

PURPOSE
Multiple myeloma (MM) is a genetically heterogeneous malignancy characterized by variable treatment responses. Although numerous drugs have been approved in recent years, the ability to predict treatment response and tailor individual therapy is limited by the absence of robust predictive biomarkers. The goal of this clinical trial was to use ex vivo, high-throughput screening (HTS) of 170 compounds to predict response among patients with relapsed or refractory MM and inform the next treatment decisions. Additionally, we integrated HTS with multi-omic analysis to uncover novel associations between in vitro drug sensitivity and gene expression and mutation profiles.
MATERIALS AND METHODS
Twenty-five patients with relapsed or refractory MM underwent a screening bone marrow or soft tissue biopsy. Sixteen patients were found to have sufficient plasma cells for HTS. Targeted next-generation sequencing was performed on plasma cell-free DNA from all patients who underwent HTS. RNA and whole-exome sequencing of bone marrow plasma cells were performed on eight and seven patients, respectively.
RESULTS
Results of HTS testing were made available to treating physicians within a median of 5 days from the biopsy. An actionable treatment result was identified in all 16 patients examined. Among the 13 patients who received assay-guided therapy, 92% achieved stable disease or better. The expression of 105 genes and mutations in 12 genes correlated with in vitro cytotoxicity.
CONCLUSION
In patients with relapsed or refractory MM, we demonstrate the feasibility of ex vivo drug sensitivity testing on isolated plasma cells from patient bone marrow biopsies or extramedullary plasmacytomas to inform the next line of therapy.
INTRODUCTION
Multiple myeloma (MM) is an incurable cancer of bone marrow plasma cells with a 4-year overall survival (OS) of 82% in the United States.1 Unlike some hematologic malignancies, MM cannot be characterized by a single-gene mutation but exhibits broad genomic heterogeneity between patients.2 In the largest study to date, 63 recurrently mutated driver genes have been reported in MM,3 making it unlikely that a single targeted therapy would be effective for all patients. Variable treatment responses confirm this hypothesis, and we currently lack predictive biomarkers to inform therapeutic decisions that can enable personalized treatment recommendations.
CONTEXT
Key Objective
Multiple myeloma (MM) is characterized by variable treatment responses, and it would be beneficial to predict which patients will respond to the numerous available drugs. The goal of this clinical trial was to determine the feasibility of ex vivo, high-throughput screening (HTS) of 170 drugs and multi-omic analysis to predict response among patients with relapsed or refractory MM and inform treatment decisions.
Knowledge Generated
Among the 25 patients screened, 16 had sufficient plasma cells for HTS and an actionable treatment result was found for all tested patients. Among the 13 patients who received assay-guided therapy, 92% achieved stable disease or better. The expression of 105 genes and mutations in 12 genes correlated with in vitro cytotoxicity.
Relevance
To our knowledge, this is the first clinical trial to demonstrate the feasibility of HTS to guide treatment decisions for patients with MM in real time.
The aim of this study was to determine the feasibility of high-throughput screening (HTS) as a precision medicine tool to inform the next line of therapy in patients with relapsed or refractory MM. Although previous studies have used HTS to evaluate sensitivity to a large panel of drugs, these studies were limited to either cell lines or archived patient samples.4-7 Our study is unique for MM in that, to our knowledge, it is the first to use HTS to inform treatment decisions in real time. We developed a Clinical Laboratory Improvement Amendments (CLIA)–approved assay to simultaneously evaluate the cytotoxic activity of 170 compounds, both US Food and Drug Administration (FDA)–approved and investigational, against plasma cells isolated from bone marrow, blood, or soft tissue biopsies that were not subjected to a freeze-thaw process, with results made available in under a week.
To gain new insight into mechanisms of drug sensitivity and resistance, we also performed a multi-omic analysis in conjunction with HTS. Using a combination of whole-exome sequencing (WES), RNA sequencing of bone marrow plasma cells, and ultradeep targeted sequencing of circulating tumor DNA (ctDNA), we integrated the genomic analysis with the functional assay using a machine learning technique to uncover novel relationships between genes and in vitro cytotoxicity.
MATERIALS AND METHODS
Study Design
The feasibility portion of this study enrolled patients from March 2018 to December 2019 (ClinicalTrials.gov identifier: NCT03389347). The primary objective was to obtain an actionable result from HTS for > 50% of patients enrolled. The secondary end point was to assess the overall response rate to the therapy informed by the HTS assay using the International Myeloma Working Group response criteria.8 Exploratory end points were to identify new agents that exhibit activity in MM that can be tested in prospective clinical trials and to correlate in vitro sensitivity with genomic sequencing.
Patients
Informed consent was obtained from all patients in accordance with protocols approved by the Institutional Review Board at the Fred Hutchinson Cancer Research Center. Eligible patients were 18 years of age or older and had relapsed or refractory MM with measurable disease. All patients had received three or more previous therapies including an immunomodulatory drug and a proteasome inhibitor. Patients were not excluded on the basis of their blood counts or organ function. All patients had an Eastern Cooperative Oncology Group of < 4 and an estimated life expectancy of > 100 days. To be eligible for HTS, patients were required to have at least 6 × 105 plasma cells isolated from a bone marrow biopsy, extramedullary plasmacytoma, or blood sample.
HTS
Mononuclear cells were separated from 5 to 10 mL bone marrow aspirates by density gradient centrifugation on Lymphocyte Separation Media (Corning, Manassas, VA). Single-cell suspensions were created from plasmacytomas by mechanical dissociation and from bone marrow core biopsies after disaggregating with a sterile mortar and pestle. Plasma cells were isolated from fresh tissue by anti-CD138 magnetic-activated cell sorting (Miltenyi Biotec, Bergisch Gladbach, Germany, Catalog No. 130-051-301). CLIA–certified HTS was performed at the Quellos High-Throughput Core Facility at the University of Washington using a platform similar to one previously validated in acute myeloid leukemia.9,10 Plasma cells were added to 384-well plates coated with a protein matrix at a density of 500-4,000 cells per well in 50 μL of Iscove’s DMEM media supplemented with l-glutamine and 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (Mediatech, Inc, Manassas, VA), 10% fetal bovine serum (HyClone, Logan, UT), and 1× penicillin-streptomycin (Gibco, Grand Island, NY) and incubated overnight to allow adhesion. One hundred seventy compounds, including both FDA-approved and investigational new drugs (Data Supplement), were dissolved in 100% dimethyl sulfoxide (DMSO) and added in eight concentrations ranging from 5 pM to 100 µM (final DMSO concentration 0.1%) in the wells in addition to a negative control (DMSO). After a 72-hour incubation at 37°C and 5% CO2, a luminescent cell viability assay (CellTiter-Glo; Promega, Madison, WI) was performed to calculate the concentration of experimental compound required to achieve 50% in vitro inhibition (IC50) by fitting data using least squares method to the standard four-parameter logistic model. Area under the curve (AUC) was determined from the integral of the dose-response curve from the overall minimum and maximum tested concentrations of compounds or drugs.
Drugs were recommended for treatment if the IC50 was ≤ 0.2 µM and less than or equal to the plasma concentration demonstrated to be safe and effective according to pharmacokinetic data reported in human clinical trials. The participant’s oncologist was provided a list of the recommended drugs on the basis of these criteria and allowed to use their discretion when selecting the final treatment to administer. Factors that influenced the oncologists’ decision included FDA approval of the recommended drugs, insurance coverage, prior treatment history, known side effect profile, and patient preference. Since monotherapy is generally not effective to treat relapsed MM, the participants' oncologists were allowed to combine the recommended drug with additional drugs, such as dexamethasone, in the final treatment plan. Participants were classified as having received assay-guided therapy if they received at least one of the recommended drugs. The drug dose approved by the FDA was used for all treatment regimens and not influenced by the results of the HTS.
Whole-Exome and mRNA Sequencing
For genomic testing, a separate bone marrow aspirate was obtained using the same biopsy needle used to collect material for HTS. Mononuclear cells were separated by density gradient centrifugation on Lymphocyte Separation Media (Corning) and cryopreserved in media containing DMSO to preserve their viability for later use. Plasma cells were isolated from cryopreserved bone marrow by anti–B-cell maturation antigen (BCMA) magnetic-activated cell sorting (Miltenyi Biotec, clone REA315). Anti-BCMA was selected since this target is more robust than CD138 for plasma cell enrichment from cryopreserved samples.11 DNA and RNA were extracted separately from both BCMA+ and BMCA− cell populations using the AllPrep Mini Kit (QIAGEN, Hilden, Germany). Whole-exome libraries were prepared (Twist Bioscience, San Francisco, CA) and sequenced (100 bp, paired-end) on a NovaSeq (Illumina, San Diego, CA) to an average depth of 230×. TruSeq mRNA libraries were prepared (Illumina) and 80 million reads (150 bp, paired-end) were sequenced on a NovaSeq.
Targeted Sequencing of Circulating Tumor DNA
On the same day as the bone marrow aspirate, 10 mL blood was collected in a cell-free DNA (cfDNA) Blood Collection Tube (Streck, La Vista, NE) to minimize cellular DNA contamination and prevent degradation of cfDNA. Plasma was separated by centrifugation, and the buffy coat was collected after lysis of red blood cells using ammonium-chloride-potassium (ACK; Gibco, Dublin, Ireland) lysis buffer. Plasma cells were isolated from the cryopreserved buffy coat using anti-BCMA magnetic beads (Miltenyi Biotec Inc). Cellular DNA was extracted from BCMA+ and BCMA− blood cells using the QIAamp DNA Mini Kit (QIAGEN). Cell-free DNA was extracted from plasma using the QIAamp MinElute ccfDNA Kit (QIAGEN). QIAseq libraries incorporating unique molecular identifiers were prepared (QIAGEN) for targeted DNA sequencing as described previously12 using a panel of 70 recurrently mutated genes in MM (Data Supplement) that includes 63 known driver genes.3 Prepared libraries were sequenced (150 bp, paired-end) on a NovaSeq to an average depth of 4,100× (refer to the Data Supplement for a description of the bioinformatic analysis).
Statistical Analysis
To identify genes correlated with in vitro drug sensitivity, elastic net regression was performed whereby the outcome variable was gene expression transcripts per million (TPM) or the presence or absence of a mutated gene and the predictor variable was AUC. Variants identified by this analysis were further refined using a two-sample Wilcoxon rank-sum test comparing AUC among mutated and nonmutated samples with two-sided α = .05. Genes mutated in only one sample were excluded because of low statistical power. Linear regression analysis was performed to assess the strength of the correlation between TPM and AUC. To account for multiple comparisons, P values were adjusted using the method of Benjamini and Hochberg. Survival curves were estimated using the Kaplan-Meier method, and the differences between subgroups were evaluated by the long-rank test. Chi-square tests were used to compare categorical variables, and t tests were used to compare numeric variables with two-sided α = .05. Clustering was performed using K-nearest neighbors. All statistical analyses were performed in R (v3.9).
RESULTS
Patient Characteristics
All patients underwent screening for trial eligibility at the Seattle Cancer Care Alliance between March 2018 and December 2019. Baseline demographics of all patients screened for the study are shown in Table 1 (N = 25). Nine were excluded because of insufficient plasma cells, and 16 patients had sufficient cells for HTS, including 13 collected from a bone marrow aspiration or biopsy and three from an extramedullary plasmacytoma.
TABLE 1.
Characteristics of Screened Patients (N = 25)

In Vitro Drug Sensitivity
Our primary end point was achieved, establishing the feasibility of HTS to inform treatment decisions in patients with relapsed or refractory MM. An actionable result was available for 100% of patients who underwent HTS, representing 64% of patients initially evaluated. Of the 16 patients who had received HTS, 13 patients received treatment guided by the assay, one elected to discontinue all therapy, one received chimeric antigen receptor T-cell therapy, and one received daratumumab (not one of the tested drugs).
The median time between sample collection and HTS was 5 days (range 4-6 days). The five drugs with the highest in vitro response measured by mean AUC were mitoxantrone, panobinostat, bortezomib, omacetaxine, and romidepsin (Fig 1). Among these, only panobinostat and bortezomib have received FDA approval for the treatment of MM. The five molecular targets associated with the highest in vitro cytotoxicity were NF-κB, survivin, IκB kinase (IKK), exportin 1 (XPO1), and histone deacetylase (Data Supplement).
FIG 1.
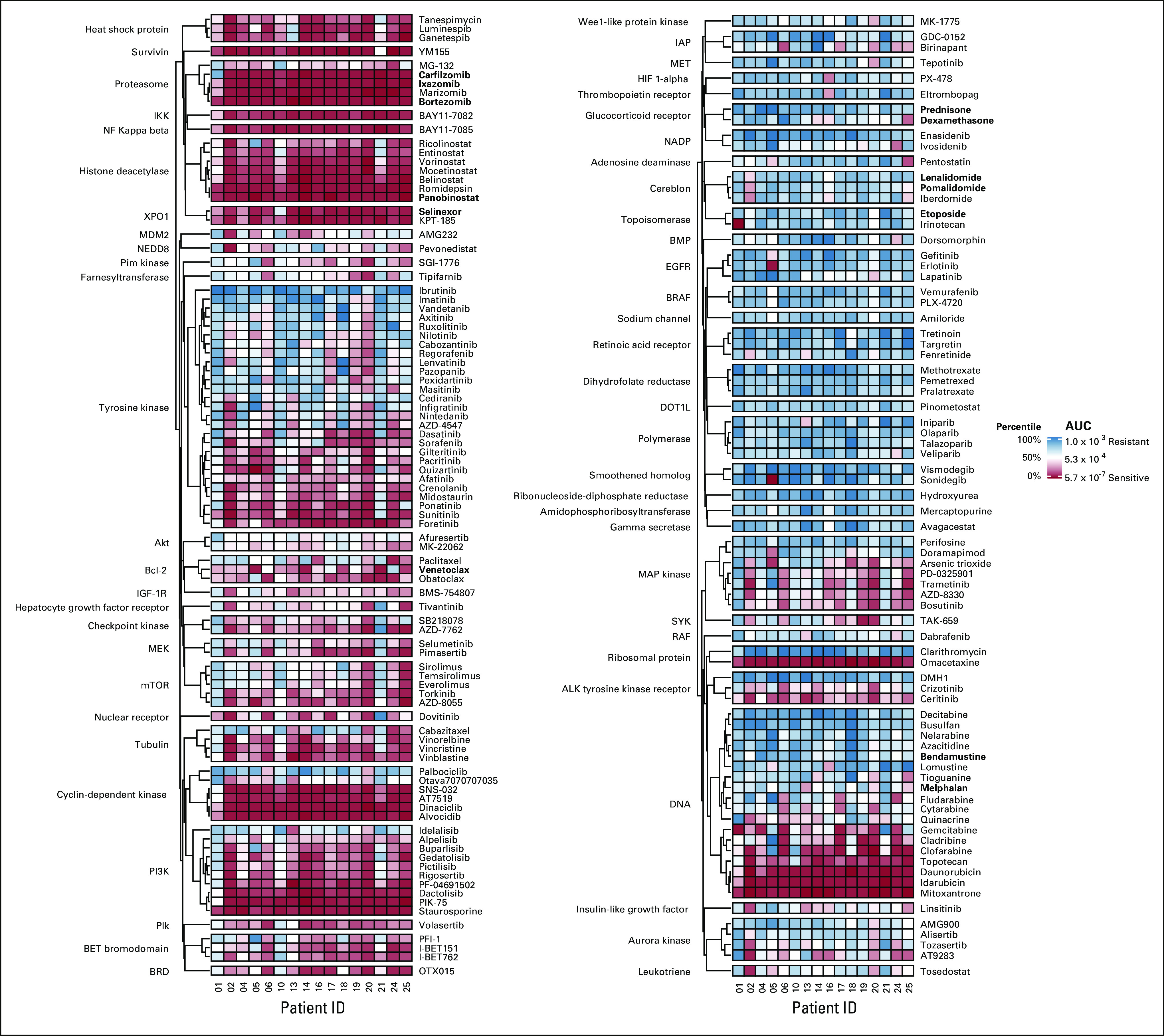
Heatmap of the AUC for all 170 drugs tested. Drugs have been grouped by their molecular target shown on the left side of the heatmap. The legend shows both the AUC and corresponding percentile rank where drugs in the 1-49th percentile are colored red indicating increased in vitro cytotoxicity relative to the drugs in the 50-100th percentile colored blue. Drugs that are recommended for treatment of MM by the National Comprehensive Cancer Network guidelines v4.2021 are bolded. AUC, area under the curve; BET, bromodomain and Extra-Terminal; BMP, bone morphogenetic proteins; BRAF, v-raf murine sarcoma viral oncogene homolog B1; BRD, bromodomain; EGFR, estimated glomerular filtration rate; HIF, hypoxia-inducible factor; IAP, inhibitors of apoptosis; IGF, insulin-like growth factor; IKK, IκB kinase; MEK, mitogen-activated protein kinase kinase; MET, mesenchymal-epithelial transition factor; MM, multiple myeloma; mTOR, mammalian target of rapamycin; NADP, nicotinamide adenine dinucleotide phosphate; I NF, nuclear factor; RAF, rapidly accelerated fibrosarcoma; SYK, spleen tyrosine kinase.
Efficacy of Assay-Guided Treatment and Correlation With Clinical Features
Among the 13 patients who received assay-guided therapy (Fig 2), the overall response rate was 46% (partial response or better) with 92% having at least stable disease (SD) (Fig 3). The number of patients demonstrating SD was 6 (46%), a partial response was achieved in 4 (31%), and a very good partial response in 2 (15%). There were no complete responses in this heavily treated cohort (median of six lines of therapy [range 3-11]; Data Supplement). The median progression-free survival for the 16 patients tested was 78 days (Fig 4A), and the median OS was 146 days (Fig 4B).
FIG 2.
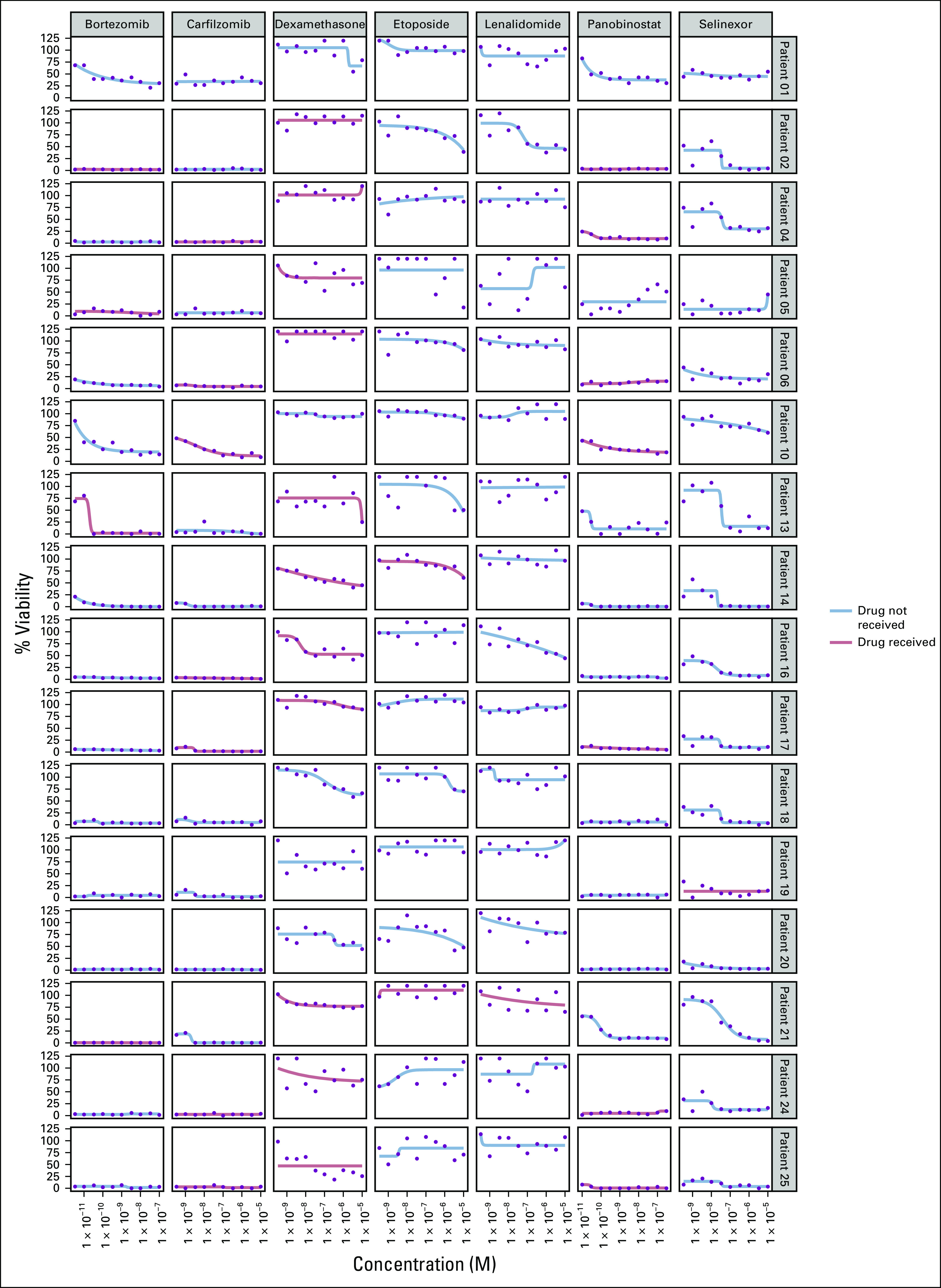
Dose-response curves for the seven tested drugs administered to the 13 patients who received assay-guided therapy. At the discretion of the treating oncologist, some patients received drugs not tested in the assay (eg, doxorubicin) or received drugs not recommended by the assay (eg, dexamethasone) but were chosen for their synergistic effect. Drugs with the lowest area under the curve have the highest in vitro cytotoxicity.
FIG 3.

Swimmers plot showing treatment response by depth and duration in relation to the chosen next line of therapy. Participants are defined as having received assay-guided therapy (blue bars) if they received one or more recommended drugs. The depth of response was measured using International Myeloma Working Group criteria. Shown to the right of the bars is the treatment received after HTS. D, dexamethasone; Dara, daratumumab; Doxil, liposomal doxorubicin; HTS, high-throughput screening; K, carfilzomib; PACE, cisplatin, doxorubicin, cyclophosphamide, and etoposide; Pano, panobinostat; PD, progressive disease; PR, partial response; R, lenalidomide; SD, stable disease; T, thalidomide; V, bortezomib; VGPR, very good partial response.
FIG 4.
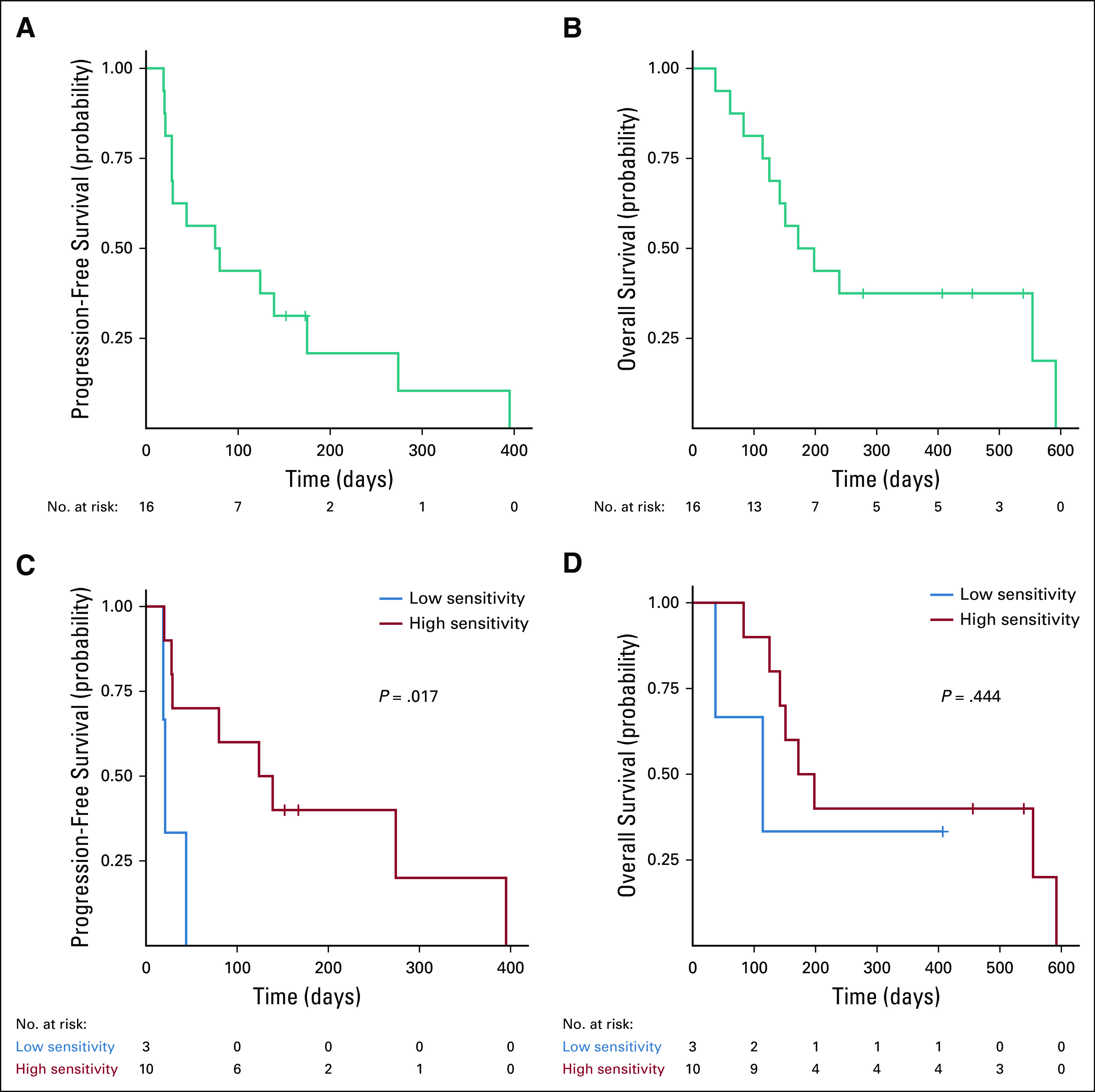
Kaplan-Meier survival analysis. (A) Progression-free survival and (B) overall survival from enrollment for all participants undergoing HTS (n = 16). (C) Progression-free survival and (D) overall survival from enrollment for participants who received assay-guided therapy (n = 13). Participants are grouped according to the mean AUC of the administered drugs. The red line denotes participants whose mean AUC corresponds to high in vitro cytotoxicity (AUC > 2 × 10−4), whereas the blue line denotes participants whose mean AUC corresponds to low in vitro cytotoxicity (AUC ≤ 2 × 10−4). Log-rank P value is shown. AUC, area under the curve; HTS, high-throughput screening.
To estimate the overall drug sensitivity for each patient’s tumor biopsy, we calculated the mean AUC of all 170 compounds tested. The median progression-free survival was 28 days for patients whose mean AUC was in the top 50th percentile (indicating a more resistant tumor) compared with 139 days for patients whose mean AUC was in the bottom 50th percentile (more sensitive tumor) (log-rank P < .042; Fig 4C). However, there was no significant difference among high versus low AUC with respect to OS (log-rank P < .151; Fig 4D).
We compared clinical features with overall drug sensitivity and identified that the absence of one or more high-risk cytogenetic abnormalities (del[17p], t(4;14), t(14;16), or 1q+) was the only feature associated with higher drug sensitivity (Fig 5A). With the exception of proteasome inhibitors, one of the most sensitive classes of drugs tested, we observed a greater in vitro cytotoxicity to drugs the patients had not previously been exposed to (Figs 5B and C). However, there was no association between AUC and the number of lines of prior therapy (Fig 5D). Wilcoxon rank-sum test identified drugs associated with sensitivity and resistance for each high-risk cytogenetic abnormality (Data Supplement) as well as the presence of any high-risk abnormality (Data Supplement).
FIG 5.
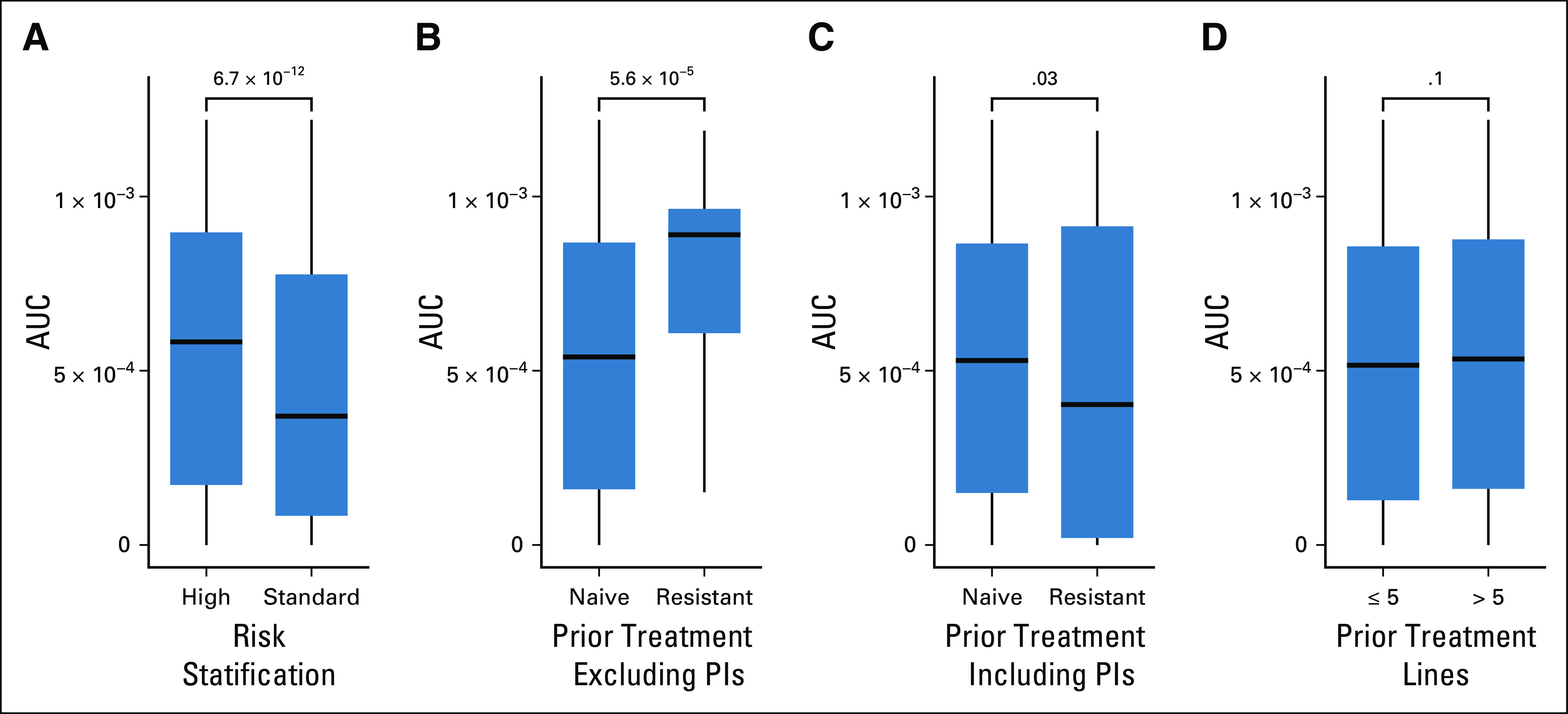
Distribution of in vitro drug response in relation to prior treatment and risk stratification. The AUC distribution of all 170 compounds split by (A) risk stratification, (B) prior treatment exposure including PIs, (C) prior treatment exposure excluding PIs, and (D) the number of prior lines of treatments. High risk is defined as patients with R-ISS stage III disease or the presence of one or more high-risk cytogenetic abnormalities detected by fluorescence in situ hybridization at diagnosis (del(17p), t(4;14), t(14;16), or 1q+). Lower AUC corresponds to increased tumor sensitivity to drugs tested. Wilcoxon rank-sum test P values are shown. AUC, area under the curve; PI, proteasome inhibitor; R-ISS, Revised International Staging System.
Correlation With Genomic Features
We investigated if DNA mutations detected in bone marrow plasma cells by WES or targeted sequencing of ctDNA within the blood correlated with AUC. For this analysis, sufficient DNA to perform ctDNA sequencing from blood was available for all patients who underwent HTS but only seven of 16 patients had sufficient leftover bone marrow plasma cells for WES (Data Supplement). We detected distinct mutations in the blood using ultradeep sequencing, which were not detectable in the bone marrow that could have been derived from extramedullary disease (Data Supplement). Elastic net regression was used to narrow the list of mutated genes associated with drug sensitivity. Only genes mutated in two or more patients were used for this analysis. The results of a Wilcoxon rank-sum test comparing AUC for each drug by gene mutation status are shown in the Data Supplement for comparisons with a P value of < .05, but none were statistically significant after adjusting for multiple comparisons. We found that mutations in seven genes associated with in vitro sensitivity to 15 drugs and mutations in five genes associated with in vitro resistance to six drugs.
To evaluate if gene expression of bone marrow plasma cells was correlated with the results of HTS, we compared normalized RNAseq values (TPM) for every expressed gene with AUC using elastic net regression. This analysis was possible for eight of the 16 patients tested since the others did not have sufficient plasma cells for RNA extraction (Data Supplement). We found that increased expression of 68 genes was correlated with in vitro cytotoxicity of 79 drugs (Data Supplement) and the decreased expression of 37 genes was correlated with in vitro cytotoxicity of 43 drugs (Data Supplement). All associations were statistically significant using linear regression analysis after adjusting for multiple comparisons (Benjamini and Hochberg adjusted P < .05).
DISCUSSION
In patients with relapsed or refractory MM, we demonstrate the feasibility of ex vivo drug sensitivity testing on isolated plasma cells from patient bone marrow biopsies or extramedullary plasmacytomas to inform the next line of therapy. To our knowledge, this is the first clinical trial to use HTS to inform the next line of therapy for patients with relapsed or refractory MM. All patients who had sufficient cells for testing had an actionable result satisfying the primary end point of our study. Results were available within a median of 5 days, giving treating physicians a clinically relevant timeframe that facilitates treatment selection and insurance approval without introducing a long delay in treatment. Additionally, we demonstrate that 92% of patients who received treatment informed by HTS were able to achieve disease control (SD or better). This was in spite of the fact that the majority of the patients tested had features of high-risk disease and had progressed after multiple lines of prior therapy.
Our assay also demonstrated that malignant plasma cells from many patients exhibit robust ex vivo sensitivity to compounds not yet evaluated in clinical trials for patients with MM, and we identified agents that warrant further investigation. These include omacetaxine, romidepsin, staurosporine, and alvocidib. Our study also confirms cytotoxicity of FDA-approved agents for relapsed MM, including panobinostat, bortezomib, ixazomib, carfilzomib, and selinexor.
In addition to using HTS as a precision medicine tool, we uncovered novel associations between genomic profiling and drug sensitivity. Since it was uncommon for multiple patient tumor cells to share the same mutated genes, we found that gene expression was more statistically robust. We used elastic net regression to predict AUC since a systematic assessment of analytical methods for drug sensitivity prediction from cancer cell lines demonstrated that method yields the most accurate results.13 A larger sample size and additional studies would be required to confirm that the associations are biological.
Our study was primarily limited by the number of plasma cells sufficient for testing. Our HTS assay requires a minimum of 6 × 105 plasma cells to test 170 compounds at eight concentrations. The cell number limitation prevented us from performing HTS in nine patients (36%). It also limited us from isolating sufficient RNA and DNA from bone marrow plasma cells for genomic analysis. A future direction would be to test a smaller array of drugs, thereby reducing the total number of cells required for HTS so that more patients may be eligible for testing.
A second limitation of our study was that HTS was performed on tumor cells only. As a result, our assay was unable to assess in vitro activity of immunotherapies whose mechanism of action is dependent on cells within the immune microenvironment such as T cells and natural killer cells. We suspect it is for this reason that the immunomodulatory drugs lenalidomide, pomalidomide, and iberdomide had surprisingly low in vitro activity. Additionally, testing both tumor and nontumor cells would have allowed us to measure differential sensitivity between benign and malignant hematopoietic cells, which would have enabled us to estimate toxicity in addition to efficacy. However, since most drugs evaluated in our assay have previously been tested in human clinical trials, the hematopoietic toxicity is already known. A potential solution to address this limitation would be to use 3D tissue-engineered bone marrow cultures and a flow cytometric readout, which has been demonstrated by others in preclinical studies.14,15
Although this study aimed to evaluate in vitro cytotoxicity of single agents, we are conducting ongoing studies to evaluate the synergy of multiple agents.16 It is well known that by exploiting multiple mechanisms of action, combination chemotherapy decreases the likelihood of tumor resistance. For example, preclinical studies have demonstrated synergistic antimyeloma activity between deacetylase inhibitors and proteasome inhibitors.17 This approach may uncover new combinations of drugs not previously evaluated that could motivate the design of future clinical trials.
Andrew J. Cowan
Stock and Other Ownership Interests: Doximity
Consulting or Advisory Role: Doximity, Sanofi, Cellectar, AbbVie
Research Funding: Bristol Myers Squibb, Janssen, AbbVie, Celgene, Nektar
Open Payments Link: https://openpaymentsdata.cms.gov/physician/274628
Damian J. Green
Consulting or Advisory Role: Juno Therapeutics, Seattle Genetics, GlaxoSmithKline, Celgene, Bristol Myers Squibb/Celgene, Janssen Oncology, Legend Biotech
Research Funding: Juno Therapeutics, Sanofi, Merck, Cellectar, Bristol Myers Squibb, Seattle Genetics, Janssen Oncology
Patents, Royalties, Other Intellectual Property: Intellectual property on pending patents related to the combination of a small molecule with BCMA-targeted CAR T-cell therapy. Royalty sharing agreement in place
Edward N. Libby
Consulting or Advisory Role: Adaptive Biotechnologies, Akcea Therapeutics
Research Funding: GlaxoSmithKline, Janssen, Genentech, BeiGene
Travel, Accommodations, Expenses: GlaxoSmithKline
Rebecca Silbermann
Consulting or Advisory Role: Sanofi/Aventis
Sylvia Chien
Employment: Omeros
Stock and Other Ownership Interests: Omeros
Ted A. Gooley
Consulting or Advisory Role: Kiadis Pharma, Pharmacyclics, Regimmune
Edus H. Warren
Stock and Other Ownership Interests: AbbVie
Pamela S. Becker
Consulting or Advisory Role: CVS Caremark, Pfizer
Research Funding: AbbVie, Amgen, Bristol Myers Squibb, JW Pharmaceutical, Novartis, Pfizer, Glycomimetics, Trovagene, Invivoscribe, Secura Bio, Cardiff Oncology
Patents, Royalties, Other Intellectual Property: Provisional patent filed: Application 62/694,874 filed July 6, 2018 entitled: “High Throughput Drug Screening of Cancer Stem Cells”
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at the American Society of Hematology Annual Meeting, Orlando, FL, December 7-10, 2019.
SUPPORT
Supported by generous private donations from Ann and Kevin Harrang, a grant from the Brotman Baty Institute, National Cancer Institute Cancer Center Support Grant No. P30 CA015704, and the Cancer Therapeutics Endowment. The Fred Hutchinson Cancer Research Center (FHCRC) Seattle Translational Tumor Research supported the development of a clinical database used in this project. The FHCRC Genomics Shared Resource and the University of Washington Northwest Genomics Center performed next-generation sequencing. The Scientific Computing Infrastructure at FHCRC was funded by Office of Research Infrastructure Programs Grant No. S10OD028685.
D.G.C. and A.J.C. equally contributed to this work.
AUTHOR CONTRIBUTIONS
Conception and design: David G. Coffey, Andrew J. Cowan, Timothy J. Martins, Damian J. Green, Jin Dai, Pamela S. Becker
Administrative support: Timothy J. Martins, Niall Curley, Sylvia Chien, Jin Dai, Edus H. Warren
Financial support: Timothy J. Martins, Jin Dai, Edus H. Warren
Provision of study materials or patients: Timothy J. Martins, Niall Curley, Damian J. Green, Edward N. Libby, Jin Dai, Pamela S. Becker
Collection and assembly of data: David G. Coffey, Andrew J. Cowan, Bret DeGraaff, Timothy J. Martins, Niall Curley, Edward N. Libby, Rebecca Silbermann, Sylvia Chien, Jin Dai, Alicia Morales, Pamela S. Becker
Data analysis and interpretation: David G. Coffey, Andrew J. Cowan, Bret DeGraaff, Timothy J. Martins, Damian J. Green, Edward N. Libby, Rebecca Silbermann, Sylvia Chien, Jin Dai, Ted A. Gooley, Edus H. Warren, Pamela S. Becker
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Andrew J. Cowan
Stock and Other Ownership Interests: Doximity
Consulting or Advisory Role: Doximity, Sanofi, Cellectar, AbbVie
Research Funding: Bristol Myers Squibb, Janssen, AbbVie, Celgene, Nektar
Open Payments Link: https://openpaymentsdata.cms.gov/physician/274628
Damian J. Green
Consulting or Advisory Role: Juno Therapeutics, Seattle Genetics, GlaxoSmithKline, Celgene, Bristol Myers Squibb/Celgene, Janssen Oncology, Legend Biotech
Research Funding: Juno Therapeutics, Sanofi, Merck, Cellectar, Bristol Myers Squibb, Seattle Genetics, Janssen Oncology
Patents, Royalties, Other Intellectual Property: Intellectual property on pending patents related to the combination of a small molecule with BCMA-targeted CAR T-cell therapy. Royalty sharing agreement in place
Edward N. Libby
Consulting or Advisory Role: Adaptive Biotechnologies, Akcea Therapeutics
Research Funding: GlaxoSmithKline, Janssen, Genentech, BeiGene
Travel, Accommodations, Expenses: GlaxoSmithKline
Rebecca Silbermann
Consulting or Advisory Role: Sanofi/Aventis
Sylvia Chien
Employment: Omeros
Stock and Other Ownership Interests: Omeros
Ted A. Gooley
Consulting or Advisory Role: Kiadis Pharma, Pharmacyclics, Regimmune
Edus H. Warren
Stock and Other Ownership Interests: AbbVie
Pamela S. Becker
Consulting or Advisory Role: CVS Caremark, Pfizer
Research Funding: AbbVie, Amgen, Bristol Myers Squibb, JW Pharmaceutical, Novartis, Pfizer, Glycomimetics, Trovagene, Invivoscribe, Secura Bio, Cardiff Oncology
Patents, Royalties, Other Intellectual Property: Provisional patent filed: Application 62/694,874 filed July 6, 2018 entitled: “High Throughput Drug Screening of Cancer Stem Cells”
No other potential conflicts of interest were reported.
REFERENCES
- 1.Paquin AR, Kumar SK, Buadi FK, et al. : Overall survival of transplant eligible patients with newly diagnosed multiple myeloma: Comparative effectiveness analysis of modern induction regimens on outcome. Blood Cancer J 8:125, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manier S, Salem KZ, Park J, et al. : Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol 14:100-113, 2017 [DOI] [PubMed] [Google Scholar]
- 3.Walker BA, Mavrommatis K, Wardell CP, et al. : Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 132:587-597, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Majumder MM, Silvennoinen R, Anttila P, et al. : Identification of precision treatment strategies for relapsed/refractory multiple myeloma by functional drug sensitivity testing. Oncotarget 8:56338-56350, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Campos CB, Meurice N, Petit JL, et al. : “Direct to Drug” screening as a precision medicine tool in multiple myeloma. Blood Cancer J 10:54, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matulis SM, Gupta VA, Neri P, et al. : Functional profiling of venetoclax sensitivity can predict clinical response in multiple myeloma. Leukemia 33:1291-1296, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva A, Silva MC, Sudalagunta P, et al. : An ex vivo platform for the prediction of clinical response in multiple myeloma. Cancer Res 77:3336-3351, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar S, Paiva B, Anderson KC, et al. : International Myeloma Working Group consensus criteria for response and minimal residual disease assessment in multiple myeloma. Lancet Oncol 17:e328-e346, 2016 [DOI] [PubMed] [Google Scholar]
- 9.Lee S-I, Celik S, Logsdon BA, et al. : A machine learning approach to integrate big data for precision medicine in acute myeloid leukemia. Nat Commun 9:42, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halpern AB, Martins TJ, Jonlin EC, et al. : Case of chemotherapy-refractory, RAS-mutated chronic myelomonocytic leukemia responsive to single-agent trametinib based on results from a high-throughput drug screen. JCO Precis Oncol 4;1367-1373, 2020 [DOI] [PubMed] [Google Scholar]
- 11.Xu C, Gu X, Padmanabhan R, et al. : smCounter2: An accurate low-frequency variant caller for targeted sequencing data with unique molecular identifiers. Bioinformatics 35:1299-1309, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coffey DG, Wu QV, Towlerton AMH, et al. : Ultradeep, targeted sequencing reveals distinct mutations in blood compared to matched bone marrow among patients with multiple myeloma. Blood Cancer J 9:77, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang IS, Neto EC, Guinney J, et al. : Systematic assessment of analytical methods for drug sensitivity prediction from cancer cell line data. Pac Symp Biocomput 63-74, 2014 [PMC free article] [PubMed] [Google Scholar]
- 14.de la Puente P, Muz B, Gilson RC, et al. : 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 73:70-84, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Walker ZJ, VanWyngarden MJ, Stevens BM, et al. : Measurement of ex vivo resistance to proteasome inhibitors, IMiDs, and daratumumab during multiple myeloma progression. Blood Adv 4:1628-1639, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Becker PS, Quach K, Gooley TA, et al. : High throughput drug synergy testing in a clinical trial of panobinostat, carfilzomib, dexamethasone to define response biomarkers for relapsed/refractory multiple myeloma. Blood 134:1902, 2019. 31778549 [Google Scholar]
- 17.Hideshima T, Richardson PG, Anderson KC: Mechanism of action of proteasome inhibitors and deacetylase inhibitors and the biological basis of synergy in multiple myeloma. Mol Cancer Ther 10:2034-2042, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]