

Abstract
Stemona alkaloids contain family members with diverse structural scaffolds. Many of them feature a γ-lactam ring embedded in their characteristic 5-7-5 fused tricyclic core. Herein, a pyrrole strategy was developed to enable the total syntheses of three Stemona alkaloids: ( ± )stemoamide, ( ± )tuberostemoamide, and ( ± )sessilifoliamide A. In these cases, a substituted pyrrole was used as the γ-lactam precursor. A sequential pyrrole oxidation and enamide reduction were realized to convert the pyrrole to the corresponding γ-lactam in those three natural products. The use of a pyrrole in an early stage of the synthesis offers the advantage of rapid construction of the key intermediates by exploiting its nucleophilicity.
Graphical Abstract
The Stemona alkaloids (also called “Bai Bu” alkaloids1a) are a large family of natural products isolated from the Stemona genus including S. tuberosa, S. japonica, and S. sessilifolia. These species are often used as antitussive herb medicines in east Asia. So far, over 215 Stemona alkaloids have been isolated. They are classified into eight groups: stenine, stemoamide, tuberostemospironine, stemonamine, parvistemoline, stemofoline, stemocurtisine, and a miscellaneous group.1 These natural products feature either a hidden pyrrolo[1,2-a]azepine or a pyrido[1,2-a]azepine core often embedded in a polycyclic skeleton. Despite the long time uses of the Stemona genus as herb medicines, biological evaluation of these purified Stemona alkaloids is quite limited. Nevertheless, the Stemona alkaloids have attracted a significant amount of synthetic attention due to their characteristic and diverse structures.2 These synthetic efforts have facilitated biological function studies of these alkaloids by providing precious materials as well as their synthetic analogs.3 Among the groups of the Stemona alkaloids, total syntheses of the stemoamide group members are quite limited. While stemoamide (1, Figure 1) itself has been a popular target for total synthesis,4 only a few more complex group members have been synthesized. Notably, stemonine (2) with a γ-butyrolactone appended on the pyrrolidine was synthesized by the groups of Williams and Chida/Sato in 20035 and 2017,6 respectively. Chida and Sato also achieved the total synthesis of saxorumamide (3) with an α,β-unsaturated γ-butyrolactone linked on the tetrahydrofuran moiety.
Figure 1.

Selected stemoamide alkaloids.
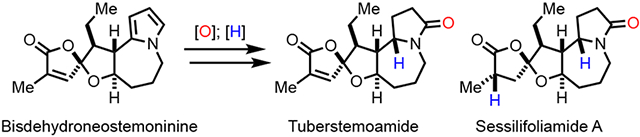
We recently took note of members of the stemoamide group with a unique oxaspirolactone moiety (4-9, Figure 1) and applied our palladium-catalyzed carbonylative spirolactonization of hydroxycyclopropanol7 to build this key structural moiety. Our initial efforts have led to the total syntheses of ( ± )bisdehydroneostemoninine(4) and ( ± ) bisdehydrostemoninine (5) with a pyrrole moiety (Figure 2A).8 The pyrrole group has significantly facilitated our synthesis by enabling the formation of two key C-C bonds due to its nucleophilicity. Meanwhile, we envisioned the possibility of synthesizing the γ-lactam or pyrrolidine-containing stemoamide group members such as tuberostemoamide (7), sessilifoliamide A (8), and stemoninine (9) by chemical manipulations of the pyrrole group. Herein, we report our total syntheses of stemoamide (1), tuberostemoamide (7), and sessilifoliamide A (8) in their racemic forms via a pyrrole oxidation strategy. Notably, while we were optimizing our syntheses, Wang and co-workers reported their elegant asymmetric total syntheses of tuberostemoamide (7), sessilifoliamide A (8), and their synthetic analogs via a completely different strategy.9 Further biological evaluation led them to identify 11,13-bis-epi-sessilifoliamide A as a butyrylcholinesterase (BChE) inhibitor for treating neurodegenerative disorders, which highlights the importance of chemical syntheses of these stemoamide alkaloids.
Figure 2.

Our previous total synthesis and current synthetic plan.
Our previous syntheses of bisdehydroneostemoninine (4) and bisdehydrostemoninine (5) used tricyclic pyrrole 10 as a key intermediate (Figure 2A), from which hydroxycyclopropanol 11 was prepared in two steps. A palladium-catalyzed carbonylative spirolactonization delivered 12 in good yield, which eventually led to bisdehydroneostemoninine (4) and bisdehydrostemoninine (5). In order to realize the conversion of bisdehydroneostemoninine (4) to tuberostemoamide (7) and sessilifoliamide A (8), a mild oxidation reaction needs to be identified to install the γ-lactam carbonyl group because the spirolactone moiety is very labile under either acidic or basic conditions (Figure 2B). While oxidation of indole to oxindole is prevalent in the literature with a broad range of reaction conditions,10 the corresponding case for pyrrole oxidation is extremely rare. Our literature resulted in one example using more than 90% pure mCPBA as the oxidant.11 The reaction presumably goes through an epoxidation of the pyrrole followed by an epoxide-carbonyl rearrangement to a dihydro-2H-pyrrol-2-one intermediate. In our case, the dihydro-2H-pyrrol-2-one intermediate (14 or 15) needs to be reduced, which is nontrivial as well. If intermediate 14 is produced, the ethyl group will sterically block the top side to make the desirable reduction more difficult.
With the above concerns in mind, we decided to use compound 10 as a model substrate, which could also lead to a total synthesis of stemoamide (1). Compound 10 was previously synthesized by us in 6 steps at gram scale from commercially available starting materials 16 (Scheme 1A). The key steps involve Clauson-Kaas pyrrole synthesis, Weinreb ketone synthesis, Luche reaction, cross metathesis, and Lewis acid-promoted tandem Friedel-Crafts cyclization and lactonization (cf. 17→10). The use of the pyrrole is essential for the success of the last tandem process due to its strong nucleophilicity. When compound 10 was subjected to the oxidation conditions with commercially available mCPBA (less than 77% purify), the reaction was quite messy with only a small amount of product 18 obtained. Further condition optimization failed to improve the reaction yield. Dimethyldioxirane (DMDO) was used as well, but only a trace amount of product was observed. We then decided to purify the commercial mCPBA by washing with buffer solution and evaporation with caution.12 The purified mCPBA gave better results. When 1,2-dichloroethane (DCE) was used as solvent at −35 °C, we were able to get consistent oxidation of 10 to 18 in 65% yield. Pd/C-catalyzed hydrogenation of 18 gave 19 in almost quantitatively yield with the desired stereochemistry at the newly generated carbon center. Regio- and stereo-selective α-methylation on the γ-butyrolactone completed the total synthesis of ( ± )stemoamide (1). The analytic data, including 1H, 13C, and HRMS of our synthetic sample, match well with the reported ones.
Scheme 1.

Approaches for the total synthesis of ( ± ) stemoamide.
We then wondered how the α-methyl group on the γ-butyrolactone will influence the oxidation and reduction steps, because for the proposed syntheses of tuberostemoamide (7) and sessilifoliamide A (8), there is an ethyl group on the tetrahydrofuran ring. Therefore, α-methylation of compound 10 was explored. Surprisingly, unlike the α-methylation of 19, in this case, a 9:1 mixture of 20 and 21 was obtained kinetically favoring 20 with undesired stereochemistry at the α-position. This result indicates that the stereochemistry of the lactam γ-position (cf. 19) is important to control the stereochemistry of the lactone α-methylation. Epimerization of 20 under the conditions of DBU in MeOH at 50 °C gave 21 in 92% yield. To our delight, the purified mCPBA oxidation worked smoothly with 21 bearing the α-methyl group. However, reduction of the double bond became problematic. The aforementioned Pd/C-catalyzed hydrogenation resulted in a 1:1 mixture of ( ± ) stemoamide (1) and its isomer. Thus, the stereochemistry of the lactone α-position does significantly influence the reduction. We then opted for a hydride reduction of the acyl iminium ion derived from 22 under mild acidic conditions with the rationale that hydride reduction could be less steric demanding, and the acyl iminium ion is highly reactive. After comprehensive investigations, we identified that the combination of NaCNBH3 with acetic acid in hexafluoro-2-propanol (HFIP) is the best for the task. A 3.6:1 mixture of ( ± )stemoamide (1) and its isomer was produced in 95% yield. Other solvents we investigated were much less effective than HFIP.13
With two different approaches to convert compound 10 to stemoamide (1) by successfully converting the pyrrole group to the corresponding γ-lactam, we began the total syntheses of tuberostemoamide (7) and sessilifoliamide A (8, Scheme 2). Following our previously reported procedures, compound 10 was advanced to bisdehydroneostemoninine (4) in six steps. To our delight, the oxaspirolactone moiety of bisdehydroneostemoninine survived the mCPBA oxidation, and γ-lactam 14 was obtained in 40% yield with 40% of starting material recovered. Our efforts to increase the conversion led to a decreased overall yield of 14. Similar to the stemoamide (1) case, with the steric effect from the ethyl group, we encountered difficulties in reducing the dihydro-2H-pyrrol-2-one with catalytic hydrogenation. When Pd/C was used in EtOH under hydrogen atmosphere, hemiaminal ethyl ether 23 was produced in almost quantitative yield. Its structure was confirmed by x-ray analysis.14 While 23 was not our desired product, it indicated that the α,β-unsaturated γ-butyrolactone could be reduced in a stereoselective manner by delivering the reducing reagent from the less hindered side to form the required stereochemistry for sessilifoliamide A (8) synthesis. Switching the solvent from EtOH to other solvents led to starting material decomposition. We then took recourse to the hydride reduction conditions established for the reduction of 22 to stemoamide (1, Scheme 1) and were able to selectively reduce the dihydro-2H-pyrrol-2-one of 14 in the presence of the α,β-unsaturated γ-butyrolactone. ( ± )Tuberostemoamide (7) was produced in 33%, which was further reduced with Pd/C-catalyzed hydrogenation in EtOH to complete the total synthesis of ( ± ) sessilifoliamide A (8). Additionally, compound 23 could be reduced to ( ± )sessilifoliamide A (8) as well by using the NaCNBH3 reduction conditions. The structural assignments for both ( ± )tuberostemoamide (7) and ( ± )sessilifoliamide A (8) were unambiguously confirmed by x-ray analysis.14
Scheme 2.

Total syntheses of ( ± )tuberostemoamide and ( ± ) sessilifoliamide A.
In summary, we have developed a pyrrole strategy to synthesize γ-lactam-containing Stemona alkaloids, including stemoamide (1), tuberostemoamide (7), and sessilifoliamide A (8) in their racemic forms. A protocol which combines a purified mCPBA oxidation with catalytic hydrogenation or NaCNBH3 reduction in HFIP was developed to achieve the task. Meanwhile, we learned that the reduction of the dihydro-2H-pyrrol-2-one intermediates derived from the corresponding mCPBA oxidation is very sensitive to the steric effect implemented by the substituents on the adjacent tetrahydrofuran ring. These syntheses together with our previous syntheses of bisdehydroneostemoninine (4), bisdehydrostemoninine (5), and their synthetic analogs, will allow us to comprehensively evaluate and understand the biological function of these Stemona alkaloids, which will be reported in due course.
Supplementary Material
ACKNOWLEDGMENT
This research was supported by NSF CAREER 1553820 and NIH R35 GM128570. The NIH P30 CA023168 is acknowledged for supporting shared NMR resources to Purdue Center for Cancer Research. The XRD data is collected on a new single crystal X-ray diffractometer supported by the NSF through the Major Research Instrumentation Program under Grant No. CHE 1625543.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures and spectra data (PDF file)
Crystallographic data for compounds 23, 7 and 8 (cif files)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Pilli RA; de Oliveira CF Nat. Prod. Rep 2000, 17, 117–127. [DOI] [PubMed] [Google Scholar]; (b) Pilli RA; Rosso GBM; de Oliveira CF Nat. Prod. Rep 2010, 27, 1908–1937. [DOI] [PubMed] [Google Scholar]; (c) Greger H Phytochem. Rev 2019, 18, 463–493. [Google Scholar]
- (2).(a) Phili RA; Rosso GB; De Oliveira MDCF Alkaloids Chem Biol 2005, 62, 77–173. [DOI] [PubMed] [Google Scholar]; (b) Alibés R; Figueredo M Eur. J. Org. Chem 2009, 2421–2435. [Google Scholar]
- (3).(a) Frankowski KJ; Setola V; Evans JM; Neuenswander B; Roth BL; Aubé J Proc. Natl. Acad. Sci. USA, 2011, 108, 6727–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McLeod MC; Singh G; Plampin JN; Rane D; Wang JL; Day VW; Aubé J Nat. Chem 2014, 6, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).(a) For a review: Brito GA; Pirovani RV Org. Prop. Proced. Int 2018, 50, 245–259. [Google Scholar]; (b) For total syntheses, see: Williams DR; Reddy JP; Amato GS Tetrahedron Lett. 1994, 35, 6417–6420. [Google Scholar]; (c) Kohno Y; Narasaka K; Bull. Chem. Soc. Jpn 1996, 69, 2063–2070. [Google Scholar]; (d) Kinoshita A; Mori M J. Org. Chem 1996, 61, 8356–8357. [Google Scholar]; (e) Jacobi PA; Lee K J. Am. Chem. Soc 1997, 119, 3409–3410. [Google Scholar]; (f) Kinoshita A; Mori M Heterocycles 1997, 46, 287–299. [Google Scholar]; (g) Jacobi PA; Lee K J. Am. Chem. Soc 2000, 122, 4295–4303. [Google Scholar]; (h) Sibi MP; Subramanian T Synlett 2004, 1211–1214. [Google Scholar]; (i) Olivo HF; Tovar Miranda R; Barragań E J. Org. Chem 2006, 71, 3287–3290. [DOI] [PubMed] [Google Scholar]; (j) Torssell S; Wanngren E; Somfai P J. Org. Chem 2007, 72, 4246–4249. [DOI] [PubMed] [Google Scholar]; (k) Bates RW; Sridhar S Synlett 2009, 1979–1981. [Google Scholar]; (l) Honda T; Matsukawa T; Takahashi K Org. Biomol. Chem 2011, 9, 673–675. [DOI] [PubMed] [Google Scholar]; (m) Wang Y; Zhu L; Zhang Y; Hong R Angew. Chem. Int. Ed 2011, 50, 2787–2790. [DOI] [PubMed] [Google Scholar]; (n) Mi X; Wang Y; Zhu L; Wang R; Hong R Synthesis 2012, 44, 3432–3440. [Google Scholar]; (o) Li Z; Zhang L; Qiu FG Asian J. Org. Chem 2014, 3, 52–54. [Google Scholar]; (p) Nakayama Y; Maeda Y; Hama N; Sato T; Chida N Synthesis 2016, 48, 1647–1654. [Google Scholar]; (q) For formal syntheses, see: Gurjar MK; Reddy DS Tetrahedron Lett. 2002, 43, 295–298. [Google Scholar]; (r) Bogliotti N; Dalko PI; Cossy J Synlett 2005, 349–351. [Google Scholar]; (s) Bogliotti N; Dalko PI: Cossy J J. Org. Chem 2006, 71, 9528–9531. [DOI] [PubMed] [Google Scholar]; (t) Bogliotti N; Dalko PI; Cossy J Synlett 2006, 2664–2666. [Google Scholar]; (u) Chavan SP; Harale KR; Puranik VG; Gawade RL Tetrahedron Lett. 2012, 53, 2647–2650. [Google Scholar]; (v) Muňoz-Bascoń J; Hernańdez-Cervantes C; Padial NM; Alvarez-Corral M; Rosales A; Rodríguez-García I Oltra JE Chem. Eur. J 2014, 20, 801–810. [DOI] [PubMed] [Google Scholar]; (w) Brito GA; Sarotti AM; Wipf P; Pilli RA Tetrahedron Lett. 2015, 56, 6664–6668. [Google Scholar]
- (5).Williams DR; Reddy JP Amato GS Tetrahedron Lett. 1994, 35, 6417–6420. [Google Scholar]
- (6).Yoritate M; Takahashi Y; Tajima H; Ogihara C; Yokayama T; Soda Y; Oishi T; Sato T; Chida N J. Am. Chem. Soc 2017, 139, 18386–18391. [DOI] [PubMed] [Google Scholar]
- (7).Davis DC; Walker KL; Hu C; Zare RN; Waymouth RM; Dai MJ J. Am. Chem. Soc 2016, 138, 10693–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma K; Yin X; Dai MJ Angew. Chem. Int. Ed 2018, 57, 15209–15212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Hou Y; Shi T; Yang Y; Fan X; Chen J; Gao F; Wang Z Org. Lett 2019, 21, 2952–2956. [DOI] [PubMed] [Google Scholar]
- (10).(a) Dalpozzo R; Bartoli G; Bencivenni G Chem. Soc. Rev 2012, 41, 7247–7290. [DOI] [PubMed] [Google Scholar]; (b) Mundal DA; Sarpong R Org. Lett 2013, 15, 4952–4955. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Xu J; Liang L; Zheng H; Chi YB; Tong R Nat. Commun 2019, 10, 4754. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hinman RL Bauman CP J. Org. Chem 1964, 29, 1206–1215. [Google Scholar]
- (11).Molinari AJ; Trybulski EJ; Bagli J; Croce S; Considine J; Qi J; Ali K; DeMaio W; Lihotze L; Cochran D Bioorg. Med. Chem. Lett 2007, 17, 5796–5800. [DOI] [PubMed] [Google Scholar]
- (12).Horn A; Kazmaier U Eur. J. Org. Chem 2018, 2531–2536. [Google Scholar]
- (13).Colomer I; Chamberlain AER; Haughey MB; Donohoe TJ Nat. Rev. Chem 2017, 1, 0088. [Google Scholar]
- (14).CCDC 1996683, 1996684, and 1996685 contains the supplementary crystallographic data for compounds 23, 7 and 8, respectively.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.