

Abstract
Introduction
Atypical teratoid rhabdoid tumor (ATRT) is a rare, often lethal brain tumor of childhood characterized by a complex epigenetic landscape amongst a simple genetic background. Recent molecular studies have defined key biologic events that contribute to tumorigenesis and molecular subtypes of ATRT.
Methods
Seminal studies on ATRT are reviewed with an emphasis on molecular pathogenesis and its relevance to novel therapeutics.
Results
In this review, we summarize the key clinicopathologic and molecular features of ATRT, completed and ongoing clinical trials and outline the translational potential of novel insights into the molecular pathogenesis of this tumor.
Conclusions
SMARCB1 loss is the key genetic event in ATRT pathogenesis that leads to widespread epigenetic dysregulation and loss of lineage-specific enhancers. Current work is defining subtype-specific treatments that target underlying molecular derangements and drive tumorigenesis.
Keywords: Atypical teratoid rhabdoid tumor, ATRT, SWI/SNF complex, epigenetics, chromatin remodeling
INTRODUCTION
Atypical teratoid rhabdoid tumor (ATRT) is a central nervous system (CNS) cancer of early childhood characterized by multi-lineage differentiation and a pathologically primitive phenotype [1, 2]. In the 1990s, seminal studies of rhabdoid tumor predisposition syndrome (RTPS) demonstrated loss of the long arm of chromosome 22 as a recurrent molecular event in rhabdoid tumors including ATRT, and further molecular analyses defined loss of SMARCB1, a core subunit of the SWI/SNF chromatin-remodeling complex (CRC), as the sole recurrent genetic event in the vast majority of ATRTs [3-5]. In stark contrast to the genetic simplicity of this tumor, recent epigenetic studies have demonstrated shared as well as subtype-specific epigenetic derangements that drive tumorigenesis, and current work is aimed at discovering how these unique changes might be exploited using novel therapeutics [6-8].
EPIDEMIOLOGY AND CLINICAL MANAGEMENT
Epidemiology and Clinical Presentation
Although ATRT only accounts for 1-2% of all pediatric CNS tumors, it is a relatively frequent malignant tumor of early childhood: three-quarters of patients with ATRT are less than three years old. ATRT accounts for about 20% of embryonal CNS tumors in this age group and up to 40 – 50% of all CNS malignancies in the first year of life [9-12]. While ATRT may rarely arise in teens and adults, the median age at diagnosis is 16 – 30 months [13-17], and there is a consistent male predominance, with a male-to-female ratio ranging from 1.1 to 2 [11, 18-20]. The most common primary location for ATRT is infratentorial, but location varies with age: posterior fossa tumors predominate in the first year of life, supratentorial tumors are more common in toddlers, and spinal tumors are more common in children 3 years of age and older [20-23]. Metastatic dissemination at initial presentation occurs in 20 – 40% of cases and has been inconsistently associated with survival [11, 14, 24-26]. In a recent meta-analysis of 130 cases of metastatic ATRT, Underiner et al reported a 3-year OS of 25% (95% CI 18 −35%) [27].
Imaging and Staging
Atypical teratoid rhabdoid tumors may arise anywhere in the CNS and should be considered as a diagnosis when evaluating any aggressive-appearing intracranial tumor in a young child. They classically present as a large, heterogeneous mass with variable evidence of necrosis, hemorrhage and peritumoral edema, and while typically intra-axial, ATRTs may also arise along cranial nerves or even within the skull base [28, 29]. Owing to their dense cellularity, ATRTs frequently demonstrate restricted diffusion on MRI. The appearance of a thick, wavy and irregularly enhancing wall surrounding a central cystic region may be more specific for ATRT compared to other tumors in young children, being present in up to 28% of cases [30-32]. Specific patterns of presentation on MRI also have associations with ATRT subtypes, which are discussed in more detail below. In one small study, ATRT-MYC tumors tended to have more pronounced peritumoral edema, and ATRT-SHH tumors were reported to be less likely to display no enhancement [30], although larger studies are needed to confirm this finding (Figure 1).
Figure 1. A spectrum of clinical imaging findings of ATRT.
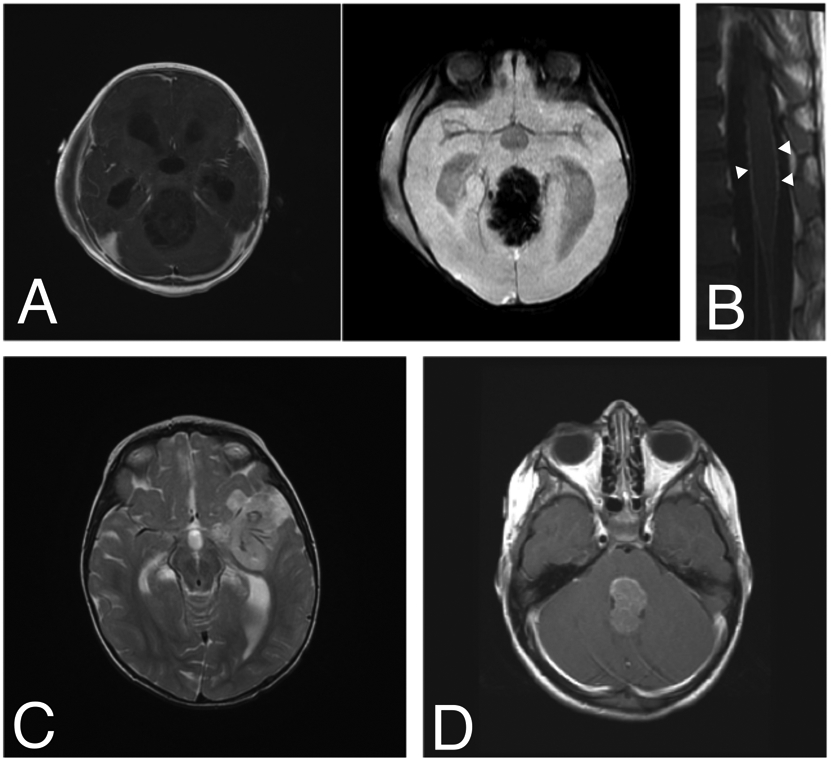
ATRT should be considered in any young child with imaging features of a highly aggressive brain tumor. A) A 9-month-old boy presented with vomiting followed quickly by coma. MRI demonstrated a 6-cm pineal-region mass with minimal, heterogeneous enhancement and abundant vascularity on susceptibility-weighted imaging. B) Diffuse leptomeningeal enhancement of the spinal cord and cauda equina were noted at the time of diagnosis (arrowheads). He underwent a gross total resection (GTR) via suboccipital craniotomy followed by ACNS0333 chemotherapy, stem cell rescue and proton beam radiotherapy with adjuvant craniospinal radiation (CSI). He currently has no evidence of disease three and a half years after diagnosis. C) A 13-month-old girl presented with coma and was found to have a 4-cm left frontotemporal, minimally enhancing mass encasing several large feeding blood vessels (T2-weighted image with contrast shown). She underwent GTR followed by adjuvant chemotherapy using the ACNS0333 protocol, stem-cell transplant and CSI. She is currently seven years since diagnosis and remains disease-free. D) A 4-year-old girl presented with nausea, vomiting and lethargy. MRI demonstrated a 3-cm, homogeneously enhancing fourth ventricular mass (T1-weighted image with contrast shown). She underwent near-total resection via suboccipital craniotomy, adjuvant chemotherapy with the SJMB-06 protocol and CSI. She is free of disease six years post-operatively.
Multiple intracranial lesions and/or the presence of extracranial lesions at presentation, especially in very young children, should raise suspicion of RTPS, which is most commonly characterized by germline mutations in SMARCB1 or, less commonly, SMARCA4 [33, 34]. The incidence of SMARCB1 germline mutations in patients with ATRT ranges 15 – 35% [26, 35], and the rate may be as high as 82% in children under one year of age. However, this estimate may be inflated by the tendency to test for germline SMARCB1 mutations in very young patients: in the most recent study with centralized, systematic testing for germline mutations using exon sequencing and multiplex ligation-dependent probe amplification (MLPA), only 17% of patients enrolled in the study were found to have a germline mutation, although this testing approach is known to miss more complex chromosomal rearrangements [6, 21, 26]. In a recent review of 26 patients with ATRT aged one year or younger in the Canadian ATRT registry, three patients (11.5%) presented with multiple MRTs with primary CNS disease, and a review of prior studies indicated a similar rate of 6.7% [21]. Given these findings, very young children (one year old or younger) in whom ATRT is suspected should undergo screening for not only disseminated CNS disease but also extra-CNS disease, particularly in the kidney, to rule out RTPS.
Clinical management
There is currently no standard-of-care treatment for ATRT. Treatment strategies have evolved toward an aggressive multimodal approach with an overall trend toward improved outcome. However, the relative contribution of each modality (surgery, high-dose chemotherapy (HDC), intrathecal [IT] chemotherapy, radiotherapy) is yet unclear and may be influenced by unique tumor biology.
ATRT tends to present as a large, highly invasive, hypervascular mass, sometimes in eloquent brain, often precluding upfront gross total resection (GTR). The rate of GTR following surgery for ATRT ranges 30 – 68%, and its impact on outcome varies by study. While initial studies indicated a significant survival benefit following GTR, this has not been borne out in recent studies, and this finding may relate to the delivery of early adjuvant radiotherapy for residual disease in some studies [13, 14, 25, 26, 36] . Nevertheless, given the limited therapeutic options for ATRT, the state-of-the-art recommendation is to achieve as complete a tumor resection as possible and pursue second-look surgery when post-operative findings demonstrate safely resectable residual tumor.
Early studies of single-modality conventional chemotherapy for infants with ATRT demonstrated no clear survival benefit [37, 38]. The first clinical trial dedicated to ATRT (rhabdomyosarcoma-like therapy IRS III) was based on conventional chemotherapy used in conjunction with IT chemotherapy and adjuvant radiotherapy. With this approach, in a small cohort of patients, objective response rate to chemotherapy prior to radiotherapy was 58%, indicating chemosensitivity in ATRT. This multimodal strategy, which used pre-radiation chemotherapy, led to a two-year 2 event-free (EFS) and overall survival (OS) of 53% and 70%, respectively (median OS not yet reported), but with significant associated toxicity [13]. Similarly, the EuroRhab study, using a registry-based regimen of conventional chemotherapy consisting of anthracycline and alkylating agents combined with intraventricular chemotherapy and age-stratified radiotherapy, was associated with a six-year EFS and OS 45% and 46%, respectively [16]. In North America, treatment strategies evolved more toward the use of HDC with stem-cell transplantation to avoid or delay the use of radiotherapy. In light of earlier observations of sustained responses in patients with ATRT [39, 40], a dedicated ATRT protocol from the Children’s Oncology Group (COG; ACNS 0333) was developed combining consolidative chemotherapy with HDC and age-stratified radiotherapy. This large trial, the only cooperative group study of its time, enrolled 65 patients and provided a four-year EFS and OS of 48% and 57%, respectively, a significant survival improvement compared to historical studies with conventional chemotherapy [26]. Slavc et al also reported significant improved outcome with the MUV-ATRT regimen with HDC, IT chemotherapy and focal radiotherapy [17]. However, the Headstart strategy based on high-dose methotrexate-based induction and myeloablative HDC failed to achieve similar outcomes, suggesting a potentially detrimental effect of prolonged induction [41]. To sort out the relative benefit of HDC, Schrey et al performed a meta-analyses of studies implementing various protocols and demonstrated a survival benefit for patients receiving HDC (median OS 19 months vs. 10.2 months; median recurrence-free survival 8 months 4.4 months) [42].
Despite the very young age of patients with ATRT, the lack of therapeutic options has necessitated that radiotherapy remain part of the contemporary arsenal against this tumor. Adjuvant radiotherapy has been used at various doses, fields and timing and remains a controversial topic. While some studies have indicated an increased risk of relapse with delayed radiation [43, 44], others have deferred or completely omitted up-front radiotherapy [15, 17]. In a Canadian registry-based study, six of the 12 long-term survivors did not receive radiation [14]. Furthermore, the ACNS0333 study, using an HDC backbone, demonstrated no negative impact on outcomes with an approach of using delayed, focal radiotherapy for most patients and avoiding craniospinal irradiation (CSI) altogether for patients older than three years of age [26]. Taken together, there are some data to suggest that radiotherapy has some efficacy against ATRT but might be delayed or deferred without a detriment to overall survival. However, these results must be interpreted with caution: as discussed below, ATRT subtypes are distinct in their age of presentation, and outcomes following radiotherapy stratified by age group may simply reflect different underlying radiosensitivity between subtypes. In fact, in ATRT-SHH tumors, withholding radiotherapy altogether appears to have no detrimental impact on overall survival [7].
Intrathecal chemotherapy (IT) has been added to conventional or HDC and/or radiotherapy regimens either for CNS prophylaxis or to treat microscopic dissemination [13, 14, 16, 17]. The most commonly used agents are methotrexate, cytarabine and topotecan. A 2009 meta-analysis demonstrated a survival benefit from IT chemotherapy in ATRT [24], but given its use as part of a complex multi-modal regimen, its contribution to survival in ATRT remains unclear. Most recently, a meta-analysis of 44 studies including 123 patients with metastatic ATRT demonstrated a benefit in OS following IT chemotherapy (three-year OS 32% vs. 22%), indicating these patients, in particular, may benefit from this approach [27].
PATHOLOGY
Atypical teratoid rhabdoid tumor is an embryonal brain tumor that is distinct in its poorly differentiated, multi-lineage phenotype that may include variable components of glial, mesenchymal and even epithelial differentiation (Figure 2A). As its name suggests, rhabdoid cells are a useful feature when identified, and are characterized by eosinophilic cytoplasm and eccentrically located nuclei with prominent nucleoli, morphologically resembling rhabdomyoblasts (Figure 2B). These rhabdoid cells are often intermingled within areas composed of cells with primitive (“small blue cell”) morphology, which often form the predominant component. The presence of rhabdoid cells and multi-lineage differentiation are unique to ATRT and help distinguish it from other embryonal tumors of the CNS [1, 2].
Figure 2. Histological features of ATRT.

A) The hypercellular tumor is usually predominantly composed of primitive-appearing cells with scant cytoplasm and hyperchromatic nuclei. Mitotic figures, apoptotic bodies and necrosis may all be readily identified. B) A subset of cells may show abundant, globular eosinophilic cytoplasm, reminiscent of rhabdoid cells. C) SMARCB1 (INI1 / BAF47 / SNF5) immunohistochemistry shows uniform loss of expression in the tumor cells, while expression is retained in endothelial nuclei serving as positive internal controls. (A, B – Hematoxylin and Eosin stain, 400x magnification; C – BAF47 clone (BD Biosciences), 200x magnification).
As a result of the primitive cell state and multi-lineage differentiation, the immunohistochemical features of ATRT are highly variable. While vimentin and epithelial membrane antigen (EMA) are expressed in nearly all tumors, nests of cells are often positive for synaptophysin, glial fibrillary acidic protein (GFAP), cytokeratin and smooth muscle actin (SMA). In light of seminal molecular studies detailed below, contemporary diagnosis is made by demonstrating absence of expression of SMARCB1 (also known as INI1, BAF47 and SNF5; Figure 2C). In the appropriate clinical and histopathologic context, diffuse absence of SMARCB1 expression is diagnostic of ATRT [2, 45]. A small proportion of ATRTs will retain expression of SMARCB1, and most SMARCB1-retained ATRTs contain mutations in SMARCA4, an ATPase subunit of the SWI/SNF complex, which are usually detectable by Sanger sequencing [46, 47].
GENETICS AND MOLECULAR IMPACT OF SMARCB1 LOSS
Seminal cytogenetic studies in the late1990s and early 2000s demonstrated recurrent loss of part or all of the long arm of chromosome 22 as a recurrent event in rhabdoid tumors [3, 4]. Analysis of overlapping areas of 22q deletions across a panel of rhabdoid tumor cell lines identified loss of Chr22q11.2, which harbors SMARCB1, as the sole shared deleted region amongst tested cell lines, and of those cell lines with structurally intact Chr22, all contained loss-of-function mutations in SMARCB1, the first indication of SMARCB1 loss as the key genetic event in rhabdoid tumor pathogenesis [5].
A large body of work over the next two decades not only reaffirmed the central role of SMARCB1 loss in ATRT but also yielded insights into the normal function of the SWI/SNF complex and shed light onto novel epigenetic mechanisms of tumorigenesis. Whole-genome sequencing studies of ATRTs have confirmed that in the vast majority of cases, the only recurrent mutation or major structural genomic event is bi-allelic inactivation of SMARCB1, either through whole-gene deletion as part of a large chromosomal event, truncating nonsense or frameshift mutation, missense mutation or (rarely) a gene fusion that leads to protein instability [13, 34, 48]. Furthermore, rhabdoid tumors are unique amongst cancer in their chromosomal stability: most rhabdoid tumors, including ATRTs, contain fewer than 10 exomic alterations, virtually none of which are shared amongst tumors [48]. While it remains to be seen whether these additional mutations are simply “passenger” mutations or somehow contribute to tumorigenesis, SMARCB1 loss is clearly the key molecular event in the pathogenesis of ATRT.
SMARCB1 is a core subunit of the SWI/SNF chromatin-remodeling complex (CRC) that is known to be necessary for normal cell differentiation and cell lineage determination. Our understanding of the normal function of the SWI/SNF complex has evolved in parallel with large-scale efforts to understand the molecular origins of ATRT and cancer broadly. By the mid-2000s, SMARCB1 loss was accepted as a key diagnostic criterion for ATRT, but a few cases were known to retain SMARCB1 expression. Using targeted sequencing, SMARCB1-retained ATRTs were found to contain loss-of-function mutations in SMARCA4 [46, 47].. Further emphasizing its role in guiding normal cellular development, a plethora of studies over the past decade has identified loss-of-function mutations in at least one SWI/SNF CRC family member in about a fifth of cancers, most commonly SMARCA4, SMARCA2, ARID1A and ARID1B, among others [49]. The striking frequency of mutations in SWI/SNF CRC family members across multiple types of cancer and specifically SMARCB1 in rhabdoid tumors highlights a critical need to understand its normal biology and tumor suppressor functions.
The intact SWI/SNF complex serves as a beacon for a host of transcription factors and chromatin-remodeling enzymes that together have far-reaching effects on cell viability, proliferation and differentiation. Recent studies have shown SMARCB1 loss leads to widespread loss of H3K27Ac at typical enhancers, stretches of DNA that facilitate transcription of nearby genes in cis that are frequently involved in cell differentiation programs [50-52]. Intact SWI/SNF complex recruits the histone acetylase (HAT) P300 to surrounding H3K27 residues, and loss of SMARCB1 precludes H3K27 acetylation [50]. Interestingly, loss of H3K27Ac in rhabdoid tumors results in selective loss of typical enhancers, which are distinguished from super enhancers by their specificity to cell-lineage differentiation programs and more focal neighboring H3K27Ac modifications [53].
In addition to H3K27Ac loss, multiple studies have identified deranged PRC2 complex-related signaling as a key molecular event in ATRT [8, 51, 54]. PRC2, via its methylase subunit EZH2, facilitates trimethylation of H3K27 (H3K27me3), a chromatin mark associated with enhancer silencing. Interestingly, integrated ChIP analyses have identified significant overlap between lost enhancers and EZH2-bound chromatin in ATRT [51]. Suppression of EZH2 abrogates tumorigenesis following SMARCB1 loss [55], and expression of EZH2 targets appears to be dependent on residual SWI/SNF function. It was previously known that SMARCA4 is necessary for tumorigenesis following SMARCB1 loss [56], and ChIP data revealed that most SMARCA4-bound targets are also bound by EZH2 [51]. Heuristically, it is easy to conceive of a yin-and-yang relationship between SWI/SNF- and PRC2-mediated enhancer modification via opposing actions at enhancers. However, re-expression of SMARCB1 in ATRT cell lines actually increases H3K27 trimethylation by an unclear mechanism [51], indicating a spatial relevance of histone modification in this epigenetic framework that relies on residual SWI/SNF complex. While more work is required to elucidate the functional interplay between the SWI/SNF chromatin-remodeling complex and PRC2, this observation has generated significant interest in EZH2 inhibitors as a potential therapy for ATRT.
In summary, SMARCB1 loss is a key feature in ATRT tumorigenesis, and this event leads to preferential loss of typical enhancers involved in normal cellular development. While much has been learned about the normal function of the SWI/SNF complex, it is important to note that composition varies widely between cell types, and our current understanding of SWI/SNF function is based on limited data from few distinct cell types. Future work must determine how cell lineage specificity of SWI/SNF composition contributes to heterogeneity in tumor subtypes [57].
MOLECULAR SUBTYPES OF ATRT
Despite the near-universal feature of SMARCB1 (and to a lesser extent, SMARCA4) loss, multiple studies have demonstrated that ATRT is comprised of multiple molecular, clinically relevant subtypes. Unsupervised hierarchical clustering analyses of gene expression and DNA methylation array data from ATRTs reveal three distinct molecular subtypes, and this finding has been reproduced across three independent cohorts [7, 58, 59]. A recent consensus statement defined these subgroups as ATRT-MYC, ATRT-SHH and ATRT-TYR[20].
Each molecular subgroup clusters around a specific clinicopathologic phenotype that may have implications on prognosis and therapeutic vulnerabilities [7, 59, 60]. ATRT-TYR tumors tend to occur in infants in the infratentorial compartment and are defined by a mesenchymal gene expression pattern centered on bone morphogenic protein (BMP) and melanogenesis signaling pathways. ATRT-SHH tumors usually occur in slightly older infants and toddlers and may occur in either the supra- or infratentorial compartment. Importantly, the defining molecular features within this subgroup appear to vary with tumor location, with molecular clustering between tumors arising in the supratentorial (ATRT-SHH-1) or infratentorial space (ATRT-SHH-2) [20]. In addition to its namesake, ATRT-SHH tumors prominently feature deranged NOTCH signaling and display a primitive neuronal gene expression pattern. Both ATRT-TYR and ATRT-SHH tumors tend to have focal aberrations in SMARCB1, whereas ATRT-MYC tumors tend to have broad deletions affecting Chr22q11.2. ATRT-MYC tumors tend to occur in older children and are characterized by dysregulated MYC signaling and over-expression of HOX cluster genes. An early study indicated that ATRT-SHH tumors may have a slight but significantly better prognosis, and although this has not been uniformly borne out in subsequent studies, the most recent study from the Children’s Oncology Group demonstrated a strong trend toward longer EFS and OS in ATRT-SHH tumors, with a 6-month EFS of 100% [26]. Importantly, defining unique molecular subgroups has facilitated the identification of logical, targeted therapeutics with subgroup-specific efficacy, as is discussed in the following section (Figure 3).
Figure 3. Enhancer landscape in ATRT and specificity to molecular subtypes.

Loss of SMARCB1 results in selective loss of typical enhancers (TEs) involved in cell lineage determination via residual activity of the SWI/SNF complex, loss of histone acetylase activity and increased relative PRC2/EZH2 activity. However, super enhancer (SE) activity is selectively retained at genes that contribute to cell proliferation and immortality in one of three specific patterns. These three molecular subgroups of ATRT are relevant to developing logical, targeted therapies and potentially prognostication.
If the genetic landscape of ATRT is so bland, why is there such molecular heterogeneity? Chromatin immunoprecipitation (ChIP) analyses demonstrate that residual super enhancer activity guides expression of key cancer-related and subgroup-specific signature genes, such as GLI2 in ATRT-SHH and MYC and HOX genes in ATR-MYC [7, 59], but the mechanism of this residual enhancer activity is unclear. It is widely held that these patterns are remnants of cell differentiation programs indicating unique cells of origin, but this remains an active area of study. Further work is needed to define the molecular mechanisms of tumorigenesis between ATRT subgroups.
NOVEL THERAPEUTICS AND FUTURE DIRECTIONS
Early in vitro studies and molecular subtyping of ATRT have led to an effort to stratify risk, refine current protocols and develop targeted therapeutics using clinical and molecular criteria [7, 25]. Using molecular data from each tumor subgroup to inform drug choice, Torchia et al performed a limited drug screen of agents targeting subgroup-specific pathways in ATRT-SHH and ATRT-MYC cell lines [6]. The results from this early drug screen demonstrate subgroup-specific therapeutic vulnerabilities that can be predicted by tumor subtype.
ATRT-SHH tumors appear to be more critically dependent on multiple targetable epigenetic regulators for survival than other tumor subtypes. While increased EZH2 activity may be a shared feature amongst multiple ATRT subtypes, it is a prominent feature in ATRT-SHH tumors, and inhibition with an EZH2 inhibitor (UNC1999) is selectively toxic to cell lines from this subgroup. Similarly, bromodomain inhibition with the experimental compound JQ1 demonstrated significant toxicity in all ATRT-SHH cell lines tested as well as some ATRT-MYC cell lines [6]. A phase-I trial using the EZH2 inhibitor tazemetostast for relapsed or refractory SMARCB1-deficient tumors is ongoing (NCT02601937). While it is tempting to speculate ATRT-SHH tumors may be responsive to SHH pathway inhibitors such as vismodegib and arsenic trioxide, the absence of mutations SHH pathway members PTCH1, SMO and SUFU, as in SHH-subtype medulloblastoma, indicate SHH signaling is likely an indirect result of SMARCB1 loss and not the dominant molecular driver in these tumors [20].
In contrast to ATRT-SHH tumors, the ATRT-MYC and ATRT-TYR subtypes display a critical dependence on receptor tyrosine kinase pathways, particularly PDGFR. The tyrosine kinase inhibitors (TKIs) dasatinib and nilotinib display selective toxicity to ATRT-MYC cell lines, and dasatinib significantly improved survival in an intracranial orthotopic xenograft model [6]. In a genome-wide CRISPR screen of eight rhabdoid tumor cell lines, cells derived from extracranial malignant rhabdoid tumors (MRTs) and ATRT-MYC tumors were found to be dependent on numerous tyrosine kinases [61]. While in vivo data from this study used MRT cell lines, there is some overlap in the DNA methylation patterns between ATRT-MYC tumors and extracranial MRTs, which raises the possibility these two tumor types may share therapeutic vulnerabilities.
Given the role of H3K27Ac loss in ATRT, histone deacetylases inhibitors (HDACis) have also attracted interest as a potential therapy across all molecular subtypes of ATRT. Torchia et al found toxicity to the HDACi LAQ824 across multiple ATRT-SHH and ATRT-MYC cell lines [6]. The HDACi vorinostat (also known as SAHA) has been shown to have efficacy against ATRT cell lines in vitro [62] and acts as a radiosensitizer in an MRT mouse model [63]. Two phase-I trials of vorinostat enrolling patients with ATRT have been completed, but results are not yet available (NCT 00217412, NCT 01076530).
SMARCB1 functions as a tumor suppressor in part by inducing cell cycle arrest in G1 phase, and SMARCB1 loss results in increased p16INK4a and Aurora Kinase A (AKA) activity, leading to cell cycle progression [64-66]. In a series of four patients with recurrent ATRT treated with alisertib, each tumor demonstrated chemotherapeutic response, and durable tumor regression was noted in two [66]. In a phase-I dose-escalation trial including 13 patients with ATRT, two patients demonstrated disease stabilization with the CDK4/6 inhibitor ribociclib [67]. Early-phase clinical trials of these agents in ATRT are ongoing (NCT03387020, NCT01076530).
In addition to small molecule inhibitors that target either specific molecular subgroups or ATRT generally, T cell-based immunotherapy is increasingly recognized as a potentially efficacious approach to treating rhabdoid tumors. Leruste et al demonstrated that rhabdoid tumors induce a robust immune response. Using a syngeneic model of ATRT-MYC in immunocompetent mice, blockade of PD-L1 led to a significant reduction in tumor growth and prolonged survival of tumor-bearing mice. Interestingly, this study also demonstrated endogenous retroviruses (ERVs) are re-expressed in rhabdoid tumors in a manner that requires SMARCB1 deficiency, defining a potential mechanism for their immunogenicity in the absence of high mutational burden [68]. Additionally, Theruvath et al demonstrated ATRTs, but not normal infant or pediatric brain, express B7-H3, a target of immunotherapies that are currently in clinical trial. Using an patient-derived xenograft model, they showed intratumoral or intraventricular injection of B7-H3-targeting chimeric-antigen receptor (CAR) T cells led to tumor regression in all animals tested [69]. Taken together, these findings indicate T cell-based immunotherapy is a promising potential treatment for rhabdoid tumors including ATRT, particularly ATRT-MYC.
The discovery of multiple molecular subtypes in ATRT has revealed numerous previously unknown therapeutic options to this highly aggressive tumor. Nevertheless, in an era when long-term survival after treatment for embryonal brain tumors is increasingly common, particular attention should be paid to the toxicity associated with aggressive multimodal regimens and novel therapeutics. Risk stratification should be further investigated by taking into account treatment response in each molecular subgroup, particularly to radiotherapy. Lessening the burden of therapy to limit the complications of neurocognitive delay, infertility and endocrinologic dysfunction should remain a priority in future clinical trials of ATRT [70].
CONCLUSION
Atypical teratoid rhabdoid tumor remains a difficult-to-treat disease that is often lethal. An aggressive multi-modal therapeutic approach that centers on maximal safe surgical resection and high-dose chemotherapy with stem-cell transplantation has significantly improved survival rates but with significant associated morbidity. Bi-allelic SMARCB1 loss is the critical genetic event in the vast majority of ATRTs, and despite their genetic homogeneity, ATRTs are an epigenetically diverse group of tumors with distinct enhancer landscapes. At least three molecular subtypes of ATRT exist, each with unique targetable pathways and potential therapeutic vulnerabilities. Ongoing clinical trials and laboratory research are focused on translating these exciting new insights into efficacious therapeutics to improve outcome in this devastating disease.
Funding:
CLN is supported by a National Institutes of Health T32 award to the Mayo Clinic Research Training Program in Neuro-Oncology (NS07494).
Footnotes
Conflicts of interest: The authors have no conflicts of interest to declare.
REFERENCES
- 1.Louis DN, Perry A, Reifenberger G, et al. (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131:803–20. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 2.Ellison D, Love S (2013) Neuropathology: A Reference Text of CNS Pathology, 3rd ed. Mosby Ltd [Google Scholar]
- 3.Sévenet N, Sheridan E, Amram D, et al. (1999) Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet 65:1342–1348. 10.1086/302639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biegel JA, Tan L, Zhang F, et al. (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467 [PubMed] [Google Scholar]
- 5.Versteege I, Sévenet N, Lange J, et al. (1998) Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature 394:203–6. 10.1038/28212 [DOI] [PubMed] [Google Scholar]
- 6.Torchia J, Golbourn B, Feng S, et al. (2016) Integrated ( epi ) -Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Article Integrated ( epi ) -Genomic Analyses Identify Subgroup-Specific Therapeutic Targets in CNS Rhabdoid Tumors. Cancer Cell 30:891–908. 10.1016/j.ccell.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torchia J, Picard D, Lafay-Cousin L, et al. (2015) Molecular subgroups of atypical teratoid rhabdoid tumours in children: an integrated genomic and clinicopathological analysis. Lancet Oncol 16:569–82 [DOI] [PubMed] [Google Scholar]
- 8.Johann PD, Erkek S, Zapatka M, et al. (2016) Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29:379–393. 10.1016/j.ccell.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 9.Woehrer A, Slavc I, Waldhoer T, et al. (2010) Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian Brain Tumor Registry, 1996-2006. Cancer 116:5725–32. 10.1002/cncr.25540 [DOI] [PubMed] [Google Scholar]
- 10.Ostrom QT, Chen Y, M de Blank P, et al. (2014) The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001-2010. Neuro Oncol 16:1392–9. 10.1093/neuonc/nou090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilden JM, Meerbaum S, Burger P, et al. (2004) Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22:2877–84. 10.1200/JCO.2004.07.073 [DOI] [PubMed] [Google Scholar]
- 12.Frühwald MC, Biegel JA, Bourdeaut F, et al. (2016) Atypical teratoid/rhabdoid tumors-current concepts, advances in biology, and potential future therapies. Neuro Oncol 18:764–78. 10.1093/neuonc/nov264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chi SN, Zimmerman MA, Yao X, et al. (2009) Intensive multimodality treatment for children with newly diagnosed CNS atypical teratoid rhabdoid tumor. J Clin Oncol 27:385–389. 10.1200/JCO.2008.18.7724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lafay-Cousin L, Hawkins C, Carret AS, et al. (2012) Central nervous system atypical teratoid rhabdoid tumours: The Canadian Paediatric Brain Tumour Consortium experience. Eur J Cancer 48:353–359. 10.1016/j.ejca.2011.09.005 [DOI] [PubMed] [Google Scholar]
- 15.Dufour C, Beaugrand A, Le Deley MC, et al. (2012) Clinicopathologic prognostic factors in childhood atypical teratoid and rhabdoid tumor of the central nervous system: a multicenter study. Cancer 118:3812–21. 10.1002/cncr.26684 [DOI] [PubMed] [Google Scholar]
- 16.Bartelheim K, Nemes K, Seeringer A, et al. (2016) Improved 6-year overall survival in AT/RT – results of the registry study Rhabdoid 2007. Cancer Med 5:1765–1775. 10.1002/cam4.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Slavc I, Chocholous M, Leiss U, et al. (2014) Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012. Cancer Med 3:91–100. 10.1002/cam4.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buscariollo DL, Park HS, Roberts KB, Yu JB (2012) Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer 118:4212–4219. 10.1002/cncr.27373 [DOI] [PubMed] [Google Scholar]
- 19.Yamasaki K, Kiyotani C, Terashima K, et al. (2020) Clinical characteristics, treatment, and survival outcome in pediatric patients with atypical teratoid/rhabdoid tumors: A retrospective study by the Japan Children’s Cancer Group. J Neurosurg Pediatr 25:111–120. 10.3171/2019.9.PEDS19367 [DOI] [PubMed] [Google Scholar]
- 20.Ho B, Johann PD, Johann PD, et al. (2020) Molecular subgrouping of atypical teratoid/rhabdoid tumors - A reinvestigation and current consensus. Neuro. Oncol 22:613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fossey M, Li H, Afzal S, et al. (2017) Atypical teratoid rhabdoid tumor in the first year of life: the Canadian ATRT registry experience and review of the literature. J Neurooncol 132:155–162. 10.1007/s11060-016-2353-0 [DOI] [PubMed] [Google Scholar]
- 22.Li D, Heiferman DM, Syed HR, et al. (2019) Pediatric primary spinal atypical teratoid rhabdoid tumor: a case series and review of the literature. J Neurosurg Pediatr 24:1–17. 10.3171/2019.4.PEDS19113 [DOI] [PubMed] [Google Scholar]
- 23.Benesch M, Nemes K, Neumayer P, et al. (2020) Spinal cord atypical teratoid/rhabdoid tumors in children: Clinical, genetic, and outcome characteristics in a representative European cohort. Pediatr Blood Cancer 67:. 10.1002/pbc.28022 [DOI] [PubMed] [Google Scholar]
- 24.Athale UH, Duckworth J, Odame I, Barr R (2009) Childhood Atypical Teratoid Rhabdoid Tumor of the Central Nervous System. J Pediatr Hematol Oncol 31:651–663. 10.1097/MPH.0b013e3181b258a9 [DOI] [PubMed] [Google Scholar]
- 25.Frühwald MC, Hasselblatt M, Nemes K, et al. (2019) Age and DNA-methylation subgroup as potential independent risk factors for treatment stratification in children with Atypical Teratoid/Rhabdoid Tumors (ATRT). Neuro Oncol. 10.1093/neuonc/noz244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reddy AT, Strother DR, Judkins AR, et al. (2020) Efficacy of high-dose chemotherapy and three-dimensional conformal radiation for atypical teratoid/rhabdoid tumor: A report from the Children’s Oncology Group trial ACNS0333. J Clin Oncol 38:1175–1185. 10.1200/JCO.19.01776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Underiner RM, Eltobgy M, Stanek JR, et al. (2020) Meta-Analysis of Treatment Modalities in Metastatic Atypical Teratoid/Rhabdoid Tumors in Children. Pediatr Neurol. 10.1016/j.pediatrneurol.2020.03.003 [DOI] [PubMed] [Google Scholar]
- 28.Kazan S, Göksu E, Mihci E, et al. (2007) Primary atypical teratoid/rhabdoid tumor of the clival region. Case report. J Neurosurg 106:308–11. 10.3171/ped.2007.106.4.308 [DOI] [PubMed] [Google Scholar]
- 29.Heuer GG, Kiefer H, Judkins AR, et al. (2010) Surgical treatment of a clival-C2 atypical teratoid/rhabdoid tumor. J Neurosurg Pediatr 5:75–9. 10.3171/2009.8.PEDS08421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowak J, Nemes K, Hohm A, et al. (2018) Magnetic resonance imaging surrogates of molecular subgroups in atypical teratoid/rhabdoid tumor. Neuro Oncol 20:1672–1679. 10.1093/neuonc/noy111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koral K, Gargan L, Bowers DC, et al. (2008) Imaging characteristics of atypical teratoid-rhabdoid tumor in children compared with medulloblastoma. Am J Roentgenol 190:809–814. 10.2214/AJR.07.3069 [DOI] [PubMed] [Google Scholar]
- 32.Au Yong KJ, Jaremko JL, Jans L, et al. (2013) How specific is the MRI appearance of supratentorial atypical teratoid rhabdoid tumors? Pediatr Radiol 43:347–354. 10.1007/s00247-012-2530-z [DOI] [PubMed] [Google Scholar]
- 33.Bruggers CS, Bleyl SB, Pysher T, et al. (2011) Clinicopathologic comparison of familial versus sporadic atypical teratoid/rhabdoid tumors (AT/RT) of the central nervous system. Pediatr Blood Cancer 56:1026–31. 10.1002/pbc.22757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hasselblatt M, Isken S, Linge A, et al. (2013) High-resolution genomic analysis suggests the absence of recurrent genomic alterations other than SMARCB1 aberrations in atypical teratoid/rhabdoid tumors. Genes Chromosom Cancer 52:185–190. 10.1002/gcc.22018 [DOI] [PubMed] [Google Scholar]
- 35.Biegel JA, Busse TM, Weissman BE (2014) SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet Part C Semin Med Genet 166:350–366. 10.1002/ajmg.c.31410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tekautz TM, Fuller CE, Blaney S, et al. (2005) Atypical teratoid/rhabdoid tumors (ATRT): Improved survival in children 3 years of age and older with radiation therapy and high-dose alkylator-based chemotherapy. J Clin Oncol 23:1491–1499. 10.1200/JCO.2005.05.187 [DOI] [PubMed] [Google Scholar]
- 37.Strother DR, Lafay-Cousin L, Boyett JM, et al. Benefit from prolonged dose-intensive chemotherapy for infants with malignant brain tumors is restricted to patients with ependymoma: a report of the Pediatric Oncology Group randomized controlled trial 9233/34. 10.1093/neuonc/not163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Von Hoff K, Hinkes B, Dannenmann-Stern E, et al. (2011) Frequency, Risk-Factors and Survival of Children With Atypical Teratoid Rhabdoid Tumors (AT/RT) of the CNS Diagnosed between 1988 and 2004, and Registered to the German HIT Database. Pediatr Blood Cancer 57:978–985. 10.1002/pbc.23236 [DOI] [PubMed] [Google Scholar]
- 39.Cohen BH, Geyer JR, Miller DC, et al. (2015) Pilot Study of Intensive Chemotherapy With Peripheral Hematopoietic Cell Support for Children Less Than 3 Years of Age With Malignant Brain Tumors, the CCG-99703 Phase I/II Study. A Report From the Children’s Oncology Group. Pediatr Neurol 53:31–46. 10.1016/j.pediatrneurol.2015.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Finkelstein-Shechter T, Gassas A, Mabbott D, et al. (2010) Atypical teratoid or rhabdoid tumors: Improved outcome with high-dose chemotherapy. J Pediatr Hematol Oncol 32:e182–e186. 10.1097/MPH.0b013e3181dce1a2 [DOI] [PubMed] [Google Scholar]
- 41.Zaky W, Dhall G, Ji L, et al. (2014) Intensive induction chemotherapy followed by myeloablative chemotherapy with autologous hematopoietic progenitor cell rescue for young children newly-diagnosed with central nervous system atypical teratoid/rhabdoid tumors: The head start III experience. Pediatr Blood Cancer 61:95–101. 10.1002/pbc.24648 [DOI] [PubMed] [Google Scholar]
- 42.Schrey D, Carceller Lechón F, Malietzis G, et al. (2016) Multimodal therapy in children and adolescents with newly diagnosed atypical teratoid rhabdoid tumor: individual pooled data analysis and review of the literature. J Neurooncol 126:81–90. 10.1007/s11060-015-1904-0 [DOI] [PubMed] [Google Scholar]
- 43.Pai Panandiker AS, Merchant TE, Beltran C, et al. (2012) Sequencing of local therapy affects the pattern of treatment failure and survival in children with atypical teratoid rhabdoid tumors of the central nervous system. Int J Radiat Oncol Biol Phys 82:1756–1763. 10.1016/j.ijrobp.2011.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen YW, Wong TT, Ho DMT, et al. (2006) Impact of radiotherapy for pediatric CNS atypical teratoid/rhabdoid tumor (single institute experience). Int J Radiat Oncol Biol Phys 64:1038–1043. 10.1016/j.ijrobp.2005.10.001 [DOI] [PubMed] [Google Scholar]
- 45.Louis DN, Perry A, Reifenberger G, et al. (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131:803–20. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 46.Hasselblatt M, Gesk S, Oyen F, et al. (2011) Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35:933–5. 10.1097/PAS.0b013e3182196a39 [DOI] [PubMed] [Google Scholar]
- 47.Bookhout C, Bouldin TW, Ellison DW (2018) Atypical teratoid/rhabdoid tumor with retained INI1 (SMARCB1) expression and loss of BRG1 (SMARCA4). Neuropathology 38:305–308. 10.1111/neup.12452 [DOI] [PubMed] [Google Scholar]
- 48.Lee RS, Stewart C, Carter SL, et al. (2012) A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest 122:2983–2988. 10.1172/JCI64400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kadoch C, Hargreaves DC, Hodges C, et al. (2013) Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet 45:592–601. 10.1038/ng.2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alver BH, Kim KH, Lu P, et al. (2017) The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat Commun 8:. 10.1038/ncomms14648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Erkek S, Johann PD, Finetti MA, et al. (2019) Comprehensive Analysis of Chromatin States in Atypical Teratoid/Rhabdoid Tumor Identifies Diverging Roles for SWI/SNF and Polycomb in Gene Regulation. Cancer Cell 35:95–110.e8. 10.1016/j.ccell.2018.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakayama RT, Pulice JL, Valencia AM, et al. (2017) SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat Genet 49:1613–1623. 10.1038/ng.3958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang X, Lee RS, Alver BH, et al. (2017) SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat Genet 49:289–295. 10.1038/ng.3746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Knutson SK, Warholic NM, Wigle TJ, et al. (2013) Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A 110:7922–7. 10.1073/pnas.1303800110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson BG, Wang X, Shen X, et al. (2010) Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell 18:316–328. 10.1016/j.ccr.2010.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xi W, Sansam CG, Thom CS, et al. (2009) Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res 69:8094–8101. 10.1158/0008-5472.CAN-09-0733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kadoch C, Crabtree GR (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: Mechanistic insights gained from human genomics. Sci Adv 1:e1500447. 10.1126/sciadv.1500447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han Z-Y, Richer W, Fréneaux P, et al. (2016) The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 7:10421. 10.1038/ncomms10421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Johann PD, Erkek S, Zapatka M, et al. (2016) Atypical Teratoid/Rhabdoid Tumors Are Comprised of Three Epigenetic Subgroups with Distinct Enhancer Landscapes. Cancer Cell 29:379–393. 10.1016/j.ccell.2016.02.001 [DOI] [PubMed] [Google Scholar]
- 60.Han Z-Y, Richer W, Fréneaux P, et al. (2016) The occurrence of intracranial rhabdoid tumours in mice depends on temporal control of Smarcb1 inactivation. Nat Commun 7:10421. 10.1038/ncomms10421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oberlick EM, Rees MG, Seashore-Ludlow B, et al. (2019) Small-Molecule and CRISPR Screening Converge to Reveal Receptor Tyrosine Kinase Dependencies in Pediatric Rhabdoid Tumors. Cell Rep 28:2331–2344.e8. 10.1016/j.celrep.2019.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kerl K, Ries D, Unland R, et al. (2013) The histone deacetylase inhibitor SAHA acts in synergism with fenretinide and doxorubicin to control growth of rhabdoid tumor cells. BMC Cancer 13:. 10.1186/1471-2407-13-286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thiemann M, Oertel S, Ehemann V, et al. (2012) In vivo efficacy of the histone deacetylase inhibitor suberoylanilide hydroxamic acid in combination with radiotherapy in a malignant rhabdoid tumor mouse model. Radiat Oncol 7:52. 10.1186/1748-717X-7-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Oruetxebarria I, Venturini F, Kekarainen T, et al. (2004) P16INK4a is required for hSNF5 chromatin remodeler-induced cellular senescence in malignant rhabdoid tumor cells. J Biol Chem 279:3807–16. 10.1074/jbc.M309333200 [DOI] [PubMed] [Google Scholar]
- 65.Betz BL, Strobeck MW, Reisman DN, et al. (2002) Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene 21:5193–203. 10.1038/sj.onc.1205706 [DOI] [PubMed] [Google Scholar]
- 66.Wetmore C, Boyett J, Li S, et al. Alisertib is active as single agent in recurrent atypical teratoid rhabdoid tumors in 4 children. 10.1093/neuonc/nov017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Geoerger B, Bourdeaut F, DuBois SG, et al. (2017) A phase I study of the CDK4/6 inhibitor ribociclib (LEE011) in pediatric patients with malignant rhabdoid tumors, neuroblastoma, and other solid tumors. Clin Cancer Res 23:2433–2441. 10.1158/1078-0432.CCR-16-2898 [DOI] [PubMed] [Google Scholar]
- 68.Leruste A, Tosello J, Ramos RN, et al. (2019) Clonally Expanded T Cells Reveal Immunogenicity of Rhabdoid Tumors. Cancer Cell 36:597–612. 10.1016/j.ccell.2019.10.008 [DOI] [PubMed] [Google Scholar]
- 69.Theruvath J, Sotillo E, Mount CW, et al. (2020) Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med 26:712–719. 10.1038/s41591-020-0821-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lafay-Cousin L, Fay-Mcclymont T, Johnston D, et al. (2015) Neurocognitive evaluation of long term survivors of atypical teratoid rhabdoid tumors (ATRT): The Canadian registry experience. Pediatr Blood Cancer 62:1265–1269. 10.1002/pbc.25441 [DOI] [PubMed] [Google Scholar]