

PURPOSE
To characterize the relationship between tumor-infiltrating lymphocytes (TIL), tumor mutational burden (TMB), and genetic alterations in microsatellite stable (MSS), microsatellite instability (MSI), or mutant POLE/POLD1 colon cancer.
MATERIALS AND METHODS
Four hundred ninety-nine resected stage I-III colon tumors treated between 2014 and 2019 were assessed for TIL; somatic mutations, copy number alterations, and structural changes in > 400 oncogenes; and MSI status.
RESULTS
Of the 499 tumors analyzed, 313 were MSS, 175 were MSI, and 11 had POLE/POLD1 pathogenic mutations. Both the percentage of tumors with a high level of TIL (≥ 4 lymphocytes per high-power field) and the median TMB differed significantly between the three phenotypes: MSS, 4.5% and 6 mutations/Mb; MSI, 68% and 54 mutations/Mb; POLE/POLD1, 82% and 158 mutations/Mb (P < .05). Within each phenotype, TMB did not vary significantly with TIL level. Among MSI tumors, the median number of frameshift indels was significantly higher in tumors with high levels of TIL (20 v 17; P = .018). In the MSS group, significantly higher proportions of tumors with high levels of TIL had mutations in the transforming growth factor-β (36% v 12%; P = .01), RAS (86% v 54%; P = .02), and Hippo (7% v 1%; P = .046) pathways; in contrast, TP53 alterations were associated with low levels of TIL (74% v 43%; P = .01).
CONCLUSION
The association between TIL, TMB, and genetic alterations varies significantly between MSI, MSS, and mutant POLE/POLD1 colon tumors. These differences may help explain tumoral immunity and lead to predictors of response to immunotherapy.
INTRODUCTION
Lymphocytes in the tumor microenvironment are thought to be critical to unlocking the potential of immunotherapy by activating the patient’s pre-existing intratumoral immunity.1,2 The association between elevated levels of tumor-infiltrating lymphocytes (TIL) and microsatellite instability (MSI) has been well-documented.3,4
CONTEXT
Key Objective
Traditionally, immunotherapy for colon cancer has been restricted to microsatellite instability (MSI) tumors, but recent studies have shown potential for response in microsatellite stable (MSS) tumors. Factors that may influence response to immunotherapy include the presence of tumor-infiltrating lymphocytes (TIL), tumor mutational burden (TBM), and certain genetic alterations in oncogenic signaling pathways. This study was aimed at characterizing the inter-relationship of TIL, tumor mutational burden, and genetic alterations in MSI, MSS, and POLE/POLD1 tumors.
Knowledge Generated
We found that TIL levels were not associated with tumor mutational burden in MSS, MSI, or POLE/POLD1 tumors but were associated with the presence of frameshift indels in MSI tumors and with alterations in the transforming growth factor-β, RAS, and Hippo pathways in MSS tumors.
Relevance
Assessment of frameshift indel load in MSI tumors and assessment of alterations in oncogenic signaling pathways in MSS tumors may help identify patients whose disease is likely to respond to immunotherapy.
The high levels of TIL in MSI cancers are hypothesized to represent an adaptive response to neoantigens resulting from mismatch repair (MMR) enzyme deficiency and accumulation of mutations.5-7 Although tumor mutational burden (TMB) has been used as a surrogate for neoantigen load,5,8,9 this framework likely oversimplifies the mechanism of TIL recruitment. Indeed, microsatellite stable (MSS) colon cancer with low TMB but high TIL and MSI colon cancer with high TMB but low TIL have been described in several studies.5,9 In a recent analysis, we found that the number of TIL per high-power field ranges greatly in both MSI and MSS colon tumors, with up to 18% of MSI tumors containing low levels of TIL and up to 36% of MSS tumors containing high levels of TIL.10
Interest in TIL has grown significantly, as they are prognostic of survival outcomes10-12 and may be predictive of response to immunotherapy in colorectal cancer.13 In this study, we used histopathologic analysis and next-generation sequencing to characterize the relationship between TIL and oncogenic signaling pathway alterations in MSI, MSS, or mutant POLE/POLD1 (POLE/D1) colon cancer.
MATERIALS AND METHODS
Study Sample
With approval from the institutional review board, institutional databases were queried for patients who underwent surgical resection of nonmetastatic colon cancer at Memorial Sloan Kettering Cancer Center (MSK) between January 2014 and December 2019, without the use of neoadjuvant therapy and with MSK-IMPACT (see below) results available (Fig 1). Demographic, clinicopathologic, and follow-up data were retrieved from the institutional databases and electronic medical records.
FIG 1.

Inclusion and exclusion of tumor samples. MSK, Memorial Sloan Kettering Cancer Center; OSH, outside hospital; pTIS, carcinoma in situ.
Pathology Analysis
Tumors were staged according to American Joint Committee on Cancer and categorized as right sided (cecum, ascending colon, or transverse colon) or left sided (descending colon, sigmoid, or rectosigmoid). The level of TIL was quantified according to a protocol proposed by the College of American Pathologists14 and as previously outlined,10,15,16 with a slight modification. Briefly, TIL within the epithelial compartment were identified on standard hematoxylin-eosin–stained slides (without specific T-cell markers). A tumor was classified as TIL-high if the mean number of lymphocytes per high-power field was ≥ 4, averaged from five high-power fields in an area determined to have the highest concentration of TIL by examination of the entire tumor.
Genomic Analysis
Tumor and normal matched DNA were analyzed with MSK-IMPACT,17 a high-throughput platform that captures somatic mutations, copy number alterations, and structural variations in exons and selected introns for 410-468 oncogenes. Microsatellite status was determined with the computational tool MSIsensor as previously described.18 MSIsensor scores ≥ 10 in colorectal and endometrioid cancers have > 99% concordance with MSI status determination by PCR and MMR status determination by immunohistochemistry.18 Briefly, based on MSIsensor analysis of MSK-IMPACT sequence reads of available microsatellite regions in each normal-tumor DNA pair, tumors were categorized as MSI if ≥ 10% of microsatellite regions were unstable and as MSS if < 10% of microsatellite regions were unstable. MSK-IMPACT was also used to identify hotspot mutations in the exonuclease domain of DNA polymerases ε (POLE) and δ1 (POLD1). The 499 tumors were then grouped into three genomic phenotypes: (1) MSI, (2) nonhypermutated MSS (MSS), and (3) hotspot POLE/D1 mutant (POLE/D1).
TMB was quantified as mutations per megabase (mt/Mb). Immunohistochemical staining for the mismatch repair proteins MLH1, MSH2, MSH6, and PMS2 was performed as previously described.15
Signaling Pathways
Somatic single-nucleotide variants (SNVs) and short insertions or deletions (indels) detected by MSK-IMPACT were analyzed in relation to genomic phenotype and TIL level. Analysis of paired tumor and normal tissue samples filters out germline single nucleotide polymorphisms.17 Genetic alterations, including insertions and deletions, that lead to altered protein structures were grouped as frameshift (FS) indels. Non-FS insertions and deletions were excluded because of presumed low frequency. Somatic mutations in designated genes in the MSK-IMPACT panel were analyzed19,20 and assigned to the 10 selected oncogenic signaling pathways as described by Sanchez-Vega et al21 (see Fig 2 and Table S3 in ref. 21). A tumor sample was considered to have an altered signaling pathway if one or more genes of the pathway had any somatic mutation. The pathway alterations were compared between the MSI, MSS, and POLE/D1 phenotypes. The mutations were also filtered for oncogenic variants using the precision oncology knowledge base OncoKB,22 and the filtered pathways were compared between the phenotypes.
Statistical Analysis
Statistical analyses were performed using IBM SPSS statistics 25 or R version 4.0. Differences in categorical variables were evaluated with the chi-square or Fisher's exact test. Medians were compared using the Mann-Whitney test. Bonferroni inequality was used to adjust for multiple comparisons. P values < .05 were considered statistically significant.
RESULTS
A total of 499 tumors (Fig 1) from 497 patients (including one synchronous tumor and one metachronous tumor) were identified: 313 MSS tumors (63%), 175 MSI tumors (35%), and 11 POLE/D1 tumors (2%). Of the 175 MSI tumors, 137 (78%) had loss of MLH1/PMS2, 18 (10%) had loss of MSH2/MSH6, 6 (4%) had loss of MSH6 only, and five (3%) had loss of PMS2 only; the remaining nine (5%) had loss of multiple proteins or no loss of proteins. Of the 11 hotspot POLE/D1 tumors, nine had POLE mutations (four with V411A/L, four with P286R, and one with S459F) and two had POLD1 mutations (one with S478N and the other with the germline mutation L474P). It is worth noting that two of the POLE/D tumors were found by MSIsensor to have acquired MMR mutations,23 which we hypothesize were secondary events.
Clinicopathologic features are listed in Table 1 in relation to cancer genomic phenotype. Patients with POLE/D1 cancer were younger, with a mean age of 47 years compared with 58 years for patients with MSS cancer (P = .013) and 66 years for patients with MSI cancer (P = .0001). Patients with POLE/D1 cancer were more likely to be male (82%) than patients with MSS cancer (53%; P = .063) or patients with MSI cancer (45%; P = .018). MSI tumors were more likely to be right sided (85%) than MSS tumors (40%; P < .00001) or POLE/D1 tumors (64%; P = .071). Compared with MSS tumors, POLE/D1 tumors were more likely to be of an early cancer stage (stage I or II) (82% v 47%; P = .023) and less likely to show evidence of lymphovascular invasion (18% v 57%; P = .011). MSI tumors were also more likely to be of stage I or II than MSS tumors (71% v 47%; P < .00001).
TABLE 1.
Clinicopathologic Characteristics in Relation to Colon Cancer Genomic Phenotype

Of the 499 tumors analyzed, 357 (72%) were TIL-low and 142 (38%) were TIL-high. The proportions of TIL-high tumors were as follows: POLE/D1, 82%; MSI, 68% (P < .00001 v MSS tumors); MSS, 4.5% (P < .00001 v POLE/D1 tumors) (Table 1). TIL level and cancer genomic phenotype were associated with TMB (Fig 2). The median TMB was 54 mt/Mb in TIL-high tumors and 6.3 mt/Mb in TIL-low tumors (P < .00001). With respect to genomic phenotype, the median TMB was 158 mt/Mb in POLE/D1 tumors, 54 mt/Mb in MSI tumors, and 6 mt/Mb in MSS tumors (P < .00001 for all comparisons) (Table 1). However, there appeared to be no relationship between TMB and TIL level of tumors within the genomic phenotype. The TMB for TIL-low and TIL-high tumors was 6.1 and 6.6 mt/Mb (P = .075) in the MSS group, 56 and 54 mt/Mb (P = .481) in the MSI group, and 203 and 158 mt/Mb (P = .909) in the POLE/D1 group (Fig 2).
FIG 2.

Association between TIL and TMB (A) for the whole study sample and (B) for the MSS, MSI, and POLE/D1 phenotypes (logarithmic scale). The outlier in the MSS TIL-high group (TMB, 151.8 mt/Mb) had a non-hotspot POLE mutation (p.M444K). MSI, microsatellite instability; MSS, microsatellite stable; TIL, tumor-infiltrating lymphocytes; TMB, tumor mutational burden.
SNVs and small indels also varied between cancer genomic phenotypes (Fig 3). The median FS indel load of 19 (range, 0-49) in MSI tumors was significantly higher than that in MSS tumors (median, 1; range, 0-4; P < .00001) and that in POLE/D1 tumors (median, 1; range, 0-23; P < .00001). Compared with MSS tumors, POLE/D1 tumors had a significantly higher number of FS indels (P = .019). POLE/D1 tumors also had higher median numbers of nonsynonymous (missense) SNVs, nonsense SNVs, and splice site mutations than MSS and MSI tumors. The median number of missense SNVs in POLE/D1 tumors was 152 (range, 33-313), compared with 35 (range, 5-342) in MSI tumors (P < .00001) and 4 (range, 0-139) in MSS tumors (P < .00001). The median number of nonsense SNVs in POLE/D1 tumors—28 (range, 6-57)—was significantly higher than that in MSI tumors (median, 3; range, 0-31; P < .00001) and that in MSS tumors (median, 1; range, 0-27; P < .00001). The median number of splice-site mutations in POLE/D1 tumors—2 (range, 1-16)—was also significantly higher than that in MSI tumors (median, 1; range, 0-22; P = .013) and that in MSS tumors (median, 0; range, 0-7; P < .00001).
FIG 3.

(A) Genetic alterations in MSS, MSI, and POLE/D1 tumors and (B) comparison of FS indel load between TIL-high MSI tumors and TIL-low MSI tumors. The data are medians with quartiles and ranges. FS, frameshift; MSI, microsatellite instability; MSS, microsatellite stable; TIL, tumor-infiltrating lymphocytes.
TIL-high MSI tumors had more FS indels than TIL-low MSI tumors (median, 20 [range, 4-49] v 17 [range, 0-49]; P = .018), mainly because of a significant difference in the numbers of FS deletions (median, 16 [range, 2-39] v 12 [range, 0-35]; P = .008). The frequency of FS indels did not differ significantly between TIL-high MSS tumors and TIL-low MSS tumors. The frequency of nonsynonymous SNVs did not differ significantly between TIL-high MSI tumors and TIL-low MSI tumors (median, 35 [range, 13-207] v 35.5 [range, 5-342]; P = .531) or between TIL-high MSS tumors and TIL-low MSS tumors (median, 5.5 [range, 2-139] v 4 [range, 0-22]; P = .064). The frequencies of nonsense, non-FS indel, and nonstop mutations did not differ significantly between TIL-high and TIL-low MSI tumors or between TIL-high and TIL-low MSS tumors.
For oncogenes in the MSK-IMPACT panel, no significant difference in the frequency of somatic mutations was found between TIL-high and TIL-low MSI tumors or between TIL-high and TIL-low MSS tumors (Data Supplement). The numbers of tumors with somatic genetic alterations in the 10 selected oncogenic signaling pathways were as follows: WNT, 465 (93%); RAS, 383 (77%); TP53, 354 (71%); phosphatidylinositol 3-kinase, 301 (60%); Notch, 255 (51%); transforming growth factor (TGF)-β, 155 (31%); Hippo, 142 (29%); cell cycle, 93 (19%); MYC, 86 (17%); nuclear erythroid 2–related factor 2, 45 (9%). The mutation frequencies for individual genes in these pathways are shown in Appendix Figure A1.
Compared with TIL-low MSS tumors, a significantly greater proportion of TIL-high MSS tumors had mutations in the TGF-β pathway (43% v 16%; P = .022) and in the cell cycle pathway (21% v 4%; P = .024) (Fig 4). TP53 pathway mutations were found in 50% of TIL-high MSS tumors, compared with 77% of TIL-low MSS tumors (P = .018). Even when the analysis was restricted to oncogenic mutations identified by OncoKB, this finding persisted. RAS pathway mutations were found in a significantly higher proportion of TIL-high MSS tumors than TIL-low MSS tumors (86% and 54%; P = .019), as were mutations in the Hippo pathway (7% v 1%; P = .046). No significant differences were found between TIL-high MSI tumors and TIL-low MSI tumors in the proportion of samples containing mutations in any of the signaling pathways examined.
FIG 4.

Proportions of (A) MSI, (B) MSS, and (C) POLE/D1 tumors, stratified by TIL level, with mutations in the indicated pathways. Shaded bars, all somatic mutations; solid bars, pathogenic mutations based on OncoKB, a precision oncology knowledge platform. *P < .05. CC, cell cycle; MSI, microsatellite instability; MSS, microsatellite stable; TIL, tumor-infiltrating lymphocytes.
DISCUSSION
Our study found a significant association between the level of TIL and the likelihood of oncogenic alterations in the TGF-β, TP53, RAS, and Hippo pathways in MSS colon tumors. Alterations in the TGF-β, RAS, and Hippo pathways were predominant in TIL-high MSS tumors, whereas alterations in the TP53 pathway were predominant in TIL-low MSS tumors. In addition, FS indels were found in a significantly higher proportion of TIL-high MSI tumors than TIL-low MSI tumors. Differences in TIL between tumors of the same genomic phenotype were not explained by differences in TMB.
Some studies have shown that alterations in the TGF-β signaling pathway can lead to decreased immune infiltrates.24 However, growing evidence indicates that dysregulation in TGF-β signaling facilitates migration of TIL,25-27 and our findings are consistent with that evidence. The bidirectional effects of TGF-β dysregulation have been pointed out previously, including suppression of tumor formation via reduction of cell proliferation and promotion of apoptosis on the one hand, while promoting tumor survival via immune evasion on the other hand.27,28 Alterations in the TGF-β signaling pathway may abrogate TGF-β–mediated immune evasion and promote TIL via CD8 activity.26-28 In mouse models, inhibition of TGF-β signaling renders colon cancer responsive to immunotherapy,29 and several clinical trials are investigating immunotherapy incorporating TGF-β inhibition for solid tumors.25,29,30
Our finding of a significantly higher proportion of TIL-low MSS tumors with alterations in the TP53 signaling pathway compared with TIL-high MSS tumors is consistent with the findings of previous studies that found an association between mutations in TP53 and suppression of immune signatures, including CD8 and immune cytolytic activity,31 possibly in connection with HLA downregulation.
Unexpectedly, oncogenic mutations in the RAS and Hippo pathways were more common in TIL-high MSS tumors than in TIL-low MSS tumors. The RAS-MAPK signaling pathway is frequently dysregulated in colon cancer, and mutations in this pathway are associated with aggressive histology, poor prognosis, and resistance to anti-EGFR therapy.28,32 Although mutations in RAS have been associated with immune exclusion,33 certain dominant oncogenic mutations of KRAS, such as G12D and G12V, may be associated with SNV-derived neoantigens.34 T-cell immunogenicity has been reported for epitopes derived from these RAS mutations,35 and immunization with these peptides can generate CD8 and CD4 T-cell responses.36,37 RAS-specific adoptive cell therapy leading to tumor regression has also been reported.38
The association between the alterations in the Hippo pathway and TIL-high MSS tumors should be interpreted carefully, as alterations in the downstream effectors of the Hippo pathway have immune-modulating effects independent of the canonical Hippo pathway.39 Furthermore, some members of the Hippo pathway are involved in a complicated interaction with the WNT pathway, which may confound the results.40,41 Upregulated WNT signaling has been reported to be associated with a reduced T-cell infiltrate in MSS colon cancer.42 However, we did not find an association between alterations in the WNT pathway and the level of TIL in MSS or MSI tumors.
We found an association between TIL and genetic alteration type in the MSI cohort. TIL-high MSI tumors had a significantly higher number of FS indels than TIL-low MSI tumors. FS mutations lead to truncated proteins with unique C-terminal sequences that generate antigenic epitopes with the potential to elicit a potent T-cell response,4,12,42-44 and indel mutations are associated with TIL density and immunogenic response in MSI colorectal tumors.5,43 In clinical responders to immune checkpoint inhibitors, the tumor generally has a high indel load.43 Combining the FS indel burden with the currently accepted MSI and TMB biomarkers12 may help identify candidates for immunotherapy.
The POLE/D1 tumors in our study had few indels but a high rate of missense mutations, which are features consistent with the loss of DNA replication fidelity.38,45,46 Although missense mutations are considered less immunogenic than FS indels, nine of the 11 POLE/D1 tumors were TIL-high, likely because of the large number of nonsense and splice site mutations.46-49 Secondarily acquired mutations in mismatch repair genes can also result in an MSI genotype with an elevated indel load,42 and in our study, two of the 11 POLE/D1 tumors were MSI, including one with biallelic inactivation of MLH1.
Although TMB was associated with both TIL and genomic phenotype, it did not differ significantly between TIL-high and TIL-low tumors within the genomic phenotype. Therefore, if TIL represent an adaptive immune response to mutation-induced neoantigens,5-7 TMB alone is likely not an adequate surrogate for meaningful neoantigen load.5,8,9
Immune editing may explain the 56 (32%) MSI tumors that were TIL-low despite high TMB. The process of elimination, equilibrium, and ultimately escape influences the immune response and promotes tumor evolution.42,50,51 Mutation in antigen-presenting machinery, such as biallelic loss of β2-microglobulin, a stabilizing subunit required for surface expression of HLA class I, is an example of such a mechanism.42,50,51 However, β2-microglobulin mutations with loss of protein expression may not be predictive of resistance to immune checkpoint inhibitors in MSI colorectal cancer.52 Regardless, we did not identify mutational differences in immune-related genes between TIL-high and TIL-low tumors. This may be attributable to an alternate mechanism of tumor escape independent of mutation pattern, lack of statistical power, or a highly diverse and complex mutational landscape in hypermutated colon cancer. For example, specific neopeptides derived from somatic mutations are rarely found in more than one patient,50 indicating that immune-evasion mutations are likely distinct.
Our study had several limitations because of its retrospective nature. The cohort’s relatively high proportion of MSI tumors was likely due to referral patterns and a bias in selecting for genetic analysis patients with MMR deficiency identified by immunohistochemistry or patients with a recurrence. In addition, some low-frequency oncogenic somatic mutations might have been missed. The association between the level of TIL and the likelihood of oncogenic alterations in the TGF-β, TP53, RAS, and Hippo pathways in MSS colon tumors should be considered a preliminary finding, since we did not adjust for multiple-hypothesis testing, and validation of these findings in an independent cohort is needed. Further validation in patients with stage IV disease would also be valuable.
In conclusion, our finding of an association between TIL and genetic alterations in colon cancer, independent of TMB, has potentially significant implications for clinical practice, especially in light of recent evidence of variations in how colon cancer responds to immunotherapy.53 These results can potentially help identify patients for whom immunotherapy is likely to be most effective.
ACKNOWLEDGMENT
We would like to thank Memorial Sloan Kettering's Molecular Diagnostics Service and Marie-Josee and Henry R. Kravis Center for Molecular Oncology for valuable support, and we would like to thank Arthur Gelmis for editing the manuscript.
Appendix
FIG A1.
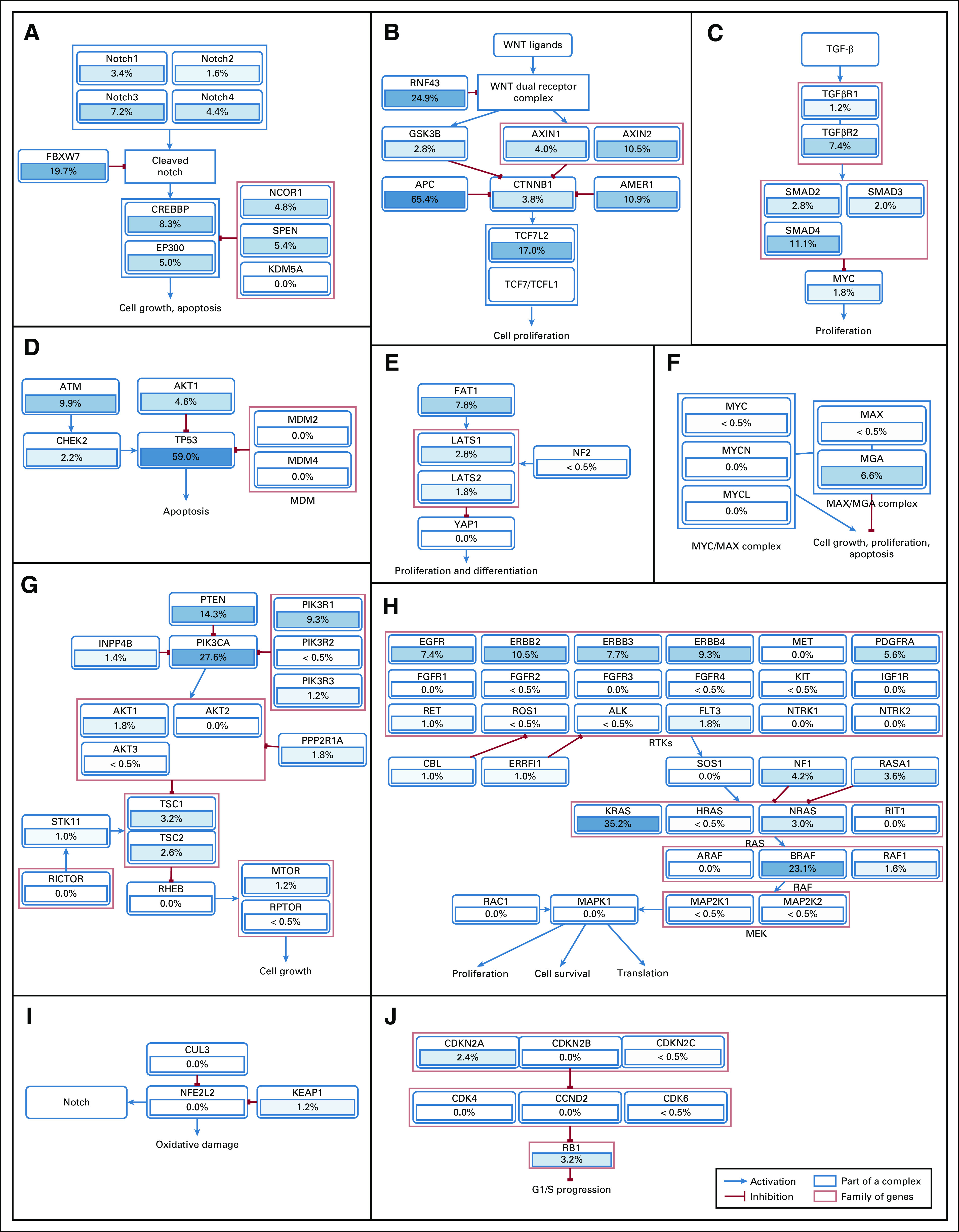
Frequencies and interactions of somatic genetic mutations in 10 selected signaling pathways (n = 499). Genetic alterations identified by the MSK-IMPACT assay were filtered by OncoKB to exclude mutations and copy number alterations of unknown significance as well as germline mutations. (A) Notch pathway. (B) WNT pathway. (C) TGF-β pathway. (D) TP53 pathway. (E) Hippo pathway. (F) MYC pathway. (G) PI3K pathway. (H) RAS pathway. (I) NRF2 pathway. (J) Cell cycle pathway.
Chad Vanderbilt
Stock and Other Ownership Interests: Paige.AI
Consulting or Advisory Role: DocDoc, Paige.AI
Other Relationship: Paige.AI
Rona Yaeger
Consulting or Advisory Role: Array BioPharma, Natera
Research Funding: Array BioPharma, Boehringer Ingelheim, Pfizer
Neil H. Segal
Consulting or Advisory Role: Bristol Myers Squibb, AstraZeneca/MedImmune, Roche/Genentech, Pieris Pharmaceuticals, Synlogic, Aduro Biotech, Kyn Therapeutics, PureTech, Gritstone Oncology, TRM Oncology, PsiOxus Therapeutics, CStone Pharmaceuticals, Immunocore, Amgen, GlaxoSmithKline, ABL Bio, Boehringer Ingelheim
Research Funding: Bristol Myers Squibb, Pfizer, Roche/Genentech, Merck, Incyte, Immunocore, AstraZeneca
Zsofia Stadler
Consulting or Advisory Role: Allergan, Genentech/Roche, Regeneron, Optos, Adverum, Novartis, Regenxbio, Gyroscope, Neurogene
Martin R. Weiser
Consulting or Advisory Role: Precisca
Research Funding: Clinical Genomics
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented at American College of Surgeons 2020 Annual Clinical Congress (virtual), October 4-8, 2020.
SUPPORT
Supported by NCI Grant No. P30 CA008748 and the John and Michelle Martello Research Fund.
DATA SHARING STATEMENT
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/po.20.00456.
AUTHOR CONTRIBUTIONS
Conception and design: Ajaratu Keshinro, Chad Vanderbilt, Mithat Gonen, Jinru Shia, Zsofia Stadler, Martin R. Weiser
Provision of study materials or patients: Canan Firat, Karuna Ganesh
Collection and assembly of data: Ajaratu Keshinro, Chad Vanderbilt, Canan Firat, Chin-Tung Chen, Rona Yaeger, Karuna Ganesh, Jinru Shia, Zsofia Stadler, Martin R. Weiser
Data analysis and interpretation: Ajaratu Keshinro, Chad Vanderbilt, Jin K. Kim, Rona Yaeger, Karuna Ganesh, Neil H. Segal, Mithat Gonen, Jinru Shia, Zsofia Stadler, Martin R. Weiser
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by the authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO’s conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Chad Vanderbilt
Stock and Other Ownership Interests: Paige.AI
Consulting or Advisory Role: DocDoc, Paige.AI
Other Relationship: Paige.AI
Rona Yaeger
Consulting or Advisory Role: Array BioPharma, Natera
Research Funding: Array BioPharma, Boehringer Ingelheim, Pfizer
Neil H. Segal
Consulting or Advisory Role: Bristol Myers Squibb, AstraZeneca/MedImmune, Roche/Genentech, Pieris Pharmaceuticals, Synlogic, Aduro Biotech, Kyn Therapeutics, PureTech, Gritstone Oncology, TRM Oncology, PsiOxus Therapeutics, CStone Pharmaceuticals, Immunocore, Amgen, GlaxoSmithKline, ABL Bio, Boehringer Ingelheim
Research Funding: Bristol Myers Squibb, Pfizer, Roche/Genentech, Merck, Incyte, Immunocore, AstraZeneca
Zsofia Stadler
Consulting or Advisory Role: Allergan, Genentech/Roche, Regeneron, Optos, Adverum, Novartis, Regenxbio, Gyroscope, Neurogene
Martin R. Weiser
Consulting or Advisory Role: Precisca
Research Funding: Clinical Genomics
No other potential conflicts of interest were reported.
REFERENCES
- 1.Battaglia S: Neoantigen prediction from genomic and transcriptomic data. Methods Enzymol 635:267-281, 2020 [DOI] [PubMed] [Google Scholar]
- 2.Pagès F, Mlecnik B, Marliot F, et al. : International validation of the consensus immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 391:2128-2139, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Kim H, Jen J, Vogelstein B, et al. : Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol 145:148-156, 1994 [PMC free article] [PubMed] [Google Scholar]
- 4.Rüschoff J, Dietmaier W, Lüttges J, et al. : Poorly differentiated colonic adenocarcinoma, medullary type: Clinical, phenotypic, and molecular characteristics. Am J Pathol 150:1815-1825, 1997 [PMC free article] [PubMed] [Google Scholar]
- 5.Giannakis M, Mu XJ, Shukla SA, et al. : Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep 15:857-865, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lichtenstern CR, Ngu RK, Shalapour S, et al. : Immunotherapy, inflammation and colorectal cancer. Cells 9:618, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tougeron D, Fauquembergue E, Rouquette A, et al. : Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Mod Pathol 22:1186-1195, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Budczies J, Seidel A, Christopoulos P, et al. : Integrated analysis of the immunological and genetic status in and across cancer types: Impact of mutational signatures beyond tumor mutational burden. Oncoimmunology 7:e1526613, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Innocenti F, Ou FS, Qu X, et al. : Mutational analysis of patients with colorectal cancer in CALGB/SWOG 80405 identifies new roles of microsatellite instability and tumor mutational burden for patient outcome. J Clin Oncol 37:1217-1227, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jimenez-Rodriguez RM, Patil S, Keshinro A, et al. : Quantitative assessment of tumor-infiltrating lymphocytes in mismatch repair proficient colon cancer. Oncoimmunology 9:1841948, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ko YS, Pyo JS: Clinicopathological significance and prognostic role of tumor-infiltrating lymphocytes in colorectal cancer. Int J Biol Markers 34:132-138, 2019 [DOI] [PubMed] [Google Scholar]
- 12.Roelands J, Kuppen PJK, Vermeulen L, et al. : Immunogenomic classification of colorectal cancer and therapeutic implications. Int J Mol Sci 18:2229, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sahin IH, Akce M, Alese O, et al. : Immune checkpoint inhibitors for the treatment of MSI-H/MMR-D colorectal cancer and a perspective on resistance mechanisms. Br J Cancer 121:809-818, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Washington MK, Berlin J, Branton P, et al. : Protocol for the examination of specimens from patients with primary carcinoma of the colon and rectum. Arch Pathol Lab Med 133:1539-1551, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shia J, Black D, Hummer AJ, et al. : Routinely assessed morphological features correlate with microsatellite instability status in endometrial cancer. Hum Pathol 39:116-125, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Shia J, Ellis NA, Paty PB, et al. : Value of histopathology in predicting microsatellite instability in hereditary nonpolyposis colorectal cancer and sporadic colorectal cancer. Am J Surg Pathol 27:1407-1417, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251-264, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Middha S, Zhang L, Nafa K, et al. : Reliable pan-cancer microsatellite instability assessment by using targeted next-generation sequencing data. JCO Precis Oncol 1, 2017. doi:10.1200/PO.17.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cerami E, Gao J, Dogrusoz U, et al. : The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2:401-404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao J, Aksoy BA, Dogrusoz U, et al. : Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6:pl1, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanchez-Vega F, Mina M, Armenia J, et al. : Oncogenic signaling pathways in The Cancer Genome Atlas. Cell 173:321-337.e10, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chakravarty D, Gao J, Phillips SM, et al. : OncoKB: A precision oncology knowledge base. JCO Precis Oncol 1, 2017. doi:10.1200/PO.17.00011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jansen AM, van Wezel T, van den Akker BE, et al. : Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet 24:1089-1092, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wasserman I, Lee LH, Ogino S, et al. : SMAD4 loss in colorectal cancer patients correlates with recurrence, loss of immune infiltrate, and chemoresistance. Clin Cancer Res 25:1948-1956, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bai X, Yi M, Jiao Y, et al. : Blocking TGF-β signaling to enhance the efficacy of immune checkpoint inhibitor. Onco Targets Ther 12:9527-9538, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunderson AJ, Yamazaki T, McCarty K, et al. : TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat Commun 11:1749, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tauriello DVF, Palomo-Ponce S, Stork D, et al. : TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554:538-543, 2018 [DOI] [PubMed] [Google Scholar]
- 28.Batlle E, Massagué J: Transforming growth factor-β signaling in immunity and cancer. Immunity 50:924-940, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koveitypour Z, Panahi F, Vakilian M, et al. : Signaling pathways involved in colorectal cancer progression. Cell Biosci 9:97, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mariathasan S, Turley SJ, Nickles D, et al. : TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554:544-548, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li L, Li M, Wang X: Cancer type-dependent correlations between TP53 mutations and antitumor immunity. DNA Repair (Amst) 88:102785, 2020 [DOI] [PubMed] [Google Scholar]
- 32.Zenonos K, Kyprianou K: RAS signaling pathways, mutations and their role in colorectal cancer. World J Gastrointest Oncol 5:97-101, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lal N, Beggs AD, Willcox BE, et al. : An immunogenomic stratification of colorectal cancer: Implications for development of targeted immunotherapy. Oncoimmunology 4:e976052, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C, Liu S, Qu R, et al. : Recurrent neoantigens in colorectal cancer as potential immunotherapy targets. Biomed Res Int 2020:2861240, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shono Y, Tanimura H, Iwahashi M, et al. : Specific T-cell immunity against Ki-ras peptides in patients with pancreatic and colorectal cancers. Br J Cancer 88:530-536, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abrams SI, Khleif SN, Bergmann-Leitner ES, et al. : Generation of stable CD4+ and CD8+ T cell lines from patients immunized with ras oncogene-derived peptides reflecting codon 12 mutations. Cell Immunol 182:137-151, 1997 [DOI] [PubMed] [Google Scholar]
- 37.Khleif SN, Abrams SI, Hamilton JM, et al. : A phase I vaccine trial with peptides reflecting ras oncogene mutations of solid tumors. J Immunother 22:155-165, 1999 [DOI] [PubMed] [Google Scholar]
- 38.Tran E, Robbins PF, Lu YC, et al. : T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med 375:2255-2262, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pan Z, Tian Y, Cao C, et al. : The emerging role of YAP/TAZ in tumor immunity. Mol Cancer Res 17:1777-1786, 2019 [DOI] [PubMed] [Google Scholar]
- 40.Varelas X, Miller BW, Sopko R, et al. : The Hippo pathway regulates Wnt/beta-catenin signaling. Dev Cell 18:579-591, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Wierzbicki PM, Rybarczyk A: The Hippo pathway in colorectal cancer. Folia Histochem Cytobiol 53:105-119, 2015 [DOI] [PubMed] [Google Scholar]
- 42.Grasso CS, Giannakis M, Wells DK, et al. : Genetic mechanisms of immune evasion in colorectal cancer. Cancer Discov 8:730-749, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fabrizio DA, George TJ Jr, Dunne RF, et al. : Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol 9:610-617, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mandal R, Samstein RM, Lee KW, et al. : Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364:485-491, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hino H, Shiomi A, Kusuhara M, et al. : Clinicopathological and mutational analyses of colorectal cancer with mutations in the POLE gene. Cancer Med 8:4587-4597, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JE, Chun SM, Hong YS, et al. : Mutation burden and I index for detection of microsatellite instability in colorectal cancer by targeted next-generation sequencing. J Mol Diagn 21:241-250, 2019 [DOI] [PubMed] [Google Scholar]
- 47.Huyghe N, Baldin P, Van den Eynde M: Immunotherapy with immune checkpoint inhibitors in colorectal cancer: What is the future beyond deficient mismatch-repair tumours? Gastroenterol Rep (Oxf) 8:11-24, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smart AC, Margolis CA, Pimentel H, et al. : Intron retention is a source of neoepitopes in cancer. Nat Biotechnol 36:1056-1058, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang F, Zhao Q, Wang YN, et al. : Evaluation of POLE and POLD1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol 5:1504-1506, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Efremova M, Finotello F, Rieder D, et al. : Neoantigens generated by individual mutations and their role in cancer immunity and immunotherapy. Front Immunol 8:1679, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picard E, Verschoor CP, Ma GW, et al. : Relationships between immune landscapes, genetic subtypes and responses to immunotherapy in colorectal cancer. Front Immunol 11:369, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Middha S, Yaeger R, Shia J, et al. : Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis Oncol 3:PO.18.00321, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chalabi M, Fanchi LF, Dijkstra KK, et al. : Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 26:566-576, 2020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
A data sharing statement provided by the authors is available with this article at DOI https://doi.org/10.1200/po.20.00456.