production of autoantibodies and T lymphocytes infiltration in kidney, liver or lungs, which can further lead to tissue damage and life threating disorders (Cai et al., 2008; Khan and Wang, 2020; Kondraganti et al., 2012; Pollard et al., 2010). Exposure to environmental agents/contaminants is known to induce/accelerate the pathogenesis of ADs (Khan and Wang, 2018; Khan and Wang, 2020; Pollard 2010). Our previous studies have shown that trichloroethene (TCE), a ubiquitous environmental contaminant, and its metabolites induce autoimmune response in experimental animals (Banerjee et al., 2020; Khan et al.,1995, 2001; Khan and Wang, 2018; Wang et al., 2019) and oxidative stress plays a critical role in TCE-mediated autoimmunity (Khan et al., 2001, Khan and Wang, 2018; Wang et al., 2013, 2019). Increased reactive oxygen species (ROS)-mediated protein modifications (carbonylation and nitration) (Wang et al., 2009), and production of lipid peroxidation-derived aldehydes [i.e., 4-hydroxynonenal (HNE) and malondialdehyde (MDA)], their protein adducts and corresponding antibodies have been shown to contribute to TCE-mediated autoimmunity (Khan et al., 2001; Wang 2008, 2012). Despite involvement of oxidative stress in TCE-mediated autoimmunity, the exact mechanism by which oxidative stress participates in inducing an inflammatory and autoimmune response, and in the pathogenesis and progression of ADs are not well understood.
The oxidative stress responsive transcription factor nuclear factor (erythroid-derived 2)-like2 (Nrf2) has emerged as a pivotal player in providing protection against TCE-mediated autoimmunity (Banerjee et al., 2020), and is known to regulate a battery of antioxidant genes such as heme oxygenase-1 (HO-1) and NAD(P)H dehydrogenase, quinone 1 (NQO1), involved in the regulation of cell homeostasis, cell growth, and functions (Ahmed et al., 2017; Cuadrado et al., 2019; Geisel et al., 2014; Liang et al., 2018; Ma et al., 2013). Previously, we have shown involvement of Nrf2 and its contribution to TCE-mediated autoimmunity in a short-term study conducted in MRL+/+ mice (Banerjee et al., 2020). However, status of Nrf2 during progressive development of SLE has not been established yet.
Mitogen-activated protein kinases (MAPKs), a family of serine/threonine protein kinases, play a crucial role in innate immunity by regulating its downstream targets, including Nrf2 and inflammatory cytokines during pathogenic infections (Jeong et al., 2017; Krementsov et al., 2013; Naidu et al., 2009). MAPKs also play an important role in sequential transduction of biological cell signals and are involved in cell proliferation, apoptosis, cell differentiation and inflammatory responses (Khan et al., 2006; Naidu et al., 2009; Sun 2009). In mammalian cells, there are three subfamilies of MAPKs (p38, JNK and ERK), which are activated in a cascade fashion. First, the oxidative stress stimuli activate p38 and JNK, followed by transmission of cellular responses to hormones and growth factors by downstream kinase ERK (Naidu et al., 2009; Sun et al., 2009). p38 MAPK pathway is known to regulate inflammatory responses and cell death (Krementsov et al., 2013; Naidu et al., 2009). T cells and macrophages isolated from an experimental autoimmune encephalomyelitis (EAE) animals showed increased phosphorylation of p38, ERK and JNK MAPKs, suggesting that phosphorylated MAPKs are involved in the initiation and progression of autoimmune response by regulating its downstream targets Nrf2 and pro-inflammatory cytokines, including IL-1, IL-6, IL-12, TNF-α and IL-17, which can then contribute to induction of inflammatory and autoimmune response (Ahmed et al., 2017; Birkner 2017; Kobayashi et al., 2016; Krementsov et al., 2013; Thalhamer et al., 2008). p38 MAPK is activated by environmental toxicants or inflammatory stimuli such as toll-like receptor (TLR) ligands or cytokines (Krementsov et al., 2013). Studies have also shown that inhibition of p38 MAPK decreases the expression of pro-inflammatory cytokines involved in the pathogenesis of ADs (Krementsov et al., 2013). The downstream MAPKs target, transcription factor Nrf2, provides protection against oxidative/xenobiotic stress, and ameliorates inflammatory response in TCE-mediated autoimmunity (Banerjee et al., 2020). Furthermore, Nrf2-deficient mice develop inflammatory and autoimmune disease phenotypes, including SLE, AIH, and multiple sclerosis (Kim et al., 2010; Kobayashi et al., 2016; Pan et al., 2011). However, it is not known if MAPKs modulate Nrf2 response to influence the TCE-mediated progressive disease development.
The present study is, therefore, focused on determining the involvement of MAPKs/Nrf2 pathways in TCE-mediated inflammatory and autoimmune responses during disease progression in MRL+/+ mice. Furthermore, the study also attempts to address if antioxidant sulforaphane (SFN) or MAPK inhibitor could provide protection against TCE-mediated inflammatory and autoimmune response.
2. Material and methods
2.1. Animals and treatments
Female MRL+/+ (MRL/MpJ) mice (5 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and maintained in the animal care facility of the University of Texas Medical Branch (UTMB) under controlled conditions of temperature and humidity with a 12-hr light/dark cycle. MRL+/+ mice are an autoimmune-prone mouse model where most autoantibodies appear several months after their birth and SLE disease appears late in the second year of their lives (Khan et al., 1995, 2001). This model thus allows evaluation of autoimmune potential of specific agents in relatively young animals. The animal protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of University of Texas Medical Branch (UTMB) and all the experiments were conducted as per National Institutes of Health (NIH) guidelines. Mice were provided standard lab chow and drinking water ad libitum and were acclimatized for a week before starting the treatments. TCE (purity 99+%, Sigma Aldrich, St. Louis, MO) was dissolved in drinking water containing 1% Alkamuls EL-620 emulsifier (Rhodia Chemicals, Cranbury, NJ). The mice, in groups of 6 each, received TCE (0.5 mg/ml) or drinking water containing emulsifier only (controls) for 24, 36 and 52 weeks to monitor specific responses during the pathogenesis and progression of the disease. Choice of TCE dose was based on previous studies (Griffin et al., 2000; Wang et al., 2007, 2012). At 24, 36 and 52 weeks following TCE treatment, the animals were euthanized under ketamine/xylazine anesthesia and blood was withdrawn. Major organs were removed, weighed, snap-frozen and stored at −80 °C for further analysis. In another set of a short-term mechanistic study, mice were randomly divided into 4 groups (n=6 per group), treated and designated as control, TCE, SFN or TCE+SFN treatment groups [TCE, 10 mmol/kg in corn oil, i.p., every 4th day; SFN (Sigma-Aldrich), 8 mg/kg in corn oil, i.p., every other day] (Guerrero-Beltrán et al., 2012; Khan 1995, 2001; Wang 2019; Zhao et al., 2010). Following 6 weeks of TCE exposure, the animals were euthanized (similarly as described above) and blood was withdrawn. Major organs were removed and weighed. Portions of liver from each group were snap-frozen for further analysis. Sera obtained after blood clotting and centrifugation were stored in small aliquots at −80 °C for further analysis.
2.2. Analysis of anti-dsDNA antibodies in MRL+/+ mice
Mouse specific ELISA kit (Alpha Diagnostic Int’l, San Antonio, TX) was used to measure the anti-dsDNA antibodies in the sera (Wang et al., 2014).
2.3. Real-time PCR
RiboPure™ RNA Purification Kit (Invitrogen, Carlsbad, CA) was used to isolate total RNA from liver tissues. cDNA was synthesized using iScript™ cDNA synthesis kit (Bio-Rad Laboratories, Hercules, CA). The primer sequencing design was done using Primer3Plus program. The primers were purchased from Integrated DNA Technologies (Coralville, IA) and mRNA expressions of Nrf2, HO-1, NF-kB, iNOS, cytokines and chemokines were determined. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous loading control (Banerjee et al., 2016). Primer sequences are provided in supplementary Table S1.
2.4. Western blotting
Nuclear proteins were extracted from liver tissues of MRL+/+ mice using NE-PER nuclear extraction reagents (ThermoFisher Scientific, Waltham, MA) following the manufacturer’s instructions, and total proteins were isolated from liver tissues using RIPA lysis buffer (Cell Signaling Technology, Danvers, MA) with 1% protease inhibitor (Sigma-Aldrich). Equal amounts of protein lysates (30 μg) were used for Western blot analyses. The immunoblots were probed with primary antibodies against Nrf2, NQO1, iNOS, pNF-κB, p38, p-p38, JNK, p-JNK, ERK, p-ERK and β-Actin (Cell Signaling Technology, Danvers, MA) (Banerjee et al., 2012; Wang et al., 2010).
2.5. Histological examination and immunohistochemical staining for CD3+ T cells
Liver and kidney tissue samples were fixed in 10% buffered formalin, dehydrated and paraffin embedded. These paraffin sections were stained with hematoxylin-eosin (H&E) (Kondraganti et al., 2012). Formalin fixed and paraffin embedded liver and kidney sections from control and TCE-treated mice were deparaffinized and then the sections were treated with target antigen retrieval solution followed by overnight incubation with polyclonal rabbit anti-mouse CD3 antibody (Abcam, Cambridge, MA) at 4°C (Kondraganti et al., 2012). Sections were incubated with secondary antibody followed by peroxidase staining, and images taken with an Olympus 1X71 microscope (Olympus, Hamburg, Germany).
2.6. Cytokine array
Cytokines in liver protein samples were measured using a mouse cytokine antibody pair-based array spotted on a membrane kit (Abcam, Cambridge, MA) according to the manufacturer’s instructions. Briefly, proteins were extracted from liver tissues of MRL+/+ mice using RIPA buffer with protease inhibitor. Equal amounts of protein (300 μg) were incubated at 4 °C overnight with the supplied membranes (blocked previously). The membranes were then washed with the washing buffer followed by incubation with biotin-conjugated anti-cytokines at 4 °C overnight. After washing, the membranes were then incubated with streptavidin-HRP at 4 °C overnight. After washing, the cytokines/chemokines on membranes were detected using chemiluminescence detection solution (Martínez-Sarrà et al., 2017).
2.7. Jurkat T cells and treatments
Human Jurkat T cells (T cells) were cultured in complete RPMI 1640 media with 10% heat inactivated FBS, 1% antibiotics (penicillin and streptomycin) and incubated at 37 °C as described previously (Banerjee et al., 2020). Cells were then treated with p38 inhibitor SB203580 (10 or 20 μM; Sigma-Aldrich) for 1 h prior to incubation with or without dichloroacetyl chloride - a highly reactive metabolite of TCE known to cause autoimmune response (DCAC, 2 mM; Sigma-Aldrich) for 24 h and the mRNA expression of TNF-α and IL-12, and protein levels of NQO1 were analyzed (He et al., 2018; Naidu et al., 2009).
2.8. Determination of protein carbonyls in the liver
Carbonyl content in the mouse liver homogenates was quantitated by Protein Carbonyl Assay kit (Cayman Chemical Co., Ann Arbor, MI) (Wang et al., 2013). The liver was homogenized (10%, w/v) in phosphate-buffered saline (pH 7.4) containing protease inhibitor cocktail. Subsequent steps were performed following the manufacturer's instructions. The carbonyl content was calculated using a molar extinction coefficient of 22,000 M−1cm−1 (Wang et al., 2013).
2.9. Statistical Analysis
The values are presented as means ± SEM. The data were analyzed using one-way or two-way analysis of variance (ANOVA) followed by Tukey-Kramer multiple comparisons test. The p values <0.05 were considered statistically significant.
3. Results
3.1. TCE exposure modulated hepatic Nrf2, NF-kB and iNOS expression
Previously we have shown TCE induces an inflammatory response in a time-dependent manner during AD pathogenesis (Kondraganti et al., 2012; Wang et al., 2009). Nrf2 maintains oxidative homeostasis by regulating antioxidant and cytoprotective genes, including HO-1 and NQO1 (Banerjee et al., 2020; Loboda et al., 2016). To investigate the role of Nrf2 in TCE-mediated inflammatory and autoimmune responses during the disease progression, we treated MRL+/+ mice with TCE for 24, 36 and 52 wks. TCE treatment led to significant decreases in Nrf2 mRNA expression in the livers at 36 and 52 wks compared to their respective controls. The decreases in Nrf2 were associated with increased mRNA expression of NF-kB (p65) and iNOS at 36 and 52 weeks, while no changes were observed at 24 wks (Fig. 1A, 1B and 1C). Protein expression patterns for Nrf2, p-65 and iNOS were similar to their mRNAs, showing decreases in Nrf2 and increases in p-p65 and iNOS at 36 and 52 wks following TCE exposure (Fig. 2A, 2B, 2C and 2D).
FIG. 1.
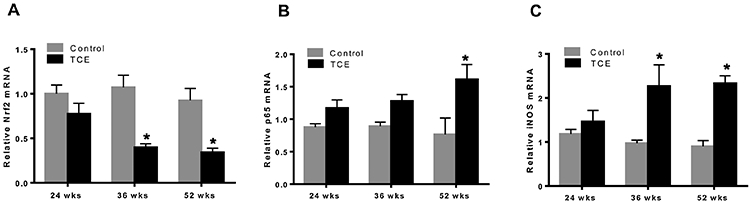
TCE exposure suppressed Nrf2, but increased NF-kB (p65) and iNOS mRNA expression in the livers of MRL+/+ mice. (A-C) mRNA expression of Nrf2, p-p65 and iNOS; n=4 in each group. *p<0.05 vs control.
FIG. 2.
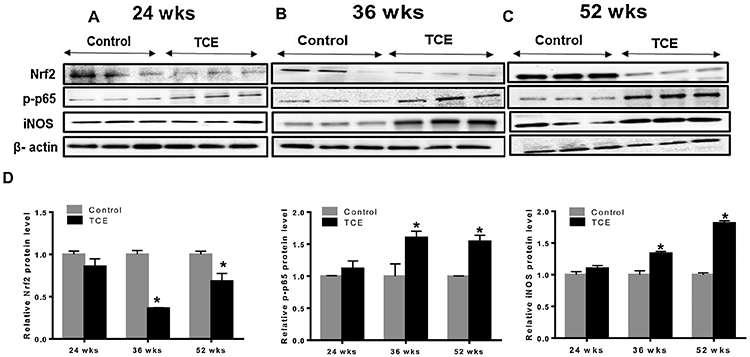
TCE exposure suppressed Nrf2, but increased NF-kB (p65) and iNOS protein expression in the livers of MRL+/+ mice. (A-C) Protein expression of Nrf2, p-p65 and iNOS. (D) Relative protein levels of Nrf2, p-p65 and iNOS; n=3 in each group. *p<0.05 vs controls.
3.2. MAPK activation (phosphorylation) following TCE exposure
MAPK subfamilies are highly linked with oxidative stress and play a major role in regulating a variety of biological processes such as gene expression, cell proliferation, differentiation, migration, inflammatory responses and cell death (Khan et al., 2006; Naidu et al., 2009). The activity of MAPKs (ERK, JNK and p38) is regulated by phosphorylation cascades (Espinosa-Diez et al., 2015). To assess the effect of TCE exposure on the activation of MAPKs, we evaluated the phosphorylated forms of ERK, JNK and p38 kinases in the livers of MRL+/+ mice treated with TCE for 24, 36 and 52 weeks. TCE exposure resulted in increased phosphorylation of p38 (p-p38), ERK (p-ERK) and JNK (p-JNK) at 36 and 52 wks, but no significant changes were observed at 24 weeks (Fig. 3A, 3B, 3C and 3D).
FIG. 3.
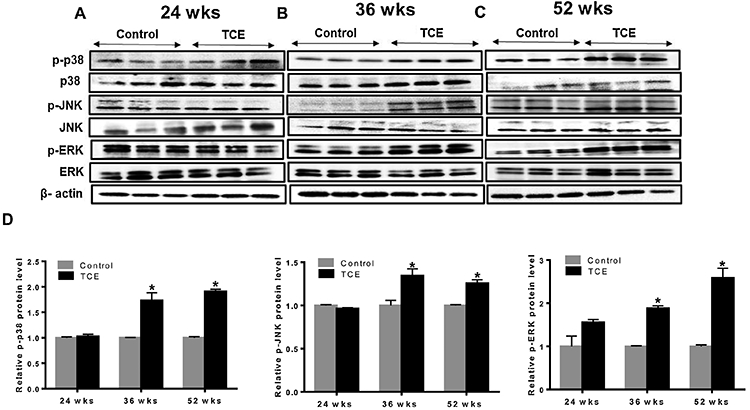
TCE-induced phosphorylation of MAPKs in the livers of MRL+/+ mice. (A-C) Protein expression of p-p38, p38, p-JNK, JNK, p-ERK and ERK. (D) Relative protein levels of p-p38, p-JNK and p-ERK were analyzed; n=3 in each group. *p<0.05 vs controls.
3.3. TCE exposure induced autoimmune response
Anti-dsDNA is a classic biomarker of SLE (Arriens et al., 2017; Ippolito et al., 2011; Pisetsky et al., 2018). To investigate TCE-mediated autoimmune response during disease progression, we, therefore, measured anti-dsDNA antibodies in the sera of mice treated with TCE for 24, 36 and 52 wks. TCE exposure led to significant increases in anti-dsDNA antibodies in mice treated with TCE for 36 and 52 wks (Fig. 4A). These findings, apart from confirming the potential of TCE in inducing an autoimmune response, are also consistent with earlier reported time-dependent responses (Wang et al., 2012).
FIG. 4.
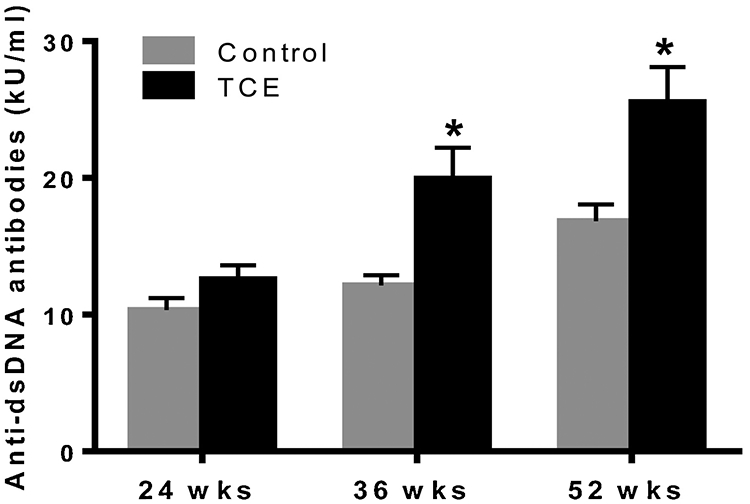
TCE treatment resulted in increased anti-dsDNA antibodies in the sera of MRL+/+ mice. Anti-dsDNA antibodies were analyzed in the sera of these mice with mouse specific ELISA kit; n=4 in each group. *p<0.05 vs controls.
3.4. Increased carbonylation of proteins (protein carbonyls) following TCE exposure
Increased carbonylation of proteins is a biomarker of oxidative protein damage (Morgan et al., 2005). To evaluate the status and potential role of protein oxidation in TCE-induced autoimmune response, we determined protein carbonyls during the progression of autoimmune response. Our results show that carbonyl content in the liver proteins was significantly increased following 36 and 52 wks of TCE exposure compared to their respective controls (Fig. 5). No changes were observed at 24 weeks of TCE exposure.
FIG. 5.
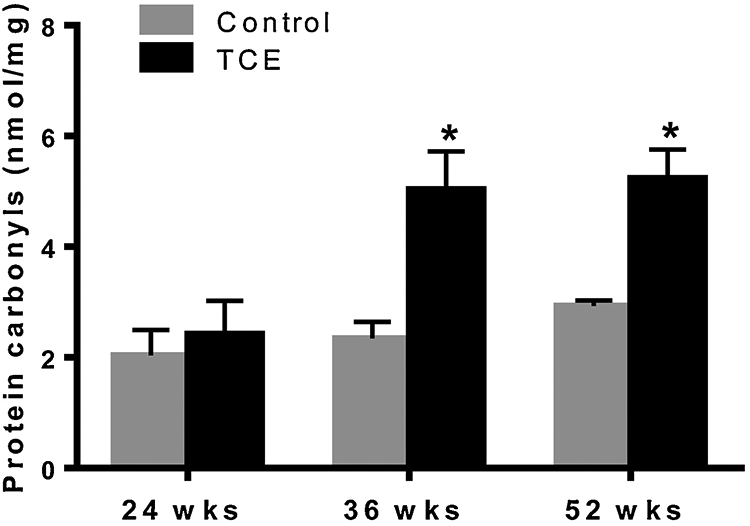
TCE exposure led to increased protein carbonyls in the livers of MRL+/+. Protein carbonyl content was analyzed in the in livers using a colorimetric assay kit; n=4 in each group. *p<0.05 vs controls.
3.5. TCE exposure leads to lymphocytic infiltration in the liver and kidney
Infiltration of immune cells in the liver or kidney is associated with AIH or SLE phenotype (Kondraganti et al., 2012; Wang et al., 2019). Based on H&E and immunohistological data, lymphocytic infiltration was seen in liver and kidney after 36 and 52 wks of TCE treatment (representative data from 52 weeks are shown in Fig. 6 a and 6. b), which is consistent with our previous findings (Kondraganti et al., 2012). CD3+ T lymphocytes were found in the renal interstitial spaces of the renal cortex [arrows; Fig. 6 b (B)] and in the periportal and central vein areas of liver [arrows; Fig. 6 b (D)].
FIG. 6.

TCE treatment induced lymphocytic infiltration in the kidneys and livers. (a; A-B and C-D) H&E sections showing lymphocyte infiltration in liver and kidney. (b; A-B) CD3+ T cells infiltration in the kidney of control and TCE-exposed mice. (b; C-D) CD3+ T cells infiltration in the liver of control and TCE-exposed mice. Arrows indicate T cell infiltration confined around interstitial spaces of the renal cortex (B) and periportal and central veins of liver (D). (20X magnification); n=4 in each group.
3.6. TCE exposure causes aberrant cytokine expression in the liver
Cytokines and chemokines play a crucial role in the development of inflammatory and ADs (Krementsov et al., 2013). We, therefore, measured cytokines in the livers at 24, 36 and 52 wks of TCE treatment. TCE exposure resulted in elevated expression of TNF-α, IL-12 (p70) and RANTES at 36 and 52 wks (Fig. 7A, 7B, 7C, 7D, 7E and 7F). The changes in these cytokines were further validated by RT-PCR analysis, which showed similar increases in their mRNA expression at 36 and 52 wks (Fig. 7G). No significant changes were observed at 24 wks of TCE exposure (Fig. 7).
FIG. 7.

TCE exposure induced cytokine/chemokine protein and mRNA expression in the livers of MRL+/+mice. (A) Representative cytokine/chemokine spot positions (in duplicate) on the membrane. (B) Relative protein levels of TNFα, IL-12 and RANTES (C) TNFα, IL-12 and RANTES mRNA expression; n = 4 in each group. *p<0.05 vs controls.
3.7. TCE-induced inflammatory response was ameliorated by blocking MAPK pathway
MAPKs play a vital role in regulating inflammatory responses and innate immunity by triggering cytokines production (Krementsov et al., 2013). Activation of MAPKs is involved in the initiation and progression of inflammatory and autoimmune response by regulating its downstream pro-inflammatory cytokines (Birkner et al., 2017; Thalhamer et al., 2008). To further determine the contribution of MAPKs, especially p38 in TCE-mediated autoimmunity, two approaches were used. The first approach in vivo utilized an antioxidant SFN along with TCE, whereas in the second approach in vitro, specific p38 inhibitor was used.
To evaluate the contribution of MAPKs in vivo, short-term studies were conducted using a relatively higher dose of TCE which can induce an autoimmune response in a relatively shorter duration (6 weeks) (Banerjee et al., 2020; Khan et al., 1995, 2001; Wang et al., 2019). MRL+/+ mice also receiving antioxidant SFN along with TCE for 6 wks, not only attenuated the TCE-mediated phosphorylation of p38 (no changes were observed for JNK; data not shown) but also suppressed the pro-inflammatory cytokines TNF-α and IL-12 production (Fig. 8A, 8B, 8C and 8D). These results thus provide evidence for a protective role of SFN against TCE-mediated oxidative stress via attenuation of p38 MAPK dysregulation. To further confirm the role of p38, we conducted in vitro studies by incubating T cells with an inhibitor of p38 (SB203580) with or without a TCE metabolite DCAC (Banerjee et al., 2020; Wang et al., 2018). Incubation of T cells with p38 inhibitor attenuated the DCAC-induced inflammatory response as evident from decreased levels of downstream cytokines TNF-α and IL-12 production (Fig. 9A and 9B). Also, p38 inhibitor ameliorated the DCAC-mediated decrease in NQO1 expression, and the response was better with the higher concentration (20 μM) of the inhibitor (Fig. 9C and 9D). These observations along with our earlier reported findings on reduction of anti-dsDNA antibodies following SFN treatment (Banerjee et al., 2020) support an important role for p38 MAPK in TCE-mediated autoimmunity.
FIG. 8.
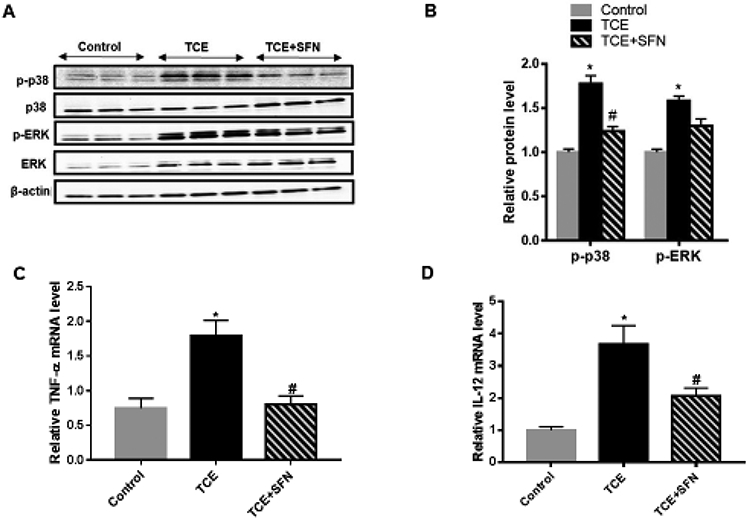
Treatment with SFN ameliorated the TCE-mediated changes in MAPKs and cytokine levels in the livers of MRL+/+ mice. (A) Mice were treated with TCE with or without SFN, and protein expression of p-p38, p38, p-ERK and ERK was determined. (B) Relative protein levels of p-p38 and p-ERK. (C-D) TNFα and IL-12 mRNA expression; n = 3-4 in each group. *p<0.05 vs controls; # p<0.05 vs TCE-treated mice.
FIG. 9.
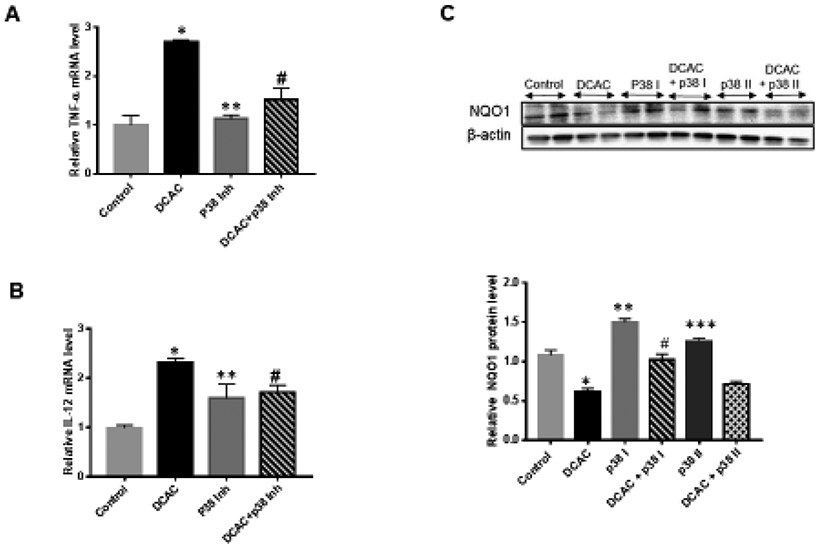
Blocking MAPK pathway with p38 inhibitor (SB203580) attenuated TCE metabolite DCAC-mediated inflammatory response in Jurkat T cells. (A-B) p38 inhibitor (10 μM) suppressed DCAC-induced increased expression of cytokines TNF-α and IL-12 mRNA. (C-D) p38 inhibitor (p38 I, 10 μM; p38 II, 20 μM) attenuated DCAC-mediated suppression of NQO1; n = 4 in each group. *p<0.05 vs control; **p<0.05 vs DCAC; # p<0.05 vs DCAC; ***p<0.05 vs DCAC ## p<0.05 vs DCAC.
4. Discussion
ADs, including SLE and AIH, are associated with immune dysregulation, increased production of autoantibodies against self-antigens and T lymphocyte infiltration in liver, kidney or lungs which can further lead to tissue damage (Khan and Wang, 2018; Khan and Wang, 2020; Pollard et al., 2010; Tsokos et al., 2016). Several intrinsic (age, sex, genetics, etc.) and extrinsic (environmental chemicals, toxicants, etc.) factors can play critical roles in the pathogenesis of ADs (Khan and Wang, 2020; Pollard et al., 2010). TCE, ubiquitous environmental contaminant, has been involved in the development of autoimmune disorders in humans (Cooper et al., 2009; Flindt-Hansen et al., 1987; Gist et al., 1995) and in experimental animals (Banerjee et al., 2020; Gilbert et al., 2009, 2014; Khan et al., 1995; Wang et al., 2007). Previously, in a short-term study, we showed that TCE exposure in MRL+/+ mice leads to downregulation of transcription factor Nrf2, which plays a critical role in cellular defense against oxidative stress (Banerjee et al., 2020). The current study was aimed to further understand the status and potential role of oxidative stress-responsive Nrf2 and MAPKs in TCE-mediated inflammatory and autoimmune responses during the disease progression, and protective mechanism of antioxidant SFN against TCE-mediated inflammatory response.
The transcription factor Nrf2 has emerged as a crucial player in protecting cells against oxidant-mediated injury by inhibiting redox-sensitive inflammatory gene expression. Nrf2 regulates battery of antioxidant and cytoprotective proteins, including HO-1, NQO1 and GSH (Banerjee et al., 2020; Jaiswal et al., 2004; Kaspar et al., 2009; Loboda et al., 2016). Nrf2-deficient mice are prone to develop disorders that are caused by oxidative stress and inflammation, leading to ADs (Li et al., 2004; Yoh et al., 2001). Nrf2 is regulated by upstream kinases such as p38 and ERK (Huang et al., 2002; Jeong et al., 2017; Rodrıguez-Ramiro et al., 2012; Sun et al., 2009). Our data in this study provides evidence that TCE exposure suppresses Nrf2 expression in a time-dependent manner. TCE exposure, on the other hand, induced the phosphorylation of upstream cell signal protein kinases MAPKs (p38, ERK and JNK). It is thus clearly evident from our findings that TCE exposure modulates MAPK and Nrf2 expressions differently, causing activation of MAPK pathway (increased phosphorylation of p38, ERK and JNK) and suppressing Nrf2 expression in a time-dependent manner. The observed changes both in Nrf2 and MAPKs following TCE exposure are remarkable as a similar time-dependent pattern in autoimmune response (ANA and anti-dsDNA antibodies) was also observed earlier (Wang et al., 2007), suggesting a clear association among these responses. Even though upstream kinases, including MAPKs, are known to modulate Nrf2 and its dependent antioxidant genes such as HO-1 and NQO1 (Naidu et al., 2009; Rodrıguez-Ramiro et al., 2012; Sun et al., 2009), precise mechanisms of their interaction in the regulation of Nrf2 is not clear. Several studies suggest that p38 MAPK negatively regulates Nrf2 activation (Huang et al., 2000; Jang et al., 2016; Keum et al., 2006; Lee et al., 2013; Yu et al., 2000). A recent study also reported that unlike ERK, p38 does not facilitate Nrf2 translocation to the nucleus, but inversely regulates Nrf2 gene induction (Wei et al., 2020). Those findings and our observation of TCE-mediated p38 MAPK activation suggest a potential mechanism for the reduced translocation of Nrf2 to the nucleus where it binds to ARE and induces the transcription of phase II detoxifying genes (Keum et al., 2006; Rodrıguez-Ramiro et al., 2012). Activation of p38 MAPK can phosphorylate Nrf2 and promote the association between Nrf2 and Keap1, thereby potentially resulting in reduced translocation of Nrf2 to nucleus (Keum et al., 2006). This is well supported by reports that SFN induces Nrf2 and suppresses p38/VCAM or NFkB expression (Chen et al., 2009; Qin et al., 2018; Zakkar et al., 2009). Interestingly, antioxidant SFN attenuated the TCE-mediated phosphorylation of p38, potentially disrupting Nrf2/Keap1 complex and allowing Nrf2 translocation to the nucleus, and thus providing protection against TCE-mediated immune response by inducing cytoprotective genes (Banerjee et al., 2020). Furthermore, our in vitro studies with p38 MAPK inhibitor showed increased expression of antioxidant NQO1, but attenuation of pro-inflammatory cytokine release from DCAC-treated T cells, also suggesting a significant role of oxidative stress-MAPK/Nrf2 axis in TCE-mediated inflammation and autoimmune response.
In a recent study, we provided evidence that crosstalk between Nrf2 and NF-κB serves as a pivotal mechanism in TCE-induced inflammatory and autoimmune response (Banerjee et al., 2020). TCE suppresses Nrf2, but increases inflammatory markers NF-κB and iNOS, leading to disturbances in the cellular redox status and causing oxidative tissue damage (Banerjee et al., 2020; Wang et al., 2007). Our findings in this study suggest that MAPKs can play an important role in TCE-mediated inflammatory response as TCE treatment resulted in increased MAPK phosphorylation as well as induction of NF-κB and iNOS expression in a time-dependent manner. MAPKs can modulate Nrf2 and NF-κB activities through phosphorylation at specific sites (Gerardi et al., 2019). In addition, MAPK p38 is also shown to play a crucial role in inflammation and transcriptional regulation of NF-κB (Saha et al., 2007), and inhibiting MAPK p38 in fact resulted in decreased transcriptional activity of NF-κB (Saha et al., 2007). Our findings in this study on MAPK, Nrf2 and NF-kB along with increases in protein carbonyls and anti-dsDNA antibodies clearly suggest a role for oxidative stress-responsive signaling pathways in TCE-mediated autoimmunity. The fact that SFN treatment resulted in attenuation of TCE-mediated anti-dsDNA antibodies (Banerjee et al., 2020), further supports a critical role of oxidative stress-responsive mechanisms.
The infiltration of immune cells into the livers, kidneys or lungs are associated with the pathogenesis and progression of ADs (Kavian et al., 2018; Kondraganti et al., 2012; Lugade et al., 2011; Wang et al., 2019). In SLE patients, it was reported that T cells increase the autoimmune response once self-tolerance is compromised and play an important role by inducing aberrant cytokine expression (Comte et al., 2015; Shlomchik et al., 2001). More importantly, Nrf2 knocked out (Nrf2−/−) mice exhibited increased T lymphocyte (CD3) infiltration in the lungs (Lugade et al., 2011). Our observation of increased T lymphocytic infiltration (in the liver and kidney) in this study and induction of pro-inflammatory cytokines in the TCE-treated livers in a time-dependent manner support a critical role of T cells in the progression of ADs.
It is well established that MAPKs play an important role in the upregulation of pro-inflammatory cytokines (Krementsov et al., 2013; Naidu et al., 2009). MAPKs are also important in the regulation of both innate and adaptive immunity by impacting cellular responses and production of cytokines that could further contribute to AD pathogenesis (Birkner et al., 2017; Lim et al., 2014). Our observation in this study that TCE induces the expression of pro-inflammatory cytokines IL-12 and TNF-α, and the chemokine RANTES in the livers in a time-dependent manner, suggests their contribution in the disease progression. These cytokines have the potential to regulate immune responses during antigen presentation and are involved in differentiation of naïve T cells, and thus present as therapeutic targets for ADs (Maldini et al., 2018; Sun et al., 2009). Interestingly, Nrf2 also controls the inflammation through transcriptional inhibition of pro-inflammatory cytokines such as IL-6 and IL-1β genes (Kobayashi et al., 2016). It is thus not surprising that Nrf2-knockout mice show higher expression of inflammatory markers such as IL-6, and TNF-α (Ahmed et al., 2017). Since p38 MAPK activation could promote the production of downstream pro-inflammatory cytokines, including IL-12 and TNF-α (Krementsov et al., 2013), blocking MAPK p38 in this study indeed led to suppression of pro-inflammatory cytokines TNF-α and IL-12 in T cells, thus presenting a potential mechanism and their involvement in TCE-mediated autoimmunity. The present study also showed that antioxidant SFN can reduce TCE-mediated p38 MAPK phosphorylation as well as expression of its downstream targets IL-12 and TNF-α. Data from our studies thus help in minimizing the knowledge gap in understanding the mechanisms by which SFN and/or p38 MAPK inhibitor could provide protection against ADs.
In conclusion, our data illustrates the status and protective role of Nrf2 in TCE-mediated inflammatory and autoimmune responses during disease progression. We also demonstrated that MAPK pathways can directly influence cytokine production in TCE-treated mice. Blocking MAPK p38 pathways and supplementation of antioxidant SFN resulted in the suppression of pro-inflammatory cytokines TNF-α and IL-12, and consequently attenuated TCE-mediated inflammatory and autoimmune responses. Our study thus provides clear evidence for the contribution of Nrf2 and MAPK (especially p38) pathways in AD pathogenesis and holds strong promise in delineating novel therapeutic targets not only against TCE-mediated inflammatory and autoimmune responses but also other ADs involving oxidative stress, in general. More in-depth studies are needed to better understand and establish therapeutic use of SFN and/or MAPK inhibitors leading to novel opportunities for the prevention and treatment of ADs.
Supplementary Material
FIG. 10.

Schematic presentation of the effects of TCE on Nrf2/MAPKs signaling pathway. TCE exposure modulated MAPKs and its downstream targets Nrf2, NF-kB and pro-inflammatory cytokines (eg. TNFα, IL-12) and resulted in a pro-inflammatory response. Antioxidant SFN supplementation and p38 MAPK inhibitor provided protection against TCE-mediated inflammation and autoimmunity by modulating Nrf2/MAPK pathways.
Highlights.
TCE- mediated dysregulation of Nrf2 signaling contributes to disease progression
TCE treatment causes increased phosphorylation of p38, ERK and JNK
TCE exposure modulates inflammatory cytokines/chemokines
Sulforaphane ameliorates TCE-induced p38 phosphorylation and inflammation
Redox-sensitive Nrf2 and MAPK signaling contribute to TCE-mediated autoimmunity
Acknowledgements
This work was supported by R01 Grants ES016302 and ES026887 from the National Institute of Environmental Health Sciences (NIEHS), NIH, and its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIEHS, NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interests.
References
- Ahmed SM, Luo L, Namani A, Wang XJ, Tang X, 2017. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta. Mol. Basis Dis 1863, 585–597. [DOI] [PubMed] [Google Scholar]
- Arriens C, Wren JD, Munroe ME, Chandra M, 2017. Systemic lupus erythematosus biomarkers: the challenging quest. Rheumatology (Oxford). 56, i32–i45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Talcott S, Safe S, Mertens-Talcott SU, 2012. Cytotoxicity of pomegranate polyphenolics in breast cancer cells in vitro and vivo: potential role of miRNA-27a and miRNA-155 in cell survival and inflammation. Breast Cancer Res. Treat 136, 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee N, Kim H, Talcott ST, Turner ND, Byrne DH, Mertens-Talcott SU, 2016. Plum polyphenols inhibit colorectal aberrant crypt foci formation in rats: potential role of the miR-143/protein kinase B/mammalian target of rapamycin axis. Nutr. Res 36, 1105–1113. [DOI] [PubMed] [Google Scholar]
- Banerjee N, Wang H, Wang G, Khan MF, 2020. Enhancing the Nrf2 Antioxidant Signaling Provides Protection Against Trichloroethene-mediated Inflammation and Autoimmune Response.Toxicol. Sci 175, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkner K, Wasser B, Loos J, Plotnikov A, Seger R, Zipp F, Witsch E, Bittner S, 2017. The Role of ERK Signaling in Experimental Autoimmune Encephalomyelitis. Int. J. Mol. Sci 18, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai P, König R, Boor PJ, Kondraganti S, Kaphalia BS, Khan MF, Ansari GAS, 2008. Chronic exposure to trichloroethene causes early onset of SLE-like disease in female MRL +/+ mice. Toxicol. Appl. Pharmacol 228, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XL, Dodd G, Kunsch C, 2009. Sulforaphane inhibits TNF-alpha-induced activation of p38 MAP kinase and VCAM-1 and MCP-1 expression in endothelial cells. Inflamm. Res 58, 513–521. [DOI] [PubMed] [Google Scholar]
- Comte D, Karampetsou MP, Tsokos GC, 2015. T cells as a therapeutic target in SLE. Lupus 24, 351–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper GS, Makris SL, Nietert PJ, Jinot J, 2009. Evidence of autoimmune-related effects of trichloroethylene exposure from studies in mice and humans. Environ. Health Perspect 117, 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Rojo AI, Wells G, Hayes JD, Cousin SP, Rumsey WL, Attucks OC, Franklin S, Levonen AL, Kensler TW, Dinkova-Kostova AT, 2019. Therapeutic targeting of the Nrf2 and Keap1 partnership in chronic diseases. Nat. Rev. Drug Discov 4, 295–317. [DOI] [PubMed] [Google Scholar]
- Desai RD, Brinton MK, 2019. Autoimmune Disease in Women: Endocrine Transition and Risk Across the Lifespan. Front. Endocrinol. Lausanne 10, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez-Pérez P, Cadenas S, Lamas S, 2015. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 6, 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flindt-Hansen H, Isager H, 1987. Scleroderma after occupational exposure to trichlorethylene and trichlorethane. Acta. Derm. Venereol 67, 263–264. [PubMed] [Google Scholar]
- Geisel J, Brück J, Glocova I, Dengler K, Sinnberg T, Rothfuss O, Walter M, Schulze-Osthoff K, Röcken M, Ghoreschi K, 2014. Sulforaphane protects from T cell-mediated autoimmune disease by inhibition of IL-23 and IL-12 in dendritic cells. J. Immunol 192, 3530–3539. [DOI] [PubMed] [Google Scholar]
- Gerardi G, Cavia-Saiz M, Rivero-Pérez D M.D., González-San ML, PilarMuñiz J, 2019. Modulation of Akt-p38-MAPK/Nrf2/SIRT1 and NF-κB pathways by wine pomace product in hyperglycemic endothelial cell line. Journal of Functional Foods 58, 255–265. [Google Scholar]
- Gilbert KM, Przybyla B Pumford NR, Han T, Fuscoe J, Schnackenberg LK, Holland RD, Doss JC, Macmillan-Crow LA, Blossom SJ, 2009. Delineating liver events in trichloroethylene-induced autoimmune hepatitis. Chem. Res. Toxicol 22, 626–632. [DOI] [PubMed] [Google Scholar]
- Gilbert KM, Reisfeld B, Zurlinden TJ, Kreps MN, Erickson SW, Blossom SJ, 2014. Modeling toxicodynamic effects of trichloroethylene on liver in mouse model of autoimmune hepatitis.Toxicol. Appl. Pharmacol 279, 284–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gist GL, Burg JR, 1995.Trichloroethylene–a review of the literature from a health effects perspective.Toxicol. Ind. Health 11, 253–307. [DOI] [PubMed] [Google Scholar]
- Griffin JM, Gilbert KM, Pumford NR, 2000. Inhibition of CYP2E1 Reverses CD4+ T-Cell Alterations in Trichloroethylene-Treated MRL+/+ Mice. Toxicol. Sci 54, 384–589. [DOI] [PubMed] [Google Scholar]
- Guerrero-Beltrán CE, Calderón-Oliver M, Pedraza-Chaverri J, Chirino YI, 2012. Protective effect of sulforaphane against oxidative stress: recent advances. Exp. Toxicol. Pathol 64, 503–508. [DOI] [PubMed] [Google Scholar]
- He T, Liu S, Chen S, Ye J, Wu X, Bian Z, Chen X, 2018. The p38 MAPK Inhibitor SB203580 Abrogates Tumor Necrosis Factor-Induced Proliferative Expansion of Mouse CD4 + Foxp3 + Regulatory T Cells. Front. Immunol 9, 1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB, 2000. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc. Natl. Acad. Sci 97, 12475–12480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HC, Nguyen T, Pickett CB, 2002. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem 277, 42769–42774. [DOI] [PubMed] [Google Scholar]
- Ippolito A, Wallace DJ, Gladman D, Fortin PR, Urowitz M, Werth V, Costner M, Gordon C, Alarcón GS, Ramsey-Goldman, Maddison P Clarke, Bernatsky S, Manzi S, Bae SC, Merrill JT, Ginzler E, Hanly JG, Nived O, Sturfel G, Sanchez-Guerrero J, Bruce I, Aranow C, Isenberg D, Zoma A, Magder LS, Buyon J, Kalunian K, Dooley MA, Steinsson K, van Vollenhoven RF, Stoll T, Weisman M, Petri M, 2011. Autoantibodies in systemic lupus erythematosus: comparison of historical and current assessment of seropositivity. Lupus 20, 250–255. [DOI] [PubMed] [Google Scholar]
- Jaiswal AK, 2004. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med 36, 1199–1207. [DOI] [PubMed] [Google Scholar]
- Jang M, Cho IH, 2016. Sulforaphane ameliorates 3-nitropropionic acid-induced striatal toxicity by activating the Keap1-Nrf2-ARE pathway and inhibiting the MAPKs and NF-κB pathways. Mol. Neurobiol 53, 2619–2635. [DOI] [PubMed] [Google Scholar]
- Jeong CB, Kang HM, Lee MC, Kim DH, Han J, Hwang DS, Souissi S, Lee SJ, Shin KH, Park HG, Lee JS, 2017. Adverse effects of microplastics and oxidative stress-induced MAPK/Nrf2 pathway-mediated defense mechanisms in the marine copepod Paracyclopina nana. Scientific Reports 7, 41323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaspar JW, Niture SK, Jaiswal AK, 2009. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med 47, 1304–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavian N, Mehlal S, Jeljeli M, Saidu NEB, Nicco C, Cerles O, Chouzenoux S, Cauvet A, Camus C, Ait-Djoudi M, Chéreau C, Kerdine-Römer S, Allanore Y, Batteux F, 2018. The Nrf2-antioxidant response element signaling pathway controls fibrosis and autoimmunity in scleroderma. Front. Immunol 9, 1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keum YS, Yu S, Chang PPJ , Yuan X, Kim JH, Xu C, Han J, Agarwal A, Kong ANT, 2006. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 66, 8804–8813. [DOI] [PubMed] [Google Scholar]
- Khan MF, Kannan S, Wang J, 2006. Activation of transcription factor AP-1 and mitogen-activated protein kinases in aniline-induced splenic toxicity. Toxicol. Appl. Pharmacol 210, 86–93. [DOI] [PubMed] [Google Scholar]
- Khan MF, Wu X, Ansari GAS, 2001. Anti-malondialdehyde antibodies in MRL+/+ mice treated with trichloroethene and dichloroacetyl chloride: possible role of lipid peroxidation in autoimmunity.Toxicol. Appl. Pharmacol 170, 88–92. [DOI] [PubMed] [Google Scholar]
- Khan MF, Wang G, 2018. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol 7, 22–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MF, Wang H, 2020. Environmental Exposures and Autoimmune Diseases: Contribution of Gut Microbiome. Front. Immunol 10, 3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MF, Kaphalia BS, Prabhakar BS, Kanz MF, Ansari GAS 1995. Trichloroethene-induced autoimmune response in female MRL +/+ mice.Toxicol. Appl. Pharmacol 134, 155–160. [DOI] [PubMed] [Google Scholar]
- Kim J, Cha YN, Surh YJ, 2010. A protective role of nuclear factor-erythroid 2-related factor-2 (Nrf2) in inflammatory disorders. Mutat. Res, 690, 12–23. [DOI] [PubMed] [Google Scholar]
- Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, Tanaka N, Moriguchi T, Motohashi H, Nakayama K, Yamamoto M, 2016. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun 7, 11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondraganti S, König R, Boor PJ, Khan S, Kaphalia BS, Khan MK, Ansari GAS, 2012. Mechanistic evaluation of trichloroethene-mediated autoimmune hepatitis-like disease in female MRL+/+ mice.The Open Toxicol. J 5, 01–10. [Google Scholar]
- Krementsov DN, Thornton TM, Teuscher C, Rincon M, 2013. The Emerging Role of p38 Mitogen-Activated Protein Kinase in Multiple Sclerosis and Its Models. Mol. Cell Biol 33, 3728–3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Khor TO, Shu L, Su ZY, Fuentes F, Kong Ah-Ng T., 2013. Dietary phytochemicals and cancer prevention: Nrf2 signaling, epigenetics, and cell death mechanisms in blocking cancer initiation and progression. Pharmacol. Ther 137, 153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Stein TD, Johnson JA, 2004. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genomics 18, 261–272. [DOI] [PubMed] [Google Scholar]
- Liang J, Jahraus B, Balta E, Ziegler JD, Hübner K, Blank N, Niesler B, Wabnitz GH, Samstag Y, 2018. Sulforaphane inhibits inflammatory responses of primary human t-cells by increasing ros and depleting glutathione. Front. Immunol 9, 02584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MX, Png CW, Tay CYB, Teo JDW, Jiao H, Lehming N, Tan KSW, Zhang Y, 2014. Differential regulation of proinflammatory cytokine expression by mitogen-activated protein kinases in macrophages in response to intestinal parasite infection. Infect. Immun 82, 4789–4801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loboda A, Damulewicz M, Pyza E, Jozkowicz A, Dulak J, 2016. Role of Nrf2/HO-1 system in development, oxidative stress response and diseases: an evolutionarily conserved mechanism. Cell Mol. Life Sci 73, 3221–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugade AA, Vethanayagam RR, Nasirikenari M, Bogner PN, Segal BH, Thanavala Y, 2011. Nrf2 regulates chronic lung inflammation and B-cell responses to nontypeable Haemophilus influenzae. Am. J. Respir. Cell Mol. Biol 45, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, 2013. Role of Nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldini CR, Ellis GI, Riley JL, 2018. CAR T cells for infection, autoimmunity and allotransplantation. Nat. Rev. Immunol 18, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Sarrà E, Montori S, Gil-Recio C, Núñez-Toldrà R, Costamagna D, Rotini A, Atari M, Luttun A, Sampaolesi M, 2017. Human dental pulp pluripotent-like stem cells promote wound healing and muscle regeneration. Stem Cell Res. Ther 8, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan PE, Sturgess AD, Davies MJ, 2005. Increased levels of serum protein oxidation and correlation with disease activity in systemic lupus erythematosus. Arthritis Rheum. 52, 2069–2079. [DOI] [PubMed] [Google Scholar]
- Naidu S, Vijayan V, Santoso S, Kietzmann T, Immenschuh S, 2009. Inhibition and Genetic Deficiency of p38 MAPK Up-Regulates Heme Oxygenase-1 Gene Expression via Nrf2. J. Immunol, 182, 7048–7057. [DOI] [PubMed] [Google Scholar]
- Pan H, Wang H, Zhu L, Mao L, Qiao L, Su X, 2011. Depletion of Nrf2 enhances inflammation induced by oxyhemoglobin in cultured mice astrocytes. Neurochem. Res 36, 2434–2441. [DOI] [PubMed] [Google Scholar]
- Pisetsky DS, Spencer DM, Lipsky PE, Rovin BH, 2018. Assay variation in the detection of antinuclear antibodies in the sera of patients with established SLE. Ann. Rheum. Dis 77, 911–913. [DOI] [PubMed] [Google Scholar]
- Pollard KM, Hultman P, Kono DH, 2010.Toxicology of Autoimmune Diseases. Chem. Res. Toxicol 23, 455–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin S, Yang C, Huang W, Du S, Mai H, Xiao J, Lü T, 2018. Sulforaphane attenuates microglia-mediated neuronal necroptosis through down-regulation of MAPK/NF-κB signaling pathways in LPS-activated BV-2 microglia. Pharmaco. Res 133, 218–235. [DOI] [PubMed] [Google Scholar]
- Rodrıguez-Ramiro I, Ramos S, Bravo L, Goya L, Martın MA, 2012. Procyanidin B2 induces Nrf2 translocation and glutathioneS-transferase P1 expression via ERKs and p38-MAPK pathwaysand protect human colonic cells against oxidative stress. Eur. J. Nutr 51, 881–892. [DOI] [PubMed] [Google Scholar]
- Saha RN, Jana M, Pahan K, 2007. MAPK p38 regulates transcriptional activity of NF-kappaB in primary human astrocytes via acetylation of p65. J. Immunol 15, 7101–7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomchik MJ, Craft JE, Mamula MJ, 2001. From T to B and back again: positive feedback in systemic autoimmune disease. Nat. Rev. Immunol 1, 147–153. [DOI] [PubMed] [Google Scholar]
- Sun Z, Huang Z, Zhang DD, 2009. Phosphorylation of Nrf2 at Multiple Sites by MAP Kinases Has a Limited Contribution in Modulating the Nrf2-Dependent Antioxidant Response. PLoS One 4, e6588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalhamer T, McGrath MA, Harnett MM, 2008. MAPKs and their relevance to arthritis and inflammation. Rheumatology (Oxford) 47, 409–414. [DOI] [PubMed] [Google Scholar]
- Tsokos GC, Lo MS, Reis PC, Sullivan KE, 2016. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol 22, 716–730. [DOI] [PubMed] [Google Scholar]
- Wang G, Cai P, Ansari GAS, Khan MF, 2007. Oxidative and nitrosative stress in trichloroethene-mediated autoimmune response.Toxicology 229,186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, König R, Ansari GAS, Khan MF 2008. Lipid peroxidation-derived aldehyde-protein adducts contribute to trichloroethene-mediated autoimmunity via activation of CD4+ T cells. Free Radic. Biol. Med 44, 1475–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Ma H, Wang J, Khan MF, 2019. Contribution of poly(ADP-ribose)polymerase-1 activation and apoptosis in trichloroethene-mediated autoimmunity. Toxicol. Appl. Pharmacol 362, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Fan X, Ansari GAS, Khan MK, 2012. Protein adducts of malondialdehyde and 4-hydroxynonenal contribute to trichloroethene-mediated autoimmunity via activating Th17 cells: dose- and time-response studies in female MRL+/+ mice.Toxicology 292, 113–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Ma H, Ansari GAS, Khan MF, 2013. N-Acetylcysteine protects against trichloroethene-mediated autoimmunity by attenuating oxidative stress. Toxicol. Appl. Pharmacol 273, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Ma H, Khan MF, 2009. Increased nitration and carbonylation of proteins in MRL+/+ mice exposed to trichloroethene: potential role of protein oxidation in autoimmunity. Toxicol. Appl. Pharmacol 237, 188–195. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Pierangeli SS, Papalardo E, Ansari GAS, Khan MF, 2010. Markers of oxidative and nitrosative stress in systemic lupus erythematosus: correlation with disease activity. Arthritis Rheum. 62, 2064–2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Wang J, Luo X, Ansari GAS, Khan MF, 2014. Nitrosative stress and nitrated proteins in trichloroethene-mediated autoimmunity. PLoS One 9, e98660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang G, Ansari GAS, Khan MF, 2018. Trichloroethene metabolite dichloroacetyl chloride induces apoptosis and compromises phagocytosis in Kupffer Cells: Activation of inflammasome and MAPKs. PLoS One 13, e0210200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang G, Liang Y, Du X, Boor PJ, Sun J, Khan MF, 2019. Redox regulation of hepatic NLRP3 inflammasome activation and immune dysregulation in trichloroethene-mediated autoimmunity. Free Radic. Biol. Med 143, 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ma N, Fan X, Yu Q, Ci X, 2020. The role of Nrf2 in acute kidney injury: Novel molecular mechanisms and therapeutic approaches. Free Radic. Biol. Med 158, 1–12. [DOI] [PubMed] [Google Scholar]
- Yoh K, Itoh K, Enomoto A, Hirayama A, Yamaguchi N, Kobayashi M, Morito N, Koyama A, Yamamoto M, Takahashi S, 2001. Nrf2-deficient female mice develop lupus-like autoimmune nephritis. Kidney Int 60, 1343–1353 [DOI] [PubMed] [Google Scholar]
- Yu R, Mandlekar S, Lei W, Fahl WE, Tan TH, Kong AN, 2000. p38 mitogen-activated protein kinase negatively regulates the induction of phase II drug-metabolizing enzymes that detoxify carcinogens. J. Biol. Chem 275, 2322–2327. [DOI] [PubMed] [Google Scholar]
- Zakkar M, Heiden KVD, Luong LA, Chaudhury H, Cuhlmann S, Hamdulay SS, Krams R, Edirisinghe I, Rahman I, Carlsen H, Haskard DO, Mason JC, Evans PC, 2009. Activation of Nrf2 in endothelial cells protects arteries from exhibiting a proinflammatory state. Comparative Study. Arterioscler.Thromb. Vasc. Biol 29, 1851–1857. [DOI] [PubMed] [Google Scholar]
- Zhao HD, Zhang F, Shen G, Li YB, Li YH, Jing HR, Ma LF, Yao JH, Tian XF, 2010. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. World J. Gastroenterol 16, 3002–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.