

Abstract
A variety of different signaling pathways are necessary for development and maintenance of the human auditory system. Normal hearing allows for the detection of soft sounds within the frequency range of 20 to 20,000 Hz, but more importantly to perceive the human voice frequency band of 250 to 6,000 Hertz. Loss of hearing is common, and is a clinically heterogeneous disorder that can be caused by environmental factors such as exposure to loud noise, infections and ototoxic drugs. In addition, variants of hundreds of genes have been reported to disrupt processes required for hearing. Noncoding regulatory variants and variants of additional genes necessary for hearing remain to be discovered as many individuals with inherited deafness are without a genetic diagnosis, despite the advent of whole exome sequencing. Here, we discuss in detail some of these deafness-causing variants of genes encoding a ligand or its receptor. Spotlighted in this review are three growth factor-receptor-pairs EDN3/EDNRB, HGF/MET and JAG/NOTCH, which individually are necessary for normal hearing. We also offer our perspective on unanswered questions, future challenges and potential opportunities for treatments emerging from molecular genetic and mechanistic studies of deafness due to these causes.
Keywords: hearing, inner ear defects, EDN3, EDNRB, HGF, MET, JAG, NOTCH
Graphical Abstract
1. INTRODUCTION
Impaired sound detection is a prevalent deficit of older adults as well as for 1 in 500 to 1 in 2,000 young children.1,2 Deficits in hearing often result from dysfunction of one of the complex, delicate structures or intricate signaling pathways in the inner ear.3 Hearing loss can be conductive, sensorineural or mixed, depending on whether the defect is in the middle ear, inner ear or both, respectively (Figure 1A). The complex structure of inner ear develops from the otic placode next to the hindbrain,4 which invaginates to form the otic vesicle (Figure 1B). This fluid-filled cyst then undergoes organogenesis to give rise to neurons of the vestibular and cochlear ganglions as well as non-sensory and sensory epithelia of the membranous labyrinth (Figure 1B). In the mature cochlea, there are three rows of sound-amplifying outer hair cells (OHCs) and a single row of inner hair cells (IHCs) responsible for mechano-chemical transduction of sound. The apical surfaces of hair cells are topped by F-actin packed stereocila (Figure 1C).5
FIGURE 1.
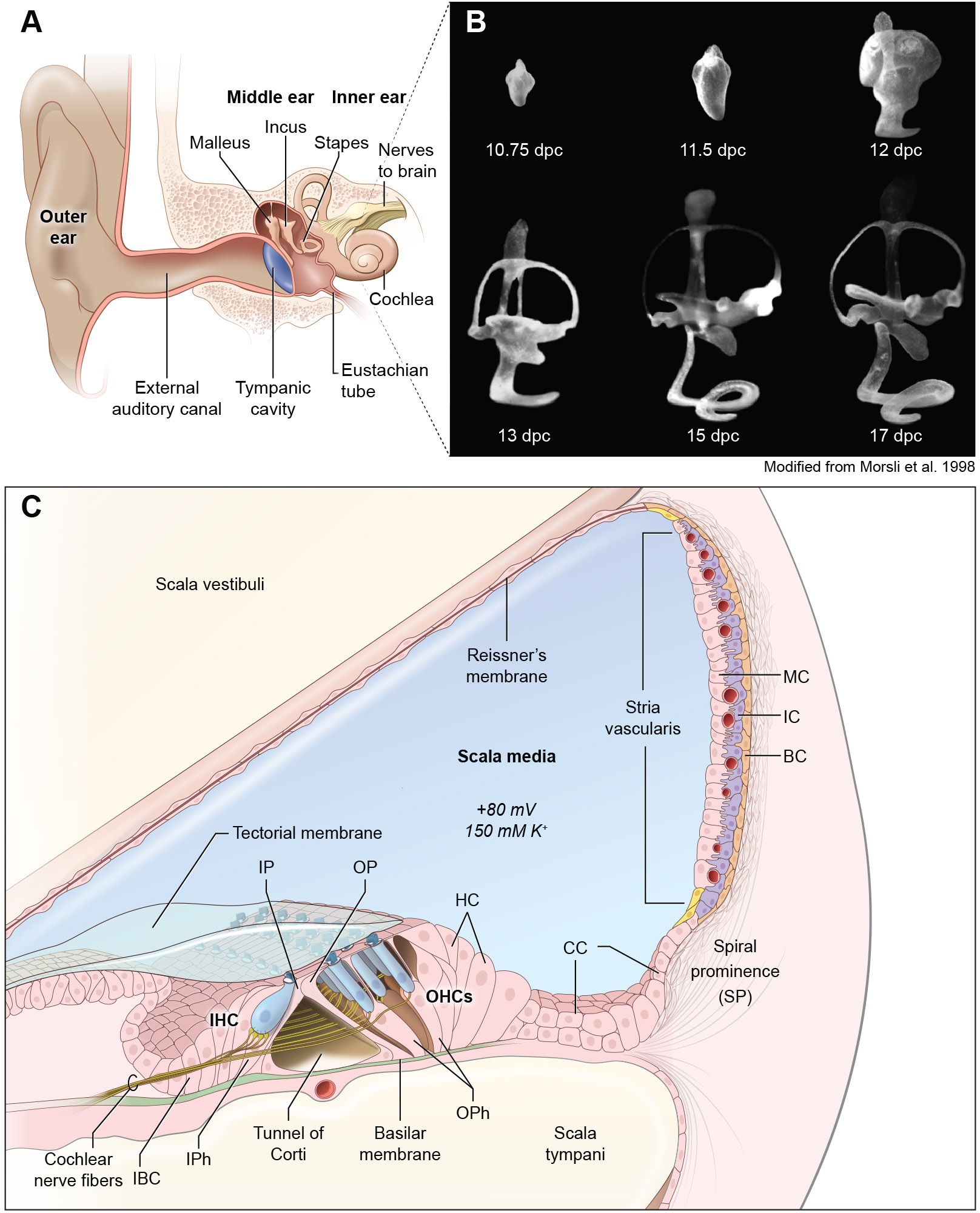
Development and structure of the ear. (A) Structure of the human ear showing its three main parts, outer, middle and inner ear. (B) Paint-filled mouse membranous labyrinths at embryonic days 10.75 days-postcoitum to postnatal day 1 (P1). Lateral views are shown. Scale bar, 200 μm. (C) Diagram of a cross-section of the cochlea. The roof of the cochlear duct is formed by two layers of flattened cells comprising Reissner’s membrane, while the base is formed by the basilar membrane, which separate the cochlear duct (scala media) from the scala vestibuli and the scala tympani. The three rows of outer hair cells, one row of inner hair cells and different types of supporting cells and the stria vascularis are shown. IHC; Inner Hair Cells, OHC, Outer Hair Cells, IP, Inner Pillar cells, OP; Outer Pillar Cells, HC; Hensen’s Cells, CC; Claudius Cells, BC; Basal Cells, IC; Intermediate cells, MC; Marginal Cells, IBC; inner border cells.
Many pathogenic variants in genes encoding growth factors, receptors, co-receptors, adaptors and effectors have been identified in analyses of hearing loss in humans (Tables 1 and 2). Receptors and ligands, for example, LGR5 and BDNF, variants of which have not been reported to cause hearing loss in humans but have important role in development of auditory system6,7 and ligand-gated ion channel receptors, such as P2XR28 are not included in this review. Our focus is on variants in genes associated with human deafness. However, confidence in the evidence for pathogenicity of variants in the deafness causing genes differs. A ClinGen Hearing Loss Gene Curation Expert Panel has concluded that some are likely not bona fide pathogenic variants,9 but rather represent coincidental co-occurrence with deafness in individual families. This appears to be the case for variants in MYO1A, MYO1C, MYO1F and TSPEAR reported in deafness.9,10
TABLE 1.
Growth factors, receptors and related proteins implicated in nonsyndromic hearing loss
| Gene | OMIM | Description; cellular location | Protein/alias | Function | HL type | Locus† |
|---|---|---|---|---|---|---|
| ADCY1 | 103072 | Adenylate Cyclase 1; transmembrane | ADCY1/AC1 | Generates the second messenger cAMP | Prelingual, sensorineural, bilateral, profound | DFNB44 |
| CCDC50 | 611051 | Coiled Coil Domain-Containing protein 50; cytoplasmic | CCDC50/YMER/C3ORF6 | Inhibits downregulation of EGFR/Effector of EGF mediated signaling | Postlingual, progressive, sensorineural, mid-frequency> | DFNA44 |
| DCDC2 | 605755 | Double Cortin Domain-Containing protein 2; cytoplasmic | DCDC2/RU2/RU2S | Inhibits WNT signaling | Prelingual, sensorineural, profound | DFNB66 |
| ELMOD3 | 615427 | ELMO/ced12 Domain-containing protein 3; cytoplasmic | ELMOD3/RBED1 | GTPase-activating protein (GAP) for small GTPases | Prelingual, mixed, severe to profound | DFNB88 |
| EPS8 | 600206 | Epidermal growth factor receptor Pathway Substrate 8; cytoplasmic | EPS8 | Elongation of stereocilia by actin remodeling | Prelingual, sensorineural, bilateral, profound | DFNB102 |
| EPS8L2 | 614988 | Epidermal growth factor receptor Pathway Substrate 8-Like protein 2; cytoplasmic |
EPS8L2 | Maintenance of stereocilia, actin remodeling | Childhood onset, progressive, bilateral, sensorineural, moderate to profound | DFNB106 |
| ESRRB | 602167 | EStrogen-Related Receptor, Beta; cytoplasmic | ESRRB/ESRL2/ERR2 | Epithelial cell fate, development of the stria vascularis | Prelingual or postlingual, sensorineural, bilateral, severe to profound | DFNB35 |
| GAB1 | 604439 | GRB2-Associated Binding protein 1; cytoplasmic | GAB1 | Mediator of HGF, IGF1 signaling | Prelingual, bilateral, sensorineural, profound | DFNB26 |
| GIPC3 | 608792 | GIPC PDZ domain-containing family, member 3; cytoplasmic | GIPC3/GAIP C-terminus-interacting protein 3 | Predicted to modulate WNT signaling | Prelingual, bilateral, sensorineural, severe to profound | DFNB15/DFNB72/DFNB95 |
| GRAP | 604330 | Growth factor Receptor-bound protein 2 (Grb2)-related Adaptor Protein; cytoplasmic | GRAP | Activates RAS signaling | Prelingual, bilateral, sensorineural, severe to profound | DFNB114 |
| HGF | 142409 | Hepatocyte Growth Factor; secreted | HGF/SF | Incorporation of neural crest cells into the stria vascularis | Prelingual, sensorineural, bilateral, moderate to severe or severe to profound | DFNB39 |
| IFNLR1 | 607404 | Interferon-Lambda Receptor 1, transmembrane | IFNLR1/ IL28RA | Functions in Jak/STAT pathway | Postlingual, sensorineural, bilateral, progressive, moderate to profound | DFNA2C |
| ILDR1 | 609739 | Immunoglobulin-Like Domain-containing Receptor 1; transmembrane and cytoplasmic | ILDR1 | Structural role at the tricellular tight junctions; receptor role in the inner ear unknown | Prelingual, sensorineural, bilateral, moderate to profound or severe to profound | DFNB42 |
| KITLG | 184745 | KIT Ligand; transmembrane and secreted | KITLG/KL/KITL/MGF/SCF/SF | Proliferation, migration from the neural crest, differentiation of melanoblasts | Prelingual, sensorineural, unilateral or asymmetric | DFNA69 |
| LRP5 | 603506 | Low density lipoprotein Receptor-related Protein 5; transmembrane | LRP5/LRP7/LR3 | Co-receptor during WNT signaling | Sensorineural, progressive, childhood onset, bilateral, low and mid frequency | -, recessive |
| MET | 164860 | Mesenchymal EpithelialTransition factor; transmembrane | MET/HGFR | Incorporation of neural crest cells into the stria vascularis | Prelingual, sensorineural, bilateral, severe | DFNB97 |
| NLRP3 | 606416 | NLR family, Pyrin domain-containing 3; cytoplasmic | NLRP3/CIAS1/NALP3/PYPAF1/ AII/AVP Receptor-Like | Leads to events which end in production of cytokine IL-1β | Postlingual, bilateral, progressive | DFNA34 |
| PDE1C | 602987 | Phosphodiesterase 1C; cytoplasmic | PDE1C/HCAM3 | Regulates levels of second messengers cAMP and cGMP | Postlingual, progressive, bilateral, mild to profound | DFNA74 |
| PTPRQ | 603317 | Protein-Tyrosine Phosphatase, Receptor-type, Q; transmembrane and cytoplasmic | PTPRQ/ PTPGMC1 | Receptor role unknown, structural role as shaft connector | Sensorineural, variable onset, progressive, bilateral, mild to moderate | DFNA73 |
| PTPRQ | 603317 | Protein-Tyrosine Phosphatase, Receptor-type, Q; transmembrane and cytoplasmic | PTPRQ/ PTPGMC1 | Receptor role unknown, structural role as shaft connector | Sensorineural, childhood onset, progressive, bilateral, moderate to profound | DFNB84A |
| ROR1 | 602336 | Receptor tyrosine kinase-like Orphan Receptor 1; transmembrane | ROR1/ NTRKR1 | Innervation of auditory hair cells in response to WNT5A signaling | Prelingual, bilateral, sensironeural, profound, auditory neuropathy, | DFNB108 |
| S1PR2 | 605111 | Sphingosine-1-Phosphate Receptor 2; transmembrane & cytoplasmic | S1PR2/EDG5/S1P2 | Development of stria vascularis | Congenital, sensorineural, bilateral, profound | DFNB68 |
DFNA, dominant hearing loss, DFNB, recessive hearing loss, OMIM, Online Mendelian Inheritance in Man (https://www.omim.org/). A comprehensive source of gene expression data in mouse and zebrafish auditory systems are available on the gEAR Portal, https://umgear.org/.
TABLE 2.
Growth factors, receptors and related proteins implicated in syndromic hearing loss
| Gene | OMIM | Description; cellular location | Protein/alias | Function | Hearing loss type | Locus; mode of inheritance |
|---|---|---|---|---|---|---|
| ACVR1 | 102576 | ACtiVin a Receptor, type I; transmembrane | ACVR1/ ACVRLK2/ ALK2 | General role as receptor for Activin, BMP or TGFB mediated SMAD signaling | Variable age of onset, conductive or sensorineural, mild to severe | Fibrodysplasia ossificans progressiva; dominant |
| ADGRV1 | 602851 | ADhesion G protein-coupled Receptor V1; transmembrane | ADGRV1/ GPR98/ MASS1/ VLGR1/ KIAA0686 | Receptor function unknown, component of stereocilia ankle links | Sensorineural, bilateral, progressive, moderate to severe | Usher syndrome 2C; recessive |
| BMP2 | 617877 | Bone Morphogenetic Protein 2, secreted | BMP2/BMP2A | Endochondral ossification | External ear malformations, conductive | Short stature, facial dysmorphism, and skeletal anomalies with or without cardiac anomalies; dominant |
| BMP2 | 617877 | Bone Morphogenetic Protein 2, secreted | BMP2/BMP2A | Endochondral ossification | Conductive | Otosclerosis |
| BMP4 | 112262 | Bone Morphogenetic Protein 4; secreted | BMP4/BMP2B/BMP2B1 | Specification of sensory and nonsensory regions of cochlea | Conductive | Otosclerosis |
| BMP4 | 112262 | Bone Morphogenetic Protein 4; secreted | BMP4/BMP2B/BMP2B1 | Specification of sensory and nonsensory regions of cochlea | Sensorineural, mild to moderate | Stickler syndrome and renal dysplasia; dominant |
| BMP7 | 112267 | Bone Morphogenetic Protein 7; secreted | BMP7/OP1 | Specifies tonotopic axis in developing inner ear | Sensorineural, bilateral, moderate | Ocular, brain, ear, palate, and skeletal anomalies; dominant |
| BRAF | 164757 | v-RAF murine sarcoma viral oncogene homologue B1; cytoplasmic, cell membrane, nuclear | BRAF/BRAF1/RAFB1 | Regulator of MAPK/ERK signaling pathway | Outer ear malformation, sensorineural | LEOPARD syndrome 3; dominant |
| DVL1 | 601365 | Dishevelled 1, cytoplasmic | DVL1/DVL | Functions downstream of WNT signaling | Outer ear malformation, conductive or sensorineural | Robinow syndrome, autosomal dominant 2; dominant |
| DVL3 | 601368 | Dishevelled 3, cytoplasmic | DVL3 | Functions downstream of WNT signaling | Rare ear malformation and hearing loss | Robinow syndrome, autosomal dominant 3; dominant |
| EDN3 | 131242 | Endothelin 3, secreted | EDN3/ET3 | Development of neural crest and its cell lineages | Sensorineural, Mild to profound, unilateral or bilateral | Waardenburg syndrome, type 4B, WS4B; dominant or recessive |
| EDNRA | 131243 | Endothelin Receptor, type A; transmembrane | EDNRA/ETA/ETRA | Development of neural crest and its cell lineages | Outer ear malformation, childhood onset, conductive, moderate | Mandibulofacial dysostosis with alopecia; dominant |
| EDNRB | 131244 | Endothelin Receptor, type B; transmembrane | EDNRB/ETB/ETBR | Development of neural crest and its cell lineages | Sensorineural, mild to profound, unilateral or bilateral | Waardenburg syndrome, type 4A, WS4A, WS1, WS2; dominant and recessive Waardenburg syndrome, type 2A, WS2A; dominant; ABCD syndrome; recessive |
| ESRRG | 602969 | EStrogen Related Receptor Gamma, nuclear | ESRRG/ERR3 | Unknown | Prelingual, sensorineural, bilateral, mild to moderate | Hearing loss with mild intellectual disability; recessive |
| FGF3 | 164950 | Fibroblast Growth Factor 3, secreted | FGF3/INT2 | Induction of inner ear fate | Outer and inner ear anomalies, prelingual, sensorineural, profound | Congenital deafness, inner ear agenesis, microtia, microdontia; recessive |
| FGF10 | 602115 | Fibroblast Growth Factor 10; secreted | FGF10 | Specification of nonsensory regions of inner ear | Outer ear malformation, conductive, sensorineural or mixed hearing loss | LADD syndrome; dominant |
| FGFR1 | 136350 | Fibroblast Growth Factor Receptor 1; transmembrane | FGFR1/FLT2/FLG | Early cell proliferation of cochlea, differentiation of sensory epithelium | Outer ear malformation in some, unilateral | Kallmann syndrome 2; dominant |
| FGFR2 | 176943 | Fibroblast Growth Factor Receptor 2; transmembrane | FGFR2/TK14 | Essential for inner ear morphogenesis | Outer ear malformations may be present, prelingual or postlingual, conductive or sensorineural or mixed, bilateral mild to severe | Apert syndrome, Crouzon syndrome CFD1; LADD syndrome; Saethre-Chotzen Syndrome, Pfeiffer syndrome type 3; Dominant |
| FGFR3 | 134934 | Fibroblast Growth Factor Receptor 3; transmembrane | FGFR3 | Controls fate of supporting cells in organ of Corti | Sensorineural or mixed, mild to moderate | CATSHL syndrome, LADD syndrome, Muenke syndrome, SADDAN; dominant or recessive |
| GDF6 | 601147 | Growth/Differentiation Factor 6; secreted | GDF6/CDMP2 | Growth of cartilage elements, SMAD signaling | Outer ear malformation, conductive or sensorineural, mild to moderate | Klippel-Feil syndrome 1; dominant multiple synostoses syndrome, dominant |
| GNAI3 | 139370 | Guanine Nucleotide-binding protein, Alpha-Inhibiting activity polypeptide 3; cytoplasmic | GNAI3/ G protein | Mediator of signaling involving G protein-coupled endothelin receptor | Outer ear malformation, conductive or sensorineural, mild to moderate | Auriculocondylar syndrome 1; dominant |
| GPRASP2 | 300969 | G Protein-coupled Receptor-Associated Sorting Protein 2; cytoplasmic | GPRASP2/GASP2 | Turnover of G-protein-Coupled Receptors | External ear malformations, congenital or progressive, bilateral, mixed hearing loss, moderate to profound | DFNX7, Deafness X-linked 7, recessive |
| GPSM2 | 609245 | G Protein Signaling Modulator 2; cytoplasmic | GPSM2/LGN/PINS | Asymmetric localization of kinocilia in postmitotic hair cells | Prelingual, sensorineural, bilateral, severe to profound | Chudley-McCullough Syndrome; recessive |
| IGF1 | 147440 | Insulin-like Growth Factor I; secreted | IGF1/IGFI/ Somatomedin C | Postnatal differentiation and maturation of cochlea and central auditory neurons | Sensorineural, prelingual, bilateral, profound | Insulin-like growth factor I deficiency; recessive |
| IGF1R | 147340 | Insulin-like Growth Factor I Receptor; transmembrane | IGF1R | Postnatal differentiation and maturation of cochlea and central auditory neurons | Bilateral, profound | Insulin-like growth factor I, resistance to; dominant |
| JAG1 | 601920 | JAGged 1; transmembrane | JAG1/JAGL1 | Patterning of the organ of Corti, outer hair cell number | Mild to severe, mixed, mid-frequency | Deafness, congenital heart defects, and posterior embryotoxon; dominant |
| KIT | 164920 | V-KIT Hardy-Zuckerman 4 feline sarcoma viral oncogene homologue; transmembrane | KIT/SCFR | Proliferation, migration from the neural crest, differentiation of melanoblasts | Prelingual, sensorineural, profound | Piebaldism (Woolf’s syndrome); dominant |
| KITLG | 184745 | KIT Ligand; trasmembrane and secreted | KITLG/KL/KITL/MGF/SCF/SF | Proliferation, migration from the neural crest, differentiation of melanoblasts | Congenital, stable unilateral and asymmetric hearing loss | Waardenburg syndrome, type 2; dominant |
| LRP2 | 600073 | Low density lipoprotein Receptor-related Protein 2; transmembrane | LRP2/ Glycoprotein 330/ megalin | Endocytic receptor, Development of ear | Outer ear malformation, sensorineural, severe to profound | Donnai-Barrow syndrome; recessive |
| LRP4 | 604270 | Low density lipoprotein Receptor-related Protein 4; transmembrane | LRP4/MEGF7 | Causes narrowing of foramina of the cranial nerves which leads to hearing loss | Hearing loss present, but no details provided | Sclerosteosis 2, recessive |
| LRP5 | 603506 | Low density lipoprotein Receptor-related Protein 5; transmembrane | LRP5/LRP7/LR3 | Co-receptor during WNT signaling | Sensorineural, | Osteosclerosis, dominant |
| MAP3K7 | 602614 | Mitogen-Activated Protein Kinase kinase kinase 7; cytoplasmic | MAP3K7/ TAK1 | Mediator of TGFB and BMP signaling | Outer and inner ear malformations, conductive. bilateral | Cardiospondylocarpofacial syndrome; dominant |
| MET | 164860 | Mesenchymal EpithelialTransition factor; transmembrane | MET/HGFR | Incorporation of neural crest cells into the stria vascularis | Prelingual, sensorineural, bilateral, severe to profound | Deafness with arthrogryposis and distinctive facial features; recessive |
| NDP | 300658 | Norrin cystine knot growth factor; secreted | NDP/Norrin | Vascularization of cochlea with particular importance in stria vascularis and spiral ganglion, binds to Frizzled receptors | Sensorineural, postlingual, childhood or adult onset, bilateral, progressive, mild to profound | Norrie Disease; X-linked recessive |
| NLRP3 | 606416 | NLR family, Pyrin domain-containing 3; cytoplasmic | NLRP3/CIAS1/NALP3/PYPAF1/Cryopyrin | Leads to events which end in production of cytokine IL-1β | Postlingual, sensorineural, bilateral, progressive, severe | CINCA syndrome, Muckle-Wells syndrome; dominant |
| NOG | 602991 | NOGgin, mouse homologue of; secreted | NOG | Inhibits signaling by GDF6, BMP, and TGFB | Stapes ankylosis, conductive, rarely sensorineural, progressive | Multiple synostoses syndrome 1; dominant |
| NOTCH2 | 600275 | Drosophila homolog of Notch, 2; transmembrane | NOTCH2 | Patterning of the organ of Corti | Childhood onset, conductive, rarely sensorineural, mild to moderate | Hajdu-Cheney syndrome; dominant |
| NOTCH3 | 600276 | Drosophila homolog of Notch, 3; transmembrane | NOTCH3 | Patterning of the organ of Corti | Outer ear malformation, conductive or mixed, bilateral, moderate | Lateral meningocele syndrome; dominant |
| NSD1 | 606681 | Nuclear receptor-binding Su-var, enhancer of zeste, and trithorax Domain protein 1; nuclear | NSD1/ARA267 | unknown | Childhood onset, otitis media, conductive | Sotos syndrome 1; dominant |
| PTPN11 | 176876 | Protein-Tyrosine Phosphatase, Nonreceptor-type, 11; cytoplasmic | PTPN11/PTP2C/SHP2 | Mediates signaling by HGF, EGF through activation of RAS/MAPK pathways | Outer ear malformation, Childhood or adult onset, sensorineural or mixed, unilateral or bilateral, mild to profound | Noonan syndrome 1, LEOPARD syndrome 1; dominant |
| RAF1 | 164760 | v-Raf-1 Rapidly Accelerated Fibrosarcoma murine leukemia viral oncogene homologue 1, cytoplasmic | RAF1/CRAF | Secondary signal transducer for receptor tyrosine kinase via RAS/MAPK pathways | Outer ear malformation, childhood or adult onset,sensorineural or mixed, unilateral or bilateral, mild to profound | Noonan Syndrome 5, LEOPARD syndrome 2; dominant |
| ROBO1 | 602430 | Roundabout, drosophila, homolog of 1; transmembrane | ROBO1/SAX3 | Axon guidance receptor | Sensorineural, profound | combined pituitary hormone deficiency; recessive |
| SEMA3E | 608166 | SEMAphorin 3E; secreted | SEMA3E/SEMAH/ KIAA0331 | Cell migration, neural crest and otic placode induction | Outer and inner ear malformations, prelingual, mixed or sensorineural, progressive, severe or profound | CHARGE syndrome; dominant |
| SLITRK6 | 609681 | SLIT- and NTRK-like family, member 6; transmembrane | SLITRK6 | Survival and innervation of sensory neurons | Sensorineural or auditory neuropathy spectrum syndrome, prelingual, bilateral moderate to profound | Deafness and myopia; recessive |
| SMAD4 | 600993 | Mothers Against Decapentaplegic, drosophila, homolog of, 4; cytoplasmic | SMAD4/MADH4/DPC4 | Mediator of signaling of BMP and TGFB superfamily members | Outer ear malformations, early onset, mixed | Myhre syndrome; dominant |
| TBL1X | 300196 | Transducin-Beta-Like 1, X-linked | TBL1X/TBL1/EBI | Component of the nuclear corepressor (NCOR) complex | Mild hearing loss | X-linked |
| TBL1Y | 400033 | Transducin-Beta-Like 1, Y-linked | TBL1Y | Component of the nuclear corepressor NCOR complex | Sensorineural, adult onset, bilateral, mild to severe | Deafness, Y-linked 2; Y-linked |
| TBL1XR1 | 608628 | Transducin-Beta-Like 1 Receptor 1; nuclear | TBL1XR1/TBLR1/IRA1/C21 | General function in WNT signaling and component of the nuclear corepressor (NCOR) complex | Outer ear malformation, prelingual, unilateral or bilateral, Conductive | Pierpont syndrome; dominant |
| TGFB1 | 190180 | Transforming Growth Factor, Beta-1; secreted | TGFB1, TGFB | Proliferation of mesenchymal and epithelial cell types | Conductive or sensorineural, bilateral, moderate | Camurati-Engelmann disease; dominant, Otosclerosis |
| TGFB1 | 190180 | Transforming Growth Factor, Beta-1; secreted | TGFB1, TGFB | Proliferation of mesenchymal and epithelial cell types | Conductive | Otosclerosis |
| THRB | 190160 | Thyroid Hormone Receptor Beta; nuclear | THRB/ERBA2/ERBAβ | Maturation of auditory system | Prelingual, sensorineural, bilateral, severe | Thyroid hormone resistance, autosomal recessive; recessive |
| THRB | 190160 | Thyroid Hormone Receptor Beta; nuclear | THRB/ERBA2/ERBAβ | Maturation of auditory system | Sensorineural, variable degree | Thyroid hormone resistance; dominant |
| TNFRSF11A | 603499 | Tumor Necrosis Factor Receptor SuperFamily, member 11A; transmembrane | TNFRSF11A/RANK/ODFR/OSTS | Role in auricular ossification, stimulates NF-kappaB signaling | Conductive or mixed, progressive, bilateral, mild to moderate | Paget disease of bone 2, early-onset; dominant |
| TNFRSF11A | 603499 | Tumor Necrosis Factor Receptor SuperFamily, member 11A; transmembrane | TNFRSF11A/RANK/ODFR/OSTS | Role in auricular ossification, stimulates NF-kappaB signaling | Prelingual, mixed, bilateral, mild to moderate, or profound | Osteolysis, familial expansile, dominant |
| TNFRSF11B | 602643 | Tumor Necrosis Factor receptor superfamily, member 11b; secreted | TNFRSF11B/OPG/OCIF | Regulates NF-kappaB signaling as a decoy receptor ligand for TNFRS11A | Sensorineural, prelingual, bilateral, profound | Paget disease of bone 5, juvenile onset; recessive |
OMIM, Online Mendelian Inheritance in Man
Here, we highlight three examples where there is robust data from human genetics and supporting data from animal models that both the receptor and its ligand, and other molecules in their signaling pathways, are necessary for hearing. When a ligand binds its receptor, a conformational change occurs in the receptor, initiating a signal propagated through second messengers.11 Different classes of receptors can affect production of many of the same second messengers, and orchestrate development and maintenance of an organism.12
2. GROWTH FACTORS RELEVANT TO THE AUDITORY SYSTEM
Ear morphogenesis, cell fate and axis formation are established and guided by the concerted action of signaling molecules including retinoic acid, sonic hedgehog (SHH), various Wingless-related integration site (WNT) proteins, fibroblast growth factor (FGF) and bone morphogenetic protein (BMP) [reviewed in 13]. Growth factors and cytokines necessary for the development of the ear are predominantly proteins and steroids, and less commonly, lipids. A rare example of a lipid messenger is sphingosine-1-phosphate (S1P) or lysosphingolipid,14 a sphingolipid that binds to its S1PR2 receptor in the inner ear, which is necessary for hearing in human and mouse.15 In addition to the classic signaling molecules involved in hearing mentioned above, NLRP3 is part of the inflammosome complex and variants cause over-production of the cytokine interleukin (IL)-1β16 result in hearing loss.17
Juxtacrine signals mediated by NOTCH pathways establish the sensory epithelium, including hair cells and supporting cells.18 In humans, WNT19 and BMP20 family members are required for development of the auditory system.21 Variants of WNT3, WNT4, WNT5A, WNT7A, BMP1, BMP2, BMP4, and BMP7 (OMIM#165330, 603490, 164975, 601570, 112264 112262, 112261, 112267) result in low set ears or external ear malformations including prominent or posteriorly rotated ears, and small external auditory canals. In a few individuals, rare variants of BMP2, BMP4, BMP7, and GDF6 (a BMP-class ligand) have been associated with syndromic deafness or surgically verified otosclerosis (Table 2). Variants of NOG encoding Noggin, which inhibits BMP signaling by blocking the ligand binding sites of the cognate receptors, also cause hearing loss in humans (Table 2). Although variants of the WNTs only lead to external ear malformations in humans,22–26 targeted deletion of their frizzled receptors cause hearing loss in mouse27 and are candidate genes for human deafness in individuals without a known cause. Norrin, the protein encoded by NDP, is a different molecule from WNT, is also a ligand of the frizzled receptor, FZ4. In humans, variants of NDP are associated with X-linked recessive Norrie Disease characterized by childhood onset blindness and various adult onset neurologic manifestations.28 About 30% to 40% of individuals with variants of NDP have a progressive increase in hearing thresholds (Table 2).
The growth factors FGF3, FGF10, HGF, IGF1, JAG1, KITLG and TGFB1 are required for development or maintenance of hearing in humans (Table 1, Table 2). Homozygous loss of function variants of FGF3 cause ear anomalies including labyrinthine aplasia, and microtia, accompanied by prelingual, sensorineural deafness.29 In mouse, FGF10 is required for development of non-sensory epithelium of the inner ear.30 Double homozygous knockouts of mouse Ffg3 and Fgf10 have diminutive otic vesicles, a more severe phenotype than that exhibited by single homozygous mutants of either gene.31 Individuals with dominantly inherited pathogenic alleles of FGF10 exhibit LADD syndrome, which is characterized by abnormalities of teeth and distal limb segments, but only 50% of cases manifest a mixed hearing loss.32 Dominant and recessive alleles associated with deafness may have reduced penetrance and variable expressivity that can be due to modifiers in the genetic background, a supposition robustly supported by observations in mouse.33,34 A few examples of genes with variants involved in syndromes where deafness is not a constant feature include DVL1, DVL3, FGFR1, KIT, MAP3K7, and NDP (Table 2). Genetic, environmental or stochastic factors responsible for reduced penetrance of deafness in syndromes due to variants of these genes remain to be discovered.
3. RECEPTORS RELEVANT TO AUDITORY SYSTEM
G-protein coupled receptors, receptor kinases and nuclear hormone receptors are three main classes of receptors, which respond to signaling molecules in the auditory system. Other types include enzymatic and non-enzymatic transmembrane proteins (Tables 1 and 2).
3.1. G-protein coupled Receptors
G-protein coupled receptors (GPCRs) have seven alpha-helical transmembrane domains with variable length intracellular N- and C-termini and intracellular and extracellular loops mediating interactions with protein partners and are coupled to multi-subunit trimeric G-proteins. Ligand binding to receptors activates multiple pathways (Figure 2A). Variants of the GPCR-encoding genes ADGRV1, EDNRA, EDNRB and S1PR2 are associated with hearing loss (Tables 1 and 2). Ligand binding activates GTP hydrolysis of the G-protein catalytic unit (Figure 2A). One consequence of G-protein coupled receptor signaling is the generation of cyclic AMP (cAMP) by activation of adenylate cyclase 1 (ADCY1) (Figure 2A). A nonsense variant of ADCY1 was reported to cause hearing loss in humans and zebrafish morphants of Adcy1b have a hearing loss due to hair cell dysfunction.35 The inner ear morphology and hearing status of mice with a targeted disruption of Adcy136 or a spontaneous retrotransposon disruption of Adcy1 (brl)37 have not been reported.
FIGURE 2.
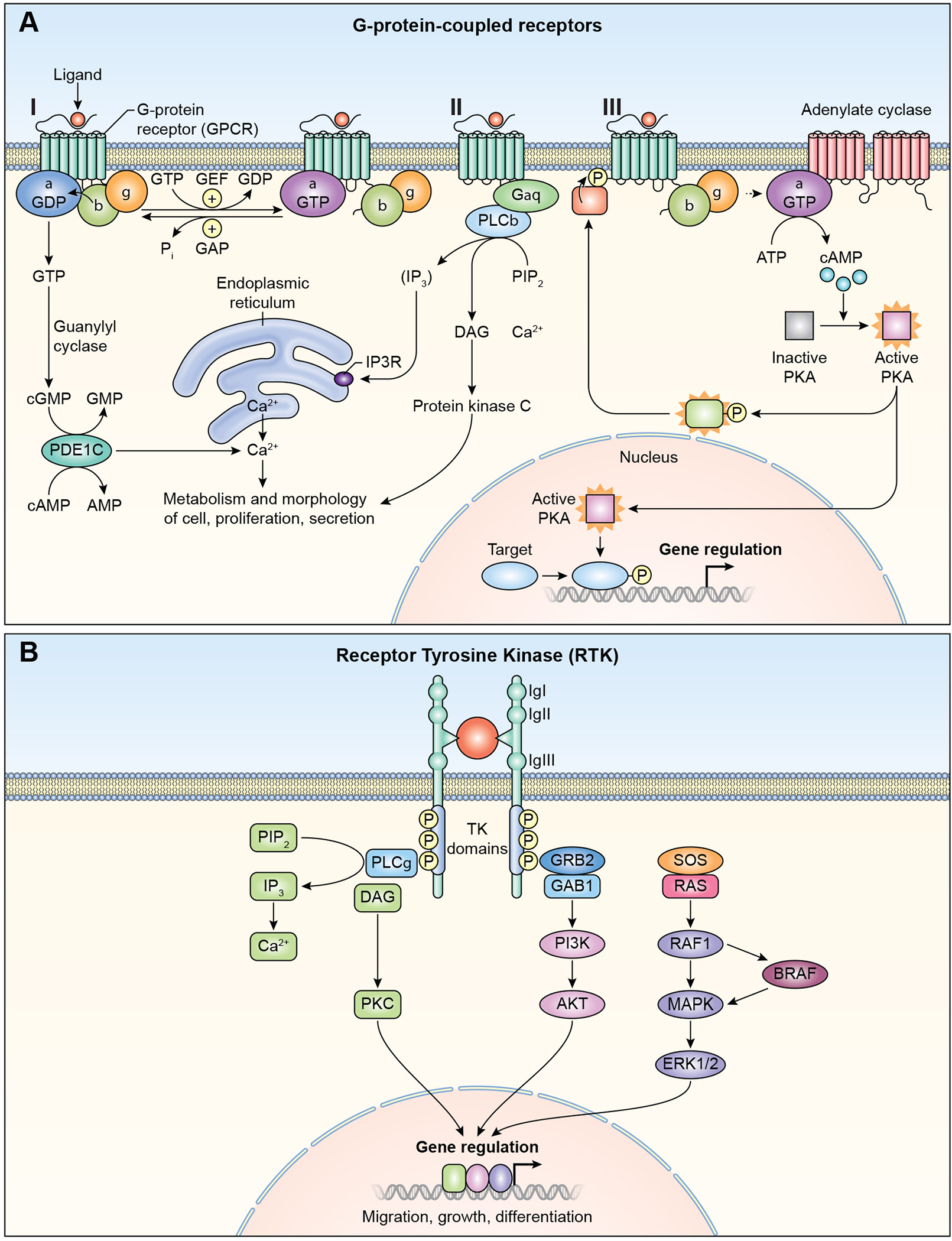
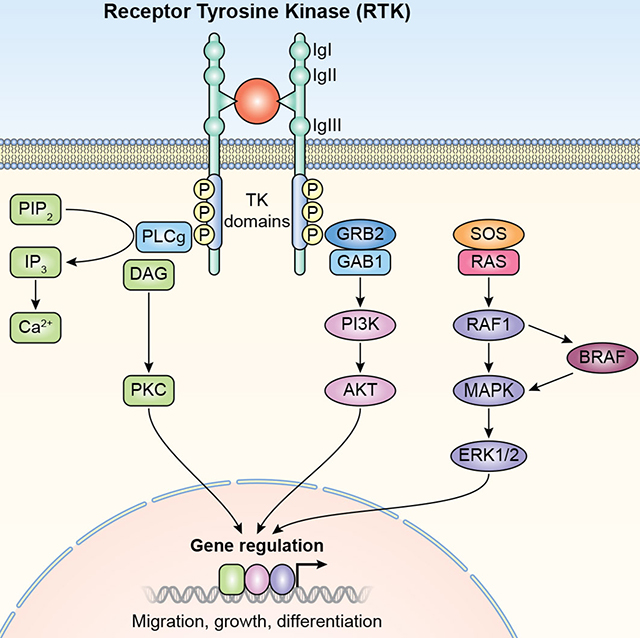
GPCR and RTK signaling pathways. (A) G-protein Coupled Receptors (GPCR). I. The receptors are coupled to the C-terminus of heterotrimeric G-proteins. G-proteins are composed of α, β, and γ subunits. GPCRs activate signaling through multiple pathways. II. Phospholipase C, (PLC), phosphatidylinositol signaling pathway. Diacyl glycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) are generated by cleavage of phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) by phospholipase C. IP3 releases calcium ions from endoplasmic reticulum. Diacylglycrol remains bound to the plasma membrane. Both diacylglycerol and calcium ions act together activate Protein Kinase C (PKC). PKC phosphorylates multiple cytoplasmic proteins that regulate cellular activity. III. Cyclic AMP signaling pathway. Cyclic AMP is a second messenger. GPCR activates adenylate cyclase, which converts ATP to cyclic AMP (cAMP). cAMP activates protein Kinase A (PKA), which then phosphorylates different proteins and transcription factors. PKA translocates to nucleus and controls gene transcription of target genes. IP3R, inositol trisphoshphate receptor, GEF; guanidine exchange factor, P; Phosphate. (B) Receptor Tyrosine Kinase (RTK). Ligand binding causes receptor dimerization and autophosphorylation. Signaling via RTK can take place through phospholipase C pathway, left side of figure (also see Figure 2A). GRB2 with other associated proteins such as GAB1, is bound to RTK. The activated receptor phosphorylates SOS1 and other guanine nucleotide exchange factors (GEF), and members of RAS, RHO and RAF, which are tethered to membranes. Signals are further propagated through the MAPK/ERK pathway to regulate gene expression. PI3K; phosphoinositide 3- kinase, Akt; Protein Kinase B.
Cyclic GMP (cGMP) is generated from GTP by guanylyl cyclase in response to ligand binding to G-protein coupled receptors. cGMP activates cGMP-dependent protein kinases. One such kinase is PRKG1, which is expressed in sensory cells and neurons of the mouse inner ear where it protects against noise induced hearing loss.38 PDE1C, which encodes phosphodiesterase 1C, and hydrolyzes cGMP and cAMP, is important for Ca+2 homeostasis (Figure 2A). Variants of human PDE1C are associated with dominantly inherited hearing loss DFNA74 (Table 1).
Regulators of G-protein coupled receptor signaling required for normal hearing include GPRASP2 and GPSM2 (Table 2). GPRASP2 regulates post-endocytic sorting of G-protein coupled receptors by binding to their C-termini.39,40 GPSM2, together with its partner GNAI3, are both expressed asymmetrically at the apical surface of hair cells, and control localization of kinocilia.41 GPSM2 interacts with the α-subunits of G-proteins, (including GNAI3), and modulates their activation.42 Additionally, GPSM2 and GNAI3 are both normally enriched in a narrow compartment at the tips of the tallest row stereocilia.43,44 A conditional deficiency of GPSM2 or GNAI3 in the mouse inner ear results in stereocilia that are shortened by ~40% and ~25%, respectively.45 Consistent with these results in mice, variants of human GPRASP2 and GPSM2 have been found to cause deafness (Table 2). The accumulation of GPSM2 and GNAI3 along with WHRN and EPS8 at the tips of stereocilia requires MYO15A, a motor protein. In the absence of MYO15A, WHRN and EPS8 fail to accumulate at the stereocilia tips. These data suggest that MYO15A transports a large complex of proteins to locations where they regulate actin polymerization dynamics of both stereocilia in post-mitotic hair cells and neuronal growth cones.45
3.1A. EDN3 and EDNRB signaling
Endothelin EDN3 is synthesized as pre-pro-endothelin and then cleaved by endothelin-converting enzyme to a 21-residue peptide.46 Endothelins are vasoconstrictors that participate in epithelial-mesenchymal interactions and are also important for melanocyte differentiation. In the inner ear, melanocytes develop from neural crest cells, some of which migrate into the intermediate cell layer of the stria vascularis and are necessary for establishing the endocochlear potential, which drives sound transduction.47 EDN3 binds to EDNRA or EDNRB, that transmit signals through Gαq/α11 G-protein subunits.47 One consequence is activation of phospholipase C which hydrolyses phoshphatidylinositol into second messengers diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3). IP3 releases Ca2+ from endoplasmic reticulum. DAG and Ca2+ together activate Protein Kinase C (PKC) (Figure 2A). PKC then activates mitogen-activated protein kinase (MAPK) pathway, phosphorylating cAMP response element binding protein (CREB).48 CREB binds to cAMP response element (CRE) DNA sequence of MITF,48 a transcription factor required for development of melanocytes.
Variants of either human EDNRB or EDN3 are associated with Waardenburg syndrome types 4A and 4B, respectively.49,50 WS4 is chacterized by sensorineural hearing loss, hair, skin and eye pigmentary abnormalities and Hirschsprung’s disease (Table 2) chacterized by a deficiency of ganglion cells of the distal colon and consequently muscles in the colon fail to peristaltically move stool leading to obstructions. EDNRB also has a distinct but unknown role for normal spiral ganglion function as Ednrb knockout mice undergo degeneration of spiral ganglion neurons.51 The time course of Ednrb expression in SGNs, exactly when SGN death occurs and also which of the three SGN subtypes (1a, 1b, or 1c) are lost in Ednrb mutants remains to be studied.
3.2. Receptor Tyrosine Kinases
Receptor Tyrosine Kinases (RTK) are single-pass transmembrane proteins with extracellular immunoglobulin-like domains and other regions of variable lengths that interact with growth factors and trigger cell growth and differentiation. The intracellular region has tyrosine kinase activity and docking sites for scaffold proteins (Figure 2B). Variants of human RTKs FGFR1, FGFR2, FGFR3, IGF1, KIT, MET and ROR1 are associated with hearing loss (Table 1, Table 2). RTKs mediate signaling through RAS/RAF/MAPK/ERK pathways (Figure 2B). Regulatory proteins of RTKs are important for signaling in the inner ear including a family of mitogen-activated protein kinase kinase kinases, BRAF and RAF1 that regulate RAS/RAF/MAPK/ERK signal transduction (Figure 2B). BRAF phosphorylates MAP3K152, and RAF1 is the principal component of MAPK pathway.53,54 The importance of BRAF and RAF1 for normal hearing is demonstrated by the fact that some of their variants are associated with LEOPARD syndromes 3 and 2 and Noonan Syndrome 5, respectively (Table 2), which are separately characterized by disparate anomalies of heart, skin, genitalia or skeleton. RAS-related GTPase RIT1 regulates p38 MAPK-dependent signaling cascades55 and variants of RIT1 (OMIM #609591) are also associated with low set ears in humans,56 pointing to the importance of RIT1 for both the morphogenesis and function of the outer ear.
Effectors of RTK signaling participating in hearing include CCDC50, EPS8, EPS8L2 among others, variants of which are associated with hearing loss DFNA44, DFNB102 and DFNB106, respectively (Table 1). CCDC50 encodes YMER, which inhibits EGFR down-regulation 57 and negatively regulates NF-kB signaling pathway.58,59 In the inner ear, EGFR signaling is important for proliferation of supporting cells.60 The epidermal growth factor receptor pathway substrate 8 (EPS8) and epidermal growth factor receptor pathway substrate 8-like protein 2 (EPS8L2) are also required for hearing.61,62 EPS8 is part of N-methyl-d-aspartate (NMDA) receptor complex,63 which controls transduction of signals from RAS to RAC. EPS8L2 acts by stimulating RAC (GTPase)-guanine nucleotide exchange factor activity of SOS1.64 In the inner ear, EPS8 and EPS8L2 are components of an electron-dense complex of proteins at the tips of hair cell stereocilia, as visualized by transmission electron microscopy. By virtue of their actin remodeling activity, they are important for elongation and maintenance of the precise lengths of stereocilia.64–68
3.2A. Hearing requires HGF and MET signaling
Hepatocyte growth factor (HGF) is secreted by mesenchymal cells, binds the MET receptor, and regulates epithelial cell development and motility by activating a variety of downstream signaling pathways.69 Like many genes, human HGF and mouse Hgf encode multiple alternative transcripts, most of which have not been well studied.70 The importance of identifying and then studying the function of each alternative transcript of a gene was recently highlighted when an alternatively spliced microexon in cytohesin 1 (CYTH1), a gene encoding a guanidine exchange factor, was shown to be necessary for spatially restricting HGF-MET signaling.71
In 40 large families from Pakistan segregating deafness, the phenotype was linked to markers for the DFNB39 locus on chromosome 7. Homozygosity was detected for either a non-coding, evolutionarily conserved three base pair deletion or an overlapping ten base pair deletion in the 3ʹ UTR of a short alternative splice isoform of HGF of unknown function.70 The presence of recognizable pathogenic variants in all of other exons and conserved sequences in the DFNB39 interval was excluded by sequencing. Yet, the possibility remains that the non-coding 3bp and 10bp deletions of HGF are in linkage disequilibrium with the real deafness-causing variant in the DFNB39 interval. The pathogenicity of these noncoding variants may be addressed by knocking in the identical 3bp or 10bp deletions in mouse Hgf. Nevertheless, there is compelling evidence from mouse that a wild type HGF expression level in the inner ear is necessary for normal hearing.70 In mouse, body-wide excessive expression of HGF from a Hgf transgene results in deafness. Additionally, a homozygous inner ear conditional knockout of Hgf causes deafness, indicating that the titer of HGF must be tightly regulated for normal hearing, and that too much or too little HGF is incompatible with normal hearing.70 During inner ear development, HGF is required for proper incorporation of neural crest cells into the intermediate cell (middle) layer of the stria vascularis (Figure 1C) in mice.72 The stria maintains an endocochlear potential of +80 to +120 millivolts and a high concentration of potassium (154 millimolar) that bathes the apical surface of hair cells and which is necessary for mechano-transduction of sound by inner hair cells (Figure 1C).
The HGF receptor MET was also demonstrated genetically to be necessary for hearing. In mouse, a complete loss of MET function results in embryonic lethality and zebrafish met morphants have reduced neuromast-derived hair cells.73 Zebrafish neuromasts resemble vertebrate inner ear sensory epithelia. In nine affected members of a human family, a predicted damaging missense variant p.(Phe841Val) of MET, located in the IPT4 domain of all MET isoforms, is associated with recessively inherited, nonsyndromic severe hearing loss.74 The IPT3 and IPT4 domains constitute a high-affinity HGF binding surface of MET. A second homozygous missense variant p.(Phe1186Cys) of MET is associated with arthrogryposis and deafness in two siblings,75 providing independent support for the conclusion that MET is required for normal hearing. It seems likely that these two damaging missense variants permit residual MET function required for embryonic development but disrupt a function of MET necessary for hearing.
The HGF-stimulated MET signaling pathway has numerous branches (Figure 2B). Variants of some of the genes encoding components of this signaling cascade are also associated with deafness in human or mouse. GAB1 is a component of a multi-subunit scaffold for various RTKs including IGF1 and MET (Figure 2B). A homozygous, hypomorphic missense variant of GAB1, p.(Gly116Glu), is associated with nonsyndromic hearing loss DFNB26 segregating in a family with eight affected individuals.76 Unexpectedly, seven normal hearing individuals in this family were also homozygous for the p.(Gly116Glu) variant. These non-penetrant individuals (but none of the affected individuals) also carry a missense variant p.(Arg544Gln) of EEF1AKNMT (also called METTL13) 76 at the dominant modifier locus DFNM1 of DFNB26 deafness. METTL13 (methyltransferase 13) is a predicted methyltransferase and is hypothesized to suppress GAB1-related deafness although the mechanism by which this might act is unknown. Interestingly, GAB1 and sprouty (SPRY2) interact with METTL13. SPRY2 down-regulates receptor tyrosine kinases and is also required for hearing in mouse.77 Taken together, it is clear that HGF/MET/GAB1/SPRY2 signaling is crucial for the auditory system, although how this is mediated is not yet fully understood.
3.3. Other Enzymatic transmembrane receptors
In addition to RTK, two types of receptors expressed in the ear have intrinsic enzymatic activity, serine/threonine kinases and receptor tyrosine phosphatases. Receptor serine/ threonine kinases are single-pass transmembrane proteins with a serine/threonine kinase domain (types I, II and III). Types I and II exist as homodimers. Type III receptors act as co-receptors by presenting the ligand to the other two classes (Figure 3A). ACVR1 encodes a type I serine/threonine kinase receptor and variants cause hearing loss in some individuals with fibrodysplasia ossificans progressiva, a connective tissue disorder (Table 2).
FIGURE 3.
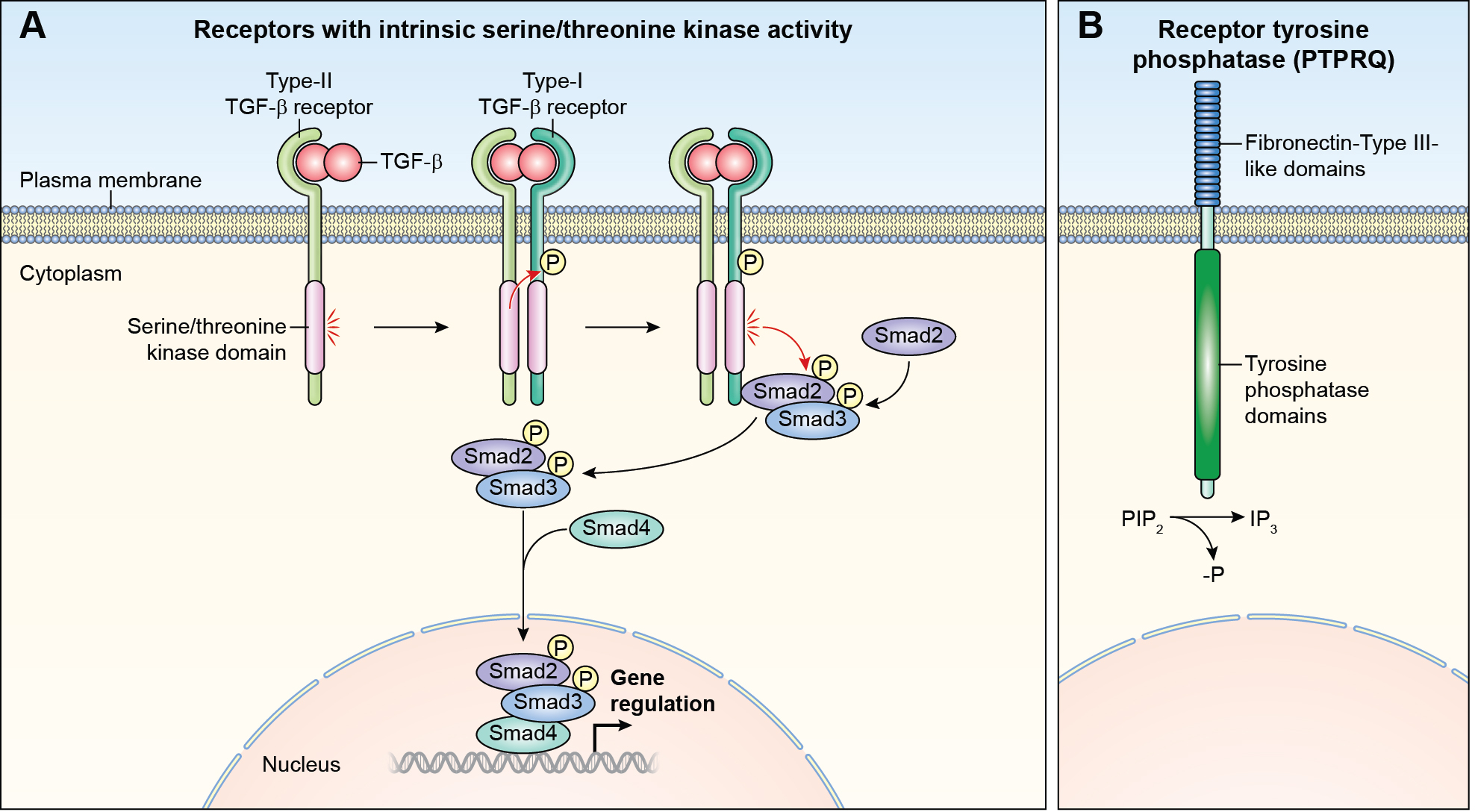
Other receptors with intrinsic enzymatic activity (A) Receptor serine/threonine kinase. A ligand of the TGFB superfamily or the BMP superfamily, binds first to a type II receptor, which in turn phosphorylates the type I receptor and leads to their dimerization. The activated receptor complex phosphorylates the SMAD family proteins. Phosphorylation of SMAD proteins disassociates them from the receptor complex. Bound SMADS translocate to the nucleus and form complexes with DNA regulatory proteins inhibiting or activating transcription of target genes. (B) Receptor tyrosine phosphatase. PTPRQ dephosphorylates phosphatidylinositol 3,4,5-trisphosphate (PIP3) to PIP2. (See Figure 2B for details of signaling via PIP2).
The receptor tyrosine phosphatases are a subclass of transmembrane protein tyrosine phosphatases (PTPase). PTPRQ is a type III receptor-like protein-tyrosine phosphatase (Figure 3B) that preferentially dephosphorylates and regulates levels of phosphatidylinositol 1,4,5-trisphosphate, PIP3,78 by dephosphorylating it to PIP2, which is further dephosphorylated to IP3 (Figure 3B). Signaling through PIP3 is important for survival, proliferation, and the subcellular architecture of diverse cellular types. Several dominant and recessive variants of human PTPRQ are associated with hearing loss in human (Table 1) and mouse.79 In the inner ear, PTPRQ contributes to hair-bundle shaft connectors, which modulate spacing of stereocilia,79 while rootlets of stereocilia develop postnatally and function to anchor stereocilia into the actin-rich cuticular plate. It remains to be determined whether PTPRQ also functions as a receptor in the inner ear.
3.4. Non-enzymatic Receptors
Transmembrane receptors with no known intrinsic enzymatic activity include ILDR1, NOTCH, TNFRS11A and SLITRK6 (Figure 4A–4D). ILDR1 is a receptor having an immunoglobulin-like extracellular domain (Figure 4A). In zebrafish, ildr1b morphants exhibit hearing loss and have a significantly reduced expression of fgf3, fgf10, and fgfr1, disrupting migration of the lateral line primordium.80 In mouse, Ildr1 is expressed in the small intestine and regulates fat-stimulated cholecystokinin secretion.81 Variants of human ILDR1 are associated with nonsyndromic deafness DFNB42 inherited as a recessive trait (Table 1), a phenotype that is recapitulated in mouse homozygous for a deletion of Ildr1.82,83 It remains to be determined if deaf human subjects with biallelic pathogenic variants of ILDR1 alleles have an additional clinically relevant phenotype involving a disruption of cholecystokinin secretion. In the mouse inner ear, ILDR1 is necessary for retention of marvel domain-containing protein 2 (MARVELD2)-originally named tricellulin encoded by TRIC- at the tricellular tight junctions.83 Human deafness-causing equivalent knock-in variants of mouse Marveld2 are also deaf.84 It is not known if there is a ligand for ILDR1 in the inner ear. Perhaps in the sensory epithelium of the inner ear, ILDR1 just functions as a structural protein at the tricellular junction between three epithelial cells.
FIGURE 4.
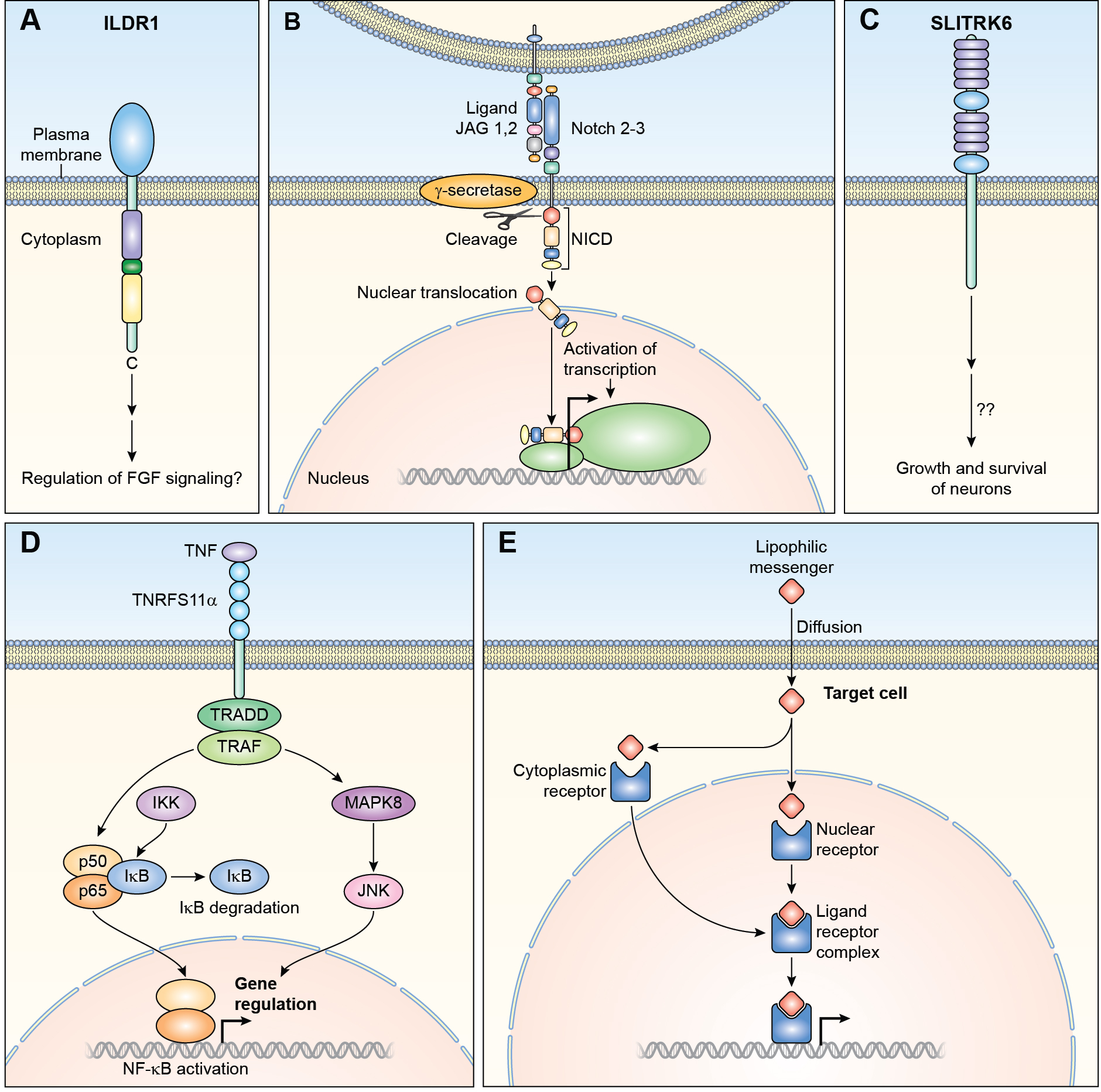
Non-enzymatic receptors (A) ILDR1 has an extracellular immunoglobin like domain and intracellular cysteine and arginine regions. Both of these are separated by a dileucine motif. Signaling through this receptor in the inner ear remains to be elucidated. (B) NOTCH receptors bind to transmembrane ligands such as JAG1. As a result of binding, NOTCH is proteolytically cleaved by γ-secretase releasing the intracellular domain of the protein (NICD), which translocates to the nucleus forming complexes with other proteins and regulate transcription of target genes. (C) SLITRK6 is a transmembrane receptor with leucine rich repeats in its extracellular domain. The mechanism of signaling through SLITRK6 is unknown. (D) TRADD and TRAFF are adapters of TNRFS receptors and participate in downstream signaling. Binding of TNF to its receptors activates either MAPK8/JNK or NF-kB (NFKB1), which is sequestered in the cytoplasm by IKB. The activation of NFKB1 by receptor binding activates a kinase (IKK) which phosphorylates IKB at specific serine residues. IKB is then ubiquitinated and degraded by the proteasome. Free NFKB1 translocates to the nucleus and regulates transcription. The second signaling pathway involves MAPK8/JNK. MAPK8 phosphorylates a number of transcription factors which modulate transcription of specific genes. (E) Ligand binding to nuclear hormone receptors cause dimerization and translocation to the nucleus. Receptor-DNA binding controls transcription of targeted genes involved in a variety of cellular activities including ion transport and proliferation. IKB; nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha, IKK; IKB Kinase.
3.4A. JAG/NOTCH signaling
Signaling through NOTCH receptors determines cell fates during development.85 NOTCH receptors interact with membrane bound ligands JAG and DLL that are present on adjacent cells (Figure 4B). Signaling induced by binding of NOTCH or JAG results in cleavage of the NOTCH receptor, which then translocates to the nucleus and alters transcription of target genes, some of which are important for hearing, such as HES1 (Figure 4B).86,87 Variants of the human genes encoding JAG1, and NOTCH2 and NOTCH3 receptors cause syndromes involving hearing loss (Table 2). In the cochlea of a Jag1 p.(Gly289Asp) heterozygous mouse, referred to as headturner, the number of outer hair cells is reduced by 33% with a slight increase in the number of inner hair cells. However, these mice are not deaf.88 In contrast, mice with a deletion of Jag1 or Notch2 limited to neural crest cells have malformed stapes and incus, and have a mild to moderate hearing loss.89
3.5. Nuclear Hormone Receptors
Nuclear hormone receptors are cytoplasmic proteins. Binding to ligand activates the receptors, which then translocate to the nucleus where they regulate target gene transcription (Figure 4E). Nuclear hormone receptors required for hearing in human include ESRRB, ESRRG and THRB (Table 1, Table 2). Studies of conditional knockout mice have shown that ESRRB controls expression of many genes encoding transporters and ion channels, such as Atp1b2, Kcnq1, Kcne1, which are all important for inner ear function,90 specifically in strial marginal cells. One consequence of the absence of ESRRB in mouse is a partial transformation of strial marginal cells into epithelial cells.90
Repressors of nuclear hormone receptors also play a role in audition. TBL1X and TBL1Y are members of WD40 repeat-containing protein family and act as repressors of nuclear hormone receptors. They are important for ear development and hearing.91,92 TBL1X and TBL1Y are components of nuclear receptor co-repressor (NCOR) complex which is required for Tri-iodothyronine (T3)-regulated gene expression.93 This regulation is important since thyroid hormone T3 and its receptor THRB play a role in cochlear development by controlling expression of genes necessary for hearing, which include KCNQ4 and SLC26A5.94
3.6. CO-RECEPTORS
Heparan sulfate proteoglycans are required for dimerization of some receptor tyrosine kinases including FGFR and EGFR.95 In addition, there are many proteins, which function as co-receptors. For example, the low-density lipoprotein (LDL) receptor-related proteins serve as co-receptors to frizzled proteins during canonical WNT signaling and their variants are associated with hearing loss.96–98 LRP5/6 is a WNT co-receptor in vertebrates,99 and Lrp5 knockdown in zebrafish with morpholinos reduces the number of supporting cells and hair cells.98 Additionally, variants of human LRP5 cause either nonsyndromic hearing loss (Table 1) or sensorineural hearing loss in patients with dominantly inherited osteosclerosis (Table 2).
4. CONCLUSION
The development of the auditory system requires multiple precisely orchestrated biochemical events. Genetic studies of hundreds of genes necessary for audition have uncovered several pathways important for normal hearing. Based initially upon studies of animal model, growth factors are now being explored for treatment of noise and ototoxic induced deafness and age related hearing loss,100–109 and some of these are currently in clinical trials.110,111 For example, topical application of IGF1 to the middle ear was used to treat sudden sensorineural hearing loss in humans, and some treated individuals showed improvements of 10 dB to 30 dB for tested frequencies as compared to controls.103,112 Hair cell regeneration in response to application of growth factors and by inhibition of specific signaling pathways is also being explored.113 For example, an inhibitor of NOTCH signaling is in phase II clinical trial for treatment of sensorineural moderate to severe hearing loss.21 As signaling pathways are involved with differentiation, proliferation, maintenance and regeneration processes, greater understanding of the in vivo ligands, receptors, protein partners and the modulation of their expression may provide opportunities to rebuild a properly patterned and functional adult human inner ear.
ACKNOWLEDGMENTS
We thank Drs. Doris Wu, Dennis Drayna and Tom Coate for ideas regarding this manuscript. TBF is supported in part by the Intramural Research Program of the National Institute on Deafness and Other Communication Disorders, National Institutes of Health, DC000039 and SN is supported by Grant 3288 from the Higher Education Commission, Islamabad, Pakistan.
Footnotes
DISCLOSURE
The authors declare no conflict of interest
DATA AVAILABILITY STATEMENT
There is no other data associated with this manuscript
REFERENCES
- 1.Tucci D, Merson MH, Wilson BS. A summary of the literature on global hearing impairment: current status and priorities for action. Otol Neurotol. 2010;31(1):31–41. [DOI] [PubMed] [Google Scholar]
- 2.Yueh B, Shapiro N, MacLean CH, Shekelle PG. Screening and management of adult hearing loss in primary care: scientific review. JAMA. 2003;289(15):1976–1985. [DOI] [PubMed] [Google Scholar]
- 3.Fritzsch B, Elliott KL. Gene, cell, and organ multiplication drives inner ear evolution. Dev Biol. 2017;431(1):3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morsli H, Choo D, Ryan A, Johnson R, Wu DK. Development of the mouse inner ear and origin of its sensory organs. J Neurosci. 1998;18(9):3327–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purves D, Augustine GJ, Fitzpatrick D, et al. , eds. Neuroscience. 2 ed: Sunderland; 2001. [Google Scholar]
- 6.Zak M, van Oort T, Hendriksen FG, Garcia MI, Vassart G, Grolman W. LGR4 and LGR5 Regulate Hair Cell Differentiation in the Sensory Epithelium of the Developing Mouse Cochlea. Front Cell Neurosci. 2016;10:186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson Chacko L, Blumer MJF, Pechriggl E, et al. Role of BDNF and neurotrophic receptors in human inner ear development. Cell Tissue Res. 2017;370(3):347–363. [DOI] [PubMed] [Google Scholar]
- 8.Lynch KJ, Touma E, Niforatos W, et al. Molecular and functional characterization of human P2X(2) receptors. Mol Pharmacol. 1999;56(6):1171–1181. [DOI] [PubMed] [Google Scholar]
- 9.DiStefano MT, Hemphill SE, Oza AM, et al. ClinGen expert clinical validity curation of 164 hearing loss gene-disease pairs. Genet Med. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peled A, Sarig O, Samuelov L, et al. Mutations in TSPEAR, Encoding a Regulator of Notch Signaling, Affect Tooth and Hair Follicle Morphogenesis. PLoS Genet. 2016;12(10):e1006369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Newton AC, Bootman MD, Scott JD. Second Messengers. Cold Spring Harb Perspect Biol. 2016;8(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heldin CH, Lu B, Evans R, Gutkind JS. Signals and Receptors. Cold Spring Harb Perspect Biol. 2016;8(4):a005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Groves AK, Fekete DM. Shaping sound in space: the regulation of inner ear patterning. Development. 2012;139(2):245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alberts B, JJohnson A, Lewis J, et al. Molecular Biology of the Cell. 6 ed. New York (NY): Garland Sciences; 2014. [Google Scholar]
- 15.Santos-Cortez RL, Faridi R, Rehman AU, et al. Autosomal-Recessive Hearing Impairment Due to Rare Missense Variants within S1PR2. Am J Hum Genet. 2016;98(2):331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hoffman HM, Wanderer AA. Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep. 2010;10(4):229–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakanishi H, Kawashima Y, Kurima K, et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci U S A. 2017;114(37):E7766–E7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basch ML, Brown RM 2nd, Jen HI, et al. Fine-tuning of Notch signaling sets the boundary of the organ of Corti and establishes sensory cell fates. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geng R, Noda T, Mulvaney JF, Lin VY, Edge AS, Dabdoub A. Comprehensive Expression of Wnt Signaling Pathway Genes during Development and Maturation of the Mouse Cochlea. PLoS One. 2016;11(2):e0148339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ma JY, You D, Li WY, Lu XL, Sun S, Li HW. Bone morphogenetic proteins and inner ear development. J Zhejiang Univ Sci B. 2019;20(2):131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Samarajeewa A, Jacques BE, Dabdoub A. Therapeutic Potential of Wnt and Notch Signaling and Epigenetic Regulation in Mammalian Sensory Hair Cell Regeneration. Mol Ther. 2019;27(5):904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niemann S, Zhao C, Pascu F, et al. Homozygous WNT3 mutation causes tetra-amelia in a large consanguineous family. Am J Hum Genet. 2004;74(3):558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandel H, Shemer R, Borochowitz ZU, et al. SERKAL syndrome: an autosomal-recessive disorder caused by a loss-of-function mutation in WNT4. Am J Hum Genet. 2008;82(1):39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Person AD, Beiraghi S, Sieben CM, et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn. 2010;239(1):327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Woods CG, Stricker S, Seemann P, et al. Mutations in WNT7A cause a range of limb malformations, including Fuhrmann syndrome and Al-Awadi/Raas-Rothschild/Schinzel phocomelia syndrome. Am J Hum Genet. 2006;79(2):402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang RN, Green J, Wang Z, et al. Bone Morphogenetic Protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1(1):87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Q, Wang Y, Dabdoub A, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116(6):883–895. [DOI] [PubMed] [Google Scholar]
- 28.Berger W, Meindl A, van de Pol TJ, et al. Isolation of a candidate gene for Norrie disease by positional cloning. Nat Genet. 1992;1(3):199–203. [DOI] [PubMed] [Google Scholar]
- 29.Tekin M, Hismi BO, Fitoz S, et al. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007;80(2):338–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Urness LD, Wang X, Shibata S, Ohyama T, Mansour SL. Fgf10 is required for specification of non-sensory regions of the cochlear epithelium. Dev Biol. 2015;400(1):59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alvarez Y, Alonso MT, Vendrell V, et al. Requirements for FGF3 and FGF10 during inner ear formation. Development. 2003;130(25):6329–6338. [DOI] [PubMed] [Google Scholar]
- 32.Shams I, Rohmann E, Eswarakumar VP, et al. Lacrimo-auriculo-dento-digital syndrome is caused by reduced activity of the fibroblast growth factor 10 (FGF10)-FGF receptor 2 signaling pathway. Mol Cell Biol. 2007;27(19):6903–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ikeda A, Zheng QY, Rosenstiel P, et al. Genetic modification of hearing in tubby mice: evidence for the existence of a major gene (moth1) which protects tubby mice from hearing loss. Hum Mol Genet. 1999;8(9):1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikeda A, Zheng QY, Zuberi AR, Johnson KR, Naggert JK, Nishina PM. Microtubule-associated protein 1A is a modifier of tubby hearing (moth1). Nat Genet. 2002;30(4):401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Santos-Cortez RL, Lee K, Giese AP, et al. Adenylate cyclase 1 (ADCY1) mutations cause recessive hearing impairment in humans and defects in hair cell function and hearing in zebrafish. Hum Mol Genet. 2014;23(12):3289–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villacres EC, Wu Z, Hua W, et al. Developmentally expressed Ca(2+)-sensitive adenylyl cyclase activity is disrupted in the brains of type I adenylyl cyclase mutant mice. J Biol Chem. 1995;270(24):14352–14357. [DOI] [PubMed] [Google Scholar]
- 37.Abdel-Majid RM, Leong WL, Schalkwyk LC, et al. Loss of adenylyl cyclase I activity disrupts patterning of mouse somatosensory cortex. Nat Genet. 1998;19(3):289–291. [DOI] [PubMed] [Google Scholar]
- 38.Jaumann M, Dettling J, Gubelt M, et al. cGMP-Prkg1 signaling and Pde5 inhibition shelter cochlear hair cells and hearing function. Nat Med. 2012;18(2):252–259. [DOI] [PubMed] [Google Scholar]
- 39.Simonin F, Karcher P, Boeuf JJ, Matifas A, Kieffer BL. Identification of a novel family of G protein-coupled receptor associated sorting proteins. J Neurochem. 2004;89(3):766–775. [DOI] [PubMed] [Google Scholar]
- 40.Moser E, Kargl J, Whistler JL, Waldhoer M, Tschische P. G protein-coupled receptor-associated sorting protein 1 regulates the postendocytic sorting of seven-transmembrane-spanning G protein-coupled receptors. Pharmacology. 2010;86(1):22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ezan J, Lasvaux L, Gezer A, et al. Primary cilium migration depends on G-protein signalling control of subapical cytoskeleton. Nat Cell Biol. 2013;15(9):1107–1115. [DOI] [PubMed] [Google Scholar]
- 42.Mochizuki N, Cho G, Wen B, Insel PA. Identification and cDNA cloning of a novel human mosaic protein, LGN, based on interaction with G alpha i2. Gene. 1996;181(1–2):39–43. [DOI] [PubMed] [Google Scholar]
- 43.Tadenev ALD, Akturk A, Devanney N, et al. GPSM2-GNAI Specifies the Tallest Stereocilia and Defines Hair Bundle Row Identity. Curr Biol. 2019;29(6):921–934 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tarchini B, Tadenev AL, Devanney N, Cayouette M. A link between planar polarity and staircase-like bundle architecture in hair cells. Development. 2016;143(21):3926–3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mauriac SA, Hien YE, Bird JE, et al. Defective Gpsm2/Galphai3 signalling disrupts stereocilia development and growth cone actin dynamics in Chudley-McCullough syndrome. Nat Commun. 2017;8:14907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurihara H, Kurihara Y, Nagai R, Yazaki Y. Endothelin and neural crest development. Cell Mol Biol (Noisy-le-grand). 1999;45(5):639–651. [PubMed] [Google Scholar]
- 47.Bondurand N, Dufour S, Pingault V. News from the endothelin-3/EDNRB signaling pathway: Role during enteric nervous system development and involvement in neural crest-associated disorders. Dev Biol. 2018;444 Suppl 1:S156–S169. [DOI] [PubMed] [Google Scholar]
- 48.Pingault V, Ente D, Dastot-Le Moal F, Goossens M, Marlin S, Bondurand N. Review and update of mutations causing Waardenburg syndrome. Hum Mutat. 2010;31(4):391–406. [DOI] [PubMed] [Google Scholar]
- 49.Edery P, Attie T, Amiel J, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet. 1996;12(4):442–444. [DOI] [PubMed] [Google Scholar]
- 50.Puffenberger EG, Hosoda K, Washington SS, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung’s disease. Cell. 1994;79(7):1257–1266. [DOI] [PubMed] [Google Scholar]
- 51.Ida-Eto M, Ohgami N, Iida M, et al. Partial requirement of endothelin receptor B in spiral ganglion neurons for postnatal development of hearing. J Biol Chem. 2011;286(34):29621–29626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brennan DF, Dar AC, Hertz NT, et al. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472(7343):366–369. [DOI] [PubMed] [Google Scholar]
- 53.de Iriarte Rodriguez R, Magarinos M, Pfeiffer V, Rapp UR, Varela-Nieto I. C-Raf deficiency leads to hearing loss and increased noise susceptibility. Cell Mol Life Sci. 2015;72(20):3983–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pandit B, Sarkozy A, Pennacchio LA, et al. Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat Genet. 2007;39(8):1007–1012. [DOI] [PubMed] [Google Scholar]
- 55.Shi GX, Andres DA. Rit contributes to nerve growth factor-induced neuronal differentiation via activation of B-Raf-extracellular signal-regulated kinase and p38 mitogen-activated protein kinase cascades. Mol Cell Biol. 2005;25(2):830–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aoki Y, Niihori T, Banjo T, et al. Gain-of-function mutations in RIT1 cause Noonan syndrome, a RAS/MAPK pathway syndrome. Am J Hum Genet. 2013;93(1):173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tashiro K, Konishi H, Sano E, Nabeshi H, Yamauchi E, Taniguchi H. Suppression of the ligand-mediated down-regulation of epidermal growth factor receptor by Ymer, a novel tyrosine-phosphorylated and ubiquitinated protein. J Biol Chem. 2006;281(34):24612–24622. [DOI] [PubMed] [Google Scholar]
- 58.Bohgaki M, Tsukiyama T, Nakajima A, et al. Involvement of Ymer in suppression of NF-kappaB activation by regulated interaction with lysine-63-linked polyubiquitin chain. Biochim Biophys Acta. 2008;1783(5):826–837. [DOI] [PubMed] [Google Scholar]
- 59.Tsukiyama T, Matsuda-Tsukiyama M, Bohgaki M, Terai S, Tanaka S, Hatakeyama S. Ymer acts as a multifunctional regulator in nuclear factor-kappaB and Fas signaling pathways. Mol Med. 2012;18:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.White PM, Stone JS, Groves AK, Segil N. EGFR signaling is required for regenerative proliferation in the cochlea: conservation in birds and mammals. Dev Biol. 2012;363(1):191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dahmani M, Ammar-Khodja F, Bonnet C, et al. EPS8L2 is a new causal gene for childhood onset autosomal recessive progressive hearing loss. Orphanet J Rare Dis. 2015;10:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Behlouli A, Bonnet C, Abdi S, et al. EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J Rare Dis. 2014;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morini R, Ferrara S, Perrucci F, et al. Lack of the Actin Capping Protein, Eps8, Affects NMDA-Type Glutamate Receptor Function and Composition. Front Mol Neurosci. 2018;11:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Offenhauser N, Borgonovo A, Disanza A, et al. The eps8 family of proteins links growth factor stimulation to actin reorganization generating functional redundancy in the Ras/Rac pathway. Mol Biol Cell. 2004;15(1):91–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Furness DN, Johnson SL, Manor U, et al. Progressive hearing loss and gradual deterioration of sensory hair bundles in the ears of mice lacking the actin-binding protein Eps8L2. Proc Natl Acad Sci U S A. 2013;110(34):13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zampini V, Ruttiger L, Johnson SL, et al. Eps8 regulates hair bundle length and functional maturation of mammalian auditory hair cells. PLoS Biol. 2011;9(4):e1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manor U, Disanza A, Grati M, et al. Regulation of stereocilia length by myosin XVa and whirlin depends on the actin-regulatory protein Eps8. Curr Biol. 2011;21(2):167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Velez-Ortega AC, Frolenkov GI. Building and repairing the stereocilia cytoskeleton in mammalian auditory hair cells. Hear Res. 2019;376:47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 2012;12(2):89–103. [DOI] [PubMed] [Google Scholar]
- 70.Schultz JM, Khan SN, Ahmed ZM, et al. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am J Hum Genet. 2009;85(1):25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ratcliffe CDH, Siddiqui N, Coelho PP, et al. HGF-induced migration depends on the PI(3,4,5)P3-binding microexon-spliced variant of the Arf6 exchange factor cytohesin-1. J Cell Biol. 2019;218(1):285–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shibata S, Miwa T, Wu HH, Levitt P, Ohyama T. Hepatocyte Growth Factor-c-MET Signaling Mediates the Development of Nonsensory Structures of the Mammalian Cochlea and Hearing. J Neurosci. 2016;36(31):8200–8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haines L, Neyt C, Gautier P, et al. Met and Hgf signaling controls hypaxial muscle and lateral line development in the zebrafish. Development. 2004;131(19):4857–4869. [DOI] [PubMed] [Google Scholar]
- 74.Mujtaba G, Schultz JM, Imtiaz A, Morell RJ, Friedman TB, Naz S. A mutation of MET, encoding hepatocyte growth factor receptor, is associated with human DFNB97 hearing loss. J Med Genet. 2015;52(8):548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Alabdullatif MA, Al Dhaibani MA, Khassawneh MY, El-Hattab AW. Chromosomal microarray in a highly consanguineous population: diagnostic yield, utility of regions of homozygosity, and novel mutations. Clin Genet. 2017;91(4):616–622. [DOI] [PubMed] [Google Scholar]
- 76.Yousaf R, Ahmed ZM, Giese AP, et al. Modifier variant of METTL13 suppresses human GAB1-associated profound deafness. J Clin Invest. 2018;128(4):1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shim K, Minowada G, Coling DE, Martin GR. Sprouty2, a mouse deafness gene, regulates cell fate decisions in the auditory sensory epithelium by antagonizing FGF signaling. Dev Cell. 2005;8(4):553–564. [DOI] [PubMed] [Google Scholar]
- 78.Oganesian A, Poot M, Daum G, et al. Protein tyrosine phosphatase RQ is a phosphatidylinositol phosphatase that can regulate cell survival and proliferation. Proc Natl Acad Sci U S A. 2003;100(13):7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goodyear RJ, Legan PK, Wright MB, et al. A receptor-like inositol lipid phosphatase is required for the maturation of developing cochlear hair bundles. J Neurosci. 2003;23(27):9208–9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sang Q, Zhang J, Feng R, et al. Ildr1b is essential for semicircular canal development, migration of the posterior lateral line primordium and hearing ability in zebrafish: implications for a role in the recessive hearing impairment DFNB42. Hum Mol Genet. 2014;23(23):6201–6211. [DOI] [PubMed] [Google Scholar]
- 81.Chandra R, Wang Y, Shahid RA, Vigna SR, Freedman NJ, Liddle RA. Immunoglobulin-like domain containing receptor 1 mediates fat-stimulated cholecystokinin secretion. J Clin Invest. 2013;123(8):3343–3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sang Q, Li W, Xu Y, et al. ILDR1 deficiency causes degeneration of cochlear outer hair cells and disrupts the structure of the organ of Corti: a mouse model for human DFNB42. Biol Open. 2015;4(4):411–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morozko EL, Nishio A, Ingham NJ, et al. ILDR1 null mice, a model of human deafness DFNB42, show structural aberrations of tricellular tight junctions and degeneration of auditory hair cells. Hum Mol Genet. 2015;24(3):609–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nayak G, Lee SI, Yousaf R, et al. Tricellulin deficiency affects tight junction architecture and cochlear hair cells. J Clin Invest. 2013;123(9):4036–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137(2):216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fischer A, Gessler M. Delta-Notch--and then? Protein interactions and proposed modes of repression by Hes and Hey bHLH factors. Nucleic Acids Res. 2007;35(14):4583–4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zheng JL, Shou J, Guillemot F, Kageyama R, Gao WQ. Hes1 is a negative regulator of inner ear hair cell differentiation. Development. 2000;127(21):4551–4560. [DOI] [PubMed] [Google Scholar]
- 88.Kiernan AE, Ahituv N, Fuchs H, et al. The Notch ligand Jagged1 is required for inner ear sensory development. Proc Natl Acad Sci U S A. 2001;98(7):3873–3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Teng CS, Yen HY, Barske L, et al. Requirement for Jagged1-Notch2 signaling in patterning the bones of the mouse and human middle ear. Sci Rep. 2017;7(1):2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chen J, Nathans J. Estrogen-related receptor beta/NR3B2 controls epithelial cell fate and endolymph production by the stria vascularis. Dev Cell. 2007;13(3):325–337. [DOI] [PubMed] [Google Scholar]
- 91.Heinen CA, Losekoot M, Sun Y, et al. Mutations in TBL1X Are Associated With Central Hypothyroidism. J Clin Endocrinol Metab. 2016;101(12):4564–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Di Stazio M, Collesi C, Vozzi D, et al. TBL1Y: a new gene involved in syndromic hearing loss. Eur J Hum Genet. 2019;27(3):466–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Astapova I, Vella KR, Ramadoss P, et al. The nuclear receptor corepressor (NCoR) controls thyroid hormone sensitivity and the set point of the hypothalamic-pituitary-thyroid axis. Mol Endocrinol. 2011;25(2):212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Winter H, Braig C, Zimmermann U, et al. Thyroid hormone receptors TRalpha1 and TRbeta differentially regulate gene expression of Kcnq4 and prestin during final differentiation of outer hair cells. J Cell Sci. 2006;119(Pt 14):2975–2984. [DOI] [PubMed] [Google Scholar]
- 95.Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19(2):117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kantarci S, Al-Gazali L, Hill RS, et al. Mutations in LRP2, which encodes the multiligand receptor megalin, cause Donnai-Barrow and facio-oculo-acoustico-renal syndromes. Nat Genet. 2007;39(8):957–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Leupin O, Piters E, Halleux C, et al. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286(22):19489–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xia W, Hu J, Liu F, et al. New role of LRP5, associated with nonsyndromic autosomal-recessive hereditary hearing loss. Hum Mutat. 2017;38(10):1421–1431. [DOI] [PubMed] [Google Scholar]
- 99.MacDonald BT, He X. Frizzled and LRP5/6 receptors for Wnt/beta-catenin signaling. Cold Spring Harb Perspect Biol. 2012;4(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lou X, Yuan H, Xie J, Wang X, Yang L, Zhang Y. Growth factors have a protective effect on neomycin-induced hair cell loss. Cell Biol Int. 2015;39(1):65–73. [DOI] [PubMed] [Google Scholar]
- 101.Khalin I, Alyautdin R, Kocherga G, Bakar MA. Targeted delivery of brain-derived neurotrophic factor for the treatment of blindness and deafness. Int J Nanomedicine. 2015;10:3245–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dinh CT, Goncalves S, Bas E, Van De Water TR, Zine A. Molecular regulation of auditory hair cell death and approaches to protect sensory receptor cells and/or stimulate repair following acoustic trauma. Front Cell Neurosci. 2015;9:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yamamoto N, Nakagawa T, Ito J. Application of insulin-like growth factor-1 in the treatment of inner ear disorders. Front Pharmacol. 2014;5:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takada Y, Beyer LA, Swiderski DL, et al. Connexin 26 null mice exhibit spiral ganglion degeneration that can be blocked by BDNF gene therapy. Hear Res. 2014;309:124–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou F, Wu P, Wang L, et al. The NGF point-injection for treatment of the sound-perceiving nerve deafness and tinnitus in 68 cases. J Tradit Chin Med. 2009;29(1):39–42. [DOI] [PubMed] [Google Scholar]
- 106.Kikkawa YS, Nakagawa T, Tsubouchi H, et al. Hepatocyte growth factor protects auditory hair cells from aminoglycosides. Laryngoscope. 2009;119(10):2027–2031. [DOI] [PubMed] [Google Scholar]
- 107.Oshima K, Shimamura M, Mizuno S, et al. Intrathecal injection of HVJ-E containing HGF gene to cerebrospinal fluid can prevent and ameliorate hearing impairment in rats. FASEB J. 2004;18(1):212–214. [DOI] [PubMed] [Google Scholar]
- 108.Amoils CP, Jackler RK, Lustig LR. Repair of chronic tympanic membrane perforations using epidermal growth factor. Otolaryngol Head Neck Surg. 1992;107(5):669–683. [DOI] [PubMed] [Google Scholar]
- 109.Wang Q, Zhao H, Zheng T, et al. Otoprotective effects of mouse nerve growth factor in DBA/2J mice with early-onset progressive hearing loss. J Neurosci Res. 2017;95(10):1937–1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Omae K, Kanemaru SI, Nakatani E, et al. Regenerative treatment for tympanic membrane perforation using gelatin sponge with basic fibroblast growth factor. Auris Nasus Larynx. 2017;44(6):664–671. [DOI] [PubMed] [Google Scholar]
- 111.Zhai SQ, Yu N, Zhu YH, Chen LW, Ma YY. Clinical efficacy of nerve growth factor in the treatment of blast-induced hearing loss: a pilot study. Eur Rev Med Pharmacol Sci. 2015;19(17):3146–3151. [PubMed] [Google Scholar]
- 112.Nakagawa T, Yamamoto M, Kumakawa K, et al. Prognostic impact of salvage treatment on hearing recovery in patients with sudden sensorineural hearing loss refractory to systemic corticosteroids: A retrospective observational study. Auris Nasus Larynx. 2016;43(5):489–494. [DOI] [PubMed] [Google Scholar]
- 113.Zheng F, Zuo J. Cochlear hair cell regeneration after noise-induced hearing loss: Does regeneration follow development? Hear Res. 2017;349:182–196. [DOI] [PMC free article] [PubMed] [Google Scholar]