

Abstract
Crohn’s disease (CD) is characterized by a chronic, progressive inflammation across the gastrointestinal tract with a series of exacerbations and remissions. A significant factor in the CD pathogenesis is an imbalance in gut microbiota composition, particularly the prevalence of Escherichia coli. In the present study, the genomes of sixty-three E. coli strains from the gut of patients with CD and healthy subjects were sequenced. In addition, eighteen E. coli-like metagenome-assembled genomes (MAGs) were reconstructed from the shotgun-metagenome sequencing data of fecal samples. The comparative analysis revealed the similarity of E. coli genomes regardless of the origin of the strain. The strains exhibited similar genetic patterns of virulence, antibiotic resistance, and bacteriocin-producing systems. The study showed antagonistic activity of E. coli strains and the metabolic features needed for their successful competition in the human gut environment. These observations suggest complex bacterial interactions within the gut which may affect the host and cause intestinal damage.
Keywords: Crohn’s disease, Escherichia coli, whole-genome sequencing, shotgun metagenome sequencing
1. Introduction
Escherichia coli is a very important facultative aerobic inhabitant of the human intestine and part of the normal flora. E. coli is among the first bacterial species to colonize the intestine during infancy, forming the human gut microbiota [1]. Imbalances in host–commensal interaction may lead to intestinal and extraintestinal infections [2]. Many works have been devoted to metagenomic investigations, discovering the changes in microbiota which promote intestinal inflammation [3,4,5,6,7,8]. Thus, a decrease in biodiversity, accompanied by an increased abundance of E. coli, was found to be associated with CD [9,10,11]. However, a single E. coli strain specific to CD has not been discovered to date.
Closely related microorganisms, especially at the strain level, occupy similar ecological niches, leading to an intense competition which can promote rapid phenotypic evolution [12]. Serotyping [13,14,15,16,17], multilocus sequence typing (MLST) [18,19], and phylotyping by Clermont’s method [20] are widely used to study the strain-level variability in E. coli. A hypothesis of the potential for O-antigens of intestinal E. coli to induce immune stimulation and play a role in the pathogenicity of IBD was formed in the 1970s. However, associations of specific O-serotypes with the disease were not found [21]. At present, conventional serotyping is being replaced by more rapid and accurate molecular typing [22,23]. High-throughput sequencing significantly expanded opportunities in genetic diversity studies into E. coli. Using the WGS technique, a number of E. coli genomes from CD patients have been characterized; however, most studies have focused on pathogenic clinical isolates [24,25,26,27,28,29]. Attempts to explain the role of E. coli in CD pathogenesis have drawn attention to adherent-invasive strains (AIEC). Research into CD has revealed the ability of E. coli to adhere to intestinal epithelial cells and survive within macrophages, which allows bacteria to move across the human intestinal barrier and potentially induce the formation of granulomas [30,31,32,33]. However, the precise mechanism of CD pathogenesis remains unclear. Studies that focus on mucosa-associated E. coli often overlook fecal commensal strains, which play an important role in the microbiota. It has been shown that the probiotic strain Nissle 1917 affects intestinal permeability and improves barrier integrity in IBD patients [34,35]. The strain outcompetes enteropathogens and reduces colitis [36,37]. However, in healthy people, a well-established microbiota prevents the integration of the probiotic strain into the existing community [38]. The replacement of pro-inflammatory E. coli with competitive strains of healthy subjects might be beneficial in reducing inflammatory activity. As another step towards a better understanding of the role of E. coli in a healthy and inflamed intestinal community, a cohort study was performed using different physiological and molecular approaches. To explore the relationships between strains within a healthy and inflamed intestine community, we isolated and thoroughly characterized fecal E. coli, and assessed their competitive potential and metabolic features.
2. Materials and Methods
2.1. E. coli Collection and Phylotyping
A total of 82 human fecal samples (40 CD patients and 42 healthy volunteers) were collected at the Kazan Federal University Hospital (Kazan, Russian) and stored at −80 °C until needed. Patients with CD were diagnosed by clinical and endoscopic examination with further histological confirmation. A complete list of inclusion/exclusion criteria for CD patients and healthy individuals has been described previously [39]. Individuals taking antibiotics six months before sample collection were excluded from the study. For shotgun metagenomic analysis, all samples were sequenced using the Illumina NextSeq 500 platform and fully characterized in our previous work [39]. The fecal samples were prepared according to the described previously protocol [40]. Briefly, the stool samples (0.1 g) were resuspended in PBS and inoculated on a selective medium (Endo agar). Up to 10 colonies from each plate were selected for species identification using matrix-assisted laser desorption/ionization (MALDI) Biotyper System (Bruker, Bremen, Germany). For DNA isolation with PureLink Genomic DNA Mini kit (Invitrogen, Thermo Scientific, Carlsbad, CA, USA), each E. coli colony was grown in the Luria-Bertani broth (LB) at 37 °C with shaking overnight. Further typing was performed through quadruplex PCR according to Clermont’s method [20], followed by in silico validation.
2.2. Whole-Genome Sequencing and Analysis
In total, of 521 cultivated colonies from 32 individuals (14 CD patients and 18 healthy controls), 97 distinct E. coli isolates were identified, then sequenced by the Illumina MiSeq platform provided by Interdisciplinary Centre for Shared Use of Kazan Federal University, and analyzed as described previously [40].
In addition, metagenome-assembled genomes (MAGs) from all 82 human fecal samples were reconstructed with metaSPAdes v. 3.14.1 [41]. Contigs longer than 1000 nucleotides were binned with Maxbin v. 2.2.7 [42]. Only 18 high-quality MAGs with a completeness of more than 80% and a contamination rate of less than 5%, as estimated by CheckM v. 1.0.11 [43], were considered for further analysis.
A phylogenetic tree was constructed using PhyloPhlAn 3.0 [44] and visualized with iTOL (https://itol.embl.de, accessed on 8 February 2021) [45]. Multidimensional scaling (MDS) plots were built on the whole-genome average nucleotide identity (ANI) distances computed with FastANI v 1.32 [46].
Serotypes were assigned using the SerotypeFinder-2.0 tool [22]. All known Clermont’s phylotypes were in silico assigned using the EzClermont tool [47] Multi-Locus Sequence Typing (MLST) was performed with MLST 2.0 [48] based on two main methods: Achtman’s scheme with internal fragments of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, recA) [49] and a Pasteur scheme consisting of eight genes (dinB, icdA, pabB, polB, putP, trpA, trpB, uidA) [50].
Nucleotide sequences were aligned to the custom virulence genes database using BLASTn (with a cut-off of 80% identity over an 80% virulence gene length, E < 10−16). Differences between the frequency of occurrence of virulence genes were calculated using Fisher’s exact test with Benjamini–Hochberg correction for multiple comparisons.
2.3. In Vitro Competition Assay
The halo assay was performed according to Ohno [51] with some modifications. The tester E. coli K-12 substr. MG1655 was inoculated into the 0.8% agar and poured onto a bottom 2% LB agar plates. A series of 10-fold dilutions of each competitor strain (starting with OD600 = 1.0) were spotted (3 μL) on the tester strain and incubated at 37 °C overnight. Each experiment was repeated twice. Zones of at least 1 mm width were considered inhibition zones.
2.4. Antimicrobial Resistance
All isolates were tested for antimicrobial susceptibility with fluoroquinolones (ciprofloxacin, levofloxacin, moxifloxacin, nalidixic acid), aminoglycosides (amikacin), carbapenems (meropenem), monobactams (aztreonam), penicillins (ampicillin), polymyxins (polymyxin B), sulfanilamides (sulfanilamide), tetracyclines (tetracycline), and cephalosporins (cefazolin, cefepime, cefotaxime, cefuroxime) using a disc-diffusion test (DDT) according to EUCAST guidelines (https://www.eucast.org/ast_of_bacteria/disk_diffusion_methodology/, accessed on 3 June 2019). The E. coli strain ATCC 25922 was used as a control. Antibiotic resistance genes in isolated strains and MAGs were predicted using the Resistance Gene Identifier (RGI) and the CARD database [52]. Differences between the frequency of occurrence of resistance genes were calculated using Fisher’s exact test with Benjamini–Hochberg correction for multiple comparisons.
2.5. Biochemical Assay
Biochemical characterization of E. coli isolates was performed using ENTEROtest 24N (Erba Lachema, Brno, Czech Republic) according to the manufacturer’s recommendations.
3. Results
3.1. Phylogenetic Analysis of E. coli Strains Isolated from CD Patients and Healthy Subjects
In the current study, we screened 82 fecal samples of 40 CD patients and 42 healthy individuals collected at the Kazan Federal University Hospital (Kazan, Russia), for which shotgun sequencing has been performed (Table 1). According to the published metagenomic data [39], a higher proportion of E. coli was found in stool samples of CD patients compared to controls. From these stool samples, E. coli strains were carefully selected and identified. Of 32 samples (14 from CD patients and 18 from healthy individuals), 97 isolates were sequenced, followed by comparative genome analysis, which revealed 33 duplicates, i.e., isolates sequenced more than once due to varying colony size and morphology. Thus, 64 strains had unique genomes, including 26 isolates from CD patients, 37 isolates from the control group, and one contaminated genome, which was filtered out. Patients with cultivated E. coli were separated into three subgroups according to disease location: ileal (n = 3), ileocolonic (n = 4), and colonic CD (n = 7).
Table 1.
Patient metadata.
| Groups | CD Patients | Healthy Controls |
|---|---|---|
| Total number of subjects | 40 | 42 |
| Age (years) | 19–61 | 20–65 |
| Gender | ||
| Female | 22 | 23 |
| Male | 18 | 19 |
| Location | ||
| Ileal | 4 | NA |
| Colonic | 22 | NA |
| Ileocolonic | 14 | NA |
| Behaviour | ||
| Non-stricturing, non-penetrating | 21 | NA |
| Stricturing | 8 | NA |
| Penetrating | 3 | NA |
| Stricturing, penetrating | 4 | NA |
| Non-penetrating, stricturing | 4 | NA |
NA: not applicable.
To comprehensively characterize fecal E. coli diversity, 18 high-quality E. coli-like MAGs (11 from CD patients and 7 from healthy controls) were reconstructed from the shotgun metagenome sequencing data of fecal samples. Four out of 18 MAGs were highly similar to the cultivated isolates from the same fecal samples, six were reconstructed from samples that failed to cultivate E. coli, and the rest were not isolated by cultivation but found in the metagenome data.
The phylogenetic analysis was performed for the full set of the cultivated 63 E. coli genomes and 18 reconstructed MAGs using reference pathogenic (enterohemorrhagic EDL933 and Sakai, APEC O1, AIEC LF82) and non-pathogenic (Nissle 1917 and K-12 substr. MG1655) strains (Figure 1, Supplementary Materials Table S1). The phylogenetic tree showed a high similarity in analyzed genomes, and disease-associated clustering of E. coli was not observed, but strains were divided according to known phylogenetic groups [19]. The phylogroups A (49%) and D (20%) represented the majority of the strains, and the rest (B1, B2, C, E, and F) were less common. Based on phylogenetic typing, it was demonstrated that strains from patients with CD were indistinguishable from those isolated from healthy volunteers.
Figure 1.


Comparative genome analysis of fecal E. coli strains isolated from patients with Crohn’s disease (CD) and healthy individuals. (a) Phylogenetic tree of 63 E. coli genomes and 18 MAGs (marked in bold) from healthy individuals (blue) and CD patients (red). Complete genomes of E. coli strains APEC O1, LF82, Sakai, EDL933, Nissle 1917, and K-12 substr. MG1655 were used as reference sequences (black). Phylogenetic groups by Clermont’s method [20] are highlighted on the inner ring. Sequence types (ST) by the Achtman’s scheme [49] and the Pasteur’s scheme [50] are marked as ST#1 and ST#2, respectively. Repeating STs are colored; (b) Multidimensional scaling (MDS) on average nucleotide identity (ANI) distance of all genomes is colored according to Clermont’s phylogroups.
In addition to phylogenetic typing, we performed MLST analysis. According to Achtman’s MLST scheme [49], 63 cultivated isolates were distributed into 36 sequence types (ST#1 in Figure 1). The most common STs were ST10 (n = 12) and ST69 (n = 11), belonging to groups A and D, respectively. Four STs were identified more than once (ST58, ST73, ST398, ST4774). Reconstructed genomes expanded this list, with three STs found (ST127, ST448, ST540). The Pasteur scheme [50] divided E. coli strains into 30 sequence types (ST#2 in Figure 1). Nine STs occurred twice or more and comprised 59% of all strains (ST2, ST3, ST34, ST5, ST87, ST390, ST520, ST661, ST835). The most frequent types among strains were ST2 (n = 9) and ST3 (n = 11) assigned to A and D groups, respectively. Two STs were identified only in MAGs (ST24, ST32). Together, in accordance with both MLST schemes, two main clusters of closely related genotypes (ST69/ST3 and ST10/ST2) from different hosts were formed.
3.2. Genetic Features of E. coli Strains of CD Patients and Healthy Individuals
It is known that intestinal commensals interact closely with the human immune system [53], and their resistance to host defense responses is partially dependent on surface structures, particularly O-antigens [54,55]. Therefore, we performed in silico serotyping, which revealed a pool of serologically distinct E. coli strains circulating in the gut community. Among the sequenced genomes and MAGs, 92% (34/37) and 86% (38/44) of E. coli were O- and H-typable in patients with CD and healthy controls, respectively. Twenty-seven different serotypes in patients with CD, and 32 in the healthy group, were discovered in total. Unrelated individuals from both cohorts shared five common serotypes, including O17/O44:H18 (n = 5), O144:H45 (n = 3), O6:H1 (n = 2), O25:H18 (n = 2), and O1:H7 (n = 2). Serotypes O8:H30 and O15:H18 occurred several times in healthy individuals, while O19:H4 and O128ac:H12 were found twice in patients with CD. The other strains displayed non-repeated combinations of O- and H-antigens. The most frequent O-antigen was O8 (8/81). The spectrum of antigens circulating in the human intestinal microbiota was discovered to be quite diverse; however, no association was found between strains of a specific serotype and CD (Supplementary Materials Table S1).
In order to assess virulence potential, the strains were screened for the presence of previously reported virulence genes [27,56,57], including genes encoding:
Hemolysins (hlyA, hlyF);
Toxins (astA, sat, vat, pic, cnf1, cvaB, cvaC, clbB);
Adhesion factors (iha, papC, papGIII, papGII, sfaS, focG, afaC, nfaE, fimH, fimC, bmaE, csgA, gafD, mat, tia, lpfA, lpfB, lpfC, tsh, ompA), capsule antigens (kpsM, kpsT, neuC);
Lipopolysaccharide biosynthesis proteins (waaL, waaV, waaW);
Flagellar antigen (fliC) and flagellar regulators (flhC, flhD, fliA);
Invasion factors (ibeA, gimB, malX);
Peyer’s patch-specific virulence factor (gipA);
Serum survival factors (iss, trat, nlpI);
CRISPR-associated proteins (cas1, cas6, cas_csy2, cas_csy3);
Porins (ompC);
Defensin resistance proteins (arlC, arlA);
Yersinia high-pathogenicity island (fyuA, ybtA, ybtP, ybtQ, ybtT);
Iron uptake systems (sitA, sitB, sitC, sitD, iucD, iutA, iroN, chuA, ireA, eitA, eitC, etsB, etsC, HRA-2);
Two-component regulatory system EnvZ/OmpR (envZ, ompR);
Stress response proteins (hfq, htrA, impK, rpoE, rpoS, dsbA);
Propanediol utilization enzyme (pduC);
Type VI secretion systems (T6SSs).
However, the comparative analysis did not reveal significant differences in the distribution of virulence genes between isolates from healthy and affected groups (Supplementary Materials Table S2). Considering the disease location, a high frequency of iha gene (bifunctional enterobactin receptor/adhesin protein) was found in strains from patients with ileitis (Fisher’s exact test, p-value adjusted with Benjamin–Hochberg correction, p ≤ 0.05) compared to colitis and ileocolitis, while the serum survival factor TraT and excisionase (p ≤ 0.05) were more frequent in patients with colitis (Figure 2). Interestingly, the most common sequence types for E. coli strains (ST10, ST69) significantly differ from other STs regarding the number of virulence genes for adhesion factors, lipopolysaccharide biosynthesis proteins, iron uptake systems, type VI secretion system, Yersinia high-pathogenicity island, capsule antigens (kpsT), serum survival factor (trat), stress response protein ImpK, and propanediol utilization enzyme (pduC) (Fisher’s exact test, p < 0.05) (Supplementary Materials Table S3). Therefore, it was supposed that the repertoire of virulence determinants may not reflect the ability of the strains to cause the disease, but may contribute to successful competition in the bacterial community. T6SSs, in particular, are known to play a role not only in bacterial adherence and invasion, biofilm formation and motility [58], but in interbacterial competition as well [59]. In this study, T6SS-2 gene cluster was identified in all E. coli genomes typed to ST3 (ST69) and belonging to phylogroup D. In one healthy- and one-CD-derived strains of phylogroup B2, both T6SS-1 and T6SS-2 systems were found. T6SS-3 locus was revealed in only one strain from a healthy individual.
Figure 2.

Frequency of occurrence of significantly different virulence genes in E. coli strains from patients with Crohn’s disease, depending on disease location (* p < 0.05, Fisher’s exact test).
Bacteriocin-producing systems benefit bacteria through their growth in a competitive environment as well. Screening of sequenced E. coli genomes revealed 28 strains (16 healthy and 12 CD-derived) containing genes for colicin and microcin production (Supplementary Materials Table S4). Microcin production systems, known for their promotion of gut colonization [60,61], were found equally in healthy and CD-derived strains. In isolated strains, the following genetic systems were identified for microcins of different classes: class I (MccC7, MccB17, MccJ25) [62], class IIa (MccV), and class IIb (MccH47, MccI47, MccM) [63]. Genes for the production of colicins (A, B, Ia, Ib, K, M, E1) were found in 23 strains (13 healthy and 10-CD derived). These isolates have the potential to acquire dominance under certain conditions. Indeed, according to CFU-counting data [40], all these strains dominated over other E. coli in the hosts. Therefore, we performed halo assays to determine the ability of the isolates to reveal antagonistic activity against other E. coli by overlaying the competitors on target bacteria K-12 substr. MG1655 (Figure 3). Only 11 isolates (four CD-derived and seven from healthy individuals) showed a phenotypic ability to inhibit the growth of the target strain.
Figure 3.
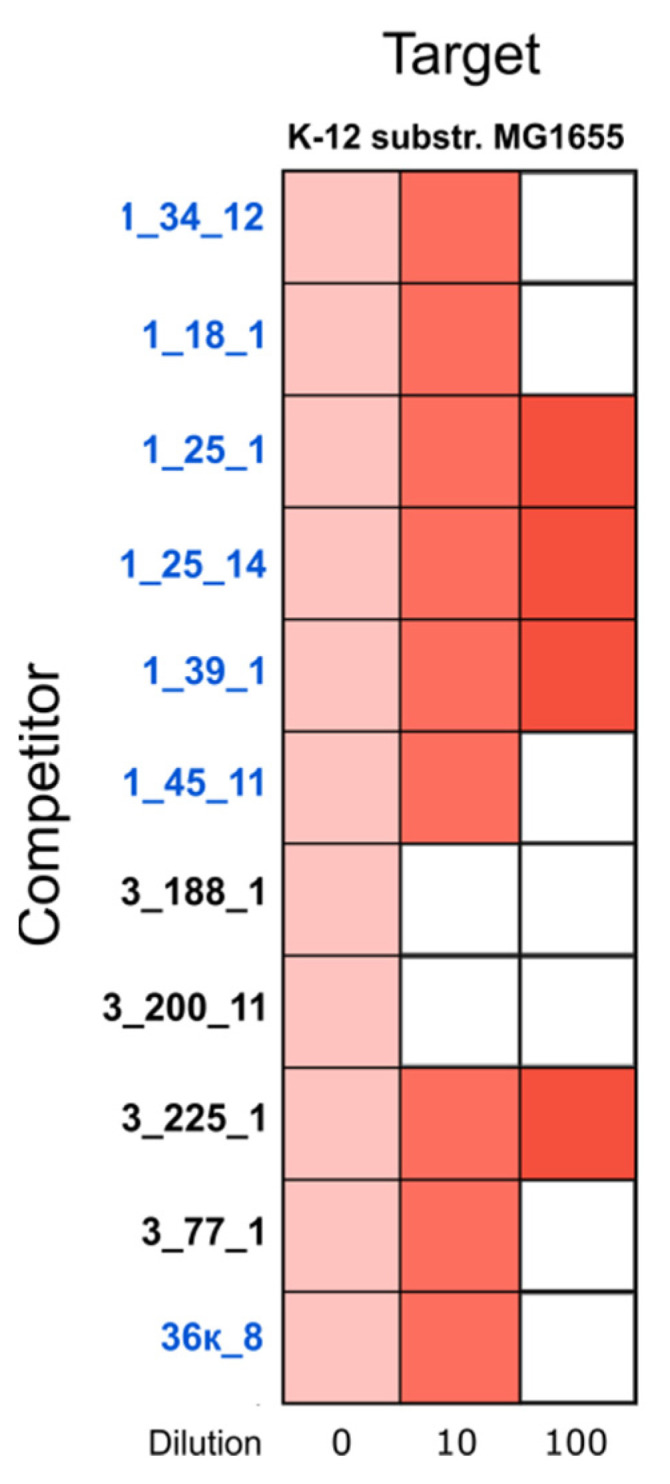
The antagonistic activity of E. coli strains from the gut against the strain K-12 substr. MG1655. Strains of healthy individuals are marked in blue. Red color indicates halo zone formation, white color—no halo. Deeper red corresponds to the lower concentration of the competitor strain.
3.3. Antibiotic Resistance and Its Genetic Determinants
Based on the disc-diffusion test, the majority of E. coli strains from CD (23/26) and healthy individuals (36/37) were non-susceptible to at least one of the tested antimicrobial drugs (Figure 4). All strains from both groups were sensitive to meropenem (carbapenems). The differences between patients with CD and healthy individuals were found in the susceptibility of the strains to amikacin and levofloxacin (Fisher’s exact test, p < 0.1), as well as to aztreonam and polymyxin (Supplementary Materials Table S5). Strains from both cohorts demonstrated non-susceptibility to 14/15 antibiotics tested, regardless of the location of intestinal inflammation. However, in one patient with ileal inflammation, both isolated strains were susceptible to all tested antibiotics. The strains that were non-susceptible to at least one agent in three or more antimicrobial categories and could be categorized as multidrug-resistant (MDR) strains [64] accounted for 14/26 (54%) of CD patients and 16/37 (43%) of individuals in the control group. Two patients with colonic inflammation harbored MDR strains which were non-susceptible to antibiotics of six different categories. The number of strains non-sensitive to ciprofloxacin, widely used for patients with active Crohn’s disease [65,66], was higher in the CD group, specifically: 4/26 (15%) in patients compared to 1/37 (3%) in controls.
Figure 4.

Susceptibility of E. coli strains from patients with Crohn’s disease (CD) and healthy individuals to the antibiotics. Numbers indicate the percent of non-susceptible strains (* p < 0.1, Fisher’s exact test).
Resistance genes predicted in silico were consistent with the phenotypic resistance revealed by the disk diffusion method (Supplementary Materials Table S6). All which were non-sensitive to tetracycline E. coli strains contained tetracycline efflux pump (tetA, tetB, tetD, tetR) genes. The inefficiency of fluoroquinolones could be due to plasmid-associated genes qnrS1 and qnrB19 and mutations in chromosome genes gyrA (S83L, D87N) and parC (S80I). The genes for class A beta-lactamases (TEM-1, TEM-150) and class D beta-lactamase OXA-1 were found in bacteria that were non-sensitive to ampicillin. The beta-lactamase TEM-150 only occurred in strains from healthy donors, not in CD patients. Although several strains carried aminoglycoside transferase genes, only one strain was detected to be non-sensitive to amikacin. Almost all E. coli strains harbored genes for AmpC type β-lactamases, which hydrolyze broad, extended-spectrum cephalosporins, genes of multidrug efflux systems MdtEF-TolC, EmrAB-TolC, EmrKY-TolC and AcrAB-TolC, MdtG and AcrD efflux pumps, and their regulators. To resist the antimicrobial activity of polymyxin, E. coli strains had genes that encode PmrF and PmrC enzymes modifying the antibiotic targets. However, only one strain from the CD patient was non-susceptible to polymyxin.
3.4. Metabolic Profiling of E. coli Strains
Intestinal commensals have adapted to the environment and developed complex ecological networks with other bacteria to acquire nutrients. Bacteria have to compete for nutrients in the gut, especially strains which belong to the same species and require similar nutrients. In this study, 63 cultivated isolates were screened for their biochemical features in order to investigate their metabolic interactions in the gut (Figure 5, Supplementary Materials Table S7).
Figure 5.

Distribution of biochemical features of E. coli strains from patients with Crohn’s disease and healthy individuals. Numbers indicate non-susceptible strains capable of utilizing corresponding carbohydrates (* p < 0.1, Fisher’s exact test).
Consistent with the biochemical assay, E. coli strains exhibited considerable variability in the utilization of diverse carbohydrates. All tested strains were capable of fermenting trehalose and hydrolyzing β-galactosides into monosaccharides. Most of the strains were lactose-positive (n = 58) and could utilize glucuronides (n = 58), melibiose (n = 59), as well as sugar alcohols such as mannitol (n = 62) and D-sorbitol (n = 58). According to our previous data on CFU counting [40], lactose-negative strains comprised a lower proportion (7.8 ± 9.3%) of E. coli compared to lactose-positive strains (53.1 ± 38.2%) (Mann–Whitney U test, p ≤ 0.01). At the same time, strains utilizing D-adonitol (n = 9) and D-arabitol (n = 9) were predominant (71.2 ± 37.5% vs. 45.9 ± 38%) within the E. coli community in human feces (Mann–Whitney U test, p < 0.1). Therefore, successful colonization with a dense and diverse intestinal microbiota is highly dependent on the ability of bacteria to compete for nutrients. Less than half of E. coli fermented raffinose (n = 25) and sucrose (n = 20), while eight strains utilized dulcitol and two bacteria could digest cellobiose. Salicin was fermented by only two CD-derived strains. Aesculin-hydrolysing E. coli accounted for 11% of all tested strains (n = 7). Only four strains (2 CD-derived and two from the controls) could use citrate on Simmons’ agar. With the exception of two healthy-derived strains, lysine was decarboxylated in all E. coli, whereas arginine and ornithine were decarboxylated in less than half of the strains (n = 39 and n = 19, respectively). Interestingly, hydrogen sulfide production was found to be associated with CD strains (n = 3, p < 0.1). The metabolic features of the strains were not associated with the disease location in patients. Such a variety of metabolic capabilities of bacteria may be associated with high inter- and interspecies competition for nutrients, contributing to intestinal colonization and resistance to elimination.
4. Discussion
In this study, we comprehensively typed 63 cultivated E. coli from CD patients with different disease locations and healthy volunteers, revealing high genetic and phenotypic heterogeneity in the strains. We found up to five E. coli strains per individual human host, with one predominant strain, which is consistent with previous studies into the diversity of the fecal E. coli [67]. To expand the understanding of strain-level diversity, we applied computational analyses of shotgun metagenome data and assembled 18 metagenome-associated E. coli genomes. The phylogenetic distribution within both groups was characterized by a majority of A and D phylogroups, followed by B1 phylogroup strains, which is inconsistent with previous reports, where A and B2 groups dominated in human and animal feces [67,68]. However, it has been shown that the proportion of B2 phylogroup strains differs between populations and may be due to geographic conditions, dietary factors or host genetic factors [69,70]. It is suggested that strains of the A, B1, and D phylogenetic groups are mostly commensal and have to acquire virulence factors to become pathogenic, while the strains of phylogroup B2 are potentially more virulent [71,72] and more frequent in IBD patients [73]. However, a previous report has shown that E. coli from patients with the inflamed and normal ileum were similarly distributed among phylogroups [74], which was confirmed in the current study. Further MLST analysis discovered the most common genotypes, ST10 and ST69, among the identified E. coli present in both cohorts. These sequence types are commonly associated with humans and food animals [75,76], and serve as intestinal colonizers, causing human infections [77,78,79]. Indeed, according to our data, strains of ST10 and ST69 genotypes harbored several virulence genes more frequently than other sequence types but were not CD-associated.
The serotype-based method illustrates the limitations in the identification of pathogenic strains associated with CD. A wide diversity of serotypes was observed in both of the studied cohorts, but none of the specific serotypes were associated with CD patients. The results of virulence gene analysis failed to distinguish the virulent strains associated with CD. These findings indicate that genomic content may not reflect the ability of the strains to cause the disease. This idea is supported by previous data demonstrating that the probiotic strain Nissle 1917 is genetically close to the uropathogenic E. coli CFT073 [80], can induce DNA double-strand breaks and chromosomal abnormalities in eukaryotic cells [81] and can cause disease under certain conditions [82]. Moreover, a recent study demonstrated that E. coli strains that inhabit the gut of healthy subjects possess genetic determinants of virulence similar to the potential pathogens [83]. Thus, there is a thin line between commensalism and pathogenicity. These virulence determinants seem to contribute to bacterial competitiveness and gut colonization, rather than directly induce the disease. It is possible that impaired tolerance to the commensal bacteria colonizing the host triggers shifts in pathogen recognition, leading to disease development. Indeed, we showed that different E. coli strains coexist in the human gut and participate in the formation of a host-specific community, but they are subject to numerous selective pressures. It is known that genetic diversity affects the ability to adapt to novel ecological conditions [84]. The ability to compete with neighbors and resist various chemicals and antibiotics is crucial for survival. In this study, bacteriocin-producing systems, conferring an advantage in terms of dominance, were observed in genomes of 26 strains. Almost half of these strains exhibited phenotypic antagonistic activity against the target strain and obtained better results than other E. coli in their hosts.
Along with bacteriocin production, the metabolic flexibility discovered in E. coli also promotes bacterial survival and intestinal colonization, but may occasionally lead to intestinal disorders. According to previous reports [39,85,86], an increased proportion of sulfate-reducing bacteria affects IBD through the overproduction of H2S, causing mucus degradation and intestinal inflammation. It is assumed that hydrogen sulfide contributes to bacterial survival in the intestine, increasing their resistance to antibiotics, and protecting against reactive oxygen species and immune-mediated killing [87,88,89]. However, at elevated concentrations, hydrogen sulfide may become toxic to the host. Although all strains possessed genes for H2S production, only three strains from one CD patient were positive in a biochemical assay, so can we speculate that these H2S-producing E. coli may contribute to an increase in total hydrogen sulfide in the gut. However, it has not yet been established whether a specific E. coli strain utilizes the same nutrients and produces gases when it grows in single culture and when it coexists with other commensal bacteria colonizing the IBD intestine.
In the current study, we observed the metabolic differences promoting fecal bacteria to successfully compete at the individual strain level. Though the fecal microbiota differs from the bacterial mucosal community, which is more directly involved in the CD pathogenesis, it may contribute to disease-associated changes in the host. An imbalance in the proportion of coexisting strains and changes in their interactions with each other and other commensals may affect hosts and disrupt intestinal homeostasis.
The main limitation of the study is that the isolated E. coli strains have a fecal origin, rather than mucous. It would be interesting to expand the collection with more samples as well as biopsies to better understand the complexity of the intestinal community. In addition, a closer look at the host–bacteria interaction may shed light on the CD pathogenesis. Taking these limitations into account, further investigations into the physiological responses of bacteria to the host are required.
5. Conclusions
This study contributes to a better understanding of the bacterial interactions in the human gut, which is crucial for revealing their role in the pathogenesis of Crohn’s disease. Whole-genome sequencing, along with metagenome-associated E. coli genome reconstruction, allows for a comprehensive characterization of the intestinal community composition and strain-level diversity, demonstrating the remarkable intra- and intraindividual genetic diversity of E. coli. We showed that different E. coli strains coexisting in the human gut of CD patients and healthy individuals exhibit similarities regarding the distribution of virulence determinants, antibiotic resistance genes, and bacteriocin-producing systems. We revealed a high metabolic flexibility in the strains, which promotes their survival and gut colonization. These observations suggest that bacterial efforts to successfully proliferate and outcompete other bacteria under intestinal conditions may implicitly influence hosts by reducing their tolerance for asymptomatic bacterial colonization and leading to disease development.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/microorganisms9061299/s1, Table S1: Typing of E. coli from CD patients and healthy individuals according to different methods. Table S2: Prevalence of virulence-associated genes in cultivated E. coli strains from CD patients and healthy individuals. Table S3: Prevalence of virulence-associated genes according to the most common sequence types (ST10, ST69) by Achtman’s scheme in all cultivated E. coli strains from CD. Table S4: Distribution of bacteriocins encoding genes identified in 28 E. coli strains from CD patients and healthy individuals. Table S5: Susceptibility to different antibiotics according to disc-diffusion assay in E. coli strains from CD patients and healthy individuals. Table S6: Antimicrobial resistance determinants identified in E. coli strains from CD patients and healthy individuals. Table S7: Prevalence of biochemical features in E. coli strains from CD patients and healthy individuals.
Author Contributions
Conceptualization, M.N.S. and T.V.G.; methodology, M.N.S., A.V.L. and D.R.K.; software, M.I.M.; validation, T.V.G.; formal analysis, M.I.M. and E.A.B.; investigation, M.N.S. and A.V.L.; resources, S.R.A., N.A.D., A.H.O. and R.A.A.; data curation, M.I.M.; writing—original draft preparation, M.N.S.; writing—review and editing, M.N.S., M.I.M., E.A.B. and A.V.L.; visualization, M.N.S. and M.I.M.; supervision, T.V.G.; project administration, T.V.G.; funding acquisition, T.V.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the subsidy allocated to Kazan Federal University for the state assignment in the sphere of scientific activities (project #0671-2020-0058) and by Russian Foundation for Basic Research (project #17-00-00433). This work is part of Kazan Federal University Strategic Academic Leadership Program.
Institutional Review Board Statement
The study was reviewed and approved by the local ethics committee of the Kazan Federal University, Kazan, Russia (dated 13 October 2017).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
Sixty-three whole-genome sequences and 18 metagenome-assembled E. coli genomes described in this paper were deposited at GenBank under BioProject accession number PRJNA560176.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Penders J., Thijs C., Vink C., Stelma F.F., Snijders B., Kummeling I., van den Brandt P.A., Stobberingh E.E. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006;118:511–521. doi: 10.1542/peds.2005-2824. [DOI] [PubMed] [Google Scholar]
- 2.Kaper J.B., Nataro J.P., Mobley H.L.T. Pathogenic Escherichia coli. Nat. Rev. Genet. 2004;2:123–140. doi: 10.1038/nrmicro818. [DOI] [PubMed] [Google Scholar]
- 3.Morgan X.C., Tickle T.L., Sokol H., Gevers D., Devaney K.L., Ward D.V., Reyes J.A., Shah S.A., LeLeiko N., Snapper S.B., et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13:R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rooks M.G., Veiga P., Wardwell-Scott L.H., Tickle T., Segata N., Michaud M., Gallini C.A., Beal C., van Hylckama-Vlieg J.E.T., Ballal S.A., et al. Gut microbiome composition and function in experimental colitis during active disease and treatment-induced remission. ISME J. 2014;8:1403–1417. doi: 10.1038/ismej.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Halfvarson J., Brislawn C.J., Lamendella R., Vázquez-Baeza Y., Walters W.A., Bramer L.M., D’Amato M., Bonfiglio F., McDonald D., Gonzalez A., et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017;2:17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vila A.V., Imhann F., Collij V., Jankipersadsing S.A., Gurry T., Mujagic Z., Kurilshikov A., Bonder M.J., Jiang X., Tigchelaar E.F., et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018;10:eaap8914. doi: 10.1126/scitranslmed.aap8914. [DOI] [PubMed] [Google Scholar]
- 7.Franzosa E.A., Sirota-Madi A., Avila-Pacheco J., Fornelos N., Haiser H.J., Reinker S., Vatanen T., Hall A.B., Mallick H., McIver L.J., et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019;4:293–305. doi: 10.1038/s41564-018-0306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Danilova N.A., Abdulkhakov S.R., Grigoryeva T.V., Markelova M.I., Vasilyev I.Y., Boulygina E.A., Ardatskaya M.D., Pavlenko A.V., Tyakht A.V., Odintsova A.K., et al. Markers of dysbiosis in patients with ulcerative colitis and Crohn’s disease. Ter. Arkhiv. 2019;91:13–20. doi: 10.26442/00403660.2019.04.000211. [DOI] [PubMed] [Google Scholar]
- 9.Mangin I., Bonnet R., Seksik P., Rigottier-Gois L., Sutren M., Bouhnik Y., Neut C., Collins M.D., Colombel J.-F., Marteau P., et al. Molecular inventory of faecal microflora in patients with Crohn’s disease. FEMS Microbiol. Ecol. 2004;50:25–36. doi: 10.1016/j.femsec.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 10.Frank D.N., Amand A.L.S., Feldman R.A., Boedeker E.C., Harpaz N., Pace N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sasaki M., Sitaraman S.V., Babbin B.A., Gerner-Smidt P., Ribot E.M., Garrett N., Alpern J.A., Akyildiz A., Theiss A.L., Nusrat A., et al. Invasive Escherichia coli are a feature of Crohn’s disease. Lab. Investig. 2007;87:1042–1054. doi: 10.1038/labinvest.3700661. [DOI] [PubMed] [Google Scholar]
- 12.Le Gac M., Plucain J., Hindré T., Lenski R.E., Schneider D. Ecological and evolutionary dynamics of coexisting lineages during a long-term experiment with Escherichia coli. Proc. Natl. Acad. Sci. USA. 2012;109:9487–9492. doi: 10.1073/pnas.1207091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kauffmann F. The serology of the coli group. J. Immunol. 1947;57:71–100. [PubMed] [Google Scholar]
- 14.Bettelheim K.A., Faiers M., Shooter R.A. Serotypes of Escherichia coli in normal stools. Lancet. 1972;300:1224–1226. doi: 10.1016/S0140-6736(72)92272-6. [DOI] [PubMed] [Google Scholar]
- 15.Orskov I., Orskov F., Jann B., Jann K. Serology, chemistry, and genetics of O and K antigens of Escherichia coli. Bacteriol. Rev. 1977;41:667. doi: 10.1128/br.41.3.667-710.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Caugant D.A., Levin B.R., Selander R.K. Genetic diversity and temporal variation in the E. coli population of a human host. Genetics. 1981;98:467–490. doi: 10.1093/genetics/98.3.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DebRoy C., Roberts E., Fratamico P.M. Detection of O antigens in Escherichia coli. Anim. Heal. Res. Rev. 2011;12:169. doi: 10.1017/S1466252311000193. [DOI] [PubMed] [Google Scholar]
- 18.Tartof S.Y., Solberg O.D., Manges A.R., Riley L.W. Analysis of a uropathogenic Escherichia coli clonal group by multilocus sequence typing. J. Clin. Microbiol. 2005;43:5860–5864. doi: 10.1128/JCM.43.12.5860-5864.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maiden M., Bygraves J.A., Feil E., Morelli G., Russell J.E., Urwin R., Zhang Q., Zhou J., Zurth K., Caugant D.A., et al. Multilocus sequence typing, a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clermont O., Christenson J.K., Denamur E., Gordon D.M. The Clermont Escherichia coli phylo-typing method revisited, improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013;5:58–65. doi: 10.1111/1758-2229.12019. [DOI] [PubMed] [Google Scholar]
- 21.Tabaqchali S., O’donoghue D.P., Bettelheim K.A. Escherichia coli antibodies in patients with inflammatory bowel disease. Gut. 1978;19:108–113. doi: 10.1136/gut.19.2.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joensen K.G., Tetzschner A.M., Iguchi A., Aarestrup F.M., Scheutz F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J. Clin. Microbiol. 2015;53:2410–2426. doi: 10.1128/JCM.00008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fratamico P.M., DebRoy C., Liu Y., Needleman D.S., Baranzoni G.M., Feng P. Advances in molecular serotyping and subtyping of Escherichia coli. Front. Microbiol. 2016;7:644. doi: 10.3389/fmicb.2016.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miquel S., Peyretaillade E., Claret L., De Vallée A., Dossat C., Vacherie B., Zineb E.H., Segurens B., Barbe V., Sauvanet P., et al. Complete genome sequence of Crohn’s disease-associated adherent-invasive E. coli strain LF82. PLoS ONE. 2010;5:e12714. doi: 10.1371/journal.pone.0012714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krause D.O., Little A.C., Dowd S.E., Bernstein C.N. Complete genome sequence of adherent invasive Escherichia coli UM146 isolated from Ileal Crohn’s disease biopsy tissue. J. Bacteriol. 2011;193:583. doi: 10.1128/JB.01290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nash J.H., Villegas A., Kropinski A.M., Aguilar-Valenzuela R., Konczy P., Mascarenhas M., Ziebell K., Torres A.G., Karmali M.A., Coombes B.K. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genom. 2010;11:667. doi: 10.1186/1471-2164-11-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camprubí-Font C., Lopez-Siles M., Ferrer-Guixeras M., Niubó-Carulla L., Abellà-Ametller C., Garcia-Gil L.J., Martinez-Medina M. Comparative genomics reveals new single-nucleotide polymorphisms that can assist in identification of adherent-invasive Escherichia coli. Sci. Rep. 2018;8:2695. doi: 10.1038/s41598-018-20843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Brien C.L., Bringer M.-A., Holt K.E., Gordon D.M., Dubois A.L., Barnich N., Darfeuille-Michaud A., Pavli P. Comparative genomics of Crohn’s disease-associated adherent-invasive Escherichia coli. Gut. 2017;66:1382–1389. doi: 10.1136/gutjnl-2015-311059. [DOI] [PubMed] [Google Scholar]
- 29.Rakitina D.V., Manolov A.I., Kanygina A.V., Garushyants S.K., Baikova J.P., Alexeev D.G., Ladygina V.G., Kostryukova E.S., Larin A.K., Semashko T.A., et al. Genome analysis of E. coli isolated from Crohn’s disease patients. BMC Genom. 2017;18:544. doi: 10.1186/s12864-017-3917-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Darfeuille-Michaud A., Neut C., Barnich N., Lederman E., Di Martino P., Desreumaux P., Gambiez L., Joly B., Cortot A., Colombel J.-F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn’s disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/S0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 31.Glasser A.-L., Boudeau J., Barnich N., Perruchot M.-H., Colombel J.-F., Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn’s disease survive and replicate within macrophages without inducing host cell death. Infect. Immun. 2001;69:5529–5537. doi: 10.1128/IAI.69.9.5529-5537.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martin H.M., Campbell B.J., Hart C., Mpofu C., Nayar M., Singh R., Englyst H., Williams H.F., Rhodes J.M. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 2004;127:80–93. doi: 10.1053/j.gastro.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 33.Carvalho F.A., Barnich N., Sauvanet P., Darcha C., Gelot A., Darfeuille-Michaud A. Crohn’s disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm. Bowel Dis. 2008;14:1051–1060. doi: 10.1002/ibd.20423. [DOI] [PubMed] [Google Scholar]
- 34.Ukena S.N., Singh A., Dringenberg U., Engelhardt R., Seidler U., Hansen W., Bleich A., Bruder D., Franzke A., Rogler G., et al. Probiotic Escherichia coli Nissle 1917 inhibits leaky gut by enhancing mucosal integrity. PLoS ONE. 2007;2:e1308. doi: 10.1371/journal.pone.0001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schultz M. Clinical use of E. coli Nissle 1917 in inflammatory bowel disease. Inflamm. Bowel Dis. 2008;14:1012–1018. doi: 10.1002/ibd.20377. [DOI] [PubMed] [Google Scholar]
- 36.Boudeau J., Glasser A.L., Julien S., Colombel J.F., Darfeuille-Michaud A. Inhibitory effect of probiotic Escherichia coli strain Nissle 1917 on adhesion to and invasion of intestinal epithelial cells by adherent–invasive E. coli strains isolated from patients with Crohn’s disease. Aliment. Pharmacol. Ther. 2003;18:45–56. doi: 10.1046/j.1365-2036.2003.01638.x. [DOI] [PubMed] [Google Scholar]
- 37.Sonnenborn U., Schulze J. The non-pathogenic Escherichia coli strain Nissle 1917–features of a versatile probiotic. Microb. Ecol. Heal. Dis. 2009;21:122–158. doi: 10.3109/08910600903444267. [DOI] [Google Scholar]
- 38.Prilassnig M., Wenisch C., Daxboeck F., Feierl G. Are probiotics detectable in human feces after oral uptake by healthy volunteers? Wien. Klin. Wochenschr. 2007;119:456–462. doi: 10.1007/s00508-007-0808-1. [DOI] [PubMed] [Google Scholar]
- 39.Sasso G.L., Khachatryan L., Kondylis A., Battey J.N.D., Sierro N., Danilova N.A., Grigoryeva T.V., Markelova M.I., Khusnutdinova D.R., Laikov A.V., et al. Inflammatory Bowel Disease—Associated Changes in the Gut: Focus on Kazan Patients. Inflamm. Bowel Dis. 2020;27:418–433. doi: 10.1093/ibd/izaa188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Siniagina M., Markelova M., Laikov A., Boulygina E., Khusnutdinova D., Kharchenko A., Misbakhova A., Grigoryeva T. Cultivated Escherichia coli diversity in intestinal microbiota of Crohn’s disease patients and healthy individuals: Whole genome data. Data Brief. 2020;28:104948. doi: 10.1016/j.dib.2019.104948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nurk S., Meleshko D., Korobeynikov A., Pevzner P.A. metaSPAdes, a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu Y.W., Tang Y.H., Tringe S.G., Simmons B.A., Singer S.W. MaxBin, an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome. 2014;2:26. doi: 10.1186/2049-2618-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parks D.H., Imelfort M., Skennerton C.T., Hugenholtz P., Tyson G.W. CheckM, assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–1055. doi: 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asnicar F., Thomas A.M., Beghini F., Mengoni C., Manara S., Manghi P., Zhu Q., Bolzan M., Cumbo F., May U., et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020;11:2500. doi: 10.1038/s41467-020-16366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Letunic I., Bork P. Interactive Tree of Life, iTOL, v4, recent updates and new developments. Nucleic Acids Res. 2019;47:W256–W259. doi: 10.1093/nar/gkz239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain C., Rodriguez-R L.M., Phillippy A.M., Konstantinidis K.T., Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018;9:5114. doi: 10.1038/s41467-018-07641-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waters N.R., Abram F., Brennan F., Holmes A., Pritchard L. Easy phylotyping of Escherichia coli via the EzClermont web app and command-line tool. Access Microbiol. 2020;2:e000143. doi: 10.1099/acmi.0.000143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Larsen M.V., Cosentino S., Rasmussen S., Friis C., Hasman H., Marvig R.L., Jelsbak L., Sicheritz-Ponten T., Ussery D.W., Aarestrup F.M., et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012;50:1355–1361. doi: 10.1128/JCM.06094-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wirth T., Falush D., Lan R., Colles F., Mensa P., Wieler L.H., Karch H., Reeves P.R., Maiden M., Ochman H., et al. Sex and virulence in Escherichia coli, an evolutionary perspective. Mol. Microbiol. 2006;60:1136–1151. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jaureguy F., Landraud L., Passet V., Diancourt L., Frapy E., Guigon G., Carbonnelle E., Lortholary O., Clermont O., Denamur E., et al. Phylogenetic and genomic diversity of human bacteremic Escherichia coli strains. BMC Genom. 2008;9:560. doi: 10.1186/1471-2164-9-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ohno M., Hasegawa M., Hayashi A., Caballero-Flores G., Alteri C.J., Lawley T.D., Kamada N., Núñez G., Inohara N. Lipopolysaccharide O structure of adherent and invasive Escherichia coli regulates intestinal inflammation via complement C3. PLoS Pathog. 2020;16:e1008928. doi: 10.1371/journal.ppat.1008928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alcock B.P., Raphenya A.R., Lau T.T.Y., Tsang K.K., Bouchard M., Edalatmand A., Huynh W., Nguyen A.-L.V., Cheng A.A., Liu S., et al. CARD 2020, antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020;48:D517–D525. doi: 10.1093/nar/gkz935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macpherson A.J., Harris N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004;4:478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 54.Miajlovic H., Smith S.G. Bacterial self-defence, how Escherichia coli evades serum killing. FEMS Microbiol. Lett. 2014;354:1–9. doi: 10.1111/1574-6968.12419. [DOI] [PubMed] [Google Scholar]
- 55.Steimle A., Autenrieth I.B., Frick J.S. Structure and function, Lipid A modifications in commensals and pathogens. Int. J. Med. Microbiol. 2016;306:290–301. doi: 10.1016/j.ijmm.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Schubert S., Picard B., Gouriou S., Heesemann J., Denamur E. Yersinia high-pathogenicity island contributes to virulence in Escherichia coli causing extraintestinal infections. Infection and immunity. Infect. Immun. 2002;70:5335–5337. doi: 10.1128/IAI.70.9.5335-5337.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chassaing B., Rolhion N., De Vallée A., Salim S.Y., Prorok-Hamon M., Neut C., Campbell B.J., Söderholm J.D., Hugot J.-P., Colombel J.-F., et al. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer’s patches via long polar fimbriae. J. Clin. Investig. 2011;121:966–975. doi: 10.1172/JCI44632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navarro-Garcia F., Ruiz-Perez F., Cataldi Á., Larzábal M. Type VI secretion system in pathogenic Escherichia coli, structure, role in virulence, and acquisition. Front. Microbiol. 2019;10:1965. doi: 10.3389/fmicb.2019.01965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Russell A.B., Peterson S.B., Mougous J.D. Type VI secretion system effectors, poisons with a purpose. Nat. Rev. Genet. 2014;12:137–148. doi: 10.1038/nrmicro3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sassone-Corsi M., Nuccio S.-P., Liu H., Hernandez D., Vu C.T., Takahashi A.A., Edwards R.A., Raffatellu M. Microcins mediate competition among Enterobacteriaceae in the inflamed gut. Nature. 2016;540:280–283. doi: 10.1038/nature20557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Massip C., Oswald E. Siderophore-Microcins in Escherichia coli, Determinants of Digestive Colonization, the First Step Toward Virulence. Front. Cell. Infect. Microbiol. 2020;10:381. doi: 10.3389/fcimb.2020.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Severinov K., Semenova E., Kazakov T. Prokaryotic Antimicrobial Peptides. Springer; New York, NY, USA: 2011. Class I microcins, their structures, activities, and mechanisms of resistance; pp. 289–308. [DOI] [Google Scholar]
- 63.Duquesne S., Destoumieux-Garzón D., Peduzzi J., Rebuffat S. Microcins, gene-encoded antibacterial peptides from enterobacteria. Nat. Prod. Rep. 2007;24:708–734. doi: 10.1039/b516237h. [DOI] [PubMed] [Google Scholar]
- 64.Magiorakos A.-P., Srinivasan A., Carey R.B., Carmeli Y., Falagas M.E., Giske C.G., Harbarth S., Hindler J.F., Kahlmeter G., Olsson-Liljequist B., et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria, an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012;18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 65.Greenbloom S.L., Steinhart A.H., Greenberg G.R. Combination ciprofloxacin and metronidazole for active Crohn’s disease. Can. J. Gastroenterol. 1998;12:53–56. doi: 10.1155/1998/349460. [DOI] [PubMed] [Google Scholar]
- 66.Wu X.W., Ji H.Z., Wang F.Y. Meta analysis of ciprofloxacin in treatment of Crohn’s disease. Biomed. Rep. 2015;3:70–74. doi: 10.3892/br.2014.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tenaillon O., Skurnik D., Picard B., Denamur E. The population genetics of commensal Escherichia coli. Nat. Rev. Genet. 2010;8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 68.Smati M., Clermont O., Le Gal F., Schichmanoff O., Jauréguy F., Eddi A., Denamur E., Picard B., Coliville Group Real-time PCR for quantitative analysis of human commensal Escherichia coli populations reveals a high frequency of subdominant phylogroups. Appl. Environ. Microbiol. 2013;79:5005–5012. doi: 10.1128/AEM.01423-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Duriez P., Clermont O., Bonacorsi S., Bingen E., Chaventré A., Elion J., Picard B., Denamur E. Commensal Escherichia coli isolates are phylogenetically distributed among geographically distinct human populations. Microbiology. 2001;147:1671–1676. doi: 10.1099/00221287-147-6-1671. [DOI] [PubMed] [Google Scholar]
- 70.Lescat M., Clermont O., Woerther P.L., Glodt J., Dion S., Skurnik D., Djossou F., Dupont C., Perroz G., Picard B., et al. Commensal Escherichia coli strains in Guiana reveal a high genetic diversity with host-dependant population structure. Environ. Microbiol. Rep. 2013;5:49–57. doi: 10.1111/j.1758-2229.2012.00374.x. [DOI] [PubMed] [Google Scholar]
- 71.Picard B., Garcia J.S., Gouriou S., Duriez P., Brahimi N., Bingen E., Elion J., Denamur E. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999;67:546–553. doi: 10.1128/IAI.67.2.546-553.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Massot M., Daubié A.-S., Clermont O., Jauréguy F., Couffignal C., Dahbi G., Mora A., Blanco J., Branger C., Mentré F., et al. Phylogenetic, virulence and antibiotic resistance characteristics of commensal strain populations of Escherichia coli from community subjects in the Paris area in 2010 and evolution over 30 years. Microbiology. 2016;162:642. doi: 10.1099/mic.0.000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petersen A.M., Nielsen E.M., Litrup E., Brynskov J., Mirsepasi H., Krogfelt K.A. A phylogenetic group of Escherichia coli associated with active left-sided inflammatory bowel disease. BMC Microbiol. 2009;9:171. doi: 10.1186/1471-2180-9-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dogan B., Scherl E., Bosworth B., Yantiss R., Altier C., McDonough P.L., Jiang Z.-D., Dupont H.L., Garneau P., Harel J., et al. Multidrug resistance is common in Escherichia coli associated with ileal Crohn’s disease. Inflamm. Bowel Dis. 2013;19:141–150. doi: 10.1002/ibd.22971. [DOI] [PubMed] [Google Scholar]
- 75.Manges A.R., Harel J., Masson L., Edens T.J., Portt A., Reid-Smith R.J., Zhanel G.G., Kropinski A.M., Boerlin P. Multilocus sequence typing and virulence gene profiles associated with Escherichia coli from human and animal sources. Foodborne Pathog. Dis. 2015;12:302–310. doi: 10.1089/fpd.2014.1860. [DOI] [PubMed] [Google Scholar]
- 76.Yamaji R., Friedman C.R., Rubin J., Suh J., Thys E., McDermott P., Hung-Fan M., Riley L.W. A population-based surveillance study of shared genotypes of Escherichia coli isolates from retail meat and suspected cases of urinary tract infections. MSphere. 2018;3:e00179-18. doi: 10.1128/mSphere.00179-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Oteo J., Diestra K., Juan C., Bautista V., Novais Â., Pérez-Vázquez M., Moyá B., Miró E., Coque T.M., Oliver A., et al. Extended-spectrum β-lactamase-producing Escherichia coli in Spain belong to a large variety of multilocus sequence typing types, including ST10 complex/A, ST23 complex/A and ST131/B2. Int. J. Antimicrob. Agents. 2009;34:173–176. doi: 10.1016/j.ijantimicag.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 78.Blanco J., Mora A., Mamani R., López C., Blanco M., Dahbi G., Herrera A., Alonso M.P., García-Garrote F., Chaves F., et al. National survey of Escherichia coli causing extraintestinal infections reveals the spread of drug-resistant clonal groups O25b, H4-B2-ST131, O15, H1-D-ST393 and CGA-D-ST69 with high virulence gene content in Spain. J. Antimicrob. Chemother. 2011;66:2011–2021. doi: 10.1093/jac/dkr235. [DOI] [PubMed] [Google Scholar]
- 79.Manges A.R., Johnson J.R. Food-borne origins of Escherichia coli causing extraintestinal infections. Clin. Infect. Dis. 2012;55:712–719. doi: 10.1093/cid/cis502. [DOI] [PubMed] [Google Scholar]
- 80.Vejborg R.M., Friis C., Hancock V., Schembri M.A., Klemm P. A virulent parent with probiotic progeny, comparative genomics of Escherichia coli strains CFT073, Nissle 1917 and ABU 83972. Mol. Genet. Genom. 2010;283:469–484. doi: 10.1007/s00438-010-0532-9. [DOI] [PubMed] [Google Scholar]
- 81.Olier M., Marcq I., Salvador-Cartier C., Secher T., Dobrindt U., Boury M., Bacquié V., Penary M., Gaultier E., Nougayrède J.-P., et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes. 2012;3:501–509. doi: 10.4161/gmic.21737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guenther K., Straube E., Pfister W., Guenther A., Huebler A. Sever sepsis after probiotic treatment with Escherichia coli NISSLE 1917. Pediatr. Infect. Dis. J. 2010;29:188–189. doi: 10.1097/INF.0b013e3181c36eb9. [DOI] [PubMed] [Google Scholar]
- 83.Raimondi S., Righini L., Candeliere F., Musmeci E., Bonvicini F., Gentilomi G., Erjavec M.S., Amaretti A., Rossi M. Antibiotic resistance, virulence factors, phenotyping, and genotyping of E. coli isolated from the feces of healthy subjects. Microorganisms. 2019;7:251. doi: 10.3390/microorganisms7080251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Touchon M., Perrin A., De Sousa J.A.M., Vangchhia B., Burn S., O’Brien C.L., Denamur E., Gordon D., Rocha E.P. Phylogenetic background and habitat drive the genetic diversification of Escherichia coli. PLoS Genet. 2020;16:e1008866. doi: 10.1371/journal.pgen.1008866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Loubinoux J., Bronowicki J.P., Pereira I.A., Mougenel J.L., Le Faou A.E. Sulfate-reducing bacteria in human feces and their association with inflammatory bowel diseases. FEMS Microbiol. Ecol. 2002;40:107–112. doi: 10.1111/j.1574-6941.2002.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 86.Attene-Ramos M.S., Nava G.M., Muellner M.G., Wagner E.D., Plewa M.J., Gaskins H.R. DNA damage and toxicogenomic analyses of hydrogen sulfide in human intestinal epithelial FHs 74 Int cells. Environ. Mol. Mutagen. 2010;51:304–314. doi: 10.1002/em.20546. [DOI] [PubMed] [Google Scholar]
- 87.Shatalin K., Shatalina E., Mironov A., Nudler E. H2S, a universal defense against antibiotics in bacteria. Science. 2011;334:986–990. doi: 10.1126/science.1209855. [DOI] [PubMed] [Google Scholar]
- 88.Mironov A., Seregina T., Nagornykh M., Luhachack L.G., Korolkova N., Lopes L.E., Kotova V., Zavilgelsky G., Shakulov R., Shatalin K., et al. Mechanism of H2S-mediated protection against oxidative stress in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2017;114:6022–6027. doi: 10.1073/pnas.1703576114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Toliver-Kinsky T., Cui W., Törö G., Lee S.J., Shatalin K., Nudler E., Szabo C. H2S, a bacterial defense mechanism against the host immune response. Infect. Immun. 2019;87:e00272-18. doi: 10.1128/IAI.00272-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sixty-three whole-genome sequences and 18 metagenome-assembled E. coli genomes described in this paper were deposited at GenBank under BioProject accession number PRJNA560176.