

Abstract
Aim
Podocyte injury plays an important role in diabetic nephropathy (DN), yet the underlying molecular mechanisms of podocyte injury in DN is not clear. Here, we investigated the role of activating transcription factor 4 (ATF4) and HO-1 in DN-induced podocyte injury.
Methods
Protein expression was measured by western blotting (WB) and immunofluorescence. Cellular apoptosis was quantified by flow cytometry. ATF4 siRNA knockdown and HO-1 overexpression in podocyte were employed to evaluate the role of ER stress in DN-induced apoptosis and autophagy response. Urinary protein levels, nephrin expression, serum creatinine and BUN were evaluated and glomerulosclerosis was quantified by Periodic Acid-Schiff staining.
Results
Expression of ATF4 was increased in podocytes exposed to serum from DN mice. ATF4 knockdown enhanced DN-induced podocyte apoptosis. HO-1 overexpression reduced the decline of DN-induced podocyte autophagy and inhibited apoptosis and the beneficial effects of HO-1 overexpression in DN were blocked by ATF4 knockdown. The diabetic mice were significantly ameliorated by HO-1 agonist hemin treatment.
Conclusions
ATF4 induces autophagy by enhancing the expression of HO-1, and inhibits podocyte apoptosis in DN. Treatment with the HO-1 agonist reduced proteinuria, apoptosis, and enhanced autophagy response, and thus improved renal function in DN mice.
Keywords: Podocyte, autophagy, HO-1, ATF4, diabetic nephropathy
Introduction
Diabetic nephropathy (DN) is a serious complication associated with diabetes. The primary indication of DN is proteinuria. DN is a risk factor for kidney disease progression and if left untreated, it can lead to end-stage renal disease (ESRD) [1,2]. Podocytes or glomerular epithelial cells are terminally specialized cells that play a critical role in forming the glomerular filtration barrier. Structural changes or injuries in podocytes are associated with renal injury resulting in proteinuria and severe renal insufficiency [3–6], and contributes to DN [7].
Autophagy is a highly conserved process that utilizes the lysosomal pathway to degrade and recover cellular proteins and to remove and recycle damaged organelles. When autophagy is activated, cell homeostasis is maintained [4,8–11]. In DN, pathogenic molecules present in the serum is shown to inhibit autophagy and induce apoptosis in podocytes [12]. Previous studies have shown that podocyte dysfunction may lead to proteinuria via activation of epithelial-to-mesenchymal transition (EMT). By blocking EMT, podocyte apoptosis can be reversed [13–15].
Endoplasmic reticulum (ER) stress is triggered by the accumulation of unfolded proteins in the cells, and thus activates the unfolded protein response (UPR) which is associated with apoptosis in podocytes. Activation of ER stress plays an important role in the development of DN [16]. Accumulating evidence has shown that ER stress also induces autophagy as a mechanism to protect the cells from entering apoptosis. UPR may alleviate misfolded protein mediated ER stress by initiating autophagy [17]. In UPR, the PERK-eIF2α-ATF4 pathway plays critical role in regulating ER stress and restores protein homeostasis PERK (Protein kinase RNA-like endoplasmic reticulum kinase) is a pivotal transducer of ER stress by phosphorylating eukaryotic initiation factor 2 α-subunit (eIF2α), resulting in promoting pro-adaptive signaling pathways by decreasing global mRNA transcription and activation of ATF4 transcription factor [18–20]. Activation of PERK-eIF2α-ATF4 signaling pathway in ER stress has been found to be associated with autophagy induction and maintenance of intracellular homeostasis. However, growing evidence suggest that impairment in autophagy during prolonged ER stress and failure of pro-adaptive responses may lead to programmed cell death or apoptosis in DN [21,22]
Heme oxygenase-1 (HO-1) is an essential enzyme in heme catabolism that has been shown to protect cells by inhibiting oxidative stress and apoptosis [23]. Previous studies have shown that ATF4 regulates HO-1 expression [24,25], however, role of ATF4/HO-1 pathway in regulating autophagy and apoptosis in DN is not defined. In the present study, we demonstrated that ATF4 regulates autophagy in DN podocytes by promoting HO-1 expression. siRNA mediated knockdown of ATF4 led to autophagy impairment and increased apoptosis in renal podocytes, which were partially rescued by mTORC1 inhibitor rapamycin and HO-1 overexpression in podocytes in vitro. Additionally, HO-1 agonist hemin treated DN mice showed marked reduction in albuminuria and renal damage accompanied by autophagy induction and reduced apoptosis in the kidney. Our present study utilizing in vitro and in vivo approaches shed light on the biology of ATF4/HO-1 pathway in regulating autophagy and apoptosis in renal podocytes in DN and suggests beneficial therapeutic effect of HO-1 agonist hemin in treating diabetic nephropathy.
Materials and methods
In vitro stuies
Mouse podocyte cells, MPC5, were purchased from the Institute of Basic Medicine, Chinese Academy of Medical Sciences (Beijing, China) (3111C0001CCC000230). Podocytes were cultured in RPMI 1640 medium (Hyclone, SH30809.01B) supplemented with 10% fetal bovine serum (FBS, 11 nM D-glucose; Gibco, 16000-044) and subcultured at a 1:4 split. For propagation, podocytes were cultured at 33 °C and supplemented with 20 U/mL recombinant murine interferon-γ (IFN-γ, sigma, I4777). Podocytes were then cultured with 1.5 mL/25 cm2 collagenase IV (Gibco, 17104-019) for one hour, and washed with 3 mL/25 cm2 phosphate-buffered saline (PBS, Solaibao, P1020) until the podocytes retracted and began to detach from the bottom of culture flask. After cultured at 37 °C for 10 days, podocytes were subcultured at a 1:2 split in RPMI 1640 medium containing 10% FBS and 5 mM D-glucose for 24 h and then seeded onto 6-well plates, adjusting the concentration to 1 × 106 cells/ml. Every well contains 0.5 mL podocytes suspension, and 1.5 mL RPMI-1640 medium supplemented with 10% fetal bovine serum. Podocytes were then cultured at 37 °C and 5% CO2 atmosphere for 24 h. Then MCP5 podocytes were treated either with 10% serum from control C57BL/KsJ dm/m mice or with 10% serum from C57BL/KsJ dm/dm DN mice for 24 h.
In vivo studies
Group sizes and treatment
Animal experiments conformed to institutional standards. Eight-week-old male db/db mice exhibiting spontaneous DN (n = 6) and control C57BL/KsJ-db/db mice (n = 12) were raised to 13 weeks and divided into three groups: blank control group (n = 6); model-db/db group (n = 6); and treatment-db/db group (n = 6). Treatments started 24 h after urine was collected at 13 weeks of age. The db/db treatment group was injected intraperitoneally with hemin (25 mg/kg body weight each time, three times a week), while the db/db model group and blank control group were injected intraperitoneally with an equivalent volume of vehicle.
Construction of HO-1 overexpression vectors
Purified HO-1 gene fragments were digested with BamHI-EcoRI and ligated into pcDNA3.1(+) vector, which resulted in a recombinant plasmid called PCDNA3.1(+)-HO-1. Competent E. coli DH5α cells were transformed with the recombinant plasmid. Colonies were grown on Luria-Bertani (LB)+Ampicillin (Amp) plates at 37 °C overnight. On the following day, monoclonal colonies were selected and inoculated in LB culture solution with Amp, and cultured in a constant temperature shaker set at 37 °C overnight. After colonies were selected, colony PCR and sequencing were performed to verify the construct.
Statement of ethics
This study was approved by the ethics committee of Zhejiang Provincial People’ Hospital. All procedures were performed according to the institutional guidelines for the care and use of laboratory animals. Mice were housed in a specific pathogen-free condition with adequate food and water. At the end of the study periods, a midline abdominal incision was performed after mice were anesthetized. Then, blood was collected from the abdominal aorta, and both kidneys were removed quickly. Finally, the mice were killed by cervical dislocation.
Measurements
Western-blotting
The protein expression of ATF4, PERK, p-PERK, elF2α, and p-elF2α in different podocyte groups was detected by Western blotting. Briefly, cell lysates were collected in lysis buffer (Biyuntian Company, P0013B) containing Protein Stabilizing Cocktail (Thermo Fisher, 89806) and transferred onto PVDF membranes. After blocking with 5% fat-free dry milk in TBS-Tween (TBST), membranes were incubated with the following primary antibodies: PERK (Affinity, AF5304, 1:1000); p-PERK (Affinity, DF7576, 1:1000); elF2α (Proteintech, 12499-1-AP, 1:1000); p-elF2α (Affinity, AF3087, 1:1000); ATF4 (Affinity, DF6008, 1:1000); caspase-3 (CST, #9662, 1:1000); HO-1 (CST, #70081, 1:800); LC3 (CST, 4108S, 1:1000); and β-actin (Cell Signaling, #4970, 1:1000). After washing with TBST, membranes were incubated at 37 °C for one hour with an HRP-labeled secondary antibody (1:5000). After washed with TBST, blots were visualized using ECL reagent mixed with stable peroxidase solution at a ratio of 1:1, which was dripped onto PVDF membranes. A Tanon-5200 gel imaging system (Tanon) was used to visualize Western blots.
Detection of cellular apoptosis by flow cytometry
After digesting with 0.25% trypsin without EDTA, podocytes were collected and centrifuged at 1500 rpm for 5 min. After discarding the supernatant, e cells were resuspended twice in chilled PBS and centrifuged for 5 min at 1500 rpm. Cells were then mixed with 300 μL Binding Buffer suspension and 5 µL Annexin V-FITC in dark for 15 min. After that, 10 µL propidium iodide (PI) solution was added and suspensions were incubated in dark for another 10 min. Apoptosis was analyzed using a BD FACSCalibur flow cytometer (BD Biosciences).
Immunofluorescence staining
Podocytes were fixed with 4% paraformaldehyde for 15 min at room temperature and washed using PBS containing 0.1% Triton X-100 (Semberg, SBJ-1141). Cells were then blocked with 5% BSA (Hyclone, SH30574.03) in PBS for one hour. Primary antibody LC3 antibody (Mitrin Biology, 14600-1-AP, 1:50) or nephrin (Santa Cruz, sc-377246, 1:100) was added to the cells and incubated at 4 °C overnight. After extensive washing with PBS, cells were incubated with secondary antibody Alexa Fluor 594-conjugated Affinipure Goat Anti-Rabbit IgG (H + L) (Jackson, 111585003, 1:100) or Cy3 AffiniPure Goat Anti-Mice IgG (H + L) (Aimeijie, A22210, 1:100) at 37 °C in dark for 1 h. Cells were then washed, counterstained by DAPI for 5 min, and photomicrographed by fluorescence microscopy or laser scanning microscopy.
Determination of urinary protein, serum creatinine, and urea levels
A fully automatic biochemical analyzer was used to determine the urinary protein of mice at 13 weeks old (24 h before treatment) and at 25 weeks old (one day before the end of treatment). Blood was collected from the abdominal aorta and sit at room temperature for 2 h. After centrifuged at 1500 rpm for 10 min, the supernatant was collected. The urine creatinine test kit (Nanjing Jiancheng, C011-1) and urea nitrogen test kit (Nanjing Jiancheng, C013-2) were used to test the specific biofluid of interest, urine, pladma, and serum according to manufacturer’s instructions.
Periodic Acid-Schiff (PAS) stain
Formalin-fixed, paraffin-embedded tissue sections were dewaxed, rehydrated, and put into 1% aqueous solution of periodic acid for 10 min. After that, tissue sections were washed in tap water and then in distilled water. After stained with Schiff reagent (Leagene, DG0005) for 10 min, tissue sections were washed by flowing water for 10 min, and then stained with Harris hematoxylin for 10 min. After incubation with 1% hydrochloric acid-ethanol for 10 s and subsequent washes, the blue colorimetric reaction was performed by incubating samples with 1% ammonia for 10 s. PAS staining was analyzed under a microscope.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining
TUNEL has become a widely used staining method to help to detect apoptotic cells in tissue sections. Briefly, apoptotic cells were detected using a TUNEL Apoptosis Detection Kit (Biobox, BA27). Apoptotic cells were evaluated by microscope.
Statistical analysis
All statistical analyses were performed using SPSS 23.0 software (Stanford University, Stanford, CA). Results are expressed as mean ± standard deviation (SD). Significance was determined by student’s t test or one-way ANOVA followed by the Duncan's multiple-range test for multiple comparisons. Non-normally distributed variables were compared using the Kruskal–Wallis test with Mann–Whitney U for post-hoc testing. All p-values are two-tailed, and p < 0.05 was considered to indicate statistical significance.
Materials
Animal models
Eight-week-old C57BL/KsJ db/db mice exhibiting spontaneous DN and control C57BL/KsJ db/m mice (22 ± 2.5 g) were provided by Changzhou Canvas Laboratory Animal Co., Ltd. (SCX (Su) 2016-0010).
Drugs and reagents
The following reagents and drugs were used in this study: RPMI-1640 medium (Hyclone, SH30809.01B); collagenase IV (Gibco, 17104-019), PBS (Solebao, P1020); bovine serum albumin (BSA, Hyclone, SH30574.03); 1% Triton x-100 (Senbega, SBJ-1141); double antibody (Beijing Xia Si Biotechnology Co., Ltd., SV30010); 0.25% Trypsin (Gibco, 15050065); rapamycin (Targetmol, T1537); hemin (Sigma, 51280-1 G); γ-IFN (Sigma, I4777); FBS (Gibco, 16000-044).
Results
In vitro studies
Serum derived from diabetic nephropathy (DN) mice caused podocyte injury and increased ATF4 expression in vitro
We examined the effect of serum derived from DN mice on podocytes ER stress in vitro. Compared to serum from healthy mice, exposure to serum from DN mice caused significant podocytes damage, increased expression of apoptosis marker cleaved caspase-3 protein, and up-regulation of p-PERK and p-eIF2α, indicating activated endoplasmic reticulum stress (Figure 1(A,B)). Additionally, expression of transcriptional activator of unfolded protein response (UPR) target genes, ATF4 was significantly increased. These results suggest that many features of ER stress including activation of PERK-eIF2α-ATF4 pathway, can be applied using this cellular model to study DN-induced podocyte injury (Figure 1).
Figure 1.

Serum from DN mice induced podocyte injury and endoplasmic reticulum stress. (A) MCP5 podocytes were treated either with 10% serum from control C57BL/KsJ dm/m mice or with 10% serum from C57BL/KsJ dm/dm DN mice for 24 h. Western blot image showing activation of PERK-eIF2α-ATF4 in ER stress signaling pathway and increased apoptosis-related molecule cleaved caspase-3 in podocytes treated with serum from DN mice as compared to control mice. (B) Densitometric quantification of protein expression from Figure 1(A). (**p < 0.01, *p < 0.05).
ATF4 knockdown promotes podocytes apoptosis in vitro
In order to determine the role of ATF4 on podocyte apoptosis, we employed gene knockdown strategy using four different ATF4-silencing small interfering RNAs, and selected the most efficient siRNA for our study (Figure 2(A)). Podocytes transfected with ATF4 siRNA displayed markedly higher cleaved caspase-3 and podocyte apoptosis when exposed to serum from DN mice as compared to negative control siRNA (Figure 2(B–E)), indicating important role of ATF4 in ameliorating podocytes apoptosis in DN.
Figure 2.

Effects of ATF4 knockdown on podocyte apoptosis when exposed to serum from DN mice in-vitro. (A) Comparison of knockdown efficiency of various ATF4 siRNA constructs and the most efficient ATF4 siRNA-3 was selected for the present study. (B) Western blot analysis showed that cleaved caspase-3 protein expression was markedly increased by ATF4 siRNA in MCP-5 cells. (C) Densitometric analysis of caspase-3 protein expression from Figure 2(B). (D) Podocyte apoptosis determined by flow cytometry showed significant increase in cell death by ATF4 siRNA treatment. (E) Quantification of apoptosis rates from Figure 2(D). siRNA-NC: Negative control siRNA. (**p < 0.01, *p < 0.05).
ATF4 silencing decreased DN serum-induced autophagy in podocytes
Autophagy plays an important role in the inhibition of podocyte injury induced by high glucose levels. While induction of autophagy can inhibit podocyte injury in the context of high glucose levels, role of eIF2α/ATF4 pathway of autophagy in podocytes of mice with diabetic nephropathy is not clear [19,26]. To investigate whether autophagy is the mechanism by which ATF4 impacts podocyte injury induced by DN mice serum, we examined the expression of autophagy markers LC3. Immunofluorescence analysis revealed that, treatment with DN mice serum significantly reduced LC3 punctate aggregates in podocytes, indicating a decrease in autophagy. ATF4 silencing led to further downregulation of LC3 punctate in podocytes, suggesting an even more pronounced reduction in autophagy. Our study further showed that treatment with mTOR inhibitor rapamycin reversed this effect, and LC3 punctate aggregates were markedly increased (Figure 3(A)). Western blot analysis showed that treatment with DN mice serum promoted cleavage of apoptosis-related caspase-3 protein and decreased the ratio of autophagy protein LC3II/I, and ATF4 silencing exacerbated these effects. Whereas Rapamycin treatment reduced the cleaved caspase-3 protein and increased the ratio of LC3II/I (Figure 3(B–D)). Apoptosis rate determined from flow cytometry analysis replicated and verified our findings of cleaved caspase-3 by western analysis (Figure 3(E,F)). Our study therefore suggests that knockdown of ATF4 promotes podocyte apoptosis by inhibiting autophagy and presumably, ATF4 positively regulates autophagy and inhibit podocytes injury.
Figure 3.

Effects of ATF4 knockdown and mTOR inhibitor rapamycin on autophagy and apoptosis in podocytes induced by serum from DN mice. (A) Immunofluorescence analysis of expression and distribution of autophagy markers protein LC3 showed significant downregulation by ATF4 siRNA in MCP-5 podocytes exposed to serum from DN mice, which was reversed by rapamycin treatment; scale indicates 10 μm. (B) Protein expression analysis supported our immunofluorescence data on LC3 downregulation by ATF4 siRNA and rescue by rapamycin. It further showed that rapamycin treatment significantly reduced cleaved caspase-3 expression. (C) Densitometric analysis of protein expression from Figure 3(B). (D) Densitometric analysis of protein expression. (E) Podocyte apoptosis was evaluated by flow cytometry. (F) Quantification of apoptosis rates. siRNA-NC: control siRNA transfected for 48 h; siRNA-ATF4: siRNA-ATF4 transfected for 48 h. RAP, Rapamycin treatment for 24 h. (**p < 0.01, *p < 0.05).
ATF4 knockdown regulates podocyte autophagy and apoptosis via HO-1
Studies showed that ATF4 regulates HO-1 expression [24], and HO-1 stimulates autophagy and inhibits podocyte apoptosis induced by high glucose [27], however role of HO-1/ATF4 axis in podocytes of diabetic nephropathy remains unknown. Our study showed that, exposure to DN mice serum caused up-regulation of HO-1, whereas HO-1 expression was decreased in podocytes after ATF4 silencing (Figure 4), suggesting a link between ATF4 and HO-1 expression in DN.
Figure 4.
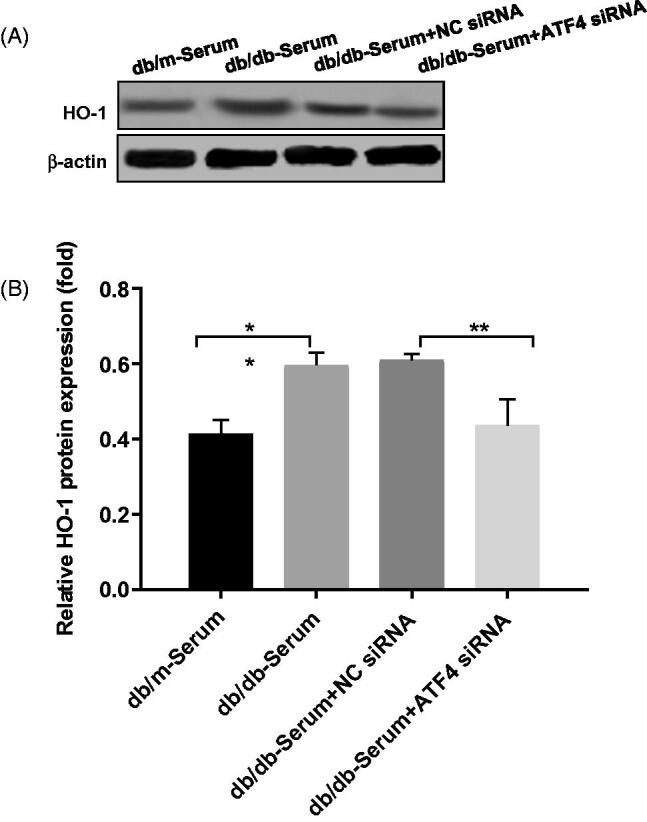
Effects of ATF4 knockdown on HO-1 expression on podocytes exposed to serum from DN mice. (A) Protein expression analysis revealed that HO-1 expression was increased by serum from DN mice, indicating increased ER stress, while ATF4 siRNA significantly reduced HO-1 expression in MCP-5 podocyte. (B) Densitometric analysis of protein expression from Figure 4(A) (**p < 0.01, *p < 0.05).
In order to verify whether ATF4 inhibits apoptosis through HO-1, we designed and synthesized a mouse-derived HO-1 sequence, constructed an overexpression vector, and verified the effect of HO-1 overexpression by western blot (Figure 5(A)). ATF4 siRNA and HO-1 overexpression vectors were transfected into podocytes. Western blot shows that HO-1 overexpression plasmid mitigated the down-regulation of HO-1 expression induced by ATF4 siRNA, and increased caspase-3 cleavage (Figure 5(B,C)). Immunofluorescence analysis showed that HO-1 overexpression is unchanged (Figure 5(D–F)). These data demonstrate that ATF4 inhibits podocyte apoptosis in DN by HO-1 regulation of autophagy.
Figure 5.

Effects of ATF4 knockdown and HO-1 overexpression on podocyte autophagy and apoptosis when subjected to serum from DN mice. (A) HO-1 overexpression was verified in MCP-5 podocytes. (B) Podocytes overexpressing HO-1 were subjected to serum from DN mice, showed significantly reduced cleaved caspase 3 expression, and co-treatment of HO-1 overexpression with ATF4 siRNA increased cleaved caspase-3 expression. (C) Densitometric analysis of protein expression from Figure 5(B). (D) Immunofluorescence analysis showed increase in autophagy LC3 protein expression by HO-1 overexpression, which was significantly reduced by ATF4 siRNA co-treatment; the scale indicates 10 μm. (E) Quantification of apoptosis rates by flow cytometry supported our western analysis of cleaved caspase-3. (F) Apoptosis was evaluated by flow cytometry. NC siRNA: control siRNA transfected for 48 h; ATF4 siRNA: siRNA-ATF4 transfected for 48 h. Vector: Empty plasmid transfected for 48 h. HO-1 OE: HO-1 overexpression plasmid transfected for 48 h. (**p < 0.01, *p < 0.05).
In vivo studies
Therapeutic effects of HO-1 agonist in mice with DN
We determined in our in vitro experiments that ATF4 inhibited podocyte apoptosis induced by serum from DN mice through autophagy induced by HO-1. Based on our initial in-vitro data showing reduced ATF4 expression promoted apoptosis and reduced HO-1 expression in podocytes exposed to serum from DN mice, we performed in-vivo study using DN mouse model to study the therapeutic effects of HO-1 agonist hemin to determine its role in podocyte apoptosis and autophagy. Previous study has shown that the expression of ATF4 were highly elevated in the db/db mice compared with that in the db/m mice [1]. In our research, we detected the expression of HO-1 and find that the expression of HO-1 was increased in DN mice, whereas hemin treatment significantly increased it. (Figure S1) Then, we evaluated urinary total protein and urinary albumin content in hemin treatment. Before the onset of the treatment, and as expected DN mice exhibited proteinuria compared to the healthy control mice. No difference in urinary total protein was found between the agonist-treated and untreated DN mice groups. At the end of the hemin treatment, we found that hemin exposure significantly decreased proteinuria as well as albuminuria compared to untreated DN mice (Figure 6(A)). Additionally, analysis of blood chemistry revealed that serum creatinine and serum urea levels were increased in DN, compared to healthy control mice, and hemin treatment significantly reduced their levels (Figure 6(B)). These data suggest beneficial therapeutic effects of HO-1 agonist hemin in alleviating renal damage in DN.
Figure 6.

Effects of HO-1 agonist hemin treatment on clinical indications of DN-induced autophagy and apoptosis in podocytes. (A) Evaluation of urinary total protein and urinary albumin showed increased content in DN mice compared to healthy controls, while hemin treatment significantly reduced proteinuria and albuminuria in DN mice. (B) Serum creatinine and urea levels were also significantly reduced by hemin treated DN mice. (C) PAS staining showed glomerular damage in DN mice and it was rescued by hemin treatment. (D) Protein expression analysis revealed that expression of critical protein in renal filtration barrier nephrin was downregulated in DN mice, and hemin treatment significantly increased the neprhin expression and densitometric analysis of protein expression. (E) TUNEL staining showed that hemin treatment significantly reduced apoptotic cells in DN mice. (F) Quantification of apoptotic cells. (G) Protein expression by Western blot. (H) Densitometric analysis of protein expression. (I)Expression and distribution of nephrin and LC3 protein were evaluated by immunofluorescence; the scale indicates 50 μm. (**p < 0.01, *p < 0.05).
We next evaluated renal tissues for glomerular damage in hemin treatment. In healthy mice, PAS staining showed that the glomerular structure of control mice was clear, and extracellular matrix and mesangial cells were normal in morphology. In DN mice, modest increase in glomerular volume, thickening of mesangial matrix, and mesangial cells proliferation were noticed. After hemin treatment, glomerular volume, mesangial matrix thickening and mesangial cell proliferation were reduced (Figure 6(C)). We also found that glomerular injury as assessed by measuring the expression of nephrin, an important renal filtration barrier protein in DN mice was downregulated significantly, and hemin treatment significantly increased nephrin expression, attenuating glomerular injury (Figure 6(D)). TUNEL assay showed that apoptosis of glomerular cells in DN mice was increased significantly, and hemin treatment significantly reduced apoptosis of glomerular cells (Figure 6(E,F)).
We further evaluated glomerular podocyte injury. Based on our data, we hypothesized that HO-1 may inhibit podocyte apoptosis induced by DN mice serum through promoting autophagy. In order to elucidate the mechanism of HO-1 action, we measured autophagy and apoptosis markers in hemin treated renal tissues used autophagy fluorescence detection and western blot. Glomerular LC3 punctate aggregates and the LC3II/I ratio in DN mice were significantly decreased compared to healthy control mice, and hemin treatment significantly increased LC3 punctate and LC3II/I ratio and inhibited apoptosis (Figure 6(G–I)). These results were consistent with the in vitro experiments and showed that hemin likely inhibits apoptosis of podocytes by inducing podocyte autophagy to alleviate renal lesions and improve renal function in this DN model.
Discussion
In this study, we used both in-vitro cellular model and in vivo mouse model to investigate the role of ATF4 and HO-1 in podocyte injury caused by DN. We demonstrated that exposure to serum from DN mice promoted podocyte injury, activated ER stress, and increased ATF4 expression. We demonstrated that siRNA-mediated knockdown of ATF4 exacerbated DN-induced podocyte apoptosis and markedly reduced autophagy response, which were partially rescued by the mTOR inhibitor rapamycin. We also showed that ATF4 knockdown significantly decreased the HO-1 expression in the podocytes. Using diabetic mouse model, we demonstrated that treatment with HO-1 agonist, hemin, markedly attenuated the DN-induced renal injury as evidenced by reduced albuminuria, ameliorated glomerulosclerosis and increased nephrin expression. The protective effect of hemin was associated with reduced renal apoptosis and enhanced autophagy response. This study highlights the protective role of ATF4 and HO-1 against DN-induced podocyte injury.
Activation of ER stress is increasingly investigated in various physiological and pathophysiological conditions and experimental evidence indicates that ER stress is involved in the glomerular damage associated with diabetic nephropathy [1]. ATF4 is a major regulator of ER stress and is a part of PERK-eIF2α-ATF4 unfolded protein response (UPR) in DN. Emerging evidence indicate that ATF4 plays critical role in regulating ER stress, autophagy and apoptosis. In our study, serum of DN mice caused significant increase in ATF4 expression, paralleled by marked induction of UPR signaling pathways and ER-induced apoptotic markers. This data indicates that serum of DN mice is sufficient to cause activation of many ER features in our cellular model. Interestingly, knockdown of ATF4 dramatically triggered podocyte apoptosis and abolished autophagy response as evidenced by increased cleaved caspase-3 expression and reduction in autophagy markers LC-3, respectively. The data are in agreement with previous report showing activation of podocyte ER stress caused by high-glucose.
Guan Bo-Jhih et al. showed that eIF2α/ATF4 signaling promotes mTORC1 activity, and demonstrated that mTORC1 is a downstream target of eIF2α/ATF4 signaling [28]. Additionally, inhibition of mTOR signaling by rapamycin is shown to prevent apoptosis and thus may regulate autophagy [4]. Supporting previous findings, our data showed that rapamycin rescued DN serum-induced apoptosis and promoted autophagy response in both non-transfected and ATF4 silenced podocytes, however recuse response was lower in ATF4 knockdown by rapamycin, consistent with the role of protective role of ATF4 in this study. Our results suggest that ATF4 is a critical player in ER stress in DN-induced podocyte injury and indicates role of mTORC1 in ATF4 mediated protective autophagy response in podocytes.
Heme oxygenase-1 (HO-1) plays pivotal role in regulating autophagy and its expression increases under stress conditions. HO-1 is shown to ameliorate oxidative stress [29] and improves renal function in DN [30]. In the present study, we investigated the role of HO-1 and ATF4 in DN-induced ER stress. We found that HO-1 expression was significantly increased in podocytes exposed to serum from DN mice suggesting activation of self-protection mechanism and ATF4 knockdown significantly reduced HO-1 expression, indicating HO-1 expression may be regulated by ATF4 in podocytes in diabetic nephropathy. This data is in agreement with previous report showing association between HO-1 expression and transcription factor ATF4 in tumor [2]. Additionally, we demonstrated that HO-1 overexpression attenuated DN-induced podocyte apoptosis and triggered autophagy response. Furthermore, ATF4 knockdown prevented the beneficial effects of HO-1 overexpression in podocytes. This data suggests that effects of ATF4 in podocyte injury are mediated at least in part via HO-1-regulated autophagy.
Using in vivo mouse model of diabetic nephropathy, we evaluated the effect of hemin, an inducer of HO-1 on renal apoptosis and autophagy. In our study, C57BL/KsJ db/db mice developed progressive proteinuria and elevated serum creatinine and BUN, as well glomerulosclerosis similar to human DN, indicating the renal dysfunction and the development of DN in our animal model. Interestingly, hemin treatment significantly attenuated albuminuria, reduced serum creatinine, BUN and ameliorated glomerular histopathology, suggesting hemin effectively prevented the development of DN. Furthermore, our results revealed that hemin alleviated number of renal apoptotic cells and markedly enhanced the expression of nephrin and autophagy marker LC-3. This data suggest that HO-1 protects DN-induced ER stress in podocyte injury by enhancing autophagy response.
Conclusions
This study provides evidence that ATF4 protects against DN-induced podocyte injury by mechanisms involving attenuating PERK-eIF2α-ATF4 pathway in ER stress mediated by mTORC1 inhibition and autophagy enhancement promoted by LC-3 upregulation. Additionally, HO-1 exerts beneficial effects in ameliorating renal apoptosis and enhanced autophagy in mouse model of DN, which subsequently improved albuminuria and glomerulosclerosis and prevents the progression of DN. This study highlights the important role of ATF4 and HO-1 in regulating renal apoptosis and autophagy, and HO-1 inducers may represent promising therapeutic strategy for preventing DN development.
Supplementary Material
Acknowledgements
We would like to thank our colleagues at Zhejiang Provincial People’ Hospital for their valuable contributions to this work.
Funding Statement
This work was supported by the Construction of Key Projects by Zhejiang Provincial Ministry under Grant [Number WKJ-ZJ-1915, WKJ-ZJ-2017]; The Zhejiang Province Chinese Medicine Modernization Program under Grant [Number 2020ZX001]; The General Project of Zhejiang Education Department under Grant [Number Y201942823]; and Clinical and Experimental Research of YSHS Granule.
Disclosure statement
The authors declare that they have no competing interests. The authors have no conflicts of interest to declare.
References
- 1.Kang MK, Park SH, Kim YH, et al. Chrysin ameliorates podocyte injury and slit diaphragm protein loss via inhibition of the PERK-eIF2α-ATF-CHOP pathway in diabetic mice. Acta Pharmacol Sin. 2017;38:1129–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leti F, Morrison E, DiStefano JK.. Long noncoding RNAs in the pathogenesis of diabetic kidney disease: implications for novel therapeutic strategies. Per Med. 2017;14:271–278. [DOI] [PubMed] [Google Scholar]
- 3.He P, Liu D, Zhang B, et al. Hepatitis B Virus X protein reduces podocyte adhesion via downregulation of α3β1 integrin. Cell Physiol Biochem. 2017;41:689–700. [DOI] [PubMed] [Google Scholar]
- 4.Jin J, Hu K, Ye M, et al. Rapamycin reduces podocyte apoptosis and is involved in autophagy and mTOR/P70S6K/4EBP1 signaling. Cell Physiol Biochem. 2018;48(2):765–772. [DOI] [PubMed] [Google Scholar]
- 5.Lin X, Zhen X, Huang H, et al. Role of MiR-155 signal pathway in regulating podocyte injury induced by TGF-β1. Cell Physiol Biochem. 2017;42:1469–1480. [DOI] [PubMed] [Google Scholar]
- 6.Huang H, You Y, Lin X, et al. Inhibition of TRPC6 signal pathway alleviates podocyte injury induced by TGF-β1. Cell Physiol Biochem. 2017;41:163–172. [DOI] [PubMed] [Google Scholar]
- 7.Tung CW, Hsu YC, Shih YH, et al. Glomerular mesangial cell and podocyte injuries in diabetic nephropathy. Nephrology (Carlton). 2018;23(Suppl 4):32–37. [DOI] [PubMed] [Google Scholar]
- 8.Ke B, Tian M, Li J, et al. Targeting programmed cell death using small-molecule compounds to improve potential cancer therapy. Med Res Rev. 2016;36:983–1035. [DOI] [PubMed] [Google Scholar]
- 9.Ouyang L, Zhang L, Liu J, et al. Discovery of a small-molecule bromodomain-containing protein 4 (BRD4) inhibitor that induces AMP-activated protein kinase-modulated autophagy-associated cell death in breast cancer. J Med Chem. 2017;60:9990–10012. [DOI] [PubMed] [Google Scholar]
- 10.Wu F, Li S, Zhang N, et al. Hispidulin alleviates high-glucose-induced podocyte injury by regulating protective autophagy. Biomed Pharmacother. 2018;104:307–314. [DOI] [PubMed] [Google Scholar]
- 11.Liu WJ, Huang WF, Ye L, et al. The activity and role of autophagy in the pathogenesis of diabetic nephropathy. Eur Rev Med Pharmacol Sci. 2018;22:3182–3189. [DOI] [PubMed] [Google Scholar]
- 12.Yasuda-Yamahara M, Kume S, Tagawa A, et al. Emerging role of podocyte autophagy in the progression of diabetic nephropathy. Autophagy. 2015;11:2385–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chitra PS, Swathi T, Sahay R, et al. Growth hormone induces transforming growth factor-beta-induced protein in podocytes: implications for podocyte depletion and proteinuria. J Cell Biochem. 2015;116:1947–1956. [DOI] [PubMed] [Google Scholar]
- 14.Cai X, Wang L, Wang X, et al. Silence of IGFBP7 suppresses apoptosis and epithelial mesenchymal transformation of high glucose induced-podocytes. Exp Ther Med. 2018;16:1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaguchi Y, Iwano M, Suzuki D, et al. Epithelial-mesenchymal transition as a potential explanation for podocyte depletion in diabetic nephropathy. Am J Kidney Dis. 2009;54(4):653–664. [DOI] [PubMed] [Google Scholar]
- 16.Tian N, Gao Y, Wang X, et al. Emodin mitigates podocytes apoptosis induced by endoplasmic reticulum stress through the inhibition of the PERK pathway in diabetic nephropathy. Drug Des Devel Ther. 2018;12:2195–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. 2017;13:681–696. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Sarkar C, Dinizo M, et al. Disrupted autophagy after spinal cord injury is associated with ER stress and neuronal cell death. Cell Death Dis. 2015;6:e1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo B, Lin Y, Jiang S, et al. Endoplasmic reticulum stress eIF2α-ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis. 2016;7:e2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gallazzini M, Pallet N.. Endoplasmic reticulum stress and kidney dysfunction. Biol Cell. 2018;110:205–216. [DOI] [PubMed] [Google Scholar]
- 21.Tsai TC, Lai KH, Su JH, et al. 7-Acetylsinumaximol B induces apoptosis and autophagy in human gastric carcinoma cells through mitochondria dysfunction and activation of the PERK/eIF2α/ATF4/CHOP signaling pathway. Mar Drugs. 2018;16(4):104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiang Q, Li F, Shi K, et al. Involvement of p38 in signal switching from autophagy to apoptosis via the PERK/eIF2α/ATF4 axis in selenite-treated NB4 cells. Cell Death Dis. 2014;5:e1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Wu S, Tang L, et al. Mesenchymal stem cells overexpressing heme oxygenase-1 ameliorate lipopolysaccharide-induced acute lung injury in rats. J Cell Physiol. 2019;234:7301–7319. [DOI] [PubMed] [Google Scholar]
- 24.Dey S, Sayers CM, Verginadis II, et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J Clin Invest. 2015;125:2592–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dias-Teixeira KL, Calegari-Silva TC, Medina JM, et al. Emerging role for the PERK/eIF2α/ATF4 in human cutaneous leishmaniasis. Sci Rep. 2017;7:17074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.B'Chir W, Maurin AC, Carraro V, et al. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong C, Zheng H, Huang S, et al. Heme oxygenase-1 enhances autophagy in podocytes as a protective mechanism against high glucose-induced apoptosis. Exp Cell Res. 2015;337:146–159. [DOI] [PubMed] [Google Scholar]
- 28.Guan BJ, Krokowski D, Majumder M, et al. Translational control during endoplasmic reticulum stress beyond phosphorylation of the translation initiation factor eIF2α. J Biol Chem. 2014;289:12593–12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao L, Kwan YW, Bulmer AC, et al. Noninvasive Real-time characterization of renal clearance kinetics in diabetic mice after receiving danshensu treatment. Oxid Med Cell Longev. 2018;2018:8267560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J, Hu L, Chen Y, et al. Sitagliptin improves renal function in diabetic nephropathy in male Sprague Dawley rats through upregulating heme oxygenase-1 expression. Endocrine. 2019;63(1):70–78. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.