

Abstract
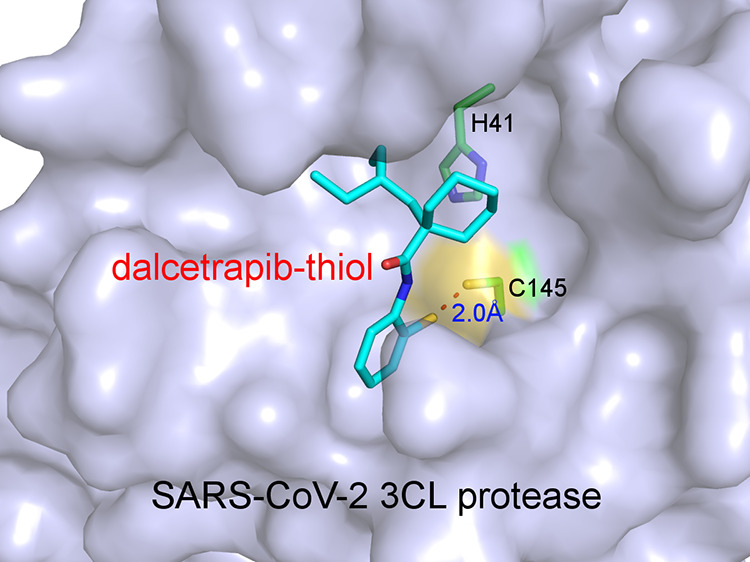
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) 3CL protease is a promising target for inhibition of viral replication by interaction with a cysteine residue (Cys145) at its catalytic site. Dalcetrapib exerts its lipid-modulating effect by binding covalently to cysteine 13 of a cholesteryl ester transfer protein. Because 12 free cysteine residues are present in the 3CL protease, we investigated the potential of dalcetrapib to inhibit 3CL protease activity and SARS-CoV-2 replication. Molecular docking investigations suggested that dalcetrapib-thiol binds to the catalytic site of the 3CL protease with a delta G value of −8.5 kcal/mol. Dalcetrapib inhibited both 3CL protease activity in vitro and viral replication in Vero E6 cells with IC50 values of 14.4 ± 3.3 μM and an EC50 of 17.5 ± 3.5 μM (mean ± SD). Near-complete inhibition of protease activity persisted despite 1000-fold dilution after ultrafiltration with a nominal dalcetrapib-thiol concentration of approximately 100 times below the IC50 of 14.4 μM, suggesting stable protease–drug interaction. The inhibitory effect of dalcetrapib on the SARS-CoV-2 3CL protease and viral replication warrants its clinical evaluation for the treatment of COVID-19.
Introduction
The severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) is the agent responsible for the COVID-19 pandemic. Its genome encodes several proteins, including the RNA-dependent RNA polymerase, main protease (3CLpro), and papain-like protease (PLpro), whose activities represent rate-limiting steps in viral replication. Upon entry of the virus in cells, its genome is released as a single-stranded positive RNA and translated into a large viral polyprotein using the host’s machinery. Following the autocatalytic cleavage of 3CLpro, the polyprotein is then cleaved at 14 different sites, 11 of these by that protease, and its recognition sequence at most of the locations was found to be Leu–Gln↓(Ser/Ala/Gly). The catalytic site of the cysteine proteases contains the Cys145–His41 dyad for 3CLpro and the classic Cys112–His273–Asp287 triad for the papain-like protease.1
Therapeutic options directly targeting the SARS-CoV-2 are presently limited to the RNA-dependent RNA polymerase inhibitor remdesivir.2 3CLpro cleaves polypeptide sequences exclusively after a glutamine residue and thus represents an ideal drug target because, to the best of our knowledge, no human host-cell protease is known with this substrate specificity.1 Peptidomimetics were designed to inhibit 3CLpro based on the structure of the binding pocket.3 The hydroxymethylketone derivative PF-008352314 inhibits 3CLpro and viral replication and is currently in the phase 1 clinical trial. The free cysteine at the catalytic site of proteases from multiple coronaviruses is located in a hydrophobic pocket and represents a potential therapeutic target.5 In addition, the coronavirus 3CL protease contains 12 free cysteine residues (i.e., not forming stable Cys–Cys bonds), thus not playing an identified role in the 3D structure of the protease.5 After the structure of 3CLpro of SARS-CoV was described,6 inhibitors were identified with EC50 values in the micromolar range.7−9 More recently, large chemical libraries have been screened in silico to identify inhibitors of this protease for the SARS-CoV-2.10,11 Potential thiol-reactive drugs targeting 3CLpro should preferably be relatively hydrophobic to enter and remain in its pocket, thus excluding several medications such as the hydrophilic captopril (log P < 1; https://hmdb.ca/metabolites/HMDB0015328).
Repurposing of currently available drugs has been proposed as a potentially efficient approach against COVID-19.12−16 Dalcetrapib is a lipid modulator that acts by binding covalently to a cysteine residue (Cys13) in a hydrophobic pocket of the target cholesteryl ester transfer protein, and this bond is a prerequisite for its pharmacological activity.17−20 Dalcetrapib-thiol also binds to a cysteine residue (Cys133) in a hydrophobic pocket of MD2, a regulator of the toll-like receptor 4 receptor signaling pathway.21 Dalcetrapib is a lipophilic thio-ester with a log P close to 7.022 (Figure 1 and Table 1). The pharmacologically active free thiol form of dalcetrapib is generated by nonspecific esterases and lipases present in body fluids such as plasma, cell homogenates, and intracellular milieu or in vitro by exposure to basic pH.23,24 The current study was conducted to test the hypothesis that dalcetrapib inhibits 3CLpro and the replication of the SARS-CoV-2.
Figure 1.

(A) Chemical structures and conversion of dalcetrapib-ester to dalcetrapib-thiol and dalcetrapib-disulfide. (B) Dalcetrapib-ester (retention time of 3.8 min) is stable under neutral or acidic conditions (black line) but is quantitatively converted into dalcetrapib-thiol (retention time, 3.1 min) in 2 mM NaOH, pH 12.6 (red line). Similar results were obtained after 30 min and 4 h incubation.
Table 1. Reference, Molecular Weight, and Log P Values for Dalcetrapib and Derivatives.
| zinc number | MW | log P | |
|---|---|---|---|
| dalcetrapib | 3976476 | 389.605 | 6.677 |
| dalcetrapib-thiol | 13813331 | 319.514 | 5.691 |
| dalcetrapib-disulfide | 114035772 | 637.12 | 11.917 |
Results
Docking of Dalcetrapib-thiol
We performed molecular docking using SwissDock25 to evaluate the potential binding modes of dalcetrapib-thiol in the active site pocket of 3CLpro. In addition to standard docking, we also used the steric-clashes alleviating receptor (SCAR) protocol by mutating the protease-reactive cysteine to glycine (C145G),26,27 which is an efficient approach to probe the possibility of covalent binding of ligands to receptors. The poses of dalcetrapib-thiol with the lowest delta G values (−8.5 kcal/mol) in both standard and SCAR protocols revealed very similar binding of this molecule in the active site of the main protease (Figure 2). In the standard protocol, dalcetrapib-thiol binds to the active site of the main protease with its carbonyl group anchored through a hydrogen bond by the main chain amide group of Glu166 (Figure 2A). Meanwhile, its aromatic ring is inserted in the S1 subsite, which recognizes the Gln residue (P1 residue) of substrate proteins, and sandwiched between Glu166 and Asn142, whereas its 2-ethylbutyl and cyclohexane moieties are snugly bound in the S2 and S4 subsites. Particularly, the van der Waals contacts with Met49, Met165, His41, and Gln189 appear to be crucial for the stabilization of dalcetrapib-thiol. The sulfur atom of dalcetrapib-thiol is 3.6 Å away from the Cys145 thiol of the protease in the standard protocol (Figure 2A,B), whereas this distance is shortened to 2.0 Å in the SCAR protocol (Figure 2C,D). This indicates the feasibility of disulfide bond formation between dalcetrapib-thiol and Cys145 of 3CLpro, and the binding pose of dalcetrapib-thiol obtained in the standard protocol could be in the trajectory toward the disulfide bond formation. The creation of a covalent bond could be facilitated by slight rotations or changes of both the aromatic ring of dalcetrapib-thiol and the thiol group of Cys145 of the main protease in order to achieve appropriate bond angles. Several ligands were bound to the active site of the main protease with their aromatic rings bound to the S1 site (e.g., PDB 5R84), as observed in our docking trials.
Figure 2.
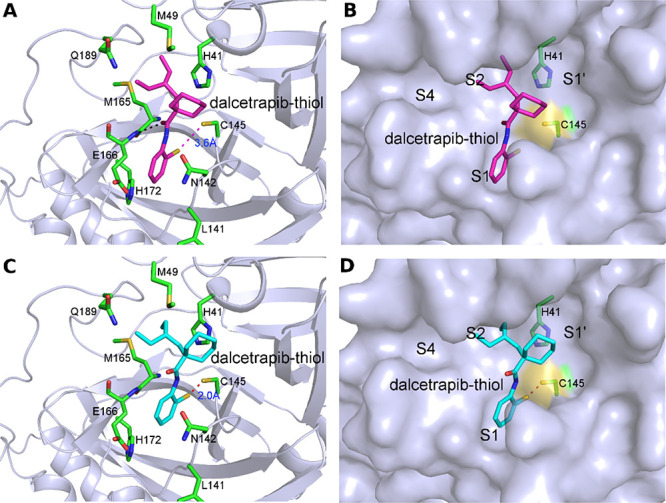
Docking of dalcetrapib-thiol in the active site of 3CLpro of the SARS-CoV-2. (A) Docking of dalcetrapib-thiol (labeled as dalcetrapib-thiol) in 3CLpro (PDB 6W63) using the standard protocol, which resulted in a distance of 3.6 Å between the thiol of dalcetrapib-thiol and Cys145. Dalcetrapib-thiol (carbon in magenta) and the surrounding residues (carbon in green) are shown in stick mode. The hydrogen bond between Dalcetrapib-thiol and the main chain amide of Glu166 is shown as a black dashed line. (B) Surface representation of (A) with the S1′, S1, S2, and S4 subsites indicated. (C) Docking of dalcetrapib-thiol to the active site of 3CLpro (PDB 6W63) using the SCAR protocol, which resulted in a distance of 2.0 Å between the thiol of dalcetrapib-thiol and Cys145. Dalcetrapib-thiol (carbon atoms in cyan) and the surrounding residues (carbon atoms in green) are shown in stick mode. (D) Surface representation of (C) with the S1′, S1, S2, and S4 subsites indicated.
Inhibition of Enzymatic Activity of 3CLpro by Dalcetrapib
We evaluated the ability of dalcetrapib to inhibit 3CLpro of the SARS-CoV-2 in vitro (Figure 3). Dalcetrapib was first de-esterified by basic hydrolysis of the ester bond to release the thiol form as described in the method section (Figure 1B). Incubation with the substrate was performed in the presence of 1 mM DTT. Some experiments where DTT was added during preincubation and protease activity, although lowered, allowed us to compute an IC50, and we observed slightly higher IC50 values (36.9 μM). Therefore, we chose to perform the preincubations in the absence of a reducing agent for the results reported in the manuscript. When dalcetrapib-thiol was incubated for 24 h in the presence of 3CLpro at room temperature before addition of the substrate, the IC50 was 14.4 ± 3.3 μM (mean ± SD) (Figure 3A). Shorter incubation of dalcetrapib-thiol for 1 h with the main protease resulted in a much higher IC50 of 75 ± 54 μM (data not shown).
Figure 3.

(A) Dalcetrapib directly inhibits SARS-CoV-2 3CLpro activity. The IC50 for dalcetrapib was determined by measuring fluorescence released from the quenched peptide exposed to the protease. Dalcetrapib was first de-esterified by basic hydrolysis of the ester bond to obtain dalcetrapib-thiol, which was then preincubated with 3CLpro for 24 h before a further 30 min preincubation in the presence of 1 mM DTT. Slopes of the relative fluorescence unit (RFU) increase were derived after the first 15 min of incubation with the peptide. Curve-fitting was performed using a GraphPad Prism four-parameter method. (B) Procedure for the assessment of the reversibility of 3CLpro inhibition by dalcetrapib. 3CLpro was incubated for 24 h with a buffer (control) or dalcetrapib-thiol produced as indicated in the method section. The incubation mixtures were then diluted or not 1000-fold through three ultrafiltration cycles to remove free dalcetrapib-thiol and concentrate 3CLpro. Samples labeled #1, #2, #3, and #4 were analyzed for 3CLpro activity (shown in panel C). (C) Assessment of the reversibility of 3CLpro inhibition by dalcetrapib. Protease activity “before” and “after” repeated dilution/filtration cycles. Dalcetrapib-thiol was preincubated with 3CLpro for 24 h at room temperature in the presence of 1 mM DTT before addition of the quenched peptide substrate. Slopes of the relative fluorescence unit (RFU) increase were derived after the first 15 min of incubation with the peptide.
The reversibility of the inhibitory activity of dalcetrapib was then evaluated. Dalcetrapib-thiol was incubated for 24 h with 3CLpro, and the solution containing both was then submitted to three cycles of dilution and filtration through a 3 kDa molecular weight cutoff membrane, thus allowing elution of free dalcetrapib-thiol (Figure 3B). Once 3CLpro was exposed to a high concentration of dalcetrapib-thiol, near-complete inhibition (97.6%) of protease activity persisted despite elution of dalcetrapib-thiol, which brought its nominal concentration down to 0.2 μM (Figure 3C), suggesting the presence of stable protease–drug interaction. Exposure to 10 mM of the reducing agents DTT or TCEP at that stage did not result in recovery of protease activity (data not shown).
When dalcetrapib-disulfide was incubated for 24 h in the presence of 3CLpro at room temperature with 1 mM DTT before addition of the substrate, the IC50 was 72.5 ± 9.1 μM (data not shown).
Inhibition of SARS-CoV-2 Replication by Dalcetrapib
SARS-CoV-2 plaque formation was linearly reduced when Vero E6 cells were preincubated with increasing concentrations of dalcetrapib for 1 h before addition of the coronavirus and during the viral replication phase resulting in an EC50 of 17.5 ± 3.5 μM (Figure 4). In contrast, the cytotoxic concentration CC50 value was greater than 1 mM. For comparison, the EC50 of the RNA-dependent RNA polymerase inhibitor remdesivir was 6.2 μM under experimental conditions used for dalcetrapib.
Figure 4.
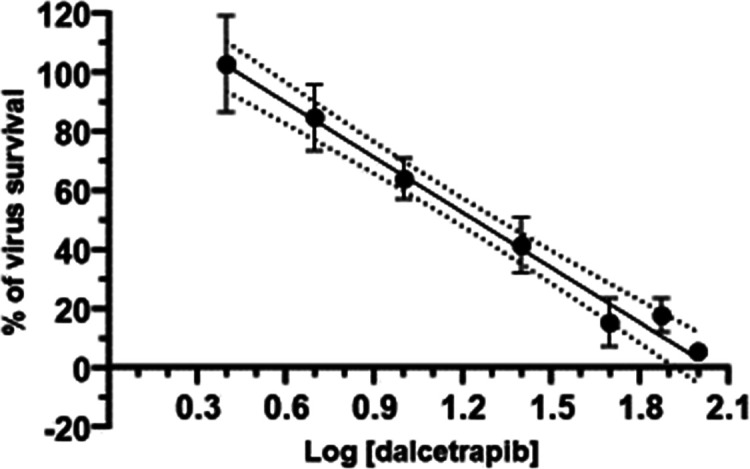
Determination of the EC50 value of dalcetrapib for the SARS-CoV-2 by the plaque reduction assay in Vero E6 cells. Virus survival in percentage is plotted against log-transformed concentrations of dalcetrapib. The dotted lines represent one SD.
When Vero E6 cells were preincubated with dalcetrapib-disulfide 1 h before addition of the coronavirus, the EC50 was greater than 100 μM (data not shown). This suggests that under the experimental conditions, the disulfide was not cell membrane-permeable, efficiently converted to the free thiol of dalcetrapib, or did not dissolve in the cell culture medium.
Discussion
Dalcetrapib was shown in the current study to bind to the catalytic site of the main 3CL protease with a delta G value of −8.5 kcal/mol and to inhibit both its activity in vitro and SARS-CoV-2 replication with IC50 values of 14.4 ± 3.3 μM and an EC50 of 17.5 ± 3.5 μM, respectively. Persistence of near-complete inhibition of protease activity despite elution of dalcetrapib-thiol suggests stable protease–drug interaction.
In our docking trials, dalcetrapib-thiol binds in the active site of the SARS-CoV-2 3CLpro with its carbonyl group anchored to the amide group of Glu166. The latter amino acid is necessary for substrate recognition, protease dimerization,28,29 and alteration of proteolytic activity. Numerous hydrophobic interactions appear to be crucial for the stabilization of dalcetrapib-thiol in the pocket. The feasibility of disulfide bond formation between dalcetrapib-thiol and Cys145 of 3CLpro is supported by the distance between their sulfur atoms (3.6 Å). This distance was shortened to 2.0 Å in the SCAR protocol.
Dalcetrapib inhibited the enzymatic activity of 3CLpro with an IC50 of 14.4 μM when dalcetrapib and the enzyme were preincubated for 24 h. The IC50 increased to 75 μM when the preincubation time was shortened to 1 h (data not shown). A similar behavior of dalcetrapib was previously observed in the inhibition of the cholesteryl ester transfer protein, with the IC50 decreasing from 1178 to 45 nM with prolongation of the incubation time from 1 to 24 h.30 This time-dependent inhibition of activity in vitro may reflect the time necessary for de-esterification of dalcetrapib and formation of stable interaction with the target protein. The persistence of almost complete inhibition of enzymatic activity once 3CLpro had been previously exposed to a high concentration of dalcetrapib despite reduction of the drug concentration down to 0.2 μM suggests stable protease–drug interaction. Thus, both covalent binding of dalcetrapib-thiol to Cys145 and hydrophobic interaction with the lipophilic binding pocket of 3CLpro5 may contribute to prevent their dissociation. The presence of 12 cysteine residues not forming Cys–Cys bonds in the amino acid sequence of 3CLpro of SARS coronaviruses is quite unusual and may have a regulatory role not yet identified.5 Several attempts to demonstrate the covalent binding of dalcetrapib-thiol to Cys145 by mass spectrometry were made. The presence of a free Cys (145) in the catalytic site as well as a total of 12 free cysteines in 3CLpro and the requirement of DTT to stabilize 3CLpro made it very difficult to demonstrate covalent binding to a single free cysteine. DTT or a similar reducing agent is required to maintain the protease activity, but dalcetrapib itself binds reversibly to DTT.31 The interaction of dalcetrapib-thiol with these free cysteines is under investigation. Regarding the selectivity of dalcetrapib-thiol, preliminary investigations suggest that the inhibition of 3CLpro by dalcetrapib-thiol is selective since the IC50 for inhibition of the second cysteine proteinase of the SARS-CoV-2, the papain-like protease,1 is 1011 μM (data not shown). This greater than 70 times selectivity for 3CLpro could be explained by the fact that the catalytic site of the main 3CLpro contains the Cys145–His41 dyad, whereas it is the classic Cys112–His273–Asp287 triad for the PLpro.
Dalcetrapib decreased plaque formation by the SARS-CoV-2 grown on Vero E6 cells linearly with increasing drug concentrations, with an EC50 of 17.5 μM. This value is in the order of magnitude of remdesivir IC50 of remdesivir (6 μM) in the same assay. Thus, inhibition of 3CLpro and SARS-CoV-2 replication supports the interaction of dalcetrapib with this critical enzyme. The general concern that formation of covalent protein adducts may result in potential safety issues has not been observed in extensive and large chronic animal and human safety studies of dalcetrapib. This may be explained by the relatively high lipophilicity of dalcetrapib, which preferentially occupies the lipophilic pocket within the protein and not exposed at the surface. In contrast, the IC50 values of dalcetrapib-disulfide for inhibition of the enzymatic activity of 3CLpro and SARS-CoV-2 replication were 72 and >100 μM, respectively. The lower IC50 obtained with dalcetrapib-thiol shows that the latter, not dalcetrapib-disulfide, is the active form against the SARS-CoV-2.
Other mechanisms could be involved in a potential beneficial effect of dalcetrapib in the prevention of SARS-CoV-2 infection. Indeed, two cysteine residues, Cys480 and Cys488, of the viral spike protein have been recently shown to be important for the binding of the spike protein to the ACE2 receptor,32 and thiol-reactive substances including dalcetrapib (JTT-705) have shown to inhibit this important interaction and prevent syncytia formation and viral entry into cells.33 Hati and Bhattacharyya34 showed that the redox status plays an important role in the stability of key disulfide bonds present in both the spike protein and the ACE2 receptor.
Extensive preclinical and clinical investigations indicate that dalcetrapib displays a satisfactory oral bioavailability and safety profile. A single dose of radiolabeled 14C-dalcetrapib 100 mg/kg administered orally to rats reaches plasma levels of 11.2 to 17.7 μM dalcetrapib equivalents at 0.5 and 2 h post-dose.35 These plasma levels are comparable to those obtained in patients receiving single doses of 1200 to 3000 mg. Dalcetrapib has been studied across a dose range of 300 to 3900 mg per day in completed and ongoing clinical trials. At 600 mg daily, the current dose was intended for long-term administration in cardiovascular patients,36 maximum plasma concentrations (tmax) of dalcetrapib-thiol are achieved 4–6 h after dosing, and the plasma half-life (t1/2) is approximately 31 h. Multiple doses of 1200 and 3900 mg achieved maximum plasma concentrations (Cmax) of between 13.8 and 22.2 μM, respectively, and plasma exposure of dalcetrapib was consistent with the dose proportionality. Dalcetrapib was safe and well-tolerated at all doses studied.
Dalcetrapib distributes in all tissues of rodents receiving multiple oral doses of 14C-dalcetrapib including the trachea, lungs, heart, and gastrointestinal tract where concentrations reach 111.6 and 68.5 μM dalcetrapib equivalents in the latter at 2 and 24 h, respectively.37 These results support the potential clinical relevance of our findings, given the observed IC50 values for dalcetrapib inhibition of 3CLpro activity and viral replication of 14.4 and an EC50 of 17.8 μM.
In rats, the percentages of an administered oral dose excreted into the feces as dalcetrapib-ester and dalcetrapib-thiol plus dalcetrapib-disulfide are 41.1 and 17.8%, respectively. The corresponding values following oral administration in monkeys are 11.6 and 22.2%.35 In humans administered with a single 600 mg dose, the majority of the drug-related material (mean of 53.4%) is excreted in feces as dalcetrapib-thiol or dalcetrapib-disulfide corresponding to a mean of 31.5% (range, 22.3–40.6%) of the administered dose. These results suggest that the intestine is exposed to high concentrations of dalcetrapib and dalcetrapib-thiol, which may contribute to potential antiviral activity of dalcetrapib at the gastrointestinal level. Because the ACE2 receptor for the SARS-CoV-2 is highly expressed in enterocytes38,39 and may be responsible for gastrointestinal involvement, high exposure of the intestinal tract to dalcetrapib may diminish virus replication locally.
The safety, pharmacokinetic properties, and potential drug interactions of dalcetrapib have been examined extensively40−42 and indicate that it may be safely administered with a large number of commonly used medications. In addition, Derks et al.43 demonstrated that dalcetrapib was safe and well-tolerated after a single dose of up to 4500 mg and multiple doses up to 3900 mg for 7 days, without QT prolongation even at these very high dosages. Dalcetrapib has been administered for 3 years to 7938 patients in a cardiovascular outcome study at a dose of 600 mg per day without major safety issues.36 Dalcetrapib currently undergoes clinical evaluation in the DalGenE phase 3 study,44 a cardiovascular outcome trial of 6149 patients with a recent acute coronary syndrome and a specific genotype in the ADCY9 gene.45 Inhibition of both polymerase and protease enzymes by remdesivir and dalcetrapib, respectively, could be envisioned in patients with severe COVID-19 as these medications exhibit complementary mechanisms of action.
Inhibition of the main 3CL protease and replication of the SARS-CoV-2 warrant the clinical evaluation of dalcetrapib as the first orally bioavailable and safe compound targeting the catalytic site of this enzyme for the treatment of COVID-19. A phase 2 clinical study is currently ongoing to determine the effect of dalcetrapib in patients with confirmed mild to moderate COVID-19, ClinicalTrials.gov identifier: NCT04676867.
Methods
Docking Methodology
Molecular docking was performed to evaluate the potential binding modes of dalcetrapib-thiol within the active site pocket of the SARS-CoV-2 main 3CL protease. To assess this, docking trials were done with SwissDock.25 For the dalcetrapib-thiol molecule, we investigated if it could be sufficiently close to react with the catalytic cysteine Cys145 forming a disulfide bond by using two strategies. The first one is a standard docking of the molecule in the catalytic site. The second uses the “steric-clashes alleviating receptor (SCAR)” protocol by mutating the receptor-reactive cysteine to glycine (C145G),26,27 which is an efficient approach to probe the possibility of covalent binding of ligands to receptors.
Inspection of all available crystal structures (152 structures as of June 17, 2020) of the SARS-CoV-2 main 3CL protease revealed noticeable conformational flexibility of the active site pocket. We used several representative structures (PDB 6LU7, 2GLX, 6W63, 5RG1, 6YB7, 7BQY, 5RH8, and 5RHA) in our docking trials. The best results were obtained using 6W63. Validation of the docking protocols was carried out by self-docking of the noncovalent inhibitor X77 in 6W63, leading to an r.m.s.d. of 0.88 Å between the best docked pose (with a delta G of −9.4 kcal/mol) and the crystallographic structure. This validates the docking with SwissDock. The 3D coordinates of dalcetrapib-thiol (ZINC000013813331) were obtained from the ZINC database (zinc.docking.org)46 in the MOL2 file format. The geometry and energy minimizations of the ligands were done with the command-line program obminimize from the OpenBabel47 package using the steepest descent algorithm and the MMFF94s force field. For the docking of dalcetrapib-thiol using the SCAR protocol, the catalytic cysteine Cys145 was mutated to a glycine prior to docking via SwissDock.
Determination of Dalcetrapib 3CLpro IC50
Proteolytic activity of 3CLpro was measured by cleavage of a fluorescently quenched peptide substrate. Dalcetrapib was first de-esterified by basic hydrolysis of its thioester bond. Dalcetrapib stock at 25 mM in ethanol was diluted to 2.5 mM in 1 mM NaOH and incubated for 30 min at 37 °C. Dalcetrapib-thiol was then diluted in an assay buffer (20 mM Tris-HCl, pH of 7.3, 100 mM NaCl, and 1 mM EDTA) with a protease for inhibition assay. Recombinant SARS-CoV-2 3CLpro (0.05 μM) (R&D Systems) in the protease assay buffer was preincubated in the presence or absence of dalcetrapib-thiol (0.2–400 μM) for 24 h at room temperature. Enzymatic reactions were then started in a black half-volume microplate by addition of 50 μM DABCYL-KTSAVLQSGFRKM-E (EDANS) (GenScript) as a substrate and incubated in protease buffer in presence of 1 mM DTT. Fluorescence (excitation, 360 nm/emission, 460 nm) was measured at room temperature every 45 s for 1 h on a Synergy H1 plate reader (Biotek). Activity was reported as the slope of the blanked linear part of the kinetic curve. Activity was estimated from duplicate measurements. We used GraphPad Prism 8.4.3 to analyze data and used inhibitor vs response (four parameters) curves to determine the values of IC50.
Testing for Reversibility of 3CLpro Inhibition by Dalcetrapib-thiol
Dalcetrapib was de-esterified as described above. Dalcetrapib-thiol was then diluted in assay buffer (20 mM Tris-HCl, pH of 7.3, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT) with protease for inhibition. The recombinant SARS-CoV-2 3CL protease (0.05 μM) (R&D Systems) in the protease assay buffer (20 mM Tris-HCl, pH of 7.3, 100 mM NaCl, 1 mM EDTA, and 1 mM DTT) was preincubated in the presence or absence of dalcetrapib-thiol (200 μM) for 24 h at room temperature. Half of the solutions were diluted 1000-fold with the assay buffer by three cycles of 10-fold dilution and concentrated back to the original volume using 3 kDa Amicon Ultracell-0.5 mL centrifugal filters (Sigma). Enzymatic reactions were then started by addition of 50 μM DABCYL-KTSAVLQSGFRKM-E (EDANS) (GenScript) as a substrate. Fluorescence (excitation, 360 nm/emission, 460 nm) was measured at room temperature every 45 s for 1 h using a Synergy H1 plate reader (Biotek). Activity was reported as the slope of the blanked linear part of the kinetic curve. Activity was estimated from duplicate measurements. We used GraphPad Prism 8.4.3 to analyze data.
Plaque Reduction Assay
Vero E6 cells were first grown in a 6-well plate at a confluency of 80–100%. On the day of infection, a viral suspension of the SARS-CoV-2 was made in MEM + 2% FBS + 1% antibiotics. The viral titer must result in about 40 plaques in nontreated wells. An aliquot of 0.4 mL of the viral suspension was added to each well, and plates were incubated at room temperature during 60 min for viral adsorption. After this step, the medium was removed, and an overlay consisting of agarose and an equal volume of each drug concentration in MEM 1X + 2% FBS + 1% antibiotics (final concentration) was added to each well in triplicate. Thus, with four plates, we could test seven concentrations of the drug plus the negative (no drug) control in triplicate. The plate was then incubated at 37 °C for 3–4 days. After incubation, the cells were fixed with 0.5 mL of 3.7% formaldehyde during 30–60 min. The agarose was then removed, and the cells were stained with crystal violet (0.8% in 50% ethanol). The number of viral plaques in each well was determined using an inverted microscope. The EC50 value, which corresponds to the concentration of a drug reducing the number of plaques by 50% compared to control wells, was then determined.
MTS Cell Cytotoxic Assay
The effect of concentrations of dalcetrapib from 1 mM to 1 μM on Vero E6 viability was assessed by incubating cultured cells in 96-well plates for 3 days in an MEM culture medium and 2% serum. Then, 10 μL of “CellTiter 96 aqueous one solution cell proliferation assay” (#G3580 Promega) that contains tetrazolium [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium] (MTS) was added to each plate and incubated for 3 h. Absorbance at 490 nm is proportional to the quantity of the formazan product and directly proportional to the number of living cells in culture.
Analysis of Dalcetrapib, Dalcetrapib-thiol, and Dalcetrapib-disulfide by UPLC
Hydrolysis of dalcetrapib-thioester to the thiol and formation of a disulfide are rapid under physiological conditions. Reliable quantitation could only be achieved by reducing all forms (thiol, disulfides, and ester) under basic conditions and analyzing total dalcetrapib-thiol after cleavage of disulfide bonds with DTT and derivatization.31 Dalcetrapib (100 μM) was incubated in neutral ethanol/water (3/1; v/v), alkaline 2 mM NaOH in ethanol/water (3/1; v/v) at pH 12.2, and acidic solutions of 2 mM HCl in ethanol/water (3/1; v/v), pH of 3.4, for 30 min and 4 h at room temperature. Incubated samples were neutralized, and 10 μL was used for ultraperformance liquid chromatography (UPLC) analysis. An analytical column CORTECS UPLC T3 1.6 μm (Waters 186008499) was used with an HPLC pump and a detector from Waters Acquity. A gradient was run from 100% mobile phase A (water/acetonitrile (1/1; v/v)) to 100% mobile phase B (water/acetonitrile (1/9; v/v)) at 0.3 mL/min between 1.5 and 5.0 min.
Acknowledgments
We thank Dr. David Kallend for constructive discussions of potential mechanisms involved in dalcetrapib antiviral activity. We thank Dr. Rezzi S. and Dr. Hodgson A. (Swiss Vitamin Institute, Lausanne Switzerland) for helpful discussion on the metabolism of dalcetrapib and metabolites.
Author Contributions
E.J.N. coordinated the research and data generated, designed the studies, and wrote the manuscript; G.B. supervised, performed the susceptibility viral replication assay, wrote methods, and reviewed the article; E.R. supervised, designed, and performed the 3CL protease assay; R.S. supervised and performed the docking investigations; V.L. designed and performed the 3CL protease assay. N.G. performed the viral replication studies; M.-E.P. performed the docking investigations; A.P. performed the analysis of dalcetrapib-ester/thiol conversion; F.L.-T. managed the research and critically reviewed and edited the manuscript; J.-C.T. supervised and managed the research and critically reviewed and edited the manuscript.
The docking and in vitro investigations of the mechanisms of inhibition of SARS-CoV-2 3CLpro by dalcetrapib-thiol were funded by DalCor Pharmaceuticals Canada.
The authors declare the following competing financial interest(s): Eric J Niesor: Dalcor Consultant; Fouzia Laghrissi-Thode: Dalcor Employee; Jean-Claude Tardif: has received research grant and honoraria from Amarin; research grant and honoraria from Astra-Zeneca, research grant, honoraria and minor equity interest from DalCor; research grant from Esperion; honoraria from HLS Therapeutics; research grant from Ionis; research grant and honoraria from Pfizer; research grant from RegenXBio; research grant and honoraria from Sanofi; research grant and honoraria from Servier; author of patents on pharmacogenomics-guided CETP inhibition and use of colchicine after myocardial infarction. Guy Boivin: None; Eric Rhéaume: None; Rong Shi:None; Véronique Lavoie : None ; Marie-Eve Picard: None; Nathalie Goyette: None;.
References
- Hartenian E.; Nandakumar D.; Lari A.; Ly M.; Tucker J. M.; Glaunsinger B. A. The molecular virology of Coronaviruses. J. Biol. Chem 2020, 12910. 10.1074/jbc.REV120.013930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yan L.; Huang Y.; Liu F.; Zhao Y.; Cao L.; Wang T.; Sun Q.; Ming Z.; Zhang L.; Ge J.; Zheng L.; Zhang Y.; Wang H.; Zhu Y.; Zhu C.; Hu T.; Hua T.; Zhang B.; Yang X.; Li J.; Yang H.; Liu Z.; Xu W.; Guddat L. W.; Wang Q.; Lou Z.; Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W.; Zhang B.; Jiang X.-M.; Su H.; Li J.; Zhao Y.; Xie X.; Jin Z.; Peng J.; Liu F.; Li C.; Li Y.; Bai F.; Wang H.; Cheng X.; Cen X.; Hu S.; Yang X.; Wang J.; Liu X.; Xiao G.; Jiang H.; Rao Z.; Zhang L.-K.; Xu Y.; Yang H.; Liu H. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 1331. 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries M.; Mohamed A. S.; Prescott R. A.; Valero-Jimenez A. M.; Desvignes L.; O’Connor R.; Steppan C.; Anderson A. S.; Binder J.; Dittmann M. Comparative study of a 3CL (pro) inhibitor and remdesivir against both major SARS-CoV-2 clades in human airway models. bioRxiv 2020, 10.1101/2020.08.28.272880. [DOI] [Google Scholar]
- Ho S. L.; Wang A. H.-J. Structural bioinformatics analysis of free cysteines in protein environments. J. Taiwan Inst. Chem. Eng. 2009, 40, 123–129. 10.1016/j.jtice.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yang M.; Ding Y.; Liu Y.; Lou Z.; Zhou Z.; Sun L.; Mo L.; Ye S.; Pang H.; Gao G. F.; Anand K.; Bartlam M.; Hilgenfeld R.; Rao Z. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 13190–13195. 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard J. E.; Elowe N. H.; Huitema C.; Fortin P. D.; Cechetto J. D.; Eltis L. D.; Brown E. D. High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chem. Biol. 2004, 11, 1445–1453. 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillaiyar T.; Manickam M.; Namasivayam V.; Hayashi Y.; Jung S. H. An Overview of Severe Acute Respiratory Syndrome-Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A.; Sellner M.; Neranjan S.; Smieško M.; Lill M. A. Potential Inhibitors for Novel Coronavirus Protease Identified by Virtual Screening of 606 Million Compounds. Int. J. Mol. Sci. 2020, 21, 3626. 10.3390/ijms21103626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji M. Potential anti-SARS-CoV-2 drug candidates identified through virtual screening of the ChEMBL database for compounds that target the main coronavirus protease. FEBS Open Bio 2020, 995. 10.1002/2211-5463.12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W.; Khan M. B.; Fischer C.; Arutyunova E.; Lamer T.; Shields J.; Saffran H. A.; McKay R. T.; van Belkum M. J.; Joyce M. A.; Young H. S.; Tyrrell D. L.; Vederas J. C.; Lemieux M. J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; Wang J. Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res. 2020, 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatansever E. C.; Yang K. S.; Drelich A. K.; Kratch K. C.; Cho C. C.; Kempaiah K. R.; Hsu J. C.; Mellott D. M.; Xu S.; Tseng C. K.; Liu W. R.. Bepridil is potent against SARS-CoV-2 in vitro. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 ( (10), ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J. M.; Monogue M. L.; Jodlowski T. Z.; Cutrell J. B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19). JAMA 2020, 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- Guy R. K.; DiPaola R. S.; Romanelli F.; Dutch R. E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. 10.1126/science.abb9332. [DOI] [PubMed] [Google Scholar]
- Okamoto H.; Yonemori F.; Wakitani K.; Minowa T.; Maeda K.; Shinkai H. A cholesteryl ester transfer protein inhibitor attenuates atherosclerosis in rabbits. Nature 2000, 406, 203–207. 10.1038/35018119. [DOI] [PubMed] [Google Scholar]
- Niesor E. J.; Magg C.; Ogawa N.; Okamoto H.; von der Mark E.; Matile H.; Schmid G.; Clerc R. G.; Chaput E.; Blum-Kaelin D.; Huber W.; Thoma R.; Pflieger P.; Kakutani M.; Takahashi D.; Dernick G.; Maugeais C. Modulating cholesteryl ester transfer protein activity maintains efficient pre-β-HDL formation and increases reverse cholesterol transport. J. Lipid Res. 2010, 51, 3443–3454. 10.1194/jlr.M008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maugeais C.; Perez A.; von der Mark E.; Magg C.; Pflieger P.; Niesor E. J. Evidence for a role of CETP in HDL remodeling and cholesterol efflux: role of cysteine 13 of CETP. Biochim. Biophys. Acta 2013, 1831, 1644–1650. 10.1016/j.bbalip.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Cunningham D.; Lin W.; Hoth L. R.; Danley D. E.; Ruggeri R. B.; Geoghegan K. F.; Chrunyk B. A.; Boyd J. G. Biophysical and biochemical approach to locating an inhibitor binding site on cholesteryl ester transfer protein. Bioconjugate Chem. 2008, 19, 1604–1613. 10.1021/bc800165n. [DOI] [PubMed] [Google Scholar]
- Manček-Keber M.; Gradisar H.; Inigo Pestaña M.; Martinez de Tejada G.; Jerala R. Free thiol group of MD-2 as the target for inhibition of the lipopolysaccharide-induced cell activation. J. Biol. Chem. 2009, 284, 19493–19500. 10.1074/jbc.M109.003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black D. M.; Bentley D.; Chapel S.; Lee J.; Briggs E.; Heinonen T. Clinical Pharmacokinetics and Pharmacodynamics of Dalcetrapib. Clin Pharmacokinet 2018, 57, 1359–1367. 10.1007/s40262-018-0656-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley D.; Young A. M.; Rowell L.; Gross G.; Tardio J.; Carlile D. Evidence of a drug-drug interaction linked to inhibition of ester hydrolysis by orlistat. J. Cardiovasc. Pharmacol. 2012, 60, 390–396. 10.1097/FJC.0b013e31826731ff. [DOI] [PubMed] [Google Scholar]
- Gross G.; Tardio J.; Kuhlmann O. Solubility and stability of dalcetrapib in vehicles and biological media. Int. J. Pharm. 2012, 437, 103–109. 10.1016/j.ijpharm.2012.07.071. [DOI] [PubMed] [Google Scholar]
- Grosdidier A.; Zoete V.; Michielin O. SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucleic Acids Res. 2011, 39, W270–W277. 10.1093/nar/gkr366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Zheng Q.; Wang Z. Potential covalent drugs targeting the main protease of the SARS-CoV-2 coronavirus. Bioinformatics 2020, 36, 3295–3298. 10.1093/bioinformatics/btaa224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ai Y.; Yu L.; Tan X.; Chai X.; Liu S. Discovery of Covalent Ligands via Noncovalent Docking by Dissecting Covalent Docking Based on a ″Steric-Clashes Alleviating Receptor (SCAR)″ Strategy. J. Chem. Inf. Model. 2016, 56, 1563–1575. 10.1021/acs.jcim.6b00334. [DOI] [PubMed] [Google Scholar]
- Chen H.; Wei P.; Huang C.; Tan L.; Liu Y.; Lai L. Only one protomer is active in the dimer of SARS 3C-like proteinase. J. Biol. Chem. 2006, 281, 13894–13898. 10.1074/jbc.M510745200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. C.; Chang G. G.; Chou C. Y. Mutation of Glu-166 blocks the substrate-induced dimerization of SARS coronavirus main protease. Biophys. J. 2010, 98, 1327–1336. 10.1016/j.bpj.2009.12.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranalletta M.; Bierilo K. K.; Chen Y.; Milot D.; Chen Q.; Tung E.; Houde C.; Elowe N. H.; Garcia-Calvo M.; Porter G.; Eveland S.; Frantz-Wattley B.; Kavana M.; Addona G.; Sinclair P.; Sparrow C.; O’Neill E. A.; Koblan K. S.; Sitlani A.; Hubbard B.; Fisher T. S. Biochemical characterization of cholesteryl ester transfer protein inhibitors. J. Lipid Res. 2010, 51, 2739–2752. 10.1194/jlr.M007468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinig K.; Bucheli F.; Kuhlmann O.; Zell M.; Pähler A.; Zwanziger E.; Gross G.; Tardio J.; Ishikawa T.; Yamashita T. Determination of dalcetrapib by liquid chromatography-tandem mass spectrometry. J. Pharm. Biomed. Anal. 2012, 66, 314–324. 10.1016/j.jpba.2012.03.056. [DOI] [PubMed] [Google Scholar]
- Khanna K.; Raymond W.; Charbit A. R.; Jin J.; Gitlin I.; Tang M.; Sperber H. S.; Franz S.; Pillai S.; Simmons G.; Fahy J. V.. Binding of SARS-CoV-2 spike protein to ACE2 is disabled by thiol-based drugs; evidence from in vitro SARS-CoV-2 infection studies. bioRxiv 2020. [Google Scholar]
- Manček-Keber M.; Hafner-Bratkovič I.; Lainšček D.; Benčina M.; Govednik T.; Orehek S.; Plaper T.; Jazbec V.; Bergant V.; Grass V.; Pichlmair A.; Jerala R. Disruption of disulfides within RBD of SARS-CoV-2 spike protein prevents fusion and represents a target for viral entry inhibition by registered drugs. FASEB J. 2021, 35, e21651 10.1096/fj.202100560R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hati S.; Bhattacharyya S. Impact of Thiol-Disulfide Balance on the Binding of Covid-19 Spike Protein with Angiotensin-Converting Enzyme 2 Receptor. ACS Omega 2020, 5, 16292–16298. 10.1021/acsomega.0c02125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takubo H.; Ishikawa T.; Kuhlmann O.; Nemoto H.; Noguchi T.; Nanayama T.; Komura H.; Kogayu M. Pharmacokinetics and disposition of dalcetrapib in rats and monkeys. Xenobiotica 2014, 44, 1117–1126. 10.3109/00498254.2014.932471. [DOI] [PubMed] [Google Scholar]
- Schwartz G. G.; Olsson A. G.; Abt M.; Ballantyne C. M.; Barter P. J.; Brumm J.; Chaitman B. R.; Holme I. M.; Kallend D.; Leiter L. A.; Leitersdorf E.; McMurray J. J.; Mundl H.; Nicholls S. J.; Shah P. K.; Tardif J. C.; Wright R. S.; dal O. I. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. 10.1056/NEJMoa1206797. [DOI] [PubMed] [Google Scholar]
- Takubo H.; Ishikawa T.; Taniguchi T.; Iwanaga K.; Nomura Y. The influence of multiple oral administration on the pharmacokinetics and distribution profile of dalcetrapib in rats. Xenobiotica 2020, 1–19. [DOI] [PubMed] [Google Scholar]
- Lee J. J.; Kopetz S.; Vilar E.; Shen J. P.; Chen K.; Maitra A. Relative Abundance of SARS-CoV-2 Entry Genes in the Enterocytes of the Lower Gastrointestinal Tract. Genes (Basel) 2020, 11, 645. 10.3390/genes11060645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamers M. M.; Beumer J.; van der Vaart J.; Knoops K.; Puschhof J.; Breugem T. I.; Ravelli R. B. G.; van Schayck P. J.; Mykytyn A. Z.; Duimel H. Q.; van Donselaar E.; Riesebosch S.; Kuijpers H. J. H.; Schippers D.; van de Wetering W. J.; de Graaf M.; Koopmans M.; Cuppen E.; Peters P. J.; Haagmans B. L.; Clevers H. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 50. 10.1126/science.abc1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks M.; Abt M.; Parr G.; Meneses-Lorente G.; Young A. M.; Phelan M. No clinically relevant drug-drug interactions when dalcetrapib is co-administered with atorvastatin. Expert Opin. Invest. Drugs 2010, 19, 1135–1145. 10.1517/13543784.2010.509342. [DOI] [PubMed] [Google Scholar]
- Derks M.; Anzures-Cabrera J.; Turnbull L.; Phelan M. Safety, tolerability and pharmacokinetics of dalcetrapib following single and multiple ascending doses in healthy subjects: a randomized, double-blind, placebo-controlled, phase I study. Clin. Drug Investig. 2011, 31, 325–335. 10.1007/BF03256931. [DOI] [PubMed] [Google Scholar]
- Derks M.; Fowler S.; Kuhlmann O. A single-center, open-label, one-sequence study of dalcetrapib coadministered with ketoconazole, and an in vitro study of the S-methyl metabolite of dalcetrapib. Clin. Ther. 2009, 31, 586–599. 10.1016/j.clinthera.2009.02.011. [DOI] [PubMed] [Google Scholar]
- Derks M.; Abt M.; Mwangi A.; Meneses-Lorente G. Lack of effect of dalcetrapib on QT interval in healthy subjects following multiple dosing. Eur. J. Clin. Pharmacol. 2010, 66, 775–783. 10.1007/s00228-010-0841-2. [DOI] [PubMed] [Google Scholar]
- Tardif J.-C.; Dubé M.-P.; Pfeffer M. A.; Waters D. D.; Koenig W.; Maggioni A. P.; McMurray J. J.; Mooser V.; White H. D.; Heinonen T.; Black D. M.; Guertin M. C.; Study design of Dal-GenE, a pharmacogenetic trial targeting reduction of cardiovascular events with dalcetrapib. Am. Heart J. 2020, 222, 157–165. 10.1016/j.ahj.2020.01.007. [DOI] [PubMed] [Google Scholar]
- Tardif J. C.; Rheaume E.; Lemieux Perreault L. P.; Gregoire J. C.; Feroz Zada Y.; Asselin G.; Provost S.; Barhdadi A.; Rhainds D.; L’Allier P. L.; Ibrahim R.; Upmanyu R.; Niesor E. J.; Benghozi R.; Suchankova G.; Laghrissi-Thode F.; Guertin M. C.; Olsson A. G.; Mongrain I.; Schwartz G. G.; Dube M. P. Pharmacogenomic determinants of the cardiovascular effects of dalcetrapib. Circ. Cardiovasc. Genet. 2015, 8, 372–382. 10.1161/CIRCGENETICS.114.000663. [DOI] [PubMed] [Google Scholar]
- Irwin J. J.; Shoichet B. K. ZINC--a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. 10.1021/ci049714+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Boyle N. M.; Banck M.; James C. A.; Morley C.; Vandermeersch T.; Hutchison G. R. Open Babel: An open chemical toolbox. Aust. J. Chem. 2011, 3, 33. 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]