

Abstract
The sigma-1 (σ1) receptor is a ‘pluripotent chaperone’ protein mainly expressed at the mitochondria–endoplasmic reticulum membrane interfaces where it interacts with several client proteins. This feature renders the σ1 receptor an ideal target for the development of multifunctional ligands, whose benefits are now recognized because several pathologies are multifactorial. Indeed, the current therapeutic regimens are based on the administration of different classes of drugs in order to counteract the diverse unbalanced physiological pathways associated with the pathology. Thus, the multi-targeted directed ligand (MTDL) approach, with one molecule that exerts poly-pharmacological actions, may be a winning strategy that overcomes the pharmacokinetic issues linked to the administration of diverse drugs. This review aims to point out the progress in the development of MTDLs directed toward σ1 receptors for the treatment of central nervous system (CNS) and cancer diseases, with a focus on the perspectives that are proper for this strategy. The evidence that some drugs in clinical use unintentionally bind the σ1 protein (as off-target) provides a proof of concept of the potential of this strategy, and it strongly supports the promise that the σ1 receptor holds as a target to be hit in the context of MTDLs for the therapy of multifactorial pathologies.
Keywords: sigma receptors, sigma-1 receptor, multi-target directed ligands (MTDLs), sigma-1 ligands, polypharmacology
1. Introduction
The most exploited paradigm in medicinal chemistry is the “one-target, one-disease”, according to which ligands are developed to act toward a single target in order to exert a beneficial effect. This kind of approach, even if largely applied, may lead to unsuccessful results. Indeed, pathologies are mostly based on tangled mechanisms that involve different kinds of targets. Thus, a single target approach could be inappropriate because of its impossibility to produce an effect on the different pathways associated with the pathology.
On the basis of this idea, a multi-target strategy has been proposed in order to hit at the same time the diverse targets involved and modulate the diverse mechanisms that underlie the pathology.
The first step of this multi-target strategy has been the co-administration of more drugs all together, with the aim of obtaining a synergic effect. Some examples of this approach are represented by current therapeutic programs. Pathologies such as hypertension, cancer, neurodegeneration, AIDS, and depression can require the administration of up to three different classes of drugs.
Although this approach has therapeutic beneficial effects, the higher the number of drugs administered, the higher the number of metabolites that could produce side effects. Moreover, the co-administration of different drugs can produce cross-resistance to the therapeutics and inconvenient pharmacokinetic profiles. With the aim to overcome such problems, the multi-target directed ligand (MTDL) approach has been introduced. This strategy is based on the administration of a single drug with multiple effects because such a drug incorporates two or more pharmacophores in a single molecule. This incorporation can produce hybrid drugs or chimeric drugs. Hybrid drugs are obtained when two or more pharmacophores are connected using a stable or metabolizable linker. These structures not only have a high molecular weight, which is detrimental for oral bioavailability, but they are also very flexible, so that binding to the targets can be reduced. On the other hand, chimeric drugs are obtained by fusing or merging two or more different pharmacophores into one. In contrast to the former type, chimeric drugs have better pharmacokinetic properties and better interaction with targets because of their rigidity [1,2].
The synthesis of MTDLs can follow a knowledge-based approach or a screening approach. The former approach is based on the use of diverse and focused libraries of molecules and relies on well-known drugs and compounds. In the screening approach instead, ligands that demonstrate a solid affinity for a certain target are explored in their activities/affinities for other targets. However, in both cases, a fine work of optimization is needed to obtain the best compromise that guarantees the interaction with all targets [3].
In this review, the authors want to discuss the use of the MTDL approach that exploits the interaction with sigma (σ) receptors. In fact, even if these receptors are still not completely understood, their involvement in several pathologies has been widely demonstrated, so that the synergy obtained by a simultaneous interaction with σ receptors and other biological targets appears as a beneficial strategy to face multifactorial pathologies.
σ Receptors were discovered in 1976, when studies performed on opioid receptors revealed the presence of proteins that do not bind naloxone and etorphine but display high affinity for (+)-pentazocine, which is still used in binding studies [4]. A milestone in the history of σ receptors has been attained when two different protein subtypes were discovered, namely σ1 and σ2 receptors [5].
2. σ1 Receptors
σ1 Receptors have been found in the central nervous system (CNS), particularly in the granular layer of the olfactory bulb, in many cortical layers, and in the dentate gyrus [6], where they exert their most important activities. A slightly lower expression has been also found in some pyramidal layers of the hippocampus, various hypothalamic nuclei, the septum, the central gray, the motor nuclei of the hindbrain, and the dorsal horn of the spinal cord [6]. At the cellular level, σ1 receptors have been found in ependymocytes, which border the ventricular compartments, and in neurons located within the CNS parenchyma [6].
In peripheral organs, σ1 receptors have been found in the gastrointestinal tract [7], vas deferens [8], in liver and kidney [9], heart [10], adrenal medulla, pituitary, testis, and ovaries [11].
The σ1 receptor is localized at the mitochondrial-associated endoplasmic reticulum (ER) membranes (MAM) in association with the binding immunoglobulin protein (BiP) in a resting state. Upon agonist stimulation, or Ca2+ depletion, the σ1 receptor dissociates from BiP and stabilizes inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) at MAM, increasing Ca2+ transfer from ER into mitochondria where adenosine triphosphate (ATP) production is facilitated [12]. Recently, it has been proposed that σ1 receptors affect store-operated Ca2+ Entry (SOCE), with agonists, such as (+)-pentazocine, increasing the SOCE activity [13].
The σ1 subtype is also able to protect against ER stress associated with reactive oxygen species (ROS), which can easily activate the inositol-requiring enzyme 1 (IRE1) at MAM. In this context, the σ1 receptor stabilizes IRE1 and enhances cellular survival, prolonging the activation of the IRE1–XBP1 signaling pathway [14]. Moreover, the interaction of σ1 with B-cell lymphoma 2 (Bcl-2) or nuclear factor erythroid 2-related factor 2 (Nrf2) pathways is able to protect against apoptosis [15]. It is now clear that the σ1 receptor, which is defined as a ‘pluripotent chaperone’ [16], interacts with a number of proteins, as recently reviewed by Schmidt and colleagues [17], although more evidence is required to ascertain a functional interaction.
This pluripotent nature of the σ1 receptor is responsible for a series of complex effects with the cell state, such as stress conditions, which can produce modifications in the σ1-mediated response to stimuli. As recently reviewed, it is likely through these protein–protein interactions that the σ1 receptor exerts its action in viral infections and also against SARS-CoV-2 [18]. It has been demonstrated that the σ1 receptor interacts with the NSP6 viral protein, and its knock-out (KO) and knock-down (KD) abate the cells’ infection by both SARS-CoV-1 and 2, prompting focused research in this direction. By contrast, the KO and KD of the σ2 subtype, which interact with the viral protein ORf9c, do not impact on cells’ infection [19].
The neuroprotective action of σ1 receptor agonists is well established, and it has been proved that this activity is exerted through different mechanisms such as intracellular Ca2+ regulation, the prevention of oxidative stress, and anti-apoptotic effects. The σ1 subtype contributes to protein homeostasis: it can stimulate neurotrophin receptor signaling and reduce protein aggregation responsible for neurodegenerative disease, and it can also activate autophagy as a protective mechanism against damage arisen by misfolded proteins. Recently, many studies proved the ability of σ1 receptors to directly or indirectly interact with receptors or enzymes with key roles in neurodegeneration, particularly in Alzheimer’s disease (AD). Importantly, in patients with AD, a reduction of σ1 receptor density [20] has been demonstrated. Some studies attributed such reduction to the E4 variant of the apolipoprotein E gene (APOE 4) [21], although some controversies still exist [22]. All these mechanisms can promote cell survival and consolidate the role of the σ1 receptor as a target for therapies against neurodegeneration [23].
From a structural point of view, the σ1 protein has a 223 amino acid sequence, conserved among vertebrates, but devoid of similarity with any other protein. The closest protein seems to be ERG2p, which is a C8-C7 sterol isomerase expressed in the yeast. The two proteins share the same steroid binding domain-like (SBDL) regions, although no isomerase activity has been shown for the σ1 receptor [24]. A breakthrough milestone in the σ1 receptor-related research is the crystal structure of the protein, which was published in 2016 [25]. Later, also the crystal structures of the σ1 protein with agonists and antagonists bound to it were disclosed [26], and important insights into the binding modes of the σ1 ligands were provided with evidence for the so far ligands-based generated models. The most important interaction between ligands and the receptor is represented by an electrostatic bond between a basic amine (present in all the high-affinity σ1 ligands) and Glu172, while the rest of the ligand is accommodated in two different hydrophobic areas, which is in line with Glennon’s pharmacophore model [27].
According to this model, the σ1 receptor pharmacophore consists of a basic amine site tolerating a little substituent (which could be H or small alkyl groups) and two hydrophobic regions: the Primary Hydrophobic Region is 6–10 Å far from the amine site, while the Secondary Hydrophobic one is 2.5–3.9 Å far from the amine. Such a model very well fits into the crystal structure, whose availability has rationalized the binding of several molecules to σ1 receptors [28].
Importantly, crystallization revealed a trimeric complex, suggesting that the protein acts through polymerization states. Each subunit is formed by a single transmembrane domain (TMD) and a large cytosolic region that contains the binding pocket for each monomer. The 223 amino acid sequence is organized into five α-helices and ten β-strands: the N-terminus passes through the membrane, forms the only TMD, and protrudes in the ER lumen. The C-terminus is a flat hydrophobic sequence associated with the cytosolic surface of the ER. Within the binding pocket, besides the ionic interaction with Glu172, σ1 receptor ligands interact with Tyr103 (through hydrogen bonds with hydrophilic residues), which provides a favorable environment for the hydrophobic bonds within the primary hydrophobic region (formed by Val84, Ala86, Trp89, Met93, Leu95, Ala98, Tyr103, Leu105, Phe107, Tyr120, Ile124, Trp164, Met170, Ile178, Thr181, Leu182, Phe184, Ala185, Thr202, and Tyr206). The secondary hydrophobic region (made of Trp89, Ser117, Tyr120, Ile124, Asp126, Phe133, Val152, His154, Thr160, Val162, Trp164, and Met170, with Ser117 and His154 exhibiting their hydrophobic side chain to the cavity) tolerates smaller groups compared to the primary one [27,28]. Interestingly, the crystal structure of the receptor bound to its ligands revealed how the agonists, in contrast to the antagonists, induce a structural shift of the α 4 helix, likely preventing the oligomerization state, supporting that the action of the σ1 receptor occurs through protein–protein interaction (PPI). Therefore, while σ1 ligands may function as allosteric modulators of PPIs, the different oligomeric structures could be responsible for the many activities performed by these versatile receptors, with agonists and antagonists differently influencing the association among protomers: agonists produce lower oligomeric structures such as monomers and dimers, while antagonists produce higher ones [29].
Overview on σ1 Receptors Ligands: Functional Activity, Proposed Endogenous Ligands, and Reference Compounds
Functional activity. Although the terms agonist and antagonist have been used to classify σ1 receptor ligands, an unambiguous definition of the functional activity (agonist versus antagonist) is often hard. As a matter of fact, both agonists (i.e., fluvoxamine and other selective serotonin reuptake inhibitors (SSRIs) [30]) and antagonists (i.e., rimcazole [31,32]) are effective in treating some pathologies such as psychosis, and there are some ligands (i.e., BMY 14802 [33,34]) that show both agonist and antagonist activities in different conditions [35].
With the aim to unequivocally ascertain agonist vs. antagonist activity, many studies have been carried out. Among them, an interesting approach was developed by Gomez-Soler and colleagues, who developed a biosensor exploiting the Fluorescence Resonance Energy Transfer (FRET) technique in living cells. The model was based on the genetic decoration of the σ1 receptor with cyan and yellow fluorescent proteins. The distance between these two portions depends on the receptor conformation, which is influenced by the nature of the ligand. In fact, while agonists make the two fluorescent portions more distant, antagonists produce a conformational change that makes the portions closer, resulting in a fluorescence emission due to the energy transfer between them [36]. Therefore, the claimed σ1 receptor agonists and antagonists studied produced decreased and increased FRET signals, respectively. Another step in the same direction was taken by Yano et al., who produced a novel receptor homomer assay based on the Bioluminescence Resonance Energy Transfer (BRET) technique. The σ1 receptor was fused in its C-terminal and N-terminal sites with NanoLuciferace (NL) (BRET donor) and the yellow fluorescent protein Venus (VN), which works as an acceptor. The most suitable fusion construct pairs were identified to evaluate the ligand-induced BRET signal, and antagonists were shown to promote higher-order homomerization [35], with results from this assay in agreement with biochemical assays. The same BRET approach was also used to evaluate the heteromerization between the σ1 receptor and BiP, with the former protein bearing NL and the latter bearing the VN portions. Again, haloperidol and (+)-pentazocine displayed an opposite trend. Nevertheless, the same authors did not use the definition ‘agonist’ or ‘antagonist’ but preferred to discuss the phenotypes of the studied molecules, as some unexpected discrepancies emerged in the ligands studied (e.g., NE100, cocaine). All in all, even if a clear distinction between the two classes is still missing, these fluorescent-based assays provide useful information about the conformational changes that underlie σ1 receptors’ mechanisms of action.
Proposed endogenous ligands. The σ1 protein has been considered an orphan receptor for a long time because of the lack of an endogenous ligand. Nevertheless, some years ago, the endogenous hallucinogen compound N,N-dimethyltryptamine (DMT) showed to protect σ1 receptors by photolabeling performed using two radioactive photoaffinity labels: 3-[125I]iodo-4-azidococaine ([125I]-IACoc), more specific for σ1 receptors and 1-N-(2′,6′-dimethyl-morpholino)-3-(4-azido-3-[125I]iodo-phenyl)propane ([125I]IAF), with affinity for both σ receptor subtypes. DMT provided a dose-dependent protection when either [125I]-IACoc or [125I]IAF was used. Moreover, DMT showed to inhibit voltage-gated Na+ ion channels in both native cardiac myocytes and heterologous cells that express σ1 receptors. This pharmacological profile suggested DMT as an endogenous agonist for the σ1 receptor (Kd = 14.75 μM) [37]. In addition to DMT, Progesterone, which binds both σ subtypes (σ1 Ki = 239 nM, σ2 Ki = 441 nM [38]), has been also proposed as an endogenous σ1 receptor antagonist able to coordinate endocrine, immune, and CNS [38,39]. This is not surprising if we consider that σ1 receptors share homology with yeast sterol isomerase [24]. However, these ligands display low σ1 receptor affinity, and their blood concentration is too low for these ligands to be considered endogenously active. Plenty of other ligands can bind σ1 receptors with high affinity. Molecular dynamic simulations at the σ1 receptor binding pocket have shown how the diverse chemical structures to which high-affinity σ1 receptor ligands belong may easily be accommodated producing different associations among protomers that lead to the different activities observed.
Reference compounds. The most important classes of ligands are briefly summarized below (Table 1):
(+)-Benzomorphans as agonists: This class of ligands has a great historical value, as (+)-N-allylnormetazocine ((+)-SKF-10047) led to distinguishing a new group of receptors different from opioid receptors [40] that were named σ, after the first letter of the compound code [41]. Moreover, (+)-Pentazocine is still a σ1 reference compound, and its tritium radiolabeled form, which shed light on the existence of two classes of σ receptors [4], is still used for σ1 receptor radioligand binding assays.
Antipsychotics as antagonists: Haloperidol has high affinity for σ1 receptors (σ1 Ki = 8 nM [9]), but the involvement of the σ1 protein in schizophrenia has not completely been demonstrated. Several investigations on σ1 receptor gene polymorphs in this context are underway [42,43].
Antidepressants as agonists: Imipramine, Fluoxetine, Fluvoxamine, and Sertraline showed moderate to high affinities for σ1 receptors (Ki values ranging from 30 to 300 nM) [44], and several σ1 receptor agonists have shown antidepressant-like effects in animal models of depression, proving the involvement of the protein in depression [45,46].
Neurosteroids as agonists/antagonists: This class contains both agonists and antagonists. Progesterone in fact is a σ1 receptor antagonist, while deoxycorticosterone, testosterone, pregnenolone–sulfate, and dehydroepiandrosterone (DHEA) can produce agonist activities. All these ligands have unconventional structures because of the absence of a basic nitrogen that determines weak affinities for σ1 receptors. Neurosteroids have been proposed as endogenous ligands for σ1 receptors, as their effect seems also to be mediated by σ1 receptor binding [39,47].
- Others: A number of other ligands have been synthesized with the aim to better understand the role of these receptors in different pathologies. The most representative examples are as follows:
- -
- -
- -
- -
- -
-
-AC915: a highly selective ligand with an antagonist profile [62];
-
-Pridopidine: initially considered a dopaminergic D2 receptor antagonist, it was later repositioned as a selective σ1 receptor agonist (see Section 3.5) [63];
- -
- -
- -
Table 1.
Representative σ1 receptor (σ1 R) ligands and their affinities for σ1 R and σ2 receptor (σ2 R).
| Name | Structure | σ1 R Ki nM |
σ2 R Ki nM |
Reference for Binding Values |
|---|---|---|---|---|
| (+)-Benzomorphans: | ||||
| (+)-SKF-10047 |
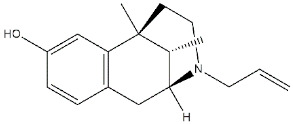
|
153 | 154335 | [9] |
| (+)-pentazocine |
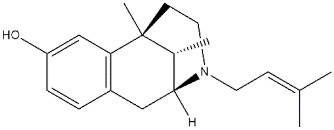
|
15.4 | 3475 | [9] |
| Antipsychotics: | ||||
| Haloperidol |
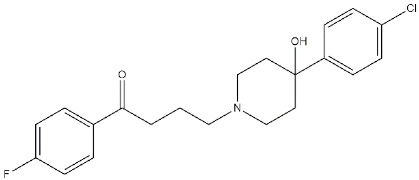
|
1.09 | 41.9 | (σ1 R) [71] (σ2 R) [9] |
| Antidepressants: | ||||
| Imipramine |
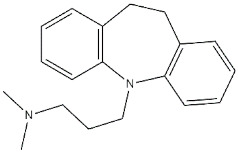
|
343 | 2107 | [44] |
| Fluoxetine |
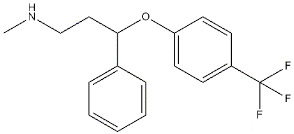
|
240 | 16100 | [44] |
| Fluvoxamine |
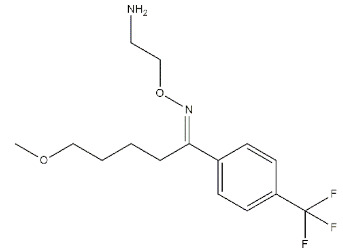
|
36 | 8439 | [44] |
| Sertraline |
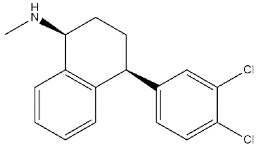
|
57 | 5297 | [44] |
| Neurosteroids: | ||||
| Progesterone |
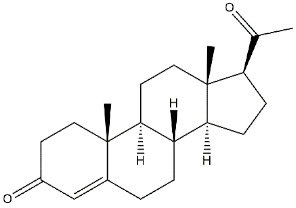
|
239 | 441 | [38] |
| Deoxycorticosterone |
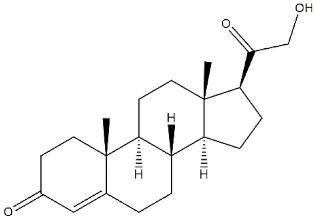
|
938 | n.d. | [39] |
| Testosterone |
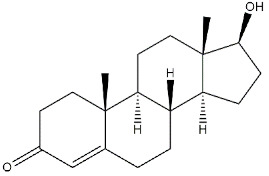
|
1014 | n.d. | [39] |
| Pregnenolone-sulphate |
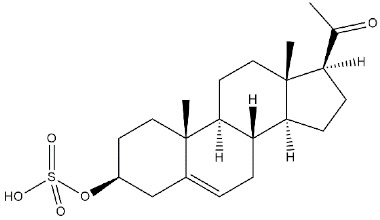
|
3196 | n.d. | [39] |
| DHEA |
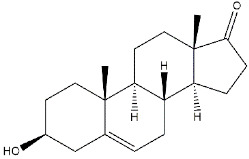
|
2.959 | n.d. | [47] |
| Others: | ||||
| SA4503 |
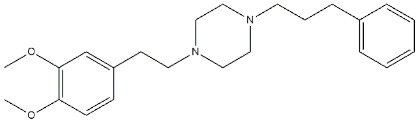
|
4.63 | 63.09 | [72] |
| PB190 |
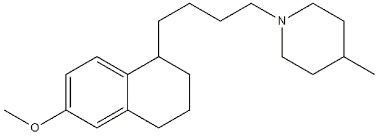
|
0.42 | 36.3 | [53] |
| PRE-084 |
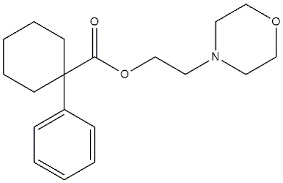
|
53.2 | 32.100 | [73] |
| NE-100 |
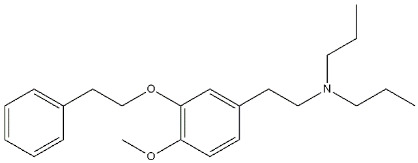
|
1.1 | 170 | [74] |
| PB212 |
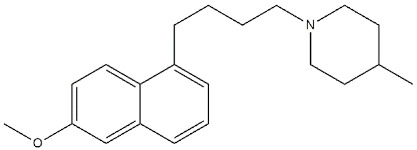
|
0.030 | 17.9 | [53] |
| AC915 |
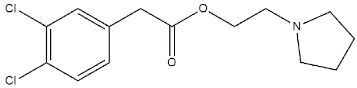
|
4.89 | >10000 | [62] |
| Pridopidine |
|
57 | 5450 | [75] |
| FTC-146 |
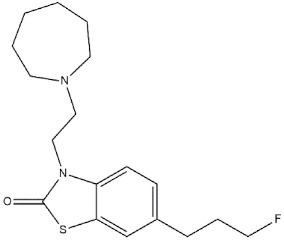
|
0.0025 | 364 | [64] |
| S1RA |
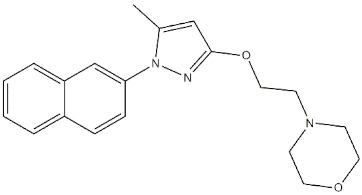
|
23.5 | >1000 | [76] |
| BD1063 |
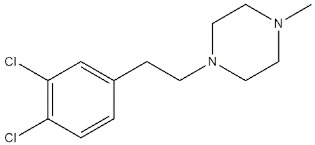
|
9.15 | 449 | [70] |
3. σ1 Receptor Ligands in a MTDLs Approach
In addition to the direct neuroprotective action of σ1 receptors, the additional modulation of targets such as opioid, N-methyl-D-aspartate (NMDA), dopaminergic, and cholinergic receptors renders the σ1 protein an intriguing target for the modulation of processes that involve these systems, prompting the development of σ1-based MTDLs for the treatment of complex and multifactorial pathologies such as AD.
3.1. MTDLs Acting at σ1 Receptor and Cholinergic System
Drug metabolism leads to metabolites that can be more potent or less potent than the parent drug. In the former case, the drug can be considered a sort of prodrug. Thus, the simultaneous presence of the two chemical entities may produce a synergistic powerful effect.
Starting from this idea, in 2010, Lecanu et al. developed pairs of drugs and the corresponding prodrugs, which act on different targets such as acetylcholinesterase (AChE) whose inhibition is a consolidated therapeutic strategy in AD, and σ1 receptors. With this aim, two series of molecules were built: hydroxyphenyl-butan-1-ones (drug) and their corresponding carbamates (prodrugs). Upon inhibition of the AChE, the prodrug releases the drug. Thus, the prodrug and drug bind and act through the σ1 receptor and AChE, while the drug exerts its antioxidant action.
Among the studied ligands, two compounds (i.e., 2,3,4-trihydroxy-phenyl)butan-1-one 1 and the corresponding N,N-dimethylcarbamoyloxy prodrug 2; Figure 1) showed the best compromise in terms of activity on the different targets. Compound 2 was able to (i) inhibit AChE with a potency comparable to reference compounds; (ii) bind σ1 receptors (high selectivity, sub-micromolar affinity, and antagonist activity); and (iii) prevent mitochondrial toxicity by inhibiting mitochondrial complex IV and V. On the other hand, compound 1, even if much less active than its bio-precursor toward σ1 receptors, was also able to attenuate reactive oxygen species (ROS), Aβ1-42-induced neurotoxicity, and mitochondrial toxicity by inhibiting complexes I, II, IV, and V.
Figure 1.

Compound (1) and (2) and their Ki and IC50 values for σ1 receptor (σ1 R) and human acetylcholinesterase (hAChE) [77].
Moreover, 2 was demonstrated to cross the blood–brain barrier (BBB) very shortly after its administration and be transformed into 1 very quickly.
Behavioral studies on animal models are needed, but preliminary data suggest that this multi-target profile can represent a novel therapeutic strategy to treat AD [77].
Many pharmacological and genetic data have proven that activation of muscarinic M1 receptors (mAChRs) attenuates symptoms of neurodegenerative pathologies [78,79], as in the case of MTDLs able to bind both M1 and σ1 receptors. The most investigated compound representative of this class is ANAVEX 2-73 (also known as Blarcamesine) (Figure 2). This compound is in advanced clinical phases for several CNS diseases such as AD, Parkinson’s disease (PD), Rett, and Fragile X Syndromes (anavex.com/#!/pipeline, accessed Apr 3, 2021). In addition to binding M1 and σ1 receptors, Blarcamesine also binds M2–M4 receptors (with micromolar affinity), Na+ channel site 2, and NMDA receptor (NMDAR) [80,81]. In this context, Fisher and co-workers, who previously developed orthosteric M1 receptors agonists (e.g., AF102B, AF267B, and AF292), extended their approach in order to target also σ1 receptors. With this aim, compound AF710B (also known as ANAVEX 3-71, Figure 2) that can activate both M1 and σ1 receptors with high potency and selectivity was identified [66]. AF710B is a positive allosteric modulator (PAM) of M1 receptor, as it improves the efficacy of carbachol, and this activity, together with a comparable agonism at the σ1 receptor can preserve synaptic elements in vitro. In vivo studies performed on trihexyphenidyl-treated rats and 3xTg-AD mice showed that AF710B can restore cognitive deficits and attenuate signs of AD phenotypes by the reduction of β-secretase 1 (BACE1) levels, GSK3β and CDK5/p25 activity (which contribute to the hyperphosphorylation of tau protein), neuroinflammation, soluble and insoluble Aβ40 and Aβ42 plaques, and tau pathology [82]. In fact, AF710B reduces the expression of the putative BACE1, so that proteolytic fragments produced by β-secretase were considerably lower in 3xTg-AD treated than in untreated mice. Moreover, tau kinases GSK3β and CDK5 take part in the mechanism of hyperphosphorylation of tau protein. In particular, in AD patients, CDK5 activator p35 is cleaved to produce the protein p25, which binds with high affinity and activates GSK3β [83]. The activation of M1 receptors produces a reduction of GSK3β expression, while the activation of σ1 prevents the formation of CDK5/p25. Therefore, the inhibition of GSK3β and CDK/p25 by AF710B, upon interaction with both M1 and σ1 receptors, results as a promising approach in the treatment of tauopathies [82].
Figure 2.

ANAVEX 3-71, 2-73 and 1-41 structures and their Ki and IC50 values for σ1 receptor (σ1 R) and muscarinic M1 receptor (M1 R) [82,84,85].
The effect of σ1 receptors in inflammation through microglia modulation has been reported [54,86,87], and AF710B was shown to reduce reactive astrocytes and activated microglia in the animals, as detected by the low levels of glial fibrillary acidic protein (GFAP) and ionized calcium-binding adapter molecule 1 (Iba-1). Notably, astrocytes and microglia are increased in number and size in AD patients.
Another study performed using AF710B on McGill-R-Thyl-APP transgenic (tg) rats revealed that this MTDL can reduce amyloid pathology and markers of neuroinflammation while increasing amyloid cerebrospinal fluid clearance and synaptic marker. The most important achievement is represented by the prolonged duration of these effects, which are maintained five weeks after the treatment is interrupted [88].
In addition to AF710B and ANAVEX 2-73, Anavex Life Sciences Corp portfolio comprehends an isomer of ANAVEX 2-73, named ANAVEX 1-41 (Figure 2) that next to the activity toward σ1 and M1 receptors, also displays activity for α1, 5-HT2, and D3 receptors [81], with an indication for the treatment of depression, stroke, and neurodegenerative diseases (anavex.com/#!/pipeline).
3.2. MTDLs Acting at σ1, 5-HT4 Receptors and AChE
In 2015, Rochais and co-workers developed the MTDL Donecopride from the combination of the AChE inhibitor Donepezil and the serotoninergic 5-HT4 receptor (5-HT4R) agonist RS67333 (Figure 3). Indeed, Donecopride inhibits AChE and exerts a partial 5-HT4R agonist activity on 5-HT4R.
Figure 3.

Donepezil, RS67333, and Donecopride structures and their Ki and IC50 values for σ receptors (σ R), σ1 receptor (σ1 R), human acetylcholinesterase (hAChE) and serotoninergic 5-HT4 receptor (5-HT4 R) [89,90,91].
Subsequent studies revealed that Donecopride has high σ1 receptor affinity (Ki = 51 nM [90]), which is in agreement with the nanomolar affinity of Donepezil for the same target [91].
This finding inspired many studies about the possible therapeutic exploitation of such combinations of activities [92,93,94,95]. Starting from these pieces of evidence, Lalut et al. postulated that the replacement of the benzene ring by an indole group, together with the insertion of a piperidine-chained spacer, could improve affinity for the σ1 receptor. Thus, N-substituted and N-unsubstituted indoles were developed, with diverse decoration on the indole and piperidine rings.
The N-unsubstituted indoles displayed a profile comparable to their lead compound in terms of affinity at 5-HT4 receptors and inhibitory activity at AChE. The most promising ligands were tested in vitro on Jukart cell membranes for the determination of the σ1 affinity, according to Ganapathy [96].
The N-benzyl derivative 3 (Figure 4) displayed the highest affinity for the σ1 receptor and the most potent inhibition toward AChE but a moderate affinity for 5-HT4 receptors. The best compromise among affinities at σ1 and 5-HT4 receptors and activity at AChE was reached by the cyclohexylmethyl derivative 4 (Figure 4), which was thus considered the lead compound of the study.
Figure 4.

Compounds (3) and (4) structures and their Ki and IC50 values σ1 receptor (σ1 R), serotoninergic 5-HT4 receptor (5-HT4 R) and human acetylcholinesterase (hAChE) [90].
This compound was tested in vivo using the dizocilpine-induced amnesia assay in mice. In the 0.1–1 mg/kg dose-range, 4 did not attenuate the dizocilpine-induced spontaneous alternation performance deficit, while at lower doses, the passive avoidance deficit was attenuated with a 40% protection. The authors concluded that the involvement of the σ1 receptor needed further explorations by including challenges with the σ1 receptor antagonist NE-100 [90].
3.3. MTDLs Acting at σ1 Receptor, AChE, BuChE, BACE1, MAO-A, MAO-B, 5-LOX, ROS, and Stem Cells
Another important class of MTDLs able to treat AD is represented by ligands developed by Estrada and co-workers in 2016. Their strategy was based on the synthesis of MTDLs able to inhibit AChE/Butyrylcholinesterase (BuChE), BACE1, and bind σ1 receptors.
For the study, the authors combined lipoic acid (LA), whose antioxidant properties [97] are exploited in AD [98], together with either the N-benzylpiperidine (NBP) moiety (featured by Donepezil, as discussed above) or the N,N-dibenzyl-N-methylamine (DBMA) moiety of the BACE1 inhibitor AP2238. This approach led to the MTDLs LA-NBPs and LA-DBMA, respectively.
More than fourteen ligands were developed, and some of them were tested for σ receptors affinity. Ki values in the sub-micromolar and sub-nanomolar range were found for σ1 receptors, and radical scavenging properties were evaluated using the Oxygen Radical Absorbance Capacity (ORAC) assay, with LA-based ligands showing good antioxidant capacities, comparable to vitamin E.
In the LA-NBP class, the lowest Ki value at the σ1 receptor was displayed by ligand (R)-5; in the LA-DBMA series, the best σ1 receptor affinity values were displayed by enantiomeric mixtures of compounds 6 and 7, which differ for the position of the amidic substituent on the central aromatic ring (3-position for compound 6 and 4-position for compound 7) (Figure 5). Both compounds showed Ki values in the nanomolar range, with no enantioselectivity.
Figure 5.

Compounds (5,6) and (7) structures and pharmacological profile for σ receptor (σ1 R and σ2 R), human acetylcholinesterase (hAChE), human butyrylcholinesterase (hBuChE), β-secretase 1 (BACE1), Oxygen Radical Absorbance Capacity (ORAC) and parallel artificial membrane assays (PAMPA-BBB) [99].
In addition, compounds 5 and 7 showed promising in vitro absorption, distribution, metabolism, excretion and toxicity (ADMET) properties, as they are able to cross the BBB by in vitro parallel artificial membrane assays (PAMPA-BBB). They also displayed low or moderate cytotoxicity in the SH-SY5Y neuroblastoma cell line (supporting their s1 agonist or partial agonist rather than antagonist activity) [99].
In order to develop a more holistic strategy to hit AD, the same group designed novel MTDLs to target monoamine oxidase A (MAO-A), monoamine oxidase B (MAO-B), lipoxygenase-5 (5-LOX), AChE/BuChE, and σ1 receptors. Additionally, ROS attenuation and mechanisms of differentiation of neural stem cells were taken into account for these novel compounds. Thus, twenty-nine new Donepezil-flavonoid hybrids (DFHs) were synthesized. The N-benzylpiperidine fragment was connected through an amide or ester linker to the 4-oxo-4H-chromene flavonoid scaffold, which is endowed with MAO-A, MAO-B, and 5-LOX inhibitory activity, as well as antioxidant, anti-inflammatory, and neurogenic properties. Moreover, the flavonoid scaffold underwent some modifications such as substitution of the 4-oxo-4H-chromene with a quinoline-4(1H)-one or quinoline and the introduction of hydroxyl/methoxy groups on the aromatic ring.
Three classes of compounds (i.e., 4-chromenone, 4-quinolone, and quinoline-2-carboxamide) were tested for their activity at AChE/BuChE, 5-LOX, and MAOs as well as for their BBB permeability and antioxidant capacities. A restricted group of ligands belonging to the 4-chromenone and 4-quinolone classes (general structure in Figure 6) was selected and tested at σ receptors, showing low nanomolar Ki values (0.98 nM > Ki > 47.4 nM) and selectivity versus the σ2 subtype. The best affinities were obtained when a single hydrogen-bond acceptor group (NO2 > NH2 > OH > OCH3) was in the 6-position of either the 4-oxo-chromene or 4-oxo-quinoline system, while the length of the linker between the amide and the N-benzylpiperidine groups had no influence on the affinity for σ1 receptors.
Figure 6.

General structures of 4-chromenone and 4-quinolone classes of ligands.
The best compromise among all studied compounds is represented by compound 8 (R = 6,7-diOMe; Z = O; X = NH and n = 2, Figure 6), which showed good neuroprotective properties: hAChE IC50 = 46 nM; hBuChE IC50 >10000 nM; PAMPA-BBB = 5.9 × 10−6 cm s−1; 5-LOX IC50 = 74.3 µM; MAO-A IC50 = 15.3 µM; MAO-B IC50 = 5.2 µM; ORAC = n.a.; σ1 R Ki = 37.4 nM; σ2 R Ki = 239 nM.
Moreover, compound 8 was shown to promote the differentiation of adult mice neural stem cells (NSC), and its predicted metabolites have been analyzed using the Derek Nexus program and shown to be not toxic. Additionally, docking by Molecular Dynamics (MD) simulation into AChE, 5-LOX, and the σ1 receptor showed low ΔGbind values in agreement with the experimentally detected interactions with the three targets. Based on these data, compound 8 was proposed as a promising candidate against AD and neurodegenerative diseases in general [100].
Based on these encouraging data, the same group developed other MTDLs targeting AChE/BuChE, BACE1, MAOs, 5-LOX, the differentiation of neural stem cells, and σ1 receptors. With this aim, the already exploited flavonoid cores (i.e., 4-chromenone or 4-quinolone) were conjugated with the N,N-dibenzyl-N-methylamine (DBMA) fragment, which was chosen for its proven interaction with the catalytic anionic site (CAS) of AChE. The two classes of 4-chromenone and 4-quinolone derivatives were tested, and the best compromise was obtained with ligands belonging to the former class. The most promising ligands (i.e., 9, 10, 11, and 12, Figure 7) were also tested at σ1 and σ2 receptors, showing comparable sub-micromolar affinities for σ1 and micromolar affinities for σ2 receptors. These results showed that functionalization in 3′ or 4′ of the central benzene ring as well as changes in the position of the methoxy/hydroxy groups on the 4-chromenone scaffold poorly influence the σ receptors’ affinity.
Figure 7.

General structure of 4-chromenone-N,N-dibenzyl-N-methylamine (DBMA) derivatives.
Despite its low affinity and selectivity for σ1 receptors (σ1 R Ki = 0.53 µM; σ2 R Ki = 1.30 µM), compound 12 represented the best compromise in terms of activity toward different targets (hAChE IC50 = 4.5 µM; hMAO-A IC50 > 100 µM; hMAO-B IC50 > 100 µM; 5-LOX IC50 = 30.4 µM; and PAMPA-BBB = 18.8 × 10−6 cm s−1). ΔGbind values obtained through docking to AChE, 5-LOX, BACE1, and σ1 were in agreement with the experimental ones, while a relatively safe profile was predicted in terms of toxicity. However, a low-confidence alert (below 67%) for human ether-a-go-go-related gene (hERG) channel inhibition was recorded, so that cardiac toxicity might be expected with this compound, which needs to be ascertained with further experimental studies. Neurogenic evaluation was performed on a primary culture of neural stem cells from the SGZ of adult rats, and compound 12 stimulated differentiation. Altogether, these data suggest compound 12 as a possible therapeutic agent that promotes brain auto-repair processes and blocks early steps of neurodegenerative cascades [101].
3.4. MTDLs Acting at σ1, NMDA Receptors, and VGCC
NMDA receptors are glutamate-gated Ca2+ permeable ion channels associated with σ1 receptors. In 1987, Quirion et al. identified the σ1 receptor as the phencyclidine (PCP) binding site [102], but further studies performed by the same group refuted the thesis and classified σ receptors as a separate class [103]. However, a strong connection between these two receptors has been found in the following years. σ1 Receptors can enhance Ca2+ influx via NMDAR, but they can also prevent toxicity induced by the excessive flux of this cation. This activity occurs through the modulation of NMDAR-associated intracellular signaling component, i.e., the neuronal Nitric Oxide Synthase (nNOS), which produces nitric oxide (NO) [104].
Low doses (100 nM) of σ1 receptor agonists enhance NMDAR function, producing a potentiation of the NMDAR response. Curiously, high doses (≥10 μM) do not exert a stronger effect. Indeed, a bell-shaped dose–response trend, which is typical of the σ1 receptor agonists-mediated action, can be observed accounting for the majority of the effects of σ1 receptor agonists on NMDAR response [105,106]. A potential explanation of this effect is that σ1 receptor agonists can directly bind NMDARs and block channel conductance [107], but further studies are needed to solve the question. Moreover, the NMDAR increased activity is fast and prolonged [108], so that interactions also with other targets may be involved [104]. Even if these mechanisms have not been completely clarified, an interaction between the σ1 receptor and NMDAR exists. In particular, as recently reviewed, σ1 receptors can associate together with GluN1, Glun2a, and Glun2b subunits [17].
Taking into consideration all these aspects, dual drugs acting on σ1 and NMDA receptors may be a promising strategy to target neurodegenerative disorders.
With the aim to discover potentialities of the aza-cage compound of the pentacyclo[5.4.0.02,6.03,10.05,9]undecane ring system (PCU), Van der Schyf et al. engaged physiological studies using sheep Purkinje fibers. These studies classified NGP1-01 (Figure 8) as the lead compound of a novel class of Ca2+ channel antagonist having a “cage” molecular structure [109].
Figure 8.

NGP-01, ANSTO-1, ANSTO-6, ANSTO-14, and ANSTO-5 structures and their Ki values for σ receptor (σ1 R and σ2 R) and N-methyl-D-aspartate receptor (NMDAR) [110,111,112].
Subsequent studies demonstrated the multi-target profile of NGP1-01 evidenced by its ability to reduce Ca2+ influx through interaction with L-type voltage-gated calcium channel (VGCC) [113] and NMDAR, with an activity comparable to the NMDAR antagonist memantine [114].
The discovery that amantadine had micromolar Ki values for σ receptors [115] led to investigate whether cage compounds could bind σ receptors. The micromolar affinity for σ1 receptors of NGP1-01 inspired the synthesis of new trishomocubanes pentacyclo[5.4.0.02,6.03,10.05,9]undecylamines and 4-azahexacyclo[5.4.1.02,6.03,10.05,9.08.11]dodecanes, such as ANSTO-1 and ANSTO-6 (Figure 8). These compounds showed good affinity for σ receptors but low affinity for NMDAR (i.e., ANSTO-1 and ANSTO-6, Figure 9) [110,111].
Figure 9.

Amantadine and memantine structures and their Ki values for σ1 receptor (σ1 R) and the phencyclidine binding site at the N-methyl-D-aspartate receptor (PCP) [115].
The subsequent SAR, 3D-QSAR, and docking studies clarified the following: (i) aza-trishomocubane derivatives have high affinity for both σ receptors; (ii) affinity for σ1 receptors is higher when the chain is longer; (iii) para- or meta-substitution with halogens is well tolerated, but the absence of substituents produces the highest selectivity and the lowest Ki value for σ1 receptors (ANSTO-14, ANSTO-5, and ANSTO-7, Figure 8) [103,104].
Nevertheless, the improved σ1 receptor affinity was accompanied by a dramatic loss in the affinity for NMDARs. Consequently, NGP1-01 can still be considered the best MTDL in the class of the cage compounds.
Deeper studies able to clarify the mechanism of the σ1 receptors-mediated antineurodegenerative activity of NGP1-01 are not available, to the best of our knowledge, but the affinity of the compound for the σ1 receptor and the antineurodegenerative properties mediated by the reduction of Ca2+ influx have been demonstrated, qualifying NGP1-01 as a potential MTDL that is useful in the treatment of neurodegenerative pathologies.
Among cage compounds, amantadine and memantine (Figure 9) deserve to be mentioned because of their well-known antagonistic activity at the phencyclidine (PCP) binding site of NMDARs [116] together with the already mentioned affinity for σ1 receptors [115]. However, while the affinity of memantine at NMDARs is around 40-fold higher than the affinity at σ1, amantadine interacts to the same extent with σ1 and NMDARs. Additionally, Peeters et al. confirmed the agonist σ1 receptor profile and shed the light on amantadine ability to positively increase dopaminergic transmission through the activation of σ1 receptors [117]. Therefore, amantadine can be considered a full-fledged MTDL.
3.5. MTDLs Acting at σ1 Receptor/Dopaminergic Transmission
σ1 Receptors are also associated with the dopaminergic transmission. BRET, coimmunoprecipitation (Co-IP), and proximity ligation assays studies demonstrated that σ1 receptors interact with dopamine (DA) D1 and D2 receptors, as well as with dopamine transporter (DAT) [17]. Beneficial effects of the σ1 chaperone function on dopaminergic transmission may be exploited in pathologies such as AD, PD, Huntington’s disease (HD), and schizophrenia.
Interactions with D1 receptors were studied by Moreno et al. that performed in vitro and ex vivo assays, which led to identify, in the presence of cocaine, the formation of a heterotrimer composed of σ1, histamine H3, and D1 receptors. Upon the formation of this trimer, the inhibition of D1 receptors was abolished, and Gs proteins were activated with an increase of cAMP, recruitment of β-arrestin, and increase in p-ERK1/2 levels [118]. As a result, σ1 antagonists have the potential to reduce cocaine effects and consequent cocaine addiction.
σ1 Receptors can also form heteromers with the D2 receptor. Biophysical, biochemical, and cell biology assays performed on mouse striatum demonstrated that the administration of cocaine can induce the formation of σ1–D2 dimers, inhibiting the downstream signaling [119].
Finally, σ1 receptors are also able to associate with DAT and reduce DAT-mediated dopamine efflux produced by methamphetamine exposure [120].
A large number of dopaminergic agents have been examined to better define their pharmacological profiles, and some of them were found to bind σ1 receptors, in accordance with a partially overlapping pharmacophore.
For a long time, Pridopidine (Figure 10) has been considered a dopaminergic antagonist because of its micromolar affinity for D2 receptors, but some in vivo data and its structural resemblance to the σ1 receptor ligands inspired Sahlholm and co-workers to perform studies that defined Pridopidine as a highly selective σ1 receptor agonist [63,121].
Figure 10.

Pridopidine structure and its Ki values for σ (σ1 R and σ2 R), dopaminergic D2 (D2 R), alpha-2c adrenergic (α2c R) and serotoninergic 5-HT1a (5-HT1a R) receptors [75].
To better understand the neuroprotective and neurorestorative role of Pridopidine, Francardo et al. assayed the drug in a mouse model of Parkinson’s disease (unilateral 6-hydroxydopamine (6-OHDA) lesion). After 5 weeks of daily administration of a low dose (0.3 mg/kg), Pridopidine produced an improvement in forelimb use and abolished the ipsilateral rotational bias typical of hemiparkinsonian animals. Moreover, the protection of nigral cell bodies, increased dopaminergic fiber density in the striatum, striatal upregulation of glial cell line-derived neurotrophic factor (GDNF), and brain-derived neurotrophic factor (BDNF) and phosphorylated extracellular signal-regulated kinases 1/2 (ERK1/2) were also observed [122]. At higher doses, Pridopidine was able to interact with other targets such as alpha-2 adrenergic α2C and serotonin 5-HT1A receptors, providing a solution to reduce L-dopa-induced dyskinesia (LID), both in PD and HD [75]. Of note, a phase III trial is in progress to evaluate the efficacy and safety of Pridopidine in HD patients (PRidopidine’s Outcome on Function in Huntington Disease, PROOF-HD, ClinicalTrials.gov Identifier: NCT04556656). Importantly, promising data of Pridopidine for the treatment of AD are emerging as the compound prevents mushroom spine loss in hippocampal cultures from APP knock-in (APP-KI) and presenilin-1-M146 V knock-in (PS1-KI) mice [123].
The ability to functionally interact with different receptors identifies Pridopidine as a MTDL, which is useful in the treatment of neurodegenerative disease.
Afobazole (Figure 11) is an anxiolytic drug designed and pharmacologically studied by FSBI “Research Zakusov Institute of Pharmacology”, Russia. This drug binds melatonin MT1 (Ki = 16 µM) and MT3 (Ki = 0.97 µM) receptors. MT3 is a regulatory site of quinone reductase 2 enzymes (NQO2), which is responsible for ROS production. For this reason, the reversible inhibition performed by Afobazole produces a cytoprotective effect.
Figure 11.

Afobazole and M-11 structures and their Ki and IC50 values for σ1 receptor (σ1 R), melatonin MT1 and MT3 receptors (MT1 R and MT3 R) and monoamine oxidase A (MAO-A), [124].
Some studies performed on Afobazole metabolites revealed that one of them, M-11, can bind the MT3 receptor (Ki = 0.39 µM) with a slightly higher affinity, while a lower affinity is associated to the MT1 receptor (Ki = 44 µM). Consequently, Afobazole activity can be considered empowered by the additive activity of its metabolite M-11 (Figure 11) [124].
Afobazole can also inhibit MAO-A, thus increasing adrenaline, noradrenaline, and dopamine levels in CNS. Additionally, it also behaves as a σ1 receptor regulator, producing a cytoprotective effect through the inhibition of ROS production [125].
The need to clarify the involvement of σ1 receptors in neurodegenerative diseases inspired some studies on the 6-OHDA-induced parkinsonism model in mice. The study revealed that the administration of Afobazole (2.5 mg/kg, i.p.) over 14 days was able to restore motor dysfunction as well as prevent decreases in dopamine in the 6-OHDA-lesioned striatum and the loss of tyrosine hydroxylase positive (TH+) neurons in the substantia nigra. Importantly, these activities were abolished by the administration of the σ1 receptor antagonist BD-1047 (3.0 mg/kg, i.p.), demonstrating that the effects of Afobazole are mediated by the σ1 receptor [126].
These pieces of evidence prove a strict connection between the dopaminergic transmission and the σ1 receptor and encourage more in-depth investigation to clarify the mechanisms involved.
With high affinity and antagonist properties at D2 receptors, Haloperidol (Figure 12) equally binds the σ1 receptor, behaving as an antagonist [127].
Figure 12.

Haloperidol and its metabolites with Ki values for σ1 and σ2 receptors (σ1 R and σ2 R) [127,128,129].
After administration, Haloperidol undergoes a complex metabolism that leads to toxic metabolites such as 4-(4-chlorophenyl)-1-[4-(4-fluorophenyl)-4-oxybutyl]pyridinium ion (HPP+) and 4-(4-(chlorophenyl)-1–4-(fluorophenyl)-4-hydroxybutyl-pyridinium (RHPP+) (Figure 12). In addition, also non-toxic metabolites have been observed (HP metabolites), such as HP metabolite II [130] and HP metabolites I and III [131] (Figure 12). Biological activities of the non-toxic HP metabolites (I, II, and III) were studied, and in particular, their affinities for σ1 receptors were evaluated. While HP metabolite III does not bind σ1 receptors, HP metabolite I exerts a reversible inhibition, and HP metabolite II exerts an irreversible inhibition at the σ1 receptor. Thus, haloperidol metabolites can reinforce its action [128].
Further studies are needed to better understand the exploitability of this pharmacological activity, but preliminary data suggest that σ1 receptor antagonists, such as Haloperidol, could be useful in blocking the acute toxicity and the rewarding effects of cocaine because of an influence on the DA release (pre-synaptic activity) and reduction of Ca2+ mobilization produced by the activation of D1 receptors (post-synaptic activity) [132]. The additional activity of Haloperidol and its active metabolites could be foreseen in the antinociceptive action occurring through the modulation of opioid receptors [133].
Moreover, studies performed by Marrazzo and colleagues revealed that HP metabolite II is also able to exert anticancer activity because of its antagonism at the σ1 receptor and agonism at the σ2 receptor [134]. A more detailed discussion of HP metabolite II anticancer potentialities is reported in Section 3.7.
3.6. MTDLs Acting at σ1 and μ-Opioid Receptors
One of the most difficult challenges in medicinal chemistry is represented by the synthesis of drugs able to manage pain for a prolonged period. In particular, chronic pain therapies are often associated with the adaptation phenomenon that is responsible for most of the side effects of these drugs, because it leads to periodic increases of the doses to be administered in order to keep the analgesia. Opioids, with their generally low therapeutic index, are still the most effective class of drugs able to relieve pain, making the need to increase the dose extremely life-threatening.
With this in mind, Garcia et al. developed a heterogeneous class of ligands with the aim to reinforce the antinociceptive activity mediated by μ-opioid receptor (MOR) agonists with an antagonist activity at the σ1 receptor that has a well-established role in the treatment of pain. Some pieces of evidence have demonstrated that the antagonist S1RA [124,127] is able to potentiate morphine antinociceptive activity without increasing side effects, so that it entered clinical trials for treating different kinds of pain [135,136].
These premises paved the way to the development of a pharmacophore model obtained by overlapping the MOR pharmacophore generated with the “Auto Pharmacophore generation” protocol (that enumerates all the pharmacophores compatible with an input molecule considering hydrogen bond acceptors and donors, hydrophobic portions, ionizable groups, and aromatic rings) within the Discovery Studio [137] with the σ1 pharmacophoric model developed by Langer [138]. This model was based on the presence of two hydrogen bond acceptors (HBA1 and HBA2), three hydrophobic portions (HYD1, HYD2, and HYD34, obtained by the fusion of two different hydrophobic portions HBA3 and HBA4 of Laggner’s pharmacophore), an aromatic ring (AR) and two positive ionizable domains (PIs).
Based on this model, several compounds were synthesized, and the 4-aryl-1-oxa-4,9-diazaspiro[5.5]undecane derivative 13 (Figure 13) provided the best results on both targets.
Figure 13.

Compounds (13–16) and their Ki and IC50 values for σ1 (σ1 R), μ-opioid (MOR) and alpha-1 adrenergic (α1AR) receptors and human ether-a-go-go-related gene channel (hERG) [139,140].
Starting from compound 13, SAR studies were performed to optimize the structure. In particular, the 2-position on the spiro[5.5]undecane was explored through the introduction of small substituents such as methyl groups (compound (R)-14, Figure 13), which increased the activity mediated by MOR and σ1 receptors, while reducing the activity at the hERG channel, which was responsible for its cardiac toxicity. Moreover, pure enantiomers were tested demonstrating that (R)-14 is 4-fold less active toward hERG and displays a 1-order of magnitude higher affinity for MOR than its (S)-counterpart. Elongation of the same alkyl group from methyl to ethyl or isopropyl produced an increase in the activity toward MOR but decreased the activity toward σ1 receptors, thus spoiling the beneficial synergy. Substitution with more polar groups produced compounds characterized by a lower affinity than compound (R)-14, but the activity at the two targets reached a more balanced compromise. Nevertheless, the reduction in lipophilicity did not improve the profile at hERG, which remained quite similar to the inhibition exerted by the more hydrophobic compound (R)-14.
Another structural exploration was conducted by introducing substituents on the phenyl ring on the 4-position of the 1-oxa-4,9-diazaspiro[5.5]undecane system. In particular, halogens and trifluoromethyl groups in ortho positions provided the best results toward the two targets, which was probably because of an increase in steric bulk. On the other hand, no improvement toward hERG inhibition was observed. Position 4 was also explored by introducing heteroaryl groups, and the best results were obtained when 2- or 3-pyridyl systems were inserted. As for the 9-position on the 1-oxa-4,9-diazaspiro[5.5]undecane system, the best results were obtained with the same groups introduced in position 4.
Among all of these compounds, 15 (Figure 13) resulted as the best compromise because of its activity toward the two targets and appreciable selectivity, with the most important achievement represented by the absence of hERG inhibition.
The promising pharmacodynamic profile, together with good solubility, moderate basicity and lipophilicity, and in vitro metabolic stability of compound 15 led the group to study its antinociceptive properties in vivo. Results from the preclinical model, based on the mechanical pressure test on a hindpaw, demonstrated a dose-dependent antinociceptive effect with an ED50 of 15 mg/kg after i.p. administration. Moreover, local administration of the compound provided promising results [139]. Further optimization of the compound was conducted with the production of new dual ligands with enhanced activity. Replacement of the aromatic ring in the 4-position of the 1-oxa-4,9-diazaspiro[5.5]undecane system with an ethyl group provided encouraging data. In fact, small alkyl groups produced a slightly reduced affinity toward MOR and σ1 receptors but considerably improved the profile toward alpha-1 adrenergic receptor (α1AR) and hERG, which was likely because of a reduced lipophilicity. As already shown by structural changes leading to compound 14, different small alkyl substituents were well tolerated at the 2-position (spirocyclopropyl, ethyl, and (R)-methyl groups), improving the affinity toward the two targets and the hERG profile. These optimization studies led to compound 16 (Figure 13), which showed good physicochemical properties, high BBB permeability, and no cytotoxic activity in the hepatocarcinoma HepG2 cells. For these reasons, compound 16 was successfully tested in the paw pressure test and in the partial sciatic nerve ligation (PSNL) model in CD1 male mice. Thus, compound 16 was selected as a Phase 1 clinical candidate in the treatment of pain [140].
3.7. σ1. Receptor and Cancer in a MTDLs Approach
The σ1 receptor is a well-established target in the neurodegenerative field. However, several cancer cell lines (e.g., lung: H69, H2019, H510; breast: MDA-MB-361, MDA-MB-435, BT20, MCF-7, T47D, MDA-MB-231, MDA-MB-468, SKBR3, BT474; prostate: PC3, LNCaP, LAPC4, C4-2, 22Rv1, VCaP, PC3, DU145; human esophageal squamous cell carcinoma: KYSE150, KYSE180, EC109; pancreas: Panc1; liver: HepG2; neuroblastoma: SK-N-BE(2)C) show high levels of σ1 proteins or SIGMAR1 transcripts. While the receptor is not an oncogene [141], σ1 receptor ligands with claimed antagonist activity exert antiproliferative and growth-inhibiting effects [142].
The involvement of the σ1 receptor in cancer prompted Riganas et al. to develop (1-adamantyl)diarylalkylamines as σ1 receptor ligands whose scaffold was based on two aromatic rings and a hydrophobic adamantane moiety, which were linked to an amine site through alkyl chains of different length. The activity of these ligands was also tested on Na+ channels, whose involvement in cancer has been proven [141,143]. Compounds from the butyl series displayed the best profile with good σ1 receptor antagonist and weak σ2 receptor agonist activities, while all the other compounds showed a lower selectivity for σ1 vs. σ2. Moreover, all of them showed from micromolar to sub-micromolar affinities for the site 2 of Na+ channels. Compound 17 (Figure 14) emerged for its cytotoxic effect in ovarian cancer cells (IGROV-1) and in vitro antiangiogenic activity in normal cell lines such as Human Umbilical Vein Endothelial Cells (HUVEC).
Figure 14.

Compound (17) structure and its IC50 values for σ1 receptor (σ1 R), σ2 receptor (σ2 R) and Na+ channel [144].
Compound 17 was characterized by high-affinity interaction with the σ2 receptor as agonist (i.e., inducer of cytotoxicity) and with the σ1 receptor as antagonist, representing the optimal compromise if anticancer agents based on σ receptors-mediated action want to be produced. Additionally, 17 was associated with an analgesic effect against neuropathic pain, which was probably due to the block of Na+ channels, thus highlighting the therapeutic potentials of the combination of these actions in one molecule [145].
Another ligand conveniently endowed with antagonism at σ1 receptors and agonism at σ2 receptors is PB28, which has been found as a potential anti SARS-CoV-2 agent, and for this reason, researchers have recently reviewed its structure–affinity/activity relationships [144,146]. This compound emerged from structure–affinity relationship (SAfiR) studies that clearly indicated how N-alkyl-piperazine connected through a methylene linker to a hydrophobic nucleus (i.e., tetralin), rather than the corresponding N-Aryl ones, addressed the σ receptors affinity, and weakened the interaction with dopamine D2 and serotonin 5-HT1A receptors [71,147]. With its sub-nanomolar affinity toward both σ receptor subtypes, and the combination of functional activities (σ1 antagonism and σ2 agonism), PB28 was tested in several cancer cells (e.g., breast, neuroblastoma, pancreas tumor cell lines) [144]. Moreover, studies performed in pancreatic cancer cell lines (i.e., Panc02, KP02, KCKO, MIA PaCa-2, BxPC-1, and Panc-1) highlighted the increase in ROS production, lysosomal membrane permeabilization (LMP), and mitochondrial superoxide production as PB28 mechanisms of action that lead to oxidative stress [148]. However, the PB28 anticancer effect both in vitro and in a preclinical model was not striking despite the promising σ binding profile. With the aim of increasing the cytotoxic activity, structural changes were made and several analogues of PB28 were synthesized, some of which were endowed with promising anticancer action. As an example, the piperidinyl analogue PB221 (Figure 15), with reduced σ receptor affinity and a more σ2-oriented profile, was thoroughly investigated in CNS tumors and displayed promising results in preclinical models [149]. Additionally, the cytotoxic activity of PB28 in resistant tumors was increased through the generation of multi-target ligands directed toward σ receptors (the σ2 receptor in particular) and P-glycoprotein, with the aim to evade the P-gp efflux whose overexpression is responsible for the drug-resistance phenomenon. While most of the compounds were active both in resistant and wild-type (wt) cancer cells [150], some MTDLs were endowed with the collateral sensitivity phenomenon that is characterized by more potent cytotoxicity in resistant rather than in the wt cells. The promising data obtained prompted to push the MTDL concept further in this context by merging the σ/P-gp scaffold with a metals chelator portion in order to stress the collateral sensitivity property thanks to the more important impact that the chelation of metals, such as iron and copper ions, has on the energy state of cells [151]. Excellent results were obtained with this class of compounds with a thiosemicarbazone structure that showed advantages in preclinical pancreatic tumor models, where the σ-mediated delivery to pancreatic tumors appeared devoid of side effects [152]. However, the σ1 involvement was not investigated in the overall effect, as focus on the σ2 subtype was given.
Figure 15.

PB28 and PB221 structures and Ki values for σ1 receptor (σ1 R) and σ2 receptor (σ2 R) [153,154].
PB28, also Haloperidol metabolite II (Figure 12), is characterized by a σ1 receptor antagonism/σ2 receptor agonism profile, and it is able to exert modest antiproliferative activity because of the increase in the Intracellular-Free Calcium Levels [Ca2+]i and apoptosis induction [155].
This evidence supported the synthesis of (R,S)-MRJF4 (Figure 16), which combined the Haloperidol metabolite II profile together with 4-phenylbutyric acid, which inhibits Histone Deacetylase (HDAC) and is involved in the transcription of genes that regulate the cell cycle, with a clear role in cancerogenesis and cancer progression.
Figure 16.

(R,S)-MRJF4 structure and Ki values for σ1 receptor (σ1 R) and σ2 receptor (σ2 R) [129].
The esterification of Haloperidol metabolite II with 4-phenylbutyric acid appeared to be detrimental for the affinity toward both classes of receptors, with a drop from nanomolar to low micromolar values. Nevertheless, antiproliferative studies demonstrated that (R,S)-MRJF4 is able to produce a more potent antiproliferative effect in LNCaP and PC3 than 4-phenylbutyric acid or Haloperidol metabolite II alone or in combination [134].
In the following years, the pure enantiomers (R)-MRJF4 and (S)-MRJF4 were studied, showing that (R)-enantiomer has a higher affinity for both σ receptors and higher anticancer activity (Figure 16) [129].
Noteworthy are some studies performed by Nieto et al. about cancer-associated pain in which paclitaxel was used to induce allodynia. This effect was observed in wild-type mice but not in the SIGMAR1 KO mice. Moreover, σ1 receptors antagonists, such as BD1063 and S1RA (Table 1), reverted paclitaxel-induced neuropathic pain in wild-type mice [156,157].
Altogether, these studies show that the antiproliferative and growth-inhibiting activity of σ1 receptor antagonists can be empowered and likely better oriented when associated to other activities. Importantly, these same antagonists can block cancer-associated pain, which is an effect probably strengthened by the block of Na+ channels, thus again supporting the MTDLs approach involving σ1 receptors also in the treatment of pain associated to cancer.
4. Conclusions and Perspectives
This review aims to point out the state of the art of MTDLs interacting with the σ1 receptor. As a result of its pluripotent chaperone activity, the σ1 receptor has the potential to interact and modulate several targets involved in a number of diseases and syndromes. The evidence that several compounds already in clinical use (e.g., Donepezil, HP, etc.) unintentionally bind the σ1 receptor has shed light on the already exploited interaction with this protein as a successful strategy in treating illnesses or associated symptoms through a MTDLs approach. Donepezil, whose interaction with σ1 receptors in vivo has been ascertained by receptor occupancy studies in the human brain [94,158], is co-administered together with Memantine through prescription medicines (i.e., Namzaric). On this basis, the rational development of MTDLs with a σ1 intentional targeting profile may lead to more important therapeutic actions. The amount of herein listed proteins, which are the object of the MTLDs development in association with the σ1 receptor, is not intended to be exhaustive. Other proteins may be taken into consideration in the MTDLs development, considering the wide range of σ1 receptor direct and indirect interactors. Cannabinoid type-2 (CB2) receptors are an example, with recent support from computational methods that point out a partial overlap of the σ1 and CB2 receptors’ pharmacophores [159]. These data strongly suggest the adoption of a merging approach for the development of dual σ1/CB2 receptors ligands for a synergistic exploitation of the pathways activated by the two targets in oncology and neurodegenerative diseases. A number of σ ligands, besides interacting at the two subtypes (see Section 3.7), are able to modulate the efflux pump P-glycoprotein (P-gp) [150,160]. This double action is worthy of being more intentionally addressed, as it may serve as a strategy to face drug-resistant tumors: the compound overcomes P-gp, which is overexpressed in many resistant tumors and exerts its cytotoxic activity (as a σ1 antagonist). Importantly, this same double action may also have a role in the treatment of CNS diseases through the bypass of the P-gp at the BBB. The waving interest in σ receptors research increased during the COVD-19 pandemic, when the proteins were found as important key host dependency factors for coronavirus infections. However, while the exploitability of the σ1 receptor as a druggable target against coronavirus still needs to be fully investigated and validated, it appears clear that this protein is involved in a plethora of pathways hampered in multifactorial CNS and cancer diseases, rendering the σ1 receptor a full-fledged target for the development of multifunctional therapeutics.
Author Contributions
Conceptualization, F.S.A. and C.A.; investigation, F.S.A.; writing—original draft preparation, F.S.A. and C.A.; writing—review and editing, F.S.A., M.N., M.C., M.L. and C.A.; supervision, C.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Bansal Y., Silakari O. Multifunctional Compounds: Smart Molecules for Multifactorial Diseases. Eur. J. Med. Chem. 2014;76:31–42. doi: 10.1016/j.ejmech.2014.01.060. [DOI] [PubMed] [Google Scholar]
- 2.Korcsmáros T., Szalay M.S., Böde C., Kovács I.A., Csermelyt P. How to Design Multi-Target Drugs: Target Search Options in Cellular Networks. Expert Opin. Drug Discov. 2007;2:799–808. doi: 10.1517/17460441.2.6.799. [DOI] [PubMed] [Google Scholar]
- 3.Morphy R., Rankovic Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005;48:6523–6543. doi: 10.1021/jm058225d. [DOI] [PubMed] [Google Scholar]
- 4.Walker J.M., Bowen W.D., Walker F.O., Matsumoto R.R., De Costa B., Rice K.C. Sigma Receptors: Biology and Function. Pharmacol. Rev. 1990;42:355–402. [PubMed] [Google Scholar]
- 5.Hellewell S.B., Bowen W.D. A Sigma-like Binding Site in Rat Pheochromocytoma (PC12) Cells: Decreased Affinity for (+)-Benzomorphans and Lower Molecular Weight Suggest a Different Sigma Receptor Form from That of Guinea Pig Brain. Brain Res. 1990;527:244–253. doi: 10.1016/0006-8993(90)91143-5. [DOI] [PubMed] [Google Scholar]
- 6.Alonso G., Phan V., Guillemain I., Saunier M., Legrand A., Anoal M., Maurice T. Immunocytochemical Localization of the Sigma(1) Receptor in the Adult Rat Central Nervous System. Neuroscience. 2000;97:155–170. doi: 10.1016/S0306-4522(00)00014-2. [DOI] [PubMed] [Google Scholar]
- 7.Roman F., Pascaud X., Chomette G., Bueno L., Junien J.L. Autoradiographic Localization of Sigma Opioid Receptors in the Gastrointestinal Tract of the Guinea Pig. Gastroenterology. 1989;97:76–82. doi: 10.1016/0016-5085(89)91418-2. [DOI] [PubMed] [Google Scholar]
- 8.Su T.P., Wu X.Z. Guinea Pig Vas Deferens Contains ρ but Not Phencyclidine Receptors. Neurosci. Lett. 1990;108:341–345. doi: 10.1016/0304-3940(90)90664-U. [DOI] [PubMed] [Google Scholar]
- 9.Hellewell S.B., Bruce A., Feinstein G., Orringer J., Williams W., Bowen W.D. Rat Liver and Kidney Contain High Densities of Sigma 1 and Sigma 2 Receptors: Characterization by Ligand Binding and Photoaffinity Labeling. Eur. J. Pharmacol. 1994;268:9–18. doi: 10.1016/0922-4106(94)90115-5. [DOI] [PubMed] [Google Scholar]
- 10.Ela C., Barg J., Vogel Z., Hasin Y., Eilam Y. Sigma Receptor Ligands Modulate Contractility, Ca++ Influx and Beating Rate in Cultured Cardiac Myocytes. J. Pharmacol. Exp. Ther. 1994;269:1300–1309. [PubMed] [Google Scholar]
- 11.Wolfe S.A.J., Culp S.G., De Souza E.B. Sigma-Receptors in Endocrine Organs: Identification, Characterization, and Autoradiographic Localization in Rat Pituitary, Adrenal, Testis, and Ovary. Endocrinology. 1989;124:1160–1172. doi: 10.1210/endo-124-3-1160. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi T., Su T.P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell. 2007;131:596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 13.Gasparre G., Abate C., Carlucci R., Berardi F., Cassano G. The σ1 Receptor Agonist (+)-Pentazocine Increases Store-Operated Ca2+ Entry in MCF7σ1 and SK-N-SH Cell Lines. Pharmacol. Rep. 2017;69:542–545. doi: 10.1016/j.pharep.2017.01.022. [DOI] [PubMed] [Google Scholar]
- 14.Mori T., Hayashi T., Hayashi E., Su T.P. Sigma-1 Receptor Chaperone at the ER-Mitochondrion Interface Mediates the Mitochondrion-ER-Nucleus Signaling for Cellular Survival. PLoS ONE. 2013;8:e76941. doi: 10.1371/journal.pone.0076941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weng T.-Y., Tsai S.-Y.A., Su T.-P. Roles of Sigma-1 Receptors on Mitochondrial Functions Relevant to Neurodegenerative Diseases. J. Biomed. Sci. 2017;24:74. doi: 10.1186/s12929-017-0380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Su T.P., Su T.C., Nakamura Y., Tsai S.Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016;37:262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt H.R., Kruse A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019;40:636–654. doi: 10.1016/j.tips.2019.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vela J.M. Repurposing Sigma-1 Receptor Ligands for COVID-19 Therapy? Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.582310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon D.E., Gordon D.E., Hiatt J., Bouhaddou M., Rezelj V.V., Ulferts S. Comparative Host-Coronavirus Protein Interaction Networks Reveal Pan-Viral Disease Mechanisms. Science. 2020;9403:eabe9403. doi: 10.1126/science.abe9403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mishina M., Ohyama M., Ishii K., Kitamura S., Kimura Y., Oda K.I., Kawamura K., Sasaki T., Kobayashi S., Katayama Y., et al. Low Density of Sigma1 Receptors in Early Alzheimer’s Disease. Ann. Nucl. Med. 2008;22:151–156. doi: 10.1007/s12149-007-0094-z. [DOI] [PubMed] [Google Scholar]
- 21.Huang Y., Zheng L., Halliday G., Dobson-Stone C., Wang Y., Tang H.-D., Cao L., Deng Y.-L., Wang G., Zhang Y.-M., et al. Genetic Polymorphisms in Sigma-1 Receptor and Apolipoprotein E Interact to Influence the Severity of Alzheimers Disease. Curr. Alzheimer Res. 2011;8:765–770. doi: 10.2174/156720511797633232. [DOI] [PubMed] [Google Scholar]
- 22.Fehér Á., Juhász A., László A., Kálmán J., Pákáski M., Kálmán J., Janka Z. Association between a Variant of the Sigma-1 Receptor Gene and Alzheimer’s Disease. Neurosci. Lett. 2012;517:136–139. doi: 10.1016/j.neulet.2012.04.046. [DOI] [PubMed] [Google Scholar]
- 23.Christ M.G., Clement A.M., Behl C. The Sigma-1 Receptor at the Crossroad of Proteostasis, Neurodegeneration, and Autophagy. Trends Neurosci. 2020;43:79–81. doi: 10.1016/j.tins.2019.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Hanner M., Moebius F.F., Flandorfer A., Knaus H.G., Striessnig J., Kempner E., Glossmann H. Purification, Molecular Cloning, and Expression of the Mammalian Sigma1-Binding Site. Proc. Natl. Acad. Sci. USA. 1996;93:8072–8077. doi: 10.1073/pnas.93.15.8072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt H.R., Zheng S., Gurpinar E., Koehl A., Manglik A., Kruse A.C. Crystal Structure of the Human σ1 Receptor. Nature. 2016;532:527–530. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmidt H.R., Betz R.M., Dror R.O., Kruse A.C. Structural Basis for σ(1) Receptor Ligand Recognition. Nat. Struct. Mol. Biol. 2018;25:981–987. doi: 10.1038/s41594-018-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glennon R. Pharmacophore Identification for Sigma-1 (σ1) Receptor Binding: Application of the “Deconstruction-Reconstruction-Elaboration” Approach. Mini-Rev. Med. Chem. 2005;5:927–940. doi: 10.2174/138955705774329519. [DOI] [PubMed] [Google Scholar]
- 28.Niso M., Mosier P.D., Marottoli R., Ferorelli S., Cassano G., Gasparre G., Leopoldo M., Berardi F., Abate C. High-Affinity Sigma-1 (σ1) Receptor Ligands Based on the σ 1 Antagonist PB212. Future Med Chem. 2019;1:2547–2562. doi: 10.4155/fmc-2019-0042. [DOI] [PubMed] [Google Scholar]
- 29.Mishra A.K., Mavlyutov T., Singh D.R., Biener G., Yang J., Oliver J.A., Ruoho A., Raicu V. The Sigma-1 Receptors Are Present in Monomeric and Oligomeric Forms in Living Cells in the Presence and Absence of Ligands. Biochem. J. 2015;466:263–271. doi: 10.1042/BJ20141321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albayrak Y., Hashimoto K. Sigma-1 Receptor Agonists and Their Clinical Implications in Neuropsychiatric Disorders. Adv. Exp. Med. Biol. 2017;964:153–161. doi: 10.1007/978-3-319-50174-1_11. [DOI] [PubMed] [Google Scholar]
- 31.Ferris R.M., Tang FL M., Chang K.J., Russell A. Evidence That the Potential Antipsychotic Agent Rimcazole (BW 234U) Is a Specific, Competitive Antagonist of Sigma Sites in Brain. J. Chem. Inf. Model. 2019;53:1689–1699. doi: 10.1016/0024-3205(86)90640-5. [DOI] [PubMed] [Google Scholar]
- 32.Gilmore D.L., Liu Y., Matsumoto R.R. Review of the Pharmacological and Clinical Profile of Rimcazole. CNS Drug Rev. 2004;10:1–22. doi: 10.1111/j.1527-3458.2004.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schoenwald R.D., Barfknecht C.F., Shirolkar S., Xia E. The Effects of Sigma Ligands on Protein Release from Lacrimal Acinar Cells: A Potential Agonist/Antagonist Assay. Life Sci. 1995;56:1275–1285. doi: 10.1016/0024-3205(95)00073-9. [DOI] [PubMed] [Google Scholar]
- 34.Taylor D.P., Eison M.S., Moon S.L., Schlemmer R.F.J., Shukla U.A., VanderMaelen C.P., Yocca F.D., Gallant D.J., Behling S.H., Boissard C.G. A Role for Sigma Binding in the Antipsychotic Profile of BMY 14802? NIDA Res. Monogr. 1993;133:125–157. [PubMed] [Google Scholar]
- 35.Yano H., Bonifazi A., Xu M., Guthrie D.A., Schneck S.N., Abramyan A.M., Fant A.D., Hong W.C., Newman A.H., Shi L. Pharmacological Profiling of Sigma 1 Receptor Ligands by Novel Receptor Homomer Assays. Neuropharmacology. 2018;133:264–275. doi: 10.1016/j.neuropharm.2018.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gómez-Soler M., Fernández-Dueñas V., Portillo-Salido E., Pérez P., Zamanillo D., Vela J.M., Burgueño J., Ciruela F. Predicting the Antinociceptive Efficacy of σ1 Receptor Ligands by a Novel Receptor Fluorescence Resonance Energy Transfer (FRET) Based Biosensor. J. Med. Chem. 2014;57:238–242. doi: 10.1021/jm401529t. [DOI] [PubMed] [Google Scholar]
- 37.Fontanilla D., Johannessen M., Hajipour A.R., Cozzi N.V., Jackson M.B., Ruoho A.E. The Hallucinogen N,N-Dimethyltryptamine (DMT) Is an Endogenous Sigma-1 Receptor Regulator. Science. 2009;323:934–937. doi: 10.1126/science.1166127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannessen M., Fontanilla D., Mavlyutov T., Ruoho A.E., Jackson M.B. Antagonist Action of Progesterone at σ-Receptors in the Modulation of Voltage-Gated Sodium Channels. Am. J. Physiol. Cell Physiol. 2011;300:C328–C337. doi: 10.1152/ajpcell.00383.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su T.P., London E.D., Jaffe J.H. Steroid Binding at σ Receptors Suggests a Link between Endocrine, Nervous, and Immune Systems. Science. 1988;240:219–221. doi: 10.1126/science.2832949. [DOI] [PubMed] [Google Scholar]
- 40.Su T.P. Evidence for Sigma Opioid Receptor: Binding of [3H]SKF-10047 to Etorphine-Inaccessible Sites in Guinea-Pig Brain. J. Pharmacol. Exp. Ther. 1982;223:284–290. [PubMed] [Google Scholar]
- 41.Matsumoto R.R., Nguyen L., Kaushal N., Robson M.J. Sigma (σ) Receptors as Potential Therapeutic Targets to Mitigate Psychostimulant Effects. Adv. Pharmacol. 2014;69:323–386. doi: 10.1016/B978-0-12-420118-7.00009-3. [DOI] [PubMed] [Google Scholar]
- 42.Ishiguro H., Ohtsuki T., Toru M., Itokawa M., Aoki J., Shibuya H., Kurumaji A., Okubo Y., Iwawaki A., Ota K., et al. Association between Polymorphisms in the Type 1 Sigma Receptor Gene and Schizophrenia. Neurosci. Lett. 1998;257:45–48. doi: 10.1016/S0304-3940(98)00797-6. [DOI] [PubMed] [Google Scholar]
- 43.Uchida N., Ujike H., Nakata K., Takaki M., Nomura A., Katsu T., Tanaka Y., Imamura T., Sakai A., Kuroda S. No Association between the Sigma Receptor Type 1 Gene and Schizophrenia: Results of Analysis and Meta-Analysis of Case-Control Studies. BMC Psychiatry. 2003;3:13. doi: 10.1186/1471-244X-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Narita N., Hashimoto K., Tomitaka S.I., Minabe Y. Interactions of Selective Serotonin Reuptake Inhibitors with Subtypes of σ Receptors in Rat Brain. Eur. J. Pharmacol. 1996;307:117–119. doi: 10.1016/0014-2999(96)00254-3. [DOI] [PubMed] [Google Scholar]
- 45.Urani A., Roman F.J., Phan V.L., Su T.P., Maurice T. The Antidepressant-like Effect Induced by Sigma(1)-Receptor Agonists and Neuroactive Steroids in Mice Submitted to the Forced Swimming Test. J. Pharmacol. Exp. Ther. 2001;298:1269–1279. [PubMed] [Google Scholar]
- 46.Skuza G., Szymańska M., Budziszewska B., Abate C., Berardi F. Effects of PB190 and PB212, New σ Receptor Ligands, on Glucocorticoid Receptor-Mediated Gene Transcription in LMCAT Cells. Pharmacol. Rep. 2011;63:1564–1568. doi: 10.1016/S1734-1140(11)70722-8. [DOI] [PubMed] [Google Scholar]
- 47.Maurice T., Roman F.J., Privat A. Modulation by neurosteroids of the in vivo (+)-[3H]SKF-10,047 binding to σ1 receptors in the mouse forebrain. J. Neurosci. Res. 1996;46:734–743. doi: 10.1002/(SICI)1097-4547(19961215)46:6<734::AID-JNR10>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 48.Matsuno K., Nakazawa M., Okamoto K., Kawashima Y., Mita S. Binding Properties of SA4503, a Novel and Selective s Receptor Agonist. Eur. J. Pharmacol. 1996;306:271–279. doi: 10.1016/0014-2999(96)00201-4. [DOI] [PubMed] [Google Scholar]
- 49.Senda T., Matsuno K., Okamoto K., Kobayashi T., Nakata K., Mita S. Ameliorating Effect of SA4503, a Novel σ1 Receptor Agonist, on Memory Impairments Induced by Cholinergic Dysfunction in Rats. Eur. J. Pharmacol. 1996;315:137–146. doi: 10.1016/S0014-2999(96)00572-9. [DOI] [PubMed] [Google Scholar]
- 50.Skuza G., Rogóz Z. A Potential Antidepressant Activity of SA4503, a Selective σ1 Receptor Agonist. Behav. Pharmacol. 2002;13:537–543. doi: 10.1097/00008877-200211000-00003. [DOI] [PubMed] [Google Scholar]
- 51.Hirano K., Tagashira H., Fukunaga K. Cardioprotective Effect of the Selective Sigma-1 Receptor Agonist, SA4503. Yakugaku Zasshi. 2014;134:707–713. doi: 10.1248/yakushi.13-00255-3. [DOI] [PubMed] [Google Scholar]
- 52.Yamashita D., Sun G.-W., Cui Y., Mita S., Otsuki N., Kanzaki S., Nibu K.I., Ogawa K., Matsunaga T. Neuroprotective Effects of Cutamesine, a Ligand of the Sigma-1 Receptor Chaperone, against Noise-Induced Hearing Loss. J. Neurosci. Res. 2015;93:788–795. doi: 10.1002/jnr.23543. [DOI] [PubMed] [Google Scholar]
- 53.Berardi F., Ferorelli S., Abate C., Pedone M.P., Colabufo N.A., Contino M., Perrone R. Methyl Substitution on the Piperidine Ring of N-[ω-(6- Methoxynaphthalen-1-Yl)Alkyl] Derivatives as a Probe for Selective Binding and Activity at the σ1 Receptor. J. Med. Chem. 2005;48:8237–8244. doi: 10.1021/jm050654o. [DOI] [PubMed] [Google Scholar]
- 54.Moritz C., Berardi F., Abate C., Peri F. Live Imaging Reveals a New Role for the Sigma-1 (σ1) Receptor in Allowing Microglia to Leave Brain Injuries. Neurosci. Lett. 2015;591:13–18. doi: 10.1016/j.neulet.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 55.Skuza G., Sadaj W., Kabziński M., Cassano G., Gasparre G., Abate C., Berardi F. The Effects of New Sigma (σ) Receptor Ligands, PB190 and PB212, in the Models Predictive of Antidepressant Activity. Pharmacol. Rep. 2014;66:320–324. doi: 10.1016/j.pharep.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 56.Su T.P., Wu X.Z., Cone E.J., Shukla K., Gund T.M., Dodge A.L., Parish D.W. Sigma Compounds Derived from Phencyclidine: Identification of PRE-084, a New, Selective Sigma Ligand. J. Pharmacol. Exp. Ther. 1991;259:543–550. [PubMed] [Google Scholar]
- 57.Maurice T., Su T.P., Parish D.W., Nabeshima T., Privat A. PRE-084, a σ Selective PCP Derivative, Attenuates MK-801-Induced Impairment of Learning in Mice. Pharmacol. Biochem. Behav. 1994;49:859–869. doi: 10.1016/0091-3057(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 58.Maurice T. Beneficial Effect of the σ1 Receptor Agonist PRE-084 against the Spatial Learning Deficits in Aged Rats. Eur. J. Pharmacol. 2001;431:223–227. doi: 10.1016/S0014-2999(01)01436-4. [DOI] [PubMed] [Google Scholar]
- 59.Okuyama S., Nakazato A. NE-100: A Novel Sigma Receptor Antagonist. CNS Drug Rev. 1996;2:226–237. doi: 10.1111/j.1527-3458.1996.tb00299.x. [DOI] [Google Scholar]
- 60.Chaki S., Tanaka M., Muramatsu M., Otomo S. NE-100, a Novel Potent σ Ligand, Preferentially Binds to Σ1 Binding Sites in Guinea Pig Brain. Eur. J. Pharmacol. 1994;251:R1–R2. doi: 10.1016/0014-2999(94)90453-7. [DOI] [PubMed] [Google Scholar]
- 61.Spinelli F., Haider A., Toscano A., Pati M.L., Keller C., Berardi F., Colabufo N.A., Abate C., Ametamey S.M. Synthesis, Radiolabelling, and Evaluation of [(11)C]PB212 as a Radioligand for Imaging Sigma-1 Receptors Using PET. Am. J. Nucl. Med. Mol. Imaging. 2018;8:32–40. [PMC free article] [PubMed] [Google Scholar]
- 62.Maeda D.Y., Williams W., Bowen W.D., Coop A. A Sigma-1 Receptor Selective Analogue of BD1008. A Potential Substitute for (+)-Opioids in Sigma Receptor Binding Assays. Bioorganic Med. Chem. Lett. 2000;10:17–18. doi: 10.1016/S0960-894X(99)00590-9. [DOI] [PubMed] [Google Scholar]
- 63.Sahlholm K., Århem P., Fuxe K., Marcellino D. The Dopamine Stabilizers ACR16 and ()-OSU6162 Display Nanomolar Affinities at the σ-1 Receptor. Mol. Psychiatry. 2013;18:12–14. doi: 10.1038/mp.2012.3. [DOI] [PubMed] [Google Scholar]
- 64.James M.L., Shen B., Zavaleta C.L., Nielsen C.H., Mesangeau C., Vuppala P.K., Chan C., Avery B.A., Fishback J.A., Matsumoto R.R., et al. New Positron Emission Tomography (PET) Radioligand for Imaging σ-1 Receptors in Living Subjects. J. Med. Chem. 2012;55:8272–8282. doi: 10.1021/jm300371c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.James M.L., Shen B., Nielsen C.H., Behera D., Buckmaster C.L., Mesangeau C., Zavaleta C., Vuppala P.K., Jamalapuram S., Avery B.A., et al. Evaluation of σ-1 Receptor Radioligand 18F-FTC-146 in Rats and Squirrel Monkeys Using PET. J. Nucl. Med. 2014;55:147–153. doi: 10.2967/jnumed.113.120261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen B., Park J.H., Hjørnevik T., Cipriano P.W., Yoon D., Gulaka P.K., Holly D., Behera D., Avery B.A., Gambhir S.S., et al. Radiosynthesis and First-In-Human PET/MRI Evaluation with Clinical-Grade [18F]FTC-146. Mol. Imaging Biol. 2017;19:779–786. doi: 10.1007/s11307-017-1064-z. [DOI] [PubMed] [Google Scholar]
- 67.Díaz J.L., Zamanillo D., Corbera J., Baeyens J.M., Maldonado R., Pericàs M.A., Vela J.M., Torrens A. Selective Sigma-1 (Sigma1) Receptor Antagonists: Emerging Target for the Treatment of Neuropathic Pain. Cent. Nerv. Syst. Agents Med. Chem. 2009;9:172–183. doi: 10.2174/1871524910909030172. [DOI] [PubMed] [Google Scholar]
- 68.Abadias M., Escriche M., Vaqué A., Sust M., Encina G. Safety, Tolerability and Pharmacokinetics of Single and Multiple Doses of a Novel Sigma-1 Receptor Antagonist in Three Randomized Phase I Studies. Br. J. Clin. Pharmacol. 2013;75:103–117. doi: 10.1111/j.1365-2125.2012.04333.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Matsumoto R.R., Bowen W.D., Tom M.A., Vo V.N., Truong D.D., De Costa B.R. Characterization of Two Novel σ Receptor Ligands: Antidystonic Effects in Rats Suggest σ Receptor Antagonism. Eur. J. Pharmacol. 1995;280:301–310. doi: 10.1016/0014-2999(95)00208-3. [DOI] [PubMed] [Google Scholar]
- 70.Matsumoto R.R., McCracken K.A., Friedman M.J., Pouw B., De Costa B.R., Bowen W.D. Conformationally Restricted Analogs of BD1008 and an Antisense Oligodeoxynucleotide Targeting σ1 Receptors Produce Anti-Cocaine Effects in Mice. Eur. J. Pharmacol. 2001;419:163–174. doi: 10.1016/S0014-2999(01)00968-2. [DOI] [PubMed] [Google Scholar]
- 71.Perrone R., Berardi F., Colabufo N.A., Leopoldo M., Abate C., Tortorella V. N-Aryl or N-Alkylpiperazine Derivatives: The Role of N-Substituent on σ1, σ2, 5-HT1A and D2 Receptor Affinity. Med. Chem. Res. 2000;10:201–207. [Google Scholar]
- 72.Lever J.R., Gustafson J.L., Xu R., Allmon R.L., Lever S.Z. σ1 and σ2 Receptor Binding Affinity and Selectivity of SA4503 and Fluoroethyl SA4503. Synapse. 2006;59:350–358. doi: 10.1002/syn.20253. [DOI] [PubMed] [Google Scholar]
- 73.Garces-Ramirez L., Green J.L., Hiranita T., Kopajtic T.A., Mereu M., Thomas A.M., Mesangeau C., Narayanan S., McCurdy C.R., Katz J.L., et al. Sigma Receptor Agonists: Receptor Binding and Effects on Mesolimbic Dopamine Neurotransmission Assessed by Microdialysis. Biol. Psychiatry. 2011;69:208–217. doi: 10.1016/j.biopsych.2010.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Colabufo N.A., Berardi F., Contino M., Perrone R., Tortorella V. A New Method for Evaluating σ2 Ligand Activity in the Isolated Guinea-Pig Bladder. Naunyn. Schmiedebergs. Arch. Pharmacol. 2003;368:106–112. doi: 10.1007/s00210-003-0777-5. [DOI] [PubMed] [Google Scholar]
- 75.Johnston T.H., Geva M., Steiner L., Orbach A., Papapetropoulos S., Savola J.M., Reynolds I.J., Ravenscroft P., Hill M., Fox S.H., et al. Pridopidine, a Clinic-Ready Compound, Reduces 3,4-Dihydroxyphenylalanine-Induced Dyskinesia in Parkinsonian Macaques. Mov. Disord. 2019;34:708–716. doi: 10.1002/mds.27565. [DOI] [PubMed] [Google Scholar]
- 76.Romero L., Zamanillo D., Nadal X., Sánchez-Arroyos R., Rivera-Arconada I., Dordal A., Montero A., Muro A., Bura A., Segalés C., et al. Pharmacological Properties of S1RA, a New Sigma-1 Receptor Antagonist That Inhibits Neuropathic Pain and Activity-Induced Spinal Sensitization. Br. J. Pharmacol. 2012;166:2289–2306. doi: 10.1111/j.1476-5381.2012.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lecanu L., Tillement L., McCourty A., Rammouz G., Yao W., Greeson J., Papadopoulos V. Dimethyl-Carbamic Acid 2,3-Bis-Dimethylcarbamoyloxy-6-(4-Ethyl-Piperazine- 1-Carbonyl)-Phenyl Ester: A Novel Multi-Target Therapeutic Approach to Neuroprotection. Med. Chem. 2012;6:123–140. doi: 10.2174/1573406411006030123. [DOI] [PubMed] [Google Scholar]
- 78.Caccamo A., Oddo S., Billings L.M., Green K.N., Martinez-Coria H., Fisher A., LaFerla F.M. M1 Receptors Play a Central Role in Modulating AD-like Pathology in Transgenic Mice. Neuron. 2006;49:671–682. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- 79.Medeiros R., Kitazawa M., Caccamo A., Baglietto-Vargas D., Estrada-Hernandez T., Cribbs D.H., Fisher A., Laferla F.M. Loss of Muscarinic M1 Receptor Exacerbates Alzheimer’s Disease-like Pathology and Cognitive Decline. Am. J. Pathol. 2011;179:980–991. doi: 10.1016/j.ajpath.2011.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vamvakidès A. [Anticonvulsant and forced swim anti-immobility effects of tetrahydro-N, N-dimethyl-2,2-diphenyl-3-furanemethanamine (AE37): Common action mechanism?] Ann. Pharm. Fr. 2002;60:88–92. [PubMed] [Google Scholar]
- 81.Vamvakidès A. [Mechanism of action of tetrahydro-N, N-dimethyl-5, 5-diphenyl-3-furanemethanamine, a putative nootropic, anti-epileptic and antidepressant compound] Ann. Pharm. Fr. 2002;60:415–422. [PubMed] [Google Scholar]
- 82.Fisher A., Bezprozvanny I., Wu L., Ryskamp D.A., Bar-Ner N., Natan N., Brandeis R., Elkon H., Nahum V., Gershonov E., et al. AF710B, a Novel M1/σ1 Agonist with Therapeutic Efficacy in Animal Models of Alzheimer’s Disease. Neurodegener. Dis. 2016;16:95–110. doi: 10.1159/000440864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chow H.M., Guo D., Zhou J.C., Zhang G.Y., Li H.F., Herrup K., Zhang J. CDK5 Activator Protein P25 Preferentially Binds and Activates GSK3β. Proc. Natl. Acad. Sci. USA. 2014;111:E4887–E4895. doi: 10.1073/pnas.1402627111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lahmy V., Meunier J., Malmström S., Naert G., Givalois L., Kim S.H., Villard V., Vamvakides A., Maurice T. Blockade of Tau Hyperphosphorylation and Aβ 1-42 Generation by the Aminotetrahydrofuran Derivative ANAVEX2-73, a Mixed Muscarinic and σ 1 Receptor Agonist, in a Nontransgenic Mouse Model of Alzheimer’s Disease. Neuropsychopharmacology. 2013;38:1706–1723. doi: 10.1038/npp.2013.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Espallergues J., Lapalud P., Christopoulos A., Avlani V.A., Sexton P.M., Vamvakides A., Maurice T. Involvement of the Sigma1 (σ1) Receptor in the Anti-Amnesic, but Not Antidepressant-like, Effects of the Aminotetrahydrofuran Derivative ANAVEX1-41. Br. J. Pharmacol. 2007;152:267–279. doi: 10.1038/sj.bjp.0707386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Behensky A.A., Yasny I.E., Shuster A.M., Seredenin S.B., Petrov A.V., Cuevas J. Stimulation of Sigma Receptors with Afobazole Blocks Activation of Microglia and Reduces Toxicity Caused by Amyloid-Β25-35. J. Pharmacol. Exp. Ther. 2013;347:458–467. doi: 10.1124/jpet.113.208348. [DOI] [PubMed] [Google Scholar]
- 87.Behensky A.A., Katnik C., Yin H., Cuevas J. Activation of Sigma Receptors with Afobazole Modulates Microglial, but Not Neuronal, Apoptotic Gene Expression in Response to Long-Term Ischemia Exposure. Front. Neurosci. 2019;13:414. doi: 10.3389/fnins.2019.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hall H., Iulita M.F., Gubert P., Flores Aguilar L., Ducatenzeiler A., Fisher A., Cuello A.C. AF710B, an M1/Sigma-1 Receptor Agonist with Long-Lasting Disease-Modifying Properties in a Transgenic Rat Model of Alzheimer’s Disease. Alzheimer’s Dement. 2018;14:811–823. doi: 10.1016/j.jalz.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 89.Rochais C., Lecoutey C., Gaven F., Giannoni P., Hamidouche K., Hedou D., Dubost E., Genest D., Yahiaoui S., Freret T., et al. Novel Multitarget-Directed Ligands (MTDLs) with Acetylcholinesterase (AChE) Inhibitory and Serotonergic Subtype 4 Receptor (5-HT4R) Agonist Activities As Potential Agents against Alzheimer’s Disease: The Design of Donecopride. J. Med. Chem. 2015;58:3172–3187. doi: 10.1021/acs.jmedchem.5b00115. [DOI] [PubMed] [Google Scholar]
- 90.Lalut J., Santoni G., Karila D., Lecoutey C., Davis A., Nachon F., Silman I., Sussman J., Weik M., Maurice T., et al. Novel Multitarget-Directed Ligands Targeting Acetylcholinesterase and σ1 Receptors as Lead Compounds for Treatment of Alzheimer’s Disease: Synthesis, Evaluation, and Structural Characterization of Their Complexes with Acetylcholinesterase. Eur. J. Med. Chem. 2019;162:234–248. doi: 10.1016/j.ejmech.2018.10.064. [DOI] [PubMed] [Google Scholar]
- 91.Kato K., Hayako H., Ishihara Y., Marui S., Iwane M., Miyamoto M. TAK-147, an Acetylcholinesterase Inhibitor, Increases Choline Acetyltransferase Activity in Cultured Rat Septal Cholinergic Neurons. Neurosci. Lett. 1999;260:5–8. doi: 10.1016/S0304-3940(98)00943-4. [DOI] [PubMed] [Google Scholar]
- 92.Maurice T., Meunier J., Feng B., Ieni J., Monaghan D.T. Interaction with σ1 Protein, but Not N-Methyl-D-Aspartate Receptor, Is Involved in the Pharmacological Activity of Donepezil. J. Pharmacol. Exp. Ther. 2006;317:606–614. doi: 10.1124/jpet.105.097394. [DOI] [PubMed] [Google Scholar]
- 93.Meunier J., Ieni J., Maurice T. The Anti-Amnesic and Neuroprotective Effects of Donepezil against Amyloid Β 25-35 Peptide-Induced Toxicity in Mice Involve an Interaction with the σ1 Receptor. Br. J. Pharmacol. 2006;149:998–1012. doi: 10.1038/sj.bjp.0706927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramakrishnan N.K., Visser A.K.D., Schepers M., Luurtsema G., Nyakas C.J., Elsinga P.H., Ishiwata K., Dierckx R.A.J.O., Van Waarde A. Dose-Dependent Sigma-1 Receptor Occupancy by Donepezil in Rat Brain Can Be Assessed with 11C-SA4503 and MicroPET. Psychopharmacology. 2014;231:3997–4006. doi: 10.1007/s00213-014-3533-2. [DOI] [PubMed] [Google Scholar]
- 95.Maurice T. Protection by Sigma-1 Receptor Agonists Is Synergic with Donepezil, but Not with Memantine, in a Mouse Model of Amyloid-Induced Memory Impairments. Behav. Brain Res. 2016;296:270–278. doi: 10.1016/j.bbr.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 96.Ganapathy M.E., Prasad P.D., Huang W., Seth P., Leibach F.H., Ganapathy V. Molecular and Ligand-Binding Characterization of the Sigma-Receptor in the Jurkat Human T Lymphocyte Cell Line. J. Pharmacol. Exp. Ther. 1999;289:251–260. [PubMed] [Google Scholar]
- 97.Castañeda-Arriaga R., Alvarez-Idaboy J.R. Lipoic Acid and Dihydrolipoic Acid. A Comprehensive Theoretical Study of Their Antioxidant Activity Supported by Available Experimental Kinetic Data. J. Chem. Inf. Model. 2014;54:1642–1652. doi: 10.1021/ci500213p. [DOI] [PubMed] [Google Scholar]
- 98.Fava A., Pirritano D., Plastino M., Cristiano D., Puccio G., Colica C., Ermio C., De Bartolo M., Mauro G., Bosco D. The Effect of Lipoic Acid Therapy on Cognitive Functioning in Patients with Alzheimer’s Disease. J. Neurodegener. Dis. 2013;2013:454253. doi: 10.1155/2013/454253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Estrada M., Pérez C., Soriano E., Laurini E., Romano M., Pricl S., Morales-García J.A., Pérez-Castillo A., Rodríguez-Franco M.I. New Neurogenic Lipoic-Based Hybrids as Innovative Alzheimer’s Drugs with σ-1 Agonism and β-Secretase Inhibition. Future Med. Chem. 2016;8:1191–1207. doi: 10.4155/fmc-2016-0036. [DOI] [PubMed] [Google Scholar]
- 100.Estrada Valencia M., Herrera-Arozamena C., de Andrés L., Pérez C., Morales-García J.A., Pérez-Castillo A., Ramos E., Romero A., Viña D., Yáñez M., et al. Neurogenic and Neuroprotective Donepezil-Flavonoid Hybrids with Sigma-1 Affinity and Inhibition of Key Enzymes in Alzheimer’s Disease. Eur. J. Med. Chem. 2018;156:534–553. doi: 10.1016/j.ejmech.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 101.Estrada-Valencia M., Herrera-Arozamena C., Pérez C., Viña D., Morales-García J.A., Pérez-Castillo A., Ramos E., Romero A., Laurini E., Pricl S., et al. New Flavonoid–N,N-Dibenzyl(N-Methyl)Amine Hybrids: Multi-Target-Directed Agents for Alzheimer’s Disease Endowed with Neurogenic Properties. J. Enzym. Inhib. Med. Chem. 2019;34:712–727. doi: 10.1080/14756366.2019.1581184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Quirion R., Chicheportiche R., Contreras P.C., Johnson K.M., Lodge D., William Tam S., Woods J.H., Zukin S.R. Classification and Nomenclature of Phencyclidine and Sigma Receptor Sites. Trends Neurosci. 1987;10:444–446. doi: 10.1016/0166-2236(87)90094-4. [DOI] [Google Scholar]
- 103.Quirion R., Bowen W.D., Itzhak Y., Junien J.L., Musacchio J.M., Rothman R.B., Su T.P., Tam S.W., Taylor D.P. A Proposal for the Classification of Sigma Binding Sites. Trends Pharmacol. Sci. 1992;13:85–86. doi: 10.1016/0165-6147(92)90030-A. [DOI] [PubMed] [Google Scholar]
- 104.Pabba M., Sibille E. Sigma-1 and N-Methyl-D-Aspartate Receptors: A Partnership with Beneficial Outcomes. Mol. Neuropsychiatry. 2015;1:47–51. doi: 10.1159/000376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bergeron R., de Montigny C., Debonnel G. Biphasic Effects of Sigma Ligands on the Neuronal Response to N-Methyl-D-Aspartate. Naunyn. Schmiedebergs. Arch. Pharmacol. 1995;351:252–260. doi: 10.1007/BF00233244. [DOI] [PubMed] [Google Scholar]
- 106.Liang X., Wang R.Y. Biphasic Modulatory Action of the Selective Sigma Receptor Ligand SR 31742A on N-Methyl-D-Aspartate-Induced Neuronal Responses in the Frontal Cortex. Brain Res. 1998;807:208–213. doi: 10.1016/S0006-8993(98)00797-5. [DOI] [PubMed] [Google Scholar]
- 107.Fletcher E.J., Church J., Abdel-Hamid K., MacDonald J.F. Blockade by Sigma Site Ligands of N-methyl-D-aspartate-evoked Responses in Rat and Mouse Cultured Hippocampal Pyramidal Neurones. Br. J. Pharmacol. 1995;116:2791–2800. doi: 10.1111/j.1476-5381.1995.tb15928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martina M., Turcotte M.E.B., Halman S., Bergeron R. The Sigma-1 Receptor Modulates NMDA Receptor Synaptic Transmission and Plasticity via SK Channels in Rat Hippocampus. J. Physiol. 2007;578:143–157. doi: 10.1113/jphysiol.2006.116178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Van der Schyf C.J., Squier G.J., Coetzee W.A. Characterization of NGP 1-01, an Aromatic Polycyclic Amine, as a Calcium Antagonist. Pharmacol. Res. Commun. 1986;18:407–417. doi: 10.1016/0031-6989(86)90162-1. [DOI] [PubMed] [Google Scholar]
- 110.Geldenhuys J.W., Van der Schyf C.J. Rationally Designed Multi-Targeted Agents Against Neurodegenerative Diseases. Curr. Med. Chem. 2013;20:1662–1672. doi: 10.2174/09298673113209990112. [DOI] [PubMed] [Google Scholar]
- 111.Kassiou M., Nguyen V.H., Knott R., Christie M.J., Hambley T.W. Trishomocubanes, a New Class of Selective and High Affinity Ligands for the Sigma Binding Site. Bioorganic Med. Chem. Lett. 1996;6:595–600. doi: 10.1016/0960-894X(96)00067-4. [DOI] [Google Scholar]
- 112.Nguyen V.H., Kassiou M., Johnston G.A.R., Christie M.J. Comparison of Binding Parameters of σ1 and σ2 Binding Sites in Rat and Guinea Pig Brain Membranes: Novel Subtype-Selective Trishomocubanes. Eur. J. Pharmacol. 1996;311:233–240. doi: 10.1016/0014-2999(96)00395-0. [DOI] [PubMed] [Google Scholar]
- 113.Geldenhuys W.J., Malan S.F., Bloomquist J.R., Marchand A.P., Van Der Schyf C.J. Pharmacology and Structure-Activity Relationships of Bioactive Polycyclic Cage Compounds: A Focus on Pentacycloundecane Derivatives. Med. Res. Rev. 2005;25:21–48. doi: 10.1002/med.20013. [DOI] [PubMed] [Google Scholar]
- 114.Kiewert C., Hartmann J., Stoll J., Thekkumkara T.J., Van Der Schyf C.J., Klein J. NGP1-01 Is a Brain-Permeable Dual Blocker of Neuronal Voltage- and Ligand-Operated Calcium Channels. Neurochem. Res. 2006;31:395–399. doi: 10.1007/s11064-005-9036-0. [DOI] [PubMed] [Google Scholar]
- 115.Kornhuber J., Schoppmeyer K., Riederer P. Affinity of 1-Aminoadamantanes for the σ Binding Site in Post-Mortem Human Frontal Cortex. Neurosci. Lett. 1993;163:129–131. doi: 10.1016/0304-3940(93)90362-O. [DOI] [PubMed] [Google Scholar]
- 116.Kornhuber J., Bormann J., Hübers M., Rusche K., Riederer P. Effects of the 1-Amino-Adamantanes at the MK-801-Binding Site of the NMDA-Receptor-Gated Ion Channel: A Human Postmortem Brain Study. Eur. J. Pharmacol. Mol. Pharmacol. 1991;206:297–300. doi: 10.1016/0922-4106(91)90113-V. [DOI] [PubMed] [Google Scholar]
- 117.Peeters M., Romieu P., Maurice T., Su T.P., Maloteaux J.M., Hermans E. Involvement of the Sigma1 Receptor in the Modulation of Dopaminergic Transmission by Amantadine. Eur. J. Neurosci. 2004;19:2212–2220. doi: 10.1111/j.0953-816X.2004.03297.x. [DOI] [PubMed] [Google Scholar]
- 118.Moreno E., Moreno-Delgado D., Navarro G., Hoffmann H.M., Fuentes S., Rosell-Vilar S., Gasperini P., Rodríguez-Ruiz M., Medrano M., Mallol J., et al. Cocaine Disrupts Histamine H3 Receptor Modulation of Dopamine D1 Receptor Signaling: σ1-D1-H3 Receptor Complexes as Key Targets for Reducing Cocaine’s Effects. J. Neurosci. 2014;34:3545–3558. doi: 10.1523/JNEUROSCI.4147-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Navarro G., Moreno E., Bonaventura J., Brugarolas M., Farré D., Aguinaga D., Mallol J., Cortés A., Casadó V., Lluís C., et al. Cocaine Inhibits Dopamine D2 Receptor Signaling via Sigma-1-D2 Receptor Heteromers. PLoS ONE. 2013;8:e61245. doi: 10.1371/journal.pone.0061245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sambo D.O., Lin M., Owens A., Lebowitz J.J., Richardson B., Jagnarine D.A., Shetty M., Rodriquez M., Alonge T., Ali M., et al. The Sigma-1 Receptor Modulates Methamphetamine Dysregulation of Dopamine Neurotransmission. Nat. Commun. 2017;8:2228. doi: 10.1038/s41467-017-02087-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sahlholm K., Sijbesma J.W.A., Maas B., Kwizera C., Marcellino D., Ramakrishnan N.K., Dierckx R.A.J.O., Elsinga P.H., Van Waarde A. Pridopidine Selectively Occupies Sigma-1 Rather than Dopamine D2 Receptors at Behaviorally Active Doses. Psychopharmacology. 2015;232:3443–3453. doi: 10.1007/s00213-015-3997-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Francardo V., Geva M., Bez F., Denis Q., Steiner L., Hayden M.R., Cenci M.A. Pridopidine Induces Functional Neurorestoration Via the Sigma-1 Receptor in a Mouse Model of Parkinson’s Disease. Neurotherapeutics. 2019;16:465–479. doi: 10.1007/s13311-018-00699-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ryskamp D., Wu L., Wu J., Kim D., Rammes G., Geva M., Hayden M., Bezprozvanny I. Pridopidine Stabilizes Mushroom Spines in Mouse Models of Alzheimer’s Disease by Acting on the Sigma-1 Receptor. Neurobiol. Dis. 2019;124:489–504. doi: 10.1016/j.nbd.2018.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kadnikov I.A., Voronin M.V., Seredenin S.B. Cytoprotective Effect of Afobazole and Its Main Metabolite M-11. Bull. Exp. Biol. Med. 2015;159:44–47. doi: 10.1007/s10517-015-2886-9. [DOI] [PubMed] [Google Scholar]
- 125.Voronin M.V., Kadnikov I.A. Contribution of Sigma-1 Receptor to Cytoprotective Effect of Afobazole. Pharmacol. Res. Perspect. 2016;4:e00273. doi: 10.1002/prp2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Voronin M.V., Kadnikov I.A., Voronkov D.N., Seredenin S.B. Chaperone Sigma1R Mediates the Neuroprotective Action of Afobazole in the 6-OHDA Model of Parkinson’s Disease. Sci. Rep. 2019;9:17020. doi: 10.1038/s41598-019-53413-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Matsumoto R.R., Pouw B. Correlation between Neuroleptic Binding to σ1 and σ2 Receptors and Acute Dystonic Reactions. Eur. J. Pharmacol. 2000;401:155–160. doi: 10.1016/S0014-2999(00)00430-1. [DOI] [PubMed] [Google Scholar]
- 128.Cobos E.J., Del Pozo E., Baeyens J.M. Irreversible Blockade of Sigma-1 Receptors by Haloperidol and Its Metabolites in Guinea Pig Brain and SH-SY5Y Human Neuroblastoma Cells. J. Neurochem. 2007;102:812–825. doi: 10.1111/j.1471-4159.2007.04533.x. [DOI] [PubMed] [Google Scholar]
- 129.Sozio P., Fiorito J., Di Giacomo V., Di Stefano A., Marinelli L., Cacciatore I., Cataldi A., Pacella S., Turkez H., Parenti C., et al. Haloperidol Metabolite II Prodrug: Asymmetric Synthesis and Biological Evaluation on Rat C6 Glioma Cells. Eur. J. Med. Chem. 2015;90:1–9. doi: 10.1016/j.ejmech.2014.11.012. [DOI] [PubMed] [Google Scholar]
- 130.Korpi E.R., Costakos D.T., Wyatt R.J. Interconversions of Haloperidol and Reduced Haloperidol in Guinea Pig and Rat Liver Microsomes. Biochem. Pharmacol. 1985;34:2923–2927. doi: 10.1016/0006-2952(85)90017-6. [DOI] [PubMed] [Google Scholar]
- 131.Usuki E., Van Der Schyf C.J., Castagnoli N. Metabolism of Haloperidol and Its Tetrahydrophyridine Dehydration Product HPTP. Drug Metab. Rev. 1998;30:809–826. doi: 10.3109/03602539808996331. [DOI] [PubMed] [Google Scholar]
- 132.Maurice T., Martin-Fardon R., Romieu P., Matsumoto R.R. Sigma1 (σ1) Receptor Antagonists Represent a New Strategy against Cocaine Addiction and Toxicity. Neurosci. Biobehav. Rev. 2002;26:499–527. doi: 10.1016/S0149-7634(02)00017-9. [DOI] [PubMed] [Google Scholar]
- 133.Guitart X., Codony X., Monroy X. Sigma Receptors: Biology and Therapeutic Potential. Psychopharmacology. 2004;174:301–319. doi: 10.1007/s00213-004-1920-9. [DOI] [PubMed] [Google Scholar]
- 134.Marrazzo A., Fiorito J., Zappal L., Prezzavento O., Ronsisvalle S., Pasquinucci L., Scoto G.M., Bernardini R., Ronsisvalle G. Antiproliferative Activity of Phenylbutyrate Ester of Haloperidol Metabolite II [(±)-MRJF4] in Prostate Cancer Cells. Eur. J. Med. Chem. 2011;46:433–438. doi: 10.1016/j.ejmech.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 135.Vidal-Torres A., De La Puente B., Rocasalbas M., Touriño C., Andreea Bura S., Fernández-Pastor B., Romero L., Codony X., Zamanillo D., Buschmann H., et al. Sigma-1 Receptor Antagonism as Opioid Adjuvant Strategy: Enhancement of Opioid Antinociception without Increasing Adverse Effects. Eur. J. Pharmacol. 2013;711:63–72. doi: 10.1016/j.ejphar.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 136.Bruna J., Videla S., Argyriou A.A., Velasco R., Villoria J., Santos C., Nadal C., Cavaletti G., Alberti P., Briani C., et al. Efficacy of a Novel Sigma-1 Receptor Antagonist for Oxaliplatin-Induced Neuropathy: A Randomized, Double-Blind, Placebo-Controlled Phase IIa Clinical Trial. Neurotherapeutics. 2018;15:178–189. doi: 10.1007/s13311-017-0572-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Discovery Studio 16. Dassault Systèmes BIOVIA; San Diego, CA, USA: 2016. [Google Scholar]
- 138.Laggner C., Schieferer C., Fiechtner B., Poles G., Hoffmann R.D., Glossmann H., Langer T., Moebius F.F. Discovery of High-Affinity Ligands of σ1 Receptor, ERG2, and Emopamil Binding Protein by Pharmacophore Modeling and Virtual Screening. J. Med. Chem. 2005;48:4754–4764. doi: 10.1021/jm049073+. [DOI] [PubMed] [Google Scholar]
- 139.García M., Virgili M., Alonso M., Alegret C., Fernández B., Port A., Pascual R., Monroy X., Vidal-Torres A., Serafini M.T., et al. 4-Aryl-1-Oxa-4,9-Diazaspiro[5.5]Undecane Derivatives as Dual μ-Opioid Receptor Agonists and σ1 Receptor Antagonists for the Treatment of Pain. J. Med. Chem. 2020;63:2434–2454. doi: 10.1021/acs.jmedchem.9b01256. [DOI] [PubMed] [Google Scholar]
- 140.García M., Virgili M., Alonso M., Alegret C., Farran J., Fernández B., Bordas M., Pascual R., Burgueño J., Vidal-Torres A., et al. Discovery of EST73502, a Dual μ-Opioid Receptor Agonist and σ1 Receptor Antagonist Clinical Candidate for the Treatment of Pain. J. Med. Chem. 2020;63:15508–15526. doi: 10.1021/acs.jmedchem.0c01127. [DOI] [PubMed] [Google Scholar]
- 141.Fulgenzi G., Graciotti L., Faronato M., Soldovieri M.V., Miceli F., Amoroso S., Annunziato L., Procopio A., Taglialatela M. Human Neoplastic Mesothelial Cells Express Voltage-Gated Sodium Channels Involved in Cell Motility. Int. J. Biochem. Cell Biol. 2006;38:1146–1159. doi: 10.1016/j.biocel.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 142.Kim F.J., Maher C.M. Sigma1 Pharmacology in the Context of Cancer. Handb. Exp. Pharmacol. 2017;244:237–308. doi: 10.1007/164_2017_38. [DOI] [PubMed] [Google Scholar]
- 143.Brackenbury W.J., Djamgoz M.B.A., Isom L.L. An Emerging Role for Voltage-Gated Na+ Channels in Cellular Migration: Regulation of Central Nervous System Development and Potentiation of Invasive Cancers. Neuroscientist. 2008;14:571–583. doi: 10.1177/1073858408320293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Abate C., Niso M., Abatematteo F.S., Contino M., Colabufo N.A., Berardi F. PB28, the Sigma-1 and Sigma-2 Receptors Modulator With Potent Anti–SARS-CoV-2 Activity: A Review About Its Pharmacological Properties and Structure Affinity Relationships. Front. Pharmacol. 2020;11:1–11. doi: 10.3389/fphar.2020.589810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Riganas S., Papanastasiou I., Foscolos G.B., Tsotinis A., Dimas K., Kourafalos V.N., Eleutheriades A., Moutsos V.I., Khan H., Margarita P., et al. New Adamantane Derivatives with Sigma Affinity and Antiproliferative Activity. Med. Chem. 2012;8:569–586. doi: 10.2174/157340612801216201. [DOI] [PubMed] [Google Scholar]
- 146.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Berardi F., Ferorelli S., Abate C., Colabufo N.A., Contino M., Perrone R., Tortorella V. 4-(Tetralin-1-Yl)- and 4-(Naphthalen-1-Yl)Alkyl Derivatives of 1-Cyclohexylpiperazine as σ Receptor Ligands with Agonist σ 2 Activity. J. Med. Chem. 2004;47:2308–2317. doi: 10.1021/jm031026e. [DOI] [PubMed] [Google Scholar]
- 148.Pati M.L., Hornick J.R., Niso M., Berardi F., Spitzer D., Abate C., Hawkins W. Sigma-2 Receptor Agonist Derivatives of 1-Cyclohexyl-4-[3-(5-Methoxy-1,2,3,4-Tetrahydronaphthalen-1-Yl)Propyl]Piperazine (PB28) Induce Cell Death via Mitochondrial Superoxide Production and Caspase Activation in Pancreatic Cancer. BMC Cancer. 2017;17:51. doi: 10.1186/s12885-016-3040-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liu C.C., Yu C.F., Wang S.C., Li H.Y., Lin C.M., Wang H.H., Abate C., Chiang C.S. Sigma-2 Receptor/TMEM97 Agonist PB221 as an Alternative Drug for Brain Tumor. BMC Cancer. 2019;19:473. doi: 10.1186/s12885-019-5700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Niso M., Abate C., Contino M., Ferorelli S., Azzariti A., Perrone R., Colabufo N.A., Berardi F. Sigma-2 Receptor Agonists as Possible Antitumor Agents in Resistant Tumors: Hints for Collateral Sensitivity. ChemMedChem. 2013;8:2026–2035. doi: 10.1002/cmdc.201300291. [DOI] [PubMed] [Google Scholar]
- 151.Pati M.L., Niso M., Ferorelli S., Abate C., Berardi F. Novel Metal Chelators Thiosemicarbazones with Activity at the σ2 Receptors and P-Glycoprotein: An Innovative Strategy for Resistant Tumor Treatment. RSC Adv. 2015;5:103131–103146. doi: 10.1039/C5RA19857G. [DOI] [Google Scholar]
- 152.Pati M.L., Niso M., Spitzer D., Berardi F., Contino M., Riganti C., Hawkins W.G., Abate C. Multifunctional Thiosemicarbazones and Deconstructed Analogues as a Strategy to Study the Involvement of Metal Chelation, Sigma-2 (σ2) Receptor and P-Gp Protein in the Cytotoxic Action: In Vitro and in Vivo Activity in Pancreatic Tumors. Eur. J. Med. Chem. 2018;144:359–371. doi: 10.1016/j.ejmech.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Abate C., Niso M., Lacivita E., Mosier P.D., Toscano A., Perrone R. Analogues of σ Receptor Ligand 1-Cyclohexyl-4-[3-(5-Methoxy-1,2,3,4- Tetrahydronaphthalen-1-Yl)Propyl]Piperazine (PB28) with Added Polar Functionality and Reduced Lipophilicity for Potential Use as Positron Emission Tomography Radiotracers. J. Med. Chem. 2011;54:1022–1032. doi: 10.1021/jm1013133. [DOI] [PubMed] [Google Scholar]
- 154.Berardi F., Abate C., Ferorelli S., Uricchio V., Colabufo N.A., Niso M., Perrone R. Exploring the Importance of Piperazine N-Atoms for σ2 Receptor Affinity and Activity in a Series of Analogs of 1-Cyclohexyl-4-[3-(5-Methoxy-1,2,3,4-Tetrahydronaphthalen-1-Yl)-Propyl]Piperazine (PB28) J. Med. Chem. 2009;52:7817–7828. doi: 10.1021/jm9007505. [DOI] [PubMed] [Google Scholar]
- 155.Brent P.J., Pang G., Little G., Dosen P.J., Van Helden D.F. The Sigma Receptor Ligand, Reduced Haloperidol, Induces Apoptosis and Increases Intracellular-Free Calcium Levels [Ca2+]i in Colon and Mammary Adenocarcinoma Cells. Biochem. Biophys. Res. Commun. 1996;219:219–226. doi: 10.1006/bbrc.1996.0208. [DOI] [PubMed] [Google Scholar]
- 156.Nieto F.R., Cendán C.M., Sánchez-Fernández C., Cobos E.J., Entrena J.M., Tejada M.A., Zamanillo D., Vela J.M., Baeyens J.M. Role of Sigma-1 Receptors in Paclitaxel-Induced Neuropathic Pain in Mice. J. Pain. 2012;13:1107–1121. doi: 10.1016/j.jpain.2012.08.006. [DOI] [PubMed] [Google Scholar]
- 157.Nieto F.R., Cendán C.M., Cañizares F.J., Cubero M.A., Vela J.M., Fernández-Segura E., Baeyens J.M. Genetic Inactivation and Pharmacological Blockade of Sigma-1 Receptors Prevent Paclitaxel-Induced Sensory-Nerve Mitochondrial Abnormalities and Neuropathic Pain in Mice. Mol. Pain. 2014;10:1–12. doi: 10.1186/1744-8069-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hashimoto K. Potential Role of the Sigma-1 Receptor Chaperone in the Beneficial Effects of Donepezil in Dementia with Lewy Bodies. Clin. Psychopharmacol. Neurosci. 2013;11:43–44. doi: 10.9758/cpn.2013.11.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Mangiatordi G.F., Intranuovo F., Delre P., Abatematteo F.S., Abate C., Niso M., Creanza T.M., Ancona N., Stefanachi A., Contino M. Cannabinoid Receptor Subtype 2 (CB2R) in a Multitarget Approach: Perspective of an Innovative Strategy in Cancer and Neurodegeneration. J. Med. Chem. 2020;63:14448–14469. doi: 10.1021/acs.jmedchem.0c01357. [DOI] [PubMed] [Google Scholar]
- 160.Abate C., Ferorelli S., Contino M., Marottoli R., Colabufo N.A., Perrone R., Berardi F. Arylamides Hybrids of Two High-Affinity σ 2 Receptor Ligands as Tools for the Development of PET Radiotracers. Eur. J. Med. Chem. 2011;46:4733–4741. doi: 10.1016/j.ejmech.2011.05.057. [DOI] [PubMed] [Google Scholar]