

Abstract
Viral infection causes a wide variety of human diseases including cancer and COVID-19. Viruses invade host cells and associate with host molecules, potentially disrupting the normal function of hosts that leads to fatal diseases. Novel viral genome prediction is crucial for understanding the complex viral diseases like AIDS and Ebola. While most existing computational techniques classify viral genomes, the efficiency of the classification depends solely on the structural features extracted. The state-of-the-art DNN models achieved excellent performance by automatic extraction of classification features, but the degree of model explainability is relatively poor. During model training for viral prediction, proposed CNN, CNN-LSTM based methods (EdeepVPP, EdeepVPP-hybrid) automatically extracts features. EdeepVPP also performs model interpretability in order to extract the most important patterns that cause viral genomes through learned filters. It is an interpretable CNN model that extracts vital biologically relevant patterns (features) from feature maps of viral sequences. The EdeepVPP-hybrid predictor outperforms all the existing methods by achieving 0.992 mean AUC-ROC and 0.990 AUC-PR on 19 human metagenomic contig experiment datasets using 10-fold cross-validation. We evaluate the ability of CNN filters to detect patterns across high average activation values. To further asses the robustness of EdeepVPP model, we perform leave-one-experiment-out cross-validation. It can work as a recommendation system to further analyze the raw sequences labeled as ‘unknown’ by alignment-based methods. We show that our interpretable model can extract patterns that are considered to be the most important features for predicting virus sequences through learned filters.
Keywords: Splice sites, Interpretable, Convolution neural network, Motif, Splicing, Learned filters
Introduction
Taxonomic classification is a vital step in metagenomic applications such as microbiome analysis, disease diagnosis, and outbreak tracking. Virology is a subfield of medicine or microbiology focuses on the characteristics of viruses like classification, structure, and evolution. Viruses are the most profuse biological entities, which infect prokaryotes (archaea and bacteria). Viruses impact on the microbial community, such as soil, ocean microbiomes, and the human gut [30]. The human gut DNA viruses extremely influence acute malnutrition and inflammatory bowel disease [46, 55]. In aquatic animals and soil habitats, viruses affect the biogeochemical functioning of their hosts [34]. Viruses replicate and infect within the cell of the human body and also alter the metabolism. A virus causes familiar infections such as cold, warts and flu and also severe diseases like COVID-19, HIV/AIDS, Ebola, and Smallpox. The human virome is the group of viruses present in or on the human body. Even though, many viruses are continuously discovered [9–12, 21, 24], still there may remain unknown viruses need to be discovered. Pathogens (viruses, bacteria) can spread very easily and quickly than before, so the risks caused by these agents are unpredictable and hard to control their expansion. As the recent outburst of Corona, Ebola, Zika, and H1N1/09 influenza A viruses caused epidemics and pandemics [7]. Viruses evolve very fast, so reliable methods for accurate virus detection are required to protect biosecurity and biosafety. Detection of unknown and divergent viruses from the metagenomics experiment datasets is a vital task in bioinformatics [14]. DNA sequences are extracted from biospecimens using Next Generation Sequencing (NGS) technologies without any prior knowledge [43]. Metagenomics, a molecular tool for sequencing the complete genome from a collection of genes. The genes are collected from a sample of material, such as tissue, blood, urine, cells, RNA, DNA, or protein, from plants, animals, or humans. A metagenomic analysis is useful in many fields like ecology, bioremediation, and biotechnology. Viral metagenomics only deals with the discovery and detection of novel viruses [9, 21, 24, 25, 31, 38, 66, 67, 69].
Literature review
Various current methods for the identification of viral genomes can be broadly classified into approaches based on alignment and machine learning. In the traditional alignment-based approach, viral sequence detection in human biospecimens is normally performed using BLAST [2]. In the first approach, the sequences are compared to known publicly available databases and classify the sequences based on the similarity index. Metagenomic datasets contain divergent virus sequences so there is no similarity at all among known database sequences. As a result, many of the sequences of viruses generated from sequencing technologies are classified as “unknown” by the NCBI BLAST [9, 38]. The most popular alignment-based techniques for viral genome classification are REGA [1, 48], USEARCH [19], and SCUEAL [49]. All these methods purely depend on the alignment score between the viral sequence being classified and the reference dataset. The major drawbacks of alignment-based approaches include, the classification performance purely depends on the selection of one of the several initial alignments and hyperparameters. These methods are expensive and their performance is unstable for divergent regions of the genome. Another tool for virus sequence detection within metagenomic sequence datasets is HMMER3 [44], which uses profile Hidden Markov Models by comparing with vFams [61] database. vFams, a database with viral family proteins was designed by Multiple Sequence Alignments (MSA) from all RefSeq viral proteins. HMMER3 detects homological viral sequences more effectively but not highly divergent ones [13] because it depends on the reference database VFams.
In the second type of approach, several methods are proposed to classify viral metagenomic sequences [3, 54]. VirSorter [57], a probabilistic tool to predict novel viruses in microbial genome data with and without reference. The model is evaluated on 3kb to 10kb sequence length metagenomic contig datasets and its performance increases with the sequence length. VirFinder [52], machine learning model to identify viral contigs based on k-mer frequency. In this model the sequence length is in the range 1kb to 5kb, which shows better results than VirSorter on small length sequences. The existing recommendation like system ViraPipe [14] used an artificial neural network and random forest by using relative synonymous codon usage frequency to improve the classification of metagenomic data into a virus and non-virus sequences. This model identified two codons (CGC and TCG) which are shown to have strong discriminative ability. These methods still confront many problems, such as their inability to extract useful hidden information from basic DNA data. Machine learning algorithms efficiency depends solely on the features that have been extracted. A machine learning model is interpretable if humans can comprehend it by observing model parameters and how it makes decisions on their own. On other hand, explainable models are too complex to understand and require additional techniques in order to follow how it works. The decision tree is a interpretable machine learning model where as the random forest is explainable [22].
Deep learning is applied for automated extraction of features due to technical advances. It is a well-known technique that has produced excellent results in the field of Natural Language Processing (NLP) [18, 64], image and video processing [36]. Deep learning applications in the bioinformatics, genomics and computational biology mostly concentrate in (i) genome sequencing and analysis [15, 32, 51, 72] (ii) classification of DNA [33, 56], chromatin [70], polyadenylation [26], and (iii) protein structure prediction [20, 62, 68, 72]. Viral genome deep classifier [23], a CNN model have been proposed for classifying viral sequences into subtypes. Long Short-Term Memory (LSTM) [58] networks have excelled in the field of NLP in recent years, especially when modelling short sentences with hundreds of words [41]. ViraMiner [65] is a model that uses Convolutional Neural Networks (CNN) to detect the viral genome sequences from human metagenomic samples. It contains pattern and frequency branches, each one is trained separately. The pattern and frequency branch achieved 0.905 and 0.917 AUC-ROC values respectively. The combination of both pattern and frequency achieved AUC-ROC of 0.923. DeepVirFinder [53] is a CNN based model used for identifying viral sequences in metagenomics. It achieved 0.93, 0.95, 0.97, and 0.98 AUC-ROC for the viral sequences of length 300, 500, 1000, and 3000 bp respectively, which infers that performance improves along with sequence length. RNN-VirSeeker [42], an LSTM based method to identify short viral sequences from CAMI and human gut metagenome datasets, with sequence length 500 bp that exhibited mean AUC-ROC of 0.9175. AUC-ROC is best metric for evaluating model performance on balanced datasets. The human metagenomic datasets are highly imbalanced so, AUC-PR is considered as the best metric. As CNN is widely criticized for their black-box design, the rational between input and output can’t be properly observable. While these existing CNN methods are used to detect viral genomes, none of the above mentioned methods performs model interpretability.
The deep learning models are non-transparent as we can’t decipher any knowledge by only peaking at neuron weights directly so, there is a demand for model expainability. In the context of deep learning the terms interpretability and explainability are often used interchangeably [60]. Recently, some explainable models are used for image analysis [60], viral genome prediction [8], and other various tasks [5, 17, 40, 50, 71]. This review [60] presents a study of the latest implementations of explainable deep learning for various medical imaging tasks along with developing a framework for clinical end-users. Two-stage CNN architecture was developed in work [5], which employs gradient based techniques to generate saliency maps for both the time dimension and the features. The human virus detection method [8] used DeepLIFT [59] to extract sub-sequences with highest contribution score and also visualized sequence logos based on mean partial Shapley values for each base at each position. The major drawback of this method is the extracted subsequences are not validated.
Contributions
In this paper, we first improve the predictive performance of viral genome sequences based on human metagenomic datasets. We demonstrate that the proposed explainable CNN model, EdeepVPP, and CNN-LSTM based model EdeepVPP-hybrid outperform both previous state-of-the-art deep learning models [42, 53, 65] and traditional machine learning models [14, 52, 57]. The EdeepVPP predicts viral sequences from novel samples with high accuracy, implying that the model can accurately predict unknown viral sequences, so it works like a recommendation system. Next, we propose a novel approach to make our models more transparent by extracting the underlying patterns that works like significant features for better classification. As a proof of concept, we validated the extracted patterns of EdeepVPP on human metagenomic datasets to the known patterns of HOCOMOCO [37] database. Unlike, most of the existing models, the proposed models are generalized, not limited to a particular family of viruses.
Proposed approach
In this section, we presented the classical introduction of CNN and LSTM, the architectures of proposed models, and metrics for evaluation.
Convolutional neural networks
CNN is one of the architectures of the neural network, used to assess visual patterns with heterogeneity from the data. CNN, a special type of multilayer neural network, which maps a fixed length input to a fixed-size output and trains with a back-propagation algorithm [39]. It contains several layers, normally one input, several hidden, one output layer, and each layer contains several neurons, and each neuron has different parameters [73]. To switch from one layer to the next, CNN stores and updates information in its filter weights after learning the relationship between input and output. Filter weights are first initialized with random uniform and then updated by back-propagation to minimize a loss (or) cost function. We used a categorical cross-entropy as the loss function is shown as follows:
| 1 |
In order to understand CNN, the non-linear activation function plays a crucial role after convolution. Sigmoid, Tanh, and ReLU (Rectified Linear Unit) are three commonly used non-linear activation functions. All these non-linear activation functions play a squashing operation. The sigmoid normalize the values between 0 and 1. The Tanh function normalizes the input values from -1 to 1. The ReLU changes negative values to zero and positive values keep the same. ReLU is simply a half-wave rectifier, the most prominent non-linear function.
| 2 |
In contrast with sigmoid and tanh(z) functions, ReLU learns much faster. The output dense layer leverages the softmax activation to measure the likelihood for each class in prediction problems. To detect patterns as features, CNNs include multiple number of filters that slide over a one-hot encoded binary vector for a sequence.
Long short-term memory
LSTMs are Recurrent Neural Network (RNN) architectures, useful to capture long term dependencies as they contain memory units which helps to remember (forget) the important (unimportant) features. It consists of a cell, an input, an output, and forget gates. The three gates monitor the flow of information into and out of the cell, and the cell remembers values over arbitrary time periods. The aim of the LSTM first layer (forget layer) is to determine which information from the cell state will be discarded.
| 3 |
The next step consists of two parts, the sigmoid layer (input gate layer) decides the values to be updated. Whereas, the tanh layer generates candidate values in the form of a vector which is added to the state.
| 4 |
| 5 |
In the update state, the new cell state is updated with forget and input gate vectors.
| 6 |
In the output gate layer, decides which part of the cell state to be contained.
| 7 |
Finally, the hidden state is calculate using output gate and current cell state.
| 8 |
Majorly the LSTM is utilized to overcome long term dependency problem.
EdeepVPP model architecture
The EdeepVPP architecture comprises one input, three one dimensional convolutions, one flattened, two dense, and one output layers that is shown in Fig. 1.A. We use conv1D that is powerful in fixed-length sequences and also in time series data for deriving visual patterns. Conv1D performs simple array operations, so the computational complexity is significantly lesser than conv2D. Due to the low computational complexities, conv1Ds are well-suited for real-time applications especially on hand-held devices like mobiles and notebooks [35]. It has been observed that gradients of the cost functions approach zero as the depth of the neural networks increases. It is difficult to train a model if the weights do not change significantly, since the weights never converge. This type of problem is called a vanishing gradient [45] that is resolved by the non-linear activation function of the ReLU [28]. The drop-out and max-pooling layers are followed by convolutional layers. Flatten converts the output of the third convolution layer to 1D array which serves as an input to the next fully connected layer.
Fig. 1.

The architecture of proposed model that is used for (A) Viral genome classification (B) To extract patterns which works like features to predict viral sequences
The convolution operation is a vital step in CNN. The first convolutional layer convolves the one-hot encoded input with 32 filters which are slide across the input genome. The filter of size 7 is stride one position at a time and the padding is set to be as ’same’ to preserve the actual size (300) of the input. These learned filters used to identify the particular patterns as features in the DNA sequence. In each convolution operation, the encoded input genome convolves with a number of k filters F={f1,f2, ..... fK}, and biases B = {b1,b2, ..... bK} are added, and each filter generates separate feature map [35].
| 9 |
where is learned filter weights at previous layer l-1, is the value after convolution operation. The non-linear activation transformation σ(.) is applied to feature maps and the same process repeated to all convolution layers.
| 10 |
ReLU activation given in equation(2), follows convolution layer to apply max(0,z) operation element-wise to generate feature maps.A dropout layer with a dropout rate of 0.2 precedes the activation layers of the ReLU, which randomly drops 20 percent of the connections in each iteration to provide regularization and to minimize over-fitting [63]. Dropout layers are accompanied by max-pooling layers of pool size 2 and slide is 2. Max-pooling calculate the maximum value for each adjacent feature map values, which is used for smoother feature activations [4]. By down-sampling process, the max-pooling decreases spatial dimensions and cost of computation and also extracts genome characteristic features and passes them on to the dense layers.
| 11 |
The first convolutional layer extracts the global characteristics along with dropout and max-pooling layers. The second and third layers are also accompanied by layers of dropout and max-pooling which extract local characteristics in the same order as the first convolution followed. The Table 1 shows the structure of the proposed model in detailed manner.
Table 1.
The detailed model structure of the proposed approach
| Step | Operation | Output dimension |
|---|---|---|
| Input Layer | One-hot encoding | 300x5 |
| Convolutional Layer 1 | Conv1D(32,7) | 300 x 32 |
| Activation(ReLU) | 300x32 | |
| Dropout(0.2) | 300x32 | |
| Max-pooling1 | 150 x 32 | |
| Convolutional Layer 2 | Conv1D(8,4) | 147 x 8 |
| Activation(ReLU) | 147 x 8 | |
| Dropout(0.2) | 147 x 8 | |
| Max-pooling1 | 73 x 8 | |
| Convolutional Layer 3 | Conv1D(8,3) | 71 x 8 |
| Activation(ReLU) | 71 x 8 | |
| Dropout(0.2) | 71 x 8 | |
| Max-pooling1 | 35 x 8 | |
| Flatten step | Flatten | 280 x 1 |
| Dense Layer1 | Dense(32) | 32 x 1 |
| Activation(ReLU) | 32 x 1 | |
| Dropout(0.2) | 32 x 1 | |
| Dense Layer2 | Dense(2) | 32 x1 |
| Activation(Softmax) | 2x1 | |
| Output Layer | Classification | Probabilities |
After completion of all stack of layers, the output of the last pooling layer transformed to ne dimensional vector and pass on to the classifier part of the model. In this part, there are two dense layers with 32 neurons in the first layer and 2 neurons in the next layer. A dropout layer is used between two dense layers. The last dense layer has a softmax activation function, which produces two probabilities belongs to either positive (true) or negative (false) target classes. A softmax function mathematically represented as below:
| 12 |
Finally, the genome sequence is classified as viral/non-viral type based on the output probability. The categorical-cross entropy loss function is given in the equation(1). After every epoch the filter weights are updated to minimize the loss function.We used Keras [27], a minimalistic, highly modular neural network library, written in Python, in our implementation of the network.
EdeepVPP-hybrid model architecture
The EdeepVPP-hybrid model consists one embedding, four one dimensional (1D) convolutions, three LSTM, two dense, and one output layers that is shown in Fig. 2. The DNA sequences consist of 5 nucleotides (A, T, C, G, N) and each nucleotide is represented as an integer. We then pass this sequence of integers which is an encoded representation of the input sequence to an embedding layer. The embedding layer transforms each input sequence into a vector representation. We set the size of vector to be 5 one for each nucleotide. If the sequence size is ’n’ then the embedded vector size is ’nx5’. EdeepVPP-hybrid is an end-to-end trainable model, that consists of four 1D convolutional layers with kernel sizes of 5, 5, 3, 5 respectively. Convolution modifies the input sequence which depends on the filter, hence the values within the filter are also trainable. The number of activation/feature maps at each convolution layers are 96, 224, 128, 224, respectively. We used 1D Max-pooling with pool size 2, that is used for down sampling the inputs given to it. The outputs of convolution layers are given towards an LSTM layer. We employed three LSTM layers with hidden units 200, 200, 150, respectively. The convolution layers mainly work as feature extractors and LSTM works as sequence predictors. The LSTM layers output is passed to the fully connected (dense) layers. We used two dense layers to increase the depth of the network which contain 896 and 448 units. The outputs of these dense layers are given to the final softmax layer to obtain the classification results. The complete network is end-to-end trainable.
Fig. 2.
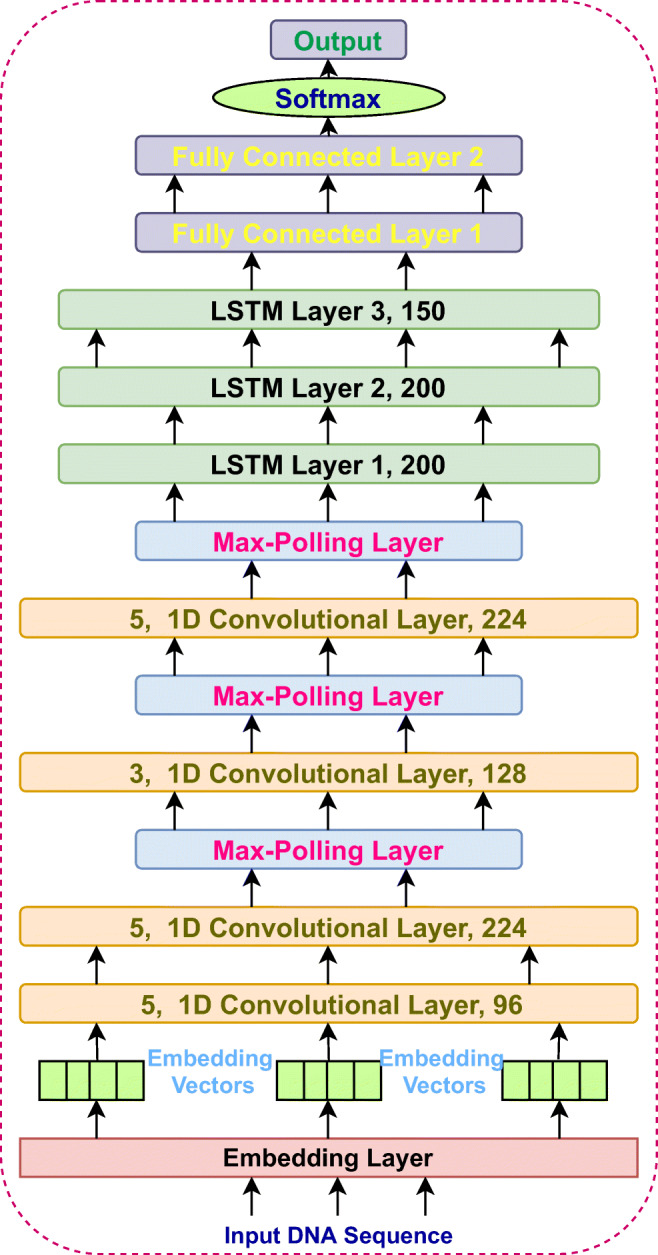
The architecture of PBVPP-hybrid model that is used for Viral genome prediction
Evaluation metrics
The proposed CNN model was assessed by using two popular classification performance metrics i.e., AUC-ROC and AUC-PR. To calculate these metrics precision (Prec), Specificity (Sp), Recall or Sensitivity (Sn),True Positive Rate (TPR), False Positive Rate (FPR), and Accuracy (Acc) are required.
| 13 |
| 14 |
| 15 |
| 16 |
| 17 |
The TP, TN, FP, FN are the number of true positive, true negative, false positive, and false negative values respectively.
Accuracy, sensitivity and specificity are sensitive to the dataset class distribution, because there are very less viral sequences than non-viral. Since the datasets used in the proposed work are extremely imbalanced, consisting of a variable number of sequences that are viral and non-viral. The AUC-ROC is better suited when both viral and non-viral samples are in similar proportions. The majority of samples would have a greater impact on the curve than the minority, which could contribute to bias. On the other hand, a precision-recall curve is largely used for the class of imbalanced problems since it does not recognize false positives and false negatives, so there is no risk of impact of majority samples, thereby providing adequate assessment.
Methods
In this section, at first we have given an overview of metagenomic datasets used in the experiments, and pre-processing of the data. Next, an overview of cross-validation and the settings used for training in the EdeepVPP model are discussed.
Datasets collection
We have collected 19 different metagenomic contig experiments, called human metagenomic datasets derived from human samples, to bespeak the EdeepVPP prediction model and to validate the model output. These datasets belongs to various patient groups, generated from next-generation sequencing, which are analyzed and labeled by PCJ-BLAST [47]. We have collected 300bp contigs for our experiments that have been described in detail in [65]. The details of human metagenomic datasets are given in the Table 2.
Table 2.
The number of viral and non viral samples in human metagenomic datasets
| Dataset | NVS∗ | NNVS+ | Dataset | NVS∗ | NNVS+ |
|---|---|---|---|---|---|
| 2011_G5 | 732 | 17713 | 2011_E2_SCC | 312 | 11368 |
| 2011_N19 | 121 | 7332 | 2013_H4 | 50 | 2227 |
| 2014_B | 111 | 3167 | 2014_E1 | 321 | 19559 |
| 2014_D3 | 0 | 422 | 2014_F1 | 896 | 13896 |
| 2014_G1 | 129 | 21822 | 2014_G6 | 62 | 4676 |
| 2014_G5 | 348 | 33230 | 2014_G7 | 22 | 773 |
| 2014_J1 | 1534 | 16644 | 2014_O | 0 | 12930 |
| 2014_N1 | 1534 | 16644 | 2014_P | 150 | 13678 |
| 2015_R1 | 1 | 14928 | 2015_F2 | 11 | 2759 |
| 2015_F | 11 | 2759 | Total | 7879 | 216727 |
∗ NVS/+NNVS-Number of Viral/Non-viral Sequences
One-hot encoding
One-hot encoding technique represents categorical data as binary vectors. DNA sequences have five bases (categories) A, C, G, T, and N. Neural networks can handle numerical data only, so the technique of one-hot encoding is used to transform DNA to binary vectors. It coverts L length DNA sequence into Lx5 matrix, where A=[1, 0, 0, 0, 0], C=[0, 1, 0, 0, 0], G=[0, 0, 1, 0, 0], T=[0, 0, 0, 1, 0], and N=[0, 0, 0, 0, 1].
Cross-validation
Cross-validation is a model quality evaluation method, which is better than the residual evaluation approach, useful to avoid overfitting and underfitting. K-fold cross-validation randomly breaks the samples of the dataset into k folds or groups of approximately equal size. Iteratively, k-1 folds at a time used as a test set and the model is tested on remaining one fold. We adopt a standard strategy to select k values like 10 to test the EdeepVPP model for different human metagenomics experiments. We also evaluated the proposed model with Leave-One-Experiment-Out (LOEO) cross-validation. We repeat the LOEO approach, by using one serum metagenomic experiment as a test set and the remaining four experiments are used to train the model. We also evaluated the model by considering 5 serum experiments as test set and remaining 14 metagenomic experiments are used to train the model.
Hyperparameter tuning
Different hyper-parameters are tuned by learning the model and the best values for parameters are selected on the basis of less validity loss. We performed random search to select optimal values for hyper-parameters. The tuned hyper-parameters are CNN filter sizes, Learning rate, and the dropout ratio, activation function and so on. The search space and the selected values for these hyper parameters are shown in the Table 3. In each fold, the neural networks was trained for only 6 epochs.
Table 3.
Hyper parameters search space and optimized values
| Hyper Parameter | Search Space | Optimal Value |
|---|---|---|
| Activation Function | ReLU,Tanh,Sigmoid | ReLU |
| Batch Size | 100,500,1000,1500 | 1000 |
| Dropout Ratio | 0.1,0.2,0.3,0.4,0.5 | 0.2 |
| # Filters | 4,8,16,32,64,128 | 32, 8, 8 |
| # Convolution Layers | 1,2,3,4,5 | 3 |
| Optimizer | ADAM, SGD | ADAM |
| Size of Filters | 32,16,8,7,6,5,4,3 | 7, 4, 3 |
| Strides | 1,2,3 | 1 |
| Type of Pooling | Average , Max | Max |
Results and discussion
In this section a systematic description of proposed models’ capability is compared with the state-of-the-art methods. We have tested our system with human metagenomic datasets in order to present the ability.
Discriminative power of the proposed models
We have observed that the human metagenomic dataset contains viral and non-viral samples that are greatly imbalanced. We shuffled these dataset sequences and created the balanced and imbalanced sets. The viral and non-viral sequences are kept in a 1:1 ratio in balanced datasets because the non-viral sequences are large in number and are selected randomly from the available sequences. Firstly, we trained the model for each human metagenomic experiment individually by using 10-fold cross-validation with 6 epochs only. Second, a 10-fold cross-validation is used on a human metagenomical dataset to verify the classifier’s impact on balanced and unbalanced datasets. The EdeepVPP model achieved 0.924 area under the ROC curve for the test set of balanced case. The proposed model achieved 0.987 AUC-PR and 0.988 AUC-ROC for an imbalanced dataset, which is a significant improvement compared to the previous models RNN-VirSeeker [42] with 0.918, DeepVirFinder [53] with 0.93, ViraMiner [65] with 0.923, VirFinder [52] with 0.893, VirSorter [57] with 0.742, and ViraPipe [14], reaching 0.79 AUC-ROC values as shown in Fig. 3. Finally, we merged all five serum datasets called human serum dataset and performed 5-fold cross validation. The proposed model achieved 0.991 AUC-ROC on human serum viral dataset.
Fig. 3.
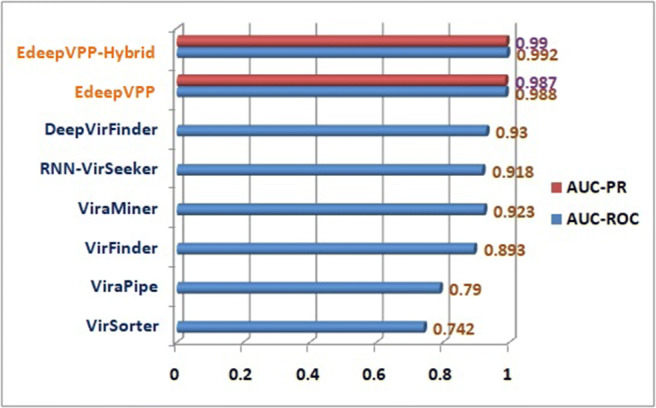
The performance comparison between VirSorter, VirFinder, ViraPipe, ViraMiner, DeepVirFinder, RNN-VirSeeker, EdeepVPP, and EdeepVPP-Hybrid with respect to AUC-ROC
The EdeepVPP model achieves AUC-PR values of 0.9881 and 0.9872 on human metagenomic and human serum datasets, respectively shown in Fig. 4. For human viral metagenomic datasets, the mean performance is stated in terms of AUC-ROC, AUC-PR, and the results are shown in Table 4.
Fig. 4.

The ROC and Precision-Recall curves for (a) human metagenomic dataset (b) human serum dataset when 10-fold and 5-fold cross-validation is used.
Table 4.
Average AUC-ROC and AUC-PR values for imbalanced human viral metagenomic datasets
| Dataset | EdeepVPP | EdeepVPP-hybrid | ||
|---|---|---|---|---|
| AUC-ROC | AUC-PR | AUC-ROC | AUC-PR | |
| 2011_G5 | 0.9792 | 0.9763 | 0.9912 | 0.9903 |
| 2011_N19 | 0.9894 | 0.9883 | 0.9940 | 0.9938 |
| 2011_E2_SCC | 0.9775 | 0.9716 | 0.9828 | 0.9798 |
| 2013_H4 | 0.9792 | 0.9712 | 0.9822 | 0.9792 |
| 2014_B | 0.9721 | 0.9685 | 0.9723 | 0.9651 |
| 2014_D3 | 0.9949 | 0.9949 | 0.9937 | 0.9937 |
| 2014_E1 | 0.9858 | 0.9803 | 0.9897 | 0.9871 |
| 2014_F1 | 0.9743 | 0.9693 | 0.9903 | 0.9890 |
| 2014_G1 | 0.9933 | 0.9923 | 0.9976 | 0.9971 |
| 2014_G5 | 0.9928 | 0.9916 | 0.9917 | 0.9897 |
| 2014_G6 | 0.9889 | 0.9851 | 0.9916 | 0.9900 |
| 2014_G7 | 0.9759 | 0.9729 | 0.9770 | 0.9708 |
| 2014_J1 | 0.9563 | 0.9512 | 0.9656 | 0.9614 |
| 2014_N1 | 0.9572 | 0.9529 | 0.9774 | 0.9750 |
| 2014_O | 0.9949 | 0.9949 | 0.9949 | 0.9949 |
| 2014_P | 0.9900 | 0.9869 | 0.9893 | 0.9848 |
| 2014_R1 | 0.9893 | 0.9737 | 0.9949 | 0.9949 |
| 2015_F | 0.9928 | 0.9916 | 0.9937 | 0.9934 |
| 2015_F2 | 0.9928 | 0.9908 | 0.9937 | 0.9931 |
| Full_DataSet | 0.9881 | 0.9872 | 0.9926 | 0.9902 |
| Serum_Dataset | 0.9912 | 0.9899 | 0.9934 | 0.9927 |
From the prevailed results, it has been stated that the values of AUC-ROC for balanced and imbalanced datasets are 92.41% and 98.81% respectively and for AUC-PR these values are 92.13% to 98.72% respectively. In particular, the imbalanced datasets have achieved high accuracy, which improves the discriminatory ability of the EdeepVPP model. For individual datasets the average AUC-ROC ranges between 95.63% and 99.49% and AUC-PR ranges between 95.12% and 99.49%.
Upon analysis of the findings, the following conclusions can be drawn.
In case of individual experiments AUC-ROC values are slightly better than AUC-PR in most of the cases.
In case of complete dataset, in imbalanced the AUC-ROC values are 6.4% and AUC-PR values are 6.6% higher than the balanced dataset, but over all our model achieved better accuracies than all the other existing methods. It shows that our model has a very strong ability to distinguish between viral and non-viral sequences in terms of discrimination.
EdeepVPP as a recommendation system
To further validate the discriminative ability of EdeepVPP, we also construct Leave-One-Experiment-Out cross-validation (LOEOCV) on the human serum dataset. The human serum dataset contains 5 metagenomic experiments derived from serum type. We trained a specific EdeepVPP model based on four metagenomic experiments data and used the remaining one to test the model. If EdeepVPP gets good performance on LOEOCV, then it implies that the EdeepVPP model predicts the novel unknown viral sequences. For each metagenomic experiment, we conduct leave-one-experiment-out evaluation process. We compared the LOEOCV results of EdeepVPP with the results of state-of-the-art existing approach ViraMiner [65] that were shown in Table 5. The results of the leave-one-out evaluation of EdeepVPP were better than the standard results of ViraMiner. These results conclude that the proposed model can better predict the novel sequences that are not involved in the training set.
Table 5.
EdeepVPP leave-one-out evaluation performance on novel human serum metagenomic contig dataset
| Left out sample set | 2014_G7 | 2014_G6 | 2014_G5 | 2014_G1 | 2011_G5 | Average |
|---|---|---|---|---|---|---|
| ViraMiner test AUC-ROC | 0.86 | 0.92 | 0.96 | 0.89 | 0.95 | 0.91 |
| EdeepVPP test AUC-ROC | 0.9771 | 0.9912 | 0.9910 | 0.9932 | 0.9603 | 0.9825 |
| EdeepVPP test AUC-PR | 0.9715 | 0.9892 | 0.9860 | 0.9921 | 0.9515 | 0.9780 |
Our model was train with divergent metagenomic contig sequences that are derived from different sample types such as skin, prostate secretion, serum, and cervix tissues. The trained EdeepVPP predicts viral sequences on the unknown test dataset, which are not included in the training set. The proposed model yields very good results in predicting viral sequences from novel samples. We trained the EdeepVPP model with 14 human metagenomic experiment datasets and tested with five serum experiment datasets. The AUC-ROC and AUC-PR values are 0.9879 and 0.9854 respectively. In Fig. 5, we visualize ROC and precision-recall curves for test set. These results imply that our model can accurately predict the unknown viral sequences.
Fig. 5.

The ROC and Precision-Recall curves when 14 metagenomic experiments as trainset and remaining five serum experiments as testset
Interpretation and visualization
It is possible to describe interpretation as the degree of comprehension of what a model does. In recent years , high precision for biological sequence classification has been provided by CNN models with complex internal implementation. In these models, a lot of effort has been made to improve the efficiency of the prediction. Acceptance of the CNN model depends not just on efficiency, but also on how the user can perceive the underlying mechanism. However, CNN models produce excellent predictive results, but they are regarded as ”black boxes” due to their complex structure. When dealing with problems with the classification of biological sequences, there is a great need for interpretability to ensure the accuracy of decisions taken by CNN models. There is a need to eliminate this ’block box’ nature and provide the transparent models to show the underlying feature extraction process. Discrimination and perception are two major advantages of the proposed model. EdeepVPP, an interpretable CNN system, is capable of extracting features for predicting viral genomes as shown in Fig. 1B.
Interpretable features (patterns) for viral genomes
After completion of training, the filters become learned filters, which means filter updates weights to optimum values. We have developed a computational step-by-step approach to identify the underlying patterns that guide the prediction of viral sequences as shown in Fig. 6.
Fig. 6.

The procedure to extract patterns by using learned filters
For the viral genome prediction, we have extracted the motifs, which are having higher activation values than the threshold (half of the highest activation value) from human metagenomic dataset. The activation value of the pattern depends on the work performed by different filters over the input sequences. Filter jobs can catch highly important structures. We have considered one motif from each filter, which has the highest activation value. The Table 6 gives the patterns one from each filter and corresponding activation values, which are highly influential patterns for detection of viral sequences in the human dataset. The motifs ACGACCG, ACGCAGT are extracted by filters 11 and 16 are biologically relevant motifs for the identification of viral sequences.
Table 6.
Top-1 motifs with the highest activation value from each filter extracted from 19 metagenomic experiments, which act as features to predict viral genomes
| Filter Id | Viral pattern | Mean activation | Activation value | Filter Id | Viral pattern | Mean activation | Activation value |
|---|---|---|---|---|---|---|---|
| 11 | ACGACCG | 0.789984 | 1.5799685 | 9 | GCTGTGA | 0.596094 | 1.1921874 |
| 16 | ACGCAGT | 0.692365 | 1.3847302 | 28 | GATTTGA | 0.587959 | 1.1759177 |
| 24 | GGGATCG | 0.678848 | 1.3576957 | 3 | GCGAGGT | 0.580528 | 1.1610559 |
| 21 | TACGGGT | 0.673739 | 1.3474776 | 7 | TCAGGTC | 0.572008 | 1.1440151 |
| 4 | AGGCGGG | 0.652724 | 1.3054485 | 19 | AAAGTCT | 0.559704 | 1.1194072 |
| 10 | AAAAATT | 0.650489 | 1.3009783 | 23 | GTCCTGA | 0.553407 | 1.1068132 |
| 18 | AGTACGA | 0.649529 | 1.2990574 | 31 | CCGTTAT | 0.549539 | 1.0990782 |
| 1 | CTTTTTT | 0.644374 | 1.2887475 | 14 | ATGGAGT | 0.548696 | 1.0973923 |
| 20 | GTCACTC | 0.634448 | 1.2688965 | 22 | GAGAAAA | 0.543563 | 1.0871266 |
| 2 | ACCTCTG | 0.632819 | 1.2656392 | 8 | ATATGTG | 0.541229 | 1.0824597 |
| 26 | CACAGTG | 0.630168 | 1.2603375 | 17 | TTGAAAT | 0.527961 | 1.0559222 |
| 5 | TGAGCTC | 0.624382 | 1.2487631 | 29 | CGTGCCC | 0.525018 | 1.0500368 |
| 32 | CTAGGCT | 0.608599 | 1.2171972 | 27 | TAACGTC | 0.518185 | 1.0363696 |
| 30 | TGGGCCG | 0.603729 | 1.2074571 | 13 | GATCCTA | 0.485952 | 0.9719047 |
| 6 | TCACATC | 0.603138 | 1.2062765 | 25 | ATGAGAG | 0.465123 | 0.9302594 |
| 12 | TCACAGC | 0.597369 | 1.2062765 |
We performed interpretability on human serum, and human metagenomic datasets. We sorted patterns by frequency and extracted top-five patterns for viral and non-viral patterns in each filter, and also top-50 patterns for viral and non-viral in overall 32 filters from human serum, and human metagenomic datasets. The top-five patterns in each filter match the patterns of the other filters. Table 7 gives all such patterns present in human 19 metagenomic contig datasets extracted with a threshold as the average of activation values. The average activation is calculated by overlapping M-L + 1 subsequences of size Lx5 of a particular sequence (M and L denote the length of the sequence and size of the filter respectively). Consider an example, in which the pattern TAAAAAA has repeated in 28 filters (1-3,7-19,21-32), and the frequency occurrence is 3229. The pattern plays a crucial role to predict the sequence as a viral one. In contrast, the pattern ACACACA is the most repeated and exists in 27 filters (6-32) with frequency 16110, which predicts the sequence as non-viral. On the basis of their repetition in several filters, the patterns are stacked together in decreasing order. Motifs (AAAAAAAAA, AAAGAAA, TTTTTTT) that are present in both viral and non-viral sequences are skipped because the impact of those patterns in viral prediction is neutral. Likewise, the patterns extracted from the human serum dataset are shown in Table 8.
Table 7.
The patterns extracted by most of the filters with a threshold as an average of all activations, which act as features to predict viral and non-viral genomes from human metagenomic datasets
| Viral pattern | Frequency | Repeated in no. of filters | Filter numbers | Non-viral pattern | Frequency | Repeated in no. of filters | Filter numbers |
|---|---|---|---|---|---|---|---|
| TAAAAAA | 3229 | 28 | 1-3,7-19,21-32 | ACACACA | 16110 | 27 | 6-32 |
| AAACAAA | 2783 | 23 | 7-29 | GAAAAAA | 8113 | 26 | 6-31 |
| TTAAAAA | 2036 | 20 | 1-20 | AAGAAAA | 4282 | 11 | 1-5, 7-12 |
| AAAAAAA∗ | 5915 | 32 | 1-32 | CACACAC | 11133 | 5 | 13-16,32 |
| AAAGAAA∗ | 3796 | 32 | 1-32 | TTTTTTT∗ | 13978 | 13 | 20-32 |
∗Neutral patterns, present in viral and non-viral genome sequences
Table 8.
Extracted patterns from 5 serum metagenomic datasets, which act as features to predict viral and non-viral genomes
| Viral pattern | Frequency | Repeated in no. of filters | Filter numbers | Non-viral pattern | Frequency | Repeated in no. of filters | Filter numbers |
|---|---|---|---|---|---|---|---|
| AAGAAAA | 1610 | 27 | 4-9, 12-32 | AAAGAAA | 2753 | 32 | 1-32 |
| TAAAAAA | 1171 | 14 | 1-2,14-22, 30-32 | ACACACA | 3446 | 17 | 16-32 |
| AAAACAA | 791 | 12 | 6-9, 14-21 | AAAAATA | 2379 | 14 | 2-15 |
| CAGAAAA | 867 | 6 | 4-9 | AAAAAAC | 1679 | 5 | 2,7-10 |
| ∗AAAAAAA | 3143 | 32 | 1-32 | ∗AAAAAAA | 18473 | 32 | 1-32 |
| ∗AAAAGAA | 3796 | 32 | 1-32 | ∗AAAAGAA | 2806 | 23 | 1,11-32 |
| ∗TTTTTTT | 1919 | 31 | 2-32 | ∗TTTTTTT | 6023 | 30 | 3-32 |
∗Neutral patterns, present in viral and non-viral genome sequences
Validation of extracted patterns
The top-50 patterns from all filters are extracted from each viral and non-viral sequences of the human metagenomic dataset. We found that some of the patterns are common for both viral and non-viral sequences that are considered as neutral as their role in classifying viral sequences are negligible. The viral patterns (except neutral) grouped and position weight matrix (PWM) is determined by measuring nucleotide frequency. PWMs are used to generate the sequence logos using WebLogo3 [16]. In the same way, the viral patterns are extracted from the human serum dataset. The logos of human metagenomic and human serum datasets are shown in Fig. 7(a) and (b) represents a motif TTTTAAT and TAAATAT respectively.
Fig. 7.

Logos of extracted viral patterns of (a) human metagenomic (b) human serum datasets. Matched motifs with logos of (a.1,a.2) human metagenomic, (b.1,b.2) human serum viral patterns in the Human and Mouse (HOCOMOCO) database
In Fig. 7(a) and (b) the size of a base in the pattern indicates the frequency probability of the corresponding nucleotide at a particular position. The MEME-Suite [6] motif comparison tool TOMTOM [29] compares one or more motifs against annotated motifs from existing databases (e.g. the database JASPAR, Human and Mouse (HOCOMOCO)). To evaluate EdeepVPP’s capability, We compared the extracted patterns of our model on human metagenomic and human serum dataset to the known patterns of HOCOMOCO [37]. We note that the learned patterns of EdeepVPP matched a large number of significant known patterns. Both the human metagenomic and human serum datasets learned motifs that were matched to 44 and 61 existing motifs, indicating that the interpretable strategy suggested in this paper establishes transparency to divulge hidden features for better classification. Figure 7(a.1, a.2) shows human metagenomic matched motifs and Fig. 7(b.1, b.2) shows human serum matched motifs in the HOCOMOCO database.
Filter visualization and ability
Filters are qualified weight vectors that play a crucial role in the identification of patterns (motifs) for classification problems. In addition to the pattern extraction, we also evaluate the robustness of EdeepVPP convolved first layer filters. Visualization of human metagenomic and human serum datasets of the first filters shown in Fig. 8, by using displayr. The X-axis of the heatmaps indicates the location of each nucleotide in the learned filter weight matrix, and the Y-axis illustrates the nucleotides (A, C, G, T, and N).
Fig. 8.

Graphical representation of heatmap of (a) human metagenomic (b) Human serum dataset learned filters
To visualize a filter of size Lx5 (L is the filter length, i.e. 7 in the first convolution layer) that contains learned weights, a heatmap is used to demonstrate the significance of bases at each place. If the extracted patterns have higher mean activation values for the filters, then the patterns have a greater impact on determining the viral sequence as true. The darker colours reflect a greater contribution of the nucleotide to that specific role.
Conclusion
The prediction of the viral genome plays a vital role in the study of complex diseases. We introduced two deep learning models, the first one is EdeepVPP, an interpretable CNN model for pattern (motif) extraction, which predicts true and pseudo viral sequences. This model consists of stack of convolution + pooling layers taking DNA sequences as an input and generating probabilities for true and false viral sequence classification as an output. The EdeepVPP module performs two tasks which are novel viral prediction and interpretability. The second model, EdeepVPP-hybrid consists of CNN and LSTM layers to identify viral genomes effectively. To evaluate the skill of the proposed models, we used 10-fold, 5-fold, leave-one-experiment-out cross-validations. The performance metrics AUC-ROC, AUC-PR are used to evaluate and compare with state-of-the-art techniques. These models outperformed all the existing viral sequence classification methods on human metagenomic and human serum data sets. Model interpretability involves three tasks, the detection of the most important patterns that lead to the identification of true and false viral sequences, the ability of the learned filter to detect these patterns, and validation of these patterns with known patterns of HOCOMOCO database. Both the human metagenomic and human serum datasets learned motifs that matched a large number of existing motifs, indicating that the interpretable strategy proposed in this work establishes transparency to reveal the hidden features for better classification. In addition, the EdeepVPP models can be expanded to predict various viral diseases such as COVID-19. We assume that the proposed CNN model EdeepVPP is capable of extracting essential features, recognizing possible viral sequences, and discovering viral-associated sequence patterns.
Biographies
Chandra Mohan Dasari
received the B.Tech in Computer Science and Engineering from Kakatiya University in the year 2009, Master of Engineering degree in Computer Science and Engineering from Jadavpur University in the year 2012. He currently works toward Ph.D. degree in Computer Science and Engineering from National Institute of Technology, Warangal, Telangana, India. His main research interests are Bioinformatics, Computational Biology, Bigdata Analytics, Machine Learning, and Deep Learning.

Raju Bhukya
has received his B.Tech in Computer Science and Engineering from Nagarjuna University in the year 2003, M.Tech degree in Computer Science and Engineering (CSE) from Andhra University in the year 2005 and Ph.D. in CSE from National Institute of Technology (NIT) Warangal in the year 2014. He is currently working as an Assistant Professor in the Department of CSE in National Institute of Technology, Warangal, Telangana, India. He is currently working in the areas of Bioinformatics and Data Mining.

Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chandra Mohan Dasari, Email: chandu.nitw44@gmail.com.
Raju Bhukya, Email: raju@nitw.ac.in.
References
- 1.Alcantara LCJ, Cassol S, Libin P, Deforche K, Pybus OG, Ranst MV, Galvao-Castro B, Vandamme A-M, De Oliveira T. A standardized framework for accurate, high-throughput genotyping of recombinant and non-recombinant viral sequences. Nucleic Acids Res. 2009;37(suppl_2):W634–W642. doi: 10.1093/nar/gkp455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Amgarten D, Braga LPP, da Silva AM, Setubal JC. Marvel, a tool for prediction of bacteriophage sequences in metagenomic bins. Front Gen. 2018;9:304. doi: 10.3389/fgene.2018.00304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angermueller C, Pärnamaa T, Parts L, Stegle O (2016) Deep learning for computational biology. Mol Syst Biol 12(7) [DOI] [PMC free article] [PubMed]
- 5.Assaf R, Schumann A (2019) Explainable deep neural networks for multivariate time series predictions. In: IJCAI, pp 6488– 6490
- 6.Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. Meme suite: tools for motif discovery and searching. Nucleic Acids Res. 2009;37(suppl_2):W202–W208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bartoszewicz JM, Seidel A, Renard BY (2020) Interpretable detection of novel human viruses from genome sequencing data. BioRxiv [DOI] [PMC free article] [PubMed]
- 8.Bartoszewicz JM, Seidel A, Renard BY (2021) Interpretable detection of novel human viruses from genome sequencing data NAR. Genom Bioinf 3(1):lqab004 [DOI] [PMC free article] [PubMed]
- 9.Bzhalava D, Ekström J, Lysholm F, Hultin E, Faust H, Persson B, Lehtinen M, de Villiers E-M, Dillner J. Phylogenetically diverse tt virus viremia among pregnant women. Virology. 2012;432(2):427–434. doi: 10.1016/j.virol.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 10.Bzhalava D, Hultin E, Mühr LSA, Ekström J, Lehtinen M, de Villiers E-M, Dillner J. Viremia during pregnancy and risk of childhood leukemia and lymphomas in the offspring: Nested case–control study. Int J Cancer. 2016;138(9):2212–2220. doi: 10.1002/ijc.29666. [DOI] [PubMed] [Google Scholar]
- 11.Bzhalava D, Johansson H, Ekström J, Faust H, Möller B, Eklund C, Nordin P, Stenquist B, Paoli J, Persson B et al (2013) Unbiased approach for virus detection in skin lesions. PloS One, 8(6) [DOI] [PMC free article] [PubMed]
- 12.Bzhalava D, Mühr LSA, Lagheden C, Ekström J, Forslund O, Dillner J, Hultin Deep sequencing extends the diversity of human papillomaviruses in human skin. Sci Rep. 2014;4:5807. doi: 10.1038/srep05807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bzhalava Z, Hultin E, Dillner J (2018) Extension of the viral ecology in humans using viral profile hidden markov models. PloS one 13(1) [DOI] [PMC free article] [PubMed]
- 14.Bzhalava Z, Tampuu A, Bała P, Vicente R, Dillner J. Machine learning for detection of viral sequences in human metagenomic datasets. BMC Bioinform. 2018;19(1):336. doi: 10.1186/s12859-018-2340-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Yi L, Narayan R, Subramanian A, Xie X. Gene expression inference with deep learning. Bioinformatics. 2016;32(12):1832–1839. doi: 10.1093/bioinformatics/btw074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crooks GE, Hon G, Chandonia J-M, Brenner SE. Weblogo: a sequence logo generator. Genome Res. 2004;14(6):1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dağlarli E (2020) Explainable artificial intelligence (xai) approaches and deep meta-learning models. In: Advances in Deep Learning. IntechOpen
- 18.Deng L, Togneri R (2015) Deep dynamic models for learning hidden representations of speech features. In: Speech and audio processing for coding, enhancement and recognition. Springer, pp 153–195
- 19.Edgar RC. Search and clustering orders of magnitude faster than blast. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 20.Eickholt J, Cheng J. Dndisorder: predicting protein disorder using boosting and deep networks. BMC Bioinform. 2013;14(1):88. doi: 10.1186/1471-2105-14-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ekström J, Bzhalava D, Svenback D, Forslund O, Dillner J. High throughput sequencing reveals diversity of human papillomaviruses in cutaneous lesions. Int J Cancer. 2011;129(11):2643–2650. doi: 10.1002/ijc.26204. [DOI] [PubMed] [Google Scholar]
- 22.Escalante HJ, Escalera S, Guyon I, Baró X, Güċlütürk Y, Güċlü U, Van Gerven M, van Lier R (2018) Explainable and interpretable models in computer vision and machine learning. Springer
- 23.Fabijańska A, Grabowski S. Viral genome deep classifier. IEEE Access. 2019;7:81297–81307. doi: 10.1109/ACCESS.2019.2923687. [DOI] [Google Scholar]
- 24.Feng H, Shuda M, Chang Y, Moore PS. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science. 2008;319(5866):1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Foulongne V, Sauvage V, Hebert C, Dereure O, Cheval J, Gouilh MA, Pariente K, Segondy M, Burguière A, Manuguerra J-C, et al. (2012) Human skin microbiota: high diversity of dna viruses identified on the human skin by high throughput sequencing. PloS one, 7(6) [DOI] [PMC free article] [PubMed]
- 26.Gao X, Zhang J, Wei Z, Hakonarson H. Deeppolya: a convolutional neural network approach for polyadenylation site prediction. IEEE Access. 2018;6:24340–24349. doi: 10.1109/ACCESS.2018.2825996. [DOI] [Google Scholar]
- 27.Inc Github. Github (2016)
- 28.Glorot X, Bordes A, Bengio Y (2011) Deep sparse rectifier neural networks. In: Proceedings of the fourteenth international conference on artificial intelligence and statistics, pp 315–323
- 29.Gupta S, Stamatoyannopoulos J, Bailey T, Stafford W (2007) Quantifying similarity between motifs genome biology [DOI] [PMC free article] [PubMed]
- 30.Hurwitz BL, U’Ren JM, Youens-Clark K (2016) Computational prospecting the great viral unknown. FEMS Microbiol Lett 363(10) [DOI] [PubMed]
- 31.Johansson H, Bzhalava D, Ekström J, Hultin E, Dillner J, Forslund O. Metagenomic sequencing of ”hpv-negative” condylomas detects novel putative hpv types. Virology. 2013;440(1):1–7. doi: 10.1016/j.virol.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 32.Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 2016;26(7):990–999. doi: 10.1101/gr.200535.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khawaldeh S, Pervaiz U, Elsharnoby M, Alchalabi AE, Al-Zubi N. Taxonomic classification for living organisms using convolutional neural networks. Genes. 2017;8(11):326. doi: 10.3390/genes8110326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura M, Jia Z-J, Nakayama N, Asakawa S. Ecology of viruses in soils: past, present and future perspectives. Soil Sci Plant Nutrition. 2008;54(1):1–32. doi: 10.1111/j.1747-0765.2007.00197.x. [DOI] [Google Scholar]
- 35.Kiranyaz S, Avci O, Abdeljaber O, Ince T, Gabbouj M, Inman DJ (2019) 1d convolutional neural networks and applications: A survey. arXiv:1905.03554
- 36.Krizhevsky A, Sutskever I, Hinton GE (2012) Imagenet classification with deep convolutional neural networks. In: Advances in neural information processing systems, pp 1097–1105
- 37.Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov R, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA et al (2018) Hocomoco: towards a complete collection of transcription factor binding models for human and mouse via large-scale chip-seq analysis, vol 46 [DOI] [PMC free article] [PubMed]
- 38.Labonté JM, Suttle CA. Previously unknown and highly divergent ssdna viruses populate the oceans. ISME J. 2013;7(11):2169–2177. doi: 10.1038/ismej.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 40.Liang H, Ouyang Z, Zeng Y, Su H, He Z, Xia S-T, Zhu J, Zhang B (2020) Training interpretable convolutional neural networks by differentiating class-specific filters. In: European Conference on Computer Vision. Springer, pp 622–638
- 41.Lipton ZC, Berkowitz J, Elkan C (2015) A critical review of recurrent neural networks for sequence learning. arXiv:1506.00019
- 42.Fu L, Miao Y, Liu Y, Hou T (2020) Rnn-virseeker: a deep learning method for identification of short viral sequences from metagenomes. IEEE/ACM Transactions on Computational Biology and Bioinformatics [DOI] [PubMed]
- 43.Meiring TL, Salimo AT, Coetzee B, Maree HJ, Moodley J, Hitzeroth II, Freeborough M-J, Rybicki EdP, Williamson A-L. Next-generation sequencing of cervical dna detects human papillomavirus types not detected by commercial kits. Virol J. 2012;9(1):164. doi: 10.1186/1743-422X-9-164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. Challenges in homology search Hmmer3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013;41(12):e121–e121. doi: 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen M (2015) Why are deep neural network hard to train; Neural networks and deep learning. Determination Press, USA
- 46.Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell. 2015;160(3):447–460. doi: 10.1016/j.cell.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nowicki M, Bzhalava D, BaŁa P. Massively parallel implementation of sequence alignment with basic local alignment search tool using parallel computing in java library. J Comput Biol. 2018;25(8):871–881. doi: 10.1089/cmb.2018.0079. [DOI] [PubMed] [Google Scholar]
- 48.Pineda-Peña A-C, Faria NR, Imbrechts S, Libin P, Abecasis AB, Deforche K, Gómez-López A, Camacho RJ, de Oliveira T, Vandamme A-M. Automated subtyping of hiv-1 genetic sequences for clinical and surveillance purposes: performance evaluation of the new rega version 3 and seven other tools. Infection Gen Evoln. 2013;19:337–348. doi: 10.1016/j.meegid.2013.04.032. [DOI] [PubMed] [Google Scholar]
- 49.Pond SLK, Posada D, Stawiski E, Chappey C, Poon AFY, Hughes G, Fearnhill E, Gravenor MB, Brown AJL, Frost SDW (2009) An evolutionary model-based algorithm for accurate phylogenetic breakpoint mapping and subtype prediction in hiv-1. PLoS Comput Biol 5(11) [DOI] [PMC free article] [PubMed]
- 50.Pope PE, Kolouri S, Rostami M, Martin CE, Hoffmann H (2019) Explainability methods for graph convolutional neural networks. In: Proceedings of the IEEE/CVF Conference on Computer Vision and Pattern Recognition, pp 10772–10781
- 51.Quang D, Xie X (2016) Danq: a hybrid convolutional and recurrent deep neural network for quantifying the function of dna sequences, vol 44 [DOI] [PMC free article] [PubMed]
- 52.Ren J, Ahlgren NA, Lu YY, Fuhrman JA, Sun F. Virfinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017;5(1):69. doi: 10.1186/s40168-017-0283-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ren J, Song K, Deng C, Ahlgren NA, Fuhrman JA, Yi L, Xie X, Poplin R, Sun F (2020) Identifying viruses from metagenomic data using deep learning. Quantit Biol:1–14 [DOI] [PMC free article] [PubMed]
- 54.Ren J, Song K, Deng C, Ahlgren NA, Fuhrman JA, Li Y, Xie X, Sun F (2018) Identifying viruses from metagenomic data by deep learning. arXiv:1806.07810 [DOI] [PMC free article] [PubMed]
- 55.Reyes A, Blanton LV, Cao S, Zhao G, Manary M, Trehan I, Smith MI, Wang D, Virgin HW, Rohwer F, et al. Gut dna viromes of malawian twins discordant for severe acute malnutrition. Proc Natl Acad Sci. 2015;112(38):11941–11946. doi: 10.1073/pnas.1514285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rizzo R, Fiannaca A, La Rosa M, Urso A (2015) A deep learning approach to dna sequence classification. In: International Meeting on Computational Intelligence Methods for Bioinformatics and Biostatistics. Springer, pp 129–140
- 57.Roux S, Enault F, Hurwitz BL, Sullivan MB. Virsorter: mining viral signal from microbial genomic data. PeerJ. 2015;3:e985. doi: 10.7717/peerj.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schmidhuber J, Hochreiter S. Long short-term memory. Neural Comput. 1997;9(8):1735–1780. doi: 10.1162/neco.1997.9.8.1735. [DOI] [PubMed] [Google Scholar]
- 59.Shrikumar A, Greenside P, Kundaje A (2017) Learning important features through propagating activation differences. In International Conference on Machine Learning. PMLR, pp 3145–3153
- 60.Singh A, Sengupta S, Lakshminarayanan V. Explainable deep learning models in medical image analysis. J Imaging. 2020;6(6):52. doi: 10.3390/jimaging6060052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skewes-Cox P, Sharpton TJ, Pollard KS, DeRisi JL (2014) Profile hidden markov models for the detection of viruses within metagenomic sequence data. PloS one, 9(8) [DOI] [PMC free article] [PubMed]
- 62.Spencer M, Eickholt J, Cheng J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinform. 2014;12(1):103–112. doi: 10.1109/TCBB.2014.2343960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava N, Hinton G, Krizhevsky A, Sutskever I, Ruslan S. Dropout: a simple way to prevent neural networks from overfitting. J Mach Learn Res. 2014;15(1):1929–1958. [Google Scholar]
- 64.Sutskever I, Vinyals O, Le Quoc V (2014) Sequence to sequence learning with neural networks. In: Advances in neural information processing systems, pp 3104–3112
- 65.Tampuu A, Bzhalava Z, Dillner J, Vicente R (2019) Viraminer: Deep learning on raw dna sequences for identifying viral genomes in human samples. PloS one, 14(9) [DOI] [PMC free article] [PubMed]
- 66.Thomas T, Gilbert J, Meyer F. Metagenomics-a guide from sampling to data analysis. Microbial Inf Exper. 2012;2(1):3. doi: 10.1186/2042-5783-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Towner JS, Sealy TK, Khristova ML, Albariño CG, Conlan S, Reeder SA, Quan P-L, Lipkin WI, Downing R, Tappero JW et al (2008) Newly discovered ebola virus associated with hemorrhagic fever outbreak in uganda. PLoS pathogens, 4(11) [DOI] [PMC free article] [PubMed]
- 68.Wang S, Weng S, Ma J, Tang Q. Deepcnf-d: predicting protein order/disorder regions by weighted deep convolutional neural fields. Int J Mol Sci. 2015;16(8):17315–17330. doi: 10.3390/ijms160817315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Willner D, Haynes MR, Furlan M, Hanson N, Kirby B, Lim YW, Rainey PB, Schmieder R, Youle M, Conrad D, et al. Case studies of the spatial heterogeneity of dna viruses in the cystic fibrosis lung. Amer J Respiratory Cell Mol Biol. 2012;46(2):127–131. doi: 10.1165/rcmb.2011-0253OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yin B, Balvert M, Zambrano D, schönhuth A, Bohte S (2018) An image representation based convolutional network for dna classification. arXiv:1806.04931
- 71.Zhang Q, Wu YN , Zhu S-C (2018) Interpretable convolutional neural networks. In: Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition, pp 8827–8836
- 72.Zhang S, Zhou J, Hu H, Gong H, Chen L, Cheng C, Zeng J. A deep learning framework for modeling structural features of rna-binding protein targets. Nucleic Acids Res. 2016;44(4):e32–e32. doi: 10.1093/nar/gkv1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zuallaert J, Godin F, Kim M, Soete A, Saeys Y, De Neve W. Splicerover: interpretable convolutional neural networks for improved splice site prediction. Bioinformatics. 2018;34(24):4180–4188. doi: 10.1093/bioinformatics/bty497. [DOI] [PubMed] [Google Scholar]