

Abstract
Nearly 15% of melanomas occur in patients with a family history and a subset of these patients have a germline mutation in a melanoma predisposing gene. CDKN2A mutations are responsible for the majority of hereditary melanoma, but many other susceptibility genes have been discovered in recent years, including CDK4, TERT, ACD, TERF2IP, POT1, MITF, MC1R, and BAP1. Additionally, melanoma risk is increased in mixed cancer syndromes caused by mutations in PTEN, BRCA2, BRCA1, RB1, and TP53. While early onset, multiple tumors, and family cancer history remain the most valuable clinical clues for hereditary melanoma, characteristic epithelioid cytology of melanocytic tumors may suggest an underlying BAP1 mutation. Herein, we review the clinical and histopathologic characteristics of melanocytic tumors associated with these germline mutations and discuss the role of genetic counseling.
Keywords: CDKN2A, germline mutation, hereditary, melanocytic nevus, melanoma
1 |. INTRODUCTION
A subset of melanoma, approximately 7% to 15%, occurs in individuals with a family history.1 The factors influencing melanoma risk in a family include shared sun exposure experiences, geographic location, skin phototype, and genetic variants.1 Approximately 22% of familial cases are caused by a mutation in a currently known single high-risk tumor predisposition gene, CDKN2A, and over half of individuals with multiple primary melanomas carry mutations in the gene.1 Melanoma may either be the dominant cancer in the family, such as in families with CDKN2A mutation, or be a part of a mixed cancer syndrome such as in families with Cowden syndrome caused by PTEN mutations. Generally, the mutations in tumor predisposition genes cause multiple tumors with earlier onset than in the general population and occur in those with a positive family history.
While only a small subset of melanoma patients have a tumor syndrome predisposing to cancer, understanding the causes of hereditary melanoma has led to the discovery of a role for several key genes, many of which are also somatically mutated in melanoma, including CDKN2A, TERT, MITF and PTEN.2 This review discusses the genetic background and clinical and histopathologic features of hereditary syndromes characterized by predisposition to melanoma, melanocytic nevi, and other melanocytic tumors. While most of these tumors are histopathologically indistinguishable from their sporadic counterparts, a subset shows characteristic features such as those caused by BAP1 germline mutations. Awareness of these syndromes will facilitate early diagnosis and improve patient care.
2 |. GERMLINE MUTATIONS
2.1 |. CDKN2A
CDKN2A is by far the most commonly mutated gene causing hereditary melanoma (Table 1).1 Germline mutations in CDKN2A increase the risk of melanoma by 65-fold.3 This tumor syndrome was first described in the 1960s by Lynch and Krush4 as familial atypical multiple mole and melanoma syndrome (FAMMM) and by Clark and colleagues as B-K mole syndrome5 or dysplastic nevus syndrome. Both groups described families with multiple clinically atypical nevi, melanomas and, in a subset of patients, pancreatic cancer. The disease gene was later identified in the 1990s as CDKN2A.6,7
TABLE 1.
Germline mutations associated with melanocytic tumors. The relevant gene and its function, clinical phenotype, and histopathological features of melanoma are summarized here.
| Genea | Gene/protein function | Phenotype | Histopathologic features or most common histopathologic subtype of melanoma |
|---|---|---|---|
| Cyclin dependent kinase inhibitor 2A (CDKN2A) | • Tumor suppressor • Two main alternate transcripts: (a) p16 inhibits CDK4 and phosphorylation of RB; (b) p14ARF inhibits HDM2 and ubiquitination of p53 |
• Melanocytic nevi, melanoma, pancreatic, upper GI, and respiratory cancers; astrocytoma, neurofibromas and schwannomas (mutation affecting p14ARF) | • Superficial spreading melanoma • Pigmentation, pagetoid scatter, and spindle cell morphology in vertical growth phase |
| Cyclin dependent kinase 4 (CDK4) | • Oncogene • Inhibits binding of p16 tumor suppressor leading to phosphorylation of RB and cell cycle progression |
• Melanocytic nevi, melanoma, pancreatic cancer | • Superficial spreading melanoma • Pigmentation and pagetoid scatter |
| Telomerase reverse transcriptase (TERT) | • Telomerase reverse transcriptase, a component of telomerase • Normally repressed in postnatal somatic cells leading to shortening of telomeres |
• Melanoma, melanocytic nevi, other reported cancers (ovarian, renal cell, bladder, breast, and bronchial cancer) | • Nodular and superficial spreading melanoma |
| Protection of telomeres 1 (POT1) | • Constituent of shelterin complex that regulates telomere processing and stability | • Melanoma, other reported cancers (glioma, chronic lymphocytic leukemia, colorectal, breast, and lung cancers) | • Superficial spreading melanoma |
| ACD shelterin complex subunit and telomerase recruitment factor (ACD) | • Constituent of shelterin complex that regulates telomere processing and stability | • Melanoma, other reported cancers (breast, ovarian, cervical, uterine, thyroid, colon, lung, renal, urinary, prostate and esophageal cancers, lymphomas and leukemias) | • Superficial spreading melanoma, and lentigo maligna melanoma |
| TERF2 interacting protein (TERF2IP) | • Constituent of shelterin complex that regulates telomere processing and stability | • Melanoma, other reported cancers (breast, ovarian, cervical, uterine, thyroid, colon, lung, renal, urinary, prostate and esophageal cancers, lymphomas and leukemias) | • Superficial spreading melanoma, and lentigo maligna melanoma |
| Melanocyte inducing transcription factor (MITF) | • A melanocytic lineage-specific transcription factor, regulating differentiation, proliferation and survival of melanocytes | • Melanoma, renal cell carcinoma • Darker hair, fair skin, and non-blue eye color |
• Amelanotic • Thicker tumors (in some populations) • Nodular melanoma |
| Melanocortin-1 receptor (MC1R) | • G protein coupled receptor for melanocyte-stimulating hormone • Controls melanogenesis and thus skin and hair color |
• Melanoma • Red hair, freckling, light skin, and UV sensitivity (loss-of-function variants) |
• Anatomic site (arms; in carriers of more than one high-risk variant) |
| BRCA1 associated protein 1 (BAP1) | • Deubiquitinating enzyme, BRCA1 binding partner • Implicated in chromatin modulation, transcriptional regulation, and DNA damage repair |
• BAP1-inactivated nevi, uveal melanoma, cutaneous melanoma, mesothelioma, and renal cell carcinoma • Other reported tumors (basal cell carcinoma, meningioma, cholangiocarcinoma, breast, lung, pancreatic, and thyroid cancer) |
BAP1-inactivated nevi: • Exclusively or predominantly intradermal • Associated common nevus component often present (a combined nevus) • Epithelioid melanocytes with round to oval vesicular nuclei and abundant amphophilic cytoplasm • May show smaller epithelioid cells without abundant eosinophilic cytoplasm • May show rhabdoid features |
Gene symbols and nomenclature obtained from the HUGO Gene Nomenclature Committee.
CDKN2A is a tumor suppressor gene encoding two transcripts, p16 and p14(ARF), that regulate two critical cell cycle pathways. p16 inhibits CDK4 and, therefore, phosphorylation of RB. p14ARF inhibits HDM2 and, therefore, ubiquitination of p53. In addition to germline mutations, CDKN2A is commonly somatically mutated in sporadic melanoma.1 Somatic biallelic inactivation of CDKN2A occurs exclusively within invasive melanomas8,9 and can be assessed with immunohistochemical staining for p16 protein. Utility and recommendations for p16 staining as a prognostic and diagnostic marker are variable.10
A family with CDKN2A mutation often includes multiple individuals with numerous atypical melanocytic nevi, sometimes more than 50.11 Patients with nevi on the buttocks or dorsal feet have the highest risk of being mutation carriers.11 Nevi are variable in appearance, ranging from banal to atypical, with large size, irregular and indistinct borders, and variable colors.1 Histologically, they exhibit features of a dysplastic nevus, including cytologic atypia, asymmetry, papillary dermal fibroplasia, lentiginous melanocytic hyperplasia, variable lymphocyte infiltration, and “shouldering” phenomenon.12
Melanoma occurs approximately 15 years earlier in CDKN2A mutation carriers than in the general population,13 with a median age of onset around 33 to 45 years compared to 53 to 61 years in the general population.14,15 Melanoma may occur in adolescence or early adulthood with some of the youngest reported cases at 13 years of age.16,17 Conversely, in a population-based series of 20 childhood melanomas, only one CDKN2A mutation was identified.18 A recent study performed in an Italian population demonstrated no difference between overall survival or melanoma-specific survival in patients with and without CDKN2A germline mutations,19 contradicting prior reports based on Swedish Cancer Registry data.3 However, these studies were performed in different populations with possible underlying differences, necessitating caution when interpreting the results.
Compared with the general population, melanomas are thinner in mutation carriers,20 possibly confounded by increased surveillance of individuals with inherited susceptibility to melanoma. Superficial spreading melanoma is most common in CDKN2A mutation carriers, including on the head and neck.21 In a case-control comparison of 81 sporadic melanomas, 123 melanoma families with the CDKN2A mutation, and 120 melanoma families without, histopathological features specific to CDKN2A mutation positive melanomas included higher pigmentation, pagetoid scatter, and spindle cell morphology in the vertical growth phase.22 No differences were found in ulceration, epidermal nesting, the presence of an associated nevus or lentigo, fibroplasia, solar elastosis, regression, or associated lymphocytes, or cell shape, cytologic grade, and mitotic figures of the radial growth phase.22 Others have found that sparser inflammation and lack of regression may be associated features.15 Similar to sporadic melanoma, somatic mutations in melanomas associated with germline CDKN2A mutation include BRAF and NRAS, although BRAF tends to be less common and NRAS mutations more common in CDKN2A germline mutation associated melanoma.23,24
Notably, a subset of patients, 28%, develops pancreatic cancer.1 Survival rates in patients with CDKN2A mutations who develop pancreatic cancer are lower compared to those without the mutation (22 vs 35 months).25 In addition, CDKN2A mutation carriers have an increased risk of upper digestive cancer and cancers involving the respiratory tract, especially in ever-smokers, suggesting that CDKN2A increases sensitivity to carcinogens in tobacco.26,27 Therefore, CDKN2A mutation carriers should be counseled about smoking cessation.
2.2 |. CDKN2A mutations affecting p14ARF transcript
Through alternatively spliced variants involving exon 1 alpha and 1 beta, CDKN2A encodes two major proteins, p16 and p14(ARF), respectively. p14(ARF) regulates the cell cycle through p53 dependent apoptotic pathways.28,29
Interestingly, families with CDKN2A mutations that affect the p14 (ARF) transcript develop tumors of the central nervous system, including astrocytoma, and nerve sheath tumors, including neurofibromas and schwannomas, in addition to melanoma (Figure 1A–C; melanoma astrocytoma syndrome).30–36 Thus far, missense mutations or deletions at exon 1 beta have been reported in at least three families.37,38 Therefore, exon 1 beta should be considered in genetic testing of hereditary melanoma, especially if neural tumors are present.
FIGURE 1.
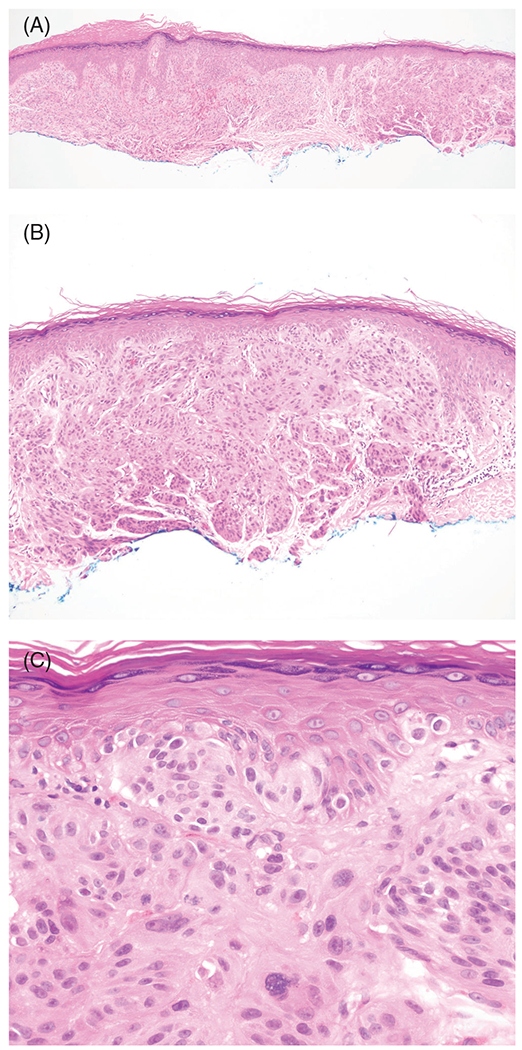
Melanoma associated with a germline deletion of exon 1B of CDKN2A gene. The patient had a history of multiple primary cutaneous melanomas and visceral metastases. A, Hemotoxylin and eosin (H&E), ×40. B, H&E, ×100. C, H&E, ×400
2.3 |. CDK4
CDK4 germline mutations are rare, with fewer than 20 families reported in the literature.39 CDK4 is an oncogene that, when mutated, inhibits binding of p16, leading to phosphorylation of RB and cell cycle progression. Similar to CDKN2A mutations, CDK4 germline mutations predispose to early onset multiple primary melanomas and increased numbers of atypical nevi.40,41 CDK4 mutation-associated melanomas often occur on the limbs, with average age of onset at 39 years (range of 18-86 years).
In a study of 17 families that included 103 individuals with melanoma, the most common histological subtype was superficial spreading melanoma.41 The average tumor thickness is 0.32 mm.12 A longitudinal study by Clark and colleagues demonstrated that a precursor nevus, either intradermal or dysplastic nevus, is often present.12 Melanocytes, sometimes heavily pigmented, are arranged as single cells and nests with upward scatter of single cells and associated with epidermal hyperplasia.12 In some tumors, epithelioid cell morphology and lesser pigmentation are noted.12
In addition to melanomas, CDK4 mutation carriers may be at higher risk of developing non-melanoma skin cancers, breast cancer, pancreatic cancer, ovarian cancer, cervical cancer, and stomach cancer, among others.1,41
2.4 |. TERT
The role of telomeres is highly implicated in tumorigenesis; germline and somatic mutations in TERT, encoding for one of the main components of telomerase, are common in human tumors, including melanoma. Somatic TERT promoter mutations are considered one of the earliest secondary mutations following BRAF and NRAS driver mutations, and are found in 30% to 70% of sporadic melanomas, especially nodular or superficial spreading melanomas.8,42,43
TERT encodes a reverse transcriptase that, together with TERC, creates a template for telomere addition, and forms the main components of telomerase. While short telomeres ultimately lead to cell senescence, longer telomeres are associated with cancer, including cutaneous melanoma.44
Germline mutations in the TERT promoter are rare but predispose to early-onset melanoma and other tumor types.45 In a study of 675 families known for high penetrance mutations (CDKN2A, CDK4, BAP1, POT1, ACD, and TERF2IP), only one family with a TERT promoter mutation was found.46 Horn et al reported TERT c.-57T(G) cosegregating with melanoma in a family characterized by early-onset melanoma diagnosed between ages 18 and 46 (mean of 34).45 Most of these individuals died within 3 years of diagnosis, suggesting the possibility of a more aggressive clinical course. Two family members were diagnosed with multiple cancers, including one with ovarian cancer and melanoma, and another with renal cell, bladder, breast, and bronchial cancer. In another family with TERT c.-57T(G) promoter mutation, seven cases of melanoma diagnosed between ages 15 and 50 were reported.46 Melanocytic nevi, basal cell carcinomas, and a bladder tumor were also reported.46
While somatic TERT promoter mutations are associated with poor prognostic factors, including increased tumor thickness, ulceration, a high mitotic rate, and lymph node metastasis, and co-occur with BRAF and CDKN2A alterations, reports on histopathologic features of melanomas associated with germline TERT mutations are lacking.47–49
2.5 |. Shelterin complex genes: POT1, ACD, and TERF2IP
The shelterin family of genes, ACD, TERF2IP, TERF1, TERF2, TINF2, and POT1, regulates telomere processing and stability. Germline mutations in ACD, TERF2IP, and POT1 cause hereditary melanoma.50 Like TERT, these genes are implicated in telomere maintenance and their mutations can increase telomere length and fragility.51 They bind to telomeric repeats thereby regulating their function.51
Aoude et al found that of 510 melanoma families, 6 families had ACD mutations and 4 had TERF2IP mutations.50 In a study of 694 patients including high-risk melanoma, 8 variants of POT1 were found exclusively in the high-risk population.52 In a large cohort of patients with first or second-degree relatives with melanoma, four families were identified with POT1 mutations (frequency of 1.7%).53 Families with POT1 mutations typically have between one and eight melanomas per family, which occur in individuals between the ages of 25 and 80.53
Of the 510 melanoma families that Aoude et al studied, most cases of ACD and TERF12P mutations were superficial spreading melanomas and lentigo maligna melanomas,50 although data are sparse. POT1 variants are typically superficial spreading melanomas.52
ACD and TERFI2P mutations also predispose individuals to breast, ovarian, cervical, uterine, thyroid, colon, lung, renal, urinary, prostate, and esophageal cancers as well as lymphomas and leukemias.1 Additionally, brain tumors are common in families with POT1 germline mutations.54 In a study of 55 families with POT1 mutations, all families had members with gliomas.54 Colorectal cancer, chronic lymphocytic leukemia, breast cancer, and small cell lung cancer are also seen.53,55,56
2.6 |. MITF
In 2005, a germline variant MITF p.E318K was shown to predispose to melanoma57,58 and later, to melanoma and renal cell carcinoma.59 This variant is present in approximately 1% of individuals of European descent and is associated with 3- to 5-fold risk of melanoma.60 However, individuals with the variant, who also have a personal or family history of pancreatic or renal cell cancer have a 31-fold or 8-fold increased risk of developing melanoma, respectively.1
As part of the Myc family of genes, MITF encodes a melanocytic-lineage-specific transcription factor that regulates the differentiation, proliferation, and survival of melanocytes.61 MITF p.E318K is a gain-of-function variant that causes impaired sumoylation of the protein, and therefore, aberrant regulation of downstream, target pathways.58 In addition, MITF stimulates hypoxia inducible factor 1A (HIF1A), part of the key pathway in renal cell carcinoma development.
Interestingly, MITF variants are associated with darker hair, fair skin, and non-blue eye color among other phenotypic characteristics60 as well as an increased nevus count, atypical nevi, depigmented nevi, and amelanotic melanomas.62 Histopathologically, nodular melanoma and thicker tumors may be more common in some populations,63 although these findings are not supported by Australian/UK data sets.58,62
2.7 |. MC1R
Variants of MC1R are relatively common, found in up to 11% of individuals,64,65 although with lower penetrance. A recent study demonstrated that 66% of a large cohort of melanoma patients were carriers of MC1R variants and 28% of the melanomas were attributable to the MC1R gene.66 Individuals with two MC1R variants have a higher melanoma risk compared to those with single variants.67 One can expect a 1.5- to 4-fold increased risk of melanoma, and a 3- to 4-fold risk of thick melanomas in MC1R variants.64,68 Overall, these variants appear to considerably contribute to melanoma burden.69
MC1R, or melanocortin-receptor 1, is a G protein coupled receptor that regulates both hair and skin pigmentation. When activated by ultraviolet (UV) radiation, the receptor is bound by alpha melanocyte-stimulating hormone, resulting in melanin upregulation within melanocytes and stimulation of DNA repair mechanisms.70 MC1R variants generally correlate with phenotypes such as red hair, freckling, UV sensitivity, and melanoma risk,70 although MC1R variants with darker phenotypes exist and also confer an increased risk of melanoma.64
In a study of 2160 patients, no significant associations were discovered between MC1R variants and histopathologic variables, including tumor thickness, Clark level, presence of mitotic figures, ulceration, pigmentation, vertical growth phase, regression, tumor infiltrating lymphocytes, or solar elastosis, although further stratification based on sun-sensitive vs sun-resistant phenotypes revealed associations with tumor thickness, presence of mitotic figures, ulceration, and tumor infiltrating lymphocytes.71 There was an association between more than one high-risk variant and predisposition to melanoma on the arms, although overall trunk was the most common anatomic site.71
2.8 |. BAP1
BAP1 is a tumor suppressor gene on 3p21 that encodes a deubiquitinating enzyme and a binding partner to BRCA1, implicated in chromatin modulation, transcriptional regulation, and DNA damage repair.72,73 Characteristic for a tumor suppressor gene, tumors show loss of heterozygosity of BAP1.74
Germline mutations in BAP1 predispose carriers to the BAP1 tumor predisposition syndrome (BAP1-TPDS), characterized by BAP1-inactivated nevi (BINs), uveal melanoma, cutaneous melanoma, mesothelioma, renal cell carcinoma, and other tumors.59,75–79 BINs are highly penetrant, present in up to 90% of mutation carriers, and typically present as multiple, skin colored or reddish-brown, dome-shaped melanocytic tumors (also or previously called BAP1-inactivlated melanocytic tumors, Wiesner nevi, BAPomas, nevoid melanoma-like melanocytic proliferations, BAP1 mutant Spitz nevi, and melanocytic BAP1-mutated atypical intradermal tumors).73,80 BINs develop early in life (median 31 years, range 10-56 years) and increase in number with age.81,82
The most common malignancy associated with this syndrome is uveal melanoma, occurring in up to 29% of patients, followed by mesothelioma (22%), cutaneous melanoma (18%), and renal cell carcinoma (9%).73,81 Additionally, basal cell carcinoma, meningioma, cholangiocarcinoma, breast cancer, lung adenocarcinoma, pancreatic cancer, and thyroid cancer have been reported.81 A quarter of patients with melanoma typically have multiple primary cutaneous melanomas.79
Morphologically, BINs feature a dome-shaped, exclusively or predominantly intradermal melanocytic tumor, with epithelioid melanocytes that have round to oval vesicular nuclei and abundant amphophilic cytoplasm with a nodular or sheet-like growth pattern (Figure 2A–C).77,78,83 An associated common nevus component is commonly present, characteristic of a combined nevus containing two or more melanocytic nevus components.84 A review of 102 BINs revealed that 69% of cases exhibit spitzoid epithelioid cytomorphology while 31% of cases had smaller epithelioid cells without abundant eosinophilic cytoplasm.78 Additionally, rhabdoid features may be present.78 In this series, 12% of BINs were associated with a germline BAP1 mutation.78 No significant differences in clinical or histopathologic features were found between tumors with a confirmed germline mutation vs tumors without, except for the presence of extensive junctional component more commonly seen with BAP1 germline mutation.78 Some lesions may exhibit atypical features, including nuclear pleomorphism, and are thus termed BAP1-inactivated melanocytomas.77 Lastly, transformation of BIN to melanoma has been documented.82
FIGURE 2.
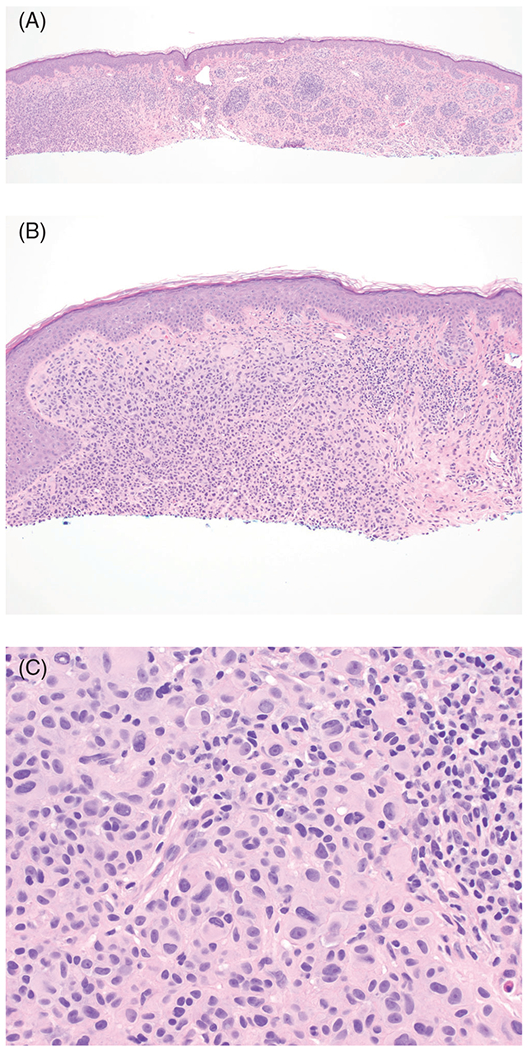
BAP1-inactivated nevus with epithelioid melanocytes. This adolescent patient had multiple BAP1-inctivated nevi. A, Hemotoxylin and eosin (H&E), ×40. B, H&E, ×100. C, H&E, ×400
Awareness of BIN and its histopathologic features will enable identification of patients and families with a high probability of germline BAP1 mutations. When pathologists encounter melanocytic tumors with epithelioid features characteristic of BIN, immunohistochemical testing for BAP1 should be considered as a screening tool for BAP1 inactivation. Because normal BAP1 protein is nuclear, cells with biallelic inactivation will show lack of nuclear staining (Figure 3A–C).81 In such cases, genetic counseling and/or testing for BAP1 germline mutation in the patient and family may be appropriate, depending on the clinical setting, i.e., multiple immunohistochemically confirmed BINs at a young age, and personal and family history of cancer.83,85,86
FIGURE 3.
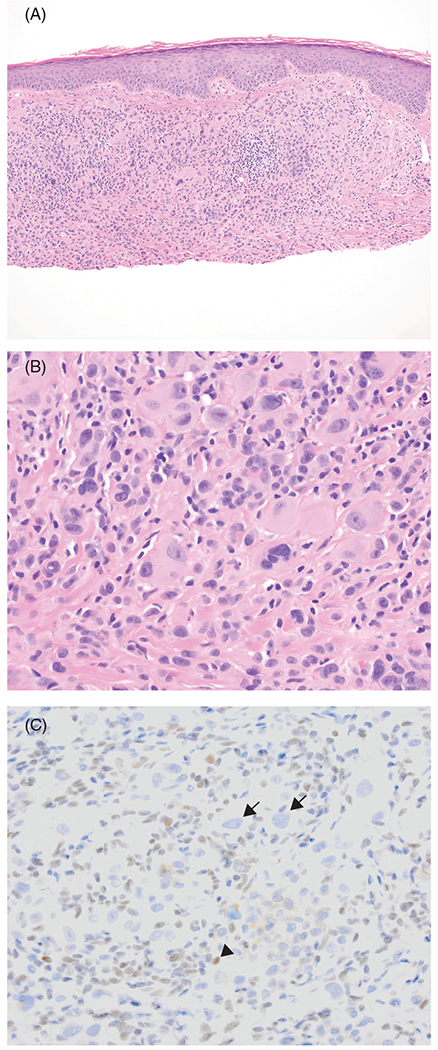
Loss of nuclear BAP1 expression in a BAP1-inactivated melanocytoma. This tumor displays nuclear pleomorphism. A, Hemotoxylin and eosin (H&E), ×100. B, H&E, ×400. C, BAP1 immunohistochemistry, ×400. Large epithelioid tumor cells have lost BAP1 expression (arrow). Lymphocytes show normal nuclear expression of BAP1 (arrowhead)
2.9 |. Mixed cancer syndromes with melanoma
Mixed cancer syndromes (or melanoma-subordinate syndromes) have an increased risk of melanoma, but lower than that of other cancers seen in the syndrome. These syndromes are caused by mutations in PTEN, TP53, BRCA1, BRCA2, and RB1, as well as xeroderma pigmentosum genes (Figure 4A–C) and are discussed in Table 2.
FIGURE 4.
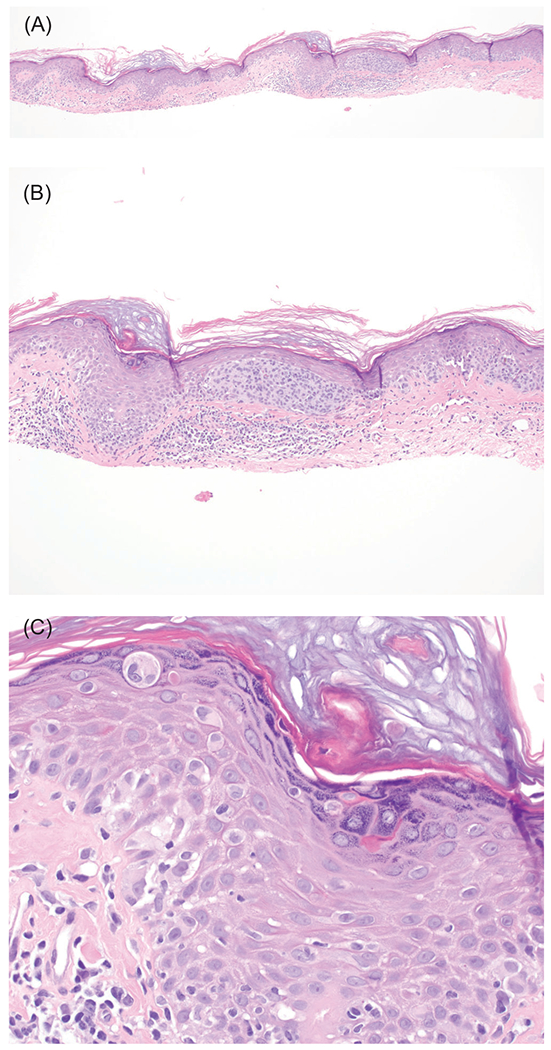
Melanoma in situ in a patient with xeroderma pigmentosum. This 22-year old patient had a history of multiple melanomas (invasive and in situ) and numerous basal cell carcinomas. A, Hemotoxylin and eosin (H&E), ×40. B, H&E, ×100. C, H&E, ×400
TABLE 2.
Melanoma-subordinate syndromes. The genes of melanoma-subordinate syndromes are summarized here, alongside the clinical phenotype or tumor spectrum90–96
| Gene | Syndrome | Phenotype and tumor spectrum |
|---|---|---|
| Phosphatase and tensin homolog (PTEN) | Cowden syndrome | • Mucocutaneous findings: trichilemmomas, acral keratoses, and oral papillomas sometimes with cobblestone appearance • Thyroid abnormalities (multinodular goiter, thyroiditis, and adenomas) • Macrocephaly • Breast cancer (most common malignancy) • Thyroid cancer • Genitourinary cancers (endometrial cancer and renal cell cancer) • GI tumors, including polyps and colorectal cancer • Neurologic tumors • Vascular tumors • Melanoma |
| Tumor protein p53 (TP53) | Li-Fraumeni syndrome | • Multiple cancers, lifetime risk of cancer 70% to 100% • Sarcomas (all types of soft tissue and bone sarcomas, including but not limited to osteosarcoma and rhabdomyosarcoma), except for Ewing sarcoma • Breast cancer • Brain tumors, including high-grade gliomas and medulloblastoma • Adrenocortical carcinoma • Melanoma |
| BRCA1 DNA repair associated (BRCA1), BRCA2 DNA repair associated (BRCA2) | Hereditary breast and ovarian cancer | • Breast and ovarian cancer • Other gynecologic cancers (Fallopian tubal, peritoneal, endometrial, and uterine papillary serous) • Pancreatic cancer • Prostate cancer • Colorectal cancer • Stomach and biliary cancer • Melanoma, including uveal melanoma (mainly BRCA2 mutation carriers) |
| RB transcriptional corepressor 1 (RB1) | Hereditary retinoblastoma | • Retinoblastoma, typically under the age of three •Pineoblastoma • Osteosarcoma • Soft tissue sarcomas • Melanoma |
| mutL homolog 1 (MLH1) mutS homolog 2 (MSH2) Epithelial cell adhesion molecule (EPCAM) mutS homolog 6 (MSH6) PMS1 homolog 2, mismatch repair system component (PMS2) |
Hereditary non-polyposis colorectal cancer (HNPCC; Lynch syndrome) | • Colorectal cancer • Endometrial cancer • Cancer of small intestine • Other genitourinary cancers (ureteral, uterine, and renal pelvis) • Melanoma |
| Damage specific DNA binding protein 2 (DDB2) ERCC excision repair 1, endonuclease non-catalytic subunit (ERCC1) ERCC excision repair 2, TFIIH core complex helicase subunit (ERCC2) |
Xeroderma pigmentosum | • Extreme sensitivity to UV light • Freckling (solar lentigines) in early childhood, poikiloderma • Skin cancers in first decade of life • Basal cell carcinoma • Squamous cell carcinoma • Melanoma • Brain tumors |
| ERCC excision repair 3, TFIIH core complex helicase subunit (ERCC3) ERCC excision repair 4, endonuclease catalytic subunit (ERCC4) ERCC excision repair 5, endonuclease (ERCC5) DNA polymerase eta (POLH) XPA, DNA damage recognition, and repair factor (XPA) |
• Lung cancer • Neurological abnormalities |
3 |. SCREENING, GENETIC TESTING, AND GENETIC COUNSELING
Screening for hereditary melanoma begins with obtaining a detailed personal and family history of cancer. As a general guide, multiple tumors of early onset are seen in hereditary cancer syndromes and the “rule of threes” can be applied: patients with a personal or family history of three or more primary melanomas and/or pancreatic cancer should be referred for genetic counseling and testing. In geographic areas with lower prevalence of melanoma, the threshold for testing is two or more primary melanomas or melanoma in situ.87 A genetic counselor or other genetics specialist can best guide the patient through this process, including education and obtaining informed consent; appropriate test selection; and post-test counseling that includes recommendations for management of extra-cutaneous cancer risks.88
Historically, individual testing of susceptibility genes was performed, but in the recent decade, next-generation sequencing technologies have enabled affordable and timely testing of multiple genes (panel testing).87 Panel testing is tailored based on the personal and family history, as thoroughly reviewed by Leachman et al.87
Generally, in mutation carriers, full-body skin exams every 6 to 12 months should be performed, and digital dermoscopy, total body photography, and further screening for visceral cancers considered as appropriate.89
4 |. SUMMARY
In conclusion, a subset of familial melanoma is caused by germline mutations in high-risk melanoma susceptibility genes, many of which are also somatically mutated in melanoma. In general, early onset, multiple tumors, and family history are clues to an underlying tumor syndrome. Pathologists can enable identification of patients at risk by recognizing BAP1-inactivated nevi. Identification of patients with a germline mutation predisposing to cancer enables genetic counseling, genetic testing of family members, and appropriate surveillance, reducing morbidity and mortality in these patients.
ACKNOWLEDGMENT
Dr Kiuru’s involvement in this article is in part supported by the Dermatology Foundation, through Career Development Award in Dermatopathology, and the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under award number K23AR074530.
Funding information
Dermatology Foundation; Foundation for the National Institutes of Health, Grant/Award Number: K23AR074530
Footnotes
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
REFERENCES
- 1.Soura E, Eliades PJ, Shannon K, Stratigos AJ, Tsao H. Hereditary melanoma: update on syndromes and management: genetics of familial atypical multiple mole melanoma syndrome. J Am Acad Dermatol. 2016;74(3):395–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reddy BY, Miller DM, Tsao H. Somatic driver mutations in melanoma. Cancer. 2017;123(S11):2104–2117. [DOI] [PubMed] [Google Scholar]
- 3.Helgadottir H, Höiom V, Tuominen R, et al. Germline CDKN2A mutation status and survival in familial melanoma cases. J Natl Cancer Inst. 2016;108(11):545–452. [DOI] [PubMed] [Google Scholar]
- 4.Lynch HT, Krush AJ. Heredity and malignant melanoma: implications for early cancer detection. Can Med Assoc J. 1968;99(1):17–21. [PMC free article] [PubMed] [Google Scholar]
- 5.Clark WH Jr, Reimer RR, Greene M, Ainsworth AM, Mastrangelo MJ. Origin of familial malignant melanomas from heritable melanocytic lesions. The B-K mole syndrome. Arch Dermatol. 1978;114(5): 732–738. [PubMed] [Google Scholar]
- 6.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8(1):15–21. [DOI] [PubMed] [Google Scholar]
- 7.Gruis NA, van der Velden PA, Sandkuijl LA, et al. Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat Genet. 1995;10(3):351–353. [DOI] [PubMed] [Google Scholar]
- 8.Shain AH, Yeh I, Kovalyshyn I, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373(20):1926–1936. [DOI] [PubMed] [Google Scholar]
- 9.Cachia AR, Indsto JO, McLaren KM, Mann GJ, Arends MJ. CDKN2A mutation and deletion status in thin and thick primary melanoma. Clin Cancer Res. 2000;6(9):3511–3515. [PubMed] [Google Scholar]
- 10.Koh SS, Cassarino DS. Immunohistochemical expression of p16 in melanocytic lesions: an updated review and meta-analysis. Arch Pathol Lab Med. 2018;142(7):815–828. [DOI] [PubMed] [Google Scholar]
- 11.Bishop JA, Wachsmuth RC, Harland M, et al. Genotype/phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J Invest Dermatol. 2000;114(1):28–33. [DOI] [PubMed] [Google Scholar]
- 12.Tucker MA, Fraser MC, Goldstein AM, et al. A natural history of melanomas and dysplastic nevi: an atlas of lesions in melanoma-prone families. Cancer. 2002;94(12):3192–3209. [DOI] [PubMed] [Google Scholar]
- 13.van der Rhee JI, Krijnen P, Gruis NA, et al. Clinical and histologic characteristics of malignant melanoma in families with a germline mutation in CDKN2A. J Am Acad Dermatol. 2011;65(2):281–288. [DOI] [PubMed] [Google Scholar]
- 14.Goldstein AM, Chan M, Harland M, et al. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006;66(20):9818–9828. [DOI] [PubMed] [Google Scholar]
- 15.Masback A, Olsson H, Westerdahl J, et al. Clinical and histopathological features of malignant melanoma in germline CDKN2A mutation families. Melanoma Res. 2002;12(6):549–557. [DOI] [PubMed] [Google Scholar]
- 16.Lynch HT, Brand RE, Hogg D, et al. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer. 2002;94(1): 84–96. [DOI] [PubMed] [Google Scholar]
- 17.Gironi LC, Colombo E, Farinelli P, et al. Germline CDKN2A mutations in childhood melanoma: a case of melanoma-pancreatic cancer syndrome. Int J Dermatol. 2015;54(12):e553–e555. [DOI] [PubMed] [Google Scholar]
- 18.Berg P, Wennberg AM, Tuominen R, et al. Germline CDKN2A mutations are rare in child and adolescent cutaneous melanoma. Melanoma Res. 2004;14(4):251–255. [DOI] [PubMed] [Google Scholar]
- 19.Dalmasso B, Pastorino L, Ciccarese G, et al. CDKN2A germline mutations are not associated with poor survival in an Italian cohort of melanoma patients. J Am Acad Dermatol. 2019;80(5):1263–1271. [DOI] [PubMed] [Google Scholar]
- 20.Aguilera P, Malvehy J, Carrera C, et al. Clinical and histopathological characteristics between familial and sporadic melanoma in Barcelona, Spain. J Clin Exp Dermatol Res. 2014;5(5):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gironi LC, Colombo E, Pasini B, et al. Melanoma-prone families: new evidence of distinctive clinical and histological features of melanomas in CDKN2A mutation carriers. Arch Dermatol Res. 2018;310(10): 769–784. [DOI] [PubMed] [Google Scholar]
- 22.Sargen MR, Kanetsky PA, Newton-Bishop J, et al. Histologic features of melanoma associated with CDKN2A genotype. J Am Acad Dermatol. 2015;72(3):496–507.e497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robert C, Rouiller J, Kannengiesser C, et al. Study of BRAF mutations in melanoma and nevi from patients with germline mutation in the CDKN2A gene. J Clin Oncol. 2006;24(18_suppl): 10075–10075. [Google Scholar]
- 24.Eskandarpour M, Hashemi J, Kanter L, Ringborg U, Platz A, Hansson J. Frequency of UV-inducible NRAS mutations in melanomas of patients with germline CDKN2A mutations. J Natl Cancer Inst. 2003;95(11):790–798. [DOI] [PubMed] [Google Scholar]
- 25.Doyle A, Kubler MM, Harris AC, et al. The impact of CDKN2A mutations on overall survival in pancreatic adenocarcinoma. J Clin Oncol. 2019;37(4_suppl):278–278.30550363 [Google Scholar]
- 26.McWilliams RR, Wieben ED, Rabe KG, et al. Prevalence of CDKN2A mutations in pancreatic cancer patients: implications for genetic counseling. Eur J Hum Genet. 2011;19(4):472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helgadottir H, Hoiom V, Jonsson G, et al. High risk of tobacco-related cancers in CDKN2A mutation-positive melanoma families. J Med Genet. 2014;51(8):545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Russell JL, Powers JT, Rounbehler RJ, Rogers PM, Conti CJ, Johnson DG. ARF differentially modulates apoptosis induced by E2F1 and Myc. Mol Cell Biol. 2002;22(5):1360–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soufir N, Lacapere JJ, Bertrand G, et al. Germline mutations of the INK4a-ARF gene in patients with suspected genetic predisposition to melanoma. Br J Cancer. 2004;90(2):503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bahuau M, Vidaud D, Jenkins RB, et al. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998;58(11):2298–2303. [PubMed] [Google Scholar]
- 31.Randerson-Moor JA, Harland M, Williams S, et al. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum Mol Genet. 2001;10(1):55–62. [DOI] [PubMed] [Google Scholar]
- 32.Kaufman DK, Kimmel DW, Parisi JE, Michels VV. A familial syndrome with cutaneous malignant melanoma and cerebral astrocytoma. Neurology. 1993;43(9):1728–1731. [DOI] [PubMed] [Google Scholar]
- 33.Azizi E, Friedman J, Pavlotsky F, et al. Familial cutaneous malignant melanoma and tumors of the nervous system. Cancer. 1995;76(9): 1571–1578. [DOI] [PubMed] [Google Scholar]
- 34.Sargen MR, Merrill SL, Chu EY, Nathanson KL. CDKN2A mutations with p14 loss predisposing to multiple nerve sheath tumours, melanoma, dysplastic naevi and internal malignancies: a case series and review of the literature. Br J Dermatol. 2016;175(4):785–789. [DOI] [PubMed] [Google Scholar]
- 35.Petronzelli F, Sollima D, Coppola G, Martini-Neri ME, Neri G, Genuardi M. CDKN2A germline splicing mutation affecting both p16ink4 and p14arf RNA processing in a melanoma/neurofibroma kindred. Genes Chromosome Cancer. 2001;31(4):398–401. [DOI] [PubMed] [Google Scholar]
- 36.Chan AK, Han SJ, Choy W, et al. Familial melanoma-astrocytoma syndrome: synchronous diffuse astrocytoma and pleomorphic xanthoastrocytoma in a patient with germline CDKN2A/B deletion and a significant family history. Clin Neuropathol. 2017;36(5): 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lesueur F, de Lichy M, Barrois M, et al. The contribution of large genomic deletions at the CDKN2A locus to the burden of familial melanoma. Br J Cancer. 2008;99(2):364–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laud K, Marian C, Avril MF, et al. Comprehensive analysis of CDKN2A (p16INK4A/p14ARF) and CDKN2B genes in 53 melanoma index cases considered to be at heightened risk of melanoma. J Med Genet. 2006;43(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bottillo I, La Starza R, Radio FC, et al. A novel germline mutation in CDK4 codon 24 associated to familial melanoma. Clin Genet. 2018; 93(4):934–935. [DOI] [PubMed] [Google Scholar]
- 40.Zuo L, Weger J, Yang Q, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12(1): 97–99. [DOI] [PubMed] [Google Scholar]
- 41.Puntervoll HE, Yang XR, Vetti HH, et al. Melanoma prone families with CDK4 germline mutation: phenotypic profile and associations with MC1R variants. J Med Genet. 2013;50(4):264–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339(6122):957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Populo H, Boaventura P, Vinagre J, et al. TERT promoter mutations in skin cancer: the effects of sun exposure and X-irradiation. J Invest Dermatol. 2014;134(8):2251–2257. [DOI] [PubMed] [Google Scholar]
- 44.Burke LS, Hyland PL, Pfeiffer RM, et al. Telomere length and the risk of cutaneous malignant melanoma in melanoma-prone families with and without CDKN2A mutations. PLoS One. 2013;8(8):e71121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horn S, Figl A, Rachakonda PS, et al. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339(6122):959–961. [DOI] [PubMed] [Google Scholar]
- 46.Harland M, Petljak M, Robles-Espinoza CD, et al. Germline TERT promoter mutations are rare in familial melanoma. Fam Cancer. 2016; 15(1):139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seynnaeve B, Lee S, Borah S, et al. Genetic and epigenetic alterations of TERT are associated with inferior outcome in adolescent and young adult patients with melanoma. Sci Rep. 2017;7(1):45704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hugdahl E, Kalvenes MB, Mannelqvist M, Ladstein RG, Akslen LA. Prognostic impact and concordance of TERT promoter mutation and protein expression in matched primary and metastatic cutaneous melanoma. Br J Cancer. 2018;118(1):98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heidenreich B, Nagore E, Rachakonda PS, et al. Telomerase reverse transcriptase promoter mutations in primary cutaneous melanoma. Nat Commun. 2014;5:3401–3401. [DOI] [PubMed] [Google Scholar]
- 50.Aoude LG, Pritchard AL, Robles-Espinoza CD, et al. Nonsense mutations in the shelterin complex genes ACD and TERF2IP in familial melanoma. J Natl Cancer Inst. 2015;107(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shi J, Yang XR, Ballew B, et al. Rare missense variants in POT1 predispose to familial cutaneous malignant melanoma. Nat Genet. 2014;46 (5):482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Müller C, Krunic M, Wendt J, von Haeseler A, Okamoto I. Germline variants in the POT1-gene in high-risk melanoma patients in Austria. G3 (Bethesda). 2018;8(5):1475–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robles-Espinoza CD, Harland M, Ramsay AJ, et al. POT1 loss-of-function variants predispose to familial melanoma. Nat Genet. 2014; 46(5):478–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bainbridge MN, Armstrong GN, Gramatges MM, et al. Germline mutations in shelterin complex genes are associated with familial glioma. J Natl Cancer Inst. 2015;107(1):384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chubb D, Broderick P, Dobbins SE, et al. Rare disruptive mutations and their contribution to the heritable risk of colorectal cancer. Nat Commun. 2016;7(1):11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Speedy HE, Kinnersley B, Chubb D, et al. Germ line mutations in shelterin complex genes are associated with familial chronic lymphocytic leukemia. Blood. 2016;128(19):2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garraway LA, Widlund HR, Rubin MA, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–122. [DOI] [PubMed] [Google Scholar]
- 58.Yokoyama S, Woods SL, Boyle GM, et al. A novel recurrent mutation in MITF predisposes to familial and sporadic melanoma. Nature. 2011; 480(7375):99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bertolotto C, Lesueur F, Giuliano S, et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature. 2011;480(7375):94–98. [DOI] [PubMed] [Google Scholar]
- 60.Berwick M, MacArthur J, Orlow I, et al. MITF E318K’s effect on melanoma risk independent of, but modified by, other risk factors. Pigment Cell Melanoma Res. 2014;27(3):485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72(7):1249–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sturm RA, Fox C, McClenahan P, et al. Phenotypic characterization of nevus and tumor patterns in MITF E318K mutation carrier melanoma patients. J Invest Dermatol. 2014;134(1):141–149. [DOI] [PubMed] [Google Scholar]
- 63.Ghiorzo P, Pastorino L, Queirolo P, et al. Prevalence of the E318K MITF germline mutation in Italian melanoma patients: associations with histological subtypes and family cancer history. Pigment Cell Melanoma Res. 2013;26(2):259–262. [DOI] [PubMed] [Google Scholar]
- 64.Kanetsky PA, Hay JL. Marshaling the translational potential of MC1R for precision risk assessment of melanoma. Cancer Prev Res. 2018;11 (3):121–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kennedy C, ter Huurne J, Berkhout M, et al. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J Invest Dermatol. 2001;117(2):294–300. [DOI] [PubMed] [Google Scholar]
- 66.Pasquali E, García-Borrón JC, Fargnoli MC, et al. MC1R variants increased the risk of sporadic cutaneous melanoma in darker-pigmented Caucasians: a pooled-analysis from the M-SKIP project. Int J Cancer. 2015;136(3):618–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Demenais F, Mohamdi H, Chaudru V, et al. Association of MC1R variants and host phenotypes with melanoma risk in CDKN2A mutation carriers: a GenoMEL study. J Natl Cancer Inst. 2010;102(20):1568–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Landi MT, Kanetsky PA, Tsang S, et al. MC1R, ASIP, and DNA repair in sporadic and familial melanoma in a Mediterranean population. J Natl Cancer Inst. 2005;97(13):998–1007. [DOI] [PubMed] [Google Scholar]
- 69.Williams PF, Olsen CM, Hayward NK, Whiteman DC. Melanocortin 1 receptor and risk of cutaneous melanoma: a meta-analysis and estimates of population burden. Int J Cancer. 2011;129(7):1730–1740. [DOI] [PubMed] [Google Scholar]
- 70.Chen S, Han C, Miao X, et al. Targeting MC1R depalmitoylation to prevent melanomagenesis in redheads. Nat Commun. 2019;10(1):877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taylor NJ, Busam KJ, From L, et al. Inherited variation at MC1R and histological characteristics of primary melanoma. PLoS One. 2015; 10(3):e0119920–e0119920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maruţić Z, Buljan M, Busam KJ. Histomorphologic spectrum of BAP1 negative melanocytic neoplasms in a family with BAP1-associated cancer susceptibility syndrome. J Cutan Pathol. 2015;42(6):406–412. [DOI] [PubMed] [Google Scholar]
- 73.Masoomian B, Shields JA, Shields CL. Overview of BAP1 cancer pre-disposition syndrome and the relationship to uveal melanoma. J Curr Ophthalmol. 2018;30(2):102–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gammon B, Traczyk TN, Gerami P. Clumped perinuclear BAP1 expression is a frequent finding in sporadic epithelioid Spitz tumors. J Cutan Pathol. 2013;40(6):538–542. [DOI] [PubMed] [Google Scholar]
- 75.Wiesner T, Obenauf AC, Murali R, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43(10): 1018–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chau C, van Doorn R, van Poppelen NM, et al. Families with BAP1-tumor predisposition syndrome in The Netherlands: path to identification and a proposal for genetic screening guidelines. Cancer. 2019;11(8):1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wiesner TMMJ, Scolyer RA. Combined naevus, including combined BAP1-inactivated naevus/melanocytoma. In: MDSR EDE, Willemze R, eds. WHO Classification of Skin Tumours. Lyon: WHO; 2018:99–101. [Google Scholar]
- 78.Garfield EM, Walton KE, Quan VL, et al. Histomorphologic spectrum of germline-related and sporadic BAP1-inactivated melanocytic tumors. J Am Acad Dermatol. 2018;79(3):525–534. [DOI] [PubMed] [Google Scholar]
- 79.Rai K, Pilarski R, Cebulla CM, Abdel-Rahman MH. Comprehensive review of BAP1 tumor predisposition syndrome with report of two new cases. Clin Genet. 2016;89(3):285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang AJ, Rush PS, Tsao H, Duncan LM. BRCA1-associated protein (BAP1)-inactivated melanocytic tumors. J Cutan Pathol. 2019;46(12): 965–972. [DOI] [PubMed] [Google Scholar]
- 81.Haugh AM, Njauw C-N, Bubley JA, et al. Genotypic and phenotypic features of BAP1 cancer syndrome: a report of 8 new families and review of cases in the literature. JAMA Dermatol. 2017;153(10): 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13(3):153–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology. 2013;45(2):116–126. [DOI] [PubMed] [Google Scholar]
- 84.Griewank KG, Müller H, Jackett LA, et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod Pathol. 2017;30(7):928–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gardner LJ, Strunck JL, Wu YP, Grossman D. Current controversies in early-stage melanoma: questions on incidence, screening, and histologic regression. J Am Acad Dermatol. 2019;80(1):1–12. [DOI] [PubMed] [Google Scholar]
- 86.Klapperich ME, Bowen GM, Grossman D. Current controversies in early-stage melanoma: questions on management and surveillance. J Am Acad Dermatol. 2019;80(1):15–25. [DOI] [PubMed] [Google Scholar]
- 87.Leachman SA, Lucero OM, Sampson JE, et al. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017;36(1):77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic. National Comprehensive Cancer Network 2019; https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf. Accessed February 2, 2020. [Google Scholar]
- 89.Badenas C, Aguilera P, Puig-Butillé JA, Carrera C, Malvehy J, Puig S. Genetic counseling in melanoma. Dermatol Ther. 2012;25(5):397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis. 2011;6(6):70–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kocabalkan O, Ozgur F, Erk Y, Gursu KG, Gungen Y. Malignant melanoma in xeroderma pigmentosum patients: report of five cases. Eur J Surg Oncol. 1997;23(1):43–47. [DOI] [PubMed] [Google Scholar]
- 92.Curiel-Lewandrowski C, Speetzen LS, Cranmer L, Warneke JA, Loescher LJ. Multiple primary cutaneous melanomas in Li-Fraumeni syndrome. Arch Dermatol. 2011;147(2):248–250. [DOI] [PubMed] [Google Scholar]
- 93.Hajkova N, Hojny J, Nemejcova K, et al. Germline mutation in the TP53 gene in uveal melanoma. Sci Rep. 2018;8(1):7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2012;12(1):68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dębniak T, Scott RJ, Górski B, et al. BRCA1/2 mutations are not a common cause of malignant melanoma in the polish population. PLoS One. 2018;13(10):e0204768–e0204768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Roesch A, Becker B, Meyer S, et al. Overexpression and hyper-phosphorylation of retinoblastoma protein in the progression of malignant melanoma. Mod Pathol. 2005;18(4):565–572. [DOI] [PubMed] [Google Scholar]